

Abstract
Yeast surface display is a proven tool for the selection and evolution of ligands with novel binding activity. Selections from yeast surface display libraries against transmembrane targets are generally carried out using recombinant soluble extracellular domains. Unfortunately, these molecules may not be good models of their true, membrane-bound form for a variety of reasons. Such selection campaigns often yield ligands that bind recombinant target but not target-expressing cells or tissues. Advances in cell-based selections with yeast surface display may aid the frequency of evolving ligands that do bind true, membrane-bound antigens. This study aims to evaluate ligand selection strategies using both soluble target-driven and cellular selection techniques to determine which methods yield translatable ligands most efficiently and generate novel binders against CD276 (B7–H3) and Thy1, two promising tumor vasculature targets. Out of four ligand selection campaigns carried out using only soluble extracellular domains, only an affibody library sorted against CD276 yielded translatable binders. In contrast, fibronectin domains against CD276 and affibodies against CD276 were discovered in campaigns that either combined soluble target and cellular selection methods or used cellular selection methods alone. A high frequency of non-target specific ligands discovered from the use of cellular selection methods alone motivated the development of a depletion scheme using disadhered, antigen-negative mammalian cells as a blocking agent. Affinity maturation of CD276-binding affibodies by error-prone PCR and helix walking resulted in strong, specific cellular CD276 affinity (Kd = 0.9 ± 0.6 nM). Collectively, these results motivate the use of cellular selections in tandem with recombinant selections and introduces promising affibody molecules specific to CD276 for further applications.
Graphical Abstract
Introduction
Advances in genomic and proteomic methods1 have increased knowledge of disease biomarkers at a rate that has outpaced the development of new molecularly targeted agents for diagnosis and therapy. Several classes of molecules can be applied to bridge this gap including engineered proteins2–4. A variety of scaffolds have shown therapeutic effectiveness as inhibitors, targeting agents for drug delivery, radioisotope carriers, and immune system engagers5 as well as diagnostic success for early disease detection, patient stratification, and treatment monitoring6.
Numerous high-throughput screening methods for selection of engineered proteins with novel specific binding activity have been applied. Most often, the discovery of ligands targeting cell surface receptors is directed using recombinantly produced soluble extracellular domains. The use of these molecules as selection targets allows for efficient screening via immobilization on solid supports7,8 or fluorescent tagging9. However, these targets are unlikely to be perfect models of full length target expressed on intact cells due to several factors including: a) improper folding of the soluble domains10–13, b) differential post-translational modification due to the production host14,15, c) presence of non-natural epitopes resulting from the biological or chemical addition of tags for purification and/or selection16, and d) possible exposure of epitopes that would not be accessible to ligands in the presence of the transmembrane domain or cell membrane. Despite selection against these molecules yielding successful, translatable engineered ligands in numerous cases17–19, many ligand engineering campaigns end in failure due to the inability of isolated soluble domain binding ligands to bind full length target expressed on intact cells. As there is no good outlet, these results are seldom reported, skewing perception of the difficulties of ligand discovery. Herein, we will use the term “translatable” to refer to ligands that bind molecular target in the genuine cellular form.
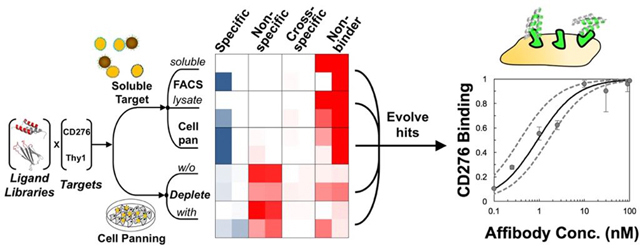
This study aims to evaluate different ligand selection methods to advance our understanding and technical ability to robustly generate ligands that bind intact, extracellularly expressed target molecules. We compare the following selection methods: 1) magnetic bead sorting7,20 and fluorescence activated cell sorting (FACS)9,20 using biotinylated soluble extracellular domains, 2) magnetic bead sorting using biotinylated soluble extracellular domains followed by FACS with detergent-solubilized cell lysate21,22, 3) magnetic bead sorting using biotinylated soluble extracellular domains followed by direct yeast panning on adherent cell monolayers22–24, 4) direct yeast panning on adherent cell monolayers, and 5) direct yeast panning on adherent cell monolayers preceded by magnetic bead depletion using biotinylated soluble proteins. Magnetic bead sorting enables very high valency (up to five million targets per 3 μm magnetic bead7) and efficiently scalable volumes. Fluorescence-activated cell sorting enables stringent quantitative analysis for fine affinity and selectivity discrimination25. Detergent-solubilized cell lysate provides complete membrane-spanning protein albeit in a modified detergent context. Direct yeast panning on adherent cell monolayers provides complete target in the full cellular context, though the nature of cell-cell (yeast-human) interactions is fundamentally different than cell-protein interactions in the other modes of selection.
The comparative analysis of selection methods is performed towards the discovery of ligands for tumor vasculature biomarkers CD276 (also known as B7–H3) and thymocyte differentiation antigen 1 (Thy1). CD276 is an immune checkpoint molecule that has both costimulatory and coinhibitory roles in T cell regulation26. It is overexpressed in a variety of cancers, including clear cell renal cell carcinoma27, cutaneous melanoma28, diffuse intrinsic pontine glioma29, hypopharyngeal squamous cell carcinoma30, prostate cancer31, ovarian cancer32, and pancreatic cancer33. Its expression is associated with progression and metastasis in several of these diseases28,30,34. Thy1 overexpression in the neovasculature of pancreatic ductal adenocarcinoma differentiates the diseased tissue from normal pancreas or chronic pancreatitis, allowing for detection of disease with superior sensitivity and specificity relative to the current standard of care35. These characteristics make both molecules attractive targets for molecular ultrasound imaging as well as other diagnostic and therapeutic applications.
This study uses two alternative ligand scaffolds, the beta-sandwich fibronectin domain36,37 and the three-helix bundle affibody38,39, whose small, single-domain architectures provide for efficient chemical conjugation and rapid physiological distribution40–43. Selections from both libraries efficiently yielded subpopulations with measurable binding activity.
Clonal characterization shows that campaigns utilizing cellular-based enrichment in part or entirely (9/16 campaigns) have a higher success rate for yielding at least one target-specific cellular binder relative to campaigns relying on completely soluble extracellular domains for enrichment (1/4 campaigns). The ability of selections using soluble extracellular domains to yield target-specific cellular binders appears to be dependent on both the target molecule and ligand library used. Selections using direct yeast panning against mammalian cell monolayers yielded a high frequency of non-target specific binders, motivating the development of a depletion scheme using disadhered antigen-negative mammalian cells as a blocking agent. This method confers a 14 ± 3-fold selectivity advantage to recovery of a dilute high-yield antigen-specific ligand from a pool containing a high-yield non-target specific ligand and a non-binding ligand.
While affinity maturation of Thy1-binding affibodies by error-prone PCR yielded a prevalently non-target specific population, maturing CD276-binding affibodies pooled from all five campaigns resulted in the isolation of a specific ligand of modest affinity (AC2, Kd = 310 ± 100 nM). Further maturation by helix-walking resulted in the successful discovery of a panel of strong, specific binding ligands to cellular CD276 (Kd = 0.9 – 20 nM).
Materials and Methods
Cells and Cell Culture
Mile Sven 1 cells stably transfected to express human CD276 (MS1-CD276)32 or human Thy1 (MS1-Thy1)35 were grown at 37 °C in a humidified atmosphere with 5% CO2 in DMEM with 4.5 g/L glucose, sodium pyruvate, and glutamine supplemented with 10% (v/v) fetal bovine serum.
Yeast surface display was performed essentially as described20. EBY100 yeast harboring expression plasmids were grown in SD-CAA medium (16.8 g/L sodium citrate dihydrate, 3.9 g/L citric acid, 20.0 g/L dextrose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids) at 30 °C with shaking. Protein expression was induced by transferring yeast cells in logarithmic phase (OD600nm<6) into SG-CAA medium (10.2 g/L sodium phosphate dibasic heptahydrate, 8.6 g/L sodium phosphate monobasic monohydrate, 19.0 g/L galactose, 1.0 g/L dextrose, 6.7 g/L yeast nitrogen base, 5.0 g/L casamino acids) and growing at 30 °C with shaking for at least 8 h. EBY100 without plasmid were grown in YPD medium (10.0 g/L yeast extract, 20.0 g/L peptone, 20.0 g/L dextrose) at 30 °C with shaking.
Naïve Library Construction and Characterization
Oligonucleotides encoding for the second-generation sitewise gradient hydrophilic fibronectin domain library44 and second-generation sitewise gradient affibody library45 were synthesized by IDT DNA Technologies. Full length amplicons for each respective library were assembled by overlap extension PCR and homologously recombined into pCT-40 yeast surface display vector, with a linker extended by 40 additional amino acids24, within yeast strain EBY100 by electroporation transformation as previously described44. Transformation efficiency was quantified by dilution plating on SD-CAA agar plates.
Full-length library construction was characterized by simultaneous labeling of the N-terminal hemagglutinin (HA) epitope and C-terminal c-Myc epitope by flow cytometry. Two million yeast were pelleted at 12,000g for 1 min, washed once with phosphate-buffered saline (PBS) with 1 g/L bovine serum albumin (PBSA), then labeled with mouse anti-c-Myc antibody 9E10 (0.5 μg/mL, BioLegend, Cat: 626802) and biotinylated goat anti-HA polyclonal antibody (2 μg/mL, Genscript, Cat: A00203) for 30 min at room temperature. Cells were washed once with 1 mL PBSA, labeled by goat anti-mouse Alexa Fluor 647 conjugate (10 μg/mL, Life Technologies, Cat: A-21235) and streptavidin Alexa Fluor 488 conjugate (2 μg/mL, Life Technologies, Cat: S11223) for 15 min at 4 °C, and washed once. Fluorescence was analyzed by flow cytometry (Accuri C6, BD Biosciences).
Magnetic Bead Selections with Soluble Extracellular Domains
Recombinant human Thy1 extracellular domain Fc fusion (Thy1-Fc) (Abcam, Cat: ab157072) was biotinylated on free amines using EZ-Link NHS-PEG4-Biotin (Thermo Fisher Scientific, Cat: 21330) with a NHS-PEG4-biotin : protein ratio of 5:1. Biotinylation was verified using matrix-assisted laser desorption ionization mass spectrometry (Sciex 5800, Applied Biosystems). Recombinant human CD276 extracellular domain (Sino Biological, Cat: 11188-H08H-B) was obtained already biotinylated from the manufacturer.
Magnetic bead selections were carried out essentially as previously described7 using 15-fold oversampling of ligand diversity at all stages. For the first round of selection, libraries were depleted of magnetic bead binders three times with streptavidin coated Dynabeads (Thermo Fisher Scientific, Cat: 11205D). Remaining yeast were incubated with CD276 or Thy1-Fc coated magnetic beads at 4 °C and washed twice with ice cold PBSA. Beads with attached cells were resuspended in SD-CAA for growth. Magnetic beads were removed using a Dynal magnet prior to the induction of protein expression for the next round of selection. For subsequent rounds, non-specific binders were depleted with streptavidin-coated magnetic beads and negative control protein-coated magnetic beads prior to enrichment with target-coated magnetic beads. Negative control targets included human IgG (Rockland Immunochemicals, biotinylated by manufacturer), glucose-6-phosphate dehydrogenase (Sigma, biotinylated by manufacturer), and a scrambled peptide of a loop from prostate stem cell antigen (United Peptide, biotinylated during peptide synthesis). Selections were carried out at room temperature and target-coated beads were washed three times with PBSA before regrowth of the attached yeast. Dilution plating on YPD plates of all negative control and target-coated bead populations was completed to quantify yield for each round.
FACS with Soluble Extracellular Domains
Fluorescence-activated cell sorting (FACS) was carried out essentially as described20. Induced yeast were simultaneously labeled with mouse anti-c-Myc antibody (9E10, BioLegend, Cat: 626802, 2.5 μg/mL) and 10–100 nM biotinylated target protein or biotinylated negative control protein for at least 30 min at room temperature. Cells were washed once with PBSA, labeled with goat anti-mouse Alexa Fluor 647 conjugate (Thermo Fisher Scientific, Cat: A-21235, 10 μg/mL) and streptavidin Alexa Fluor 488 conjugate (Thermo Fisher Scientific, Cat: S-11223, 2 μg/mL) for 15 min at 4 °C, and washed with 1 mL PBSA. Cells with the highest binding : ligand display ratio (AlexaFluor488:AlexaFluor647) were sorted using a FACSAria II (BD Bioscience).
FACS Selections with Detergent Solubilized Cell Lysates
Detergent-solubilized cell lysates were prepared essentially as described22. MS1-Thy1 and MS1-CD276 cells were grown to 70–90% confluence in 75 cm2 tissue culture-treated flasks. Culture medium was removed and the cells were washed once with 5 mL PBS. Cells were detached by trypsin-EDTA treatment for 4–7 minutes, quenched with serum-containing culture medium, and centrifuged at 500g for 3 min. Pelleted cells were washed three times with ice cold PBS and pelleted at 300g for 3 min at 4 °C. Washed cells were resuspended in PBS with 0.5 mg/mL fresh sulfo-NHS-biotin (Thermo Fisher Scientific, Cat: 21217), rotated for 30 min at room temperature, and washed twice with ice cold PBSA to quench and remove excess biotin. Cells were resuspended in 100–200 μL FACS lysis buffer (PBS with 1% (v/v) Triton X-100, 2 mM EDTA, and 1× cOmplete protease inhibitor cocktail (Roche)) and incubated with rotation at 4 °C for 15 min. Cell debris was pelleted at 15,000g for 30 min at 4 °C and removed. Induced yeast were washed once with PBSA, then incubated with cell lysate and mouse anti-c-Myc antibody (2.5 μg/mL) simultaneously for 2 h at 4 °C with rotation. Yeast were washed with 1 mL ice cold PBS containing 1% (v/v) Triton-X 100 and then with 1 mL ice cold PBSA. Cells were incubated with goat anti-mouse Alexa Fluor 647 conjugate (10 μg/mL) and streptavidin Alexa Fluor 488 conjugate (2 μg/mL) at 4 °C for 15 min and washed with 1 mL ice cold PBSA. Cells with the highest binding : ligand display ratio (AlexaFluor488:AlexaFluor647) were sorted using a FACSAria II.
Yeast Cell Panning Selections
Cell panning selections were carried out essentially as described24. Mammalian cells were grown in 6-well plates to approximately 90% confluence. Culture medium was removed, and cells were washed three times with ice cold PBSA with 1 mM CaCl2 and 0.5 mM Mg2SO4 (PBSACM). For the first round of selection, 2.4×109 yeast (three-fold diversity of fibronectin library, six-fold diversity of affibody library) were washed once with ice cold PBSACM, resuspended to 1×108 yeast/mL in ice cold PBSACM, and applied to mammalian cells in 1 mL aliquots dropwise. Cells were incubated without shaking for 2 h at 4 °C and unbound yeast were removed by aspiration. Cells were washed with 1 mL ice cold PBSACM four times with 25 gentle tilts and 5 nutations and one time with 10 nutations. Bound yeast were recovered by scraping cell monolayers and resuspending them in SD-CAA growth medium. Yield was quantified by dilution plating on YPD plates. For each subsequent round, at least 15-fold of the recovered yield were washed and resuspended to no more than 1×108 yeast/mL in ice cold PBSACM. Yeast were panned, in parallel, against one target-positive and two target-negative cell lines.
Clonal Characterization of Sorted Populations by Yeast-Cell Panning
Forty-eight colonies from each selection campaign, obtained by plating yeast populations on SD-CAA, were picked and resuspended in 1 mL SG-CAA in deep-well 96-well plates. Plates were covered and grown at 30 °C with shaking for at least 8 h.
Target-positive and target-negative mammalian cells were grown to approximately 80% confluence in 24-well plates. Cells were washed 3 times with ice cold PBSACM. 250 μL of induced clonal yeast was added dropwise directly to one well of target-positive and one well of target-negative mammalian cells. Cells were incubated without shaking for at least 2 h at 4 °C. Cells were washed with 250 μL ice cold PBSACM twice with 25 gentle tilts and 5 nutations and once with 10 nutations. Yeast binding was visualized using EVOS FL Cell Imaging System (Thermo Fisher Scientific) at 40× total magnification.
Individual clone binding strength was categorized as −, +, ++, or +++ through counting associated yeast in a random microscope field. Clones were characterized as “-” if fewer than 15 yeast were observed, “+” if 15 to 50 yeast were observed, “++” if greater than 50 yeast were observed but mammalian cells were still visible, and “+++” if yeast were the dominant organism seen in the frame.
DNA Sequencing
Plasmid DNA from yeast clones that bound target-positive but not target-negative cells was recovered by zymoprep of 200 μL of each individual clone. Ligand sequences were amplified in 50 μL PCR mixtures containing 2 μL zymoprep DNA, 1× Phusion High Fidelity buffer, 0.5 μM each of primers W5 and W346, 0.2 mM dNTP mixture, and 2.5 U Phusion polymerase (New England Biolabs). PCR products were purified by agarose gel electrophoresis and Sanger sequenced with GeneAmp5 primer (5’-CGACGATTGAAGGTAGATACCCATACG-3’) (Eurofins MWG Operon).
Error-Prone PCR of Fibronectin and Affibody Domains
Random mutation of fibronectin domains and affibodies was performed essentially as described46 by error-prone PCR with nucleoside analogs47. Zymoprepped plasmid DNA was mutated by error-prone PCR of full fibronectin domain or affibody genes using primers W5/W3, fibronectin loops using primers BCHPEP5/BCHPEP3, DEHPEP5/DEHPEP3, and FGHPEP5/FGHPEP3, and affibody helices using primers ABY1F-b/ABY1R and ABY2F/ABY2R-b (Supplemental Table 1). PCR products were purified by agarose gel electrophoresis, amplified in four 200 μL PCR mixtures, concentrated by ethanol precipitation, and resuspended in 30 μL buffer E several hours before electroporation. Mutated sublibraries were homologously recombined with linearized pCT-Gene (cut with NdeI, PstI-HF, and BamHI-HF), pCT-40-FnHP-Loop (cut with SmaI, NcoI-HF, and NdeI) for fibronectin loop shuffling, or pCT-40-Helix (cut with SmaI, NcoI-HF, and NdeI) for affibody helix shuffling in EBY100 yeast by electroporation transformation as described44. Transformation efficiency was quantified by dilution plating on SD-CAA plates.
Expression Plasmids for Depletion Model
CD276-specific or non-specific affibody clones were chosen from populations selected to bind either soluble CD276 extracellular domain or MS1-CD276 cells with relative binding strength defined with a phase microscopy assay. Clones HS (high-yield specific) and LS (low-yield specific) bind MS1-CD276 cells as ‘+++’ and ‘+’, respectively while binding MS1-Thy1 as ‘-’ when assessed by microscopy. Clones HN (high-yield non-target specific) and LN (low-yield non-target specific) bind both MS1-CD276 and MS1-Thy1 as ‘+++’ and ‘+’, respectively. Clones HS and LS were cloned into pCT-40 vector by NheI and BamHI restriction sites. Non-target specific affibody clones HN and LN were cloned into pCT-40V48 vector (pCT-40 with the MYC tag replaced by a V5 tag) by NheI and BamHI restriction sites. Affibody clone A524, an affibody with no known binding partner, was cloned into pCT-40E48 vector (pCT-40 with the MYC tag replaced by an E-tag) by NheI and BamHI restriction sites.
Determination of Optimal Incubation Time for Cellular Selections
MS1-CD276 were grown to approximately 90% confluence in 12-well plates. Culture medium was removed and cells were washed three times with 500 μL ice cold PBSACM (PBS with 1 g/L bovine serum albumin, 1 mM CaCl2, 0.5 mM Mg2SO4).
5×107 yeast expressing CD276-specific affibody clone HS or LS were pelleted at 8,000g for 1 min, washed once with ice cold PBSACM, and resuspended in 500 μL ice cold PBSACM. Yeast were then applied to MS1-CD276 monolayers dropwise and incubated at 4 °C without shaking for 15 min, 30 min, 45 min, 60 min, or 120 min. Cells were washed 5 times with 500 μL ice cold PBSACM as described24. Briefly, cells were tilted 25 times and rotated 5 times for the first four washes and rotated 10 times only for the fifth wash. 500 μL SD-CAA was added dropwise to the washed wells, and cells were recovered by scraping. Yeast recovery was quantified by dilution plating on YPD plates (10 g/L yeast extract, 20 g/L peptone, 20 g/L dextrose, 16 g/L agar).
Depletion of Non-specific Clones with Sequential Depletion
Mixtures 0.2 ± 0.1% CD276-specific, 3 ± 2% non-specific, and 97 ± 2% non-binding affibody-displaying yeast were generated and compositions were quantified by flow cytometry analysis. Yeast were pelleted and washed once with ice cold PBSACM, then resuspended to a concentration of 1×108 yeast/mL in ice cold PBSACM.
MS1-Thy1 and MS1-CD276 were grown to approximately 90% confluence in 12-well plates. Culture medium was removed and cells were washed three times with 500 μL ice cold PBSACM. 500 μL of yeast mixture were added to MS1-Thy1 monolayers dropwise and incubated at 4 °C for 15 min without rotation. The binding buffer containing unbound yeast was collected. Monolayers were washed four times as described in the previous section, with unbound yeast collected and pooled. The collected yeast were concentrated into 500 μL ice cold PBSACM and applied to the next washed MS1-Thy1 monolayer. This process was repeated for zero, two, four, or six depletion steps against MS1-Thy1 monolayers. Recovered yeast were then applied to an MS1-CD276 monolayer and incubated at 4 °C for 120 min. Monolayers were washed five times, and bound yeast were recovered by scraping as before. Yeast recovery was quantified by dilution plating on YPD plates. Recovered yeast were grown in 5 mL SD-CAA at 30 °C with shaking. Protein expression was induced by resuspending a portion of the outgrowth in SG-CAA at OD600nm<1 and growing these yeast for at least eight hours at 30 °C with shaking.
Mixture compositions were quantified by flow cytometry analysis. Yeast were pelleted and washed once with 1 mL PBSACM. Yeast were then labeled with 20 μL goat anti-c-myc FITC conjugate (Bethyl Laboratories, Cat: A190–104F, 2 μg/mL), goat anti-V5 FITC conjugate (Bethyl Laboratories, Cat: A190–119F, 2 μg/mL), or goat anti-E-tag FITC conjugate (Bethyl Laboratories, Cat: A190–132F, 2 μg/mL) for 20 min at room temperature. Yeast were then pelleted and washed once with PBSACM. Fluorescence was analyzed using an Accuri C6 (BD Biosciences). Enrichment ratios were determined by dividing the percentage positive for one tagged construct by the total percentage of induced yeast.
Depletion of Non-specific Clones by Pre-Blocking with Disadhered Mammalian Cells
MS1-Thy1 were grown to approximately 90% confluence in a 75 cm2 tissue culture-treated flask. Culture medium was removed and cells were washed once with 5 mL PBS. Cells were disadhered by trypsin-EDTA treatment for 6 min, then quenched by the addition of serum-containing culture medium. Cells were then pelleted at 500g for 3 min, trypsin-containing culture medium was removed, and cells were resuspended in fresh culture medium for counting using a Countess II FL (Thermo Fisher Scientific).
Mixtures of 0.3 ± 0.1% CD276-specific, 1.9 ± 0.8% non-specific, and 98 ± 0.7% non-binding affibody-displaying yeast were generated, and compositions were quantified by flow cytometry analysis. Yeast were pelleted and washed with ice cold PBSACM and resuspended to 5×107 yeast in 500 μL ice cold PBSACM containing 1×106 MS1-Thy1. Yeast and MS1-Thy1 were incubated at 4 °C with rotation for 2 hours. After incubation, samples were added dropwise to washed MS1-CD276 monolayers in 12-well plates and incubated at 4 °C without rotation for 15 min. Monolayers were washed five times with 500 μL ice cold PBSACM as described above. Bound yeast were recovered by scraping. Yeast recovery was quantified by dilution plating on YPD plates. Recovered yeast were grown, protein expression was induced, and final mixture composition was determined by flow cytometry as described above.
Helix Walking Library Construction
CD276 rational mutagenesis libraries were constructed using an analogous method to CDR-walking49–51. The first affibody helix was diversified at seven sites using degenerate oligonucleotides while retaining the parental sequence in the second helix. Separately, the second affibody helix was diversified at six sites using degenerate oligonucleotides while retaining the parental first helix. The diversified oligonucleotide for one helix and parental oligonucleotide for the other helix were assembled by overlap extension PCR and homologously recombined into pCT-Helix yeast surface display vector within yeast strain EBY100 by electroporation transformation. Transformation efficiency was quantified by dilution plating on SD-CAA agar plates. Full-length library construction was characterized by flow cytometry as previously described.
Protein Production
Gel-purified PCR amplicons were digested by NheI-HF and BamHI-HF and ligated into a pET-22b vector containing a C-terminal His6 tag (Novagen, EMD Millipore) using T4 DNA ligase (New England Biolabs). Plasmids were transformed via heat-shock into T7 Express E. coli (New England Biolabs) and plated on lysogeny broth (LB) (10.0 g/L tryptone, 5.0 g/L yeast extract, 10.0 g/L sodium chloride) agar plates containing kanamycin (50 mg/L). Clones were verified by Sanger sequencing of plasmids recovered by bacterial miniprep (Epoch Life Science).
E. coli were grown to saturation in 5 mL LB containing kanamycin at 37 °C with shaking. Cultures were diluted to OD600 = 0.03 with 100 mL LB in 250 mL baffled culture flasks. At OD600 = 0.5 – 1.0, protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside overnight at 30 °C with shaking. Cells were pelleted at 3220g for 20 min, resuspended in bacterial lysis buffer (50 mM sodium phosphate (pH 8.0), 0.5 M sodium chloride, 5% glycerol, 5 mM CHAPS, and 25 mM imidazole supplemented with protease inhibitor) and subjected to 4–5 freeze-thaw cycles. Insoluble cell debris was removed by centrifugation at 12,000g for 10 min followed by filtration (0.2 μm). Protein was purified by metal affinity chromatography on 2 mL of Cobalt HisPur Resin (Thermo Fisher Scientific) or HisPur Cobalt Resin Spin Columns (Thermo Fisher Scientific) according to the manufacturer’s protocol. Eluted fractions were pooled and buffer exchanged by HPLC prior to lyophilization. Lyophilized protein was resuspended in PBS and protein concentration was quantified by absorbance at 280 nm52. Protein identity was verified by MALDI-TOF-MS using a 5800 MALDI/TOF-MS (AB-Sciex).
Affinity Titration of Affibody Domains
Detached MS1-Thy1 and MS1-CD276 cells were washed and individually labelled with varying concentrations of purified affibody for 25 minutes at 4 °C with rotation unless additional time was needed to approach binding equilibrium. Cells were pelleted at 500g for 3 minutes and washed with 1 mL of ice cold PBSACM prior to labelling with 50 uL anti-His6 FITC conjugate (Abcam ab1206; 10 μg/mL) for 25 minutes at 4 °C. Cells were again pelleted and washed with 1 mL of ice cold PBSACM. Fluorescence was analyzed using an Accuri C6. The dissociation constant was calculated by non-linear least squares regression using a 1:1 binding model.
Thermal Stability of Affibody Domains
Secondary structure, midpoint of thermal denaturation (Tm), and protein refolding was determined using circular dichroism spectroscopy performed on a Jasco-815 spectrophotometer. Purified and lyophilized protein was resuspended in PBS to a concentration of 20 μM and placed in a 1 mm path length quartz cuvette. Ellipticity was measured between 200 and 260 nm wavelengths at 20 °C before and after heating to 98 °C in order to observe secondary structure of the folded affibody. Samples were then heated at 1 °C/min from 20 to 98 °C while the ellipticity was monitored at 220 nm. The midpoint of thermal denaturation was calculated by non-linear least squares regression using a two-state protein unfolding model.
Results
Twenty ligand selection campaigns were carried out using two naïve ligand scaffold libraries – fibronectin domain and affibody – to discover binders to two vascular biomarkers – CD276 and Thy1 – via five selection approaches. Three approaches used soluble extracellular domains immobilized on magnetic beads for initial sorting followed by recombinant extracellular domain FACS selections, detergent-solubilized lysate FACS, or yeast cell panning selections added as translatable sorts after sufficient monovalent affinity for soluble domains was established (Figure 1). Two approaches used panning on adherent mammalian cells with or without depletion of non-specific binders with streptavidin-coated magnetic beads. Iterative selections and affinity maturation were performed until monovalent binding detectable by flow cytometry was observed in the FACS-based schemes or strong yield enrichment were observed in the adherent cell panning schemes.
Figure 1. Ligand discovery methods.

An affibody library and a fibronectin domain library were sorted for ligands that bound CD276 or Thy1 specifically. Libraries were sorted by five different schemes: 1) magnetic bead selection with recombinant extracellular domains followed by FACS with recombinant extracellular domains, 2) magnetic bead selection followed by FACS with detergent solubilized cell lysate, 3) magnetic bead selection followed by cell panning selection, 4) cell panning selection with magnetic bead depletion, and 5) cell panning selection.
Ligand Selections using Soluble Extracellular Domain Driven Methods
Ligand selections using magnetic bead sorting and FACS with only soluble extracellular domains successfully generated ligands that exhibit binding at 100 nM target concentrations (Figure 2A–D). Ligands from both campaigns isolated against CD276 appear to have high specificity for their targets, showing no cross-reactivity with streptavidin, the reagent used for both immobilization and fluorescent labeling in all sorts. Ligands evolved against Thy1-Fc, despite depletion during magnetic bead selection with biotinylated human IgG exclusively, do still show a significant amount of cross-reactivity for human IgG, suggesting that depletion was not complete enough. This phenomenon is similarly prevalent in the fibronectin and affibody populations. Through stringent gating, ligands that appear to bind Thy1-Fc differentially were isolated but were not further validated prior to clonal screening.
Figure 2. Enriched ligands evaluated for soluble extracellular domain and detergent-solubilized cell lysate binding.

Yeast-displayed ligand populations were enriched for binders against CD276 and Thy1 using soluble extracellular domains immobilized on magnetic beads followed by FACS with either soluble extracellular domains or detergent-solubilized cell lysates. Binding specificity for each enriched population was assessed by comparison to negative controls. For FACS with soluble extracellular domains, yeast were labeled with 100 nM soluble CD276 or Thy1-Fc extracellular domains (blue) or irrelevant negative control proteins (300 nM streptavidin for CD276 or 100 nM human IgG for Thy1-Fc; orange) (A-D). For selection of ligands with detergent-solubilized cell lysates against CD276, yeast were labeled with MS1-CD276 lysate (blue) or MS1-Thy1 lysate (orange). For selection of ligands with detergent-solubilized cell lysates against Thy1, yeast were labeled with MS1-Thy1 lysate (blue) or MS1-CD276 lysate (orange) (E-H).
The use of detergent-solubilized cell lysate FACS as a translatable sort was largely not beneficial to isolation of target-specific binders (Figure 2E–H). The Thy1-targeted affibody campaign did not yield substantial binding while the Thy1-targeted fibronectin campaign yielded minimal Thy1 binding and comparable cross-reactivity. Enrichment of affibodies to CD276 yielded frequent binders but prevalent cross-reactivity. The CD276-targeted fibronectin population was likewise cross-reactive albeit with some preferential CD276 binding.
The introduction of cellular panning selections after enrichment on target-conjugated magnetic beads allowed for isolation of cellular target specific binding in several cases (Figure 3A–D). Single-trial ligand selections against CD276, initially via recombinant CD276-coated magnetic beads and then via adherent MS1-CD276 cell panning, showed a strong yield preference for MS1-CD276 cells relative to MS1-Thy1 cells (3% vs. 0.3% for fibronectin after three rounds and 7% vs. 0.1% for affibody after a single round of panning). Nominal yield preference was observed for both ligand campaigns selected against Thy1-Fc, with reasonable yield for fibronectins and negligible yield of affibodies.
Figure 3. Enriched ligands evaluated for cellular binding.

Yeast-displayed ligand populations were enriched through sequential rounds of selection for binding against CD276 and Thy1 using cell panning methods. Rec. + Panning indicates recombinant target-coated magnetic bead selections followed by cellular panning. Depleted Panning and Panning indicate cellular panning with or without, respectively, depletion of non-specific binders via streptavidin-coated magnetic beads. Binding specificity for each enriched population was assessed by cell panning. Yeast were panned for binders on monolayers of target-positive MS1 cells (blue) or target-negative MS1 cells as a negative control (orange). In the depleted panning case, recovery of yeast from the first (white) and second (gray) magnetic beads was also quantified. Data represent single-run analyses during the course of each discovery campaign.
Ligand Selections using Cell Panning Driven Methods
The enrichment-only selection strategy for adherent cellular panning yielded enrichment of ligands after three rounds of selection in all cases (Figure 3I–L). For fibronectin-based campaigns, yield preference was not observed for target-expressing cells relative to target negative cells. Nominal yield preference was observed for both affibodies selected against MS1-Thy1 (2.6% vs. 2.0%) and MS1-CD276 (2.8% vs. 1.8%). These outcomes are reasonable given the use of a naïve library, which likely contains binders to a variety of common cellular epitopes.
Depletion selections using streptavidin-coated magnetic beads were employed as a facile way to attempt to remove non-specific ligands. Unfortunately, the recovery of yeast during magnetic bead depletions was minimal across all campaigns and did not result in yield preference for target-expressing cells relative to target-negative cells after three rounds of selection (Figure 3 E–H). Notably, the populations exhibit substantially higher yields on mammalian cell panning (both target-positive and target-negative) than on streptavidin-coated magnetic bead selections.
Cellular Target Specificity and Relative Binding Strength Characterization
While the previous analyses provide valuable binding characterization of the enriched populations, ligand discovery relies on the identification of individual clones with the desired function. Thus, characterizations of cellular target specificity and relative binding strength of isolated clones were carried out using a clonal cell panning microscopy assay (Figure 4). Forty-eight clones from each enriched population were grown, induced for yeast display, and evaluated for binding to target-expressing and target-negative cells via yeast display / adherent mammalian cell panning. Binding strength was characterized by a relative count of yeast present in a microscope field (Figure 4B). Clones were characterized as “hits” if ligand-displaying yeast substantively bound target-expressing cells and did not appreciably bind target-negative cells. Ligands characterized in this way have previously translated to binding cellular targets as soluble ligands53. Unsuccessful clones, or “misses”, have multiple phenotypes: binding to both target-expressing and target-negative cells (not specific for target of interest), binding only to target-negative cells (counterspecific), and non-binding to target-expressing and target-negative cells (non-binding). Clones that specifically bound to target-expressing cells were sequenced to assess the diversity of the selected populations.
Figure 4. Clonal assessment of specificity for cellular target by yeast-displayed cell panning.

(A) Forty-eight individual clones from each sorted population were panned for binding to target-expressing and target-negative MS1 cells and characterized by phase microscopy. Binding specificity was characterized as described in the text. Relative binding strength was classified by yeast density observed in a random microscopy field. Sequence diversity of specific binders was determined by Sanger sequencing random hits. Each box contains results for four scaffold/target pairs as detailed in the legend at the right. (B) Representative images of yeast displaying “+++”, “++”, “+”, and “-” clones are shown.
Fibronectins selected against both soluble Thy1-Fc and CD276 ectodomains, as well as affibodies selected against Thy1-Fc, failed to yield any cell-binding ligands, with all attempts falling into the non-binding category. In contrast, affibodies selected against soluble CD276 showed a hit rate of 47/48 with various binding strengths. This population also retained ample diversity with 3 unique sequences out of 5 analyzed. Similarly, when selections with detergent-solubilized cell lysates were added to soluble target magnetic selections, only affibodies selected against CD276 yielded hits, albeit with weaker relative binding strength than the analogous population selected with soluble target methods only. This result is contrary to expectation as the concentration of soluble CD276 used for FACS (100 nM) is less stringent than the estimated CD276 concentration in the detergent-solubilized cell lysate (~33 nM). It is possible that the avidity afforded by multiple proteins present in individual detergent micelles encouraged the recovery of ligands with lower binding affinity, yielding this observation. These same clones would not be recovered using soluble CD276 for FACS, as the target would be monovalent. This hypothesis is supported by the lack of overlap in sequences recovered from both campaigns, albeit with a small sample size.
The addition of cellular selections to soluble target methods yielded hits from both the fibronectin and affibody populations selected against CD276. The fibronectin domains bound weakly and have converged on a single sequence whereas the affibodies exhibit diversity of both binding strength and sequence. Cellular selections were not able to salvage cell-binding activity from either the fibronectin or affibody populations selected against soluble Thy1-Fc.
Selections for ligands using cell panning with magnetic bead depletion yielded hits for fibronectin against CD276 as well as affibody against CD276. These hits generally had weak to moderate binding strength with high sequence diversity. Conversely to the methods that started with recombinant target-coated magnetic beads, the major source of misses was a lack of desired target specificity, with all populations having at least 20/48 within this category. Similarly, direct cell panning without depletion yielded hits for affibody against both Thy1 and CD276. These populations generally showed weak to moderate binding, high sequence diversity, and a high frequency of undesired specificity. In total, 8 of 12 campaigns that involved adherent cell panning yielded specific hits whereas 2 of 8 campaigns without adherent cell panning yielded specific hits.
Depletion of non-specific binders against adhered mammalian cells
The ability to recover cellular binders via adherent mammalian cell panning that were not enriched via recombinant target approaches motivates further study. Yet, the abundance of enriched ligands that were not specific for the desired target warrants development of improved selection methods. One identified potential avenue for decreasing the prevalence of non-target specific ligands was sequential depletion against target-negative mammalian cell monolayers. Unfortunately, the previously demonstrated effective incubation time for cellular selections is two hours22,24, which limits the number of sequential depletion steps that can be completed in a day. However, optimization of incubation time has not been reported. To determine if a reduced incubation time can be comparably effective, two CD276-specific affibody clones (named HS and LS for high- and low-yielding in the clonal panning assay) were panned against MS1-CD276 with 15, 30, 45, 60, or 120 min. incubation times. Both clones showed yields that were effectively invariant with time, with the slope of the recovery versus incubation time plot being essentially zero in both cases (0.5×104 ± 2×10−4 for LS and 0.45×10−3 ± 1×10−3 for HS) (Figure 5). Thus, 15 min was the chosen duration for depletion steps for the remainder of the study.
Figure 5. Optimization of incubation time for yeast-displayed ligand enrichment.

Yeast displaying affibody clones LS (A) or HS (B) were panned for binding to adherent MS1-CD276 with varying incubation times. Recoveries are presented as the mean ± error deviation of 7–12 trials.
Sequential Depletion of Non-Specific Binders Against Adhered Mammalian Cells
The ability to deplete non-specific binders while retaining specific binders was then experimentally investigated. Mixtures of CD276-specific, non-specific, and non-binding ligands were sorted by depleting with 0, 2, 4, or 6 sequential exposures to MS1-Thy1 cells followed by an enrichment step against MS1-CD276 (Figure 6). For the mixture of high-yield binders (clones HS, HN (high-yield non-specific), and A5 (non-binder)), the 0-depletion case yielded similar enrichment ratios for clones HS (24 ± 20) and HN (29 ± 7). The use of sequential depletion steps, on average, conferred an enrichment advantage to the CD276-specific HS clone relative to the non-specific HN (Figure 6A), but these differences largely lack statistical significance (p=0.08 for 2 depletions, p=0.03 for 4 depletions, and p=0.3 for 6 depletions). As additional depletion steps were added, the enrichment ratio of both clones HS and HN remained essentially unchanged (p>0.05 for all comparisons). However, it was difficult to draw conclusions from this data set because the enrichments after mammalian cell depletion were moderately inconsistent. For example, the average enrichment ratio of clone HS with 2 depletion steps was 140 ± 130. The enrichment ratios that yield this average were 220, 260, 270, 24, 20, and 17. Importantly, the final three trials listed essentially matched the average enrichment ratio of clone HN under this condition (24 ± 2), which suggested that there was no advantage conferred to clone HS despite the two depletion steps in these instances. Yet, no depleted experiment was ever appreciably less enriched than the non-depleted samples suggesting the potential for benefit over multiple iterations. Results using the low-yield binder mixture (LS, LN, and A5) showed similar performance (Figure 6B). Thus, sequential depletions exhibited potential efficacy for high-yielding clones, although performance was variable.
Figure 6. Sequential depletion of non-specific binders with mammalian cell monolayers.

Mixtures of high-yield (A) or low-yield (B) CD276-specific, non-specific, and non-binding yeast were subjected to selection with 0, 2, 4, or 6 depletion steps followed by a single enrichment step. All individual enrichment ratios are shown for 3–10 trials as well as the average.
Ligand Selections Using Cellular-Based Depletion
As an alternative to sequential depletion against mammalian cell monolayers, a pre-blocking strategy using disadhered mammalian cells was employed. In this approach, yeast mixtures are incubated with disadhered target-negative mammalian cells for 2 hrs prior to introduction of the whole mixture to a target-positive mammalian cell monolayer. Application of this scheme to a high-yield binder mixture conferred a considerable enrichment advantage to CD276-specific clone HS (120 ±32) relative to non-specific clone HN (12 ± 10), resulting in a 14 ± 6-fold selectivity in favor of clone HS (Figure 7A). This was a significantly higher selectivity (p=0.0002) than the non-depletion scheme (0.7 ± 0.6), the current standard in the field. However, application of the same strategy to the low-yield mixture resulted in a weaker selectivity advantage of 7 ± 10 for CD276-specific clone LS relative to non-specific clone LN (Figure 7B). This selectivity was nominally improved, but not definitively better, than the non-depletion case (0.2 ± 0.1, p=0.07).
Figure 7. Depletion of non-specific binders with mammalian cell pre-blocking.

Mixtures of high-yield (A) or low-yield (B) CD276-specific, non-specific, and non-binding yeast were incubated with either CD276-negative disadhered MS1-Thy1 cells (mammalian cell pre-block) or buffer only (no depletion) followed by incubation with MS1-CD276 cell monolayers. Enrichment ratios for CD276-specific (black), non-specific (gray), or non-binding (white) clones are shown as the mean ± standard deviation of 6–12 trials. (C). Specific (black) or non-target-specific (grey) clones were subjected to selection with or without mammalian cell pre-blocking for a total of 13 clone. Each point represents the yield of a single well of selection using pre-blocking normalized by a corresponding well selected without pre-blocking. Each clone was panned either in duplicate or triplicate.
To further investigate this trend, we panned 13 clones previously assayed for their cellular specificity (five CD276-selected specific clones, four non-target specific ligands isolated from selections against MS1-CD276, and four non-target specific ligands isolated from selections against MS1-Thy1) in parallel with and without pre-blocking. Non-specific clones were effectively depleted relative to specific binders (p = 0.006; Figure 7C). For a majority of clones, pre-blocking by target-negative mammalian cells was able to deplete the polyspecific clones (p<0.05 for 6 of 8 clones; median yield ratio = 0.33) without altering the recovery of specific ligands (p>0.05 for 5 of 5 clones; median yield ratio = 0.93) compared to panning without pre-blocking (Figure 7C). Thus, pre-blocking non-specific ligands with suspended non-target mammalian cells provides a compelling approach to improve yeast cell panning on adherent mammalian cells.
Directed Evolution of CD276-binding Affibodies
The preceding sections elucidate and enhance the ability to discover and select ligands against cellular targets using yeast surface display. To further exemplify this ability and to engineer synthetic ligands for targeting cancer vasculature, we also performed directed evolution of affibodies targeting CD276. Collectively across the selection campaigns, a diverse set of specific CD276-binding affibodies were identified. These clones were pooled for further directed evolution to select mutants with improved affinity amenable to preclinical development. DNA from this pooled population was mutated via error-prone PCR on the whole gene and on shuffled helices. The resulting diversified population was subjected to one round of cell panning, one round of FACS with 5 nM recombinant CD276, and one round of FACS with cell lysate. DNA sequencing of the enriched affibodies revealed a dominant clone hereafter termed AC2. Notably, this clone was also identified in sequencing binders from the unmutated soluble, recombinant target selection (Figure 4). AC2 was produced and purified from E. coli and evaluated for binding to MS1-CD276 cells. Despite isolation by binding to low concentrations of antigen in the yeast-displayed context, affinity titration experiments with soluble AC2 revealed a weak binding affinity (310±100 nM) to MS1-CD276 (Figure 10).
Figure 10. Deep sequencing of the merged library after enrichment reveals substantially improved mutants compared to parental.

(A) Top unique sequences of the merged library listed by number of reads. Parental AC2 is supplied as a reference. Blue lettering indicates diversified residues in the helix-walking libraries and dashes indicate parental amino acids at the indicated position. Clone names are supplied for sequences that were pursued for characterization. (B) Sitewise amino acid enrichments for the merged library. Amino acid frequencies were calculated by grouping, counting, and quad-root dampening identical sequences. Values shown are change in amino acid frequency in sorted populations compared to theoretical amino acid diversity of the naïve library. Amino acids not allowed by library design are shown in grey, except in cases where they are substantially enriched or depleted.
Prior work evolving antibodies, Gp2 domains, and fibronectin type III domains has shown success in isolating binders by loop-walking49–51 in which each structural segment of the binding paratope is independently evolved and then improved segments are merged. To perform an analogous maturation of the AC2 affibody, combinatorial libraries were created by diversifying helix one while retaining parental helix two and vice versa. The genetic diversity was designed to include the parental amino acid and chemically homologous amino acids accessible with degenerate codons (Supplemental Figure 2). The diversity of each library was limited to 5×107 to retain high confidence of sampling any sequence through yeast surface display by FACS. Each sub-library was subjected to a single magnetic bead sort, with non-target depletion, to eliminate non-binders and non-target-specific binders, followed by FACS with 5 nM soluble extracellular CD276 and then FACS with 150 nM solubilized MS1-CD276 cell lysate. Analysis showed an increase in the binding to CD276-positive lysate with minimal cross-reactivity towards target-negative lysate (Figure 8A). Deep sequencing of the resulting populations showed a relatively diverse first helix with at least four mutations from parental AC2 in the top sequences (Supplemental Figure 3). Sites K9 and W18 were conserved, while I10 was either conserved or mutated to homologous valine. F11 and G14 tolerated numerous options including the parental residue, while V13 and Y17 strongly favored two hydrophobic residues other than parental. Conversely, the second helix library retained less diversity, with parental being the dominant clone.
Figure 8. Evolved populations of yeast-displayed ligands show increasing binding to CD276 lysate while maintaining target specificity.

(A) Yeast populations collected after preliminary sorting on recombinant target beads (first column) or final sorting on target-positive lysate (second column) were labelled with 150 nM target cell lysate and analyzed for binding by flow cytometry. Yeast collected after sorting on target-positive lysate (third column) were labelled with 150 nM target-negative cell lysate and analyzed for specificity by flow cytometry. Substantial binding improvement can be observed between preliminary sorting and the final population, with low cross-reactivity to target-negative lysate. (B) A mixture of the merged library and triple-sorted single-helix library was labelled with 0.5 nM target lysate (left) or 50 nM target-negative lysate (right) and analyzed for binding by flow cytometry. The population shows significantly binding to target-positive lysate with minimal cross-reactivity to target-negative lysate.
DNA from the diversified helix from both libraries was extracted and merged to create a library with both helices diversified. The merged library and final single-helix library populations were combined and sorted stringently on FACS with 0.5 nM target-positive cell lysate. Analysis of this final library showed significant binding to CD276 cell lysate relative to control Thy1 lysate (Figure 8B).
Clonal Sanger sequencing returned three unique sequences; AC9 (5 of 8 sequences), AC12 (2 of 8 sequences), and AC16 (1 of 8 sequences). Consistent with the single-helix enrichment data, while each clone possessed mutations in the first helix, all clones retained the parental genotype in the second helix (Figure 9). Affinity titration of each of these clones revealed strong specific binding (KD = 18 ± 4 nM for AC9, KD = 0.9 ± 0.6 nM for AC12, and KD = 20 ± 4 nM for AC16) (Figure 9A and Figure 9B), an increase of 15–300× compared to parental AC2. Notably, these clones did not strongly bind to MS1-Thy1 cells relative to MS1-CD276 cells (Supplemental Figure 4). The thermal stability, secondary structure, and refoldability of all affibody variants was evaluated by circular dichroism spectroscopy. The thermal stability of all clones was substantially increased (Tm = 61.5 ± 0.1 °C for AC9, Tm = 62.4 ± 0.1 °C for AC12, and Tm = 58.5 ± 0.1 °C for AC16) relative to AC2 (48.6 ± 0.1 °C) (Figure 9D and Supplemental Figure 5). As expected, the secondary structure was shown to be predominately alpha-helical (Figure 9C and Supplemental Figure 6). Additionally, both parental AC2 and AC12 were tested for refolding by cooling after thermal denaturation. Both AC2 and AC12 showed almost no change in its CD spectra after cooling, indicating that they were likely refoldable (Figure 9C and Supplemental Figure 6).
Figure 9. Characterization of parental and evolved CD276-binding Affibodies.

(A) affibody variants AC2, AC9, AC12, and AC16 were characterized for their binding affinity (KD). Blue lettering indicates diversified residues in the helix-walking libraries. (B) Purified affibody variants AC2 (upper-left), AC9 (upper-right), AC12 (lower-left), and AC16 (lower-right) were used to label MS1-CD276 cells at the indicated concentrations. Binding was quantified by flow cytometry. The best-fit estimate of KD and 68% confidence interval are indicated by solid and dashed lines, respectively. (C) Purified affibody AC12 was analyzed by circular dichroism spectroscopy in triplicate between 200 and 260 nm wavelengths before (solid) and after (dashed) thermal denaturation and cooling. (D) Purified affibody AC12 was scanned at a wavelength of 220 nm during heating from 20 to 98 °C (1 °C/min). The midpoint of thermal denaturation (Tm) was calculated by linear least-squares regression using a two-state protein unfolding model.
To expand upon Sanger sequencing, the final library was also deep sequenced (Figure 10). While the first helix of the merged library showed similar trends in enrichment to the single-helix version of the library, a dominant amino acid appeared to have emerged at each site with the exception of F11 and G14. Notably, while the preferred amino acids in the merged G14 were not drastically altered from the single-helix counterpart, isoleucine emerged as the predominant residue of F11, when it was previously less prevalent in the single-helix library. As well, the top sequences once again showed a lack of diversity in the second helix, with parental being the favored phenotype. However, an interesting exception to this trend existed in the second most prevalent sequence, which possessed the parental first sequence and a number of mutations in the second helix not seen elsewhere.
Discussion
High-throughput screening methods have facilitated the discovery of proteins with novel binding activity, but difficulties translating these interactions from the laboratory targets to cellular targets have hampered progress in meeting clinical demands. The majority of ligand selection campaigns for membrane-bound targets – which represent a broadly important class of molecules – are performed with purified soluble domains, which are not necessarily good models of their corresponding full length antigens and can thus yield pools of binders with high affinity and specificity for the soluble domain but no activity toward the cellular antigen. Yeast surface display methods for direct cellular selections22–24 and fluorescence-activated cell sorting with detergent-solubilized cell lysates21,22 have been developed to sort for ligands with activity toward full length, cellular antigens, but the efficacy of all of these methods for ligand development has yet to be directly compared. We compared combinations of five selection techniques using two different naïve ligand libraries and two clinically relevant targets to determine the most efficient method for selection of proteins with specific activity toward cellular antigens. Hybrid selection techniques proved to yield the highest frequency of successful campaigns.
Campaigns conducted against only purified extracellular domains of Thy1 and CD276 failed, at this point in the discovery process, to yield ligands that bound cellular antigens in three out of four cases. It remains possible that mutation and/or additional selections would have yielded functional ligands. In all three failed campaigns, some level of non-specificity was observed at levels detectable with flow cytometry analysis. The Thy1 used in this study was an Fc fusion, and despite aggressive depletion with human IgG during magnetic bead selections, the presence of this tag allowed for the enrichment of clones that bind human IgG instead of their intended target (Figure 2). Interestingly, although it did not bind other reagents, the fibronectin population selected against CD276 also bound the soluble extracellular domain of MET (Supplemental Figure 1), a target it had never been exposed to during sorting. These case studies illustrate the importance of both careful consideration in reagent selection and sort design, as incomplete depletion or depletion with irrelevant antigens can allow specificity problems to persist. In contrast, the affibody population sorted against soluble CD276 returned a high frequency of diverse clones that specifically bound cellular CD276 with various binding strengths, suggesting that the CD276 used in this study contains some unknown number of translatable epitopes that could be accessed with the helical surface paratope library more efficiently than the loop paratope library.
The addition of FACS with detergent-solubilized cell lysates after magnetic bead selections with soluble extracellular domains was not able to salvage translatable binding ligands from any of the three failed campaigns (Figure 2E–H and Figure 4A). It is unclear whether lysate quality or target affinity prevented the isolation of a minority population of translatable binders in these selections. However, affibodies sorted against soluble CD276 were able to be isolated from two rounds of FACS sorting, albeit with weaker binding strength than the corresponding population isolated by soluble target selection methods only.
Direct cellular selections conducted after soluble target-based enrichment were able to enrich a population of CD276-specific fibronectin domains that consisted of a single translatable clone family (Figure 3B and Figure 4A). This clone was likely a minority component in the bead sorted population as it was not isolated without cellular enrichment. However, its existence in the bead sorted population does confirm that at least one translatable epitope exists in the soluble CD276 extracellular domain that is accessible by a loop paratope. Cellular selections also isolated a wide variety of CD276-binding affibodies. Interestingly, none of the affibodies isolated from bead sorting followed by cellular selection showed sequence overlap with those isolated from soluble target methods alone. This likely has to do with the nature of the affinity-based FACS conducted prior to characterization of clones isolated with soluble target only. Unmodified cellular selections provide minimal affinity pressure48, so affibodies isolated with this method likely fall in the population of clones with weaker affinity for both soluble and cellular CD276. Even cellular selections were not able to isolate cell-binding ligands from the fibronectin and affibody populations selected against soluble Thy1-Fc, suggesting that any translatable epitopes in this protein were not accessible by either paratope type.
Collectively, the use of cellular selections yielded hits in three out of four campaigns attempted. In contrast with the soluble domain-driven selection methods, which predominantly suffered from non-binders at the cellular level, the major source of “misses” for cellularly selected clones was non-specificity in all cases. Because the cell surface contains a wide array of macromolecules, including proteins, lipids, and polysaccharides, non-specific ligands have many options for binding partners. A subset of non-specific ligands potentially bind a specific antigen that is expressed on both target-expressing and target-negative cell lines. Others could bind macromolecules non-selectively through hydrophobic interactions, allowing them high avidity for enrichment on any cell type. In an attempt to deplete these non-specific binders, a set of campaigns utilized magnetic bead depletions prior to enrichment on mammalian cell monolayers. Population level quantification of recovery showed that this type of depletion was insufficient, recovering sub-1% quantities of yeast displaying non-specific ligands in all attempts (Figure 3). Indeed, when individual clones were assessed, the non-specificity problem persisted essentially identically to the case of cellular selection without depletion (Figure 4A). This result suggests that non-specificity has different characteristics depending on which selection method is used. Importantly, not all clones determined as “hits” in the clonal cell panning assay showed antigen specificity. Three affibodies against Thy1 from the cellular selection only arm showed non-specificity when produced as soluble proteins, suggesting some unquantified level of inconsistency between the yeast displayed and soluble versions of the proteins.
To decrease the prevalence of non-specific clones in cellular selections, multiple potential depletion techniques were explored. Sequential depletion, which has previously shown selectivity enhancement in panning for antibody fragments that bind glioblastoma stem-like cells54, resulted in inconsistent enrichment of target specific binders from these mixtures, which could be a result of non-specific yeast-mammalian cell interactions that are not ligand mediated or sequestration of yeast in the corners of the plate wells (Figure 6). It is known from previous studies that there is a baseline level of background recovery of approximately 0.1% of the yeast input24. Loss of the rare CD276-specific yeast through this mechanism would be impactful on their ability to be recovered. This effect may be compounded through each additional depletion step, potentially explaining the generally decreasing trend in enrichment of CD276-specific binders as more depletion steps are added. It is unclear, however, why the enrichment ratio of the non-specific binding yeast remains essentially unchanged with increasing depletion. It is possible that the yield of a non-specific binder against target negative cells may have been lower than expected, disfavoring depletion. Overall, these results suggest that sequential depletion against target negative mammalian cell monolayers will not confer the enrichment advantage to target specific binders in a practical setting.
Initial studies using depletion with disadhered mammalian cells yielded encouraging results in the high-yield case (Figure 7). If continued through multiple rounds, the 14 ± 6-fold selectivity advantage for the CD276-specific binder relative to the non-specific binder would propagate, accomplishing this study’s goal of non-specific binder depletion. The reduced ability to accomplish the same selectivity with the low-yield binder mixture was a surprising result. It appeared, based on the 0-depletion data, that the low-yield non-specific binder enriched more strongly against target positive cells than the low-yield CD276-specific binder. This could be the cause for the lack of selectivity, as any non-specific binder that is not depleted by the pre-blocking strategy has a 6-fold higher enrichment ratio than the CD276-specific binder. When 13 additional clones were tested with and without pre-blocking, a selective advantage to antigen-specific binders was observed.
The ability to strongly deplete the high-yield non-specific binders while being unable to deplete low-yield non-specific binders can still yield a functional improvement in selections from naïve libraries. It has been shown that strong- and mid-affinity binders can quickly out-enrich weaker binders in an EGFR-binding model system48. With the depletion of high-yield non-specific binders, the high- and mid-yield specific binders will have the opportunity to similarly out-enrich the remaining low-yield non-specific binders that could not be effectively depleted. Although the depletion of non-specific binders will not be complete in this regime, it will confer an important incremental improvement for cellular selections using yeast surface display libraries.
Towards the preclinical development of a synthetic ligand for CD276, we aimed to further enhance the affinity of existing CD276 binders by evolving the hits from all affibody campaigns towards CD276 via error-prone PCR and aggressive sorting. Despite this, sequenced clones were revealed to be a single sequence, AC2, which was found when the unmutated recombinant-selected population was sequenced (Supplemental Table 2). This indicates that random mutation was unsuccessful in producing a functional clone with higher binding affinity to CD276 cell-lysate. Given the moderate affinity of AC2, it is possible that the sparse sampling of sequence space created by error-prone PCR was insufficient to produce a substantially improved clone over AC2.
As an alternate approach, helix-walking, analogous to CDR-walking or loop-walking, was pursued as a mutagenesis strategy to create a more focused library with a higher proportion of functional clones. Two libraries were created in which a single helix was mutated while the other was constrained to parental AC2. Given the relative success of translating affibodies matured against recombinant CD276, a preliminary bead and FACS sort was conducted with recombinant target to deplete nonfunctional and nonspecific molecules under low affinity pressure. Further sorting with cell lysate FACS showed little cross-reactivity relative to previous sorts conducted on the naïve library and isolated a wide variety of CD276-binding affibodies in the helix one library (Supplemental Figure 3). The helix two library returned parental as the most prevalent sequence, with very little sequence diversity observed, indicating that far fewer beneficial mutations in the second helix exist.
Once merged and more aggressively sorted, the final population yielded a variety of sequences more prevalent than parental (Figure 9). For the most part, all sequences more prevalent than parental contained no mutations in the second helix, with the notable exception of the second most prevalent sequence, which contained several second helix mutations. This sequence warrants further study as to whether combination with any of the observed first helix mutations would provide further affinity, but is outside the scope of this study.
To confirm the increased affinity of the population, several top sequences were chosen at random and tested for binding affinity and thermal stability, which displayed substantial improvement over the parental CD276-binding affibody.
Conclusion
In conclusion, the current study advances the understanding of the advantages and disadvantages of multiple yeast-displayed ligand selection techniques, which can lead to the identification of more robust ligand selection strategies. Ligands isolated from soluble extracellular domain-driven campaigns can yield surprising non-specificity despite aggressive depletion, and ultimately not translate to cellular binding. Cellular selections can be employed as a follow-up to the use of soluble targets to enrich clones that bind translatable domains, but these are not present and accessible in all target-ligand pairings. Cellular selections from naïve libraries often lead to non-specificity, but depletion using disadhered mammalian cells – more so than soluble control proteins – shows promising results. A lead affibody against CD276 was isolated through these methods, which can be utilized for diagnostic and therapeutic benefit in future work.
Supplementary Material
Acknowledgements
This work was funded by the National Institutes of Health (R01 EB023339) and the American Cancer Society (130418-RSG-17-110-01-TBG). We appreciate assistance from the University of Minnesota Flow Cytometry Core.
Footnotes
Supporting Information
Assessment of fibronectin specificity to soluble CD276; affibody helix walking library design; deep sequencing the enriched CD276-binding population; cellular specificity, stability, and secondary structural analysis of CD276 binders; oligonucleotide sequences used in error-prone PCR; clonal Sanger sequencing of initial selection campaigns and depletion system
References
- (1).Mäbert K; Cojoc M; Peitzsch C; Kurth I; Souchelnytskyi S; Dubrovska A Cancer Biomarker Discovery: Current Status and Future Perspectives. Int. J. Radiat. Biol 2014, 90 (8), 659–677. [DOI] [PubMed] [Google Scholar]
- (2).Stern L; Case B; Hackel B Alternative Non-Antibody Scaffolds for Molecular Imaging of Cancer. Curr. Opin. Chem. Eng 2013, 2, 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Banta S; Dooley K; Shur O Replacing Antibodies: Engineering New Binding Proteins. Annu. Rev. Biomed. Eng 2013, 15, 93–113. [DOI] [PubMed] [Google Scholar]
- (4).Binz HK; Amstutz P; Plückthun A Engineering Novel Binding Proteins from Nonimmunoglobulin Domains. Nat. Biotechnol 2005, 23 (10), 1257–1268. [DOI] [PubMed] [Google Scholar]
- (5).Leader B; Baca QJ; Golan DE Protein Therapeutics: A Summary and Pharmacological Classification. Nat. Rev. Drug Discov 2008, 7 (1), 21–39. [DOI] [PubMed] [Google Scholar]
- (6).James ML; Gambhir SS A Molecular Imaging Primer: Modalities, Imaging Agents, and Applications. Physiol. Rev 2012, 92 (2), 897–965. [DOI] [PubMed] [Google Scholar]
- (7).Ackerman M; Levary D; Tobon G; Hackel B; Orcutt KD; Wittrup KD Highly Avid Magnetic Bead Capture: An Efficient Selection Method for de Novo Protein Engineering Utilizing Yeast Surface Display. Biotechnol. Prog 2009, 25 (3), 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McCafferty J; Griffiths a D.; Winter G; Chiswell DJ Phage Antibodies: Filamentous Phage Displaying Antibody Variable Domains. Nature 1990, 348 (6301), 552–554. [DOI] [PubMed] [Google Scholar]
- (9).Boder ET; Wittrup KD Yeast Surface Display for Screening Combinatorial Polypeptide Libraries. Nat. Biotechnol 1997, 15, 553–557. [DOI] [PubMed] [Google Scholar]
- (10).Baneyx F; Mujacic M Recombinant Protein Folding and Misfolding in Escherichia Coli. Nat. Biotechnol 2004, 22 (11), 1399–1408. [DOI] [PubMed] [Google Scholar]
- (11).Gasser B; Saloheimo M; Rinas U; Dragosits M; Rodríguez-Carmona E; Baumann K; Giuliani M; Parrilli E; Branduardi P; Lang C; Porro D; Ferrer P; Tutino M; Mattanovich D; Villaverde A Protein Folding and Conformational Stress in Microbial Cells Producing Recombinant Proteins: A Host Comparative Overview. Microb. Cell Fact 2008, 7 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mazzei GJ; Edgerton MD; Losberger C; Lecoanet-Henchoz S; Graber P; Durandy A; Gauchat JF; Bernard A; Allet B; Bonnefoy JY Recombinant Soluble Trimeric CD40 Ligand Is Biologically Active. Journal of Biological Chemistry 1995, pp 7025–7028. [DOI] [PubMed] [Google Scholar]
- (13).Singer E; Landgraf R; Horan T; Slamon D; Eisenberg D Identification of a Heregulin Binding Site in HER3 Extracellular Domain. J. Biol. Chem 2001, 276 (47), 44266–44274. [DOI] [PubMed] [Google Scholar]
- (14).Gomord V; Faye L Posttranslational Modification of Therapeutic Proteins in Plants. Curr. Opin. Plant Biol 2004, 7 (2), 171–181. [DOI] [PubMed] [Google Scholar]
- (15).Demain AL; Vaishnav P Production of Recombinant Proteins by Microbes and Higher Organisms. Biotechnol. Adv 2009, 27, 297–306. [DOI] [PubMed] [Google Scholar]
- (16).Kruziki MA; Bhatnagar S; Woldring DR; Duong VT; Hackel BJA 45-Amino-Acid Scaffold Mined from the PDB for High-Affinity Ligand Engineering. Chem. Biol 2015, 22 (7), 946–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hackel BJ; Ackerman ME; Howland SW; Wittrup KD Stability and CDR Composition Biases Enrich Binder Functionality Landscapes. J. Mol. Biol 2010, 401 (1), 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Friedman M; Nordberg E; Höidén-Guthenberg I; Brismar H; Adams GP; Nilsson FY; Carlsson J; Ståhl S Phage Display Selection of Affibody Molecules with Specific Binding to the Extracellular Domain of the Epidermal Growth Factor Receptor. Protein Eng. Des. Sel 2007, 20 (4), 189–199. [DOI] [PubMed] [Google Scholar]
- (19).Horak E; Heitner T; Robinson MK; Simmons HH; Garrison J; Russeva M; Furmanova P; Lou J; Zhou Y; Yuan Q; Weiner LM; Adams GP; Marks JD Isolation of ScFvs to in Vitro Produced Extracellular Domains of EGFR Family Members. Cancer Biother. Radiopharm 2005, 20 (6), 603–613. [DOI] [PubMed] [Google Scholar]
- (20).Chen TF; de Picciotto S; Hackel BJ; Wittrup KD Engineering Fibronectin-Based Binding Proteins by Yeast Surface Display. Methods Enzymol 2013, 523, 303–326. [DOI] [PubMed] [Google Scholar]
- (21).Cho YK; Shusta EV Antibody Library Screens Using Detergent-Solubilized Mammalian Cell Lysates as Antigen Sources. Protein Eng. Des. Sel 2010, 23 (7), 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tillotson BJ; Cho YK; Shusta EV Cells and Cell Lysates: A Direct Approach for Engineering Antibodies against Membrane Proteins Using Yeast Surface Display. Methods 2013, 60 (1), 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang XX; Shusta EV The Use of ScFv-Displaying Yeast in Mammalian Cell Surface Selections. J. Immunol. Methods 2005, 304 (1–2), 30–42. [DOI] [PubMed] [Google Scholar]
- (24).Stern LA; Schrack IA; Johnson SM; Deshpande A; Bennett NR; Harasymiw LA; Gardner MK; Hackel BJ Geometry and Expression Enhance Enrichment of Functional Yeast-Displayed Ligands via Cell Panning. Biotechnol. Bioeng 2016, 113(11), 2328–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).VanAntwerp JJ; Wittrup KD Fine Affinity Discrimination by Yeast Surface Display and Flow Cytometry. Biotechnol. Prog 2000, 16 (1), 31–37. [DOI] [PubMed] [Google Scholar]
- (26).Hofmeyer KA; Ray A; Zang X The Contrasting Role of B7–H3. Proc. Natl. Acad. Sci 2008, 105 (30), 10277–10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Crispen PL; Sheinin Y; Roth TJ; Lohse CM; Kuntz SM; Frigola X; Houston Thompson R; Boorjian SA; Dong H; Leibovich BC; Blute ML; Kwon ED Tumor Cell and Tumor Vasculature Expression of B7–H3 Predict Survival in Clear Cell Renal Cell Carcinoma. Clin. Cancer Res 2008, 14 (16), 5150–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang J; Chong KK; Nakamura Y; Nguyen L; Huang SK; Kuo C; Zhang W; Yu H; Morton DL; Hoon DSB B7–H3 Associated with Tumor Progression and Epigenetic Regulatory Activity in Cutaneous Melanoma. J. Invest. Dermatol 2013, 133 (8), 2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhou Z; Luther N; Ibrahim GM; Hawkins C; Vibhakar R; Handler MH; Souweidane MM B7–H3, a Potential Therapeutic Target, Is Expressed in Diffuse Intrinsic Pontine Glioma. J. Neurooncol 2013, 111 (3), 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Katayama A; Takahara M; Kishibe K; Nagato T; Kunibe I; Katada A; Hayashi T; Harabuchi Y Expression of B7–H3 in Hypopharyngeal Squamous Cell Carcinoma as a Predictive Indicator for Tumor Metastasis and Prognosis. Int. J. Oncol 2011, 38 (5), 1219–1226. [DOI] [PubMed] [Google Scholar]
- (31).Yuan H; Wei X; Zhang G; Li C; Zhang X; Hou J B7–H3 over Expression in Prostate Cancer Promotes Tumor Cell Progression. J. Urol 2011, 186 (3), 1093–1099. [DOI] [PubMed] [Google Scholar]
- (32).Lutz AM; Bachawal SV; Drescher CW; Pysz MA; Willmann JK; Gambhir SS Ultrasound Molecular Imaging in a Human CD276 Expression-Modulated Murine Ovarian Cancer Model. Clin. Cancer Res 2014, 20 (5), 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yamato I; Sho M; Nomi T; Akahori T; Shimada K; Hotta K; Kanehiro H; Konishi N; Yagita H; Nakajima Y Clinical Importance of B7–H3 Expression in Human Pancreatic Cancer. Br. J. Cancer 2009, 101 (10), 1709–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhao X; Li DC; Zhu XG; Gan WJ; Li Z; Xiong F; Zhang ZX; Zhang GB; Zhang XG; Zhao H B7–H3 Overexpression in Pancreatic Cancer Promotes Tumor Progression. Int. J. Mol. Med 2013, 31 (2), 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Foygel K; Wang H; Machtaler S; Lutz AM; Chen R; Pysz M; Lowe AW; Tian L; Carrigan T; Brentnall TA; Willmann JK Detection of Pancreatic Ductal Adenocarcinoma in Mice by Ultrasound Imaging of Thymocyte Differentiation Antigen 1. Gastroenterology 2013, 145 (4), 885–894.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Koide A; Bailey CW; Huang X; Koide S The Fibronectin Type III Domain as a Scaffold for Novel Binding Proteins. J. Mol. Biol 1998, 284 (4), 1141–1151. [DOI] [PubMed] [Google Scholar]
- (37).Lipovšek D Adnectins: Engineered Target-Binding Protein Therapeutics. Protein Eng. Des. Sel 2011, 24 (1–2), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nord K; Nilsson J; Nilsson B A Combinatorial Library of an α-Helical Bacterial Receptor Domain. Protein Eng 1995, 8 (6), 601–608. [DOI] [PubMed] [Google Scholar]
- (39).Lofblom J; Feldwisch J; Tolmachev V; Carlsson J; Stahl S; Frejd FY Affibody Molecules: Engineered Proteins for Therapeutic, Diagnostic and Biotechnological Applications. FEBS Lett 2010, 584 (12), 2670–2680. [DOI] [PubMed] [Google Scholar]
- (40).Baum RP; Prasad V; Müller D; Schuchardt C; Orlova A; Wennborg A; Tolmachev V; Feldwisch J Molecular Imaging of HER2-Expressing Malignant Tumors in Breast Cancer Patients Using Synthetic 111In- or 68Ga-Labeled Affibody Molecules. J. Nucl. Med 2010, 51 (6), 892–897. [DOI] [PubMed] [Google Scholar]
- (41).Hackel BJ; Kimura RH; Gambhir SS Use of 64 Cu-Labeled Fibronectin Domain with EGFR-Overexpressing Tumor Xenograft: Molecular Imaging. Radiology 2012, 263 (1), 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Case BA; Kruziki MA; Stern LA; Hackel BJ Evaluation of Affibody Charge Modification Identified by Synthetic Consensus Design in Molecular PET Imaging of Epidermal Growth Factor Receptor. Mol. Syst. Des. Eng 2018, 3 (1), 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Stern LA; Case BA; Hackel BJ Alternative Non-Antibody Protein Scaffolds for Molecular Imaging of Cancer. Curr. Opin. Chem. Eng 2013, 2 (4), 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Woldring DR; Holec PV; Zhou H; Hackel BJ High-Throughput Ligand Discovery Reveals a Sitewise Gradient of Diversity in Broadly Evolved Hydrophilic Fibronectin Domains. PLoS One 2015, 10 (9), e0138956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Woldring DR; Holec PV; Stern LA; Du Y; Hackel BJ A Gradient of Sitewise Diversity Promotes Evolutionary Fitness for Binder Discovery in a Three-Helix Bundle Protein Scaffold. Biochemistry 2017, 56 (11), 1656–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hackel BJ; Kapila A; Wittrup KD Picomolar Affinity Fibronectin Domains Engineered Utilizing Loop Length Diversity, Recursive Mutagenesis, and Loop Shuffling. J. Mol. Biol 2008, 381 (5), 1238–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Zaccolo M; Williams DM; Brown DM; Gherardi E An Approach to Random Mutagenesis of DNA Using Mixtures of Triphosphate Derivatives of Nucleoside Analogues. J. Mol. Biol 1996, 255 (4), 589–603. [DOI] [PubMed] [Google Scholar]
- (48).Stern LA; Csizmar CM; Woldring DR; Wagner CR; Hackel BJ Titratable Avidity Reduction Enhances Affinity Discrimination in Mammalian Cellular Selections of Yeast-Displayed Ligands. ACS Comb. Sci 2017, 19 (5), 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Barbas CF; Hu D; Dunlop N; Sawyer L; Cababa D; Hendry RM; Nara PL; Burton DR In Vitro Evolution of a Neutralizing Human Antibody to Human Immunodeficiency Virus Type 1 to Enhance Affinity and Broaden Strain Cross-Reactivity. Proc. Natl. Acad. Sci 1994, 91 (9), 3809–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kruziki MA; Sarma V; Hackel BJ Constrained Combinatorial Libraries of Gp2 Proteins Enhance Discovery of PD-L1 Binders. ACS Comb. Sci 2018, 20 (7), 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lipovšek D; Lippow SM; Hackel BJ; Gregson MW; Cheng P; Kapila A; Wittrup KD Evolution of an Interloop Disulfide Bond in High-Affinity Antibody Mimics Based on Fibronectin Type III Domain and Selected by Yeast Surface Display: Molecular Convergence with Single-Domain Camelid and Shark Antibodies. J. Mol. Biol 2007, 368 (4), 1024–1041. [DOI] [PubMed] [Google Scholar]
- (52).Gill SC; von Hippel PH Calculation of Protein Extinction Coefficients from Amino Acid Sequence Data. Anal. Biochem 1989, 182 (2), 319–326. [DOI] [PubMed] [Google Scholar]
- (53).Wang XX; Cho YK; Shusta EV Mining a Yeast Library for Brain Endothelial Cell-Binding Antibodies. Nat. Methods 2007, 4 (2), 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Zorniak M; Clark PA; Umlauf BJ; Cho Y; Shusta EV; Kuo JS Yeast Display Biopanning Identifies Human Antibodies Targeting Glioblastoma Stem-like Cells. Sci. Rep 2017, 7 (1), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.