

Abstract
Glioblastoma is an aggressive brain tumor that carries a poor prognosis. The tumor’s molecular and cellular landscapes are complex and their relationships to histologic features routinely used for diagnosis are unclear. Here we present the Ivy Glioblastoma Atlas, an anatomically-based transcriptional atlas of human glioblastoma that aligns individual histologic features with genomic and gene expression patterns, thus assigning a molecular significance to the most important morphologic hallmarks of glioblastoma. The atlas and its clinical and genomic database are freely accessible online data resources that will serve as a valuable platform for future investigations of glioblastoma pathogenesis, diagnosis, and treatment.
Glioblastoma is the most common and the most lethal malignant brain tumor (1). Even for patients receiving aggressive treatment, the median survival is 12–15 months (2). The tumors evolve rapidly as they acquire new mutations; the resultant increase in intratumor genomic heterogeneity leads to the development of drug resistance, which limits the long-term efficacy of therapies (3, 4). Two large-scale efforts aimed at characterizing the genomic alterations in human glioblastoma are The Cancer Genome Atlas (TCGA), which is a catalog of multi-omics data, including genomics, transcriptomic, DNA methylomics, proteomics, etc. (5, 6), and REpository for Molecular BRAin Neoplasia DaTa (REMBRANDT), which also includes multiple data domains (7). These efforts helped clarify the role of genomic alterations in the pathogenesis of glioblastoma but were not designed to address intratumor heterogeneity. Subsequent studies addressed heterogeneity spatially within bulk tumor or at the single cell level (4, 8–12), but we lack the systematic understanding of the molecular heterogeneity of this tumor as it relates to the anatomical heterogeneity framed by the variable combination of the classical histological features of glioblastoma, which include tumor infiltration, endothelial cell proliferation, and necrosis. This notion is underscored by the empirical pathology-guided selection of samples typically applied for molecular studies. Here, we report the Ivy Glioblastoma Atlas (http://glioblastoma.alleninstitute.org/), a comprehensive pathology-molecular map of glioblastoma, to guide the unbiased assignment of key molecular alterations to each of the known anatomical features of glioblastoma. By systematically determining the genomic alterations and gene expression profiles of each anatomic feature, we have generated a molecular-pathology encyclopedia of glioblastoma. The atlas will be invaluable for the accurate deconvolution of individual anatomical states within any new tumor, therefore providing unique information for the comprehensive diagnostic characterization of glioblastoma heterogeneity.
To create the atlas, we surveyed the anatomic features by in situ hybridization (ISH), analyzed their transcriptomes by laser microdissection (LMD) and RNA sequencing (RNA-Seq), and validated the feature specific, gene expression enrichment of newly-identified markers by ISH (Fig. 1). We created a clinical and genomic database (http://ivygap.org/) for the 41-patient cohort (table S1) whose tumors (n=42) were evaluated to create the atlas. We describe gene sets whose expression is enriched in the anatomic features, measurements of intra- and inter-tumor heterogeneity, and a molecular subtype classification of transcriptomic samples from our atlas and The Cancer Genome Atlas (TCGA). Together, these two on line resources constitute the Ivy Glioblastoma Atlas Project (Ivy GAP).
Fig. 1. Data generation, analysis, and presentation pipeline for the Ivy Glioblastoma Atlas Project.

(A) Clinical data were collected for the Ivy cohort of 41 patients. (B) Tissue preparation required en bloc resection and formation of tissue blocks with custom L bars. (C) Two studies, Anatomic Feature Based Profiling and Cancer Stem Cell Marker Based Profiling, provided a framework for the ISH surveys, LMD/RNA-Seq experiments, and ISH validations. (D) Informatics included image registration, ontology development, and anatomic feature prediction based on a novel machine learning (ML) analysis of histological data. Search tools support queries of the data set by tumor, tumor block, and gene expression filtered by anatomic feature, molecular subtype, and clinical information. Searchable manual labels delineating the laser microdissections for 270 RNA-Seq samples from the two studies overlay the histology images. The atlas is equipped with image viewers that resolve the histology at 0.5μm/pixel, a transcriptome browser, an application programming interface, and help documentation. The database has detailed longitudinal clinical information and MRI time courses (table S1). (E) This free resource is made available as part of the Ivy Glioblastoma Atlas Project (Ivy GAP). (http://glioblastoma.alleninstitute.org/) via the Allen Institute data portal (http://www.brain-map.org), the Ivy GAP Clinical and Genomic Database (http://ivygap.org/) via the Swedish Neuroscience Institute (http://www.swedish.org/services/neuroscience-institute), and The Cancer Imaging Archive (https://wiki.cancerimagingarchive.net).
To identify gene sets with enriched expression in each anatomic feature (fig. S1), we used LMD to isolate RNA from the leading edge (LE), infiltrating tumor (IT), cellular tumor (CT), pseudopalisading cells around necrosis (PAN), and microvascular proliferation (MVP). In total, we isolated 122 samples from 3 different blocks per tumor in 8–10 tumors. In consultation with a neuropathologist, we manually drew outlines (LMD guidelines) for each of the anatomic features on images of histologically-stained tissue sections. Three additional neuropathologists independently validated the LMD guide lines, and the results showed excellent concordance (table S2). Differential gene expression analysis revealed a total of 3627 genes that had enriched expression in LE, CT, PAN, and MVP samples (Fig. 2A, table S3). Multidimensional scaling demonstrated that samples from these four features were largely distinct, whereas IT appeared to fall on a continuum between LE and CT (Fig. 2B). Gene Ontology enrichment analysis of gene sets with enriched expression in anatomic features (Fig. 2C) confirmed and extended previous reports (13, 14). In general, samples from the same anatomic feature, whether derived from the same or different tumors, were more similar to each other than to other samples from the same tumor (Fig. 2D), and within a given anatomic feature, inter-tumor heterogeneity exceeded intra-tumor heterogeneity (fig. S2).
Fig. 2. Gene expression in anatomic features.
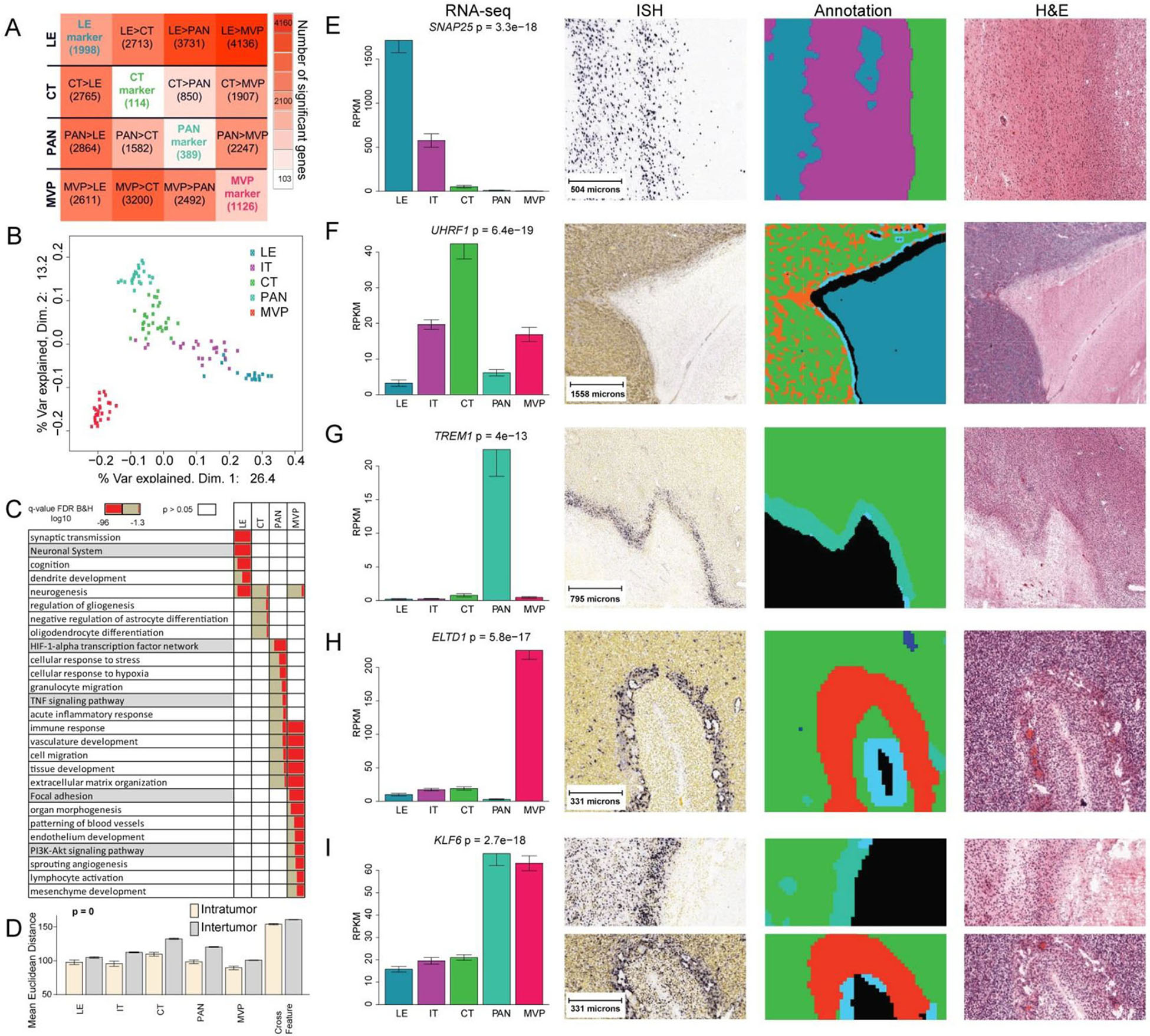
(A) Differential expression matrix based on genes identified in the 122 anatomic feature RNA-Seq samples isolated in triplicate from 8–10 tumors. Values are numbers of genes, of the total 3627, whose expression is enriched in the row feature relative to the column feature (FDR<0.01, fold change >2; P<0.1, BH corrected). Values on diagonal are numbers of genes with higher expression in one feature compared with all other features (i.e. top marker genes). (B) Multidimensional scaling of all genes reflects anatomic specificity. (C) Gene ontology enrichment analysis. LE and CT were enriched for gene ontology terms related to neuronal systems and glial cell differentiation, respectively, whereas PAN was associated with stress, hypoxia, and immune responses, and MVP was related to angiogenesis, immune regulation, and response to wounding. (D) Mean Euclidean distance within and between tumors based on hierarchical clustering of all genes in all 122 anatomic feature RNA-Seq samples grouped by anatomic feature (fig. S1; fig. S2). Cross Feature measures variance between anatomic features. (E-I) Representative marker genes showing RNA-Seq expression levels for features isolated by LMD, representative ISH, ML annotations for ISH and H&E, and H&E adjacent to ISH. LE (blue), IT (purple), CT (green), PNZ (light blue), PAN (turquoise), HBV (orange), MVP (red/magenta).
We selected 31 genes with enriched expression in anatomic features for further analysis by ISH, and found that 27 showed at least partial agreement and 22 showed good agreement between RNA-Seq and ISH assessments of enrichment in PAN, CT, or MVP (table S4) (Fig. 2E–I). Assessing enrichment of gene expression by ISH required that we calculate the overlap between the expression pattern and our machine learning (ML) annotations for each anatomic feature, which we validated using (i) ML-determined rates of accuracy and precision (table S5); (ii) an inter-neuropathologist test to establish agreement on definitions of anatomic features (fig. S1, tables S6, S7); and (iii) neuropathology concordance analyses (tables S8–table S11).
To characterize intra-tumor genetic heterogeneity across anatomic features, we assessed RNA-Seq derived copy number changes in the features and compared them to the DNA level copy number variations (CNVs) (12) from the corresponding bulk tumor (fig. S3; table S12). The CT and PAN samples consistently showed gene expression changes corresponding to the CNVs, whereas LE samples did not as LE samples by definition consist largely of non-neoplastic cells and hence would not harbor the CNVs. On the other hand, MVP samples showed some gene expression changes corresponding to the CNVs indicating a mixture of tumor and non-neoplastic cells. To evaluate the distribution of somatic mutations targeting key glioblastoma genes within the different anatomic features of this tumor, we used RNA-Seq to call Single Nucleotide Variants (SNVs) in eight genes (TP53, PTEN, EGFR, ATRX, IDH1, NF1, PIK3R1, PIK3CA) known to harbor recurrent and functionally important mutations in glioblastoma across anatomic features for tumors where there was at least one sample available from each of the LE, CT, PAN and MVP features (fig. S4; table S13). We detected somatically mutated alleles in RNA from CT, PAN and MVP samples, but only the wild-type variants in LE samples (fig. S4A). The ratio of mutant to wild-type expression was least for MVP relative to CT and PAN samples (fig. S4B). Some of the SNVs occurring across anatomic features were corroborated by ISH data (table S1). Together, the copy number and mutation analyses indicated that LE samples largely consist of non-neoplastic cells, CT and PAN samples comprise largely tumor cells and MVP samples have a mixture of tumor and non-neoplastic cells. The observed intra-tumor heterogeneity in copy number and mutation profiles is consistent with previous studies (8, 9). Only 3 tumors from our 41-patient cohort harbored the R132H mutation in isocitrate-dehydrogenase 1 (IDH1) (table S1); thus, there was insufficient statistical power for analysis of this mutation by anatomic feature. We did not identify any mutation associated with a particular anatomic feature that predicted overall survival better than the promoter methylation status of the MGMT gene in the bulk tumor (fig. S5A,B) (15).
Finally, we developed an admixture model using a 293 gene signature matrix (table S14) for computational decomposition of bulk tumor samples into four anatomic features (LE, CT, PAN, and MVP), and classified the 122 anatomic feature RNA-Seq samples on the basis of histology, admixture (table S14), molecular subtype (6) , and cell type gene expression signature (table S15) enrichment (fig. S6A–D; table S16). Several genes exhibited differential expression across known molecular subtypes of glioblastoma within each anatomic feature (fig. S7A–C). Enrichment of the cell type gene expression signatures in the anatomic features was consistent with Gene Ontology enrichment analyses (Fig. 2C). The correlation between the anatomic feature gene sets and molecular subtypes (table S16) is broadly consistent with results of previous studies (8, 9). When we applied our admixture model to 167 RNA-Seq samples of the TCGA data, we observed similar patterns (fig. S8A–C; table S16).
This atlas and the associated database for clinical and genomic data will serve as a valuable platform for developing and testing new hypotheses related to the pathogenesis, diagnosis, and treatment of glioblastoma. We note that investigators are already leveraging this resource (16–33). In one study, Miller et al. (22) used the atlas to prioritize potential druggable targets based on relationships to tumor microenvironment signatures. We envision use of the Ivy GAP dataset in preclinical studies where investigators identify the cells that drive tumor growth, and target their resident anatomic features for the preferred drug delivery route to maximize the therapeutic effect, as demonstrated by Yu et al.(31).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Allen Institute founders, P. G. Allen and J. Allen, for their vision, encouragement, and support. We thank B. Aronow, B. Bernard, D. Ghosh, L. Hood, C. Hubert, J. Lathia, B. Lin, J. Olson, N. Sanai, I. Shmulevich, Q. Tian, and I. Ulasov for providing lists of genes for putative cancer stem cell markers. We thank J. Rich for his critical review of the manuscript and helpful comments. We thank N. Hansen from Swedish Research Institute for help with patient consent and clinical data collection. We thank T. Crossley for help with the ivygap.org website. We thank P. Sonpatki for help with neuropathology evaluation forms. We thank B. Facer and N. Stewart for artistic and administrative assistance, respectively.
Funding:This project was supported by The Ben and Catherine Ivy Foundation. R.C.R. was supported by NINDS R01 NS091251 and NCI R01 CA136808 grants (to R.C.R.), and A.I. was supported by R01CA178546, U54CA193313, R01CA179044, R01CA190891, R01NS061776 and The Chemotherapy Foundation (to A.I.).
Dedication: This project is dedicated to G.D.F., a dedicated and talented neurosurgeon, as well as visionary in glioblastoma research, who passed away during the course of the study.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: A Materials Transfer Agreement was executed on 20 May 2010 between the Allen Institute for Brain Science and Swedish Health Services to govern the transfer of human tissue between the two institutions, consistent with the approved IRB protocol and consent form. Requests for tissue should be addressed to R.B.P. Tissue accrued in the study will be shared with the scientific community depending on the availability, requested amount, and proposed study plan. Requests for tissue sent to Swedish Health Services will be reviewed for merit, IRB consent, and scientific value on a case by case basis. Separate tables of the Supplementary Materials are available at www.sciencemag.org/content/. The RNA-Seq and copy number data are publically available at Gene Expression Omnibus through GEO series accession number GSE107560. The MRIs are available at The Cancer Imaging Archive (https://wiki.cancerimagingarchive.net/display/Public/Ivy+GAP). The atlas image and RNA-Seq FPKM data are available as part of the Ivy Glioblastoma Atlas Project (http://glioblastoma.alleninstitute.org/) via the Allen Institute data portal (http://www.brain-map.org). The detailed clinical data are available through the Ivy GAP Clinical and Genomic Database (http://ivygap.org/) via the Swedish Neuroscience Institute (http://www.swedish.org/services/neuroscience-institute).
REFERENCES AND NOTES
- 1.Ostrom QT et al. , CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology. 19(S5), 1–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R et al. , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Frattini V et al. , The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet 45, 1141–1149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer M et al. , Single cell-derived clonal analysis of human glioblastoma links functional and genomic heterogeneity. Proc. Natl. Acad. Sci. U.S.A. 112, 851–856 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas Research Network, Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhaak RG et al. , Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Madhavan S et al. , Rembrandt: helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. 7, 157–167 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sottoriva A et al. , Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. U.S.A 110, 4009–4014 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A et al. , Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 15, 530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazor T et al. , DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 28, 307–317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JK et al. , Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nature Genetics 49, 594–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel AP et al. , Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pen A, Moreno MJ, Martin J, Stanimirovic DB, Molecular markers of extracellular matrix remodeling in glioblastoma vessels: microarray study of laser-captured glioblastoma vessels. Glia 55, 559–572 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Dong S et al. , Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J. of Neuropathology and Experimental Neurology 64, 948–955 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegi ME et al. , MGMT gene silencing and benefit from temozolomide in glioblastoma. New Engl. J. Med 352, 997–1003 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Muller S et al. , Single-cell sequencing maps gene expression to mutational phylogenies in PDGF- and EGF-driven gliomas. Molecular Systems Biology 12, 889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mineo M et al. , The long non-coding RNA HIF1A-AS2 facilitates the maintenance of mesenchymal glioblastoma stem-like cells in hypoxic niches. Cell Rep. 15, 2500–2509 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheerathodi M et al. , The cytoskeletal adapter protein spinophilin regulates invadopodia dynamics and tumor cell invasion in glioblastoma. Mol. Cancer Res. 14, 1277–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh D et al. , TGFbeta-Responsive HMOX1 expression is associated with stemness and invasion in glioblastoma multiforme. Stem Cells 34, 2276–2289 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollak J et al. , Ion channel expression patterns in glioblastoma stem cells with functional and therapeutic implications for malignancy. PLOS One 12, e0172884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin X et al. , Targeting glioma stem cells through combined BMI1 and EZH2 inhibition. Nature Med. 23, 1352–1361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller TE et al. , Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 547, 355–359 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Godlewski J et al. , MicroRNA signatures and molecular subtypes of glioblastoma: the role of extracellular transfer. Stem Cell Rep. 8, 1497–1505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Q et al. , Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller S et al. , Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biology 18, 234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantanhede IG, de Oliveira JRM, PDGF family expression in glioblastoma multiforme: data compilation from Ivy Glioblastoma Atlas Project database. Scientific Rep. 7, 15271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otani Y et al. , Fibroblast growth factor 13 regulates glioma cell invasion and is important for bevacizumab-induced glioma invasion. Oncogene 37, 777–786 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Mills BN, Albert GP, Halterman MW, Expression profiling of the MAP kinase phosphatase family reveals a role for DUSP1 in the glioblastoma stem cell niche. Cancer Microenvironment 10, 57–68 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SY, Kim JK, Jeon HY, Ham SW, Kim H, CD133 Regulates IL-1beta signaling and neutrophil recruitment in glioblastoma. Molecules and Cells 40, 515–522 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansson E et al. , CD44 interacts with HIF-2alpha to modulate the hypoxic phenotype of perinecrotic and perivascular glioma cells. Cell Rep. 20, 1641–1653 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Yu D et al. , Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. U.S.A 114, E6147–E6156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwiatkowski SC et al. , Neuropilin-1 modulates TGFbeta signaling to drive glioblastoma growth and recurrence after anti-angiogenic therapy. PLOS One 12, e0185065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khagi S, Miller CR, Putting “multiforme” back into glioblastoma: intratumoral transcriptome heterogeneity is a consequence of its complex morphology. Neuro-Oncology 19, 1570–1571 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hothi P et al. , High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget 3, 1124–1136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lein ES et al. , Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Culling CFA, Allison RT, Barr WT, “Haemotoxylin and its counterstains” in Cellular Pathology Technique. (Butterworths, London, ed. 4, 1985), pp. 160–161. [Google Scholar]
- 37.Rozen S, Skaletsky H, Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol 365–86 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Sorzano CO, Thevenaz P, Unser M, Elastic registration of biological images using vector-spline regularization. IEEE Trans Biomed Eng 52, 652–663 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Criminisi A, Shotton J, “Decision forests for computer vision and medical image analysis” in Advances in Computer Vision and Pattern Recognition (Springer, 2013), pp. 368. [Google Scholar]
- 40.Criminisi A et al. , Regression forests for efficient anatomy detection and localization in computed tomography scans. Med. Image Anal. 17, 1293–1303 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Pathak SD, Chalana V, Haynor DR, Kim Y, Edge-guided boundary delineation in prostate ultrasound images. IEEE Trans. Med. Imaging 19, 1211–9 (2000). [DOI] [PubMed] [Google Scholar]
- 42.Cohen J, A coefficient of agreement for nominal scales. Educational and Psychological Measurement 20, 37–46 (1960). [Google Scholar]
- 43.Orringer DA et al. , Rapid intraoperative histology of unprocessed surgical specimens via fibre-laser-based stimulated Raman scattering microscopy. Nat. Biomed. Eng 1, 0027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer LR et al. , The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 41, D64–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aronesty E, ea-utils: “Command-line tools for processing biological sequencing data” 2011) https://github.com/ExpressionAnalysis/ea-utils.
- 46.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN, RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller JA et al. , Improving reliability and absolute quantification of human brain microarray data by filtering and scaling probes using RNA-Seq. BMC Genomics 15, 154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kadota K, Nishiyama T, Shimizu K, A normalization strategy for comparing tag count data. Algorithms Mol. Biol 7, 5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Storey JD, Bass AJ, Dabney A, Robinson D, qvalue: Q-value estimation for false discovery rate control. R package version 2.2.2 (2015). http://github.com/jdstorey/qvalue. [Google Scholar]
- 52.Newman AM et al. , Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Bardes EE, Aronow BJ, Jegga AG, ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 37, W305–311 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olshen AB, Venkatraman ES, Lucito R, Wigler M, Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng L et al. , Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chase A et al. , TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica 95, 20–26 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cerami E et al. , The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cancer Genome Atlas Research Network et al. , Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. New Engl. J. Med 372, 2481–2498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bao ZS et al. , RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 24, 1765–1773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shah N, Schroeder B, Cobbs C, MGMT methylation in glioblastoma: tale of the tail. Neuro-Oncol. 17, 167–168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y et al. , Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Darmanis S et al. , A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. U.S.A 112, 7285–7290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meyer J et al. , PCR- and restriction endonuclease-based detection of IDH1 mutations. Brain Pathol. 20, 298–300 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoshimoto K et al. , Development of a real-time RT-PCR assay for detecting EGFRvIII in glioblastoma samples. Clin. Cancer Res. 14, 488–493 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.