

Abstract
Contrast agents that go beyond qualitative visualization and enable quantitative assessments of functional tissue performance represent the next generation of clinically useful imaging tools. An optimized and efficient large-scale synthesis of the cationic iodinated contrast agent (CA4+) is described for imaging articular cartilage. Contrast enhanced CT (CECT) using CA4+ reveals significantly greater agent uptake of CA4+ in articular cartilage compared to similar anionic or nonionic agents, and CA4+ uptake follows Donnan Equilibrium. The CA4+ CECT attenuation obtained from imaging ex vivo human hip cartilage correlates with the glycosaminoglycan (GAG) content, equilibrium modulus, and coefficient of friction: key indicators of cartilage functional performance and osteoarthritis stage. Finally, preliminary toxicity studies in a rat model show no adverse events and a pharmacokinetics study documents a peak plasma concentration at 30 minutes after dosing with the agent no longer present in vivo at 96 hours via excretion in the urine.
Keywords: Computed tomography, iodinated contrast agent, cationic contrast agent, compressive modulus, osteoarthritis, glycosaminoglycan
Graphical Abstract
Introduction
X-ray computed tomography (CT) is a well-established imaging modality that captures virtual slices of biological structures non-invasively, and is commonly employed to diagnose disease and trauma, to guide interventional or therapeutic procedures, and to monitor the effectiveness of therapy. The quality of CT images for diagnostic purposes largely depends on differences in the X-ray attenuation of the tissue of interest relative to its surrounding environment – known as image contrast. CT provides the best contrast between neighboring tissues that have different densities and composition. CT is less effective at distinguishing between contiguous soft tissues (for example, synovial fluid and articular cartilage), both of which are composed largely of water. When natural image contrast is insufficient for clear differentiation of tissues, imaging agents are employed to afford a temporary enhancement of the image quality.1–9
Several FDA approved contrast agents are iodinated water-soluble anionic or nonionic (Figure 1) small molecules (<2000 g/mol),10 which are often used for imaging of the vasculature. First-generation iodinated contrast agents possessed anionic polar groups to increase solubility, but ionic moieties are prone to interactions with cell membranes and other biological structures as well as possess intrinsically high osmolality.11,12 In order to overcome these issues, second-generation contrast agents employed hydroxyl and amide functional groups, that readily hydrogen bond, to promote water solubility. Additionally, multiple iodinated aromatic rings are fused together in order to reduce osmolality while maintaining high administered iodine content. Though several contrast agents (ionic, nonionic and one/two aromatic ring structures) are approved for clinical use and produced worldwide, new contrast agents with high water solubility, low viscosity and osmolality, and reduced toxicity continue to be of interest.13–29 Contrast agents that go beyond qualitative visualization and enable quantitative assessments of functional tissue performance represent the next, third-generation of contrast agents. One clinical area where such contrast agents can have a significant impact is in the diagnosis of soft tissue-based diseases like osteoarthritis (OA).
Figure 1.

Chemical structures of the cationic (CA4+) as well as commercially available negatively charged (ioxaglate) and neutral (iodixanol) contrast agents.
OA affects over 27 million Americans and this patient population is projected to grow to over 40 million adults in the US by the year 2030.30–32 Especially prevalent in the knee, hip, and hand, OA produces pain, lifestyle limitations, and significant loss of productivity in its later stages. Unfortunately, clinicians are not able to detect the disease in its earliest stages, when the affected cartilage tissue may still be salvageable. Currently, OA is diagnosed in the clinic based on symptoms such as pain, swelling, and abnormal joint function including stiffness, locking, and reduced mobility. To assist in diagnosis, plain radiographs (two-dimensional projection images) of the affected joints are obtained to evaluate structural changes such as joint space narrowing, the presence of osteophytes, or that of subchondral bone sclerosis.33–36
These symptoms and radiographic findings appear late in the disease, which leaves clinicians with few treatment options. Unfortunately, there is currently no definitive treatment for OA, other than joint replacement surgery, but early diagnosis may provide opportunities for interventions that can ameliorate disease progression rather than treating the late-stage symptoms. A recognized biomarker for early OA is loss of glycosaminoglycans (GAGs) in the cartilage extracellular matrix (ECM).37–45 The GAGs impart an anionic fixed charge density, which contributes to maintaining fluid pressurization, tissue compressive stiffness, and lubricity. Thus, methods that can detect and quantify GAG concentration, distribution, and changes thereof in cartilage, are in immediate need.
Today, commercially available anionic contrast agents (e.g., iothalamate,46 ioxaglate47), originally developed for cardiovascular applications, are being investigated for imaging cartilage tissue. These anionic contrast agents partition into cartilage in inverse proportion to GAG content due to electrostatic repulsion between the negatively charged GAGs and the contrast agent. The disadvantages of using an anionic probe to detect negatively charged GAGs include requiring high concentrations to be applied in order to produce sufficient contrast-to-noise ratios for quantitative imaging and weak to moderate correlations between CT attenuation and GAG content. Recently, we reported cationic contrast agents for imaging of ex vivo41,48 and in vivo49 cartilage. These cationic agents perform superior to anionic agents, and show high sensitivity for quantifying GAG content41,48 as well as equilibrium compressive modulus and coefficients of friction50 in ex vivo bovine articular cartilage. Our approach belongs to a larger strategy of using electrostatic interactions to detect proteins, polysaccharides, and nucleic acid,51 to create self-healing polymer networks,52,53 or to assemble supramolecular assemblies of non-clinical interest.54–56
Herein, we report the synthesis, characterization, and analysis of the hexa-iodinated cationic contrast agent (CA4+) for sensitive and quantitative ex vivo imaging of human hip cartilage, and key initial data required for subsequent translation of this agent to the clinic. Specifically, we describe the optimized large-scale synthesis and characterization of CA4+. Next, the uptake of the cationic contrast agent is evaluated in bovine articular cartilage in comparison to commercially available anionic and neutral iodinated contrast agents. Donnan equilibrium modeling is then applied to the cationic contrast agents to describe their mode of uptake into articular cartilage. Subsequently, CA4+ is evaluated for assessment of GAG content, stiffness, and frictional performance in human cadaveric hip articular cartilage. Finally, results from a preliminary toxicity study of CA4+ in a rat model are described along with results from a pharmacokinetics study using the 14C radio labeled CA4+.
Results & Discussion
Large scale synthesis and chemical characterization of the CA4+ cationic iodinated contrast agent
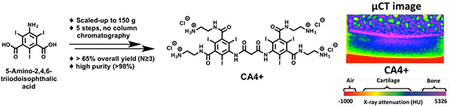
Establishment of an efficient and reliable large-scale synthesis of CA4+ is required for pre-clinical development and further evaluation of CA4+ for additional ex vivo and in vivo imaging studies. Previously, we reported the small scale (<5 g starting material) synthesis of CA4+,45 but not the large scale (>150 g starting material) synthesis. The procedure for the large-scale synthesis of CA4+ is shown in Scheme 1. The contrast agent CA4+ 5 was synthesized from commercially available 5-amino-2,4,6-triiodisophthalic acid 1 in five steps in approximately 68% overall yield with a straightforward workup involving washing, precipitation and filtration, without requiring column chromatography purification. Each reaction step was repeated at least three times and yields are reported as Average ± SD (N ≥ 3).
Scheme 1.

Large scale synthesis of the chloride salt of CA4+ 5.
First, 5-amino-2,4,6-triiodisophthaloyl chloride 2 was synthesized by refluxing the mixture of 1 in thionyl chloride (19 equiv.) and catalytic dimethyl formamide (DMF) for 6–7 hours. The reaction mixture was then precipitated in ice-cold water and filtered to obtain a yellow solid. This solid was then dissolved in ethyl acetate, washed (basic and brine wash), concentrated to a minimum amount (not till dryness) and filtered again to obtain 2 as light yellow powder in 94.2 ± 1.4 % yield. The solid obtained was sufficiently pure (single spot on TLC) to carry out the next step without further purification. This procedure represents an improvement over the previously reported procedure.45 Specifically, the quantity of thionyl chloride, used as solvent and reactant, was reduced from 40 to 19 equivalents. DMF was used as catalyst and the reaction time was reduced from overnight to 6–7 h. We also explored an alternative procedure to minimize the usage of thionyl chloride, where compound 2 was synthesized using only four equivalents of thionyl choride in the solvent ethylene dichloridewith (see SI for details). Although we successfully synthesized 2 using both procedures, the workup is more convenient and simple using thionyl chloride as the reagent and solvent.
Next, 2 was coupled with malonyl chloride (exactly 0.5 equiv.) in anhydrous THF (fresh bottle and transferred through cannula) at 50 °C for overnight. The reaction mixture was cooled to room temperature and then left for precipitation at −20 °C overnight. The tetrakis(acylchloride) intermediate 3 was obtained as a white solid by filtration followed by a quick wash with cold THF (cooled +4 °C for 30–40 minutes prior to use). The mother liquor was concentrated, left for re-precipitation at −20 °C and filtered. Use of dry THF, exactly 0.5 equiv. of malonyl chloride, and maintaining the reaction under an inert atmosphere were key parameters to obtain a high yield and single spot by TLC (89.4 ± 2.7% yield). The tetrakis(Boc-protected) precursor 4 was then synthesized by coupling 3 with tert-butyl-N-(2-aminoethyl)carbamate in anhydrous dimethylacetamide as a solvent and diisoproplyethylamine as an HCl scavenger, generated during the course of the reaction. After overnight stirring at 50 °C, the reaction mixture was precipitated in 1 mol/L aqueous HCl, filtered and the resulting white solid cake was washed with water thrice to remove any trace solvents. After air-drying for 2–3 hours (in the filtration funnel with continuous vacuum) the cake was further dried by lyophilization to obtain 4 as a white powder in 89.7 ± 2.2% yield. The Boc-group deprotection was then carried out by dissolving 4 in dichloromethane:trifluoroacetic acid (1:1). After 2 hours, the reaction mixture was precipitated in diethyl ether and filtered to afford the trifluoroacetic acid salt of CA4+ (CA4+ TFA salt) 5a as a white powder. After air-drying for 2–3 hours the white powder was further dried under high vacuum overnight to remove trace diethyl ether. The trifluoroacetate counter ion was exchanged with chloride ion (Cl¯) by repeated dissolution in 3 mol/L aqueous hydrochloric acid (2X) and precipitation in acetone. The crude product was dissolved in minimum quantity of 0.1 mol/L aqueous HCl and precipitated by drop wise addition into a large excess of acetone (constant stirring) to yield the chloride salt of CA4+ 5 as a white powder in high yield (92.3 ± 3.2% yield, two steps) and characterized by 1H NMR (Figure S9). The complete exchange of trifluoroacetate ion with chloride ion was confirmed by 19F NMR spectrometry. A sharp single peak was observed at −73.73 ppm in the 19F NMR spectra for CA4+ TFA salt (Figure S7), 5a, corresponding to the trifluoroacetate ion, while no peak was observed in 19F NMR spectra for chloride salt of CA4+ 5 (Figure S11). All the compounds were characterized by 1H, 13C NMR (Figure S1-S11) and high-resolution mass spectroscopy. The purity of the CA4+ 5 was determined by analytical reverse-phase HPLC with 1 mL/min flow rate of 95/5 water/ACN (isocratic) and found to be >98%. CA4+ 5 was detected as a single peak at 245 nm with a retention time of 2.5 minutes (Figure S12). HR-MS (ESI-TOF) for C27H33I6N10O6 [M+H]+ was observed at 1354.7031 (calculated 1354.6847 [M+H]+) (Figure S13). The overall improvements in the synthetic procedure are in time, amounts of reactants, purification, and yield, and these benefits are summarized in Table S1. Using the above procedure, we have synthesized more than 1 kg of CA4+ 5 during the last year for use in ex vivo and in vivo cartilage imaging experiments.
The shelf life of CA4+ 5 as a powder at room temperature and in aqueous solution at 4 ºC (12 or 24 mgI/mL, pH=7.4, 400 mOsm/kg) was determined by HPLC after one year. The HPLC trace for both samples after one year was identical, with a single peak at the same retention time, to that of the HPLC trace after synthesis. Thus, CA4+ 5 as a powder or in aqueous 4 ºC can be stored for one year, prior to use.
Uptake of the cationic contrast agent (CA4+) into articular cartilage
Diffusion analysis of the positively charged contrast agent (CA4+) over time into bovine cartilage was carried out and compared with the diffusion of commercially available negatively charged (ioxaglate) and nonionic (iodixanol) contrast agents (Figure 1). The cationic, anionic and nonionic contrast agents were evaluated over a period of 48 hours using high-resolution micro computed tomography (μCT). The equilibrium time (time to reach 95% of the maximum CECT attenuation) was calculated from Equation 1, where α is the CECT attenuation of the cartilage at diffusion equilibrium and τ is the diffusion time constant (the time required to reach 63.2% of the maximum attenuation, α).
| (Equation 1). |
The diffusion time course (Figure 2(A)) and uptake percentage (Figure 2(B)) for each contrast agent were compared. The time required for CA4+ to reach diffusion equilibrium was substantially longer than for ioxaglate or iodixanol. CA4+ reached equilibrium (95% of the maximum CECT attenuation) in 37.5 hours compared to 11.7 hours for iodixanol and 9.1 hours for ioxaglate (Table S2 & S3). Additionally, CA4+ was more highly taken up by cartilage (~400% of the exposed concentration relative to < 100% for ioxaglate and iodxanol), as might be expected, given that CA4+ is electrostatically attracted to the cartilage’s anionic fixed charge density. For the charged contrast agents, equilibrium distribution corresponded to the location and density of the anionic GAGs (Figures 3 and S14). Ioxaglate, with an overall −1 charge, distributed inversely to GAG content. Since the highest concentration of GAGs are present in the deep cartilage layer, attenuation due to ioxaglate penetration was mainly seen in the superficial and middle zones. Iodixanol, which is nonionic, also distributed primarily throughout the middle and superficial zone of cartilage, with more agent in the superficial (likely reflecting its higher free water content). The equilibrium contrast agent distribution of CA4+ was markedly different, distributing directly proportionally to GAG content—primarily in the deep zone of cartilage. This partitioning difference likely explains the longer time required to reach diffusion equilibrium, since CA4+ diffusion proceeds throughout the full depth of the cartilage, including into the deep layer.
Figure 2.

Diffusion trajectories of cationic (CA4+), anionic (ioxaglate), and nonionic (iodixanol) contrast agents in bovine articular cartilage. (A) Average CECT attenuation values (reported in Hounsfield Units, HU) over the 48-hour time course of diffusion into the cartilage of bovine osteochondral plugs. Each point is reported as the average of six samples (mean ± standard deviation). Each contrast agent was fit to a nonlinear diffusion model (Equation 1). The arrow marks the time when 95% of the equilibrium attenuation is reached. (B) Equilibrium CECT attenuation (normalized by concentration) for each contrast agent in bovine osteochondral plugs (*p<0.0001).
Figure 3.

μCT images of a bovine osteochondral plug visualized in color maps. All images are taken of the same plug cross section. The color map ranges from air in red (each image bottom), to cartilage in yellow, green and blue, & bone in dark blue and purple (each image top). The contrast agents diffuse into cartilage over time and the color turns from yellow to green to blue indicating higher CT attenuation.
Donnan equilibrium modeling of cationic contrast agent uptake in cartilage
The uptake of cationic molecules in articular cartilage could be mediated by binding interactions, nonspecific electrostatic interactions (characterized by Donnan equilibrium theory), or both. In order to elucidate the mechanism of CA4+ uptake in articular cartilage, two groups of bovine cartilage plugs (N=3 each) were serially equilibrated in escalating concentrations of CA4+ and imaged using μCT. Donnan equilibrium theory was used to calculate the partitioning of sodium ion given the measured partitioning of the cationic contrast agent, and the results were plotted and fit with nonlinear least squares (Figure 4). Donnan equilibrium theory predicts that the partition coefficients (KCA) for CA4+ will follow the partition coefficient for Na+ (KNa+) via the power law relation shown in Equations 2 & 5 (see experimental section, where Z is the total charge on the contrast agent). The nonlinear least-squares fits for the partition coefficient of cations in bovine cartilage plugs predicted Z = 3.71 for CA4+ (R2 = 0.55, and R2 = 0.95, respectively (Figure 4). The results indicate that CA4+ contrast uptake followed closely with the Donnan equilibrium theory prediction of a molecule bearing +3.7 charge, which is close to the formal charge of +4.
Figure 4.

Donnan equilibrium modelling showing the partition coefficient of CA4+ as a function of sodium ion partition coefficient. Nonlinear least-squares best fits suggest a valence (Z) on the CA4+ as Z = 3.71, compared to the theoretical value (Z = 4).
Cationic contrast agents provide quantification of biochemical and biomechanical measures of human cartilage quality
To assess whether CECT using CA4+ produces CECT attenuations that correlate with GAG content, compressive stiffness, and coefficients of friction of human ex vivo cartilage, twelve osteochondral plugs were extracted from the femoral heads of cadaveric human hips. The plugs were assessed for compressive stiffness using a 4-step stress-relaxation regimen to determine equilibrium compressive modulus, and torsional coefficients of friction were measured using a rotational plug-on-platen regimen. Subsequently, each plug was serially diffused to equilibrium in ioxaglate or CA4+ (24 mgI/mL) at concentrations chosen to produce similar tissue contrast (i.e., ioxaglate at ~6x the concentration of CA4+; 144 mgI/mL) and imaged using μCT. Finally, the GAG content of the cartilage plugs was determined following our published procedures.49 Previous reports showed that lower concentrations of ioxaglate (<100 mgI/mL) provide poorer correlations between CECT attenuations and GAG, hence we used a high concentration for this study.38,39
Comparing CECT attenuation to biochemical content, 79% of the variation in μCT attenuation was attributable to variations in GAG content (R2 = 0.79, p < 0.001) for ioxaglate and 89% of the variation in μCT was attributable to variations in GAG content (R2 = 0.89, p < 0.001) for CA4+ (Figure 5(A)). Relating the CECT data to tissue stiffness, 82% of the variation in μCT attenuation was attributable to variations in equilibrium compressive modulus (R2 = 0.82, p < 0.001) for ioxaglate and 54% of the variation was attributable to variations in modulus (R2 = 0.54, p = 0.01) for CA4+ (Figure 5(B)). CECT attenuation correlated less strongly, and not always statistically significantly, to coefficients of friction μkinetic, μstatic, and μstatic_equilibrium (R2 = 0.31, 0.44, and 0.19 respectively for ioxaglate, Figure 6(A); R2 = 0.48, 0.54, and 0.31 respectively for CA4+, Figure 6(B)). The coefficients of determination for coefficients of friction are weaker in this experiment than those previously reported,50 perhaps reflecting the heterogeneity of the human articular cartilage used compared to young bovine cartilage.
Figure 5.

CECT attenuation correlates strongly with GAG content (A) and equilibrium compressive modulus (B) using both CA4+ and ioxaglate.
Figure 6.

CECT attenuation correlates moderately with coefficients of friction for ioxaglate (A) and CA4+ (B).
Given these results, both contrast agents, at the concentrations used here, reflect the GAG content, equilibrium compressive modulus, and to a more limited extent coefficients of friction, using high resolution imaging at diffusion equilibrium. Importantly, a lower administered dose of CA4+, compared to ioxaglate, can be used. These data are similar to a previous study using bovine osteochondral plugs, and support those findings while extending the proof-of-concept to human cartilage tissue.50 The correlation between attenuation and E is weaker than reported for young bovine cartilage and this may reflect the tissue type, and additional studies with human cartilage tissues are needed. These results are important for establishing the feasibility of using CECT as a clinical tool, since it explores CECT for quantifying cartilage functional performance in human cartilage, which can be different in several ways compared to bovine.33,57–59
The next step in the development of this CECT technique will be the imaging of an intact ex vivo human hip in a clinical scanner. CECT imaging of the hip would be valuable in a clinical setting, since MRI of the hip has unique shortcomings such as magnetic field inhomogeneity in the pelvic region due to large and variable body habitus.60,61 Beforehand, a number of considerations will need to be addressed. First, the maximum spatial resolution of current clinical CT instruments is relatively low compared to μCT scanners. For the thin cartilage (< 1.5 mm) in the human femoral head, the cartilage may be difficult to distinguish from the surroundings and both partial-volume artifacts will likely have a larger impact on obtaining quantitative data. Second, although femoral head cartilage is thin, it is not clear how much diffusion time will be sufficient to reach diffusion equilibrium. This needs to be more thoroughly investigated, as does the impact of incomplete diffusion on contrast agents’ ability to assess cartilage health and functional performance. Recent evidence suggests that for ioxaglate, pre-equilibrium contrast agent partitioning can provide quantitative measures of cartilage quality, but the same has not been evaluated for CA4+.62 Importantly as well, the human hip contains very tightly conforming cartilage surfaces, which may impede and bias the penetration of contrast agents injected intra-articularly in vivo. Thus, optimization of the administration procedure will be required.
Assessment of CA4+ safety, pharmacokinetics, excretion, and tissue distribution in a rat model
A maximum tolerated dose study was performed using N=4 groups of n=6 Sprague-Dawley rats each (3 male, 3 female). Escalating volumetric doses of 24 mgI/mL 14C-labeled CA4+ from 50–200 μL in 50 μL increments were administered via intra-articular injection. The animals showed minimal weight loss (from 0.25% to 4.7%, 1.8 ± 1.4% (mean ± standard deviation)) from the initial body weight. No adverse observations were noted at necropsy, and terminal complete blood count (CBC) found no dose-related abnormal changes. At euthanasia (day 8) nearly all of the animals had high potassium, total protein, albumin, and globulin levels (Table S4) compared to controls/normal values (Table S6 & S7). Male rats were also found to have high phosphorus, glucose, and alkaline phosphatase levels (Table S5) compared to normal levels (Table S6 & S7). No clinical chemistry changes were proportional to dosage given. Also, no animal died prior to scheduled necropsy.
Nine additional animals were used for toxicokinetic analysis, where animals were administered 100 μL of the agent at 2X concentration intra-articularly in one randomly chosen rat stifle joint. Blood was drawn at four post-dose time points per animal range from 5 min to 144 hr. Body weights decreased in most of the animals (from 1.85% to 8.99%, avg 3.79 ± 2.93% body weight loss from initial pre-dose weight) while two animals gained weight. No adverse clinical observations nor abnormal findings were noted during gross necropsy.
For the pharmacokinetics and tissue distribution study, a radio labeled CA4+ (14C-labeled CA4+, 5*) was synthesized by coupling two 5-amino-2,4,6-triiodisophthaloyl moieties with 14C- labeled malonyl chloride and following the same synthetic steps described in Scheme 1. The radiochemical purity for the 14C-labeled CA4+ 5* (>99%) was determined by HPLC with 1 mL/min flow rate of 0.05 % TFA in water (isocratic). 14C-labeled CA4+ 5* (with specific activity 5.82 μCi/mg) was detected as a single peak with a retention time of 6.71 minutes (see SI). Next, twenty additional rats (6 females and 14 males) were dosed intravenously or intra-articularly with 24 mgI/mL of CA4+. Two groups (Group 1 and Group 2) were dosed intravenously with 100 μL of 14C-labeled CA4+ 5* and one group (Group 3) was dosed intra-articularly in one randomly selected stifle joint with 50 μL of 14C-labeled CA4+ 5*. There were three females and three males (n = 6) in Group 1, eight males (n = 8) in Group 2, and three females and three males (n = 6) in Group 3. Approximately 1 hour post-dose, one animal from Group 1 was dead. A gross necropsy was performed on the animal and no macroscopic abnormalities were noted. Approximately 5 hours post-dose, all surviving animals were observed to be normal. Clinical observations for all animals for the remainder of the study were normal.
Blood draws at pre-dose, 0.5, 1, 3, 8, 48, and 96 hours post-dose revealed that plasma concentrations for both IV and IA injections peaked at 0.5 h for both males and females, with females having a higher observed plasma concentration (Figure 7(A)). At the 1 h time point, plasma concentrations were still relatively high for both groups, with females having a higher observed plasma concentration. Plasma concentrations were observed to decline rapidly for both groups, females and males, from the 3 h time point to the 96 h time point. At the 96 h time point, 14C labeled CA4+ was almost completely eliminated from plasma for both groups, females and males. For both the IV and IA administration, the Tmax was 0.5 h indicating peak plasma concentrations (Cmax) occurred 0.5 h after dosing. At 96 h post-dose, the observed levels were similar to the levels at the pre-dose time point. The bioavailability of IA dosing in comparison to IV bolus (100% bioavailability) was 58.01%.
Figure 7.

(A) IV and IA plasma concentrations (male and female). (B) Kidney, liver, and lung concentrations (1, 8 and 24 h post-dose). (C) Urine and feces concentrations.
The results from the PK study show >95% plasma clearance in 24h, and yet CA4+ slowly diffuses into the cartilage, as described above. The rate of plasma clearance and diffusion into the cartilage are critical parameters for successful use of this technique. However, the uptake study was done in bovine cartilage plugs while the PK study was done in rats. The uptake of CA4+ in thin murine cartilage is significantly faster than thick bovine cartilage (1 hr to penetrate fully, versus 24 hrs – unpublished results).
The concentration of agent was highest in the kidneys compared to the lungs and liver, with radioactivity levels starting at 100 nCi/g at the 1 h time point and plateauing at approximately 350 nCi/g at the 24 h time point (Figure 7(B)). Excreted agent was observed primarily in urine, with radioactivity levels peaking at approximately 60 nCi/g at the 8 h time point and declining to approximately 15 nCi/g at the 24 h time point (Figure 7(C)). The combination of high kidney uptake and high urine activity suggests a primarily renal excretion route for the agent. However, persistent high activity suggests that CA4+ is, to some extent, being retained within kidney tissue, likely owing to its high (+4) positive charge. Whether this has an influence on renal function or whether the CA4+ might be similarly retained in other tissues with high membrane potential (e.g., myocardium) is the subject of future studies. Additionally urine was not continuously collected and analyzed, and, thus, we were unable to determine the amount of injected compound excreted in the urine. In contrast to renal excretion, radioactivity in the feces was relatively low with approximately 0.08 nCi/g at the 8 h time point and approximately 2.22 nCi/g at the 24 h time point (Figure 7(C)). The radioactivity was also low for both liver and lung. Levels for the liver peak at approximately 6 nCi/g at the 1 h time point, and plateau at approximately 5 nCi/g at the 8 and 24 h time point. Levels for the lung peak at approximately 5 nCi/g at the 1 h time point, and declined to approximately 1 nCi/g at the 8 and 24 h time point.
Conclusions
The synthesis of CA4+ is accomplished in excellent yield over 5 steps with high purity (>98 %), and can be scaled to 100 grams in the laboratory. Studies elucidating the binding of contrast agents in cartilage, including modeling of the mechanism of uptake, reveal that the cationic agent CA4+ is highly taken up in articular cartilage compared to anionic and nonionic contrast agents, and partitions throughout cartilage according to electrostatic interactions following Donnan equilibrium theory. However, the time to reach diffusion equilibrium for CA4+ is slower compared to the nonionic and anionic contrast agents. CECT using the cationic agent affords attenuations that are indicative of GAG content and compressive stiffness, and moderately indicative of coefficients of friction. These data, along with promising indicators of tolerability in vivo in a rat model, offer promise for the continued development of CECT using CA4+ as a diagnostic imaging tool for early OA. Importantly as well, diagnosis and monitoring of early OA may provide clinicians and researchers with a heretofore unavailable opportunity to develop better therapies via an imaging technique that enables quantitative evaluation and serial monitoring.
Experimental Section
Large scale synthesis and chemical characterization of cationic iodinated contrast agent General experimental methods
Unless otherwise mentioned, all other chemicals were purchased from commercial sources (Sigma-Aldrich, Acros and AK Scientific Inc.) and used without further purification. Anhydrous THF over molecular sieves and anhydrous DMA were purchased and transferred in the reaction vessel through cannula. All reactions were performed under a nitrogen atmosphere and at room temperature unless otherwise noted. All reactions were monitored by analytical thin-layer chromatography (TLC) on aluminium pre-coated plates of silica gel 60G F254 with detection by viewing under UV light. NMR spectra were recorded by Agilent 500 MHz VNMRS spectrometer with a Varian ultra-shielded magnet at 500 MHz for 1H, 470 MHz for 19F, 125 MHz for 13C. 1H NMR chemical shifts were assigned relative to the signal of a residual protonated solvent: DMSO-d6 δ 2.50, CDCl3 δ 7.26. 13C NMR chemical shifts were assigned relative to the signal of a residual protonated solvent: DMSO δ 39.52, CDCl3 δ 77.16. Peak multiplicities were denoted by s (singlet); bs (broad singlet), m (multiplet). High resolution mass spectra were obtained on a Waters QTOF spectrometer. The yield recorded in each experiment was the isolated yield. The purity of the final compound was determined to be ≥ 95% by analytical high pressure liquid chromatography (HPLC).
5-Amino-2,4,6-triiodoisophthaloyl chloride 2.
The commercially available (AK Scientific Inc. #E824 (previous #69824)) 5-amino-2,4,6-triiodoisophthtalic acid (100 g, 178.94 mmol, 1 equiv.) was suspended in thionyl chloride (250 mL, 3.427 mol, 19.1 equvi.) in 1 L round bottom flask to form grey insoluble mixture. Subsequently, DMF (0.5 mL, as a catalyst) was added. The reaction mixture was refluxed for 6–7 h. As the reaction proceeded, the material solubilized to produce a yellow (deep yellow/brown) soluble, albeit cloudy, solution. The reaction mixture was cooled to RT, precipitated into ice-cold water (1500 mL). The aqueous mixture was stirred for 20–25 min and filtered and washed with water to obtaine yellow solid. The yellow solid was transferred into a 2 L erlenmeyer flask, dissolved in 1500 mL ethyl acetate, to which was added saturated NaHCO3 solution (300 mL). The mixture was stirred for 20–25 min. The aqueous layer was discarded and organic layer was washed again with saturated NaHCO3 solution (300 mL), and brine (300 mL). The organic layer was separated, dried over Na2SO4, filtered and evaporated to minimum amount (not till dryness) on rotary evaporator (30 °C bath temperature) and filtered again. The air-dried solid was further dried under high vacuum. Product 2 was isolated as light yellow powder. Yield: 100.5 g (94.2±1.4 %). Rf = 0.28 (1:4 EtOAc:Hexane). 13C NMR (125 MHz, CDCl3): δ in ppm 82.8 ((C=O)-C=C(-I)-C(C=O)= aromatic ring carbon), 85.4 (-C(-I)=C(-NH)-C(-I)= aromatic ring carbon), 148.7 (=C(-NH)- aromatic ring carbon), 156.1 (=C(-C=O)- aromatic ring carbon), 169.6 (-C(=O)-Cl). The product 2 was sufficiently pure (single spot on TLC) and was taken on to the next step without further purification.
5,5’-[Malonylbis(azanediyl)]bis(2,4,6-triiodoisophthaloyl dichloride) 3.
5-amino-2,4,6-triiodoisophthaloyl chloride 2 (80 g, 134.29 mmol, 1 equiv.) was dissolved in anhydrous THF (640 mL, 8 mL/g of 2), under N2. Malonyl chloride (6.54 mL, 67.14 mmol, 0.5 equiv.) was added dropwise at room temperature. The reaction was heated to 50 °C and allowed to stir overnight. The solution was cooled to room temperature and then left to precipitate at −20 °C overnight. White solid was filtered and quickly washed with cold THF twice (2 × 250 mL, cooled by keeping Erlenmeyer flask, containing THF, either in ice-water bath or refrigerator (+4 °C) for 30–40 minutes). The mother liquor was concentrated (up to 30 %) on rotary evaporator and then again allowed to precipitate at −20 °C overnight, and filtered. Product 3 was isolated as white solid. The reaction was repeated thrice and average overall yield was 75.6 g (89.4±2.7 %). Rf = 0.52 (1:1 EtOAc:Hexane). 13C NMR (125 MHz, CDCl3): δ in ppm 41.4 (-C(=O)-CH2-C(=O)-), 88.4 ((C=O)-C=C(-I)-C(C=O)= aromatic ring carbon), 97.7 (-C(-I)=C(-NH)-C(-I)= aromatic ring carbon), 141.3 (=C(-NH)- aromatic ring carbon), 149.3 (=C(-C=O)- aromatic ring carbon), 164.0 (-NH-C(O)-CH2-C(=O)-NH), 169.3 (-C(=O)-Cl). The product 3 was sufficiently pure (single spot on TLC) and was taken on to the next step without further purification.
Tetra-tert-butyl [({5,5’-[malonylbis(azanediyl)]bis(2,4,6-triiodoisophthaloyl)}tetrakis(azanediyl))tetrakis(ethane-2,1-diyl)]tetracarbamate 4.
Intermediate 3 (70 g, 55.55 mmol, 1 equiv.) was dissolved in anhydrous dimethyl acetamide (210 mL, 3 mL/g of 3) as a clear, slightly yellowish solution under N2. Next, N,N-diisopropylethylamine (67.8 mL, 389.8 mmol, 7 equiv.) was added dropwise at room temperature. The reaction was cooled to 0 °C, followed by dropwise addition of tert-butyl-N-(2-aminoetyl)carbamate (38.6 mL, 24.45 mmol, 4.4 equiv.). The ice-water bath was removed after 30 minutes and reaction mixture was heated to 50 °C overnight. The reaction mixture was precipitated into 1 N HCl in water (1750 mL), forming a white solid. The white solid was filtered and washed thoroughly with water (3 × 600 mL). The filtration and washing process took several hours. The solid cake was dried to completion by lyophilisation overnight to obtain 4 as a white powder. Yield: 87.4 g (89.7±2.2 %). 1H NMR (500 MHz, DMSO-d6): δ in ppm 1.38 (s, 36H, -NH-C(=O)-O-(-CH3)3), 3.15–3.23 (m, 16H, -NH-CH2-CH2-NH-), 3.51 (s, 2H, -C(=O)-CH2-C(=O)-), 6.75 (bs, 4H, -NH-C(=O)-O-(-CH3)3), 8.46–8.75 (m, 4H, -C(=O)-NH-CH2-CH2-NH-), 10.20 (s, 2H, -NH-C(=O)-CH2-C(=O)-NH-); 13C NMR (125 MHz, DMSO): δ in ppm 28.2 (-C(CH3)3), 37.5, 38.8, (-NH-CH2-CH2-NH3+), 42.4 (-C(=O)-CH2-C(=O)-), 77.8 (-O-C-(CH3)3), 90.3 ((C=O)-C=C(-I)-C(C=O)= aromatic ring carbon), 99.1 (-C(-I)=C(-NH)-C(-I)= aromatic ring carbon), 142.7 (=C(-NH)- aromatic ring carbon), 150.0 (=C(-C=O)- aromatic ring carbon), 155.5 (-NH-C(=O)-O-(CH3)3), 164.8 (-NH-C(O)-CH2-C(=O)-NH), 169.3 (-NH-C(=O)-ring). HR-MS (ESI-TOF): [M+H]+ theoretical m/z = 1776.8770, observed m/z = 1777.2998. The product 4 was sufficiently pure and was taken on to the next step without further purification.
5,5’-[Malonylbis(azanediyl)]bis[N1,N3-bis(2-aminoethyl)-2,4,6-triiodoisophthalamide], trifluoroacetate salt (CA4+ TFA salt) 5a.
Compound 4 (85 g, 48.44 mmol) was mixed with 1:1 mixture of DCM:TFA (300 mL, 3.5 mL/g of 4). Immediately, bubbling was observed. Over the course of an hour, 4 was completely dissolved. The reaction was continued for one more hour to ensure the complete Boc-deprotection. The reaction mixture was then precipitated in diethyl ether (1200 mL, 4 times the volume of DCM:TFA mixture) to afford a white solid. The solid product was then filtered, washed with diethyl ether (2 × 350 mL) and dried under vacuum to yield 5a as a white powder. Yield: 86.4 g (98.6±1.3 %) 1H NMR (500 MHz, DMSO): δ in ppm 3.00 (s, 8H, -NH-CH2-CH2-NH3+), 3.41–3.53 (m, 10H, -C(=O)-CH2-C(=O)-, -NH-CH2-CH2-NH3+), 7.99 (bs, 12H, -NH3+), 8.66–8.86 (m, 4H, -C(=O)-NH-CH2-CH2-NH3+), 10.19–10.30 (m, 2H, -NH-C(=O)-CH2-C(=O)-NH-); 19F NMR (470 MHz, DMSO-d6): δ in ppm −73.72 (s, -CF3-C(=O)-); 13C NMR (125 MHz, DMSO): δ in ppm 36.6 (-NH-CH2-), 37.4 (-CH2-NH3+), 42.5 (-C(=O)-CH2-C(=O)-), 90.3 ((C=O)-C=C(-I)-C(C=O)= aromatic ring carbon), 99.2 (-C(-I)=C(-NH)-C(-I)= aromatic ring carbon), 99.4 -C(-I)=C(-NH)-C(-I)= aromatic ring carbon), 109.5, 115.9, 118.2 (-CF3-CO-), 142.9 (=C(-NH)- aromatic ring carbon), 149.5 (=C(-C=O)- aromatic ring carbon), 158.1 (–CF3-C(=O)O-), 164.8 (-NH-C(=O)-CH2-C(=O)-NH), 169.5 (-NH-C(=O)-ring). HR-MS (ESI-TOF): [M+Na]+ theoretical m/z = 1354.6847, observed m/z = 1354.6752.
5,5’-[Malonylbis(azanediyl)]bis[N1,N3-bis(2-aminoethyl)-2,4,6-triiodoisophthalamide], chloride salt (CA4+) 5.
Trifluoroacetate ion was exchanged with chloride counter ion (Cl-) by dissolving 5a (150 g, 82.8mmol) in 3 N HCl (450 mL), and subsequent precipitation in acetone (4500 mL). The process was repeated two times to achieve complete conversion of trifluoroacetate salt 5a into chloride salt 5. At last, the crude product was dissolved in 0.1 N HCl (350 mL), treated with activated charcoal (20 g), filtered and washed twice with 50 mL of 0.1 N HCl. The solution (350 mL + 50 mL + 50 mL = 450 mL) was precipitated by dropwise addition into acetone (4500 L) with constant vigorous stirring. Drop-by-drop addition and constant vigorous stirring are necessary for formation of fine precipitates. The solution was filtered, washed with 14:1 acetone:water mixture and dried under high vacuum to obtain 5 as a white powder. Yield: 116.7 g (93.8±2.1 %). 1H NMR (500 MHz, DMSO): δ in ppm 2.97 (bs, 8H, -NH-CH2-CH2-NH3+), 3.49–3.66 (m, 10H, -C(=O)-CH2-C(=O)-, -NH-CH2-CH2-NH3+), 8.30 (bs, 12H, -NH3+), 8.71–8.81 (m, 4H, -C(=O)-NH-CH2-CH2-NH3+), 10.21–10.35 (m, 2H, -NH-C(=O)-CH2-C(=O)-NH-); 19F NMR (470 MHz, DMSO-d6): no peak observed; 13C NMR (125 MHz, DMSO): δ in ppm 36.5 (-NH-CH2-), 37.4 (-CH2-NH3+), 42.2 (-C(=O)-CH2-C(=O)-), 90.6 ((C=O)-C=C(-I)-C(C=O)= aromatic ring carbon), 100.0 (-C(-I)=C(-NH)-C(-I)= aromatic ring carbon), 142.8 (=C(-NH)- aromatic ring carbon), 149.4 (=C(-C=O)- aromatic ring carbon), 164.8 (-NH-C(O)-CH2-C(=O)-NH), 169.5 (-NH-C(=O)-ring). HR-MS (ESI-TOF): [M+H]+ theoretical m/z = 1354.6847, observed m/z = 1354.7031. The purity of 5 salt was determined by analytical reverse-phase HPLC. Briefly, a Varian ProStar HPLC pump and UV-vis detector with Hamilton C18 HxSil 5μm 250×4.6 mm column was used with 1 mL/min flow rate of 95/5 water/ACN (isocratic). Product 5 was detected as a single peak at 245 nm with a retention time of 2.3 minutes (Figure S12). The purity of the CA4+ 5 was found to be >98%. Shelf life of CA4+ 5 powder and aqueous solution of CA4+ 5 (12 or 24 mgI/mL, pH=7.4, 400 mOsm/kg), was determined by recording HPLC profile after one year. Both, CA4+ 5 powder and its aqueous solution remain unaltered (eluted as single peak at the same retention time) when stored for one year in refrigerator (+4 ºC). CA4+ 5 powder can be stored unaltered even at room temperature for a year.
A cationic contrast agent (CA4+) is highly taken up in articular cartilage
Contrast Agent Solutions
Solutions of 80 mgI/mL iodixanol (Visipaque, Medline Industries, Inc., Mundelein, Illinois) and 80 mgI/mL ioxaglate (Hexabrix, Guerbet, Bloomington, IN) were prepared by dilution into Nanopure water (Barnstead Nanopure Thermo Scientific, Waltham, MA) and balanced to 400 ± 20 mOsm/kg. The solution of 12 mgI/mL CA4+ (Contrast Agent with 4+ charge) was prepared by adding 1.182 g of dry concentrate to 50 mL of Nanopure water, balanced to pH 7.0 (perpHecT LogR meter, model 310, Thermo Scientific, Waltham, MA) with 4.0 M sodium hydroxide (Fisher Scientific, St. Louis, MO) and balanced to 400 ± 20 mOsm/kg. The parameters of these solutions are listed in Table 1, where the molecular weight is listed of the ionized species.
Table 1:
Contrast agent solution parameters.
| Contrast Agent | Ionic MW [g/mol] | Concentration (mgl/mL) | Charge |
|---|---|---|---|
| CA4 | 1354 | 12 | +4 |
| odixanol | 1550 | 80 | 0 |
| loxaglate | 1269 | 80 | −1 |
The osmolality of solutions was balanced with sodium chloride (Fisher Scientific, Pittsburgh, PA) and measured with a freezing point osmometer (The Advanced Osmometer, Advanced Instruments, Inc., Norwood, MA). A preservative cocktail containing 5mM ethylenediaminetetraacetic acid (EDTA), 5mM benzamidine HCl, 5x Antibiotic-Antimycotic (Anti-Anti #15240096, Life Technologies, Carlsbad, CA) was used in solutions containing ex vivo osteochondral samples to prevent nonspecific degradation of the cartilage. The mixture was added by 100-fold dilutions of 500mM, 500mM and 500x stock solutions, respectively. Osteochondral plugs were allowed to recover for at least 12 hours in 400 ± 20 mOsmol/kg saline at 4 °C between each of the mechanical tests, micro computed tomography (μCT) imaging and the 1,9 dimethylmethylene blue (DMMB) assay for GAG content.
7mm Osteochondral Bovine Plugs
Six osteochondral plugs (7 mm diameter) were cored from the femoral groove of the stifle joint from a freshly slaughtered, skeletally mature cow (36–54 months, Research 87, Boylston, MA) using a diamond-tipped cylindrical cutter (Catalog #102080, Starlite Industries, Rosemont, PA) irrigated with 0.9% saline at room temperature. The samples were frozen at −20°C in 400 ± 20 mOsmol/kg saline with the inhibitor mixture.
Electrochemical modelling of cationic contrast agent uptake in cartilage emphasizes electrostatic interactions
Previous studies using cationic contrast agents have demonstrated high contrast uptake into cartilage. The mechanism for this high contrast uptake could be purely electrostatic (i.e. high concentrations of mobile cations evolve in the tissue due to the anionic fixed charge density contributed by immobilized GAGs), or a stronger interaction (more akin to receptor binding) could be present between the cationic agent and moieties inside the cartilage ECM. If the latter occurs, determining a binding constant is an important step for characterizing the interaction and for further developing the quantitative CECT technique. Receptor-ligand binding studies are very commonly conducted by incubating a receptor with a wide range of ligand concentrations to determine a dissociation constant, Kd, which is an indicator of the binding strength between the receptor-ligand pair.63–65 Thus, this technique was used to evaluate the binding affinity between cationic contrast agents (CA4+, bearing +4 charge) and bovine cartilage ECM. Additionally, small cations in cartilage are also affected by electrostatic attraction to anionic GAGs, which could also explain the high partitioning of cationic contrast agents.
The cationic contrast agent CA4+ was prepared as six serial dilutions in deionized water containing 2, 4, 8, 16, 32, and 64 mgI/mL. Six serial dilutions of ioxaglate were prepared (Hexabrix-320®, Mallinckrodt, Hazelwood, MO) at the same iodine concentrations. All dilutions contained a preservative cocktail of protease inhibitors, antibiotics, and antimycotics to prevent tissue degradation over the course of the experiment (5 mM EDTA, 5 mM Benzamidine HCl, Sigma-Aldrich, St. Louis, MO; 1x Gibco® Antibiotic-Antimycotic, Life Technologies, Carlsbad, CA). The pH of each solution was adjusted to pH 7.4 with sodium hydroxide, and sodium chloride was added to each dilution to achieve a solution iso-osmolar to synovial fluid (400 mOsmol/kg). Nine bovine osteochondral plugs (7 mm diameter) were harvested from one bovine femoral condyle to obtain samples with a narrow variation in GAG content. All nine samples were imaged with μCT prior to contrast agent immersions (baseline scan). Three plugs (n=3) were randomly chosen for CA4+ immersions, and each was immersed in a large reservoir (4 mL) of 2 mgI/mL CA4+ for 24 hours and then imaged. Following imaging, the plugs were immersed in the next dilution in order of concentration (4 mgI/mL CA4+) for 24 hours and then imaged similarly. This protocol of serial immersions was followed repeatedly to collect CECT imaging data for CA4+ at all six concentrations. The remaining six plugs were assigned to ioxaglate (n=3) immersions, and an identical protocol was followed for each contrast agent. The contrast agent solutions were also scanned in a similar geometry as the plugs in order to produce a standard conversion curve between contrast agent concentration (mgI/mL) and CT attenuation (Hounsfield Units). After plug immersion and scanning for the highest concentrations (64 mgI/mL), all nine plugs were washed in copious saline for 24 hours and prepared for DMMB assay. Briefly, the cartilage was removed from the subchondral bone, weighed, and lyophilized to determine wet and dry cartilage masses. Each cartilage specimen was digested in papain buffer and the GAG content was assessed using the DMMB assay (see SI).
Images of the cartilage and underlying subchondral bone were acquired using a micro computed tomographic imaging system (μCT40, Scanco Medical AG, Brüttisellen, Switzerland) at an isotropic voxel resolution of 36 μm, 70 kVp tube voltage, 113 μA current and 300 ms integration time. The CT data sets were imported into image processing software (Analyze®, AnalyzeDirect, Overland Park, KS) and the cartilage was segmented using a semi-automatic contour based segmentation algorithm. The mean cartilage x-ray attenuation values using the Hounsfield Scale were obtained by averaging attenuation values for all cartilage tissue over the entire segmented volume. Then, the non-contrast (baseline) attenuation was subtracted from all contrast immersion attenuations, and the standard curve was used to convert CECT attenuation to contrast concentration inside the cartilage tissue. Linear regression analysis was used to express the intra-tissue contrast concentration as a linear function of the contrast reservoir concentration for plugs immersed in ioxaglate. For CA4+, a modified Langmuir isotherm was fit to the data to illustrate the nonlinear relationship between reservoir concentration and intra-tissue concentration (MATLAB®, MathWorks, Natick, MA). This model allowed an EC50 value to be calculated.
In order to determine if the cationic contrast agents partitioned in cartilage according to the predications of Donnan equilibrium theory, a previously published method was implemented in which the partition coefficient of contrast agent, KCA4+, was plotted as a function of the predicted sodium ion partition coefficient, KNa+. The partition coefficients for CA4+ was calculated as the ratio between the intra-tissue concentration at equilibrium and the bath concentration. The partition coefficient for Na+ was calculated using the law of electroneutrality and Donnan equilibrium theory using custom MATLAB code. That is, Donnan equilibrium requires that:66
| Equation 2 |
Where is the intra-tissue concentration of contrast agent, is the bath contrast concentration, is the intra-tissue sodium concentration, is the bath sodium concentration, is the intra-tissue chloride concentration, and is the bath chloride concentration. Since charge neutrality requires that the sum of all charges inside the cartilage tissue are a net of zero,
| Equation 3 |
Where FCD is the intra-tissue (negative) fixed charge density contributed primarily by the GAGs. Combining Equations 1 & 2 and solving for allows the partition coefficients to be calculated:
| Equation 4 |
was each plotted as a function of according to their relationship shown in Equation 1,67 where:
| Equation 5 |
and a nonlinear least-squares method was used to estimate the net charge (Z) on the CA4+ (MATLAB®, MathWorks).
Cationic contrast agents provide quantification of biochemical and biomechanical measures of human cartilage quality
Two human hemi-pelvis specimens (ages 40 and 61, female and male respectively) were obtained and thawed at 4°C (Med-Cure, Portland, OR). Six osteochondral plugs (7 mm diameter) were harvested from each of the two femoral heads using a diamond tipped cylindrical cutter under constant irrigation (Figure 8). Each plug was rigidly clamped in a mechanical testing apparatus (Enduratec3230, BOSE, Eden Prairie, MN) and a compressive pre-load of 5 N was applied between the cartilage surface and a nonporous UHWPE platen to ensure complete contact between the cartilage and platen. While immersed in saline, each plug was subjected to 4 incremental 5% compressive strain steps (5 μm/sec) in unconfined compression with stress relaxation (45 minutes) intervening between strain steps. A collection rate of 10 Hz was used to record the force and displacement data, and a linear fit to stress vs. strain at each equilibrium was used to calculate the equilibrium compressive modulus for each cartilage specimen.40,50 Following recovery overnight in saline, each plug was subjected to torsional friction by applying an 18% compressive strain against a polish aluminum platen (5 μm/sec), relaxing for 70 minutes, and rotating 720° at 5 °/sec (an effective velocity of 0.3 mm/sec) to derive static and kinetic coefficients of friction.50,68 Force, displacement, torque, and rotational data were collected at 10 Hz. Coefficients of friction (μ) were calculated as follows:
| Equation 6 |
Figure 8.

Osteochondral cores (7 mm diameter) were harvested from various locations around the femoral head surfaces.
N = normal force, and:
μstatic: the maximum value of μ for the first 10° of rotation
μstatic_equilibrium: computed using the maximum value of T from the first 10° of rotation and N as the normal force at the end of the last relaxation period
μkinetic: the average value of μ during the second 360° of rotation
After recovering the plugs overnight in saline, they were immersed in ioxaglate solution (80 mgI/mL) for 24 hours and then CECT imaged (μCT40, Scanco Medical AG, Brüttisellen, Switzerland) at an isotropic voxel resolution of 36 μm, 70 kVp, 114 μA, and 300 ms integration. The plugs were rinsed of the ioxaglate in saline for 24 hours, immersed in CA4+ solution (12 mgI/mL) for 24 hours and then re-imaged. After rinsing the plugs in saline for 24 hours to remove the CA4+, the GAG content (as a % of wet tissue mass) of each specimen was measured using the 1,9-dimethylmethylene blue assay.
For the μCT plug images, the cartilage was segmented from the subchondral bone using a semi-automatic contour based algorithm. The mean x-ray linear attenuation coefficient of each cartilage plug was converted to Hounsfield Units (HU) by normalizing to that of water scanned in a similar geometry at the same settings on the μCT40. Univariate linear regression (SPSS v17.0, IBM, Armonk, NY) was performed to evaluate the relationship between μCT attenuation (HU), GAG content (% of wet weight), equilibrium compressive modulus (E,MPa), and coefficients of friction (μstatic, μstatic_equilibrium, and μkinetic).
Toxicity evaluation in a rat model suggests safety of CA4+ for intra-articular injection
Synthesis of the radiolabeled CA4+ was performed by incorporating the 14C into the malonic diamide bridging group to minimize the potential for metabolic cleavage from the hexa-iodo, diaryl core structure (Scheme 2). The synthesis proceeded smoothly and the product was prepared in purity and quantity needed for the toxicology program.
Scheme 2.

Synthesis of 14C-labeled CA4+ 5*.
Intra articular Maximum Tolerance Dose (MTD) and Toxicokinetic (TK) in rats. A MTD and TK study was performed by Toxikon in accordance with all relevant guidelines and applicable study protocol/outline.
Maximum Tolerance Dose (MTD):
The study design consisted of four dose groups as shown in Table 2. Each group received a different dose of test article intra-particularly. Animals were observed twice daily for clinical signs of toxicity. Findings were recorded as they were observed. Body weights were recorded once daily for all animals. Terminal (fasted) body weights were recorded prior to scheduled necropsy on Day 8. Clinical pathology investigations of hematology and clinical chemistry were performed on all MTD study animals at termination (Day 8). Animals were deprived of food overnight for the collection of clinical pathological parameters. Blood samples were collected for terminal clinical pathology via a peripheral blood vessel or cardiac blood draw in K2EDTA collection tubes. The serum fraction of the blood was prepared from blood collected in tubes with no anticoagulant for clinical chemistry analysis.
Table 2:
Group animal assignment (MTD).
| Animal Assignment | ||
|---|---|---|
| Group # | Dose (μL) | # of Animals |
| 1 | 50 | 3M / 3F |
| 2 | 100 | 3M / 3F |
| 3 | 150 | 3M / 3F |
| 4 | 200 | 3M / 3F |
| M = Male F = Female | ||
Toxicokinetic (TK) Analysis:
A new group of animals was administered test article at 100 μL of 2X concentration intra-articularly (determined according the MTD results) in one stifle chosen randomly using appropriate software. Approximately 0.5 mL of blood was drawn from a peripheral vein or other appropriate site pre-dose, and post-dose at the time point describe Table 3.
Table 3:
Blood drawn time point (TK).
| TK Sampling | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Animal # | Pre-Dose | 5 Min. | 30 Min. | 1h | 3h | 8h | 24h | 48h | 72h | 96h | 120h | 144h |
| 1 | x | x | x | x | ||||||||
| 2 | x | x | x | x | ||||||||
| 3 | x | x | x | x | ||||||||
| 4 | x | x | x | x | ||||||||
| 5 | x | x | x | x | ||||||||
| 6 | x | x | x | x | ||||||||
| 7 | x | x | x | x | ||||||||
| 8 | x | x | x | x | ||||||||
| 9 | x | x | x | x | ||||||||
h = Hours
Pharmacokinetic, Excretion, And Tissue Distribution of Radioactivity In Rats Following A Single Intravenous Or Intra-Articular Dose Of 14C-CA4+.
The purpose of this study was to determine the pharmacokinetics, excretion, and tissue distribution of 14C-labeled CA4+ 5* following intravenous or intra-articular administration of a single dose of 14C-labeled CA4+ 5* in Sprague Dawley rats. Although this study was Non-GLP, it was conducted according to the accredited Quality System in effect at Toxikon, including ISO/IEC 17025, 2005, General Requirements for the Competence of Testing and Calibration Laboratories. Toxikon’s Quality System also encompasses the general principles and practices of GxP regulations, specifically GLPs and GMPs. Number and Species: 30 Sprague-Dawley rats (Rattus norvegicus) were included in the study file. Data for the 20 dosed animals is included in the report. Sex: 21 males and 9 females (females were non-pregnant and nulliparous); 14 males and 6 females were dosed. Two groups (Group 1 and Group 3) were dosed intravenously with 100 μL of Formulation 1 of 14C-labeled CA4+ 5* and one group (Group 4) was dosed intra-articularly with 50 μL of Formulation 2 of 14C-labeled CA4+ 5* (see group animal assignment in table 3). There were three females and three males (n = 6) in Group 1, eight males (n = 8) in Group 3, and three females and three males (n = 6) in Group 4. All animals were dosed once on Day 1 intravenously or intra-articularly in one stifle (selected randomly). Approximately 0.5 mL of blood was drawn via retro-orbital venipuncture from animals in Groups 1 and 4 at pre-dose, 0.5, 1, 3, 8, 48, and 96 hours post-dose. Blood was collected in K2EDTA collection tubes and processed to plasma via centrifugation. These samples were stored at −80 ± 12°C until analysis by liquid scintillations counting.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to gratefully acknowledge support in part from the National Institutes of Health (R01GM098361 and R43AR063563), the T32 Pharmacology Training grant (5T32GM008541–14; JDF), and Boston University.
ABBREVIATIONS
- ACN
acetonitrile
- Boc
tetr-butyloxycarbonyl protecting group
- DCM
dichloromethane
- DMA
dimethylacetamide
- DIPEA
N,N-diisopropylethylamine
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TLC
thin layer chromatography
Footnotes
Competing Interest
MWG is a co-founder of Ionic Pharmaceuticals and MW was an employee of Ionic Pharmaceuticals. Ionic Pharmaceuticals paid for the radiolabeled study. AG is President of BICL, LLC and Consultant to Sanofi Aventis, GE Healthcare, AstraZeneca, TissueGene, OrthoTrophix, Pfizer, and MerckSerono.
REFERENCES
- (1).Hallouard F; Anton N; Choquet P; Constantinesco A; Vandamme T Iodinated Blood Pool Contrast Media for Preclinical X-Ray Imaging Applications – A Review. Biomaterials 2010, 31 (24), 6249–6268. [DOI] [PubMed] [Google Scholar]
- (2).Hasebroock KM; Serkova NJ Toxicity of MRI and CT Contrast Agents. Expert Opin. Drug Metab. Toxicol 2009, 5 (4), 403–416. [DOI] [PubMed] [Google Scholar]
- (3).Kün-Darbois J-D; Manero F; Rony L; Chappard D Contrast Enhancement with Uranyl Acetate Allows Quantitative Analysis of the Articular Cartilage by microCT: Application to Mandibular Condyles in the BTX Rat Model of Disuse. Micron 2017, 97, 35–40. [DOI] [PubMed] [Google Scholar]
- (4).Lee N; Choi SH; Hyeon T Nano-Sized CT Contrast Agents. Adv. Mater 2013, 25 (19), 2641–2660. [DOI] [PubMed] [Google Scholar]
- (5).Mattrey RF; Aguirre DA Advances in Contrast Media Research. Acad. Radiol 2003, 10 (12), 1450–1460. [DOI] [PubMed] [Google Scholar]
- (6).Torchilin VP PEG-Based Micelles as Carriers of Contrast Agents for Different Imaging Modalities. Adv. Drug Delivery Rev. 2002, 54 (2), 235–252. [DOI] [PubMed] [Google Scholar]
- (7).Ravi Kumar MNV Handbook of Particulate Drug Delivery; American Scientific Publishers: Stevenson Ranch, Ca., 2006. [Google Scholar]
- (8).Faraj KA; Cuijpers VMJI; Wismans RG; Walboomers XF; Jansen JA; van Kuppevelt TH; Daamen WF Micro-Computed Tomographical Imaging of Soft Biological Materials Using Contrast Techniques. Tissue Eng. Part C Methods 2009, 15 (3), 493–499. [DOI] [PubMed] [Google Scholar]
- (9).Karhula SS; Finnilä MA; Lammi MJ; Ylärinne JH; Kauppinen S; Rieppo L; Pritzker KPH; Nieminen HJ; Saarakkala S Effects of Articular Cartilage Constituents on Phosphotungstic Acid Enhanced Micro-Computed Tomography. PLoS One 2017, 12 (1), e0171075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lusic H; Grinstaff MW X-Ray-Computed Tomography Contrast Agents. Chem. Rev 2013, 113 (3), 1641–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Aspelin P Why Choice of Contrast Medium Matters. Eur. Radiol. Suppl 2006, 16 (S4), D22–D27. [Google Scholar]
- (12).Hunt CH; Hartman RP; Hesley GK Frequency and Severity of Adverse Effects of Iodinated and Gadolinium Contrast Materials: Retrospective Review of 456,930 Doses. AJR. Am. J. Roentgenol 2009, 193 (4), 1124–1127. [DOI] [PubMed] [Google Scholar]
- (13).Christiansen C X-Ray Contrast Media--an Overview. Toxicology 2005, 209 (2), 185–187. [DOI] [PubMed] [Google Scholar]
- (14).Pillai KMR; Diamantidis G; Duncan L; Ranganathan RS Heterocyclic Nonionic X-Ray Contrast Agents. III. The Synthesis of 5-[4-(Hydroxymethyl)-2-Oxo-3-Oxazolidinyl]-2,4,6-Triiodo-1,3-Benzenedicarboxamide Derivatives. J. Org. Chem 1994, 59 (6), 1344–1350. [Google Scholar]
- (15).Böhle F; Carretero JM; González L; Martín JL New Iodinated Contrast Agents. The Physicochemical and Biological Properties of Asymmetric Dimers. Invest. Radiol 1994, 29 Suppl 2, S264–6. [DOI] [PubMed] [Google Scholar]
- (16).Ranganathan RS; Arunachalam T; Song B; Mantha S; Ogan M; Wedeking P; Yost F; Jagoda E; Tweedle M Evaluation of N,N’-bis-Dimethyldiatrizoic Acid Analogs as Liver Imaging Agents. Acad. Radiol 1998, 5 Suppl 1, S23–7. [DOI] [PubMed] [Google Scholar]
- (17).Marinelli ER; Arunachalam T; Diamantidis G; Emswiler J; Fan H; Neubeck R; Pillai KMR; Wagler TR; Chen C-K; Natalie K; Soundararajan N; Ranganathan RS Heterocyclic Nonionic X-Ray Contrast Agents V: A Facile Conversion of 2-Tetrahydrofuroamides into α-Hydroxy-δ-Valerolactams and a General Synthesis of Lactams Conjugated to 2,4,6-Triiodoisophthalamides. Tetrahedron 1996, 52 (34), 11177–11214. [Google Scholar]
- (18).Newington IM; Humphries G; Lasbistes N; Morisson-Iveson V; Nairne J; Passmore J; Thanning M; Wistrand L-G; Wynn D The Synthesis and Evaluation of Trimeric X-Ray Contrast Agents. Tetrahedron Lett. 2011, 52 (24), 3065–3067. [Google Scholar]
- (19).Wynn DG; Humphries G; Morisson-Iveson V; Nairne J; Newington IM; Passmore J; Wistrand L-G The Synthesis and Evaluation of Unsymmetrical Dimeric X-Ray Contrast Agents. Tetrahedron Lett. 2011, 52 (24), 3068–3071. [Google Scholar]
- (20).Namasivayam S; Kalra MK; Torres WE; Small WC Adverse Reactions to Intravenous Iodinated Contrast Media: A Primer for Radiologists. Emerg. Radiol 2006, 12 (5), 210–215. [DOI] [PubMed] [Google Scholar]
- (21).Aspelin P; Aubry P; Fransson S-G; Strasser R; Willenbrock R; Berg KJ Nephrotoxic Effects in High-Risk Patients Undergoing Angiography. N. Engl. J. Med 2003, 348 (6), 491–499. [DOI] [PubMed] [Google Scholar]
- (22).Davidson C; Stacul F; McCullough PA; Tumlin J; Adam A; Lameire N; Becker CR Contrast Medium Use. Am. J. Cardiol 2006, 98 (6A), 42K–58K. [DOI] [PubMed] [Google Scholar]
- (23).Chai CM; Karlsson JOG; Almén T Incidence of Ventricular Fibrillation during Left Coronary Arteriography in Pigs: Comparison of a Solution of the Nonionic Dimer Iodixanol with Solutions of Five Different Nonionic Monomers. Acta Radiol. 2008, 49 (2), 150–156. [DOI] [PubMed] [Google Scholar]
- (24).Arunachalam T; Fan H; Pillai KMR; Ranganathan RS Heterocyclic Nonionic X-Ray Contrast Agents. 4. The Synthesis of Dihydro-2(3H)-Furanylidenamino, 5-Oxo-1-Pyrrolidinyl, and 5-Oxo-4-Morpholinyl Derivatives by an Intramolecular Iodocyclization Approach. J. Org. Chem 1995, 60 (14), 4428–4438. [Google Scholar]
- (25).Riefke B; González L; Carretero J; González C; Jimenéz I; Alguacil LF; Böhle F; Martin F; Martin JL ICJ 3393: A New Lodinated Nonionic Low Osmolal Dimer with Low Viscosity as Contrast Agent for X-Ray. Acad. Radiol 2002, 9 Suppl 1, S178–81. [DOI] [PubMed] [Google Scholar]
- (26).Sovak M; Terry R; Abramjuk C; Faberová V; Fiserova M; Laznicek M; Leuschner J; Malinak J; Zahradnik P; Masner O; Seligson A Iosimenol, a Low-Viscosity Nonionic Dimer: Preclinical Physicochemistry, Pharmacology, and Pharmacokinetics. Invest. Radiol 2004, 39 (3), 171–181. [DOI] [PubMed] [Google Scholar]
- (27).Fernandez Cabezudo MJ; Petroianu G; Al-Ramadi B; Langer RD Iosimenol, a New Non-Ionic Dimeric Contrast Medium, Does Not Induce Immunoreactivity in the Popliteal Lymph Node Assay. Br. J. Radiol 2007, 80 (957), 713–718. [DOI] [PubMed] [Google Scholar]
- (28).Chai C-M; Rasmussen H; Eriksen M; Hvoslef A-M; Evans P; Newton BB; Videm S Predicting Cardiotoxicity Propensity of the Novel Iodinated Contrast Medium GE-145: Ventricular Fibrillation during Left Coronary Arteriography in Pigs. Acta Radiol. 2010, 51 (9), 1007–1013. [DOI] [PubMed] [Google Scholar]
- (29).Wistrand L-G; Rogstad A; Hagelin G; Roed L; Oulie I; Gram A; Evans P; Rasmussen H; Grant D; Iveson P; Newton B; Thaning M GE-145, a New Low-Osmolar Dimeric Radiographic Contrast Medium. Acta Radiol. 2010, 51 (9), 1014–1120. [DOI] [PubMed] [Google Scholar]
- (30).Lawrence RC; Felson DT; Helmick CG; Arnold LM; Choi H; Deyo RA; Gabriel S; Hirsch R; Hochberg MC; Hunder GG; Jordan JM; Katz JN; Kremers HM; Wolfe F Estimates of the Prevalence of Arthritis and Other Rheumatic Conditions in the United States. Part II. Arthritis Rheum. 2008, 58 (1), 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cheng YJ; Hootman JM; Murphy LB; Langmaid GA; Helmick CG CDC. Prevalence of Doctor-Diagnosed Arthritis and Arthritis-Attributable Activity Limitation --- United States, 2007−−2009. MMWR 2010, 59 (39), 1261–1265. [PubMed] [Google Scholar]
- (32).McNeil JM; Binette J CDC. Prevalence of Disabilities and Associated Health Conditions Among Adults --- United States, 1999. MMWR 2001, 50 (7), 120–125. [PubMed] [Google Scholar]
- (33).Mow VC; Huiskes R Basic Orthopedic Biomechanics and Mechanobiology, 3rd ed.; Lippincott Williams & Wilkins: Philadelphia, 2005. [Google Scholar]
- (34).Lohmander LS What Can We Do about Osteoarthritis? Arthritis Res. 2000, 2 (2), 95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Guermazi A; Hayashi D; Roemer FW; Felson DT Osteoarthritis A Review of Strengths and Weaknesses of Different Imaging Options. Rheum. Dis. Clin. NA 2013, 39, 567–591. [DOI] [PubMed] [Google Scholar]
- (36).Guermazi A; Hayashi D; Roemer F; Felson DT; Wang K; Lynch J; Amin S; Torner J; Lewis CE; Nevitt MC Severe Radiographic Knee Osteoarthritis E Does Kellgren and Lawrence Grade 4 Represent End Stage Disease? E the MOST Study. Osteoarthr. Cartil 2015, 23, 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Silvast TS; Jurvelin JS; Aula AS; Lammi MJ; Töyräs J Contrast Agent-Enhanced Computed Tomography of Articular Cartilage: Association with Tissue Composition and Properties. Acta Radiol. 2009, 50 (1), 78–85. [DOI] [PubMed] [Google Scholar]
- (38).Palmer AW; Guldberg RE; Levenston ME Analysis of Cartilage Matrix Fixed Charge Density and Three-Dimensional Morphology via Contrast-Enhanced Microcomputed Tomography. Proc. Natl. Acad. Sci. U. S. A 2006, 103 (51), 19255–19260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kallioniemi AS; Jurvelin JS; Nieminen MT; Lammi MJ; Töyräs J Contrast Agent Enhanced pQCT of Articular Cartilage. Phys. Med. Biol 2007, 52 (4), 1209–1219. [DOI] [PubMed] [Google Scholar]
- (40).Bansal PN; Joshi NS; Entezari V; Grinstaff MW; Snyder BD Contrast Enhanced Computed Tomography Can Predict the Glycosaminoglycan Content and Biomechanical Properties of Articular Cartilage. Osteoarthr. Cartil 2010, 18 (2), 184–191. [DOI] [PubMed] [Google Scholar]
- (41).Bansal PN; Joshi NS; Entezari V; Malone BC; Stewart RC; Snyder BD; Grinstaff MW Cationic Contrast Agents Improve Quantification of Glycosaminoglycan (GAG) Content by Contrast Enhanced CT Imaging of Cartilage. J Orthop Res 2011, 29 (5), 704–709. [DOI] [PubMed] [Google Scholar]
- (42).Piscaer TM; Waarsing JH; Kops N; Pavljasevic P; Verhaar JAN; van Osch GJVM; Weinans H In Vivo Imaging of Cartilage Degeneration Using microCT-Arthrography. Osteoarthritis Cartilage 2008, 16 (9), 1011–1017. [DOI] [PubMed] [Google Scholar]
- (43).Cockman MD; Blanton CA; Chmielewski PA; Dong L; Dufresne TE; Hookfin EB; Karb MJ; Liu S; Wehmeyer KR Quantitative Imaging of Proteoglycan in Cartilage Using a Gadolinium Probe and microCT. Osteoarthritis Cartilage 2006, 14 (3), 210–214. [DOI] [PubMed] [Google Scholar]
- (44).Xie L; Lin ASP; Guldberg RE; Levenston ME Nondestructive Assessment of sGAG Content and Distribution in Normal and Degraded Rat Articular Cartilage via EPIC-microCT. Osteoarthritis Cartilage 2010, 18 (1), 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Joshi NS; Bansal PN; Stewart RC; Snyder BD; Grinstaff MW Effect of Contrast Agent Charge on Visualization of Articular Cartilage Using Computed Tomography: Exploiting Electrostatic Interactions for Improved Sensitivity. J. Am. Chem. Soc 2009, 131 (37), 13234–13235. [DOI] [PubMed] [Google Scholar]
- (46).Entezari V; Bansal PN; Stewart RC; Lakin BA; Grinstaff MW; Snyder BD Effect of Mechanical Convection on the Partitioning of an Anionic Iodinated Contrast Agent in Intact Patellar Cartilage. J. Orthop. Res 2014, 32 (10), 1333–1340. [DOI] [PubMed] [Google Scholar]
- (47).Yoo HJ; Hong SH; Choi J-Y; Lee IJ; Kim SJ; Choi J-A; Kang HS Contrast-Enhanced CT of Articular Cartilage: Experimental Study for Quantification of Glycosaminoglycan Content in Articular Cartilage. Radiology 2011, 261 (3), 805–812. [DOI] [PubMed] [Google Scholar]
- (48).Bansal PN; Stewart RC; Entezari V; Snyder BD; Grinstaff MW Contrast Agent Electrostatic Attraction rather than Repulsion to Glycosaminoglycans Affords a Greater Contrast Uptake Ratio and Improved Quantitative CT Imaging in Cartilage. Osteoarthr. Cartil 2011, 19 (8), 970–976. [DOI] [PubMed] [Google Scholar]
- (49).Stewart RC; Bansal PN; Entezari V; Lusic H; Nazarian RM; Snyder BD; Grinstaff MW Contrast-Enhanced CT with a High-Affinity Cationic Contrast Agent for Imaging Ex Vivo Bovine, Intact Ex Vivo Rabbit, and in Vivo Rabbit Cartilage. Radiology 2013, 266 (1), 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lakin BA; Grasso DJ; Shah SS; Stewart RC; Bansal PN; Freedman JD; Grinstaff MW; Snyder BD Cationic Agent Contrast-Enhacned Computed Tomography Imaging of Cartilge Correlates with the Compressive Modulus and Coefficient of Friction. Osteoarthr. Cartil 2013, 21, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Fechner LE; Albanyan B; Vieira VMP; Laurini E; Posocco P; Pricl S; Smith DK Electrostatic Binding of Polyanions Using Self-Assembled Multivalent (SAMul) Ligand Displays – Structure–activity Effects on DNA/heparin Binding. Chem. Sci 2016, 7 (7), 4653–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Appel EA; Tibbitt MW; Greer JM; Fenton OS; Kreuels K; Anderson DG; Langer R Exploiting Electrostatic Interactions in Polymer–Nanoparticle Hydrogels. ACS Macro Lett. 2015, 4 (8), 848–852. [DOI] [PubMed] [Google Scholar]
- (53).Hunt JN; Feldman KE; Lynd NA; Deek J; Campos LM; Spruell JM; Hernandez BM; Kramer EJ; Hawker CJ Tunable, High Modulus Hydrogels Driven by Ionic Coacervation. Adv. Mater 2011, 23 (20), 2327–2331. [DOI] [PubMed] [Google Scholar]
- (54).Lin X; Navailles L; Nallet F; Grinstaff MW Influence of Phosphonium Alkyl Substituents on the Rheological and Thermal Properties of Phosphonium-PAA-Based Supramolecular Polymeric Assemblies. Macromolecules 2012, 45 (23), 9500–9506. [Google Scholar]
- (55).Wang Q; Mynar JL; Yoshida M; Lee E; Lee M; Okuro K; Kinbara K; Aida T High-Water-Content Mouldable Hydrogels by Mixing Clay and a Dendritic Molecular Binder. Nature 2010, 463, 339–343. [DOI] [PubMed] [Google Scholar]
- (56).Wathier M; Lakin BA; Bansal PN; Stoddart SS; Snyder BD; Grinstaff MW A Large-Molecular-Weight Polyanion, Synthesized via Ring-Opening Metathesis Polymerization, as a Lubricant for Human Articular Cartilage. J. Am. Chem. Soc 2013, 135 (13), 4930–4933. [DOI] [PubMed] [Google Scholar]
- (57).Taylor SD; Tsiridis E; Ingham E; Jin Z; Fisher J; Williams S Comparison of Human and Animal Femoral Head Chondral Properties and Geometries. Proc. Inst. Mech. Eng. H 2012, 226 (1), 55–62. [DOI] [PubMed] [Google Scholar]
- (58).Athanasiou KA; Rosenwasser MP; Buckwalter JA; Malinin TI; Mow VC Interspecies Comparisons of in Situ Intrinsic Mechanical Properties of Distal Femoral Cartilage. J. Orthop. Res. Off. Publ. Orthop. Res. Soc 1991, 9 (3), 330–340. [DOI] [PubMed] [Google Scholar]
- (59).Athanasiou KA; Agarwal A; Muffoletto A; Dzida FJ; Constantinides G; Clem M Biomechanical Properties of Hip Cartilage in Experimental Animal Models. Clin. Orthop. Relat. Res 1995, 316, 254–266. [PubMed] [Google Scholar]
- (60).Kamel IR; Merkle EM Body MR Imaging at 3 Tesla; Cambridge University Press, 2011. [Google Scholar]
- (61).Krishnamurthy R; Pednekar A; Kouwenhoven M; Cheong B; Muthupillai R Evaluation of a Subject Specific Dual-Transmit Approach for Improving B1 Field Homogeneity in Cardiovascular Magnetic Resonance at 3T. J. Cardiovasc. Magn. Reson. Off. J. Soc. Cardiovasc. Magn. Reson 2013, 15 (1), 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Stewart RC; Honkanen JTJ; Kokkonen HT; Tiitu V; Saarakkala S; Joukainen A; Snyder BD; Jurvelin JS; Grinstaff MW; Töyräs J Contrast-Enhanced Computed Tomography Enables Quantitative Evaluation of Tissue Properties at Intrajoint Regions in Cadaveric Knee Cartilage. Cartilage 2016, doi: 10.1177/1947603516665443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Pollard TD A Guide to Simple and Informative Binding Assays. Mol. Biol. Cell 2010, 21 (23), 4061–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).de Jong LAA; Uges DRA; Franke JP; Bischoff R Receptor–ligand Binding Assays: Technologies and Applications. J. Chromatogr. B 2005, 829 (1–2), 1–25. [DOI] [PubMed] [Google Scholar]
- (65).Hulme EC; Trevethick MA Ligand Binding Assays at Equilibrium: Validation and Interpretation. Br. J. Pharmacol 2010, 161 (6), 1219–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Donnan FG The Theory of Membrane Equilibria. Chem. Rev 1924, 1, 73–90. [Google Scholar]
- (67).Byun S; Tortorella MD; Malfait AM; Fok K; Frank EH; Grodzinsky AJ Transport and Equilibrium Uptake of a Peptide Inhibitor of PACE4 into Articular Cartilage Is Dominated by Electrostatic Interactions. Arch Biochem Biophys 2010, 499 (1–2), 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Schmidt TA; Gastelum NS; Nguyen QT; Schumacher BL; Sah RL Boundary Lubrication of Articular Cartilage: Role of Synovial Fluid Constituents. Arthritis Rheum. 2007, 56 (3), 882–891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.