

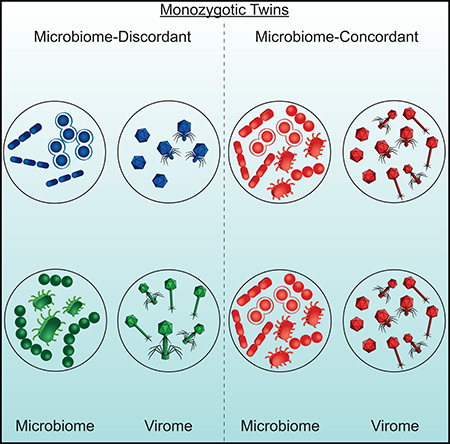
SUMMARY
The virome is one of the most variable components of the human gut microbiome. Within twin-pairs, viromes have been shown to be similar for infants but not for adults, indicating that as twins age and their environments and microbiomes diverge, so do their viromes. The degree to which the microbiome drives the vast virome diversity is unclear. Here, we examine the relationship between microbiome and virome diversity in 21 adult monozygotic twin pairs selected for high or low microbiome concordance. Viromes derived from virus-like particles are unique to each individual, are dominated by Caudovirales and Microviridae, and exhibit a small core that includes crAssphage. Microbiome-discordant twins display more dissimilar viromes compared to microbiome-concordant twins, and the richer the microbiomes, the richer the viromes. These patterns are driven by bacteriophages, not eukaryotic viruses. Collectively, these observations support a strong role of the microbiome in patterning for the virome.
eTOC blurb:
The virome remains a relatively unexplored component of the microbiome. Moreno-Gallego and Chou et al., examined the viromes of monozygotic twins to ask how microbiome diversity relates to virome diversity, without host genetic variables. Twin pairs were sorted by high or low microbiome concordance, which revealed a correlated virome relatedness.
GRAPHICAL ABSTRACT
INTRODUCTION
The human gut microbiome is composed of a vast diversity of bacterial cells, along with a minority of archaeal and eukaryotic cells, together forming a very dense microbial ecosystem (1011 −1012 cells per gram of feces) (Sender et al., 2016). The cells of the microbiome and the constituents of the virome (between 109 to 1012 virus-like particles (VPLs) per gram of feces) are in about equal proportion (Castro-Mejía et al., 2015; Hoyles et al., 2014; Ogilvie and Jones, 2017; Reyes et al., 2010). The virome is primarily composed of bacteriophages and prophages, and it also includes rarer eukaryotic viruses and endogenous retroviruses (Breitbart et al., 2003; Minot et al., 2011; Reyes et al., 2010). Currently, the majority of phages have no matches in databases and their hosts remain to be elucidated. Matching phages to their hosts is challenging: for instance, the host of the most common human gut phage, crAssphage, has only recently been identified as Bacteroides spp. (Shkoporov et al., 2018; Yutin et al., 2018). In addition to the identification of hosts, other questions remain as to the factors most important in shaping the virome, and how predictive the cellular fraction of the microbiome can be of the virome.
The temporal population dynamics of phages and their hosts might be expected to be linked. Indeed, population oscillations of viruses and their bacterial hosts are described for aquatic systems, where they indicate that viruses play a key role in regulating bacterial populations (Suttle, 2007; Thingstad, 2000; Thingstad et al., 2014; Weitz and Dushoff, 2008). But such patterns of predator/prey dynamics are not typical for the human gut virome and microbiome (Minot et al., 2011; Reyes et al., 2013; Rodriguez-Brito et al., 2010; Rodriguez-Valera et al., 2009). (For clarity, from here on we use ‘microbiome’ to refer to cellular fraction of the microbiome, e.g., mostly bacterial cells.) Nonetheless, the virome and microbiome do display some common patterns of diversity across hosts, such as high levels of interpersonal differences and relative stability over time (Reyes et al., 2010). The microbiome tends to be more similar for related individuals compared to unrelated individuals, possibly due to shared dietary habits, which drive similarity between microbiomes (Cotillard et al., 2013; David et al., 2014). In accord, diet has been associated with virome diversity, quite possibly through diet effects on the microbiome (Minot et al., 2011). In infants, twin comparisons have revealed viromes to be more similar between co-twins than between unrelated individuals (Lim et al., 2015; Reyes et al., 2015). This pattern was not observed in adult twins (Reyes et al., 2010), possibly due to divergence of their microbiomes. The degree to which the microbiome itself drives patterns of virome diversity across hosts has been difficult to assess due to confounding factors such as host relatedness.
Here, we focus on adult monozygotic (MZ) twin gut microbiomes to explore further the relationship between microbiome and virome diversity. By studying the viromes of MZ twin pairs, we control for host genetic relatedness. Although MZ twin pairs generally have more similar microbiomes compared to dizygotic (DZ) twin pairs or unrelated individuals, MZ twins nevertheless can display a large range of within-twin-pair microbiome diversity (Goodrich et al., 2014). We previously generated fecal microbiome data for twin pairs from the TwinsUK cohort (Goodrich et al., 2014), and based on this information we selected twin pairs either highly concordant or highly discordant for their microbiomes. We generated viromes from virus-like particles obtained from the same samples from which the microbiomes were derived. Results indicate that microbiome diversity and virome diversity measures are positively associated.
RESULTS
Selection of microbiome-concordant and discordant monozygotic twin pairs –
We selected twin pairs with a similar body mass index (BMI), whose microbiomes were either concordant or discordant for microbiome between-sample diversity (β-diversity) based on previously obtained 16S rRNA gene data. The adult co-twins in this study did not share a household and we assume that other environmental variability was similar across twin pairs. We determined the degree of concordance or discordance between co-twins’ microbiomes based on three β-diversity distance metrics: Bray-Curtis, weighted UniFrac and unweighted UniFrac (See STAR Methods). As expected, the β-diversity measures were correlated (Pearson pairwise correlation coefficient > 0.4). Based on the distribution of pairwise distance measures, we selected 21 MZ twin pairs from the boundaries of all three distributions (Figure 1A), while maintaining a balanced distribution of age and BMI across the set (Table S1). Within the 21 selected twin pairs, the microbiomes of microbiome-concordant co-twins were, as expected, more similar to each other than microbiomes of microbiome-discordant co-twins (p = 6.31×10−12). The microbiomes of the discordant co-twins differed compositionally at all taxonomic levels, particularly at the phylum level, with Firmicutes and Bacteroidetes, the two dominant phyla, contributing the most to the variation between co-twins (Figure 1B and 1C).
Figure 1. Microbiome discordance in twin pairs.

(A) The β-diversity measures of the microbiotas of 354 monozygotic twin pairs from a previous study (Goodrich et al., 2014) are shown. Each dot represents the β-diversity of a pair of twins, measured by the weighted UniFrac (x-axis), unweighted UniFrac (z-axis), and Bray-Curtis (y-axis) β-diversity metrics. The plane is the least squared fitted plane Bray-Curtis ~ Weighted UniFrac + Unweighted UniFrac. A subset of twin pairs with concordant microbiotas (blue) and discordant microbiotas (orange) were chosen from the two edges. Black dots indicate the samples used for virome and whole fecal metagenome comparison. (B) Comparison of the taxonomic profiles (relative abundance) at the Phylum level for the 21 MZ twin pairs concordant (1–9) or discordant (10–21) for their microbiotas. (C) Differences in the relative abundances for the major phyla for concordant (blue points, n=9) and discordant (orange points, n=12) twin pairs. Mann-Whitney’s U test. *** p < 0.0005, * p = 0.055.
Shotgun metagenomes of VLPs –
We isolated VLPs from the same fecal samples that had been used for 16S rRNA gene diversity profiling (See STAR Methods). DNA extracted from VLPs was used in whole genome amplification followed by shotgun metagenome sequencing (See STAR Methods). A first library (“large-insert-size library”) was selected with an average insert size of 500 bp (34,325,116 paired reads in total; 817,265 ± 249,550 paired reads per sample after quality control) and used for de novo assembly of viral contigs. Smaller fragments with an average insert size of 300bp were purified in a second library (“small-insert-size library”) and sequenced. The resulting pair-end reads were merged into 25,324,163 quality filtered longer reads to increase mapping accuracy (602,956 ± 595,444 merged reads per sample) (See STAR Methods) (Table S2).
Identification of putative bacterial contaminants –
Viromes prepared and sequenced from VLPs may be contaminated with bacterial DNA (Roux et al., 2013). However, given that phages are major agents of horizontal gene transfer and that temperate viruses often comprise up to 10% of bacterial genomes in a prophage state, removal of potential bacterial contamination risks also removing viral reads. To assess bacterial DNA contamination, we mapped virome reads against a set of 8,163 fully assembled bacterial genomes. Our strategy consisted of evaluating the coverage along the length of each genome (in bins of 100Kb), and those genomes with a median coverage greater than 100 were considered contaminants. Reads mapping to short regions were considered to be prophages or horizontally transferred genes and retained (See STAR Methods) (Figure 2A). Reads mapping to genomes determined to be potential contaminants were removed from further analyses.
Figure 2. Bacterial contamination in VLP preparations.

(A) Heatmaps of VLP reads from a single sample (4A) mapping to bacterial genomes before (upper) and after (lower) the removal of reads determined as contaminants. Genomes are sorted by length and split in bins of 100,000 bp. Bacterial genomes with a median coverage greater than 100 were considered as contaminants. (B) Cladogram based on the NCBI taxonomy of the 65 genomes identified as contaminants across all VLP extractions. (Right) Spearman rank correlation coefficient (rho) between the abundance of the bacterial genomes in the VLP extractions and 16S rRNA gene profile from the microbiome. (Left) Total abundance of each bacterial genome added across all individuals.
We identified 65 bacterial genomes as potential contaminants, with 1% ± 1.125 (average ± std) of reads per sample mapping to those bacterial genomes (Table S2). The majority (37/68) belonged to the Firmicutes phylum; at the species level, Bacteroides dorei, B. vulgatus, Ruminococcus bromii, Faecalibacterium prausnitzii, B. xylanisolvens, Odoribacter splanchnicus and B. caecimuris (in that order) were detectable in at least 50% of the samples (Table S2). If the most abundant bacterial species in the microbiome are the most likely sources of contamination, then their relative abundance as contaminants should correspond to their relative abundances in the microbiome. However, we observed no significant correlation between the relative abundances of taxa represented in the contaminant DNA and in the microbiomes (Figure 2B).
Functional profiles support viral enrichment in VLP purifications –
To assess the functional content of the viromes, we annotated the “short-insert-size library” raw reads using the KEGG annotation of the Integrated Gene Catalog (IGC) (Li et al., 2014) (See STAR Methods). In line with previous reports (Breitbart et al., 2008; Minot et al., 2011; Reyes et al., 2010), the majority of reads (85.43 ± 5.74%) from our VLP metagenomes mapped to genes with unknown function (Figure 3A).
Figure 3. Comparison of the gene content of whole fecal metagenomes and viromes.

(A) The relative abundance of KEGG categories in whole fecal metagenomes and viromes, including all hits to IGC genes, regardless of the annotation. (B) Heatmap of the relative abundance of the second level of KEGG categories in whole fecal metagenomes and viromes, excluding the IGC genes with unknown annotation. A.V.: Additional viromes; A.M.: Additional microbiomes (whole genome extractions). Intra-class coefficient (ICC) for A.M. = 0.99; ICC for A.V. = 0.85; ICC concordant-microbiome co-twins = 0.69; ICC discordant-microbiome cotwins = 0.68.
To further verify that sequences were derived from VLPs and not microbiomes generally, we conducted an internal check in which we generated and compared additional metagenomes from VLPs and bulk fecal DNA for an additional 4 individuals (2 twin pairs; Figure 1A). As expected, the functional profiles of viromes and microbiome-metagenomes derived from the same samples were dissimilar. Virome reads that mapped to annotated genes were enriched in two categories: Genetic Information Process (48.87% ± 12.12) and Nucleotide Metabolism (17.59% ± 8.81), compared to 24.31% ± 1.28 and 5.47% ± 0.4 for the microbiome-metagenome, respectively (Figure 3B). Most of the other functional categories present in the bacterial metagenomes were essentially absent from the viromes. Furthermore, the functional annotations of the viromes showed greater between-sample variability than the microbiomes and a lower intraclass correlation coefficient (Figure 3B).
Viromes are unique to individuals –
We assembled reads from the “large-insert-size library” resulting in a total of 107,307 contigs ≥ 500 nt (max: 79,863 nt; mean 1,186nt ± 1,741; Figure S1). To assess the structure and composition of the viromes, a matrix of the recruitment of reads against dereplicated contigs was built (See STAR Methods). The recruitment matrix included 14,584 contigs that were both long (> 1,300 nt) and well covered (> 5X); these are referred to as ‘virotypes’ (Figure S1). Analysis of the recruitment matrix showed that each individual harbored a unique set of virotypes: 3,415 virotypes (23.41% of total) were present in only one individual; 413 virotypes (2.83%) were present in at least 50% of the individuals; only 18 virotypes (0.1%) were present in all individuals.
Twins with concordant microbiomes share virotypes –
We checked for virotypes shared between twins and observed that co-twins did not share more virotypes than unrelated individuals (p = 0.074). We then assessed microbiome-concordant and discordant twin pairs separately: twins with a discordant microbiome did not share more virotypes that unrelated individuals (p = 0.254), whereas twins with a concordant microbiome did share more virotypes than unrelated individuals (p = 0.048). Furthermore, we also found that twins with a concordant microbiome shared more virotypes than twins with a discordant microbiome (p = 0.015; Figure S2).
Bacteriophage dominance of the gut virome –
In order to characterize the taxonomic composition of the virome, we attempted to annotated all 66,446 dereplicated and well covered contigs (Figure S1) using a voting system approach that exploited the information in both the assembled contigs and their encoding proteins (See STAR Methods). In addition, we performed a custom annotation on two highly abundant gut-associated bacteriophage families: (i) the crAssphage (Dutilh et al. 2014; Yuting et al. 2018) and (ii) the Microviridae families (Székely and Breitbart 2016). For this, we used profile Hidden Markov Models (HMMs) to search for crAssphage (dsDNA viruses) and Microviridae (ssDNA viruses) contigs (See STAR Methods).
Using HMMs allowed us to identify distant homologs, which we then incorporated into a phylogenetic tree with known reference sequences to confirm the annotation and better resolve the taxonomy. We annotated 108 contigs (19 crAssphage, 90 Microviridae), validated the family assignment of 68 contigs, and assigned a subfamily to 97 contigs without previous subfamily assignment. For the Microviridae, only 11 contigs had a previous taxonomic assignment, all belonging to the Gokushovirinae: we confirmed these and 23 more as Gokushovirinae, 54 as Alpavirinae and 1 contig as Pichovirinae (Figure S3A). For the crAssphage, 11 contigs were clustered with the original crAssphage, 3 contigs grouped with the reference Chlamydia phage, and 5 contigs grouped with the reference IAS virus (Figure S3B).
After collating the voting system annotation and the HMM annotation, a total of 12,751 contigs (29,62%) were taxonomically assigned (Figure S1). Viromes were dominated by bacteriophages with only 6.42% of contigs annotated as Eukaryotic viruses. As expected, most of the contigs (96.98%) were dsDNA viruses, while only 2.43% of contigs were annotated as ssDNA viruses. Caudovirales was the most abundant Order, with its three main families represented: Myoviridae (20.22% ± 4.83), Podoviridae (10.54% ± 3.27), and Siphoviridae (35.25% ± 7.19). The crAssphage family constituted on average 13.26% (± 12.24%) of the contigs, reaching a maximum contribution of 55.80% in one virome, and Microviridae represented 3.87% ± 2.57 of the viromes. Interestingly, we observed that Phycodnaviridae exceeded 1% of average abundance (1.77% ± 1.12; Figure 4A) and that contigs related to any nucleocytoplasmic large DNA viruses (NCLDV) had a mean relative contribution of 3.99% ± 2.22. The 18 contigs present in all samples included 10 annotated as crAssphage, 2 annotated as “unclassified Myoviridae”, 2 “unclassified Caudovirales”, 1 classified as Microviridae, and 3 unclassified. Within a defined taxonomic profile for each sample, we looked for differences in composition between viromes at all taxonomic levels for concordant and discordant twin-pairs. There were no significant differences between groups for any taxa at the Order and Family levels, including crAssphage and Microviridae families (Figure 4B).
Figure 4. Virome composition.

Comparison of the taxonomic profiles at the Family level for the 21 MZ twin pairs concordant (1–9) or discordant (10–21) for their microbiomes. (A) The viral family composition of the MZ twins. (B) Differences of the relative abundances of each family for concordant (blue points, n=9) and discordant (orange points, n=12) twin pairs.
We used CRISPR spacer mapping and the microbe-versus-phage (MVP) database (Gao et al., 2018) to predict hosts for virotypes and taxonomically characterized contigs (See STAR Methods). As host annotation was directed to bacteriophages, we did not gain any information for contigs annotated as Eukaryotic viruses. These approaches allowed us to identify putative hosts for 910 contigs. Within these 910 contigs, only one was previously annotated as crAssphage, and as expected, its host was inferred to be a member of Bacteroidetes. In total we identified 1,280 bacterial putative host strains, including 187 species from 87 genera over several phyla; most of them from Firmicutes (92), followed by Bacteroidetes (41) and Proteobacteria (38). The median number of host for each contig was 1 (IQR=1–2), while the median number of phages per host, at the strain level, was 2 (IQR=1–3) (Figure S4).
Virome diversity correlates with microbiome diversity –
To assess the relationship between virome and microbiome diversity, we examined the within-samples diversity (α-diversity) and β-diversity of the viromes using three different layers of information that we recovered from the sequence data: i) virotypes, ii) taxonomically annotated contigs, and iii) annotated genes from short reads (Figure S1).
Alpha-diversity –
α-diversities of the microbiome and the virome were positively correlated in two of the three layers of information used to test the correlation (virotypes and taxonomy annotated contigs but not genes; Figure 5A). We used annotated contigs to ask about the α-diversity within subgroups of viruses: (ssDNA eukaryotic, dsDNA eukaryotic, ssDNA bacteria and dsDNA bacteria). Our results show that the diversity of eukaryotic viruses does not correlate with the microbiome α-diversity. In contrast, bacteriophages and microbiome α-diversity were positively correlated, for both ssDNA or dsDNA bacterial viruses (Figure 5B).
Figure 5. Bacteriophages diversity correlates with microbiome diversity but eukaryotic viruses diversity does not.

(A) Correlation of Shannon α-diversity of viromes to Shannon α-diversity of microbiomes (n=42). Virotypes: Pearson correlation coefficient = 0.406, m = 0.3, p = 0.007, R2 = 0.165; Taxonomy: Pearson correlation coefficient = 0.389, m = 0.25, p = 0.010, R2 = 0.151; Genes: Pearson correlation coefficient = 0.105, m = 0.11, p = 0.506, R2 = 0.011 (B) Correlation of the Shannon α-diversity of the virome, calculated from contigs annotated as ssDNA eukaryotic viruses, ssDNA phages, dsDNA eukaryotic viruses, and dsDNA phages, to Shannon α-diversity of the microbiome (n=42). ssDNA eukaryotic viruses: Pearson correlation coefficient = 0.027, m = 0.034, p = 0.863, R2 = 0.000751; ssDNA bacteriophages: Pearson correlation coefficient = 0.394, m = 0.35, p = 0.009, R2 = 0.155; dsDNA eukaryotic viruses: Pearson correlation coefficient = 0.143, m = 0.15, p = 0.368, R2 = 0.020; dsDNA bacteriophages: Pearson correlation coefficient = 0.400, m = 0.25, p = 0.008, R2 = 0.16.
Beta-diversity –
We observed that concordant twins had lower virome βdiversity compared to discordant twins using Hellinger distances (Figure 6); the mean binary Jaccard distance and Bray-Curtis dissimilarity of viromes also showed the same trend (Figure S5A and S5B). Similar to what we observed with α-diversity, regardless of the layer of information used, the mean Hellinger distance of viromes within MZ twin pairs with concordant microbiomes was significantly lower than that of MZ twin pairs with discordant microbiomes (p < 0.04, Mann-Whitney’s U test) (Figure 6). We did not observe significant differences in β-diversity when concordant twins or discordant twins were split by sex (p > 0.05, Mann-Whitney’s U test). Still, any inference about sex influence is limited as the number of individuals per group is halved. Furthermore, a similar significant positive correlation was observed between microbiome and virome β-diversity when using the annotated contigs. This relationship was driven by the bacteriophages (p = 0.009, Mann-Whitney’s U test), but not the eukaryotic viruses (p = 0.243, Mann-Whitney’s U test).
Figure 6. Virome Beta-diversity patterns mirror microbiome Beta-diversity.

Box plots show the distribution of Hellinger distances for microbiomes and viromes, according to the three different layers of information recovered (virotypes, genes, and taxonomy), for concordant co-twins (solid blue, n=9), discordant co-twins (solid orange, n=12), unrelated samples within the concordant co-twins (blue outline, n=144), and unrelated samples within the discordant co-twins (orange outline, n=264). Significant differences between means (Mann-Whitney’s U test, p < 0.020) are denoted with different letters.
Finally, we compared the virome and microbiome pairwise distances among related (co-twins) and unrelated individuals. The pairwise distance matrices showed a positive correlation between virome and microbiome β-diversity measures not only within twin pairs (Pearson correlation coefficient > 0.50) but also generally across all individuals (Pearson correlation coefficient > 0.25; p < 0.003, Mantel test; Figure S5C). These results show that regardless of genetic relatedness between hosts, individuals with more similar microbiomes harbour more similar viromes.
DISCUSSION
Co-twins, like other siblings, generally have more similar gut microbiomes within their twinships compared to unrelated individuals (Lee et al., 2011; Palmer et al., 2007; Tims et al., 2013; Turnbaugh et al., 2009; Yatsunenko et al., 2012). Moreover, MZ twins have overall more similar microbiomes than DZ twins, although at a whole-microbiome level this effect is small and primarily driven by a small set of heritable microbiota (Goodrich et al., 2014, 2016). Within a population of MZ twin pairs, however, the range of within-twin pair differences in the microbiomes can be as great as for DZ twins (Goodrich et al., 2014). We took advantage of the large spread in β-diversity for MZ co-twins to select co-twins that were either highly concordant or discordant for their gut microbiomes. Our analysis of their viromes showed that despite the high variation in the gut viromes between individuals, and regardless of host relatedness, the more dissimilar their microbiomes, the more dissimilar their viromes. This pattern was driven by the bacteriophage component of the virome.
By choosing MZ twins from a distribution of β-diversities in the microbiomes, we removed host genetic relatedness as a variable possibly impacting the virome. Previous studies of the viromes and microbiomes of infant twin pairs showed that the microbiomes and viromes of co-twins were more similar than those of unrelated individuals, suggesting shared host genotype and/or environment were key (Lim et al., 2015; Reyes et al., 2015). In contrast, an early study of the virome of adult twins showed that adult co-twins did not have more similar viromes than unrelated individuals (Reyes et al., 2010); however, in light of the current study’s results, this was likely a power issue. Indeed, in our dataset we observed that regardless of whether twins were concordant or discordant for their microbiomes, co-twins had more similar viromes (virotypes and taxonomy) than unrelated individuals.
The previously reported greater virome similarity in young compared to adult twins has been related to the fact that infants have a greater shared environment compared to adult twins (Lim et al., 2015), particularly in terms of their diet. Minot et al., have also shown that individuals on the same diet have more similar gut viromes than individuals on dissimilar diets (Minot et al., 2011). It is well established that diet is a strong driver of daily microbiome fluctuation (Claesson et al., 2012; David et al., 2014; De Filippo et al., 2010; Wu et al., 2011), so the effect of diet on the virome is likely mediated by the microbiome. However, we did not control for diet, so it is possible that the microbiome discordance that we observe was caused by co-twins eating differently around the time of sampling. Regardless of what underlies the variance in microbiome concordance, it is strongly associated with virome concordance.
The relationship between virome richness and microbiome richness had not previously been directly addressed in adults. We observed that the α-diversity of the microbiome and the virome were positively correlated using two of the three layers of information describing virome diversity. Specifically, this pattern was observed for virotypes and taxonomy but not for genes. However, since virome genes were observed to be enriched in only two categories, Genetic Information Processing and Nucleotide Metabolism, we would not expect differences in diversity of virome genes between subjects. The taxonomic annotation layer showed that the bacteriophage component of the virome, not the eukaryotic viruses, was driving this α-diversity correlation pattern.
The positive relationship between virome and microbiome α-diversity suggests that a greater availability of hosts drives a greater diversity of viruses. These observations are in accordance with “piggyback the winner” model, which posits that in a dense environment, phages opt for a lysogenic cycle and multiply with their hosts (Knowles et al. 2016). Indeed, longitudinal studies of the human gut virome have reported genes associated with lysogeny, low mutation rate over time in temperate-like contigs, and long-term stability of the virome, suggesting preference for a lysogenic cycle (Minot et al., 2013; Reyes et al., 2010). Nevertheless, phage predation has been acknowledged as an important factor for the maintenance of highly diverse and efficient ecosystems (Rodriguez-Valera et al., 2009) and may play a role in the maintenance of diversity in a rapidly changing ecosystem as the human gut (David et al., 2014). Short scale time-series analyses of virome-microbiome interactions, along with a better understanding of the lysogenic-lytic switch in viral reproduction, would help to interpret the observed patterns in the human gut virome.
The composition of the viromes described here was similar to what has been previously reported for adult fecal viromes (Minot et al., 2011, 2013; Reyes et al., 2010). From the annotated fraction of the virome, the order Caudovirales and its families Siphoviridae, Myoviridae, and Podoviridae, along with crAssphage, were the dominant phages in all samples. Manrique et al. have summarized the phage colonization of the infant gut as follows: the eukaryotic viruses first dominate the newborn gut, followed by the Caudovirales, and by 2.5 years of age the Microviridae start to dominate (Manrique et al., 2017). We did observe abundant Microviridae in our sample set, but the Caudovirales were the dominant group. Age was not related to patterns of diversity in the set of adult subjects studied here.
Despite the high diversity and uniqueness of each virome described here, we nonetheless recovered a core virome among the subjects: 18 contigs were present in all samples. More than half of these contigs were annotated as crAssphage, consistent with recent reports that this phage is widespread (Dutilh et al., 2014; Manrique et al., 2016; Yarygin et al., 2017). Other shared virotypes in our dataset were classified as Myoviridae and Microviridae. We also recovered contigs mapping to representative families of the nucleocytoplasmic large DNA viruses (NCLDV), Phycodnaviridae and Mimiviridae. These types of viruses are increasingly reported as members of the human gut virome (Colson et al., 2013; Halary et al., 2016). A core set of bacteriophages consisting of nine representatives, including crAssphage, has previously been reported for the human gut (Manrique et al., 2016). Widely shared virotypes may indicate the wide sharing of specific hosts between individuals, or that these viruses have a broad host range within the human microbiome.
Our use of the HMMs to annotate viral contigs allowed a deep exploration into the taxonomic content of the virome. We annotated a diversity of contigs beyond what was revealed from comparisons to public databases, and also confirmed those annotations. Because each type of virus (e.g., family) requires its own HMM, we applied this method to a few key groups. When applied to the crAssphage, the HMM retrieved contigs that grouped only with sequences derived from fecal viromes and not with sequences from other environments (e.g., terrestrial or marine). This suggests that although crAssphage is a diverse group of bacteriophages, its diversity in the human gut is restricted to sequences related to the reference crAssphage genome (Dutilh et al., 2014), the IAS virus reference (Shkoporov et al., 2018), or Chlamydia bacteriophage (Yutin et al., 2018). We also applied HHM to the family Microviridae, which are single strand DNA bacteriophages. We were able to confirm the presence of diverse members of Gokushovirinae and Alpavirinae subfamilies. Although there is evidence that described Alpavirinae genomes constitute a third group of the Microviridae family (Krupovic and Forterre 2011; Roux et al. 2012), they correspond to prophages, which makes it difficult to integrate them into the taxonomy of the International Committee on Taxonomy of Viruses (ICTV), thus, no contigs were annotated as Alpavirinae prior to application of the HMM profiles.
For each taxonomic group of viruses, there is a corresponding set of bacterial hosts. From the 16S rRNA gene diversity data we used to select the twin pairs, it is clear which bacteria phyla contribute the most to the differences in the microbiomes of concordant and discordant twins. But unlike for bacterial, we were not able to discern such clear patterns by order or family in the virome. Indeed, most of the bacteriophage diversity is grouped in just one order, Caudovirales, and its three families Myoviridae, Podoviridae and Siphoviridae. Representatives of these families can infect unrelated hosts (Barylski et al., 2017). Thus, we wouldn’t necessarily expect specific orders or families of viruses to show the patterns observed in the bacterial phyla.
Finally, we noted an interesting pattern of complete bacterial genome coverage for select bacterial species. As these putative contaminants were not the most abundant members of the microbiome, they are unlikely to represent random contamination of bulk DNA. Why certain bacterial genomes showed such high coverage is unclear. One possibility is that we are observing the host species range of transposable phages. Phages such as the Mu phage randomly integrate into the host genome (Taylor, 1963), amplify by successive rounds of replicative transposition, and then can package any section of their host’s genome (Hulo et al., 2011; Toussaint and Rice, 2017). Intriguingly, several of the contaminants detected here (e.g., B. vulgatus, B. dorei, F. prausnitzii and B. thetaiotaomicron) have also been reported as contaminants in other human gut virome studies (Minot et al., 2011; Roux et al., 2013), which could indicate host-specificity of Mu phages. Alternative explanations include vesicle production, gene transfer agents and/or generalized transduction processes (Biller et al., 2014; McDaniel et al., 2010; Minot et al., 2011). Further comparisons of whole bacterial genomes recovered in diverse virome datasets may help shed light on their source, particularly if the same bacterial species are recovered across multiple studies.
Prospectus –
Our results show that gut microbiome richness and diversity correlate to virome richness and diversity, and vice-versa. The mechanics underlying this association remain to be resolved for the human gut. This relationship may be useful to take into consideration when designing future studies of the virome and the factors that affect it. Baseline microbiome diversity may be important to balance between groups, for instance, prior to assessing the diversity of the virome.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ruth Ley (ruth.ley@tuebingen.mpg.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fecal Samples –
Fecal samples used in this study were obtained as part of previous studies (Goodrich et al., 2014; Jackson et al., 2016). From 16S rRNA gene diversity previously measured for 354 monozygotic twin pairs whose fecal samples were received between January 28th 2013 and July 14th 2014 (Goodrich et al., 2014), we selected 11 concordant and 13 discordant MZ co-twins based on three microbiota β-diversity distances within twin pairs: unweighted UniFrac, weighted UniFrac (Lozupone et al., 2007) and Bray-Curtis (Bray and Curtis, 1957). Twins pairs in the concordant and the discordant groups were selected to be balanced between those two groups for sex, age, BMI, and BMI difference within a twin pair (Table S1). Twins within the concordant group ranged in age from 23 to 77 years old and included 5 men and 4 women, while those in the discordant group ranged in age from 29 to 81 years old with 5 men and 7 women. All work involving the use of these previously collected samples was approved by the Cornell University IRB (Protocol ID 1108002388).
METHOD DETAILS
Isolation of virus-like particles (VLPs) from human fecal samples –
VLP isolation procedures were based on the previously described protocols (Gudenkauf et al., 2014; Minot et al., 2013). For VLP isolation, ~0.5 g of fecal sample was resuspended by vortexing for 5–10 minutes in 15 ml PBS, previously filtered through 0.02 μm filter (Whatman). The homogenates were centrifuged for 30 min at 4,500 xg, and the supernatant was filtered through 0.22 μm polyethersulfone (PES) Express Plus Millipore Stericup (150 ml) to remove cell debris and bacterial-sized particles. The filtrate was then concentrated on a Millipore Amicon Ultra-15 Centrifugal Filter Unit 100K to ~1 ml. The concentrate was transferred to 5 Prime Phase Lock Gel and incubated with 200 μl chloroform for 10 min at room temperature. After being centrifuged for 1 min at 15,000 ×g, the aqueous layer was transferred to a new microcentrifuge tube, and was treated with Invitrogen TURBO DNase (14 U), Promega RNase One (20 U) and 1 μl Benzonase Nuclease (E1014 Sigma Benzonase® Nuclease) at 37 °C for 3 hr (Gudenkauf and Hewson, 2016; Reyes et al., 2012). After incubation, 0.04 volumes 0.5 M EDTA was added to each sample. The sample was then stored at −80 °C before further processing.
Viral DNA shotgun sequencing –
The viral DNA was extracted with PureLink® Viral RNA/DNA Mini Kit from Invitrogen™. Each viral DNA sample was then amplified using GenomePlex® Complete Whole Genome Amplification (WGA2) Kit from Sigma-Aldrich (Gudenkauf and Hewson, 2016). Two blank controls were included in this step, but very low yield precluded library construction. The amplified product was then fragmented with Covaris S2 Adaptive Focused Acoustic Disruptor with the parameters set as follows: the duty cycle set at 10%, cycle per burst 200, intensity 4 and duration 60 seconds. Each viral sequencing library was prepared following Illumina TruSeq DNA Preparation Protocol with one unique barcode per sample. All barcoded libraries were pooled together. Half of the pool was size selected by BluePippin (Sage Science, Beverly, MA, USA) to enrich fragments with longer inserts (425 bp to 875 bp including the adapters). Both pools, the “large-insert-size library” and the “short-insert-size library”, were sequenced in independent lanes on an Illumina HiSeq 2500 instrument, operating in Rapid Run Mode with 250 bp paired-end chemistry at the Cornell Biotechnology Resource Center Genomics Facility.
Whole fecal metagenome shotgun sequencing –
The genomic DNA was isolated from an aliquot of ~100 mg from each sample using the PowerSoil® - htp DNA isolation kit (MoBio Laboratories Ltd, Carlsbad, CA). Each sequencing library was then prepared following Illumina TruSeq DNA Preparation Protocol with 500 ng DNA using the gel-free method, 14 cycles of PCR, and with one unique barcode per sample. Sequencing was performed on an Illumina HiSeq 2500 instrument in Rapid Run mode with 2×150 bp paired-end chemistry at the Cornell Biotechnology Resource Center Genomics Facility.
Assessment of Bacterial Contamination –
A set of 8,163 finished bacterial genomes was retrieved from the NCBI FTP on 21 February 2017. Reads per sample were mapped against this bacterial genomes dataset using Bowtie2 v.2.2.8 (Langmead and Salzberg, 2012) with the following parameters: --local --maxins 800 - k=3. Genome coverage per base was calculated considering only reads with a mapping quality above 20 using view and depth Samtools commands v.1.5 (Li et al., 2009). Next, genome coverage was averaged for 100Kbp bins. We observed that evenly covered genomes had a median bin coverage of at least 100; those genomes with a median bin coverage greater than 100 were considered as contaminants. The reads mapping to those genomes were removed. Bacterial genomes can have one or more prophage(s) in their genomes (Munson-McGee et al., 2018); bursting events of those prophages can occur, generating several VLPs. As a conservative measure to avoid the loss of reads originating from prophages and not the bacterial genome per se, bins with a coverage over three standard deviations of the bacterial mean coverage were also identified and catalogued as prophages-like regions. Reads mapping to potential contaminant genomes were tagged as “contaminants” and removed from further analysis while reads mapping to high coverage bins were tagged as “possible prophages”.
A matrix of the abundance of each potential contaminant per sample was built using an in-house Python script and normalized by RPKM. In parallel, from Goodrich et al. data (Goodrich et al., 2014), the relative abundance of each OTU was recovered and summarized at the species level using summarize_taxa.py qiime script. The Spearman rank order correlation between relative abundances of contaminants and their corresponding 16S rRNAs data was calculated for species in both sets.
Functional profiles –
The joined and trimmed reads from the “short-insert-size library” were mapped onto Integrated Gene Catalogs (IGC), an integrated catalog of reference genes in the human gut microbiome (Li et al., 2014) by BLASTX using DIAMOND v.0.7.5 (Buchfink et al., 2015) with maximum e-value cutoff 0.001, and maximum number of target sequences to report set to 25.
After the mapping onto IGC, an abundance matrix was generated using an inhouse Python script. The matrix was then annotated according to the KEGG annotation of each gene provided by IGC. The annotated abundance matrix was rarefied (subsampling without replacement) to 2,000,000 read hits per sample. The KEGG functional profile was then generated using QIIME 1.9 (Quantitative Insights Into Microbial Ecology) (Caporaso et al., 2010) using the command summarize_taxa_through_plots.py. The Intraclass Correlation Coefficient of the functional profiles for each group (additional microbiomes, additional viromes, viromes of concordant-microbiome samples and viromes of discordant-microbiome samples) was calculated using the Psych R package.
De-novo assembly –
Reads from the “large-insert-size library” that remain paired (forward and reverse) after the trimming step were assembled using the Integrated metagenomic assembly pipeline for short reads (InteMAP) (Lai et al., 2015) with insert size 325 bp ± 100 bp. Each sample was assembled separately. After the first run of assembly, all clean reads were mapped to the assembled contigs using Bowtie2 v.2.2.8 (Langmead and Salzberg, 2012) with the following parameters: --local --maxins 800. The pairs of reads that aligned concordantly at least once were then submitted for the second run of assembly by InteMAP. Contigs larger than 500 bp from all samples were pooled together and compared all vs all, using an in-house Perl script. From this analysis, it was possible to identify potential circular genomes, and to dereplicate contigs that were contained in over 90% of their length within another contig.
The recruitment of reads to the dereplicated metagenomic assemblies was used to build an abundance matrix, applying a filter of coverage and length as recommended in Roux et al. (Roux et al., 2017). Reads (not tagged as contaminants in the previous step) were mapped to dereplicated contigs using Rsubread v.1.28.0 (Liao et al., 2013). Mapping outputs were parsed using an in-house Python script into an abundance matrix that was normalized by reads per kilobase of contig length per million sequenced reads per sample (RPKM) and transformed to Log10(x+1), x being the normalized abundance. Contigs with a normalized coverage bellow 5× were excluded. Finally, a filter on contig length was applied to obtain virotypes. A length threshold was chosen as the elbow of the decay curve generated when plotting the number of contigs as a function of length, which occurred at a length of 1,300 bp.
HMM annotation –
Independent HMM profiles were built to identify crAss-like contigs and Microviridae contigs. To build the HMM-crAsslike profile, sequences for the Major Capsid Protein (MCP) of the proposed crAss-like family (Yutin et al., 2018) were retrieved from ftp.ncbi.nih.gov/pub/yutinn/crassphage_2017/. Multiple sequence alignments (MSA) were done using MUSCLE v.3.8.31(Edgar, 2004) and inspected using UGENE v.1.31.0 (Okonechnikov et al., 2012); positions with more than 30% of gaps were removed. Finally, the HMM-crAsslike profile was built using hmmbuild from the HMMER package v.3.1b2 (http://hmmer.org/) (Eddy, 1998). For the Microviridae case, all HMM-profiles for the viral protein 1 (VP1) developed by Alves et al. (Alves et al., 2016) were adopted.
Predicted proteins of the assembled contigs were queried for matching the HMM-profiles using hmmsearch (Eddy, 1998). Matching proteins with an e-value below 1×10−5 were considered as true homologs but only proteins between the size rank of the reference proteins (crAsslike MCP: 450–510 residues; Microviridae: 450–800 residues), a coverage of at least 50% and a percentage of identity of at least 40% to at least one reference sequence were used for further analysis. Coverage and identity percentages were determined with a BLASTp search of the true homologues against the reference sequences.
True homologues passing the filters mentioned above were used in phylogenetic analysis. Reference and homologous sequences were aligned using MUSCLE v.3.8.31 and sites with at least 30% of gaps were removed using UGENE v.1.31.0. A maximum-likelihood (ML) phylogenetic analysis was done using RAxML v.8.2.4 (Stamatakis, 2014), the best model of evolution was obtained with prottest v.3.4.2 (Darriba et al., 2011) and support for nodes in the ML trees were obtained by bootstrap with 100 pseudoreplicates.
Taxonomic profiles –
To infer the taxonomic affiliation of the assembled VLPs, genes were predicted from all assembled contigs larger than 500 bp using GeneMarkS v.4.32 (Besemer et al., 2001). The amino acid sequence of the predicted genes was then used in a BLASTp search against the NR NCBI viral database using DIAMOND v.0.7.5 (Buchfink et al., 2015) with maximum e-value cutoff 0.001 and maximum number of target sequences to report set to 25. Using the BLASTp results, the taxonomy of each gene was assigned by the lowest-common-ancestor algorithm in MEtaGenome ANalyzer (MEGAN5) v.5.11.3 (Huson et al., 2011) with the following parameters: Min Support: 1, Min Score: 40.0, Max Expected: 0.01, Top Percent: 10.0, Min-Complexity filter: 0.44. Independently, the taxonomy annotation of each contig was obtained using CENTRIFUGE v.1.0.4 (Kim et al., 2016) against the NT NCBI viral genomes database. The final taxonomic annotation of each contig was then assigned using a voting system where the taxonomic annotation of each protein and the CENTRIFUGE annotation of the contig were considered as votes. With all the possible votes for a contig, an N-ary tree was built and the weight of each node was the number of votes including that node. The taxonomic annotation of a contig will be the result of traversing the tree passing through the heaviest nodes with one consideration: if all children-nodes of a node have the same weight the traversing must be stopped. The taxonomic profile was considered as a subset of the recruitment matrix containing all contigs annotated either by the voting system or annotated through the HMM profiles (see above).
Prediction of phage-host interaction –
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) were identified using the PilerCR program v.1.06 (Edgar, 2007) from the same set of 8,163 bacterial used to asses the bacterial contamination. Spacers within the expected size of 20 bp and 72 bp (Horvath and Barrangou, 2010) were used as queries against virotypes and taxonomically annotated contigs using BLASTn (v.2.6.0+) with short query parameters (Camacho et al., 2009). Matches covering at least 90% of the spacer and with an e-value < 0.001 were considered to be CRISPR spacer-virus associations. Additionally, virotypes and taxonomically annotated contigs were mapped against the representatives genomes of the viral clusters in the MVP database (Gao et al., 2018) using LAST-959 (Kiełbasa et al., 2011). As viral clusters in MVP comprise sequences that have at least 95% identity along at least 80% of their lengths, only matches that fulfill those constraints were kept. The host(s) of a contig was determined from its matching viral cluster.
Diversity indexes –
The Shannon diversity index within-samples (α-diversity) and the Hellinger distance within co-twins (β-diversity) were calculated using diversity and vegdist functions of Vegan R package for all three abundance matrices generated (function, taxonomy and read recruitment matrices). Correlations between virome α-diversity and microbiome α-diversity were measured using the Pearson correlation coefficient. Correlations between viromes β-diversity and the microbiomes β-diversity were computed with a the Mantel test using the Pearson correlation coefficient. Additionally, the β-diversity between concordant MZ co-twins was compared to the β-diversity between discordant MZ co-twins; p values were calculated using a Mann-Whitney U test.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of twins/individuals in each group (Figure 1C, Figure 4B, Figure 6, Figure S5.A and Figure S5.B) or the number of comparisons (Figure 5, Figure S2 and Figure S5C) is denoted using “n”; p values were obtained using Mann-Whitney U test or Mantel test using the Python library “scipy”; correlation coefficients were measured as the Pearson correlation coefficient using the Python library “scipy”; alpha and beta-diversity metrics were calculated with the R package “vegan”; Intraclass coefficient was calculated using the R package “psych”; maximum-likelihood phylogenetic analysis was done using RAxML.
Supplementary Material
Table S1. Related to Figure 1. Additional information pertaining to the 21 selected MZ twin pairs (metadata), and counts of viromes reads and contigs per sample.
Table S2. Related to Figure 2. Median bin coverage of bacterial genomes by VLP reads per sample.
Highlights.
The gut virome is highly unique to each individual and dominated by bacteriophages
Gut microbiome diversity, within and between subjects, is mirrored in their viromes
These patterns of diversity are driven by bacteriophages, not by eukaryotic viruses
Microbiome abundances and diversity is predicative of virome richness and diversity
ACKNOWLEDGEMENTS
We thank Laura Avellaneda-Franco for helpful discussions. This work was supported by the Max Planck Society, by grants from the NIH (NIDDK RO1 DK093595 and DP2 OD007444), and by a Fellowship in Science and Engineering to RL from the David and Lucile Packard Foundation. The TwinsUK cohort is supported by the Wellcome Trust; European Community’s Seventh Framework Programme (FP7/2007–2013); National Institute for Health Research (NIHR)- funded BioResource, Clinical Research Facility and Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
Jupyter notebooks and scripts describing the data analysis process are available on GitHub at https://github.com/leylabmpi/TwinsUK_virome
The sequence data have been deposited in the European Nucleotide Archive under the study accession number PRJEB29491.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alves JMP, de Oliveira AL, Sandberg TOM, Moreno-Gallego JL, de Toledo MAF, de Moura EMM, Oliveira LS, Durham AM, Mehnert DU, Zanotto PM de A, et al. (2016). GenSeed-HMM: A tool for progressive assembly using profile HMMs as seeds and its application in Alpavirinae viral discovery from metagenomic data. Front. Microbiol 7, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barylski J, Enault F, Dutilh BE, Schuller MBP, Edwards RA, Gillis A, Klumpp J, Knezevic P, Krupovic M, Kuhn JH, et al. (2017). Genomic, proteomic, and phylogenetic analysis of spounaviruses indicates paraphyly of the order Caudovirales. bioRxiv. doi: 10.1101/220434 [DOI]
- Besemer J, Lomsadze A, and Borodovsky M (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biller SJ, Schubotz F, Roggensack SE, Thompson AW, Summons RE, and Chisholm SW (2014). Bacterial vesicles in marine ecosystems. Science 343, 183–186. [DOI] [PubMed] [Google Scholar]
- Bray JR, and Curtis JT (1957). An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr 27, 326–349. [Google Scholar]
- Breitbart M, Hewson I, Felts B, Mahaffy JM, Nulton J, Salamon P, and Rohwer F (2003). Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol 185, 6220–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbart M, Haynes M, Kelley S, Angly F, Edwards RA, Felts B, Mahaffy JM, Mueller J, Nulton J, Rayhawk S, et al. (2008). Viral diversity and dynamics in an infant gut. Res. Microbiol 159, 367–373. [DOI] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Mejía JL, Muhammed MK, Kot W, Neve H, Franz CMAP, Hansen LH, Vogensen FK, and Nielsen DS (2015). Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 3, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, Harris HMB, Coakley M, Lakshminarayanan B, O’Sullivan O, et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184. [DOI] [PubMed] [Google Scholar]
- Colson P, Fancello L, Gimenez G, Armougom F, Desnues C, Fournous G, Yoosuf N, Million M, La Scola B, and Raoult D (2013). Evidence of the megavirome in humans. J. Clin. Virol 57, 191–200. [DOI] [PubMed] [Google Scholar]
- Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, et al. (2013). Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, and Posada D (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, and Alm EJ (2014). Host lifestyle affects human microbiota on daily timescales. Genome Biol. 15, R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, and Lionetti P (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U. S. A 107, 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GGZ, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, et al. (2014). A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun 5, 4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2007). PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao NL, Zhang C, Zhang Z, Hu S, Lercher MJ, Zhao X-M, Bork P, Liu Z, and Chen W-H (2018). MVP: a microbe–phage interaction database. Nucleic Acids Res. 46, D700–D707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell JT, et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, Spector TD, Bell JT, Clark AG, and Ley RE (2016). Genetic determinants of the gut microbiome in UK Twins. Cell Host Microbe 19, 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudenkauf BM, and Hewson I (2016). Comparative metagenomics of viral assemblages inhabiting four phyla of marine invertebrates. Frontiers in Marine Science 3, 23. [Google Scholar]
- Gudenkauf BM, Eaglesham JB, Aragundi WM, and Hewson I (2014). Discovery of urchin-associated densoviruses (family Parvoviridae) in coastal waters of the Big Island, Hawaii. J. Gen. Virol . 95, 652–658. [DOI] [PubMed] [Google Scholar]
- Halary S, Temmam S, Raoult D, and Desnues C (2016). Viral metagenomics: are we missing the giants? Curr. Opin. Microbiol 31, 34–43. [DOI] [PubMed] [Google Scholar]
- Horvath P, and Barrangou R (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. [DOI] [PubMed] [Google Scholar]
- Hoyles L, McCartney AL, Neve H, Gibson GR, Sanderson JD, Heller KJ, and van Sinderen D (2014). Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol 165, 803–812. [DOI] [PubMed] [Google Scholar]
- Hulo C, de Castro E, Masson P, Bougueleret L, Bairoch A, Xenarios I, and Le Mercier P (2011). ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. 39, D576–D582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh H-J, Weber N, and Schuster SC (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res. 21, 1552–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiełbasa SM, Wan R, Sato K, Horton P, and Frith MC (2011). Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Song L, Breitwieser FP, and Salzberg SL (2016). Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles B, Silveira CB, Bailey BA, Barott K, Cantu VA, Cobián-Güemes AG, Coutinho FH, Dinsdale EA, Felts B, Furby KA, et al. (2016). Lytic to temperate switching of viral communities. Nature 531, 466–470. [DOI] [PubMed] [Google Scholar]
- Lai B, Wang F, Wang X, Duan L, and Zhu H (2015). InteMAP: Integrated metagenomic assembly pipeline for NGS short reads. BMC Bioinformatics 16, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sung J, Lee J, and Ko G (2011). Comparison of the gut microbiotas of healthy adult twins living in South Korea and the United States. Appl. Environ. Microbiol 77, 7433–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol 32, 834–841. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ES, Zhou Y, Zhao G, Bauer IK, Droit L, Ndao IM, Warner BB, Tarr PI, Wang D, and Holtz LR (2015). Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med 21, 1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, and Knight R (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 73, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrique P, Bolduc B, Walk ST, van der Oost J, de Vos WM, and Young MJ (2016). Healthy human gut phageome. Proc. Natl. Acad. Sci. U. S. A . 113, 10400–10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrique P, Dills M, and Young MJ (2017). The human gut phage community and its implications for health and disease. Viruses 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel LD, Young E, Delaney J, Ruhnau F, Ritchie KB, and Paul JH (2010). High frequency of horizontal gene transfer in the oceans. Science 330, 50. [DOI] [PubMed] [Google Scholar]
- Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, and Bushman FD (2011). The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 21, 1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, and Bushman FD (2013). Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. U. S. A . 110, 12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson-McGee JH, Peng S, Dewerff S, Stepanauskas R, Whitaker RJ, Weitz JS, and Young MJ (2018). A virus or more in (nearly) every cell: ubiquitous networks of virus-host interactions in extreme environments. ISME J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie LA, and Jones BV (2017). The human gut virome: form and function. Emerging Topics in Life Sciences 1, 351–362. [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M, and UGENE team (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. [DOI] [PubMed] [Google Scholar]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, and Brown PO (2007). Development of the human infant intestinal microbiota. PLoS Biol. 5, e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, and Gordon JI (2010). Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Semenkovich NP, Whiteson K, Rohwer F, and Gordon JI (2012). Going viral: next-generation sequencing applied to phage populations in the human gut. Nat. Rev. Microbiol 10, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Wu M, McNulty NP, Rohwer FL, and Gordon JI (2013). Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. U. S. A 110, 20236–20241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Blanton LV, Cao S, Zhao G, Manary M, Trehan I, Smith MI, Wang D, Virgin HW, Rohwer F, et al. (2015). Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. U. S. A 112, 11941–11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Brito B, Li L, Wegley L, Furlan M, Angly F, Breitbart M, Buchanan J, Desnues C, Dinsdale E, Edwards R, et al. (2010). Viral and microbial community dynamics in four aquatic environments. ISME J. 4, 739–751. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Valera F, Martin-Cuadrado A-B, Rodriguez-Brito B, Pasić L, Thingstad TF, Rohwer F, and Mira A (2009). Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 7, 828–836. [DOI] [PubMed] [Google Scholar]
- Roux S, Krupovic M, Debroas D, Forterre P, and Enault F (2013). Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biol. 3, 130160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Emerson JB, Eloe-Fadrosh EA, and Sullivan MB (2017). Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 5, e3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, and Milo R (2016). Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell 164, 337–340. [DOI] [PubMed] [Google Scholar]
- Shkoporov A, Khokhlova EV, Brian Fitzgerald C, Stockdale SR, Draper LA, Paul Ross R, and Hill C (2018). ΦCrAss001, a member of the most abundant bacteriophage family in the human gut, infects Bacteroides. bioRxiv. doi: 10.1101/354837 [DOI] [PMC free article] [PubMed]
- Stamatakis A (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttle CA (2007). Marine viruses--major players in the global ecosystem. Nat. Rev. Microbiol 5, 801–812. [DOI] [PubMed] [Google Scholar]
- Taylor AL (1963). Bacteriophage-induced mutation in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 50, 1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thingstad TF (2000). Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr 45, 1320–1328. [Google Scholar]
- Thingstad TF, Våge S, Storesund JE, Sandaa R-A, and Giske J (2014). A theoretical analysis of how strain-specific viruses can control microbial species diversity. Proc. Natl. Acad. Sci. U. S. A 111, 7813–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tims S, Derom C, Jonkers DM, Vlietinck R, Saris WH, Kleerebezem M, de Vos WM, and Zoetendal EG (2013). Microbiota conservation and BMI signatures in adult monozygotic twins. ISME J. 7, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint A, and Rice PA (2017). Transposable phages, DNA reorganization and transfer. Curr. Opin. Microbiol 38, 88–94. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz JS, and Dushoff J (2008). Alternative stable states in host–phage dynamics. Theor. Ecol 1, 13–19. [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarygin K, Tyakht A, Larin A, Kostryukova E, Kolchenko S, Bitner V, and Alexeev D (2017). Abundance profiling of specific gene groups using precomputed gut metagenomes yields novel biological hypotheses. PLoS One 12, e0176154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N, Makarova KS, Gussow AB, Krupovic M, Segall A, Edwards RA, and Koonin EV (2018). Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat Microbiol 3, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 1. Additional information pertaining to the 21 selected MZ twin pairs (metadata), and counts of viromes reads and contigs per sample.
Table S2. Related to Figure 2. Median bin coverage of bacterial genomes by VLP reads per sample.