


Abstract
Motor enzymes that process nucleic-acid substrates play vital roles in all aspects of genome replication, expression, and repair. The DNA and RNA nucleobases are known to affect the kinetics of these systems in biologically meaningful ways. Recently, it was shown that DNA bases control the translocation speed of helicases on single-stranded DNA, however the cause of these effects remains unclear. We use single-molecule picometer-resolution nanopore tweezers (SPRNT) to measure the kinetics of translocation along single-stranded DNA by the helicase Hel308 from Thermococcus gammatolerans. SPRNT can measure enzyme steps with subangstrom resolution on millisecond timescales while simultaneously measuring the absolute position of the enzyme along the DNA substrate. Previous experiments with SPRNT revealed the presence of two distinct substates within the Hel308 ATP hydrolysis cycle, one [ATP]-dependent and the other [ATP]-independent. Here, we analyze in-depth the apparent sequence dependent behavior of the [ATP]-independent step. We find that DNA bases at two sites within Hel308 control sequence-specific kinetics of the [ATP]-independent step. We suggest mechanisms for the observed sequence-specific translocation kinetics. Similar SPRNT measurements and methods can be applied to other nucleic-acid-processing motor enzymes.
INTRODUCTION
Nucleic-acid-processing motor enzymes such as helicases, polymerases and translocases that move along DNA or RNA play vital roles in genome replication, expression and maintenance (1–6). The nucleic-acid (NA) sequence has been shown to regulate important properties of some of these enzymes. For example, RNA polymerase transcriptional elongation and pausing are controlled by specific DNA sequences (7–9). Helicases are known to have sequence-dependent DNA unwinding kinetics due to the energetic differences between AT and GC base pairs (10–12). Recent reports have demonstrated that the DNA helicases Hel308 and UvrD translocate on single-stranded DNA (ssDNA) in a sequence-dependent manner (13,14), however the mechanism for this sequence-dependence is unclear. To understand the role that DNA sequence plays in sequence-dependent translocation on ssDNA by helicase Hel308, we used Single-molecule Picometer Resolution Nanopore Tweezers (SPRNT, (15,16)), a high-throughput technology with both high spatiotemporal resolution and the ability to directly reveal the NA sequence that is passing through a motor enzyme. Here we use SPRNT to determine the relationship between kinetic parameters and the DNA sequence in Hel308.
In SPRNT, a single Mycobacterium smegmatis porin A (MspA) nanopore (17) in a phospholipid bilayer forms the only electrical connection between two electrolyte solutions (Figure 1A). A voltage applied across the nanopore causes ions to flow through the nanopore, establishing a measurable ion current. Negatively charged NA bound to a motor enzyme is attracted into the nanopore by the electric field until the enzyme comes to rest on the rim of the nanopore. The enzyme walks along the NA, causing the NA to move through the nanopore. The NA bases in the nanopore reduce the ion current flowing through the nanopore depending on the nucleotide sequence in the nanopore, enabling nanopore sequencing (18–20). The ion current also provides a precise time-record of enzyme position along the NA (Figure 1B). During SPRNT, the electric force on the DNA results in a force (∼0.2 pN/mV, resulting in ∼35 pN force at 180 mV voltage) on the enzyme which can be with, or against the enzyme motion (15). While it is possible that this force affects helicase kinetics, we observed that Hel308 is independent of the applied voltage over the range of ∼30–65 pN (13). SPRNT is the only single-molecule technology which simultaneously reads out the NA sequence and provides high-resolution measurements of enzyme motion on NA (<1 Å spatial resolution at 1 ms state duration, (15,16)), making this technology an ideal method for understanding how NA sequence affects motor enzyme kinetics.
Figure 1.

(A) A single DNA molecule (black) bound to a Hel308 helicase (blue) is drawn into the MspA pore (yellow) by the voltage applied with electrodes (gray). Ion current is measured and used to monitor the progression of a Hel308 helicase along the DNA strand. X represents the distance between the nanopore constriction and the site of sequence dependent effects in the helicase. (B) Position versus time trace for a single Hel308 molecule, with backwards steps indicated. The DNA sequence in MspA nanopore is shown with the associated DNA position. This curve is constructed from the current vs. time data by a non-linear transformation function as described in (13,15,16). (C) DNA design. While Hel308 walks along a 21-nt DNA test sequence (red) that we change to alter Hel308’s kinetics, the heteropolymer measuring sequence (blue) is pulled through the MspA constriction, yielding a high-contrast series of ion current states on which kinetic measurements are made. N is any of A, C, G and T. X in this sequence refers to an abasic site, not to be confused for the distance measurement in panel A.
We previously used SPRNT to characterize how the ATPase cycle of the superfamily 2 DNA helicase Hel308 from Thermoccocus gammatolerans coordinates translocation on a single-stranded DNA (ssDNA) substrate (13). Hel308 is a 3′ to 5′ translocase/helicase found in archaea and eukaryotes and is thought to be involved in restarting stalled replication machinery (21,22). We found that Hel308 pulled DNA through the MspA nanopore in two steps per nucleotide translocated (13,15). We hypothesized these sub-nt movements of the DNA to be the result of conformational changes of Hel308. The kinetics of one of these steps are [ATP]-dependent while the kinetics of the other are [ATP]-independent. These two steps thus represent two separate stages of the Hel308 hydrolysis cycle, with the [ATP]-dependent step involved in ATP and ADP binding, and the [ATP]-independent step in ATP hydrolysis. The [ATP]-independent step corresponds to a closed conformation of Hel308, and ATP hydrolysis results in a relative change in binding energies of the RecA folds to different sites on the DNA, generating forward motion of the helicase. We found, surprisingly, that the state dwell-times, as well as the propensity of Hel308 to step backwards, varied substantially along a heterogenous DNA substrate for both [ATP]-dependent and [ATP]-independent states, which we hypothesized to be due to DNA interacting base-specifically with Hel308 (13). Here, we investigate the effects of DNA sequence on ssDNA translocation by Hel308 helicase by examining 1256 single-molecule SPRNT recordings of Hel308 over 26 different short ssDNA substrates of varying DNA sequence (Supplementary Table S1).
MATERIALS AND METHODS
Pore establishment
A single M2-NNN MspA nanopore was established in a 1,2-di-O-phytanyl-sn-glycero-3-phosphocholine (DOPHPC) lipid bilayer using methods that have been well established (16). Lipids were ordered from Avanti Polar Lipids.
DNA preparation
A complementary DNA strand was annealed to the template DNA strand so that the template has a free 5′ end, and an 8-base 3′ overhang. Hel308 binds to this 8-base overhang, and can begin to unwind dsDNA in solution, meaning that an event can start at any location along the DNA molecule, although most experiments begin near the start of the DNA sequence (Supplementary Figure S1). To prevent accumulation of ADP in bulk we perfused new reagents every 45 minutes. The 5′ end of the template is drawn into the pore, dissociating the complement. If a Hel308 is bound to the DNA it will translocate from 3′ to 5′, drawing the ssDNA out of the pore. While in principle it is possible for DNA to go through the pore in the 3′ orientation, no such events were observed.
The DNA sequences for the template strands are listed in Supplementary Table S1.
The DNA sequence for the complementary strand is:
5′ CCTGCATGAGAATGCGATAGTGAGATTTTTTTTTTTTTTTTTTTTZ 3′, where ‘Z’ is a cholesterol tag to facilitate binding of the DNA into the bilayer to increase the interaction rate of DNA with the nanopore. This complement was used for each sequence in this study. Supplementary Figure S2 shows a depiction of the DNA+enzyme+nanopore configuration at the start of each experiment.
Proteins
Hel308 from Thermococcus gammatolerans EJ3 (accession number WP_015858487.1) and M2-NNN MspA (accession number CAB56052.1) were prepared as described previously (15).
Operating conditions: All experiments were run at 400 mM KCl, with 10 mM HEPES at pH 8.0 and 10 mM MgCl2. Once a single M2-NNN MspA nanopore was established, a buffer with the above conditions along with 1 mM ATP was perfused to the cis well. ATP was ordered from Sigma Aldrich. The perfusion is done to maintain constant concentrations in the reaction volume. DNA, DTT, and Hel308 were added to final concentrations of 5 nM, 1 mM and 50 nM, respectively. All experiments were performed at elevated temperature 37°C as described in (13). We show in the supplementary data that the kinetics of Hel308 translocation are independent of [KCl] down to 100 mM and independent of the applied voltage down to 60 mV (∼12 pN, Supplementary Figures S3 and S4). Because of the large excess in Hel308 to DNA, we considered the possibility that we could have multiple helicases on the same DNA molecule, which could influence Hel308 kinetics. We measured the distribution of helicase velocities and found it to be well-described by a single-gaussian distribution (P = 0.37, KS test). If there were a significant contribution to the kinetics from multiple-Hel308-bound DNAs, then we would expect the distribution of velocities to be bimodal, which we do not observe.
Data acquisition
Data was acquired with custom labview software on an Axopatch 200B amplifier at 50 kHz, and downsampled by averaging to 5 kHz.
Raw Data Analysis: Ion-current versus time raw traces were converted to position versus time traces as described in (13,15,16). Dwell-times were determined by aligning ion-current segments to the consensus sequence as in (13).
Experiment design
In SPRNT, the DNA sequence in the narrowest part of the MspA nanopore (termed ‘the constriction’) determines the ion current flowing through the nanopore, while the DNA sequence in the helicase (X nucleotides away from the constriction, Figure 1A) determines the stepping kinetics (Figure 1A). Thus, in order to interpret the results of these experiments, we must first determines the registration distance, X, in nucleotides, between the MspA nanopore constriction and the nucleotides in Hel308 that are responsible for sequence-dependent kinetics. Therefore, we observe how kinetic parameters change as we change the DNA substrate passing through Hel308.
We designed DNA strands with two distinct regions: first a heteropolymer that produces an easily recognizable ion-current signal, maximizing the sensitivity of SPRNT (‘measuring sequence’, Supplementary Figure S5), and second, a variable region where test DNA sequences reside within Hel308 when the heteropolymer sequence is in the MspA constriction (‘test sequence’, Figure 1C., Supplementary Table S1). We measured two kinetic quantities in these experiments: (1) the distribution of dwell times at each DNA position in the heteropolymer and (2) the probability the enzyme moves backwards at each DNA position, where we define a backwards step as a motion of the enzyme that corresponds to DNA movement from 5′ to 3′ through MspA (as opposed to a forwards step that corresponds to DNA movement from 3′ to 5′, Figure 1B). Because the [ATP]-independent step is rate limiting at saturating [ATP] and causes >95% of Hel308 backwards steps, we focus mostly on analysis of the [ATP]-independent step, with brief comments on the [ATP]-dependent step.
RESULTS
Hel308 translocation over homopolymer sequences & dinucleotide repeats
We first analyzed Hel308 translocation over 21-nucleotide-long homopolymer sequences of adenine (A), cytosine (C), and thymine (T) to develop a basic understanding for how DNA sequence affects translocation kinetics (Supplementary Table S1). Due to secondary structure formation, we did not test homopolymer guanine (G), however we examined a G-rich strand below. We found that the average dwell-time of [ATP]-independent steps was approximately constant over many nucleotides for each of the homopolymers, but different between each of the three sequences (tA = 70 ± 2 ms, tC = 55 ± 3 ms and tT = 44 ± 1 ms, Figure 2A). However, after ∼20 nt (positions 20–25, Figure 2A), the dwell times of each of the three sequences exhibited a common pattern, indicating that Hel308 had exited the ‘test sequence’ and begun walking along the ‘measuring sequence’ common to each of the DNA strands that we used in this experiment.
Figure 2.

(A) Average dwell-time of forwards steps versus DNA position for Hel308 translocation on homopolymer sequences of adenine (left), cytosine (middle) and thymine (right). The light blue line plotted in each panel is the average taken over DNA positions 0–16. The shaded boxes indicate the DNA positions with identical kinetics between each sequence, suggesting that Hel308 is translocating over the ‘measuring sequence’ identical to each of the DNA sequences used in this study. (B) Probability of a backwards step vs. DNA position for Hel308 translocation on homopolymer sequences of adenine (left), cytosine (middle) and thymine (right). The light blue line plotted in each panel is the average taken over DNA positions 0–16. (C) Average dwell-time of forwards steps vs. DNA position for Hel308 translocation over dinucleotide repeats of GT (far left), AG (middle left), CA (middle right) and TC (far right). (D) Probability of a backwards step vs. DNA position for Hel308 translocation over dinucleotide repeats of GT (far left), AG (middle left), CA (middle right) and TC (far right). Each of these plots are time ordered, so that 3′ is on the left and 5′ is on the right. Gaps are DNA positions where an ion-current state was similar to an adjacent state. These gaps are slightly different between the homopolymer (panels A and B) and dinucleotide experiments (panels C and D) because the experiments used slightly different measuring sequences (supplementary discussion, Supplementary Figure S5, Supplementary Table S1). All error bars are SEM.
Next, we examined the probability of a backwards step vs. DNA position for Hel308 translocation over the homopolymer sequences (Figure 1B). We found that the probability of a backwards step for adenine and thymine homopolymers to be 15 ± 1% and 19 ± 1%, respectively, however, Hel308 rarely backstepped on the cytosine homopolymer (2.4 ± 0.7%). Again, after ∼20 nt, the probability of a backwards steps was similar for each of the three sequences. This is not surprising since the dwell-times and the probability of backward steps should only depend on sequence within the enzyme and not on the nucleotide type in the constriction of the MspA nanopore (13).
In addition to homopolymer test sequences, we performed experiments of Hel308 translocation over all possible dinucleotide repeats that would not form hairpin structures (AG, CA, TC and GT, Supplementary Table S1). Figure 2C shows the average dwell-time vs. DNA position for each dinucleotide test sequence. As expected, the dwell-times alternates with a 2-nt periodicity, consistent with sequence-dependent effects. Again, after ∼20 nt, the dwell times for each sequence exhibit the same pattern as shown in Figure 1A for the homopolymer test sequence. Figure 2D shows the probability of a backwards step versus DNA position for each of the four dinucleotide test sequences. The backwards step probability also alternates with 2-nt periodicity. In three of four dinucleotide test sequences the dwell-times are correlated with the probability of a backwards step (GT, CA and TC), whereas in the other sequence (AG) the dwell-times are anti-correlated with the probability of a backwards step, which could imply different mechanisms for Hel308 backwards steps that depend on the DNA sequence in Hel308. Interestingly, the two sequences with cytosine show that Hel308 rarely backsteps at every other nucleotide position. In the two dinucleotide sequences containing adenine we see that the probability of a backwards step at every other nucleotide position is larger than was observed in the homopolymer adenine sequence (29 ± 2% in CA and 57 ± 2% in AG). This suggests that more than one nucleotide in Hel308 is contributing to the observed kinetics, and that adenine promotes backstepping.
Determining the registration between MspA nanopore constriction and Hel308 by DNA sequence substitution
To determine the registration distance, X, we measured Hel308 translocation over a heteropolymer test sequence (Figure 3A, black). We observed that at DNA position 9 on this DNA sequence Hel308 backsteps >50% of the time. Based on our results above suggesting that adenine adjacent to cytosine and guanine encourage Hel308 backstepping (Figure 2) we identified the sequence 3′…AGAC… 5′ as being a possible candidate for causing the large probability of a backwards step. To test that this DNA sequence was responsible for the observed kinetics, we used our knowledge that cytosine seems to reduce backstepping and changed the DNA sequence AGAC sequentially to ACAC, ACCC and CCCC. Figure 3 shows the probability of a backwards step versus DNA position for each of these four sequences. We found that by offsetting the DNA sequence by X = 20 nt compared to the sequence measured within the MspA nanopore constriction, that Hel308 had significantly reduced backstepping at the location of each introduced cytosine (positions 7, 8, 9 in Figure 3), while backstepping at other positions was mostly unchanged. In fact, with the registration set at 20 nt, the probability of a backwards step at each DNA position with a cytosine at X = 20 nt was <5%, whereas when other nucleotides are 20 nt away from the MspA nanopore constriction, the probability of a backwards step is larger. The dwell-times also change at the same DNA positions as the backstepping kinetics (Supplementary Figures S6-S7, supporting discussion). These results suggest a registration distance of X = 20 nt between the MspA nanopore constriction and the nucleotides that are primarily responsible for sequence-dependent kinetics within Hel308.
Figure 3.

Probability of a backwards step versus DNA position for sequential mutations to a heteropolymer test sequence. Three bases suspected of causing the large probability of a backwards step at position 9 (top, black) were sequentially mutated to cytosine: (top) AGAC to ACAC (middle) ACAC to ACCC (bottom) ACCC to CCCC. The DNA sequence has been offset 20 nt compared to the nucleotides within the MspA constriction so that changes in the DNA sequence match up with the most significant changes in the observed probability of a backwards step. The introduction of each cytosine causes the probability of a backwards step to decrease compared to the original ‘AGA’ sequence. Each of these plots are time ordered, so that 3′ is on the left and 5′ is on the right. Gaps are DNA positions where an ion-current state was similar to an adjacent state. All error bars are SEM.
Analysis of guanine rich sequences
To understand the effects of guanine on Hel308 kinetics, we analyzed Hel308 translocation over a guanine-rich sequence. Interestingly, we found that in many cases the probability of a backwards step was small when a guanine was present 20 nt away from the MspA nanopore constriction. However, three consecutive guanines from X = 19 nt to X = 21 nt away from the MspA constriction produced large probability of a backwards steps (50 & 70% for two GGG motifs, Supplementary Figure S8), suggesting that at least three nucleotides affect sequence-dependent kinetics.
Motivated by variations in the homopolymer and dinucleotide experiments such as the long dwell-time states in the dinucleotide AG experiment at positions 18 and 20 (Figure 2C), we performed an experiment in which we varied the sequence context surrounding a single ‘GGG’ motif to determine if other nucleotide positions affected Hel308 kinetics. Figure 4 shows the probability of a backwards step versus DNA position for two test DNA sequences, one of the form 3′…AAAGGGCCC…5′ and another of the form 3′ …CCCGGGAAA… 5′. We found that when the central guanine was at X = 20 nt, the probability of a backwards step for the first test sequence was 36 ± 3% while for the second test sequence it increased to 86 ± 7%, suggesting that the DNA sequence context surrounding X = 20 nt away from MspA also affects translocation kinetics.
Figure 4.

Probability of a backwards step vs. DNA position for two sequences, one of the form 3′ …AAAGGGCC… 5′ (red) and one of the form 3′ …CCCGGGAAA… 5′ (blue). Changing the context around the ‘GGG’ motif causes the probability of a backwards step to change from 36% to 86% at the location of the central guanine suggesting additional sequence context also contributes to the observed backstepping behavior. Each of these plots are time ordered, so that 3′ is on the left and 5′ is on the right. Gaps are DNA positions where an ion-current state was similar to an adjacent state. All error bars are SEM.
Hel308 translocation over heteropolymers
To determine which additional sites in Hel308 are responsible for sequence-dependent kinetics (Figure 5A), we analyzed Hel308 translocation over ten 21-nt-long heteropolymer sequences (Supplementary Table S1). Figure 5B shows the probability of a backwards step vs. DNA position, given the identity of the Xth nucleotide away from the MspA constriction. For example, the blue data point at X = 20 nt is the average probability of a backwards step given that an adenine was located 20 nt away from MspA’s constriction. As expected based on the above experiments, we see that when adenine is at X = 20 nt the probability of a backwards step is large compared to the mean, and when cytosine is at X = 20 nt the probability of a backwards step is significantly reduced compared to the mean. In addition, this plot reveals that when cytosine is 17 nt away from the MspA constriction the probability of a backwards step is also reduced, consistent with our observation that cytosines to the 5′ side of a ‘GGG’ motif led to a reduction in the probability of a backwards step (Figure 4).
Figure 5.

(A) Schematic illustrating the registration (X) between DNA bases within the MspA constriction and DNA bases within Hel308 helicase. (B) Average probability of a backwards step vs. position above the MspA constriction (X), given that the nucleotide at position X is adenine (blue), cytosine (green), guanine (yellow) or thymine (red). The black line is the average taken over all data. (C) Average dwell-time vs. X given the nucleotide at position X. The black line is the average taken over all data.
Figure 5C shows the average dwell-time of Hel308 forwards steps vs. DNA position, again conditioned on the Xth nucleotide away from the constriction. We see that at X = 20 nt there is a slight decrease in dwell-time due to cytosine, but at X = 17 nt cytosine leads to an increase in the dwell-time compared to the mean. Thymine at X = 17 nt leads to a decreased dwell-time, consistent with our observation that the dwell-times of Hel308 translocation over the thymine homopolymer were shorter than either adenine or cytosine.
Analysis of the [ATP]-dependent step
While we have focused primarily on the [ATP]-independent step, we also examined the [ATP]-dependent step. As above, we plotted the average dwell-time vs. DNA position conditioned on the Xth nucleotide away from the MspA nanopore constriction (Supplementary Figure S9). We found no obvious relation between single DNA positions and the dwell-time, however statistical analysis using the dwell-time distributions of homopolymers and dinucleotide repeats suggests that that the DNA sequence does affect the [ATP]-dependent step (supporting discussion). This suggests that several DNA bases are responsible for sequence-dependent behavior in the [ATP]-dependent step. A study involving far more sequence contexts will be necessary to elucidate the cause of sequence-dependent kinetics in the [ATP]-dependent step.
DISCUSSION
We applied SPRNT to measure the effects of ssDNA base composition on translocation kinetics of the helicase Hel308. The ability of SPRNT to resolve sub-nucleotide-long DNA motion at millisecond time scales and to correlate the kinetics with the DNA composition within the enzyme reveals detailed sequence-dependent behavior of Hel308. Direct observation of such sequence-dependent activity was beyond the capabilities of other single-molecule techniques. We found that sequence-dependent kinetics in the Hel308 helicase are primarily determined by the nucleotides at two sites in Hel308, at X = 17 nt and X = 20 nt relative to the nanopore MspA constriction. Fully understanding the mechanism for sequence-dependence requires a detailed look at how and where the DNA interacts with amino-acid residues in Hel308.
Hel308 helicase has five major domains (23): the two RecA folds which are present in SF1 and SF2 helicases and generate directed motion by an inch-worm mechanism (3,24), a winged-helix domain which promotes binding to duplex DNA (25), a ratchet domain that interacts with DNA bases via hydrogen bonding and stacking interactions at multiple sites with the DNA, and a helix-loop-helix domain that is autoinhibitory to unwinding activity (26). The crystal structure of Hel308 from Archaeoglobus fulgidus conjugated with DNA shows that Hel308 interacts directly with the DNA bases at ∼10 different sites (23). The residues that interact with the DNA bases are conserved between Hel308 from T. gammatolerans that we used in this study and A. fulgidus, (Supplementary Figure S10, supporting discussion). Figure 6 depicts the contacts between the Hel308 and the template DNA sequence. The two RecA folds and winged-helix domain interact primarily with the DNA backbone, whereas the ratchet domain 4 interacts with the bases themselves at several different sites (labeled by +3, +5, +6 Figure 6). The presence of base-specific interactions in the ratchet domain offers a possible explanation for the sequence-dependent kinetics, because the interaction energies between those amino acids and the DNA are likely to be modified by the base content. These conserved residues are potential mutation sites that could be used to determine which amino-acid residues affect sequence-dependent kinetics.
Figure 6.
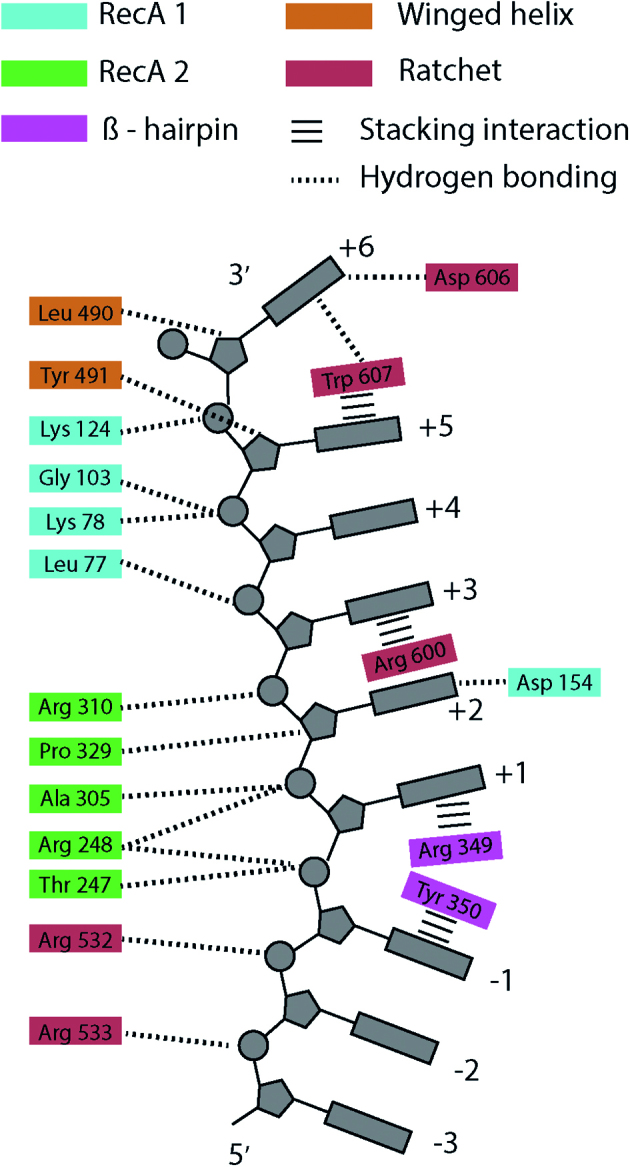
A cartoon depiction of the suspected contacts between Hel308 amino-acid residues and the template DNA based on comparison of Hel308 from Thermoccocus gammatolerans and the crystal structure of Hel308 from Archaeoglobus fulgidus (23). Hel308 amino acid residues are colored according to the protein domain: RecA domain 1 (blue), RecA domain 2 (green), Beta-hairpin motif (pink), Winged-helix domain 3 (orange), Ratchet domain 4 (red). Dotted lines are hydrogen bonding interactions, and horizontal bars are stacking interactions. The contacts shown on the left interact with the sugar-phosphate backbone, whereas the contacts on the right interact with the bases themselves. The DNA structure is distorted between sites –1 and +1 by the Beta hairpin, and between the +5 and +6 sites by the Ratchet domain. The DNA 3′ end is at the top and the 5′ end at the bottom.
An alternative explanation for sequence-dependent stepping is that steric effects of the bases within the helicase modify the relative binding energies of the RecA folds to the DNA substrate. The primary effects of sequence on Hel308 kinetics occur at X = 17 nt and X = 20 nt. The three nucleotide separation between these sites is commensurate with the separation of the RecA domain's contacts along the DNA backbone (23). In Figure 6 this could correspond to the nucleotide positions labeled by +1 and +4. These are the positions at which the motif IV helix in RecA domain 1 and the motif Ia helix in RecA domain 2 contact the DNA. During ATP-induced conformational changes of Hel308, the relative binding energies of these helices are believed to result in the inchworm-like motion of the helicase along the DNA. It is possible that steric effects of the bases within the helicase core affect these interactions, and thereby influence translocation kinetics. In future work, we will mutate positions in Hel308 that contact the DNA to elucidate the mechanism of sequence-specific translocation. We can then construct curves similar to those shown in Figure 5 for a library of Hel308 mutants, and determine which mutations lead to changes in the kinetics at X = 17 nt and X = 20 nt.
Noting the observed sequence-dependent translocation kinetics by Hel308 and UvrD (14), the heterogeneity of DNA, and the large number of contacts between SF1 and SF2 helicases and the DNA (23,27), it seems likely that many NA motor enzymes will be shown to have sequence-dependent kinetics. Whether these effects in helicases are due to direct interactions between the helicase and DNA bases or through some other mechanism are still to be determined. Furthermore, the biological role that such sequence dependence plays in helicases remains unclear. In order to fully understand motor enzyme kinetics, it is crucial that such sequence-specific effects are well understood and quantified. We have demonstrated here that SPRNT is an ideal technique for determining the effects of NA sequence on an enzyme's kinetic parameters. SPRNT can be applied in a similar manner to many motor enzyme systems to shed light into the mechanisms by which NA sequence regulates enzyme behavior.
DATA AVAILABILITY
Raw and processed data are publicly available in figshare DOI: 10.6084/m9.figshare.7355126.v1.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Illumina for providing Hel308.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health, National Human Genome Research Institute [R01HG005115]. Funding for open access charge: National Human Genome Research Institute [R01HG005115].
Conflict of interest statement. J.M.C., A.H.L., H.B., J.H.G. and the University of Washington have filed a provisional patent on the SPRNT technology.
REFERENCES
- 1. Fairman-Williams M.E., Guenther U.-P., Jankowsky E.. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010; 20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Byrd A.K., Raney K.D.. Superfamily 2 helicases. Front. Biosci. Landmark Ed. 2012; 17:2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lohman T.M., Tomko E.J., Wu C.G.. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat. Rev. Mol. Cell Biol. 2008; 9:391–401. [DOI] [PubMed] [Google Scholar]
- 4. Goodman M.F. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu. Rev. Biochem. 2002; 71:17–50. [DOI] [PubMed] [Google Scholar]
- 5. Tsai M.-D. How DNA polymerases catalyze DNA replication, repair, and mutation. Biochemistry. 2014; 53:2749–2751. [DOI] [PubMed] [Google Scholar]
- 6. Jonkers I., Lis J.T.. Getting up to speed with transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 2015; 16:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vvedenskaya I.O., Vahedian-Movahed H., Bird J.G., Knoblauch J.G., Goldman S.R., Zhang Y., Ebright R.H., Nickels B.E.. Interactions between RNA polymerase and the “core recognition element” counteract pausing. Science. 2014; 344:1285–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee A., Gabizon R., Vahedian-Mohaved H., Ebright R.H., Bustamante C.. RNA polymerase translocation in processive transcription elongation and Pausing: Dynamics, Force-Dependence, and modulation by Sequence-Specific RNAP-DNA interactions. Biophys. J. 2017; 112:211a. [Google Scholar]
- 9. Larson M.H., Mooney R.A., Peters J.M., Windgassen T., Nayak D., Gross C.A., Block S.M., Greenleaf W.J., Landick R., Weissman J.S.. A pause sequence enriched at translation start sites drives transcription dynamics in vivo. Science. 2014; 344:1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng W., Arunajadai S.G., Moffitt J.R., Tinoco I., Bustamante C.. Single–base pair unwinding and asynchronous RNA release by the hepatitis C virus NS3 helicase. Science. 2011; 333:1746–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qi Z., Pugh R.A., Spies M., Chemla Y.R.. Sequence-dependent base pair stepping dynamics in XPD helicase unwinding. Elife. 2013; 2:e00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carter A.R., Seaberg M.H., Fan H.-F., Sun G., Wilds C.J., Li H.-W., Perkins T.T.. Sequence-dependent nanometer-scale conformational dynamics of individual RecBCD–DNA complexes. Nucleic Acids Res. 2016; 44:5849–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Craig J.M., Laszlo A.H., Brinkerhoff H., Derrington I.M., Noakes M.T., Nova I.C., Tickman B.I., Doering K., de Leeuw N.F., Gundlach J.H.. Revealing dynamics of helicase translocation on single-stranded DNA using high-resolution nanopore tweezers. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:11932–11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomko E.J., Lohman T.M.. Modulation of Escherichia coli UvrD Single-Stranded DNA translocation by DNA base composition. Biophys. J. 2017; 113:1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derrington I.M., Craig J.M., Stava E., Laszlo A.H., Ross B.C., Brinkerhoff H., Nova I.C., Doering K., Tickman B.I., Ronaghi M. et al.. Subangstrom single-molecule measurements of motor proteins using a nanopore. Nat. Biotechnol. 2015; 33:1073–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laszlo A.H., Derrington I.M., Gundlach J.H.. MspA nanopore as a single-molecule tool: From sequencing to SPRNT. Methods. 2016; 105:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Butler T.Z., Pavlenok M., Derrington I.M., Niederweis M., Gundlach J.H.. Single-molecule DNA detection with an engineered MspA protein nanopore. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:20647–20652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kasianowicz J.J., Brandin E., Branton D., Deamer D.W.. Characterization of individual polynucleotide molecules using a membrane channel. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:13770–13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manrao E.A., Derrington I.M., Laszlo A.H., Langford K.W., Hopper M.K., Gillgren N., Pavlenok M., Niederweis M., Gundlach J.H.. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol. 2012; 30:349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laszlo A.H., Derrington I.M., Ross B.C., Brinkerhoff H., Adey A., Nova I.C., Craig J.M., Langford K.W., Samson J.M., Daza R. et al.. Decoding long nanopore sequencing reads of natural DNA. Nat. Biotechnol. 2014; 32:829–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guy C.P., Bolt E.L.. Archaeal Hel308 helicase targets replication forks in vivo and in vitro and unwinds lagging strands. Nucleic Acids Res. 2005; 33:3678–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tafel A.A., Wu L., McHugh P.J.. Human HEL308 localizes to damaged replication forks and unwinds lagging strand structures. J. Biol. Chem. 2011; 286:15832–15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Büttner K., Nehring S., Hopfner K.-P.. Structural basis for DNA duplex separation by a superfamily-2 helicase. Nat. Struct. Mol. Biol. 2007; 14:647–652. [DOI] [PubMed] [Google Scholar]
- 24. Singleton M.R., Dillingham M.S., Wigley D.B.. Structure and mechanism of helicases and Nucleic Acid Translocases. Annu. Rev. Biochem. 2007; 76:23–50. [DOI] [PubMed] [Google Scholar]
- 25. Northall S.J., Buckley R., Jones N., Penedo J.C., Soultanas P., Bolt E.L.. DNA binding and unwinding by Hel308 helicase requires dual functions of a winged helix domain. DNA Repair. 2017; 57:125–132. [DOI] [PubMed] [Google Scholar]
- 26. Richards J.D., Johnson K.A., Liu H., McRobbie A.-M., McMahon S., Oke M., Carter L., Naismith J.H., White M.F.. Structure of the DNA repair helicase Hel308 reveals DNA binding and autoinhibitory domains. J. Biol. Chem. 2008; 283:5118–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Velankar S.S., Soultanas P., Dillingham M.S., Subramanya H.S., Wigley D.B.. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999; 97:75–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data are publicly available in figshare DOI: 10.6084/m9.figshare.7355126.v1.