

Abstract
Human rabies is almost invariably fatal, and globally it remains an important public health problem. Our knowledge of rabies pathogenesis has been learned mainly from studies performed in experimental animal models, and a number of unresolved issues remain. In contrast with the neural pathway of spread, there is still no credible evidence that hematogenous spread of rabies virus to the central nervous system plays a significant role in rabies pathogenesis. Although neuronal dysfunction has been thought to explain the neurological disease in rabies, recent evidence indicates that structural changes involving neuronal processes may explain the severe clinical disease and fatal outcome. Endemic dog rabies results in an ongoing risk to humans in many resource-limited and resource-poor countries, whereas rabies in wildlife is important in North America and Europe. In human cases in North America, transmission from bats is most common, but there is usually no history of a bat bite and there may be no history of contact with bats. Physicians may not recognize typical features of rabies in North America and Europe. Laboratory diagnostic evaluation for rabies includes rabies serology plus skin biopsy, cerebrospinal fluid, and saliva specimens for rabies virus antigen and/or RNA detection. Methods of postexposure rabies prophylaxis, including wound cleansing and administration of rabies vaccine and human rabies immune globulin, are highly effective after recognized exposure. Although there have been rare survivors of human rabies, no effective therapy is presently available. Therapeutic coma (midazolam and phenobarbital), ketamine, and antiviral therapies (known as the “Milwaukee protocol”) were given to a rabies survivor, but this therapy was likely not directly responsible for the favorable outcome. New therapeutic approaches for human rabies need to be developed. A better understanding of basic mechanisms involved in rabies pathogenesis may be helpful in the development of potential new therapies for the future.
Keywords: lyssavirus, pathogenesis, rabies virus, viral encephalitis
Introduction
Rabies is an ancient disease and globally continues to be an important public health problem, with about 75,000 human deaths per year, mostly in Asia and Africa. Sadly, many of the deaths occur in children. In resource-limited and resource-poor countries, endemic dog rabies, sustained by dog-to-dog transmission of the rabies virus, results in an ongoing risk of transmission to humans due to dog bites. Rabies in wildlife is a problem in North America and in Europe. Highly effective prophylactic therapy has been developed for administration after recognized exposures, but may not be administered for a variety of reasons. Basic research on rabies, usually involving animal models, has provided important information for our understanding of the disease. However, unresolved issues remain, and further research is needed in order to understand better the basic mechanisms involved in the disease. This information may prove to be helpful in the development of novel therapeutic approaches, because current approaches have proved to be ineffective. Human rabies remains almost an invariably fatal disease.
Overview of rabies pathology and pathogenesis
In rabies there are mild inflammatory changes in the central nervous system. Neurons are the neural cell type predominantly infected by the rabies virus, and there are few degenerative changes in neurons. Infected neurons may contain eosinophilic inclusions in the cytoplasm, called Negri bodies. Negri bodies are most prominent in large neurons (eg, Purkinje cells) and ultrastructurally are composed of large aggregates of granulofilamentous matrix material and variable numbers of viral particles.1
In the vast majority of cases, the rabies virus enters the body via a bite from an infected animal. The virus is present in the saliva and is deposited in subcutaneous tissues or muscles, or in other tissues of the host. Bat bites normally result in much more superficial sites of viral entry than bites from carnivores. The rabies virus less commonly can enter a host via a scratch or be related to a mucosal exposure. Aerosols generated in a bat cave containing millions of bats or related to laboratory accidents may very rarely allow viral entry into the host by an olfactory pathway. Finally, rabies has also been transmitted by transplantation of tissues, including corneas in eight recipients and a vascular conduit in one, and also by organ transplantation from donors with rabies in the US2,3 and Germany.4
Our knowledge of the pathways of viral spread has been learned from experimental studies performed in animal models of rabies. There is a long incubation period in rabies, typically lasting 20–90 days and rarely exceeding a year or more. The rabies virus glycoprotein binds to the nicotinic acetylcholine receptor,5 which is located on the postsynaptic membrane at the neuromuscular junction. The rabies virus must traverse the synapse to reach the presynaptic nerve terminal. Tsiang performed studies in a rat model of rabies with administration of colchicine using elastomer cuffs around the sciatic nerve in order to inhibit fast axonal transport, which was found to inhibit the spread of rabies virus.6 The rabies virus travels within axons of peripheral nerves by retrograde fast axonal transport. The rabies virus has been used as a neuroanatomical tracer in order to define circuits of synaptically linked neurons in rodents and primates, and these studies have shown that axonal transport of the rabies virus occurs exclusively in the retrograde direction.8,9 These tracing studies have also shown that the motor pathway to the spinal cord is the primary pathway of spread, but the role of a sensory pathway, with initial infection in local dorsal root ganglia neurons, remains an unsettled issue, particularly in mice and in models using the footpad route of inoculation.7,8
Once the rabies virus infects spinal cord neurons, dissemination proceeds quickly throughout the central nervous system by fast axonal transport along neuroanatomical pathways. Many neuronal cell types are infected in a widespread distribution in the central nervous system, whereas infection of non-neuronal cells, including astrocytes,9 occurs much less commonly. The brain infection results in behavioral changes, likely due to infection of neurons in limbic areas, and this facilitates transmission by biting in rabies vectors. Subsequently, there is spread of the rabies virus away from the central nervous system (centrifugal spread) along neuronal pathways, particularly involving the parasympathetic nervous system, to multiple organs, including the heart, gastrointestinal tract, adrenal medulla, skin, and salivary glands. Salivary gland infection is important in rabies vectors because the rabies virus is secreted in high titer in the saliva, which allows transmission to additional hosts by biting. There is infection involving cardiac ganglia and in the myocardium,10 and, in some cases, there may be a myocarditis with clinical manifestations.11–14 A skin biopsy is a useful antemortem diagnostic test in humans because the rabies virus antigen may be detected in nerves adjacent to hair follicles,15,16 and/or the rabies virus RNA may be detected in the skin biopsy specimen using reverse transcription-polymerase chain reaction (RT-PCR) amplification.17
The most detailed studies of events that take place during the incubation period in a natural model of rabies were performed by Charlton et al18 in a skunk model. A Canadian isolate of street (wild-type) rabies virus obtained from skunk salivary glands was inoculated into hindlimb abductor digiti quinti muscles of skunks. They were killed at serial time points, and tissues, including abductor digiti quinti muscles, local spinal dorsal root ganglia, and spinal cord, were collected for studies using immunohistochemical staining and RT-PCR amplification for detection of the rabies virus antigen and RNA, respectively. The studies showed that viral genomic RNA was frequently present in the inoculated muscle (found in four of nine skunks), but not in either spinal ganglia or the spinal cord, when skunks were sacrificed during the incubation period at 62–64 days postinoculation. Immunohistochemical studies performed prior to the development of clinical disease showed evidence of infection of extrafusal muscle fibers and occasional fibrocytes at the site of inoculation. We can conclude that the rabies virus is present at or near the site of the bite during most of the incubation period. The infection of muscle fibers may be a critical pathogenetic step for the virus to gain access to the peripheral nervous system.
Unfortunately, no similar studies have been performed in a bat model of rabies in which the exposures are much more superficial involving the epidermis, dermis, and/or subcutaneous tissues. The skin is richly innervated and it is likely that the rabies virus gains access to small sensory or autonomic nerves, but no information is available about the sequential steps involved in this process during the incubation period. The bat rabies virus variant most commonly responsible for human rabies in North America is associated with silver-haired (Lasionycteris noctivagans)/Eastern Pipistrelle (Perimyotis subflavus) bats.19 Experimental studies on the silver-haired bat virus indicate that the virus replicates well at lower than normal body temperatures (34°C) and is associated with higher infectivity in cell types present in the dermis, including fibroblasts and epithelial cells, than with the coyote street virus.20 Hence, the silver-haired bat virus is likely well adapted for efficient local replication in the dermis, which could explain the success of this variant.
Does the rabies virus invade the central nervous system from the blood?
The traditional view is that the rabies virus spreads within its hosts exclusively by spread in a neural pathway within neurons or neuronal processes, including dendrites and axons. The possibility that the rabies virus may also spread by a hematogenous route has been considered for a number of years. Lodmell et al21 addressed this question by asking whether viral RNA is ever present in blood during rabies virus infection. This group experimentally infected mice by the intramuscular route and assayed blood for the detection of rabies virus RNA using an RT-PCR assay. They found that viral RNA was present in the blood of 30/32 (94%) mice from between one hour and two days after inoculation. This finding was attributed to trauma to blood vessels at the site of inoculation with leakage of the inoculated virus into the blood circulation. Later viral RNA was detected in 21/25 (84%) mice that developed clinical signs of rabies and were exsanguinated 2–4 days after the onset of paralysis. Detectable levels of neutralizing antibody were also present in the sera of 11/21 (52%) clinically ill mice with blood positive for viral RNA. This study showed that viral RNA was detected after the development of central nervous system disease, which, unfortunately, does not address the issue of whether the bloodstream is a pathway of spread to the central nervous system. Detection of viral RNA does not necessarily indicate the presence of infectious virus and may reflect leakage of noninfectious viral RNA originating in infected central nervous system tissues into the bloodstream. In the introduction to this report, the authors clearly indicate that they were not asking whether the rabies virus might invade the central nervous system from the blood, which is the important question that remains unanswered.
In another study, Preuss et al22 attempted “to elucidate the pathogenetic relevance of hematogenous rabies virus spread”. They inoculated mice intramuscularly in the gastrocnemius muscle or intravenously into the tail vein with 106 focus-forming units of silver-haired bat rabies virus variant. After intramuscular inoculation, there was initial infection of spinal cord motor neurons and brain stem neurons with subsequent spread to forebrain neurons. After intravenous inoculation, the silver-haired bat rabies virus initially infected hypothalamic nuclei that are connected by neurosecretory fibers to the neurohypophysis (posterior pituitary) and median eminence, which are circumventricular organs that have an incomplete blood–brain barrier. These findings suggest that hematogenous spread of the silver-haired bat rabies virus leads to retrograde invasion of the central nervous system at the neurovascular interface of the hypothalamus–pituitary system. The authors used these data to support their speculation that the rabies virus might reach the brain by a hematogenous route after organ transplantation because of the presumed long time period that it would take to establish neural connections between host and grafted tissues for classical spread by neural routes. However, it is not clear at all that intact neural connections would be necessary, because viral uptake may occur directly from an injured infected neuron or neuronal process in the organ or tissue from the donor to another neuron or neuronal process in the recipient. In support of this hypothesis is the fact that corneal transplantation has resulted in the transmission of rabies in at least eight cases.23 It is highly unlikely that corneal transplantation would be associated with large intravenous doses of the rabies virus. It is not at all surprising that experimentally large intravenous viral doses would initiate brain infection at sites in which the blood–brain barrier is relatively poorly developed (eg, circumventricular organs). However, this is a highly unnatural situation that does not reflect what likely occurs during natural infection. It is doubtful that natural infection is associated with large quantities of infectious virus entering the circulation. Hence, no strong evidence has been provided that hematogenous spread plays a significant role in the spread of the rabies virus to the central nervous system either under natural conditions or with organ transplantation.
Mechanisms for neuronal dysfunction and death
Natural rabies is normally characterized by severe neurologic signs and a fatal outcome with relatively mild neuropathologic changes in the central nervous system consisting of mild inflammatory changes with few degenerative neuronal changes. The observed histopathological changes do not explain the severe clinical disease and the usual fatal outcome. These findings have given support to the concept that neuronal dysfunction must play an important role in producing the disease.24 A variety of experimental studies in rabies virus infection have investigated potential abnormalities in neurotransmission involving acetylcholine,25–27 serotonin,28,29 and γ-amino-n-butyric acid.30 Abnormalities of uncertain significance were found, but no fundamental defects were demonstrated that explain neuronal dysfunction in rabies.
Dysfunction of ion channels has been shown in rabies virus-infected cultured mouse neuroblastoma NA cells with the whole-cell patch clamp technique.31 The infection reduced the functional expression of voltage-dependent sodium channels and inward rectifier potassium channels, and there was a decreased resting membrane potential reflecting membrane depolarization. No change in the expression of delayed rectifier potassium channels was demonstrated, indicating that nonselective dysfunction of ion channels had not occurred. The reduction in sodium channels and inward rectifier potassium channels could prevent infected neurons from firing action potentials and generating synaptic potentials, resulting in functional impairment.
Koprowski et al have hypothesized that nitric oxide neurotoxicity may mediate neuronal dysfunction in rabies.32 Induction of inducible nitric oxide synthase mRNA and increased brain levels of nitric oxide33 have been demonstrated in rabies virus-infected rodents. However, it is not clear whether these elevated levels played any significant role in producing neuronal dysfunction, and the role of nitric oxide in rabies pathogenesis needs further study. Excitotoxicity has been studied in rabies virus-infected primary neuron cultures and in an experimental mouse model of rabies without any supporting evidence that excitotoxicity plays an important role in neuronal dysfunction or death.34
Early in vivo studies by Prosniak et al35 in mice using subtraction hybridization showed that infection with fixed rabies virus resulted in downregulation of about 90% of genes at more than four-fold lower levels in comparison with the normal brain. In contrast, only about 1.4% of genes became upregulated, including genes involved in regulation of cell metabolism, protein synthesis, and growth and differentiation. Using proteomic profiling on brain homogenates in mice infected with the street rabies virus, Dhingra et al36 found that the expression of proteins involved in ion homeostasis was altered (H+ ATPase and Na+/K+ ATPase were upregulated and Ca2+ ATPase were downregulated), and there was downregulation of proteins relevant to synaptic physiology, which are involved in docking and fusion of synaptic vesicles to the presynaptic membrane. These abnormalities could all a play a role in neuronal dysfunction.
Neurotropic viruses may cause cell death by either apoptosis or necrosis.37–39 Necrosis is associated with energy failure, whereas apoptosis depends on synthesis of macromolecules and requires energy. Each of these forms of cell death is associated with typical morphologic features. The challenge virus standard strain of fixed rabies virus has been observed to induce apoptotic cell death in rat prostatic adenocarcinoma cells,40 mouse neuroblastoma cells,41 and mouse embryonic hippocampal neurons.42 Prominent apoptotic death of neurons has been observed in the brains of mice of various ages that were inoculated intracerebrally with challenge virus standard,40,41,43 whereas neuronal death is not prominent after peripheral inoculation of adult animals.24,43,44 Attenuated viruses are stronger inducers of neuronal apoptosis in cultured primary neurons and also in mice after peripheral inoculation than are more virulent virus strains.42,45 Contrary to previous reports,46 we have recently provided strong evidence that neuronal apoptosis does not play an important role in human rabies.47 Hence, in rabies virus infection there are complex mechanisms involved in the death or survival of neurons both in vitro and in animal models using different viral strains and routes of inoculation, but neuronal cell death is not prominent in natural rabies. Neuronal apoptosis is a host mechanism employed to limit viral spread and does not serve a fundamental role in the pathogenesis of rabies.24
During the prodromal period of rabies, patients may experience pain, paresthesias, or itching at the site of the wound (often healed at the time), which likely reflects involvement of local sensory (eg, dorsal root) ganglia.23 There is neuronal infection with inflammatory changes associated with degenerative neuronal changes in dorsal root ganglia in human rabies and in most experimental models of rabies.48 The neuronal degeneration is much greater than what is seen in the central nervous system in the mouse model with footpad inoculation of challenge virus standard48 and is highly inflammatory, which suggests the possibility of an immune-mediated process. The neuronal degeneration lacks morphologic features of necrosis or morphologic or biochemical features of apoptosis. In gangliocytes there is an early “axotomy response”, and later the gangliocytes contain numerous autophagic compartments. At an advanced stage of degeneration, there are partially membrane-bound empty vacuoles in gangliocytes, indicating that autophagy is an important mechanism involved in this degenerative neuronal process.
New insights concerning neuronal processes
Li et al49 initially suggested that the degeneration of neuronal processes and disruption of synaptic structures may form the basis for neuronal dysfunction in rabies virus infection. They showed severe destruction and disorganization of neuronal processes in silver-stained hippocampal sections from mice infected intracerebrally with the pathogenic N2C strain of the rabies virus.
Our laboratory has recently examined morphological changes in neurons, with an emphasis on the structural integrity of neuronal processes, following hindlimb foot-pad inoculation of transgenic mice expressing yellow fluorescent protein in a subpopulation of neurons, which facilitates the visualization of morphological details of dendrites, axons, and presynaptic nerve terminals.43,50 In these mice, yellow fluorescent protein expression is driven in a subpopulation of neurons, resulting in strong fluorescent signals in dendrites, axons, and presynaptic nerve terminals.50 In this model, conventional histopathology showed mild inflammatory changes without significant degenerative neuronal changes. However, at late clinical time points, with the development of severe clinical neurological disease, fluorescence microscopy showed marked abnormalities, especially beading and/or swelling involving dendrites and axons of layer V cortical pyramidal neurons, severe involvement of axons in the brainstem and the inferior cerebellar peduncle, and severe abnormalities affecting axons of cerebellar mossy fibers (Figure 1). Toluidine blue-stained resin sections and electron microscopy showed vacuolation in cortical neurons that corresponded to swollen mitochondria and vacuolation in the neuropil of the cerebral cortex. Axonal swellings were observed. Vacuolation was also observed ultrastructurally in axons and presynaptic nerve endings. These morphological changes are sufficient to explain the severe clinical disease and fatal outcome and provide strong evidence that the fundamental defect in rabies virus-infected neurons may involve neuronal processes, which is not apparent on routine histopathological studies.
Figure 1.
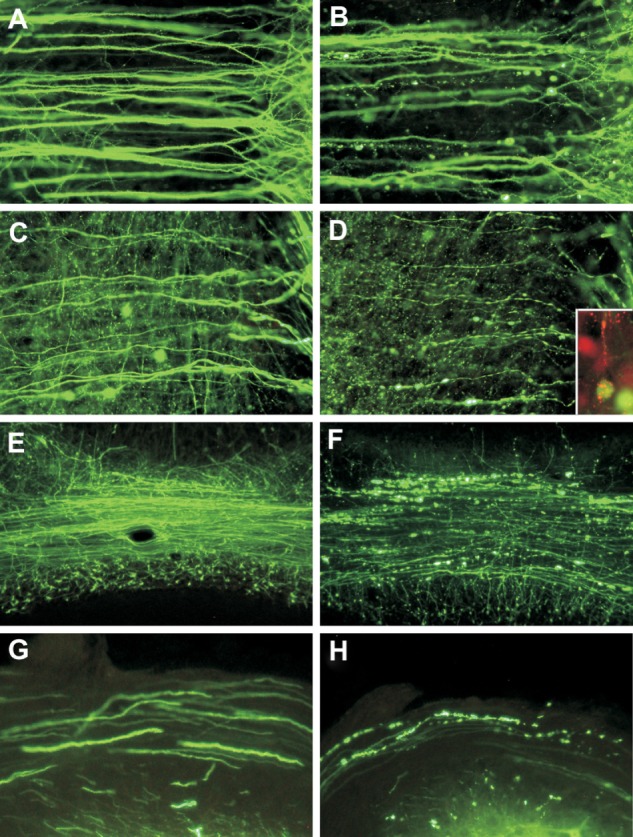
Fluorescence microscopy showing dendrites (A and B) and axons (C and D) of layer V pyramidal neurons in the cerebral cortex of mock-infected (A and C) and moribund challenge virus standard-infected (B, D, and D inset) yellow fluorescent protein mice.43 In infected mice, beading is observed in a minority of dendrites (B), and more axons are involved (D). There are no abnormalities in the dendrites (A) or axons (C) of mock-infected mice. Axons in mock-infected mice are slightly varicosed (C), which is characteristic of these fibers. Fluorescence microscopy shows rabies virus antigen (red) in the perikaryon and dendrite of a yellow fluorescent protein-expressing neuron (D inset). Morphology of the cerebellar mossy fibers of mock-infected (E) and moribund challenge virus standard-infected yellow fluorescent protein mice (F). Mossy fiber axons in the cerebellar commissure of moribund mice show severe beading (F), whereas no abnormalities were observed in mock-infected mice (E). Axons in the inferior cerebellar peduncles are normal in mock-infected mice (G) and show marked beading in challenge virus standard-infected moribund mice (H). A–D) ×230; D) inset, ×220; E, F) ×80; G, H) ×350.
Copyright © 2010, American Society for Microbiology. Adapted with permission from Scott CA, Rossiter JP, Andrew RD, Jackson AC. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome in experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol. 2008;82:513–521.43
We have studied mechanisms for the neuronal process degeneration in vitro using cultured adult mouse sensory dorsal root ganglion neurons because these neurons are relatively permissive to rabies virus infection, which allows detailed evaluation of changes involving their neurites (axons). We have evaluated immunostaining in challenge virus standard-infected and mock-infected cultures of dorsal root ganglion neurons derived from adult mice for neuron specific β-tubulin, rabies virus antigen, and amino acid adducts of 4-hydroxy-2-nonenal (4-HNE), which is a marker of lipid peroxidation and hence oxidative stress.51 Neuronal viability (determined by using trypan blue exclusion), terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, and axonal growth were also assessed in the cultures. Challenge virus standard infected up to about half of the cultured dorsal root ganglion neurons, similar to the findings of other investigators. Neuronal viability and TUNEL staining were similar in challenge virus standard-infected and mock-infected dorsal root ganglion neurons. There were significantly more 4-HNE-labelled puncta at two and three days postinfection in challenge virus standard-infected cultures than in mock infection (Figure 2). Axonal outgrowth was reduced at these time points in challenge virus standard infection versus mock-infected cultures. Axonal swellings with 4-HNE-labelled puncta were also associated with aggregations of actively respiring mitochondria, and there is recent evidence that 4-HNE directly impairs mitochondrial function in cultured dorsal root ganglion neurons.52 In addition to our findings in rabies virus infection, oxidative stress has been recognized to be an important component of experimental acute encephalitis caused by herpes simplex virus type 1 in mice53–55 and also plays a role in neurode-generation in a variety of diseases, including Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, and diabetic neuropathy.56–62
Figure 2.
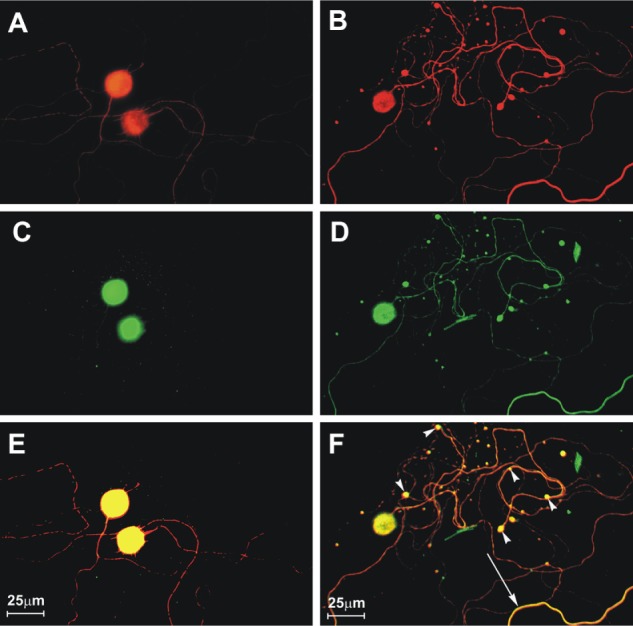
Challenge virus standard infection, but not mock infection, induces formation of 4-hydroxy-2-nonenal (4-HNE)-labeled axonal swellings.51 Fluorescence microscopy showing mock-infected (A, C, and E) and challenge virus standard-infected (B, D, and F) dorsal root ganglion neurons at 72 hours postinfection. β-tubulin (A and B) is a marker of dorsal root ganglion neuronal cell bodies and axons (red) and expression of β-tubulin in challenge virus standard-infected neurons (B) showed multiple axonal swellings but a lack of axonal swellings in mock-infected neurons (A). 4-HNE (green) was poorly expressed in the axons of mock-infected dorsal root ganglion neurons (C) but showed greater expression in the axons of challenge virus standard-infected neurons (D) and accumulation in regions with axonal swellings (D). In challenge virus standard-infected neurons, merging of signals for β-tubulin and 4-HNE (yellow) showed there was strong expression of these elements in axons, both with axonal swellings (arrowheads) and without axonal swellings (arrow, F), but not in mock infected neurons (E).
Copyright © 2010, American Society for Microbiology. Adapted with permission from Jackson AC, Kammouni W, Zherebitskaya E, Fernyhough P. Role of oxidative stress in rabies virus infection of adult mouse dorsal root ganglion neurons. J Virol. 2010;84:4697–4705.51
Epidemiology
Worldwide, there are at least 55,000 fatal human cases of rabies each year,63 and perhaps as many as 75,000 cases per year. Unfortunately, children share a disproportionately high burden of the disease. The vast majority of cases occur in developing countries, especially in Asia and Africa, and are related to endemic dog rabies where there is dog-to-dog transmission of the rabies virus, and many humans are at risk of exposure due to dog bites. For a variety of economic and cultural reasons, effective methods of controlling dog rabies have not been employed in these locations. In contrast, in North America, most human cases of rabies are caused by bat rabies variants, and the incidence has increased over the past two decades (Table 1). The majority (60%) of human cases of rabies due to bat variants do not give a history of a bat bite or scratch, and 33% have no history of contact with bats at all (Table 1). Hence, the absence of a history of an animal bite or even the absence of a history of contact with bats is common in association with human rabies cases. Many insectivorous bats, including silver-haired bats and Eastern Pipistrelle bats, are small and their bites may not be recognized, resulting in the failure to initiate effective prophylactic therapy after an exposure. The rabies virus variant found in silver-haired and Eastern Pipistrelle bats is presently responsible for most human cases of rabies in the US19 and Canada. Brazilian (also called Mexican) free-tail bats (Tadarida braziliensis) are medium-sized bats that have endemic rabies and occasionally transmit the rabies virus to humans. Vampire bats also transmit the rabies virus to humans and cattle in Latin America. Some bat species, including big brown bats (Eptesicus fuscus) and little brown bats (Myotis lucifugus), are frequently infected by rabies virus variants, but only rarely transmit the infection to humans.
Table 1.
Indigenously acquired cases of human rabies from bats in the US and Canada (1950–2009)*
| Occurrence of cases by decade | Cases (n) |
|---|---|
| 1950–1959 | 7 |
| 1960–1969 | 1 |
| 1970–1979 | 9 |
| 1980–1989 | 3 |
| 1990–1999 | 20 |
| 2000–2009 | 20 |
| Total | 60 |
| Type of case | |
| Bite or scratch | 23 (38.3%) |
| Direct contact with no recognized bite | 11 (18.3%) |
| House exposure but no direct contact | 6 (10.0%) |
| No history of bat contact | 20 (33.3%) |
| Total | 60 |
Terrestrial rabies in the US is caused by rabies virus variants that cause rabies in a variety of carnivore species in particular geographical regions (Figure 3). Raccoons have endemic rabies throughout the Eastern US, with over 2300 laboratory diagnosed cases per year with passive surveillance.64 Raccoon rabies has spread slowly north from Florida from the 1940s and currently involves all states in the Eastern US, and incursions have occurred across the Canadian border into Ontario, Quebec, and New Brunswick. Human rabies is very rarely due to the raccoon rabies virus variant, with only a single documented case to date.65 Raccoon bites and other exposures are usually well recognized, and appropriate postexposure rabies prophylactic therapy is usually initiated promptly, which may account for the rare cases of transmission to humans. Endemic skunk rabies is present in the Midwestern US and California and also in the prairie provinces of Canada. Endemic rabies in red and gray foxes is now uncommon in the US, and rabies in red foxes has been well controlled in Canada and Europe with the use of oral immunization programs.66 Worldwide, a variety of other animals may be vectors of rabies, including mongooses, jackals, coyotes, and wolves. Most human rabies exposures actually come from domestic animals, particularly from companion animals such as dogs and cats. All mammals are considered to be potentially susceptible to rabies. Rodents are not reservoirs of the rabies virus. Small rodents, including mice, rats, chipmunks, squirrels, gerbils, hamsters, and guinea pigs, and lagomorphs (eg, rabbits and hares) are rarely infected with the rabies virus and have not been known to transmit rabies to humans.67 Woodchucks account for the majority of reported cases of rabies in rodents.68 Rabies virus variants can be identified using RT-PCR assay and sequencing or by characterization using monoclonal antibodies. These methods are especially useful in identifying the source of infection in human cases, particularly when there is no history of an animal exposure.
Figure 3.
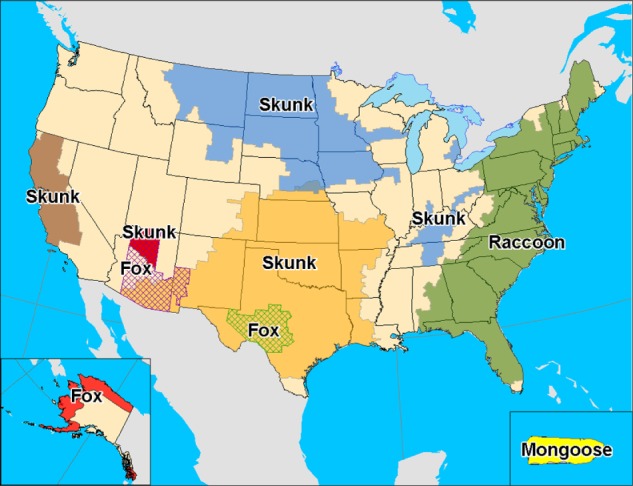
Distribution of the major rabies virus variants among wild terrestrial reservoirs in the US and Puerto Rico in 2008 (from Centers for Disease Control and Prevention [USA]).64
In 2004 there were four iatrogenic rabies cases in Texas resulting from transplantation of organs and a vascular conduit,2,3 and three cases occurred in Germany in 2005 due to organ transplantation4 (Table 2). In addition, there are eight well documented cases of rabies transmitted by corneal transplantation23 and also three cases with corneal transplantation from donors with rabies in which the recipients did not develop rabies.69,70 Clinical screening of organ and tissue donors is important, but laboratory screening for rabies is problematic because of time limitations, a variety of logistical issues, and difficulties associated with false positive results, which would lead to wastage of valuable organs in a situation in which there is a shortage of available organs for transplantation.
Table 2.
| Gender/age (years) | Organ transplanted | Onset of clinical rabies post-transplantation | |
|---|---|---|---|
| Donor in US | Male/20 | – | – |
| Recipient 1 | Male/53 | Liver | 21 days |
| Recipient 2 | Female/50 | Kidney | 27 days |
| Recipient 3 | Male/18 | Kidney | 27 days |
| Recipient 4 | Female/55 | Iliac artery segment | 27 days |
| Donor in Germany | Female/26 | – | – |
| Recipient 1 | Female/46 | Lung | 6 weeks |
| Recipient 2 | Male/72 | Kidney | 5 weeks |
| Recipient 3 | Male/47 | Kidney and pancreas | 5 weeks |
Copyright © 2010, Elsevier Academic Press. Adapted with permission from Jackson AC. Human disease. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007.23
Clinical overview
In North America and Europe, rabies diagnosis is often not made until relatively late in the clinical course or after death, in part because physicians are not familiar with the disease and may not recognize even typical clinical features of rabies. In the US and Canada, the history of an animal bite is commonly absent in fatal cases, particularly because bat bites from insectivorous bats are small and may not be recognized71 or may be attributed to insects or other causes. There may be a history of exposure to bats but no history of direct bat contact. Rabies usually develops from 20 to 90 days after an exposure. Rarely, the incubation period can be as short as a few days or more than a year after the exposure. Prodromal symptoms are nonspecific and may last for a few days and include fever, chills, malaise, fatigue, insomnia, anorexia, headache, anxiety, and irritability. The earliest and more specific symptoms suggesting rabies in about half of patients likely reflect infection and inflammatory changes in local dorsal root ganglia.72 Patients experience pain, paresthesias, or pruritus at or close to the bite site, which has often healed by the time of symptom onset.
There are two clinical forms of rabies, ie, an encephalitic form (furious) in 80% of cases and a paralytic form (dumb) in 20%. The underlying pathogenetic mechanisms of these clinical forms remain poorly defined,24 but it is likely that paralytic rabies reflects a greater burden of disease in the spinal cord, spinal nerve roots, and peripheral nerves, whereas encephalitic rabies reflects greater involvement in the brain. In the encephalitic form, patients have episodes of generalized arousal or hyperexcitability, which are separated by lucid periods.73 Patients may have aggressive behavior, confusion, and hallucinations. Fever is common, and signs of autonomic dysfunction, including hypersalivation, sweating, piloerection, and priapism, may be present. About 50%–80% of patients develop hydrophobia, which is the most classical clinical manifestation of rabies. Patients may initially experience pain in the throat or have difficulty swallowing. On attempts to swallow, they experience contractions of the diaphragm and other inspiratory muscles, which last for about 5–15 seconds. Subsequently, the sight, sound, or even mention of water (or any liquids) may trigger the spasms. A draft of air on the skin may have the same effect (aero-phobia). The disease may progress through paralysis, coma, and multiple organ failure, and eventually it causes death. Encephalitis caused by other viruses is generally associated with earlier impairment of consciousness with less prominent early evidence of brain stem involvement.
In paralytic rabies, flaccid muscle weakness develops early in the course of the disease, often beginning in the bitten extremity and spreading to the other extremities and facial muscles. Sphincter involvement, pain, and sensory disturbances also occur. Hydrophobia is unusual, although bulbar and respiratory muscles become involved later in the course of the illness. Patients with paralytic rabies usually survive longer than patients with the encephalitic form of disease. Paralytic rabies may be misdiagnosed as an inflammatory polyneuropathy (eg, Guillain-Barré syndrome) or a spinal cord disorder.
Cardiopulmonary disorders are the most common medical complications of rabies. Sinus tachycardia is very common and the heart rate is often greater than expected for the degree of fever.74 A variety of cardiac arrhythmias may occur, including wandering atrial/nodal pacemaker, sinus bradycardia, and supraventricular or ventricular ectopic beats/arrhythmias, as well as heart failure, hypotension, and cardiac arrest.72,74 The cardiac manifestations probably reflect infection involving the autonomic nervous system (eg, cardiac ganglia) or myocardium,10 and there may be an associated myocarditis.11–13 Respiratory complications include hyperventilation, hypoxemia, respiratory depression with apnea, atelectasis, and aspiration pneumonia.72 Either hyperthermia or hypothermia may be present, reflecting hypothalamic involvement in the infection. Gastrointestinal hemorrhage is a common complication.75 Endocrine complications include inappropriate secretion of antidiuretic hormone and diabetes insipidus.72,76
Laboratory diagnosis of rabies
Imaging studies do not provide definitive evidence of rabies, and laboratory investigations are necessary to confirm a clinical suspicion of rabies. Computed tomographic scans of the head are usually normal. Magnetic resonance imaging may show normal studies,77,78 and increased signals in gray matter areas have been observed.74–76 Increased signals on T2-weighted images in the medulla and pons were observed in a case associated with a bat rabies virus variant in the US.79
Cerebrospinal fluid analysis often becomes abnormal in rabies. Anderson et al found a cerebrospinal fluid pleocytosis in 59% of cases during the first week of illness and in 87% after the first week.80 The white blood cell count is usually less than 100 cells/µL and mononuclear cells predominate. The protein level may be mildly elevated and the glucose level is usually normal. Serum neutralizing antibodies against the rabies virus are not usually present in unimmunized patients until after the 10th day of the illness.72 Early in the illness, the rabies virus may very occasionally be isolated from the saliva or cerebrospinal fluid.80 A skin biopsy may confirm a diagnosis of rabies during life.16 Rabies virus antigen may be detected in tissue sections from skin biopsies. A full thickness punch skin biopsy (usually 5–6 mm in diameter) containing hair follicles (minimum of 10) can be obtained from the posterior region of the neck at the hairline. Multiple sections of the biopsy specimen should be prepared with immunostaining for rabies virus antigen, which is usually performed using the direct fluorescent antibody technique. Detection of antigen in corneal impression smears is less sensitive than in skin biopsies16 and is frequently not used. Diagnosis of rabies using a brain biopsy is usually unnecessary, but postmortem brain tissues can be assessed for rabies virus antigen and viral isolation. Small amounts of rabies virus RNA may be detected in saliva, skin biopsies, cerebrospinal fluid, and brain tissues using RT-PCR assay. This technique has proved to be an important advance in the diagnosis of rabies. Out of 20 human rabies cases diagnosed antemortem in the US between 1980 and 1996, rabies virus RNA was detected by RT-PCR assay in saliva from all 10 patients who submitted to the test, including three who had negative viral isolation from saliva.19 A recent study showed a higher specificity (100%) and higher sensitivity (≥98%) of RT-PCR (using a heminested protocol) on skin biopsy specimens than on saliva specimens. However, saliva is much easier to collect, and RT-PCR assay sensitivity using saliva was found to be 100% when three successive samples were analyzed.17 Negative tests for rabies virus antigen or RNA do not completely exclude rabies unless they are performed on brain tissues. Multiple specimens may need to be collected and tested to confirm a diagnosis of rabies.
Prevention of human rabies
Human rabies can be very effectively prevented, and animal control is an important component in reducing the public health risk to humans. Dog rabies can be eliminated by well established control methods and routine dog rabies vaccination programs. Wildlife rabies is more difficult to control and often requires well organized campaigns, which may include oral immunization methods used in North America and Europe. Control of bat rabies remains a difficult challenge. Anticoagulants are used for the control of vampire bat rabies in Latin America.
Rabies can be very effectively prevented after recog-nized rabies exposures if current guidelines by the Centers for Disease Control and Prevention67 and World Health Organization,63 which are available on the Morbidity and Mortality Weekly Report (http://www.cdc.gov/mmwr/) and World Health Organization (http://www.who.int/en/) websites, respectively, are observed. A decision must be made after an animal exposure whether or not to initiate postexposure rabies prophylaxis. This will depend on the nature of the exposure, the species of the animal involved, and the local epidemiological situation. After a human is bitten by a dog, cat, or ferret, the animal should be captured, confined, and observed for a period of at least 10 days and then examined by a veterinarian prior to its release. If the animal is a stray or unwanted, or if signs of rabies are present or develop during the observation period, the animal should be killed and the head transported under refrigeration for a laboratory examination. The brain should be examined for the presence of the rabies virus, usually with the fluorescent antibody technique and viral isolation using cell culture or mouse inoculation. Because the incubation period for animals other than dogs, cats, and ferrets is uncertain, they should be killed immediately after an exposure and the head submitted for a laboratory examination. If the result is negative, one may safely conclude that the animal’s saliva did not contain the rabies virus, and if immunization has been initiated then it should be discontinued. If an animal escapes after an exposure, it should be considered rabid unless information from public health officials indicates that this is unlikely, and rabies prophylaxis should be initiated. The physical presence of a bat may warrant postexposure prophylaxis when a person (such as a small child or sleeping adult) is unable to report reliably contact that could have resulted in a bite,67 although there is controversy about this recommendation because of the low risk involved.81
After a decision has been made to initiate postexposure rabies prophylaxis, attention must be given to all three components, ie, wound cleansing, active immunization with rabies vaccine, and passive immunization with human rabies immune globulin in a previously unimmunized individual.67 Active immunization is achieved with four doses of a modern cell culture vaccine (recently reduced from five doses82), including purified chick embryo cell culture vaccine or human diploid cell vaccine (administered intramuscularly in the deltoid muscle on days 0, 3, 7, and 14); passive immunization is performed with human rabies immune globulin at a dose of 20 IU/kg with local infiltration into and around the wound(s), and any remainder of the dosage should be given intramuscularly at a site distant from vaccine administration. Pre-exposure rabies immunization may also be performed for individuals at risk of rabies exposure.67
Management of human rabies
There is no established therapy for human patients with rabies. There have been seven well documented rabies survivors, and all but one of them received the rabies vaccine before the onset of their illness.23,83 Only two survived with good neurological outcomes and both were infected by bat rabies virus variants,84,85 which suggests that these variants may be less virulent than canine rabies variants. An approach to human rabies management was recently published in order to help physicians who face this difficult problem.86 The article recommended using a combination of therapies and discussed the pros and cons of using a variety of specific agents.
There are situations where a decision may be made to embark on an aggressive approach to therapy with the goal of recovery, and such an approach will require the full resources of a critical care unit and have a high risk of failure. The following should be considered “favorable” factors for initiating an aggressive therapeutic approach: therapy with dose(s) of rabies vaccine prior to the onset of illness; young age; a healthy and immunocompetent individual; rabies due to a bat rabies variant (bat rabies viruses may be less neurovirulent than canine or other variants that are responsible for most human cases of rabies); early presence of neutralizing antirabies virus antibodies in serum and cerebrospinal fluid (occurs in less than 20% of all patients with rabies); and mild neurological disease at the time of initiation of therapy.
In 2004, a 15-year-old rabies patient, who had not received a rabies vaccine prior to the onset of clinical disease, survived.84 She was bitten on a finger by a bat and did not seek medical attention or receive treatment at that time. About one month after the bite, she developed typical clinical features of rabies, and neutralizing antirabies virus antibodies were detected in her sera and cerebrospinal fluid. Her therapy included therapeutic (induced) coma using intravenous midazolam for seven days. A burst-suppression pattern on her electroencephalogram was maintained, and supplemental phenobarbital was given. She also received therapy with ketamine and antiviral therapy, including ribavirin and amantadine. She improved and was discharged from hospital with neurologic deficits, and she subsequently had progressive neurologic improvement resulting in only mild neurologic deficits.87 This is the first documented survivor who had not received a rabies vaccine prior to the onset of clinical rabies. As discussed in the accompanying editorial, it is unknown whether therapy with one or more specific agents played an important role in the favorable outcome of this patient.88 However, since that time, there have been more than 20 cases in which the main components of the Milwaukee protocol have been used and fatal outcomes have resulted.89 Most concerning is the abundant presence of rabies virus antigen90,91 and high numbers of rabies virus RNA copies4,91 in the postmortem brains of therapeutic failures, including one patient who had complete loss of neurons in the cerebral cortex at autopsy.90 These reports provide abundant evidence that the therapy has failed to produce viral clearance from the brain. New human rabies therapeutic approaches are needed rather than continuing to repeat this flawed therapy. The induction of coma per se has not been shown to be useful in the management of infectious diseases of the nervous system, and there is no evidence supporting this approach in rabies or other viral encephalitides.89,92 Therapeutic coma should not become a routine therapy for the management of rabies.
Conclusion
Human rabies remains an important international public health problem, particularly related to endemic rabies in Asia and Africa. We have learned much about the pathogenesis of rabies from experimental studies performed in animal models, although some unresolved or controversial issues remain. The diagnosis of rabies needs to be considered even in the absence of a known animal exposure. Appropriate diagnostic laboratory testing should be performed in order to confirm a diagnosis of rabies, including rabies serology and testing of specimens for the detection of rabies virus antigen and RNA. Rabies can be very effectively prevented after recognized exposures. Once human rabies develops, there is no effective therapy. Efforts must be made to develop new therapies in the future based on an improved understanding of rabies pathogenesis.
Footnotes
Disclosure
The author has no relevant affiliations or financial involvement with any organization or entity having a financial interest in or financial conflict with the subject matter or materials discussed in this work.
References
- 1.Rossiter JP, Jackson AC. Pathology. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007. [Google Scholar]
- 2.Burton EC, Burns DK, Opatowsky MJ, et al. Rabies encephalomyelitis: clinical, neuroradiological, and pathological findings in 4 transplant recipients. Arch Neurol. 2005;62:873–882. doi: 10.1001/archneur.62.6.873. [DOI] [PubMed] [Google Scholar]
- 3.Srinivasan A, Burton EC, Kuehnert MJ, et al. Transmission of rabies virus from an organ donor to four transplant recipients. N Engl J Med. 2005;352:1103–1111. doi: 10.1056/NEJMoa043018. [DOI] [PubMed] [Google Scholar]
- 4.Maier T, Schwarting A, Mauer D, et al. Management and outcomes after multiple corneal and solid organ transplantations from a donor infected with rabies virus. Clin Infect Dis. 2010;50:1112–1119. doi: 10.1086/651267. [DOI] [PubMed] [Google Scholar]
- 5.Lentz TL, Burrage TG, Smith AL, Crick J, Tignor GH. Is the acetyl-choline receptor a rabies virus receptor? Science. 1982;215:182–184. doi: 10.1126/science.7053569. [DOI] [PubMed] [Google Scholar]
- 6.Tsiang H. Evidence for an intraaxonal transport of fixed and street rabies virus. J Neuropathol Exp Neurol. 1979;38:286–296. doi: 10.1097/00005072-197905000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Jackson AC, Reimer DL. Pathogenesis of experimental rabies in mice: an immunohistochemical study. Acta Neuropathol. 1989;78:159–165. doi: 10.1007/BF00688204. [DOI] [PubMed] [Google Scholar]
- 8.Coulon P, Derbin C, Kucera P, Lafay F, Prehaud C, Flamand A. Invasion of the peripheral nervous systems of adult mice by the CVS strain of rabies virus and its avirulent derivative AvO1. J Virol. 1989;63:3550–3554. doi: 10.1128/jvi.63.8.3550-3554.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson AC, Phelan CC, Rossiter JP. Infection of Bergmann glia in the cerebellum of a skunk experimentally infected with street rabies virus. Can J Vet Res. 2000;64:226–228. [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson AC, Ye H, Phelan CC, et al. Extraneural organ involvement in human rabies. Lab Invest. 1999;79:945–951. [PubMed] [Google Scholar]
- 11.Ross E, Armentrout SA. Myocarditis associated with rabies: report of a case. N Engl J Med. 1962;266:1087–1089. doi: 10.1056/NEJM196205242662105. [DOI] [PubMed] [Google Scholar]
- 12.Cheetham HD, Hart J, Coghill NF, Fox B. Rabies with myocarditis: two cases in England. Lancet. 1970;1:921–922. doi: 10.1016/s0140-6736(70)91048-2. [DOI] [PubMed] [Google Scholar]
- 13.Raman GV, Prosser A, Spreadbury PL, Cockcroft PM, Okubadejo OA. Rabies presenting with myocarditis and encephalitis. J Infect. 1988;17:155–158. doi: 10.1016/s0163-4453(88)91767-7. [DOI] [PubMed] [Google Scholar]
- 14.Metze K, Feiden W. Rabies virus ribonucleoprotein in the heart. N Engl J Med. 1991;324:1814–1815. doi: 10.1056/NEJM199106203242515. [DOI] [PubMed] [Google Scholar]
- 15.Bryceson ADM, Greenwood BM, Warrell DA, et al. Demonstration during life of rabies antigen in humans. J Infect Dis. 1975;131:71–74. doi: 10.1093/infdis/131.1.71. [DOI] [PubMed] [Google Scholar]
- 16.Warrell MJ, Looareesuwan S, Manatsathit S, et al. Rapid diagnosis of rabies and post-vaccinal encephalitides. Clin Exp Immunol. 1988;71:229–234. [PMC free article] [PubMed] [Google Scholar]
- 17.Dacheux L, Reynes JM, Buchy P, et al. A reliable diagnosis of human rabies based on analysis of skin biopsy specimens. Clin Infect Dis. 2008;47:1410–1417. doi: 10.1086/592969. [DOI] [PubMed] [Google Scholar]
- 18.Charlton KM, Nadin-Davis S, Casey GA, Wandeler AI. The long incubation period in rabies: delayed progression of infection in muscle at the site of exposure. Acta Neuropathol. 1997;94:73–77. doi: 10.1007/s004010050674. [DOI] [PubMed] [Google Scholar]
- 19.Noah DL, Drenzek CL, Smith JS, et al. Epidemiology of human rabies in the United States, 1980 to 1996. Ann Intern Med. 1998;128:922–930. doi: 10.7326/0003-4819-128-11-199806010-00012. [DOI] [PubMed] [Google Scholar]
- 20.Morimoto K, Patel M, Corisdeo S, et al. Characterization of a unique variant of bat rabies virus responsible for newly emerging human cases in North America. Proc Natl Acad Sci U S A. 1996;93:5653–5658. doi: 10.1073/pnas.93.11.5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lodmell DL, Dimcheff DE, Ewalt LC. Viral RNA in the bloodstream suggests viremia occurs in clinically ill rabies-infected mice. Virus Res. 2006;116:114–118. doi: 10.1016/j.virusres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Preuss MA, Faber ML, Tan GS, et al. Intravenous inoculation of a bat-associated rabies virus causes lethal encephalopathy in mice through invasion of the brain via neurosecretory hypothalamic fibers. PLoS Pathogens. 2009;5:e1000485. doi: 10.1371/journal.ppat.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson AC. Human disease. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007. [Google Scholar]
- 24.Jackson AC. Pathogenesis. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007. [Google Scholar]
- 25.Jackson AC. Cholinergic system in experimental rabies in mice. Acta Virol. 1993;37:502–508. [PubMed] [Google Scholar]
- 26.Tsiang H. Neuronal function impairment in rabies-infected rat brain. J Gen Virol. 1982;61:277–281. doi: 10.1099/0022-1317-61-2-277. [DOI] [PubMed] [Google Scholar]
- 27.Dumrongphol H, Srikiatkhachorn A, Hemachudha T, Kotchabhakdi N, Govitrapong P. Alteration of muscarinic acetylcholine receptors in rabies viral-infected dog brains. J Neurol Sci. 1996;137:1–6. doi: 10.1016/0022-510x(95)00275-7. [DOI] [PubMed] [Google Scholar]
- 28.Ceccaldi P-E, Fillion M-P, Ermine A, Tsiang H, Fillion G. Rabies virus selectively alters 5-HT1 receptor subtypes in rat brain. Eur J Pharmacol. 1993;245:129–138. doi: 10.1016/0922-4106(93)90120-x. [DOI] [PubMed] [Google Scholar]
- 29.Bouzamondo E, Ladogana A, Tsiang H. Alteration of potassium-evoked 5-HT release from virus-infected rat cortical synaptosomes. Neuro Report. 1993;4:555–558. doi: 10.1097/00001756-199305000-00023. [DOI] [PubMed] [Google Scholar]
- 30.Ladogana A, Bouzamondo E, Pocchiari M, Tsiang H. Modification of tritiated g-amino-n-butyric acid transport in rabies virus-infected primary cortical cultures. J Gen Virol. 1994;75:623–627. doi: 10.1099/0022-1317-75-3-623. [DOI] [PubMed] [Google Scholar]
- 31.Iwata M, Komori S, Unno T, Minamoto N, Ohashi H. Modification of membrane currents in mouse neuroblastoma cells following infection with rabies virus. Br J Pharmacol. 1999;126:1691–1698. doi: 10.1038/sj.bjp.0702473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koprowski H, Zheng YM, Heber-Katz E, et al. In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic disease. Proc Natl Acad Sci U S A. 1993;90:3024–3027. doi: 10.1073/pnas.90.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hooper DC, Ohnishi ST, Kean R, Numagami Y, Dietzschold B, Koprowski H. Local nitric oxide production in viral and autoimmune diseases of the central nervous system. Proc Natl Acad Sci U S A. 1995;92:5312–5316. doi: 10.1073/pnas.92.12.5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weli SC, Scott CA, Ward CA, Jackson AC. Rabies virus infection of primary neuronal cultures and adult mice: failure to demonstrate evidence of excitotoxicity. J Virol. 2006;80:10270–10273. doi: 10.1128/JVI.01272-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prosniak M, Hooper DC, Dietzschold B, Koprowski H. Effect of rabies virus infection on gene expression in mouse brain. Proc Natl Acad Sci U S A. 2001;98:2758–2763. doi: 10.1073/pnas.051630298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhingra V, Li X, Liu Y, Fu ZF. Proteomic profiling reveals that rabies virus infection results in differential expression of host proteins involved in ion homeostasis and synaptic physiology in the central nervous system. J Neurovirol. 2007;13:107–117. doi: 10.1080/13550280601178226. [DOI] [PubMed] [Google Scholar]
- 37.Griffin DE, Hardwick JM. Perspective: virus infections and the death of neurons. Trends Microbiol. 1999;7:155–160. doi: 10.1016/s0966-842x(99)01470-5. [DOI] [PubMed] [Google Scholar]
- 38.Allsopp TE, Fazakerley JK. Altruistic cell suicide and the specialized case of the virus-infected nervous system. Trends Neurosci. 2000;23:284–290. doi: 10.1016/s0166-2236(00)01591-5. [DOI] [PubMed] [Google Scholar]
- 39.Fazakerley JK, Allsopp TE. Programmed cell death in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:95–119. doi: 10.1007/978-3-662-10356-2_5. [DOI] [PubMed] [Google Scholar]
- 40.Jackson AC, Rossiter JP. Apoptosis plays an important role in experimental rabies virus infection. J Virol. 1997;71:5603–5607. doi: 10.1128/jvi.71.7.5603-5607.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Theerasurakarn S, Ubol S. Apoptosis induction in brain during the fixed strain of rabies virus infection correlates with onset and severity of illness. J Neurovirol. 1998;4:407–414. doi: 10.3109/13550289809114539. [DOI] [PubMed] [Google Scholar]
- 42.Morimoto K, Hooper DC, Spitsin S, Koprowski H, Dietzschold B. Pathogenicity of different rabies virus variants inversely correlates with apoptosis and rabies virus glycoprotein expression in infected primary neuron cultures. J Virol. 1999;73:510–518. doi: 10.1128/jvi.73.1.510-518.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott CA, Rossiter JP, Andrew RD, Jackson AC. Structural abnormalities in neurons are sufficient to explain the clinical disease and fatal outcome in experimental rabies in yellow fluorescent protein-expressing transgenic mice. J Virol. 2008;82:513–521. doi: 10.1128/JVI.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reid JE, Jackson AC. Experimental rabies virus infection in Artibeus jamaicensis bats with CVS-24 variants. J Neurovirol. 2001;7:511–517. doi: 10.1080/135502801753248097. [DOI] [PubMed] [Google Scholar]
- 45.Jackson AC, Rasalingam P, Weli SC. Comparative pathogenesis of recombinant rabies vaccine strain SAD-L16 and SAD-D29 with replacement of Arg333 in the glycoprotein after peripheral inoculation of neonatal mice: less neurovirulent strain is a stronger inducer of neuronal apoptosis. Acta Neuropathol. 2006;111:372–378. doi: 10.1007/s00401-005-0006-z. [DOI] [PubMed] [Google Scholar]
- 46.Juntrakul S, Ruangvejvorachai P, Shuangshoti S, Wacharapluesadee S, Hemachudha T. Mechanisms of escape phenomenon of spinal cord and brainstem in human rabies. BMC Infect Dis. 2005;5:104. doi: 10.1186/1471-2334-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jackson AC, Randle E, Lawrance G, Rossiter JP. Neuronal apop-tosis does not play an important role in human rabies encephalitis. J Neurovirol. 2008;14:368–375. doi: 10.1080/13550280802216502. [DOI] [PubMed] [Google Scholar]
- 48.Rossiter JP, Hsu L, Jackson AC. Selective vulnerability of dorsal root ganglia neurons in experimental rabies after peripheral inoculation of CVS-11 in adult mice. Acta Neuropathol. 2009;118:249–259. doi: 10.1007/s00401-009-0503-6. [DOI] [PubMed] [Google Scholar]
- 49.Li XQ, Sarmento L, Fu ZF. Degeneration of neuronal processes after infection with pathogenic, but not attenuated, rabies viruses. J Virol. 2005;79:10063–10068. doi: 10.1128/JVI.79.15.10063-10068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng G, Mellor RH, Bernstein M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 51.Jackson AC, Kammouni W, Zherebitskaya E, Fernyhough P. Role of oxidative stress in rabies virus infection of adult mouse dorsal root ganglion neurons. J Virol. 2010;84:4697–4705. doi: 10.1128/JVI.02654-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akude E, Zherebitskaya E, Roy Chowdhury SK, Girling K, Fernyhough P. 4-Hydroxy-2-nonenal induces mitochondrial dysfunction and aberrant axonal outgrowth in adult sensory neurons that mimics features of diabetic neuropathy. Neurotox Res. 2010;17:28–38. doi: 10.1007/s12640-009-9074-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valyi-Nagy T, Olson SJ, Valyi-Nagy K, Montine TJ, Dermody TS. Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology. 2000;278:309–321. doi: 10.1006/viro.2000.0678. [DOI] [PubMed] [Google Scholar]
- 54.Milatovic D, Zhang Y, Olson SJ, et al. Herpes simplex virus type 1 encephalitis is associated with elevated levels of F2-isoprostanes and F4-neuroprostanes. J Neurovirol. 2002;8:295–305. doi: 10.1080/13550280290100743. [DOI] [PubMed] [Google Scholar]
- 55.Schachtele SJ, Hu S, Little MR, Lokensgard JR. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J Neuroinflammation. 2010;7:35. doi: 10.1186/1742-2094-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med. 2004;10(Suppl):S18–S25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 57.Dexter DT, Carter CJ, Wells FR, et al. Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem. 1989;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 58.Pedersen WA, Fu W, Keller JN, et al. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol. 1998;44:819–824. doi: 10.1002/ana.410440518. [DOI] [PubMed] [Google Scholar]
- 59.Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer’s disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 60.Giasson BI, Duda JE, Murray IV, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 61.Zherebitskaya E, Akude E, Smith DR, Fernyhough P. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: role of glucose-induced oxidative stress. Diabetes. 2009;58:1356–1364. doi: 10.2337/db09-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chowdhury SK, Zherebitskaya E, Smith DR, et al. Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes. 2010;59:1082–1091. doi: 10.2337/db09-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.World Health Organization . WHO Expert Consultation on Rabies: First Report. Geneva: World Health Organization; 2005. [PubMed] [Google Scholar]
- 64.Blanton JD, Palmer D, Rupprecht CE. Rabies surveillance in the United States during 2009. J Am Vet Med Assoc. 2010;237:646–657. doi: 10.2460/javma.237.6.646. [DOI] [PubMed] [Google Scholar]
- 65.Silverstein MA, Salgado CD, Bassin S, et al. First human death associated with raccoon rabies – Virginia, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:1101–1103. [PubMed] [Google Scholar]
- 66.Rosatte RC, Tinline RR, Johnston DH. Rabies control in wild carnivores. In: Jackson AC, Wunner WH, editors. Rabies. 2nd ed. London: Elsevier Academic Press; 2007. [Google Scholar]
- 67.Manning SE, Rupprecht CE, Fishbein D, et al. Human rabies prevention – United States, 2008: recommendations of the advisory committee on immunization practices. MMWR Morb Mortal Wkly Rep. 2008;57(No. RR-3):1–28. [PubMed] [Google Scholar]
- 68.Childs JE, Colby L, Krebs JW, et al. Surveillance and spatiotemporal associations of rabies in rodents and lagomorphs in the United States, 1985–1994. J Wildl Dis. 1997;33:20–27. doi: 10.7589/0090-3558-33.1.20. [DOI] [PubMed] [Google Scholar]
- 69.Vetter JM, Frisch L, Drosten C, et al. Survival after transplantation of corneas from a rabies-infected donor. Cornea. 2010 Sep 15; doi: 10.1097/ICO.0b013e3181e4572a. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 70.Sureau P, Portnoi D, Rollin P, Lapresle C, Chaouni-Berbich A. Prevention of inter-human rabies transmission after corneal graft. C R Seances Acad Sci III. 1981;293:689–692. French. [PubMed] [Google Scholar]
- 71.Messenger SL, Smith JS, Rupprecht CE. Emerging epidemiology of bat-associated cryptic cases of rabies in humans in the United States. Clin Infect Dis. 2002;35:738–747. doi: 10.1086/342387. [DOI] [PubMed] [Google Scholar]
- 72.Hattwick MAW. Human rabies. Public Health Rev. 1974;3:229–274. [Google Scholar]
- 73.Warrell DA. The clinical picture of rabies in man. Trans R Soc Trop Med Hyg. 1976;70:188–195. doi: 10.1016/0035-9203(76)90037-7. [DOI] [PubMed] [Google Scholar]
- 74.Warrell DA, Davidson NM, Pope HM, et al. Pathophysiologic studies in human rabies. Am J Med. 1976;60:180–190. doi: 10.1016/0002-9343(76)90427-7. [DOI] [PubMed] [Google Scholar]
- 75.Kureishi A, Xu LZ, Wu H, Stiver HG. Rabies in China: recommendations for control. Bull World Health Organ. 1992;70:443–450. [PMC free article] [PubMed] [Google Scholar]
- 76.Bhatt DR, Hattwick MAW, Gerdsen R, Emmons RW, Johnson HN. Human rabies: diagnosis, complications, and management. Am J Dis Child. 1974;127:862–869. doi: 10.1001/archpedi.1974.02110250088013. [DOI] [PubMed] [Google Scholar]
- 77.Sing TM, Soo MY. Imaging findings in rabies. Australas Radiol. 1996;40:338–341. doi: 10.1111/j.1440-1673.1996.tb00415.x. [DOI] [PubMed] [Google Scholar]
- 78.Mrak RE, Young L. Rabies encephalitis in a patient with no history of exposure. Hum Pathol. 1993;24:109–110. doi: 10.1016/0046-8177(93)90070-w. [DOI] [PubMed] [Google Scholar]
- 79.Pleasure SJ, Fischbein NJ. Correlation of clinical and neuroimaging findings in a case of rabies encephalitis. Arch Neurol. 2000;57:1765–1769. doi: 10.1001/archneur.57.12.1765. [DOI] [PubMed] [Google Scholar]
- 80.Anderson LJ, Nicholson KG, Tauxe RV, Winkler WG. Human rabies in the United States, 1960 to 1979: epidemiology, diagnosis, and prevention. Ann Intern Med. 1984;100:728–735. doi: 10.7326/0003-4819-100-5-728. [DOI] [PubMed] [Google Scholar]
- 81.Huot C, De SG, Duval B, Maranda-Aubut R, Ouakki M, Skowronski DM. The cost of preventing rabies at any cost: post-exposure prophylaxis for occult bat contact. Vaccine. 2008;26:4446–4450. doi: 10.1016/j.vaccine.2008.06.076. [DOI] [PubMed] [Google Scholar]
- 82.Rupprecht CE, Briggs D, Brown CM, et al. Use of a reduced (4-dose) vaccine schedule for postexposure prophylaxis to prevent human rabies: recommendations of the advisory committee on immunization practices. MMWR Recomm Rep. 2010;59:1–9. [PubMed] [Google Scholar]
- 83.Ministerio da Saude in Brazil Rabies, human survival, bat – Brazil: (Pernambuco) [Accessed Oct 30, 2010]. ProMED-mail 20081114 3599 Available at: http://www.promedmail.org/
- 84.Willoughby RE, Jr, Tieves KS, Hoffman GM, et al. Survival after treatment of rabies with induction of coma. N Engl J Med. 2005;352:2508–2514. doi: 10.1056/NEJMoa050382. [DOI] [PubMed] [Google Scholar]
- 85.Hattwick MAW, Weis TT, Stechschulte CJ, Baer GM, Gregg MB. Recovery from rabies: a case report. Ann Intern Med. 1972;76:931–942. doi: 10.7326/0003-4819-76-6-931. [DOI] [PubMed] [Google Scholar]
- 86.Jackson AC, Warrell MJ, Rupprecht CE, et al. Management of rabies in humans. Clin Infect Dis. 2003;36:60–63. doi: 10.1086/344905. [DOI] [PubMed] [Google Scholar]
- 87.Hu WT, Willoughby RE, Jr, Dhonau H, Mack KJ. Long-term follow-up after treatment of rabies by induction of coma. N Engl J Med. 2007;357:945–946. doi: 10.1056/NEJMc062479. [DOI] [PubMed] [Google Scholar]
- 88.Jackson AC. Recovery from rabies. N Engl J Med. 2005;352:2549–2550. doi: 10.1056/NEJMe058092. [DOI] [PubMed] [Google Scholar]
- 89.Jackson AC. Update on rabies diagnosis and treatment. Curr Infect Dis Rep. 2009;11:296–301. doi: 10.1007/s11908-009-0044-0. [DOI] [PubMed] [Google Scholar]
- 90.McDermid RC, Saxinger L, Lee B, et al. Human rabies encephalitis following bat exposure: failure of therapeutic coma. Can Med Assoc J. 2008;178:557–561. doi: 10.1503/cmaj.071326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hunter M, Johnson N, Hedderwick S, et al. Immunovirological correlates in human rabies treated with therapeutic coma. J Med Virol. 2010;82:1255–1265. doi: 10.1002/jmv.21785. [DOI] [PubMed] [Google Scholar]
- 92.Wilde H, Hemachudha T, Jackson AC. Viewpoint: management of human rabies. Trans R Soc Trop Med Hyg. 2008;102:979–982. doi: 10.1016/j.trstmh.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 93.De Serres G, Dallaire F, Cote M, Skowronski DM. Bat rabies in the United States and Canada from 1950 through 2007: human cases with and without bat contact. Clin Infect Dis. 2008;46:1329–1337. doi: 10.1086/586745. [DOI] [PubMed] [Google Scholar]
- 94.Pue HL, Turabelidze G, Patrick S, et al. Human rabies – Missouri, 2008. MMWR Morb Mortal Wkly Rep. 2009;58:1207–1209. [PubMed] [Google Scholar]