

Abstract
The purpose of our study was to test the hypothesis that prenatal tobacco smoking exposure (PSE) could modulate the association of genetic variants with ADHD. A community based case-control study was conducted among Chinese children and 168 ADHD patients and 233 controls were recruited by using combination diagnosis of DSM-IV, SNAP-IV and semi-structured clinical interview. Logistic regression analysis was performed to estimate the effect of prenatal tobacco smoking exposure and genotype frequencies on ADHD susceptibility individually by adjustment for potential confounders. Multiplicative and additive interaction analysis were performed to evaluate the interactions between risk genes and PSE with regard to ADHD. Prenatal tobacco smoke exposure was a significant risk factor of ADHD even after adjusted for other potential confounders. ADRA2A rs553668, DRD2 rs1124491 and SLC6A4 rs6354 were identified to be associated with ADHD. A significant multiplicative and additive gene-environment interactions were observed between the PSE and the ADRA2A rs553668 in relation to ADHD and ADHD-ODD. The risk of the genetic variants in ADHD was increased significantly if the child had prenatal tobacco exposure. The genetic risk for ADHD could be influenced by the presence of environmental risks. The environmental and the genetic risks are not distinct to each other. More gene-environment interaction studies were needed to reveal the etiology of ADHD.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a chronic neurodevelopmental disorder characterized by developmentally inappropriate level of inattentiveness and hyperactivity. It is one of the most frequently diagnosed disorders among children and adolescents, with the pooled prevalence of 7.2%1 worldwide and 6.26%2 in China according to the most recent meta-analysis. Previous studies reveal that individuals affected by ADHD frequently lead to various social functional impairment, such as social communication disorder, bad peer relationships, academic performance difficulties3, develop personality disorder, substance abuse and even criminality4,5.
As a complex multi-factorial disease, exact etiology of ADHD is still elusive. The biologic basis for ADHD is thought to be largely genetic6,7. The genetic architecture of ADHD is complex and numerous single nucleotide polymorphisms (SNPs) of genes involved in the regulation of neurotransmitter systems are identified to be associated with ADHD8–10.Among them, genes involved in the dopaminergic, serotonergic and noradrenergic pathways are the most common candidate genes for ADHD studies7,11–13.
Adding to genetic influence, environmental factors are assumed to be co-involved in the etiology of ADHD14. Some prenatal and perinatal factors are reported to be associated with ADHD, such as younger maternal pregnant age15, caesarean, induced delivery, preterm birth, low birth weight16, and mother smoking during pregnancy. With regard to prenatal smoking, it is constantly identified to be a risk factor for offspring hyperactivity symptom and ADHD14,17–19. Evidence from epidemiological researches suggests that maternal cigarette smoking and environmental tobacco exposure during pregnancy are all in association with high risk of ADHD. Hyunjoo Joo20 and his colleagues have described that independent and combined exposure to pre- and postnatal secondhand smoke were associated with ADHD, particularly with the hyperactivity symptom domain. A large population-based birth cohort have provided that maternal smoking during pregnancy was associated with children hyperactivity symptom19. Although large numbers of studies demonstrated a relationship between maternal smoking exposure during pregnancy and ADHD, most of previous studies have investigated the effect of smoking exposure only and there were few studies on gene–environment interactions which were more likely to play the real role in the etiology of ADHD21.
Importantly, the environmental risk is not distinct to the genetic influence21, but it may be required to uncover the genetic liability14. The studies about the G ene× Environment studies of gene and smoking exposure on ADHD are limited. A gene-environment interplay study reports that the co-exist group of prenatal smoking exposure and 10 R/10 R genotype of DAT gene showed a higher mean scores of hyperactivity-impulsivity and inattentiveness than other groups22. But the findings remain speculative since the participants are lack of a clinical diagnosis of ADHD, and the ADHD scores of the participants was derived from the Conners’ Parent Rating Scale which will lead to the overdiagnosis of the ADHD.
Developing of novel technologies have made it more common in identifying genetic risk factors of ADHD, however, proved the genetic architecture of ADHD to be far more complex, since the genetic influences expressed only additional risks to disease susceptibility23. Therefore, gene and environment interaction research will be an inevitable way to explore the etiologic mechanisms of ADHD. However, studies about the joint effects of gene and environmental factors on ADHD is limited14. To fill the knowledge gap, we conducted the case-control study to elucidate the potential synergistic effect of the prenatal tobacco exposure during pregnancy and children genetic variants on children diagnosed ADHD and its symptom domains.
Results
Characteristics of participants
The study involved 168 ADHD patients and 233 controls. A higher proportion of males was noted with the 5.2:1 boys/girls ratio. Compared with the controls, ADHD children were more likely to have younger mother (p < 0.05). There were no significant difference between ADHD patients and controls in terms of family history of neural system diseases, parental marital status, preterm birth (pregnancy < 37weeks), Low birth weight(birth weight <2500 g) and delivery mode, levels of blood lead and postnatal tobacco exposure. Meanwhile, ADHD children scored higher of SNAP-IV scales (Inattention domain, hyperactive domain and ODD domain) and of CBCL total scores than controls (Table 1).
Table 1.
Characteristics of ADHD children and healthy controls.
| ADHD | Controls | p-value | |
|---|---|---|---|
| Gender, N(%) | 0.200 | ||
| Male | 141(83.93) | 183(78.54) | |
| Female | 27(16.07) | 50(21.46) | |
| Age(year), Mean (SD) | 8.55(1.62) | 8.60(1.76) | 0.760 |
| Family history of neural system diseases, N(%) | 0.570 | ||
| Yes | 2(1.19) | 1(0.43) | |
| No and Unknown | 166(98.81) | 232(99.57) | |
| Parental marital status, N(%) | 0.245 | ||
| Marries | 148(88.10) | 214(91.85) | |
| Single, separated, divorced or widowed | 16(11.90) | 14(8.15) | |
| Pregnancy age of mother(y), mean(S.D.) | 26.54(4.19) | 27.57(5.11) | 0.028 |
| Low birth weight(<2500 g), N(%) | 12(7.14) | 17(7.30) | 0.953 |
| Preterm birth(<37 weeks), N(%) | 17(10.12) | 21(9.01) | 0.695 |
| Delivery mode, N(%) | 0.254 | ||
| Cesarean section | 50(29.76) | 57(24.47) | |
| Vaginal delivery | 118(70.24) | 176(75.54) | |
| Breast feeding, N(%) | 101(60.11) | 146(62.66) | 0.161 |
| Intelligence Quotient, mean (S.D.) | 98.50(12.07) | 98.90(13.02) | 0.730 |
| SNAP-IV rating scales, mean(S.D) | |||
| Inattention scores | 1.95(0.49) | 0.64(0.42) | <0.001 |
| Hyperactivity scores | 1.40(0.73) | 0.42(0.36) | <0.001 |
| Oppositional defiant scores(ODD) | 1.04(0.69) | 0.45(0.42) | <0.001 |
| CBCL scales, mean(S.D) | 44.87(25.07) | 18.91(18.89) | <0.001 |
| Blood lead (ug/dL), mean (S.D.) | 3.58(1.59) | 3.26(1.69) | 0.846 |
| Male | 3.65(1.61) | 3.57(1.79) | 0.345 |
| Female | 3.64(1.17) | 3.63(1.16) | 0.722 |
| Inattention | 3.45(1.58) | 3.36(1.69) | 0.345 |
| Hyperactivity/impulsivity | 3.64(1.58) | 3.63(1.71) | 0.721 |
| ADHD-ODD(N = 40) | 3.71(1.59) | 3.63(1.70) | 0.111 |
| Prenatal tobacco exposure, N(%) | 58(34.52) | 53(22.74) | 0.010 |
| Postnatal tobacco exposure, N(%) | 53(31.54) | 55(23.61) | 0.087 |
| Drinking during pregnancy, N(%) | 4(2.4) | 6(2.6) | 0.896 |
p-value was calculated using chi-squared test if percentages are compared, or t test if means are compared.
Associations between the ADRA2A, DRD2, and SLC6A4 genetic variants and ADHD susceptibility
A total of 22 SNPs were detected. The genotype distributions of SLC6A4 rs25533 and rs2020923, and SLC6A3 rs2975226 in the controls deviated from Hardy–Weinberg equilibrium (P < 0.05). Thus they were excluded from further analysis. Consequently, a total of 19 SNPs remained in the subsequent analysis
In individual SNP association analysis, ADRA2A rs553668, DRD2 rs1124491 and SLC6A4 rs6354 were nominally significant at the uncorrected 0.05 significance level with adjustment for age and gender. Among these SNPs, ADRA2A rs553668G allele was associated with an increased risk of ADHD in the additive model, and SLC6A4 rs6354 (GG + GT) was associated with an increased risk of ADHD, whereas DRD2 rs1124491 (AA) was associated with a decreased risk of ADHD. After FDR adjustment, SLC6A4 rs6354 remained statistically significant (P = 0.019, Table S1).
As the Table 2 illuminated, the detected SNPs were not associated with all symptom domains of ADHD. After adjusting for age and gender, individuals carrying the ADRA2A rs553668 GG or AG genotype showed an increased risk of ADHD compared with those carrying AA genotype (adjusted OR = 1.76, 95% CI = 1.12–2.77, P = 0.013), but the SNP was not associated with ADHD-ODD. Additionally, individuals who carried DRD2 rs1124491 AG or GG genotype displayed a risk association with inattention symptom compared to AA genotype (adjusted OR = 2.22, 95% CI = 1.11–4.46, P = 0.022). Compared with those carrying TT genotype, the risk association with ADHD-ODD was increased significantly in individuals carrying the SLC6A4 rs6354 GT or GG genotype (adjusted OR = 2.18, 95% CI = 1.04–4.56, P = 0.034), but no association was observed in other ADHD symptom domains. The ORs and 95% CIs changed slightly in different adjustment model (Model 1, 2, 3, Table 2).
Table 2.
Association Between risk genotypes of ADRA2A, DRD2 and SLC6A4 genes and ADHD with Symptom Domains.
| Symptom domain(N) | Model 1 | Model 2 | Model 3 | |||
|---|---|---|---|---|---|---|
| OR | (95% CI) | OR | (95% CI) | OR | (95% CI) | |
| ADRA2A rs553668a | ||||||
| All ADHD(160) | 1.76* | (1.12–2.77) | 2.07* | (1.27–3.37) | 2.00* | (1.23–3.23) |
| Inattention(141)d | 1.91* | (1.19–3.07) | 2.05* | (1.26–3.36) | 2.19* | (1.32–3.63) |
| Hyperactivity/impulsivity(78)e | 2.18* | (1.21–3.94) | 2.28* | (1.24–4.18) | 2.37* | (1.29–4.34) |
| ODD(39)f | 1.53 | (0.72–3.25) | 1.79 | (0.82–3.91) | 2.07 | (0.92–4.66) |
| Control(225) | 1 | 1 | 1 | |||
| DRD2 rs1124491b | ||||||
| All ADHD(163) | 2.22† | (1.11–4.46) | 2.11† | (1.01–4.06) | 2.38† | (1.33–5.83) |
| Inattention(143)d | 2.11† | (1.01–4.06) | 2.42† | (1.15–5.11) | 2.11† | (1.03–4.39) |
| Hyperactivity/impulsivity(78)e | 1.73 | (0.76–3.94) | 1.83 | (0.79–4.26) | 1.81 | (0.78–4.19) |
| ODD(36)f | 1.58 | (0.52–4.77) | 1.78 | (0.57–5.59) | 1.63 | (0.53–5.00) |
| Control(222) | 1 | 1 | 1 | |||
| SLC6A4 rs6354c | ||||||
| All ADHD(167) | 1.73† | (1.07–2.78) | 1.51 | (0.91–2.51) | 1.55 | (0.93–2.59) |
| Inattention(153) d | 1.58 | (0.97–2.56) | 1.40 | (0.84–2.42) | 1.42 | (0.83–2.43) |
| Hyperactivity/impulsivity(85)e | 1.62 | (0.91–2.88) | 1.56 | (0.85–2.84) | 1.48 | (0.80–2.73) |
| ODD(39)f | 2.18† | (1.04–4.56) | 2.38† | (1.09–5.19) | 2.46† | (1.10–5.50) |
| Control(222) | 1 | 1 | 1 | |||
aThe ORs and 95% CIs were estimated the GG/AG vs AA genotype of ADRA2A rs553668; bThe ORs and 95% CIs were estimated the GG/AG vs AA genotype of DRD2 rs1124491: cThe ORs and 95% CIs were estimated the GT/GG vs TT genotype of SLC6A4 rs6354.d: all the patients with SNAP-IV inattention subscale score ≥ 1.6.e: all the patients with SNAP-IV hyperactivity subscale score ≥ 1.6.f: all the ADHD patients with SNAP-IV ODD subscale score ≥ 1.6.
The ORs and 95% CIs were estimated by logistic regression model. Model 1:adjusted for participants age and gender; Model 2: variables in model 1plus pregnancy age of mother, family history of nervous system diseases, low birth weight, preterm birth; Model 3, variables in model 2 plus the blood lead concentration and postnatal tobacco smoke exposure.
*p < 0 0.01, †p < 0.05.
Prenatal tobacco smoke exposure (PSE) as a risk factor for ADHD
The prenatal tobacco smoke exposure (PSE) showed a risk association with ADHD and all symptom domains, and slight change of the OR and 95% CI was observed in different adjustment model (Model1, 2, 3). The strong association was shown in ODD symptom domain (OR = 2.80, 95% CI = 1.39–5.66) and in the hyperactivity symptom domain (OR = 2.12, 95% CI = 1.23–3.60, Table 3).
Table 3.
Association between the prenatal tobacco smoke exposure and ADHD with the symptom domains.
| Symptom domain(N) | Model 1 | Model 2 | Model 3 | |||
|---|---|---|---|---|---|---|
| OR | (95% CI) | OR | (95% CI) | OR | (95% CI) | |
| All ADHD(N = 168) | 1.77a | (1.14–2.76) | 1.90a | (1.19–3.05) | 1.75b | (1.07–2.94) |
| Inattention(N = 154)a | 1.76b | (1.16–2.77) | 1.88b | (1.16–3.04) | 1.71b | (1.01–2.90) |
| Hyperactivity(N = 86)b | 2.12a | (1.23–3.60) | 2.29a | (1.31–4.00) | 2.34a | (1.23–4.47) |
| ODD(N = 40)c | 2.80a | (1.39–5.66) | 2.94a | (1.41–6.12) | 3.15a | (1.38–7.21) |
| Control(N = 233) | 1 | 1 | 1 | |||
aall the patients with SNAP-IV inattention subscale score ≥ 1.6. ball the patients with SNAP-IV hyperactivity subscale score ≥ 1.6. call the ADHD patients with SNAP-IV ODD subscale score ≥ 1.6.
The ORs and 95% CIs were estimated for PSE using logistic regression model. Model 1: adjusted for participants age and gender; Model 2: variables in model 1plus pregnancy age of mother, family history of nervous system diseases, low birth weight, preterm birth; Model 3, variables in model 2 plus the blood lead concentration and postnatal tobacco smoke exposure.
ap < 0.01,bp < 0.05.
Interaction between prenatal tobacco smoking exposure and genetic variants
The MDR analysis revealed that the three-factor model including rs553668, rs1124491 and prenatal tobacco smoking exposure was the best predictor for ADHD risk (Table 4).
Table 4.
MDR analyses of the gene-environment interactions between SNP rs553668, rs1124491, rs6354and risk factors in ADHD risk.
| Model | TBA | CV consistency | P value |
|---|---|---|---|
| ADRA2A rs553668 | 0.5215 | 8/10 | 0.018 |
| ADRA2A rs553668, DRD2 rs1124491 | 0.5946 | 9/10 | 0.000 |
| ADRA2A rs553668, DRD2 rs1124491,PSE | 0.6310 | 10/10 | 0.000 |
| ADRA2A rs553668,PSE | 0.6120 | 8/10 | 0.000 |
PSE: prenatal tobacco smoking exposure, CV consistency, cross-validation consistency. TBA: testing balance accuracy.
Furthermore, we calculated the multiplicative and additive interactions between ADRA2A rs553668, DRD2 rs1124491, SLC6A4 rs6354 and prenatal tobacco smoking exposure after adjustment of age of the child, gender, pregnancy age of mother, and postnatal smoking exposure.
A significant additive interaction between PSE and ADRA2A rs553668 polymorphisms is detected. The AP due to interaction was 0.48(95% CI = 0.09–0.87), and RERI was 1.88(95% CI = 0.06–4.12), Meanwhile, multiplicative interaction was displayed in this group (Pmul = 0.003). The multiplicative and additive interactions were found in all symptom domain of ADHD. However, no multiplicative and additive interactions were detected between DRD2 rs1124491 polymorphism and PSE, nor between SLC6A4 rs6354 polymorphism and PSE (Table 5).
Table 5.
Interactions between PSE and genetic variants on child ADHD risk
| PSE | Genotype | Cases, N(%) | Controls, N(%) | OR(95% CI) | P mul | AP(95% CI) | RERI(95% CI) |
|---|---|---|---|---|---|---|---|
| ADRA2A rs553668 | |||||||
| No | AA | 26(16.25) | 61(27.11) | 1 | 0.003 | 0.48(0.09–0.87) | 1.88(0.06–4.12) |
| Yes | AA | 14(8.75) | 23(10.22) | 1.36(0.58–3.22) | |||
| No | AG + GG | 76(47.50) | 115(51.11) | 1.65(0.93–2.90) | |||
| Yes | AG + GG | 44(27.50) | 26(11.56) | 3.54(1.73–7.25) | |||
| DRD2 rs1124491 | |||||||
| No | AA | 7(4.27) | 27(12.16) | 1 | 0.175 | −0.55(−1.84–0.73) | −3.34(−11.42–4.74) |
| Yes | AA | 99(60.36) | 145(65.32) | 5.34(1.64–4.46) | |||
| No | AG + GG | 6(3.66) | 6(2.70) | 3.99(1.46–0.89) | |||
| Yes | AG + GG | 52(31.71) | 44(19.82) | 6.04(2.07–7.65) | |||
| SLC6A4 rs6354 | |||||||
| No | TT | 74(44.31) | 147(63.91) | 1 | 0.234 | −0.80(−2.42–0.82) | −1.37(−3.39–0.65) |
| Yes | TT | 44(26.35) | 39(16.96) | 2.13(1.20–3.78) | |||
| No | TG + GG | 34(20.36) | 32(13.91) | 1.97(1.10–3.52) | |||
| Yes | TG + GG | 15(8.98) | 12(5.22) | 1.79(0.75–4.28) | |||
Pmul was calculated using the multiplicative interaction term in the logistic regression analysis.
The ORs, 95% CIs, AP and RERI were adjusted by age, gender, pregnancy age of mother, and postnatal smoking exposure.
Figure 1 described the ORs and the 95% CIs for the only unfavorable genotype exposure, the only prenatal tobacco smoking exposure and the joint effect of them with regard to ADHD and all symptom domains. Most of the risk genotypes showed an increased risk association with ADHD when co-existed with PSE compared with individuals without PSE. The risk association of ADHD in ADRA2A rs553668 GG or AG carrier without PSE was moderately increased (OR = 1.65, 95% CI = 0.93–2.90, P = 0.084), compared with AA carrier without PSE. But the OR for the rs553668 GG or AG carriers with PSE, was significantly increased to 3.54(95% CI: 1.73–7.25, P = 0.001 Table S2). The group of DRD2 rs1124491 AG/GG genotype with PSE also showed a significant association with ADHD compare with whose without PSE (OR = 6.04, 95% CI = 2.07–17.65, P = 0.001, Table S3). And SLC6A4 rs6354 GT/GG genotype carriers with PSE also showed an increased association in hyperactivity and ADHD-ODD symptoms (hyperactivity domain: OR = 2.92, 95% CI = 1.10–7.75, p = 0.031; ODD domain: OR = 4.42 95% CI = 1.32–14.79, p = 0.016.Table S4).
Figure 1.
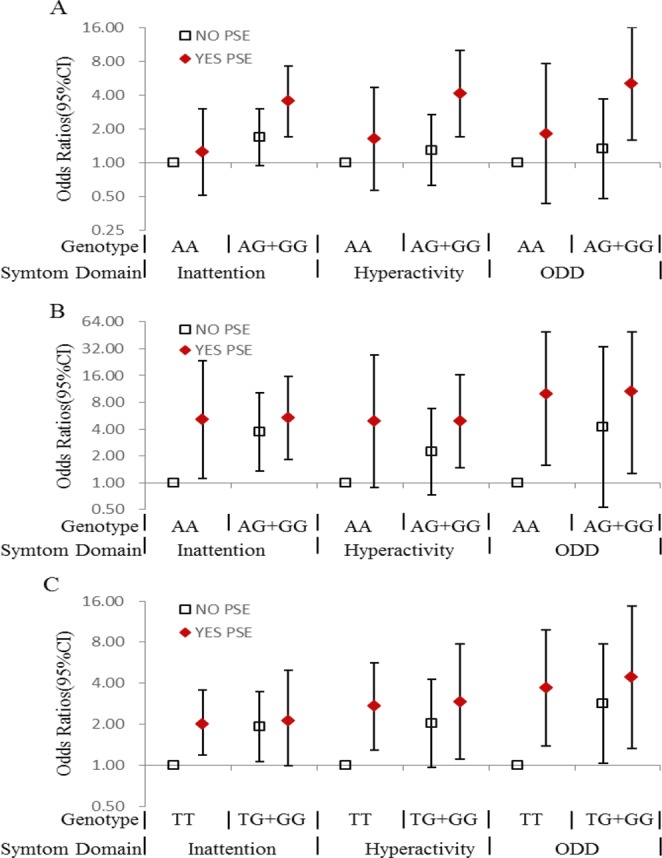
Prenatal tobacco exposure and genetic variant in association with ADHD and its symptom domains. (A) ADRA2A gene (rs553668) variant and prenatal tobacco exposure in association with ADHD and its symptom domains; (B) DRD2 gene (rs1124491) and prenatal tobacco exposure in association with ADHD and its symptom domains: (C) SLC6A4 gene (rs6354) and prenatal tobacco exposure in association with ADHD and its symptom domains.
Discussion
The current case-control study demonstrated a significantly association of prenatal tobacco exposure with all ADHD subtype and ADHD concurring with ODD(ADHD-ODD) in children, especially a strong association with hyperactivity and ADHD-ODD, and the effect size did not diminished even after adjusted by potential environmental influencing covariates. In addition, the study found three SNPs of ADRA2A, DRD2 and SLC6A4 genes associated with ADHD, but the association disappeared in some ADHD subtypes after adjusted by potential environmental covariates. Interestingly, when grouped with prenatal tobacco exposure, however, the association of the genetic variants and ADHD reappeared although there are no multiplicative and additive interaction between DRD2, SLC6A4 genes and PSE.
With regard to the findings in the genetic analysis, the results of risk association with ADHD in the children carrying ADRA2A rs553668 GG or AG genotype were in concordance with the findings of several studies, and indicated that rs553668, being known as the DraI site in the 3′-untranslated region of ADRA2A gene, A-to-G variant increased the risk of ADHD24,25. Meanwhile, the findings showed the DRD2 rs1124491 in the significant association with inattention type of ADHD. DRD2 gene (Dopamine Receptor D2 gene), encodes the D2 subtype of the dopamine receptor. A population-based birth cohort have reported an association of DRD2 rs1124491 with ADHD in males and low persistence in female ADHD patients26,27. Moreover, a common variant SLC6A4 rs6354 was identified to be in association with ADHD-ODD. SLC6A4 rs6354 located in the untranslated(exon 2) 5′-UTR region of SLC6A4 gene—a potential module of exonic splicing enhancer or silencer by bioinformatics prediction— and may affect the expression level or exonic splicing of serotonin28. Evidences show that one repeat length polymorphism-STin2 located in the untranslated exon 2 of SLC6A4 gene may be significantly associated with major depressive disorder and suicide29.
A limited number of G × E studies have shown that genetic factors interact with maternal smoking to heighten risk for ADHD. Anita Thapar30 and his colleagues found maternal smoking during pregnancy was shown a significant association with offspring ADHD symptom scores in twins study after adjustment by genetic factors, although the main effect of cigarette exposure was small. One gene-environment interaction study evaluated maternal smoking and the DAT gene. In the cases with co-exist of maternal smoking during pregnancy and 10 R/10 R genotype of DAT gene, the mean score of hyperactivity-impulsivity and inattentiveness derived from the Conners’ Parent Rating Scale were higher than other groups22. The findings in the current study infer that the susceptibility of ADHD for the unfavorable ADHD genotypes carriers might increase when the children having the history of in-utero exposure to cigarette smoking.
However, the molecular mechanisms responsible for the interaction remain unknown. Nicotine, the main psychoactive component of tobacco, can accumulated in human brain fast when they are smoking31. The neurotoxic effects of nicotine exposure on human fetal brain development have not been studied directly. However, a large number of animal studies have found that nicotine has obvious neurotoxic effects on fetal brain development32. Animal models confident that the nicotine can readily cross the placental barrier into fetal circulation, and the blood-brain barrier, which protects the adult brain from toxic chemicals, is not fully formed until about six months after birth33. So, nicotine can cross the blood-brain barrier and specifically binds to nicotinic acetylcholine receptors (nAChRs) in the fetal brain34, and nAChRs have an important role in regulating synaptic plasticity and brain development. Concerning of animal models, chronic nicotine exposure to animal brain developmentally leads to long-term changes of decreasing presynaptic nAChRs function, which cause altering nicotine-stimulated release of those neurotransmitters and the altering continues for a long time even after nicotine exposure has been removed35,36. And neurotransmitters dysregulation of acetylcholine, dopamine and serotonin are assumed to be the reason of ADHD21. The decreased presynaptic nAChRs function may increase the risks of dysfunctions of dopaminergic, serotonergic and noradrenergic neurotransmission caused by genetic variants. That may explain the strong joint effects of prenatal tobacco exposure and genetic variants on all ADHD subtypes. But the exact reasons of the increasing risks require more extensive research.
Lead is a heavy metal that exists in the environment in nature, and it is a most widely used neurotoxicants. Many studies have shown that high blood lead levels (≥10 µg/dL) are harmful to children neurodevelopment37,38. Moreover, evidences support that longer term low level exposure (<10 µg/dL) associated with intellectual impairment39,40. Unfortunately, the effects of lead exposure on ADHD are less clear. In the current study, no statistically significant association was found between low blood lead levels (<10 µg/dL) and diagnosed ADHD and all the symptoms. A growing body of evidence now support that low level exposure (<5 µg/dL) also associated with cognitive impairment, suggesting that there is no threshold for developmental neurotoxicity37,41,42. Stephani Kim5 and his colleagues examined the relationship between blood lead levels and ADHD in children, and found that the odds ratios for the risk of ADHD is 4.63 (95% CI: 1.36–15.72) in the “≥2 mg/dL” group and the odds ratios increased to 7.25 (1.66–31.67) in the “≥3 mg/dL” group. However, some studies reported that the association between blood lead levels and ADHD did not exhibit a dose-response relationship43.
To the best of our knowledge, this paper is the first G × E interaction research about SNPs and prenatal tobacco smoke exposure in association to ADHD and all the subtypes. The major strength of this study is the standardized procedures of ADHD assessment in children and the precise and rigorous diagnosis21, aiming to minimize the overdiagnosis or underdiagnosis. However, several limitations have to be considered during interpretation of the findings. The major limitation of this study is that the small sample size has decreased the power to detect the effects of the SNPs and the gene-environment interactions. One inherent methodologic problem in this case-control design is that the retrospective assessment of PSE might introduce systematic error in the calculation of the association between PSE and ADHD because of recall bias. In order to diminish the recall bias, we recruited the new diagnosed patients from the community survey. Although the regression model controlled for important confounding variables such as age, gender, mother and father education, and postnatal smoke exposure status, it did not take into account other possible environmental pollutants, such as manganese, mercury, arsenic and bisphenol A and phthalates5,44,45, which can be a focus of future research. Psychological and social environment are also the important for child psychological development, but those did not measured in our study. This is a weakness of the study. The participants in our study were enrolled from one city of China, which limited the results interpretation in general populations. Finally, its cross-sectional nature limits explication about the causal effects of G × E interactions. Future research may focus on a longitudinal study to confirm the causality.
Methods
Participants
This study was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, and all methods were performed in accordance with the approved guidelines and regulations. Informed consent was obtained from all participants and their parents.
Children who were diagnosed for ADHD by pediatric psychiatrists were recruited as the cases in the Liuzhou Women and Children Healthcare Hospital, and 6–12-year-old healthy children from 2 elementary schools in Liuzhou City were also recruited as the controls. The exclusion criteria for both groups were: 1) a history of mental retardation, autism spectrum disorders, bipolar disorder, language disorders or learning disabilities, pervasive developmental disorder; 2) a history of brain disease, seizure disorder, or other neurological disorders; 3) intelligence quotient (IQ) < 70; and 4) the presence of any chronic physical disease; 5) a blood lead concentration ≥10(µg/dL).
Permissions were given for the eligible patients and controls to participate in the study, and those children whose parents could not be contacted or denied consent for participation were excluded. As a result, a total of 168 cases and 233 controls (6–12 years of age) were recruited.
Assessments of children ADHD
The status of both the case and control subjects were confirmed with a semi-structured clinical interview with the parents and children that was conducted according to the DSM-IV ADHD Rating Scale by a pediatric psychiatrist. Additionally, diagnoses were made that included both in categories and severity ratings using the 26-item Chinese version of SNAP-IV scale46. The 4-point response is scored 0–3 (Not at All = 0, Just A Little = 1, Quite a Bit = 2, and Very Much = 3) on SNAP-IV scale. Subscale scores are calculated by summing the scores on the items in the specific symptom domains (inattention, hyperactive and ODD), in which average score 0–1 means no features, 1.1–1.5 means subclinical, 1.6–2.0 means significant, and >2.1 means very severe. We defined oppositional defiant disorder as the ODD symptom domain score ≥1.6. The Child Behavior Checklist (CBCL) scale was conducted to evaluate child potential behavior problems and the Wechsler Intelligence Scale for Children (WISC) was used to evaluate full-scale IQ of all participants. All instruments used in the current study are validated Chinese versions.
Measurement of blood lead concentrations
To measure lead concentrations in blood, 2–3 mL of whole blood was drawn from each child and kept it in heparin-containing tubes. Blood lead level in whole blood was measured using graphite furnace atomic absorption spectrometry (Spectral AA-240FS, Varian, American). The limit of detection for blood lead was 0.1 µg/dL. None of the blood samples showed below the limit of detection.
DNA extraction and genotyping
Genomic DNA was extracted from peripheral blood leukocytes using the TIANamp® Blood DNA Kit DP318 (TIANGEN BIOTECH CO., LTD., Beijing, China).
The candidate genes within the dopamine, norepinephrine and serotonin neurotransmitter pathways were selected based on recent findings47, and included 7 genes (i.e., SLC6A3, SLC6A4, DRD2, ANKK1, DRD4, ADRA2A, SNAP25). The screening procedure for the candidate single nucleotide polymorphism(SNP) was as follow: First, we searched the F-SNP database (http://compbio.cs.queensu.ca/F-SNP) to retrieve information about the predictive functions of the SNPs within these genes and those of potential effects on splicing, transcription, translation, and post translation processes were selected. Second, we filtered the SNPs by the MAF of CHB (minor allele frequency for Han Chinese in Beijing) in the database of 1000 genomes (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) and selected SNPs with MAFs > 5% for the next step. Third, we tested the linkage disequilibrium (LD) among selected SNPs using SNAP Pairwise LD (http://www.broadinstitute.org/mpg/snap/ldsearchpw.php), and only one was reserved if several SNPs were in strong LD with each other (r2 ≥ 0.80). Consequently, a total of 16 SNPs were retained. High-quality genotyping was performed on the Sequenom MassARRAY platform (San Diego, USA) by BIO MIAO BIOLOGICAL TECHNNOLOGY (Beijing, China) following the standard experimental procedures from the manufacturer. In addition, samples with a call rate <80% or Hardy-Weinberg equilibrium (HWE) < 0.05 in the control group were removed.
Determination of tobacco exposure
Tobacco exposure status was obtained from the children’s parents by a questionnaire. The questions were constructed in two parts: mother tobacco exposure and child secondhand smoke exposure. Firstly, we asked whether the mother smoked before pregnancy or not (>100 cigarettes) and whether she smoked during pregnancy. If yes, we then asked how many cigarette expensed per day. Only three of the mothers reported being an ex-smoker or having smoked during pregnancy. Besides, we inquired whether or not she was exposed to secondhand smoke during pregnancy at home or in the workplace and how long she stayed in the smoking circumstance per day/week. Secondly, we investigated whether any family members smoked at home after the child birth and how many cigarette the members expensed per day/week.
Confounders and Covariates
A face-to-face questionnaire survey was administered for their parents by a trained interviewer to collect socio-demographic variables, prenatal and perinatal characteristics. Socio-demographic variables included age, gender, and parental marital status (categorized as married, or single, separated, divorced and widowed). Prenatal and perinatal characteristics included birth weight, pregnancy age of mother, preterm birth and the mode of delivery (cesarean section or vaginal delivery). The family history of neural system diseases and ADHD were asked additionally, but no participant declared of ADHD family history in their families.
Statistical Analysis
Differentials in the distribution of socio-demographic variables, prenatal and perinatal characteristics between patients and controls were compared by chi-squared test or t test, when appropriate. Hardy-Weinberg equilibrium (HWE) for genotypes was calculated in controls by a goodness-of-fit chi-squared test. Logistic regression analysis was used to estimate the effect of PSE and genotype frequencies on ADHD susceptibility by computing odds ratios (ORs) and 95% confidence intervals (95% CIs). Four inheritance models (codominant, dominant, additive and recessive models) were estimated for each SNP. Meanwhile, the statistical power to detect the effects of the SNPs was calculated by Power v3.0.048,49, and for SNPs with MAF of 0.11(rs6354), 0.43(rs1124491), and 0.44(rs553668), for example, we calculated that the power for our sample size to detect an OR of 1.50 was 43.6, 74.1 and 74.8%, respectively.
The Multifactor Dimensionality Reduction (MDR) 2.0 beta 8.1 program (https://sourceforge.net/projects/mdr/) was used to assess the gene-environment interactions between risk genes and the environmental risks. The details analytic protocol was described in the previous study50,51. Furthermore, multiplicative and additive interactions were computed. Multiplicative interaction was evaluated by the interaction term of logistic regression model. Synergy index (SI), attributable proportion due to interaction (AP), and relative excess risk due to interaction (RERI) and their corresponding 95% CIs were calculated by additive interaction analysis, as suggested by Tomas Andersson52. If there is no additive interaction, RERI and AP are equal to 0, or SI are equal to 1. The multiplicative and additive interaction analyses were adjusted for age of the child, gender, pregnancy age of mother, and postnatal smoking exposure. Since a higher proportion of males and younger ADHD children(<9 years old) was noted in our study.
All analysis were performed using SPSS version 22.0 software (SPSS Inc., Chicago, Illinois, USA). All tests were two-tailed, and statistical significance was defined at P < 0.05.
Supplementary information
Acknowledgements
We thank the study participants and their parents. We also acknowledge the collaboration of the staff of the Liuzhou maternal and child care hospital. This work was supported by the Fundamental Research Funds for the Central Universities (Grand number:lzujbky-2017–87).
Author Contributions
All authors in the manuscript who have made substantial contributions to the work. Yanni Wang designed the study did the statistical analysis, and drafted the initial manuscript. Dan Hu helped to design the study, collected the data. Wenjing Chen assisted with data collection, Hongli Xue did the lab work. Yukai Du designed the study, directed the statistical analysis, and reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-40850-w.
References
- 1.Thomas R, Sanders S, Doust J, Beller E, Glasziou P. Prevalence of attention-deficit/hyperactivity disorder: a systematic review and meta-analysis. Pediatrics. 2015;135:e994–1001. doi: 10.1542/peds.2014-3482. [DOI] [PubMed] [Google Scholar]
- 2.Wang T, et al. Prevalence of attention deficit/hyperactivity disorder among children and adolescents in China: a systematic review and meta-analysis. BMC Psychiatry. 2017;17:32. doi: 10.1186/s12888-016-1187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin J, et al. The divergent impact of COMT Val158Met on executive function in children with and without attention-deficit/hyperactivity disorder. Genes, brain, and behavior. 2016;15:271–279. doi: 10.1111/gbb.12270. [DOI] [PubMed] [Google Scholar]
- 4.Harpin VA. The effect of ADHD on the life of an individual, their family, and community from preschool to adult life. Arch Dis Child. 2005;90(Suppl 1):i2–7. doi: 10.1136/adc.2004.059006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim S, et al. Lead, mercury, and cadmium exposure and attention deficit hyperactivity disorder in children. Environ Res. 2013;126:105–110. doi: 10.1016/j.envres.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faraone SV, et al. Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry. 2005;57:1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Faraone SV, Mick E. Molecular genetics of attention deficit hyperactivity disorder. The Psychiatric clinics of North America. 2010;33:159–180. doi: 10.1016/j.psc.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L, et al. Polygenic transmission and complex neuro developmental network for attention deficit hyperactivity disorder: genome-wide association study of both common and rare variants. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2013;162B:419–430. doi: 10.1002/ajmg.b.32169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brookes K, et al. The analysis of 51 genes in DSM-IV combined type attention deficit hyperactivity disorder: association signals in DRD4, DAT1 and 16 other genes. Molecular Psychiatry. 2006;11:934–953. doi: 10.1038/sj.mp.4001869. [DOI] [PubMed] [Google Scholar]
- 10.Lasky-Su J, et al. Genome-wide association scan of quantitative traits for attention deficit hyperactivity disorder identifies novel associations and confirms candidate gene associations. American journal of medical genetics. Part B, Neuropsychiatric genetics: the official publication of the International Society of Psychiatric Genetics. 2008;147B:1345–1354. doi: 10.1002/ajmg.b.30867. [DOI] [PubMed] [Google Scholar]
- 11.Lundwall RA, Dannemiller JL. Genetic contributions to attentional response time slopes across repeated trials. BMC neuroscience. 2015;16:66. doi: 10.1186/s12868-015-0201-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bissonette GB, Roesch MR. Editorial: Neural Circuitry of Behavioral Flexibility: Dopamine and Related Systems. Frontiers in behavioral neuroscience. 2016;10:6. doi: 10.3389/fnbeh.2016.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ettinger U, Merten N, Kambeitz J. Meta-analysis of the association of the SLC6A3 3′-UTR VNTR with cognition. Neuroscience and biobehavioral reviews. 2016;60:72–81. doi: 10.1016/j.neubiorev.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 14.Swanson JM, et al. Etiologic subtypes of attention-deficit/hyperactivity disorder: brain imaging, molecular genetic and environmental factors and the dopamine hypothesis. Neuropsychol Rev. 2007;17:39–59. doi: 10.1007/s11065-007-9019-9. [DOI] [PubMed] [Google Scholar]
- 15.Hvolgaard Mikkelsen S, Olsen J, Bech BH, Obel C. Parental age and attention-deficit/hyperactivity disorder (ADHD) International journal of epidemiology. 2016;46:409–420. doi: 10.1093/ije/dyw073. [DOI] [PubMed] [Google Scholar]
- 16.Halmoy A, Klungsoyr K, Skjaerven R, Haavik J. Pre- and perinatal risk factors in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry. 2012;71:474–481. doi: 10.1016/j.biopsych.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Ruisch, I. H., Dietrich, A., Glennon, J. C., Buitelaar, J. K. & Hoekstra, P. J. Maternal substance use during pregnancy and offspring conduct problems: A meta-analysis. Neurosci Biobehav Rev, 10.1016/j.neubiorev.2017.08.014 (2017). [DOI] [PubMed]
- 18.Goodwin RD, et al. Serious Psychological Distress and Smoking During Pregnancy in the United States: 2008–2014. Nicotine Tob Res. 2017;19:605–614. doi: 10.1093/ntr/ntw323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kotimaa AJ, et al. Maternal Smoking and Hyperactivity in 8-Year-Old Children. Journal of the American Academy of Child & Adolescent Psychiatry. 2003;42:826–833. doi: 10.1097/01.CHI.0000046866.56865.A2. [DOI] [PubMed] [Google Scholar]
- 20.Joo H, et al. Secondhand Smoke Exposure and Low Blood Lead Levels in Association With Attention-Deficit Hyperactivity Disorder and Its Symptom Domain in Children: A Community-Based Case-Control Study. Nicotine Tob Res. 2017;19:94–101. doi: 10.1093/ntr/ntw152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thapar A, Cooper M. Attention deficit hyperactivity disorder. The Lancet. 2016;387:1240–1250. doi: 10.1016/s0140-6736(15)00238-x. [DOI] [PubMed] [Google Scholar]
- 22.Kahn RS, Khoury J, Nichols WC, Lanphear BP. Role of dopamine transporter genotype and maternal prenatal smoking in childhood hyperactive-impulsive, inattentive, and oppositional behaviors. The Journal of Pediatrics. 2003;143:104–110. doi: 10.1016/s0022-3476(03)00208-7. [DOI] [PubMed] [Google Scholar]
- 23.Asherson P, Consortium I. Attention-Deficit Hyperactivity Disorder in the post-genomic era. European child & adolescent psychiatry. 2004;13(Suppl 1):I50–70. doi: 10.1007/s00787-004-1006-6. [DOI] [PubMed] [Google Scholar]
- 24.Unal D, Unal MF, Alikasifoglu M, Cetinkaya A. Genetic Variations in Attention Deficit Hyperactivity Disorder Subtypes and Treatment Resistant Cases. Psychiatry Investig. 2016;13:427–433. doi: 10.4306/pi.2016.13.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho SC, et al. Possible association of the alpha-2A-adrenergic receptor gene with response time variability in attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:957–963. doi: 10.1002/ajmg.b.30725. [DOI] [PubMed] [Google Scholar]
- 26.Nyman ES, et al. ADHD Candidate Gene Study in a Population-Based Birth Cohort: Association with DBH and DRD2. Journal of the American Academy of Child & Adolescent Psychiatry. 2007;46:1614–1621. doi: 10.1097/chi.0b013e3181579682. [DOI] [PubMed] [Google Scholar]
- 27.Nyman ES, et al. Sex-specific influence of DRD2 on ADHD-type temperament in a large population-based birth cohort. Psychiatr Genet. 2012;22:197–201. doi: 10.1097/YPG.0b013e32834c0cc8. [DOI] [PubMed] [Google Scholar]
- 28.Lovejoy EA, Scott AC, Fiskerstrand CE, Bubb VJ, Quinn JP. The serotonin transporter intronic VNTR enhancer correlated with a predisposition to affective disorders has distinct regulatory elements within the domain based on the primary DNA sequence of the repeat unit. The European journal of neuroscience. 2003;17:417–420. doi: 10.1046/j.1460-9568.2003.02446.x. [DOI] [PubMed] [Google Scholar]
- 29.Rao S, et al. Resequencing three candidate genes discovers seven potentially deleterious variants susceptibility to major depressive disorder and suicide attempts in Chinese. Gene. 2017;603:34–41. doi: 10.1016/j.gene.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Anita Thapar, et al. Maternal Smoking During Pregnancy and Attention Deficit Hyperactivity Disorder Symptoms in Offspring. Am J Psychiatry. 2003;160:1985–1989. doi: 10.1176/appi.ajp.160.11.1985. [DOI] [PubMed] [Google Scholar]
- 31.Rose JE, et al. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci USA. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King, E., Campbell, A., Belger, A. & Grewen, K. Prenatal Nicotine Exposure Disrupts Infant Neural Markers of Orienting. Nicotine Tob Res, 10.1093/ntr/ntx177 (2017). [DOI] [PMC free article] [PubMed]
- 33.Luck W, Nau H, Hansen R, Steldinger R. Extent of nicotine and cotinine transfer to the human fetus, placenta and amniotic fluid of smoking mothers. Developmental pharmacology and therapeutics. 1985;8:384–395. doi: 10.1159/000457063. [DOI] [PubMed] [Google Scholar]
- 34.Carter S, Paterson J, Gao W, Iusitini L. Maternal smoking during pregnancy and behaviour problems in a birth cohort of 2-year-old Pacific children in New Zealand. Early Hum Dev. 2008;84:59–66. doi: 10.1016/j.earlhumdev.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 35.Damborsky JC, Griffith WH, Winzer-Serhan UH. Neonatal nicotine exposure increases excitatory synaptic transmission and attenuates nicotine-stimulated GABA release in the adult rat hippocampus. Neuropharmacology. 2015;88:187–198. doi: 10.1016/j.neuropharm.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gold AB, Keller AB, Perry DC. Prenatal exposure of rats to nicotine causes persistent alterations of nicotinic cholinergic receptors. Brain Res. 2009;1250:88–100. doi: 10.1016/j.brainres.2008.10.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y, et al. Association between blood lead levels (<5 mug/dL) and inattention-hyperactivity and neurocognitive profiles in school-aged Korean children. Sci Total Environ. 2010;408:5737–5743. doi: 10.1016/j.scitotenv.2010.07.070. [DOI] [PubMed] [Google Scholar]
- 38.Nicolescu R, et al. Environmental exposure to lead, but not other neurotoxic metals, relates to core elements of ADHD in Romanian children: performance and questionnaire data. Environ Res. 2010;110:476–483. doi: 10.1016/j.envres.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Lanphear BP. & Bearer, C. F. Biomarkers in paediatric research and practice. Arch Dis Child. 2005;90:594–600. doi: 10.1136/adc.2003.048819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heyer DB, Meredith RM. Environmental toxicology: Sensitive periods of development and neurodevelopmental disorders. Neurotoxicology. 2017;58:23–41. doi: 10.1016/j.neuro.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 41.Nigg J, Nikolas M, Burt SA. Measured gene-by-environment interaction in relation to attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2010;49:863–873. doi: 10.1016/j.jaac.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho SC, et al. Effect of environmental exposure to lead and tobacco smoke on inattentive and hyperactive symptoms and neurocognitive performance in children. J Child Psychol Psychiatry. 2010;51:1050–1057. doi: 10.1111/j.1469-7610.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 43.Huang S, et al. Childhood Blood Lead Levels and Symptoms of Attention Deficit Hyperactivity Disorder (ADHD): A Cross-Sectional Study of Mexican Children. Environ Health Perspect. 2016;124:868–874. doi: 10.1289/ehp.1510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu W, et al. S100beta in heavy metal-related child attention-deficit hyperactivity disorder in an informal e-waste recycling area. Neurotoxicology. 2014;45:185–191. doi: 10.1016/j.neuro.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 45.Hu D, et al. Associations of phthalates exposure with attention deficits hyperactivity disorder: A case-control study among Chinese children. Environ Pollut. 2017;229:375–385. doi: 10.1016/j.envpol.2017.05.089. [DOI] [PubMed] [Google Scholar]
- 46.Swanson JM, et al. Clinical Relevance of the Primary Findings of the MTA: Success Rates Based on Severity of ADHD and ODD Symptoms at the End of Treatment. Journal of the American Academy of Child & Adolescent Psychiatry. 2001;40:168–179. doi: 10.1097/00004583-200102000-00011. [DOI] [PubMed] [Google Scholar]
- 47.Kamradt, J. M., Nigg, J. T., Friderici, K. H. & Nikolas, M. A. Neuropsychological performance measures as intermediate phenotypes for attention-deficit/hyperactivity disorder: A multiple mediation analysis. Development and psychopathology, 1–14, 10.1017/S0954579416000195 (2016). [DOI] [PMC free article] [PubMed]
- 48.Foppa I, Spiegelman D. Power and sample size calculations for case-control studies of gene-environment interactions with a polytomous exposure variable. American journal of epidemiology. 1997;146:596–604. doi: 10.1093/oxfordjournals.aje.a009320. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Closas M, Lubin JH. Power and sample size calculations in case-control studies of gene-environment interactions: comments on different approaches. American journal of epidemiology. 1999;149:689–692. doi: 10.1093/oxfordjournals.aje.a009876. [DOI] [PubMed] [Google Scholar]
- 50.Hahn LW, Ritchie MD, Moore JH. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics (Oxford, England) 2003;19:376–382. doi: 10.1093/bioinformatics/btf869. [DOI] [PubMed] [Google Scholar]
- 51.Das M, et al. Role of gene-gene/gene-environment interaction in the etiology of eastern Indian ADHD probands. Progress in neuro-psychopharmacology & biological psychiatry. 2011;35:577–587. doi: 10.1016/j.pnpbp.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 52.Andersson T, Alfredsson L, Källberg H, Zdravkovic S, Ahlbom A. Calculating measures of biological interaction. European Journal of Epidemiology. 2005;20:575–579. doi: 10.1007/s10654-005-7835-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.