

Lung infections associated with pneumonia or cystic fibrosis caused by Pseudomonas aeruginosa or other bacteria result in significant morbidity and mortality, in part owing to the development of multidrug resistance, also against last-resort antibiotics. Lytic bacteriophages — viruses that specifically kill bacteria — can reduce lung-associated infections, yet their clinical use is hindered by difficulties in delivering active phage to the deep lung. Here, we show that phage-loaded polymeric microparticles deposit throughout the lung via dry powder inhalation and that they deliver active phage. Phage-loaded microparticles effectively reduced P. aeruginosa infections and the associated inflammation in wild-type and cystic-fibrosis-transmembrane-conductance-regulator knockout mice, and rescued the mice from pneumonia-associated death. The polymeric microparticles might constitute a clinically translatable therapy for eradicating hospital-acquired lung infections and infections associated with cystic fibrosis.
Approximately 5.6 million cases of community-acquired pneumonia occur annually in the United States of America (USA), and it is the most common cause of death from infectious disease1. Another 200,000 cases of healthcare-associated pneumonia result in 36,000 annual deaths in the USA2. The lungs of cystic fibrosis (CF) patients (~29,000 annually3) also become colonized by pathogenic bacteria resulting in chronic infection and inflammation4. Pseudomonas aeruginosa is the main causative agent of morbidity and mortality in lung infections5,6. Antibiotic therapy is limited by dosing and multi-drug resistance4,5,7, and even last-resort antimicrobials8 are ineffective in eradicating infections9–12.
Phage are promising agents for the treatment of bacterial infections due to their ability to infect and lyse bacteria, replicate, and degrade biofilm matrix13–17. Because the mechanisms by which phage target bacteria are unrelated to the action of antibiotics, phage therapy is effective against multi-drug resistant bacteria18,19. The use of mixtures of phage reduces the development of resistance against phage therapy20. Phage are specific to one, or a few, closely related target bacterial species, and do not infect the commensal microflora of the patient21, which can be severely perturbed by antibiotics. In addition, phage have been shown to produce less endotoxin upon bacterial lysis compared to antibiotics such as β-lactams22. Phage can be engineered to deliver biofilm-degrading enzymes, increase antibiotic efficacy, or provide alternative antibacterial strategies23–26. Recently, compassionate phage treatment of two patients with antibiotic-resistant bacteria in Europe and USA shows the translational potential, efficacy and safety of phage therapy27,28. Several studies have shown the potential of phage therapy to reduce lung bacterial infections29–38; a recent study showed that phage and the immune system act synergistically to clear pathogenic lung infections39. However, the use of nebulizers or intranasal administration in these studies limit phage stability, clinical translation, and patient compliance. To overcome these limitations, dry powder formulations of bacteriophage with or without excipients have also been tested40–45. However, rapid loss of phage activity due to exposure to harsh fabrication conditions, lack of a suitable carrier for deep lung delivery, and proof of efficacy in animal models are major challenges for clinical translation of phage therapy when used without suitable delivery vehicles. Here, we describe engineered phage-loaded microparticles (phage-MPs) for pulmonary administration via dry powder inhalation to deliver therapeutic doses of active phage to the site of infection and demonstrate significant reductions in bacterial counts and enhanced survival of the host. This therapeutic formulation is stable at room temperature and can be translated to conventional inhalers for ease of administration and increased patient compliance.
For pulmonary delivery, particles must have an aerodynamic diameter (daer) of ~1–5 µm for efficient deposition to the deep lung46. The daer is related to the physical diameter d and density ρ of the particle . For polymeric particles (ρ ≈ 1.0 g cm−3), particle diameter should be ~1–5 µm. However, at this size range, alveolar macrophages rapidly clear particles47, thereby reducing the therapeutic effect. To avoid clearance, particle diameter should exceed 5 µm, beyond the uptake capability of alveolar macrophages. This can be accomplished by using larger particles that are porous to reduce their density. Furthermore, large particles have lower aggregation compared to smaller particles and allow for improved aerosolization for dry powder formulations46. Such porous particles have been used for delivery of various therapeutics48–52, but their use for delivery of phage has not been reported.
Results
Phage-MPs effectively kill bacteria.
We prepared hollow poly(lactic-co-glycolic acid) (PLGA) MPs via water-oil-water double emulsion (Fig. 1a-b). Porous particles were generated by including ammonium bicarbonate as an effervescent in the inner aqueous phase53. By tuning processing parameters, we generated highly porous MPs of appropriate size (d = 8.0 ± 4.5 µm) and density (~ 0.3 g cm−3) to yield a daer of 2–5 µm (Fig. 1c-d, Supplementary Fig. 1).
Figure 1. Porous MPs deliver active phage and are effective against P. aeruginosa.
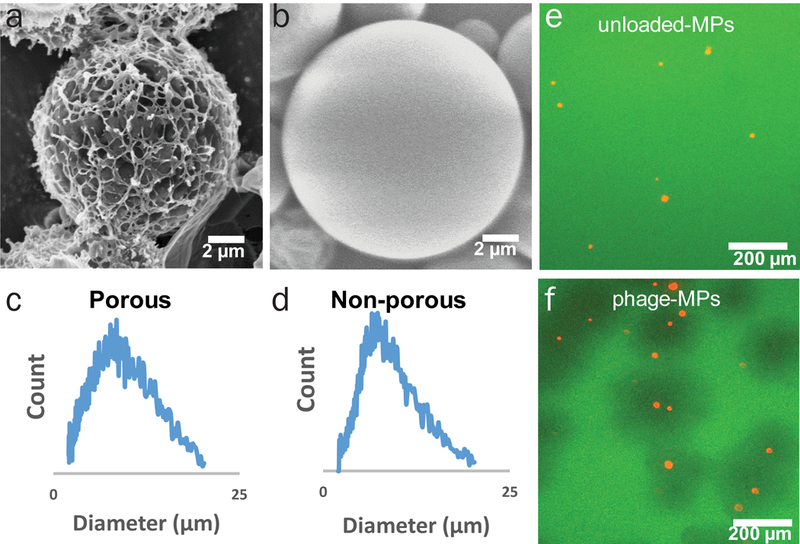
Scanning electron micrograph of (a) porous PLGA MPs and (b) non-porous PLGA MPs. Size distribution of (c) porous PLGA MPs and (d) non-porous PLGA MPs. Fluorescence microscopy images of green fluorescent protein expressing PAO1-GFP with (e) unloaded and (f) phage-loaded, fluorescent MPs (loaded with DiI stain).
Several lytic phage against P. aeruginosa (Supplementary Table 1) were amplified, purified by liquid chromatography, and loaded onto MPs by incubation in phage-containing solution. In this system, the phage is not encapsulated within the MPs but is deposited on the surface of the MPs after the MPs are synthesized. This approach minimizes any loss in phage activity from the solvents used for MP fabrication, and no loss of phage activity was observed upon loading. MPs were loaded with a defined mixture of 3–5 different phage to increase infectivity and bactericidal activity and reduce the probability for the development of resistance. Porous MPs provide large surface area for phage adsorption, and phage loading was 2.6 ± 0.2 plaque forming units (PFU) per particle or ~2.6 × 106 PFU mg−1 MPs. Endotoxin levels for the phage-MP formulation were 0.078 ± 0.003 EU/mg, two orders of magnitude below the 20 EU/device limit stipulated by FDA. As expected, these larger porous phage-MPs (8.0 µm diameter) exhibited reduced internalization by macrophages compared to non-porous, smaller (1 µm diameter) phage-particles (Supplementary Fig. 2). To test the bactericidal activity of phage-loaded MPs, phage-MPs or empty MPs were plated onto lawns of fluorescent P. aeruginosa PAO1-GFP and incubated for 16 h at 37oC. Efficient lysis of bacteria was observed around phage-MPs as demonstrated by zones devoid of fluorescence, whereas control MPs had no effect on PAO1-GFP levels (Fig. 1e-f). Notably, the killing zone for phage-MPs extended beyond the particle area, indicating that phage propagated radially outwards from the MPs, demonstrating that phage delivered from MPs can infect, replicate, propagate, and kill target bacteria beyond those in direct contact with the MPs. We next tested whether phage-MPs can infect and kill bacteria in synthetic sputum to model the viscous mucus in the lung. Synthetic sputum was prepared54 and growth curves in the presence and absence of phage-MPs were monitored. No growth was observed in presence of phage-MPs while bacteria rapidly grew in control conditions (Supplementary Fig. 3). This result demonstrates that phage-MPs can infect and effectively kill bacteria in viscous model sputum. A major challenge for treatment of lung-associated infections is the formation of biofilms by P. aeruginosa5,55. We tested the ability of phage-MPs to lyse bacterial biofilms. Treatment with phage-MPs resulted in effective killing of P. aeruginosa in biofilms (Fig. 2).
Figure 2. Phage-MPs are effective against P. aeruginosa biofilms.

Images of PA103 biofilms stained with SYTO 9 (green, live cells) and propidium iodide (red, dead cells) 24 h after treatment with (a-c) MPs only or (d-f) phage-MPs. g, Quantification of mean pixel intensity for live/dead bacteria using ImageJ. Measurements were taken from independent samples (n=4 for empty-MPs and n=5 for phage-MPs, mean ± SD, two-tailed t test with Welch’s correction).
We prepared dry powder formulations of phage-MPs by lyophilizing phage-loaded MPs suspended in lactose. Dry powder formulations of phage-MPs were tested for 16 days and only showed a slight loss of titer when stored at room temperature (Supplementary Fig. 4). Such dry powder formulations can be easily administered to patients using inhalers, have a long shelf life, and increase patient compliance. We next performed studies to determine release kinetics of phage from dry powder MP formulations. An initial burst release of approximately 10–15% of loaded phage was observed followed by very slow release (Supplementary Fig. 5), consistent with stable deposition of the phage on the particle surface and the slow degradation of the PLGA polymer used56. Attempts to characterize long-term release kinetics were not successful as this requires incubation of in aqueous buffer and there is loss of phage activity over time in aqueous environment at 37 °C.
Inhaled phage-MPs distribute throughout the murine lung.
For delivery to the mouse lung, lyophilized powder was mixed with inhalation-grade lactose and aerosolized using a mouse dry powder insufflator. We first evaluated the safety and clearance of MPs delivered via endotracheal intubation. Intravital imaging with near-infrared dye-labeled MPs showed that MPs were cleared from the lungs of healthy mice within 18 h (Supplementary Fig. 6). Histological sections at 1 and 7 days post-delivery demonstrated minimal leukocyte infiltration and normal lung tissue structure (Supplementary Fig. 7). No differences were observed in tissue histology between lungs receiving MP-lactose powder and control (no MPs) lactose powder.
We next evaluated the distribution of fluorescent MP-lactose dry powder after insufflation via endotracheal intubation. Organs were explanted immediately after delivery and imaged for total fluorescence. All fluorescence was localized to the lungs (Fig. 3a-b), and fluorescent MPs were found throughout lung tissue (Fig. 3c). These results demonstrate targeting of MPs to the lung as well as distribution throughout the lung via dry powder inhalation. To quantify phage delivery, we delivered phage-MPs or phage alone (no MPs) via insufflation. High phage titers (104-105 PFU/mg of tissue) were recovered from lungs treated with phage-MPs, whereas no phage were recovered from lungs receiving free phage (Supplementary Fig. 8a). These results demonstrate that delivery of phage using MPs significantly enhances the delivery of active phage to the lung. We also examined the deep lung distribution of phage-loaded porous and non-porous MPs. Phage were delivered as previously described and the deep lungs were harvested. Significantly higher deposition of phage in the deep lung was observed for delivery with porous MPs compared to non-porous MPs (Supplementary Fig. 8b).
Figure 3. Dry powder formulations of porous MPs for lung delivery.
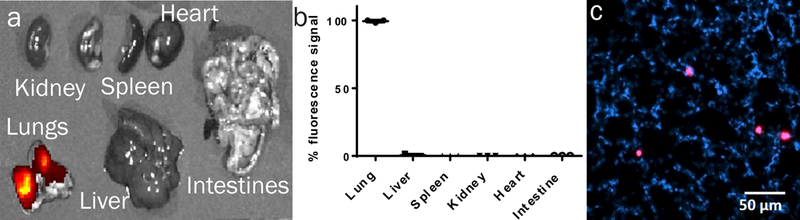
a, Fluorescence images of explanted organs immediately after MPs administration. b, Quantification of fluorescence signal from organs after fluorescent MPs administration (n=3 mice, mean ± SD). Measurements were taken from distinct samples. c, Fluorescence imaging of lung cryosection after MP insufflation; nuclei stained with DAPI, MPs labeled magenta. Shown is a representative image from a total of nine images taken. Data were collected on male healthy (non-infected) C57BL/6 mice.
An attractive attribute of phage compared to other antimicrobials is the ability to persist and replicate at infection sites until the pathogen is cleared. We established an acute model of lung infection using different PAO1-GFP doses and evaluated the persistence of bacteria at 24 h post-inoculation in healthy mice (Supplementary Fig. 9). A dose of 5×106 CFU PAO1-GFP was selected for subsequent experiments. Phage-MPs were delivered to both control and PAO1-GFP-infected lungs, and 3 orders of magnitude higher phage counts were detected in PAO1-GFP-infected lungs compared to control (uninfected) lungs at 0 h (Supplementary Fig. 8) and 18 h post-delivery (Supplementary Fig. 10). This result demonstrates that phage delivered to the lung via MPs can infect and amplify in vivo only in the presence of target bacteria.
Phage-MPs treatment clears infection from mice lungs.
We tested the ability of phage-MPs to reduce acute bacterial lung infections in wild-type mice. Phage-MPs and controls were delivered via insufflation to PAO1-GFP-infected mice, and lungs were assayed for bacterial and phage counts after 24 h (Fig. 4a-b). Delivery of phage-MPs reduced bacterial counts by an order of magnitude (p=0.0203) compared to bacteria + phage group whereas free phage (same dose as phage-MPs) delivery had no effect on bacterial counts (p>0.12) compared to animals treated with bacteria alone. Counts of phage active against P. aeruginosa were elevated for both free phage and phage-MPs. Importantly, treatment of infected mice with phage-MPs rescued 100% of subjects from pneumonia-associated death compared to only 13% survival for untreated mice over six days (Fig. 4c). These results demonstrate that phage-loaded MPs effectively reduce P. aeruginosa lung infections from wild-type mice and rescue mice from pneumonia-associated death.
Figure 4. Phage-loaded porous MPs reduce bacteria in the lungs of mice.
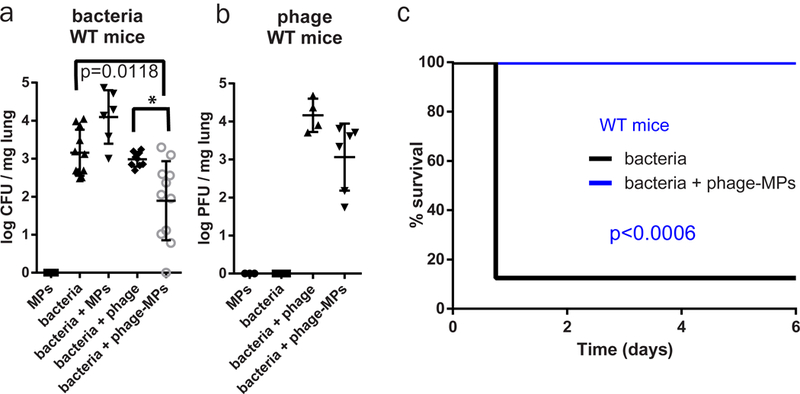
Antibacterial efficacy of free phage and phage-MPs was examined in a mouse model of acute lung infection with P. aeruginosa PAO1-GFP. After 24 h of treatment, (a) bacterial load (n=3 mice for MPs; n=11 mice for bacteria; n=6 mice for bacteria+MPs; n=9 mice for bacteria+phage; n=11 mice for bacteria+phage-MPs, mean ± SD) and (b) phage load (n=3 mice for MPs; n=6 mice for bacteria; n=4 mice for bacteria+phage; n=6 mice for bacteria+phage-MPs, mean ± SD) in lung homogenate were quantified. Data were pooled from two independent experiments. For (a), one-way Kruskal-Wallis non-parametric test was used to detect statistical differences followed by Dunn’s multiple comparison test with adjustment for multiple comparisons. * p = 0.0203. c, Percent survival of PAO1-GFP-inoculated mice that were untreated or treated with phage-MPs (n=8 mice/group). The Mantel-Cox test was used to detect differences between survival. Data were collected on male C57BL/6 mice.
We also examined the effects of phage-MPs on P. aeruginosa infection in the gut-corrected CF transmembrane conductance regulator (CFTR) knockout (CFKO) mouse57. This model mimics several aspects of the CF human condition, including increased pathogenesis from P. aeruginosa lung infections58,59. A dosing study with PAO1-GFP identified a suitable dose to obtain an acute infection (Supplementary Fig. 11). A dose of 2×106 CFU PAO1-GFP was used; this dose is lower than that needed for wild-type mice in the earlier experiment, suggesting that CFTR-KO mice have reduced capacity to clear bacteria from the lungs. PAO1-GFP infection and delivery of phage-MPs were performed as described earlier. Delivery of phage-MPs dramatically reduced bacterial counts in the lungs by three orders of magnitude, near the assay detection limit (Fig. 5a-b). In contrast to wild-type mice, treatment with phage alone also reduced the bacterial burden in the lung of CFTR-KO mice, but significant levels of bacteria were still present. This modest efficacy could be the result of higher phage:bacteria ratio as the bacteria dose delivered to CFTR-KO mice is lower than that for wild-type mice. Nevertheless, when comparing free phage to phage-MPs, use of phage-MPs resulted in ~1.5 orders of magnitude lower bacteria counts compared to free phage. Phage were detected in lung tissue for both phage-MPs and free phage. We assessed bacteria levels in the liver of these mice to determine whether the infection spread to other organs. Remarkably, no mice treated with phage-MPs showed any detectable bacteria in the liver (0 out of 7 mice), whereas >70% of untreated mice (5 out of 6 mice) or mice treated with free phage (5 out of 7 mice) had measurable levels of bacteria in the liver (Fig. 5c). Histological analyses revealed massive infiltration of host immune cells in untreated infected lungs (Fig. 5d). Infected mice treated with free phage displayed moderate levels of cell infiltration (Fig. 5e). Importantly, infected mice treated with phage-MPs had low levels of immune cell infiltration (Fig. 5f), indicating that this treatment reduces the bacterial load as well as infection-associated inflammation. Immunostaining analysis on lung sections confirmed these observations (Supplementary Fig. 12).
Figure 5. Phage-loaded porous MPs reduce bacteria in the lungs of CFTR knockout mice.
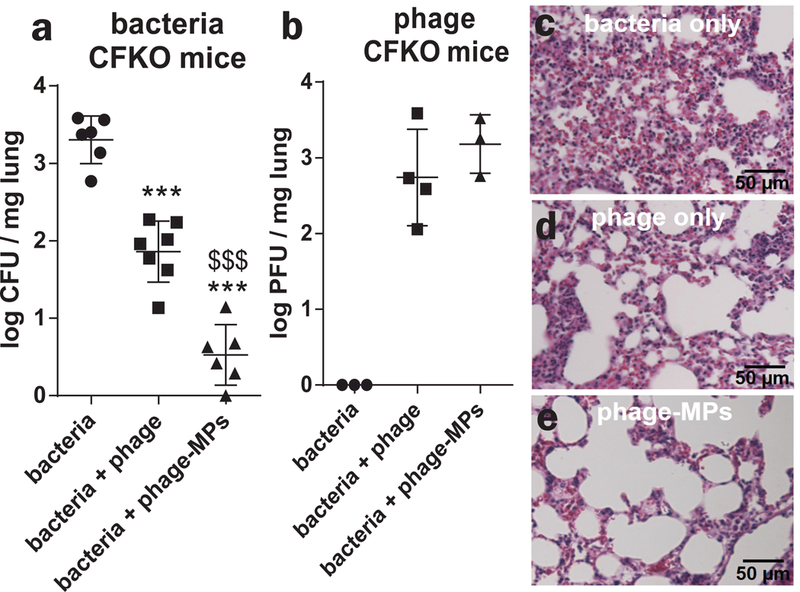
Antibacterial efficacy of free phage and phage-MPs was examined in a CFTR knockout mouse model of acute lung infection with P. aeruginosa (PAO1-GFP). After 18 h of treatment, (a) bacterial load (n=6 mice for bacteria and bacteria+phage-MPs; n=7 for bacteria+phage, mean ± SD) and (b) phage load (n=3 mice for bacteria and bacteria+phage-MPs, 4 mice for bacteria+phage, mean ± SD) in lung homogenate were quantified. For (a-b), data were pooled from two independent experiments. One-way ANOVA was used to detect statistical differences followed by Tukey’s multiple comparison test with adjustment for multiple comparisons. *** p < 0.0001 vs. bacteria control, $ $ $ p < 0.0001 vs. bacteria+phage group. Data were collected on both male and female CFTR knockout mice. Measurements were taken from distinct samples. Images of histological sections of mouse lung stained with H&E at 18 h after treatment with (c) bacteria only, (d) bacteria + phage and (e) bacteria + phage-MPs. These are representative images from a total of five images taken for each group.
Phage-MPs kill clinical bacterial strains.
To evaluate the clinical potential of the phage-MPs formulation, we screened phage-MPs against clinical strains of bacteria isolated from hospital-associated lung infections and CF patients. The phage-MPs formulation was effective against all acute infection clinical strains and five out of six clinical strains from CF patients tested in vitro, including antibiotic-resistant strains (CFBR 337 and CFBR 505 are tobramycin-resistant) (Supplementary Table 2). We then evaluated the in vivo efficacy of phage-MPs against PA103 (a historical strain derived from the sputum of a patient60) in mice. A dosing study with PA103 identified a dose (1.6×105 CFU) suitable to obtain an acute infection in wild-type mice (Supplementary Fig. 13). To achieve high phage titers against PA103, our phage library was screened against PA103 and three different phage were chosen and loaded onto the MPs (Supplementary Table 3). PA103 infection and delivery of phage-MPs were performed as described earlier. Delivery of phage-MPs effectively reduced bacterial counts in the lungs showing efficacy against a patient-derived bacterial strain (Fig. 6a-b). To test whether direct deposition of phage onto MPs, instead of simple co-delivery of phage and MPs, is required for effective in vivo bacterial clearance, a physical mixture of phage and unloaded MPs with inhalation-grade lactose was evaluated. In contrast to effective bacteria reduction with phage-MPs, a physical mixture of phage and unloaded MPs did not reduce bacteria load in the lung (Fig. 6a), demonstrating that only phage that is directly deposited on the MPs kills bacteria in vivo. We also tested 10 bacterial colonies recovered from phage-MP treated mice for development of phage resistance against the stock phage mixture during the 18 h of incubation in the mouse lung. All of the recovered colonies were still sensitive to phage-MPs.
Figure 6. Phage-MPs significantly reduce infection by a clinically derived strain of bacteria in wild-type mice.
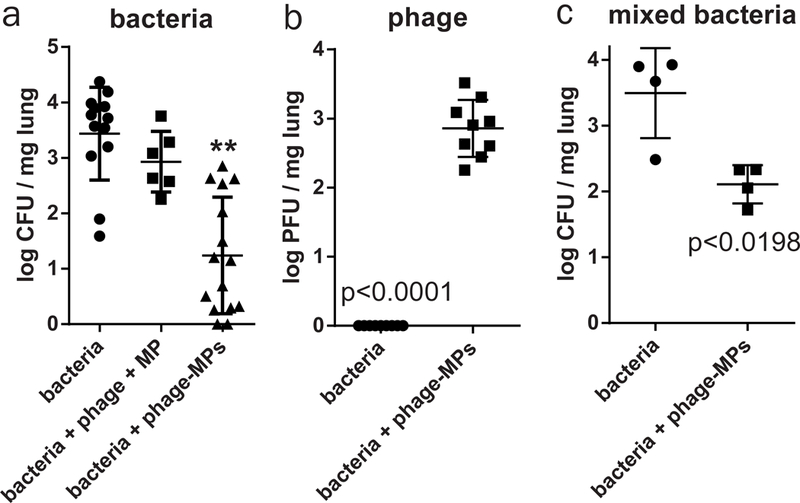
Antibacterial efficacy of phage-MPs was examined in a mouse model of lung infection with the clinical strain of P. aeruginosa (PA103). After 18 h of treatment, (a) bacterial load (n=13 mice for bacteria, 6 mice for bacteria+phage+MP, and 15 mice for bacteria+phage-MPs, mean ± SD) and (b) phage load (n=9 mice/group, mean ± SD) in lung homogenate were quantified. Data were pooled from two independent experiments. Data were collected on both male and female wild-type CFTR littermates. c, Antibacterial efficacy of phage-MPs was examined in a mouse model of lung infection with a mixture of P. aeruginosa strains (PAO1-GFP and PA103). After 18 h of treatment, bacterial load (n=4 mice/group, mean ± SD) in lung homogenate was quantified. Data were collected on male wild-type C57BL/6 mice. For (a) one-way ANOVA was used to detect statistical differences followed by Holm-Sidak’s multiple comparison test with adjustment for multiple comparisons. **p < 0.0001 vs. bacteria control and p = 0.001 vs. bacteria + phage + MP groups. For (b,c), a two-tailed Welch’s t test was used to detect statistical differences. Measurements were taken from distinct samples.
Phage-MPs are effective against infection from multiple strains.
Another challenge for antimicrobial therapy against P. aeruginosa infection is the presence of distinct populations and their diversification in the patient lung 61,62. To test whether phage-MP treatment can be effective against infection consisting of multiple strains of bacteria, we infected mice with a mixture of PAO1-GFP (2.5×106 CFU) and PA103 (8×104 CFU) strains. Phage listed in Supplementary Tables 1 and 3 were mixed and loaded on MPs. Treatment with phage-MPs effectively reduced total bacterial counts in the lungs, showing efficacy against infections comprised of simultaneous infection by two strains of P. aeruginosa (Fig. 6c). We also tested 10 bacterial colonies of each strain recovered from phage-MP-treated mice for development of phage resistance against the stock phage mixture and all recovered bacteria were still susceptible to phage-MP treatment.
Finally, to evaluate the efficacy of phage therapy after repeated administration, we delivered empty-MPs and phage-MPs in naïve mice. After 21 days, mice in both groups were infected with PA103 and phage-MPs treatment was performed. Pre-exposure with phage-MPs did not affect the efficacy of treatment when compared to no pre-exposure control group (Fig. 7). Furthermore, no antibody titers in blood (0/5 mice) were detected against phages at any time point tested (day 0, 21 and 22). This result indicates that pre-exposure to phage-MPs via dry powder delivery did not reduce its functional performance.
Figure 7. Pre-treatment with phage-MPs does not reduce functional efficacy of the treatment to clear bacteria from mouse lungs.
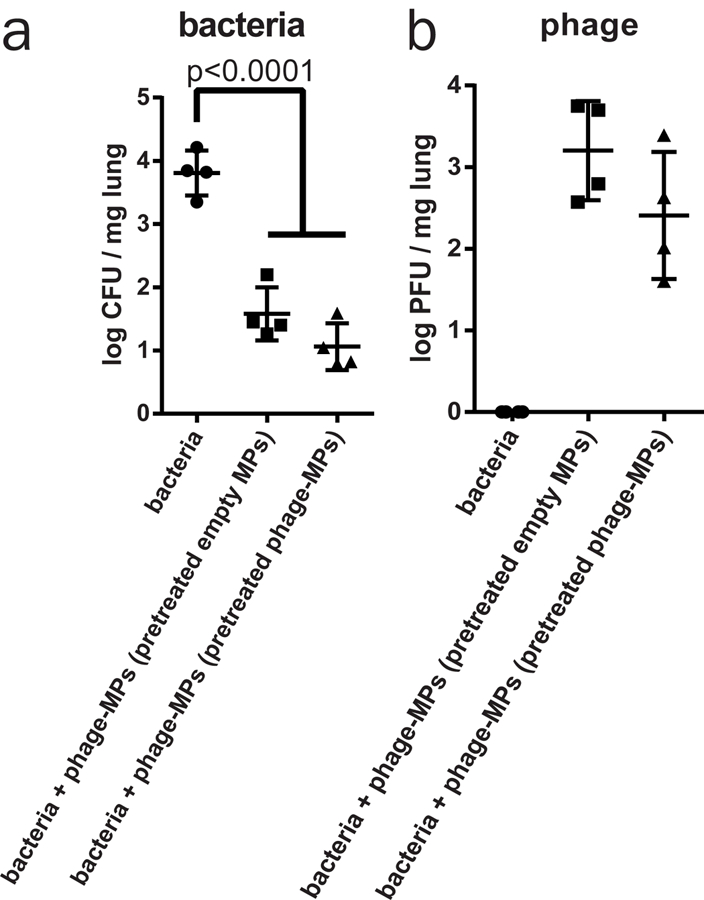
Antibacterial efficacy of phage-MPs was examined in a mouse model of lung infection after 21 days of pre-treatment with phage-MPs or empty MPs with PA103. After 18 h of treatment, (a) bacterial load (n=4 mice/group, mean ± SD) and (b) phage load (n=4 mice/group, mean ± SD) in lung homogenate were quantified. Data were collected on both male wild-type C57BL/6 mice. One-way ANOVA followed by Tukey’s multiple comparison tests were used to detect statistical differences.
Discussion
We demonstrate that phage can be effectively delivered to lungs using polymeric MPs and MP-delivered phage retain its activity to kill host bacteria. Phage-loaded MPs significantly reduced P. aeruginosa infections and associated inflammation from healthy and CF mice and rescued mice from pneumonia-associated death. Importantly, phage-MPs were effective against clinically derived strains, including antibiotic-resistant strains, as well as lung infections caused by two strains of bacteria. The acute infection murine model used in our study lacks biofilm formation, mucus plugging, and genetic, phenotypic and spatial diversification of bacterial strains 61,62. Nonetheless, we show that phage-MPs were effective against clinically derived strains from acute infections and CF patients and were able to kill bacteria growing in biofilms. Interestingly, when we delivered free phage to the healthy lung as a dry powder formulation and immediately assayed the lungs, we did not recover any active phage from the lung homogenate. This result is in contrast to prior studies 29–34, where free phage administration was shown to be effective against bacterial infections. This discrepancy could be due to differences in the mode of administration. All previous reports have used intra-nasal administration of phage in liquid, whereas we used dry powder formulation of free phage. Another reason for this discrepancy could be that the titers delivered for free phage in our experiment (~106 PFU/mg MPs) were significantly lower compared to previous reports (109-1010 PFU). For the acute lung infection model in healthy and CF mice examined in this study, phage-only treatment had modest efficacy in reducing bacterial burden even though active phage was recovered. In contrast, phage-MP treatment significantly reduced bacterial burden, rescued mice from pneumonia-associated death, and prevented bacterial colonization of the liver. We attribute this enhanced bactericidal efficacy for phage-MPs to improved delivery to the lung afforded by the engineered MPs. Indeed, direct deposition of phage onto the MPs was required for effective in vivo bactericidal activity as physical mixtures of phage and unloaded MPs had no effects.
Bacteria recovered from the lungs of infected mice treated with phage-MPs were tested for their susceptibility against phage-MPs, and all test samples were still susceptible to phage infection and lysis. Although this result suggests lack of phage resistance, more in-depth analysis in future studies is necessary. The present analysis focused on acute lung infection in healthy and CF murine models; future studies will test the efficacy of dry powder inhalation-based phage-MPs therapy in more challenging chronic infection models. Phage therapy (via intra-nasal route) has been recently been shown to be effective against P. aeruginosa chronic infections63. The mice used in our experiments are expected to have several commensal bacterial species 64,65. Although we did not investigate the effect of phage treatment on other resident species of bacteria in mice lungs, our treatment was able to reduce the counts of pathogenic bacteria from the whole lung microbiome. Even though phage are highly specific for their host species, it will be important for future studies to evaluate the effect of phage treatment on the entire lung microbiome, especially how targeting a single organism impacts the remaining organisms in that community. A concern with phage therapy is that immune responses to the phage could result in elevated, undesirable inflammation and immune reactions that could reduce phage activity. Indeed, intraperitoneal administration of phage resulted in high titer phage-specific antibodies in the blood 66. In the present study, we did not observe any adverse or elevated inflammation to phage-MP treatment nor the presence of phage-specific antibodies in the blood of treated mice. In addition, pre-exposure of phage-MPs did not result in reduction of its functional efficacy to subsequent bacterial challenge. These differences in host responses to phage and phage activity could be due to different routes of administration (IP vs. lung insufflation), phage dose delivered, and/or the use of MPs as phage delivery carriers. Further studies are needed to establish whether adverse immune responses, antibody production, and/or reduction in phage efficacy arise when phage-MPs are administered several times. Nevertheless, dry formulations of phage-MPs represent a promising alternative to antibiotic regimens, which are severely limited by ineffective dosing and emergence of antibiotic resistance. The ability to deliver active phage using engineered biomaterials as dry powder formulations provides significant clinical benefits for lung infections, including longer drug stability, ease of administration with fewer side effects, and increased patient compliance.
Methods:
PLGA MPs:
MPs were prepared by double emulsion of water in oil in water. 200 mg PLGA (RG503H, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 7 mL dichloromethane (DCM). Ammonium bicarbonate (ABC, Sigma-Aldrich) was used as an effervescent to create porous MPs in 1 mL of internal aqueous phase at 4% w v-1. The concentration of ABC can be varied to tune the porosity and density of MPs. The solution was homogenized at 3000 rpm for 2 min and then mixed with 50 mL of 1% poly(vinyl alcohol) (PVA) (Sigma-Aldrich) and homogenized for 2 min. The final emulsion was added to 100 mL 1% PVA and stirred to evaporate all DCM for 5 h. MPs were washed with deionized water 4 times to remove PVA and free polymer and lyophilized for 16 h. Fluorescent MPs were prepared by adding 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) or 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindotricarbocyanine iodide (DiR) dye (Thermo Scientific, Rockford, IL, USA) (5 mg dye per 200 mg PLGA) in DCM.
Particle size and density:
MPs size distribution was characterized using a Multisizer 3 Coulter counter. To determine density of MPs, 1.0 mg lyophilized MPs were suspended and counted in the Coulter counter. Total volume of MPs in 1.0 mg was calculated by using the average diameter obtained from the Coulter counter. Density was calculated by theoretical calculation based on mass = density*volume.
Scanning electron microscopy of MPs:
For scanning electron microscopy (SEM, LEO 1550), an MP suspension (5 μL of 1 mg mL−1 solution) was dispensed on a SEM stub, air dried for 2 h at room temperature, and sputter coated with 5 nm of gold to make the sample conductive. Imaging was done with a 5 KeV electron beam.
Fluorescent plaque imaging:
PLGA MPs were loaded with phage, washed, and various dilutions were plated with PAO1-GFP on trypticase soy agar (TSA) plates. Plates were incubated for 18 h at 37oC and imaged on a Nikon-C2 laser scanning confocal microscope with a 10X objective. PAO1-GFP was excited with 405 nm laser and emitted light was collected through a 485/35 nm filter cube. Particles were excited with 561 nm laser and emitted light was collected through a 595/50 nm filter cube.
Phage amplification:
Phage (Supplementary Tables 1 and 3, described previously 67) were amplified in liquid culture of 25% trypticase soy broth (TSB) (Becton Dickinson, Sparks, MD, USA). Phage were added to an exponential phase, shaking culture of their respective bacterial host at 107–108 CFU mL−1 and incubated at 37oC until lysis occurred (culture visibly cleared). The resulting lysate was clarified further by adding 2% (v v−1) chloroform, decanting centrifugation, and filtration of the supernatant (0.2 µm pore membrane).
Phage purification:
For phage purification, an anion-exchange chromatography column CIMs QA-8f mL Tube Column (BIA Separations, Ljubljana, Slovenia) was used 68. The column was attached to an AKTA™ FPLC system (GE Healthcare, Little Chalfont, UK) with P900 pump system and analyzed with UNICORN™ 5.01 software. Phage lysate was diluted 50% with Tris buffer (40 mM Tris-HCl, pH 7.5) and loaded onto the column. Phage were then washed with Tris buffer (20 mM Tris-HCl, pH7.5). For elution, a gradient was used by linearly mixing an increasing amount of 1.5 M NaCl, 20 mM Tris-HCl solution with 20 mM Tris-HCl buffer and collected using a fraction collector. Phage were then dialyzed with 0.1 M phosphate buffer supplemented with 2 mM MgSO4, passed through an endotoxin removal column and stored at 4oC.
Active phage counts:
Preparations were counted for active phage with plaque assays using the traditional top agar overlay method 69.
Phage loading on particles:
4 mg MPs were suspended in a solution of phage mixture (~1010-1011 phage mL−1) for 4 h with mild shaking. MPs were then washed 3 times with PBS. To determine phage loading levels on MPs, ΦPaer14 was loaded on the particles. 500 µL of phage-MP and 500 µL of chloroform were mixed for 20 min at room temperature to dissolve the PLGA. Free-phage standards were run in parallel to determine the effects of chloroform on phage stability. Samples were centrifuged at 500g for 5 minutes to separate the organic and aqueous phases. Phage titers were performed on the aqueous phase. Particles were enumerated using a hemocytometer and the number of phage per particles were calculated using the following formula ((PFU/mL) * adjustment) / (particles/mL), where the adjustment was determined by dividing PFU/mL of a phage standard by the PFU/mL of the same standard exposed to chloroform for 20 min as described above.
Dry powder preparation:
To prepare dry powder formulations, 4.0 mg of washed phage-MP were suspended in 100 µL solution of lactose (40 mg mL−1) resulting in a 1:1 ratio of particle:lactose and lyophilized for 24 h. For free phage dry powder formulation, phage titers were adjusted to the amounts of phages loaded on the MPs, suspended in 100 µL solution of lactose (40 mg mL−1) resulting in a 1:1 ratio of particle:lactose and lyophilized for 24 h. For stability testing, the dry powder mixture was stored at room temperature. Phage activity in the lyophilized powder was tested using the phage enumeration method described above on PA103. For in vivo work, lyophilized powder was mixed with 32 mg inhalation grade lactose (Respitose ML006, DFE Pharma) resulting in a 1:10 final ratio of particle:lactose. Powder was vortexed for 1 h at room temperature (VWR mini vortexer; speed 7).
Phage-MP endotoxin quantification:
Lyophylized phage-MPs (0.5 mg) were washed by centrifugation in 40 mL of endotoxin-free water as per the FDA Guidance for Industry Pyrogen and Endotoxins Testing (www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm314718). Endotoxin was then quantified using a LAL chromogenic endotoxin quantification kit (Thermo Scientific, Rockford, IL, USA) per the manufacturer’s instructions.
Phage release from particles:
Lyophilized phage-MPs were suspended in PBS and total PFU were enumerated using the top agar overlay method. For phage release, phage-MPs were centrifuged at 500g for 5 min and the supernatant was assayed for PFU.
In vitro phage-MP phagocytosis:
RAW264.7 macrophages were cultured in DMEM supplemented with 4.5 g/L glucose and 10% FBS in 24 well plates at 37 °C and 5.0% CO2 in a humidified incubator. Fluorescent DiI labeled particles were synthesized as described above. Smaller particles (0.94 ± 0.59 µm) were generated by modifying the homogenization speed to 10,000 rpm. The PAO1 phage mixture was loaded on the MPs. Following loading, MPs of different sizes were added to tissue culture media at a concentration of 0.25 mg/mL and incubated with cells for 2 h. Cells were washed 5 times with PBS to remove excess particles, fixed, and stained with DAPI and phalloidin-iFluor488 (abcam, Cambridge, MA). Samples were imaged using a Nikon-C2 laser scanning confocal microscope through a 10X objective. Three images of each well were acquired. Macrophage phagocytosis was evaluated in ImageJ by performing maximum intensity Z projections and thresholding the green (phalloidin) channel followed by particle tracking analysis (minimum size of 20 pixels) to identify the cell border. Fluorescent blue (DAPI) and red (MP) images were then overlaid and MPs falling inside cell borders containing a nucleus were counted as having been phagocytosed by cells. The total number of cells within an image field was quantified using the ComDet (v.0.3.6) plugin. The percent of phagocytic cells was computed by dividing the number of phagocytic cells in an image by the total number of cells in the image.
In vitro growth of bacteria in synthetic CF sputum:
Synthetic CF sputum was prepared as described previously54. 50 µL culture of PA103 was grown for 16 h in synthetic CF sputum and was used to inoculate 3 mL of the sputum medium in presence or absence of 0.1 mg of phage-MPs. Cultures were grown at 37oC and regular optical density measurements were taken to monitor the bacterial growth.
In vitro activity of phage-MPs against bacterial biofilms:
To form biofilms, 5 µL culture of PA103 was grown for 16 h in 25% TSB and was used to inoculate 500 µL of 25% TSB in each well of a 48 well plate and allowed to grow for 24 h at 37oC. Biofilms were carefully washed with PBS and incubated with 0.01 mg/mL of MPs or phage-MPs for 18 h at 37oC in 25% TSB. Biofilms were then washed and stained with LIVE/DEAD® BacLight™ (Thermo Scientific, Rockford, IL, USA) (3uL dye mixture per mL of bacterial medium) and imaged on a Nikon-C2 laser scanning confocal microscope with a 10X objective. SYTO9 (live cell stain) was excited with 488 nm laser and emitted light was collected through a 525/50 nm filter cube. Propidium iodide (to stain cells with compromised membrane that are considered dead or dying) was excited with 561 nm and emitted light was collected through a 595/50 nm filter cube. To prevent bleed through of fluorescence signal, each fluorophore was excited and emitted light collected in series. Image from each well was analyzed for mean pixel intensity in live and dead channel using ImageJ software (NIH, Bethesda, MD, USA). Ratio of mean intensity of live/dead was plotted.
In vitro activity of phage-MPs against clinical strains of bacteria:
To test activity of phages loaded on the MPs, a 5 µL drop of phage-MPs at 4 mg mL−1 was spotted onto a lawn of respective bacteria (Supplementary Table 2) and allowed to grow for 16 h at 37oC. Formulation was defined as effective if the spot was visually cleared of any bacterial growth after 16 h incubation.
Bacterial inoculum preparation:
Colonies were re-suspended in sterile phosphate-buffered saline (PBS) and the optical density (OD) was measured at 600 nm. For each OD value, serial dilutions were plated on ampicillin-fortified (100 µg mL−1) LB agar plates for 24 h at 37°C with atmospheric CO2. Colonies were counted and plotted against their respective initial OD values. Bacterial challenges were performed with P. aeruginosa expressing green fluorescence protein (PAO1-GFP) or the clinically derived bacteria strain PA103 (ATCC 29260). Bacteria were grown on ampicillin-fortified (100 µg mL−1) LB agar plates for 24 h at 37°C with atmospheric CO2. Colonies were removed from the plate with an inoculation loop and re-suspended in sterile PBS. OD measurements were taken of the solution and adjusted to achieve the required concentration based on previously determined OD growth curve for the respective bacteria.
Care and use of mice:
All experiments were conducted in accordance with Institutional Animal Care and Use Committee-approved protocols at the Georgia Institute of Technology and Emory University. All animals were housed in specific pathogen-free housing with 12 h:12 h light:dark cycles and had food and water ad libitum. The gut-corrected CF transmembrane conductance regulator (CFTR) knockout mouse Cftrtm1Unc Tg(FABPCFTR)-1Jaw/J57 (C57BL/6 and FVB/Nj background) was used in some experiments. These CFTR-KO mice are heterozygous for whole-body knockout of murine CFTR and have human CFTR expression regulated by expression of the fatty acid-binding protein promoter. As such, expression of human CFTR is limited to the small and large intestine with subsequent improved viability and no dietary restrictions or special dietary supplementation compared to the whole-body knockout. C57BL/6 or wild-type littermates of CFTR-KO mice were utilized as wild-type mice. Both female and male mice were used (randomized) for experiments involving CFTR knockout and littermates. Only male mice were used for experiments involving C57BL/6 mice. All animals were used within the age range of 8–14 weeks.
In vivo bacterial challenge:
Mice were placed under general anesthesia with an intraperitoneal injection of ketamine/xylazine (100 mg kg−1 and 10 mg kg−1, respectively). Dry powder phage-MPs (1.0 mg) were delivered to lungs via a Dry Powder Insufflator™ model DP-4M (Penn-Century, Wyndmoor, PA, USA). Bacteria were inoculated using a Microsprayer® Syringe Assembly (model MSA-250-M, Penn-Century) by injecting 50 µL of appropriate OD of bacterial solution through an endotracheal tube. Successful lung delivery was confirmed by change in respiratory status of the animal. Mice were placed under a warming lamp and allowed to recover from general anesthesia. Eighteen to twenty-four hours following the intratracheal instillation of bacteria, mice were euthanized by intraperitoneal overdose of pentobarbital (150 mg kg−1, Euthasol) for organ collection. For assaying phage counts in tissues, organs were explanted, weighed and homogenized in sterile PBS. Homogenates were serially diluted and cultured on LB agar plates fortified with ampicillin (100 µg mL−1) to select for PAO1-GFP or PA103. Homogenate serial dilutions were also tittered for phages on the inoculating bacteria, either PAO1-GFP or PA103. Culture plates were incubated at 37°C and atmospheric CO2 and bacterial colonies or plaques were counted after 16 h incubation. For mixed infections, colony morphology was used to distinguish the different strains of bacteria. The detection limit of colonies was defined as 10 colonies in undiluted lung homogenate plates to prevent counting any lung tissue fragments as false positive.
Immune response against phages:
Mice were given no treatment or dry powder formulations of phage-MPs and empty-MPs. After 21 days, all mice were challenged with bacteria and were either treated with phage-MPs or left untreated. Mice were euthanized after 18 hours and assayed for phage and bacteria as described above. For all time-points (day 0, 21 and 22), serum was collected via cheek bleed. To assay for the presence of antibodies, phage were incubated in a 96 well plate for 1 h and washed multiple times with 0.05% v/v Triton-X-100 in PBS. Samples were then incubated in 1% w/v casein blocker (Life Technologies). Samples were then treated with serum collected at each time-point (diluted 1:10 in PBS) for 1 h. Wells were then incubated with alkaline phosphatase (ALP)-conjugated anti-mouse antibody (1 µg/mL) (Jackson Immunoresearch, 715–055-151). Following several washes, samples were incubated with 5-methyl umbelliferyl phosphate substrate (60 µg/mL) and fluorescence read at excitation/emission of 360 nm/465 nm on a HTS 7000 Plus plate reader (Perkin Elmer, Akron, Ohio, USA).
Phage resistance:
Individual colonies recovered from phage-MPs treated mice were suspended in PBS and plated on TSA plates. A 10 µL drop of original phage mixture was spotted on soft agar seeded with the appropriate host strain and allowed to grow for 16 h at 37oC. Bacteria were defined as susceptible to phage mixture if the spot was cleared of any bacterial growth after incubation for 16 h.
Live animal imaging:
Hair at the dorsum was removed and the animal was imaged using 750 nm excitation/800 nm emission with an IVIS Spectrum CT system (Perkin Elmer, Waltham, MA, USA) or a Fluorescence Molecular Tomography imaging system (Perkin Elmer). Fluorescence intensity was analyzed using the manufacturer’s software.
Lung histology:
Lungs were harvested at necropsy and fixed in 10% neutral buffered formalin for 16 h, washed with PBS and dehydrated in 70% ethanol and embedded in paraffin. Sections (5 µm) were cut using a Microm 355H microtome and stained with hematoxylin and eosin. For immunohistochemistry, sections were treated with 0.05% trypsin for 10 min at 37°C, blocked with goat serum and stained with rabbit anti-P. aeruginosa antibody (Abcam ab68538, Cambridge, MA, USA). Slides were washed, blocked with goat serum and then stained with AlexaFluor 555-conjugated goat anti-rabbit IgG (Thermo Scientific, A21428) and DAPI. For lung cryosection images, fluorescent microparticles were delivered and mice were immediately euthanized. Lungs were harvested and mounted in OCT embedding compound and flash frozen in liquid nitrogen. 10–20 µm thick sections were cut using a cryostat (Leica CM3050 S). The slides were dried at room temperature for 30 mins, washed with PBS and stained with DAPI.
Statistical analysis:
All experiments were performed on independent biological replicates. Sample size for each experimental group and the statistical test used to determine significant differences among groups are reported in the appropriate figure legend. Non-parametric tests were used if the data did not meet the assumptions of t-tests or ANOVA. For in vitro experiments, sample size was not pre-determined, and all samples were included in the analysis. For animal experiments, sample size was determined based on power calculations to detect 20% differences among means using variances from previous/pilot experiments. All animals were used for analysis unless the mice died or had to be euthanized because of pre-defined euthanization criteria (significant weight loss, unresponsive to external stimuli) according to IACUC-approved protocols. The investigators were not blinded to outcome assessment.
Data Availability:
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information. The data in the figures are available from the corresponding author upon request.
Supplementary Material
Acknowledgements.
This work was funded by the National Institutes of Health (R01 AR062920 [AJG], F30 AR069472 [CTJ], S10 OD016264 [AJG]) and a research partnership between Children’s Healthcare of Atlanta and the Georgia Institute of Technology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Centers for Disease Control and Prevention. Use of trade names and commercial sources is for identification only and does not imply endorsement by the Public Health Service or the US Department of Health and Human Services. We thank Dr. Randy Hunt for his advice and discussions over the course of the project. We also thank DFE Pharma for providing inhalation grade lactose Respitose ML006. Clinical strains were obtained from the Clinical and Translational Research Core of the CF@LANTA RDP Center, funded by the CF Foundation (MCCART15R0). PA103 was a gift from Dr. Joanna Goldberg at Emory University.
Footnotes
Competing Interests
The authors declare no competing interests.
References:
- 1.Niederman MS Challenges in the management of community-acquired pneumonia: the role of quinolones and moxifloxacin. Clin Infect Dis 41 Suppl 2, S158–166 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Klevens RM et al. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep 122, 160–166 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cystic Fibrosis Foundation Patient Registry 2015 Annual Data Report (2015). [Google Scholar]
- 4.Cohen TS & Prince A Cystic fibrosis: a mucosal immunodeficiency syndrome. Nat Med 18, 509–519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciofu O, Tolker-Nielsen T, Jensen PO, Wang H & Hoiby N Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv Drug Deliv Rev 85, 7–23 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Magill SS et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med 370, 1198–1208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiner LM et al. Antimicrobial-Resistant Pathogens Associated With Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect Control Hosp Epidemiol 37, 1288–1301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johansen HK, Moskowitz SM, Ciofu O, Pressler T & Hoiby N Spread of colistin resistant non-mucoid Pseudomonas aeruginosa among chronically infected Danish cystic fibrosis patients. J Cyst Fibros 7, 391–397 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Gould IM The epidemiology of antibiotic resistance. Int J Antimicrob Agents 32 Suppl 1, S2–9 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Gomez MI & Prince A Opportunistic infections in lung disease: Pseudomonas infections in cystic fibrosis. Curr Opin Pharmacol 7, 244–251 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Ciofi Degli Atti M et al. An outbreak of extremely drug-resistant Pseudomonas aeruginosa in a tertiary care pediatric hospital in Italy. BMC Infect Dis 14, 494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sprenger M & Fukuda K Antimicrobial resistance. New mechanisms, new worries. Science 351, 1263–1264 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Donlan RM Preventing biofilms of clinically relevant organisms using bacteriophage. Trends Microbiol 17, 66–72 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Servick K Drug development. Beleaguered phage therapy trial presses on. Science 352, 1506 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Geier MR, Trigg ME & Merril CR Fate of bacteriophage lambda in non-immune germ-free mice. Nature 246, 221–223 (1973). [DOI] [PubMed] [Google Scholar]
- 16.Bruttin A & Brüssow H Human volunteers receiving Escherichia coli phage T4 orally: a safety test of phage therapy. Antimicrob. Agents Chemother 49, 2874–2878 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes KA, Sutherland IW, Clark J & Jones MV Bacteriophage and associated polysaccharide depolymerases – novel tools for study of bacterial biofilms. J Appl Microbiol 85, 583–590 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Biswas B et al. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect Immun 70, 204–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chanishvili N, Chanishvili T, Tediashvili M & Barrow PA Phages and their application against drug-resistant bacteria. J Chem Technol Biotechnol 76, 689–699 (2001). [Google Scholar]
- 20.Wright A, Hawkins CH, Änggård EE & Harper DR A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin Otolaryngol 34, 349–357 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Cho I & Blaser MJ The human microbiome: at the interface of health and disease. Nat Rev Genet 13, 260–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dufour N, Delattre R, Ricard JD & Debarbieux L The Lysis of pathogenic Escherichia coli by bacteriophages releases less endotoxin than by beta-lactams. Clin Infect Dis 64, 1582–1588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu TK & Collins JJ Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA 104, 11197–11202 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bikard D et al. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol 32, 1146–1150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Citorik RJ, Mimee M & Lu TK Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat Biotechnol 32, 1141–1145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yosef I, Manor M, Kiro R & Qimron U Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci USA 112, 7267–7272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schooley RT et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob Agents Chemother 61, e00954–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jennes S et al. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury—a case report. Critical Care 21, 129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pabary R et al. Antipseudomonal bacteriophage reduces infective burden and inflammatory response in murine lung. Antimicrob Agents Chemother 60, 744–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Debarbieux L et al. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J Infect Dis 201, 1096–1104 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Morello E et al. Pulmonary bacteriophage therapy on Pseudomonas aeruginosa cystic fibrosis strains: first steps towards treatment and prevention. PLoS One 6, e16963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golshahi L, Seed KD, Dennis JJ & Finlay WH Toward modern inhalational bacteriophage therapy: nebulization of bacteriophages of Burkholderia cepacia complex. J Aerosol Med Pulm Drug Deliv 21, 351–360 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Hraiech S, Bregeon F & Rolain JM Bacteriophage-based therapy in cystic fibrosis-associated Pseudomonas aeruginosa infections: rationale and current status. Drug Des Devel Ther 9, 3653–3663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang M et al. Therapeutic effect of the YH6 phage in a murine hemorrhagic pneumonia model. Res Microbiol 166, 633–643 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Sahota JS et al. Bacteriophage delivery by nebulization and efficacy against phenotypically diverse Pseudomonas aeruginosa from cystic fibrosis patients. J Aerosol Med Pulm Drug Deliv 28, 353–360 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Saussereau E et al. Effectiveness of bacteriophages in the sputum of cystic fibrosis patients. Clin Microbiol Infec 20, O983–990 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Saussereau E & Debarbieux L Bacteriophages in the experimental treatment of Pseudomonas aeruginosa infections in mice. Adv Virus Res 83, 123–141 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Alemayehu D et al. Bacteriophages phiMR299–2 and phiNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. MBio 3, e00029–00012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roach DR et al. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host & Microbe 22, 38–47.e34 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Golshahi L, Lynch KH, Dennis JJ & Finlay WH In vitro lung delivery of bacteriophages KS4-M and PhiKZ using dry powder inhalers for treatment of Burkholderia cepacia complex and Pseudomonas aeruginosa infections in cystic fibrosis. J Appl Microbiol 110, 106–117 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Puapermpoonsiri U, Spencer J & van der Walle CF A freeze-dried formulation of bacteriophage encapsulated in biodegradable microspheres. Eur J Pharm Biopharm 72, 26–33 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Matinkhoo S, Lynch KH, Dennis JJ, Finlay WH & Vehring R Spray-dried respirable powders containing bacteriophages for the treatment of pulmonary infections. J Pharm Sci 100, 5197–5205 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Vandenheuvel D et al. Feasibility of spray drying bacteriophages into respirable powders to combat pulmonary bacterial infections. Eur J Pharm Biopharm 84, 578–582 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Leung SSY et al. Effects of storage conditions on the stability of spray dried, inhalable bacteriophage powders. Int J Pharm 521, 141–149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meenach SA et al. Design, physicochemical characterization, and optimization of organic solution advanced spray-dried inhalable dipalmitoylphosphatidylcholine (DPPC) and dipalmitoylphosphatidylethanolamine poly(ethylene glycol) (DPPE-PEG) microparticles and nanoparticles for targeted respiratory nanomedicine delivery as dry powder inhalation aerosols. Int J Nanomedicine 8, 275–293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edwards DA et al. Large porous particles for pulmonary drug delivery. Science 276, 1868–1871 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Champion JA, Walker A & Mitragotri S Role of particle size in phagocytosis of polymeric microspheres. Pharm Res 25, 1815–1821 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ungaro F et al. Insulin-loaded PLGA/cyclodextrin large porous particles with improved aerosolization properties: in vivo deposition and hypoglycaemic activity after delivery to rat lungs. J Control Release 135, 25–34 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Ungaro F et al. Engineering gas-foamed large porous particles for efficient local delivery of macromolecules to the lung. Eur J Pharm Sci 41, 60–70 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Dhanda DS, Tyagi P, Mirvish SS & Kompella UB Supercritical fluid technology based large porous celecoxib-PLGA microparticles do not induce pulmonary fibrosis and sustain drug delivery and efficacy for several weeks following a single dose. J Control Release 168, 239–250 (2013). [DOI] [PubMed] [Google Scholar]
- 51.d’Angelo I et al. Large Porous Particles for Sustained Release of a Decoy Oligonucelotide and Poly(ethylenimine): Potential for Combined Therapy of Chronic Pseudomonas aeruginosa Lung Infections. Biomacromolecules 17, 1561–1571 (2016). [DOI] [PubMed] [Google Scholar]
- 52.De Stefano D et al. A decoy oligonucleotide to NF-kappaB delivered through inhalable particles prevents LPS-induced rat airway inflammation. Am J Respir Cell Mol Biol 49, 288–295 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Yang Y et al. Development of highly porous large PLGA microparticles for pulmonary drug delivery. Biomaterials 30, 1947–1953 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Diraviam Dinesh S Artificial Sputum Medium doi: 10.1038/protex.2010.212 (2010). [DOI] [Google Scholar]
- 55.Safdar N, Crnich CJ & Maki DG The pathogenesis of ventilator-associated pneumonia: its relevance to developing effective strategies for prevention. Respir Care 50, 725–739; discussion 739–741 (2005). [PubMed] [Google Scholar]
- 56.Makadia HK & Siegel SJ Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3, 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou L et al. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science 266, 1705–1708 (1994). [DOI] [PubMed] [Google Scholar]
- 58.Poschet JF et al. Molecular basis for defective glycosylation and Pseudomonas pathogenesis in cystic fibrosis lung. Proc Natl Acad Sci USA 98, 13972–13977 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pier GB Role of the cystic fibrosis transmembrane conductance regulator in innate immunity to Pseudomonas aeruginosa infections. Proc Natl Acad Sci USA 97, 8822–8828 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu PV The roles of various fractions of Pseudomonas aeruginosa in its pathogenesis: II. Effects of lecithinase and protease. J Infect Dis 116, 112–116 (1966). [DOI] [PubMed] [Google Scholar]
- 61.Foweraker JE, Laughton CR, Brown DF & Bilton D Phenotypic variability of Pseudomonas aeruginosa in sputa from patients with acute infective exacerbation of cystic fibrosis and its impact on the validity of antimicrobial susceptibility testing. J Antimicrob Chemother 55, 921–927 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Winstanley C, O’Brien S & Brockhurst MA Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol 24, 327–337 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waters EM et al. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 72, 666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh N, Vats A, Sharma A, Arora A & Kumar A The development of lower respiratory tract microbiome in mice. Microbiome 5, 61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barfod KK et al. The murine lung microbiome changes during lung inflammation and intranasal vancomycin treatment. Open Microbiol J 9, 167–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biswas B et al. Bacteriophage therapy rescues mice bacteremic from a clinical isolate of vancomycin-resistant Enterococcus faecium. Infect Immun 70, 204–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehman SM & Donlan RM Bacteriophage-mediated control of a two-species biofilm formed by microorganisms causing catheter-associated urinary tract infections in an in vitro urinary catheter model. Antimicrob Agents Chemother 59, 1127–1137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adriaenssens EM et al. CIM((R)) monolithic anion-exchange chromatography as a useful alternative to CsCl gradient purification of bacteriophage particles. Virology 434, 265–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adams MH Bacteriophages (Interscience Publishers, 1959). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its supplementary information. The data in the figures are available from the corresponding author upon request.