

Abstract
The ability to efficiently modify the genome using CRISPR technology has rapidly revolutionized biology and genetics and will soon transform medicine. Duchenne muscular dystrophy (DMD) represents one of the first monogenic disorders that has been investigated with respect to CRISPR-mediated correction of causal genetic mutations. DMD results from mutations in the gene encoding dystrophin, a scaffolding protein that maintains the integrity of striated muscles. Thousands of different dystrophin mutations have been identified in DMD patients, who suffer from a loss of ambulation followed by respiratory insufficiency, heart failure, and death by the third decade of life. Using CRISPR to bypass DMD mutations, dystrophin expression has been efficiently restored in human cells and mouse models of DMD. Here, we review recent progress toward the development of possible CRISPR therapies for DMD and highlight opportunities and potential obstacles in attaining this goal.
Keywords: muscular dystrophy, skeletal muscle, CRISPR, dystrophin
INTRODUCTION
More than 800 monogenic disorders result in degeneration or dysfunction of skeletal muscle (1). Among the most severe is Duchenne muscular dystrophy (DMD), which is caused by mutations in the X-linked dystrophin gene, a massive gene spanning ∼2.3 megabases (2). DMD is the most common lethal monogenic disorder, and it primarily affects boys owing to their single X chromosome. The incidence of the disease is estimated at 1:5,000 boys worldwide. Approximately two-thirds of DMD mutations are inherited by the sons of mothers who are unknowing carriers of dystrophin mutations, and the remaining cases result from spontaneous germline mutations (3). More than 3,000 different dystrophin mutations have been shown to cause DMD, raising significant challenges for the development of gene correction therapies that might be applicable to large cohorts of patients rather than single individuals. Roughly two-thirds of dystrophin mutations involve exon deletions that disrupt the dystrophin open reading frame (ORF), and point mutations or duplications account for the rest.
DMD is typically diagnosed in the first few years of life due to muscle weakness and progresses to loss of ambulation in the early teens (4). Degeneration of skeletal muscle leads to skeletal deformities and respiratory insufficiency, and cardiomyopathy ultimately causes death in the third decade of life. A subset of patients also displays cognitive impairment. Becker muscular dystrophy (BMD) is a relatively mild form of the disease caused by in-frame deletions of the dystrophin gene. Patients with BMD exhibit a spectrum of phenotypes ranging from relatively mild muscle weakness and ambulation throughout life to severe muscle dysfunction resembling DMD (5). A major clinical goal has been to convert DMD to BMD through gene replacement or gene editing.
Since the discovery of the dystrophin gene in 1987 (2), there have been myriad efforts to develop therapies to delay disease progression and enhance muscle function in DMD patients. Various pharmacologic approaches, oligonucleotide-mediated exon skipping to bypass mutant exons, my-oblast and stem cell transfer, and gene replacement to provide truncated forms of dystrophin have been advanced in recent years, and numerous clinical trials are under way to assess their efficacy (6). While some therapies have shown transient benefits, the majority treat the consequences and symptoms of the disease rather than the underlying genetic cause, and none have yet restored long-term expression of the dystrophin protein.
Most recently, gene editing has been explored as a possible means of permanently removing genetic mutations that cause DMD, thereby restoring production of the missing dystrophin protein. No clinical trials have yet been initiated for the correction of DMD by gene editing, but the field is moving rapidly and represents a potentially transformative therapy unlike any other. Here we review recent progress toward gene editing as a means of eliminating the genetic mutations responsible for DMD and forestalling disease progression.
DYSTROPHIN
Dystrophin functions in muscle as a mechanical link to tether the actin cytoskeleton to the inner surface of the sarcolemma (7). Lack of dystrophin in skeletal muscle results in fragility of the sarcolemma, impaired intracellular signaling, myocyte necrosis, inflammatory infiltration, and, eventually, replacement of muscle with fibrotic and fatty tissue (8). During DMD progression, satellite cells, an injury-responsive stem cell population beneath the muscle basal lamina, becomes activated and fuses with damaged myofibers. Ultimately, this regenerative cell population becomes depleted and muscle degeneration ensues (9). In contrast, the adult heart lacks a robust stem cell population, so the absence of dystrophin in the heart ultimately results in myocyte dysfunction, consequent loss of pump function, and fatal cardiomyopathy (10).
The dystrophin gene encompasses 79 exons that are spliced together to give rise to a 427-kDa protein (Figure 1). The splicing patterns of these exons are conserved from mice to humans, allowing studies in mice to be directly extrapolated to the human gene. The encoded dystrophin protein contains four functional domains: an actin-binding domain at the N terminus that anchors the protein to the cytoskeleton; a central region containing 24 spectrin-like repeats that forms the rod domain, which is interrupted by four hinge domains; a cysteine-rich domain that binds β-dystroglycan; and a C-terminal domain that binds dystrobrevin and syntrophins, mediating sarcolemma localization.
Figure 1.
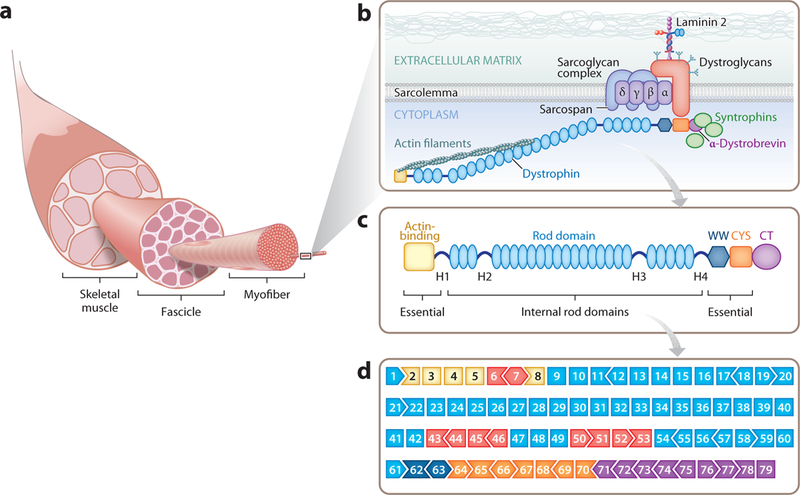
(a) Skeletal muscle is composed of thousands of multinucleated myofibers. Myofibers are held together in groups called fascicles. (b) The dystrophin-glycoprotein complex (DGC) resides on the sarcolemma of the myofiber and acts as an anchor. The N terminus of dystrophin connects with actin filaments and the C terminus interacts with the DGC, providing stability and integrity to the muscle cell. (c) Dystrophin protein structure. The N terminus of dystrophin contains the primary actin-binding domain, whereas the C terminus contains the dystroglycan-, dystrobrevin-, and syntrophin-binding sites. The N and C termini are essential for dystrophin function. The central rod domain acts like a spring between the two ends. The 24 spectrin-like repeats in the rod domain can be shortened to create a functional but less flexible dystrophin. (d ) The exon structure of the dystrophin gene, showing the 79 exons. The open reading frame (ORF) compatibility is shown by the shape of the adjacent exons. The exons are color coded to match the major functional dystrophin protein domains in panel c. The exons within the mutational hotspot regions are indicated in red. Adapted from Reference 21.
Dystrophin is a component of the dystrophin-glycoprotein complex (DGC), which anchors the cytoskeleton to the extracellular matrix and stabilizes the muscle membrane in response to contractions (Figure 1b). The DGC also contains α-dystroglycan, an extracellular peripheral membrane protein and a receptor for laminin-2, which links the DGC to the extracellular matrix; β-dystroglycan, which binds dystrophin; four sarcoglycans (α-, β-, γ-, and δ-); and sarcospan, which contributes to the stabilization of the complex. In addition, the spectrin-like repeats in the central rod domain of dystrophin bind neuronal nitric oxide synthase, targeting it to the sarcolemma.
The full-length form of dystrophin expressed in muscle tissues contains 3,684 amino acids. Interestingly, due to the modular structure of the dystrophin protein domains, internally truncated forms of dystrophin can retain function. In fact, alternatively spliced dystrophin products are found in muscle tissue of mild and asymptomatic BMD patients (11). Microdystrophin, a 1,001-amino-acid truncated form of dystrophin, which lacks the redundant regions of the rod domain and contains the minimal functional regions of the protein, is currently in clinical trials as a gene therapy for DMD (12, 13).
Although >3,000 different mutations have been shown to cause DMD, the majority of these mutations are positioned within so-called hotspots of the dystrophin gene and cluster around specific exons (Figure 1d). It has been estimated that ∼60% of DMD patients could benefit from gene editing strategies that skip the out-of-frame exons within these hotspots, and up to 80% of the patient population could potentially benefit if additional less common mutations were corrected. However, the remaining patients, who lack large genomic regions that include essential domains of the dystrophin protein, would not be treatable by gene editing and are therefore candidates for other gene replacement therapies.
GENOME EDITING
Clustered regularly interspaced short palindromic repeats (CRISPR) was first identified as a system for bacterial immunity in which segments of viral DNA are incorporated into the genome and subsequently transcribed into RNAs that can cooperate with the Cas9 endonuclease to recognize future viral pathogens and mediate their destruction (14–16). In recent years, the CRISPR system has been adapted as a tool that can edit the genome of nearly any organism. The CRISPR system involves two components: a single guide RNA (sgRNA) with complementarity to any sequence in the genome, and the Cas9 endonuclease, which associates with the sgRNA at the genomic target sequence. Cas9 from Streptococcus pyogenes (SpCas9) is the most commonly used enzyme, which cuts DNA adjacent to the protospacer adjacent motif (PAM) NAG or NGG (14–16). Cas9 protein from Staphylococcus aureus (SaCas9) uses the PAM motif NNGRR, which is more complex and limits the potential target sequences for gene editing (17). Another endonuclease smaller than SpCas9 is Cpf1 from Lachnospiraceae bacterium (LbCpf1), which requires a PAM sequence of 5′-TTTN-3′ (18). These and other types of Cas9 proteins offer more options for CRISPR editing site selection (19, 20). Gene editing can also be achieved using zinc-finger nucleases and transcription activator-like effector nucleases. We refer the reader to another article for consideration of these approaches (21).
Gene editing can occur through any of three pathways depending on the proliferative status of the cell, the presence or absence of an exogenous DNA template, and DNA sequence homologies surrounding the DNA sequence being targeted. In proliferative cells, when Cas9, sgRNA, and a DNA template are provided, gene editing can occur through homology-directed repair (HDR), which results in replacement of the targeted genomic region by the exogenous DNA template. Since this pathway is restricted to proliferating cells, it might be applicable to satellite cells, but it cannot be readily deployed in differentiated skeletal or cardiac myocytes. In the absence of an exogenous DNA template, a sgRNA can direct Cas9 to introduce a double-stranded break (DSB) in DNA, which is subsequently repaired through an imprecise process known as nonhomolo-gous end-joining (NHEJ), resulting in insertions and deletions (indels). This type of editing has been especially effective in deleting splice donor or acceptor site sequences in out-of-frame exons, thereby allowing restoration of the ORF of the dystrophin gene. Fortuitously, one of the PAM sequences of Cas9, NAG, corresponds to the universal splice acceptor site sequence, thus enabling delivery of Cas9 to the splice acceptor of any exon and skipping of that exon through creation of an indel. In a variation of NHEJ, referred to as microhomology-mediated end joining, specific deletions can be introduced into a targeted genomic region flanked by regions of short homology, which recombine in a precise way. An unexpected but potentially highly useful recent discovery is that NHEJ editing with one sgRNA, a process referred to as single-cut CRISPR, results preferentially in the incorporation of a single nucleotide at the DSB (22). This has been attributed to the creation of a one-nucleotide overhang at the site of DNA cleavage by Cas9, which is filled by a DNA polymerase and ligated (23). For exons that are out of frame by a single nucleotide, this type of gene editing thus allows efficient reframing of the protein.
Aside from the devastating clinical consequences of DMD and the lack of effective long-term therapy (24), multiple features of the disease render it amenable to gene editing as a therapeutic strategy. First, the modular structure of the rod domain of dystrophin makes it possible to delete mutant exons in this region of the gene and restore the ORF. Second, the location of the dystrophin gene on the X chromosome means that affected boys harbor only one mutant allele that needs to be corrected, and there are no concerns about inadvertently disrupting a wild-type copy of the gene. Third, only a minor fraction of normal dystrophin expression levels needs to be restored to achieve therapeutic benefit. This contrasts with disorders in which near-normal levels of a missing protein need to be produced or in which complete elimination of a toxic protein is required to achieve therapeutic efficacy. Moreover, because skeletal muscle is a syncytium in which hundreds of nuclei can share a common cytoplasm, genetic correction of even a small fraction of muscle nuclei can allow production of dystrophin and its distribution throughout the myofibers. In studies of mosaic mice generated by germline editing of a dystrophin mutation, we estimated that as little as 15% genetic correction was sufficient to restore dystrophin expression to normal levels in nearly all myofibers (25).
EDITING DUCHENNE MUSCULAR DYSTROPHY MUTATIONS IN PATIENT-DERIVED INDUCED PLURIPOTENT STEM CELLS
The breakthrough discovery of induced pluripotent stem cells (iPSCs) has transformed our prospects for disease modeling in vitro. The use of disease-specific iPSCs provides surrogate models of human diseases, platforms for drug discovery, and possible stem cell–based cell re-placement therapies. Easily accessible cell types, such as peripheral blood mononuclear cells, skin fibroblasts, or cells from urine samples, can be collected from a patient and reprogrammed to stem cells and their derivatives, effectively recapitulating the patient’s disease and genetic background in a culture dish (26, 27). The advantages of modeling diseases by using human iPSCs are the indefinite self-renewal capacity and pluripotency of these stem cells. In comparison to animal models of disease, patient-derived iPSCs provide an expandable and unlimited source for testing potential treatments or studying underlying disease mechanisms. Many human iPSC lines have been established to model muscular dystrophies, including DMD (27–30), facioscapulohumeral muscular dystrophy (31, 32), limb-girdle muscular dystrophy (26, 33, 34), and myotonic dystrophy (35). For DMD, multiple patient-derived iPSC lines with different types of mutations have been used to model disease-related gene expression, evaluate mutation-dependent variability, and test potential therapeutic strategies (27, 29).
Applications of gene editing that can be performed in human iPSC disease models include gene knockouts (36), insertion of transgenes (37), and repair of disease-relevant mutations (30). Differentiation of patient-derived iPSCs also provides a platform of cell types for CRISPR/Cas9-mediated gene editing. Thus, gene editing in iPSCs has become a standard research tool in regenerative medicine and human disease modeling.
CRISPR/Cas9-mediated gene editing has been applied to correct a variety of DMD mutations found in human myoblasts and patient-derived iPSC lines. Correction approaches for DMD include permanent exon removal, exon skipping, exon reframing, and exon knock-in (27, 38, 39). Functional dystrophin gene restoration has been demonstrated by CRISPR/Cas9 editing in iPSCs derived from DMD patients with exon deletions, exon duplications, and point mutations (27, 30, 39, 40). This includes the mutational hotspot regions in the repetitive rod domain and the essential actin-binding domains of dystrophin. Recently, CRISPR/Cpf1-mediated correction of DMD mutations in patient-derived iPSCs was also demonstrated (18). Corrected DMD patient-derived iPSCs can be used to model functional improvement and study disease-related mechanisms, for example, calcium handling in DMD dilated cardiomyopathy (39), and to monitor how a modified version of dystrophin functions in a human genetic background of DMD.
An important and often ignored challenge of iPSC technology is the variability between individual iPSC lines in their differentiation properties and phenotypes caused by variations in genetic background and reprogramming methods used to generate the cell lines. These intrinsic variations between individual iPSC lines can hinder the detection of subtle phenotypes or lead to misinterpretation of disease-relevant phenotypes (41, 42). To address this issue, isogenic pairs of disease-specific and control iPSCs that differ exclusively at specific DMD mutations have been generated using CRISPR technology and used to control for variation and defining differences in the muscle contractile profile (27).
It has been proposed that human iPSCs be used as a source for cell-based replacement transplantation therapy in other diseases, such as age-related macular degeneration (43). However, iPSC transplantation is not a feasible therapy for DMD owing to inability to access all the muscles of the body, low efficacy of engraftment of muscles, and the recognized genomic instability of iPSCs.
MUTATION CORRECTION BY CRISPR/CAS9 GENE EDITING IN VIVO
Animal Models of Duchenne Muscular Dystrophy
To date, more than 60 spontaneous or engineered DMD animal models have been reported (44). Murine and canine models have been favored to elucidate pathogenic mechanisms and develop new therapeutic approaches in DMD. The most widely used DMD mouse model is the mdx mouse, with a spontaneous nonsense point mutation (cytosine-to-thymine transition) in exon 23 that leads to loss of dystrophin expression (45, 46). The mdx mouse exhibits a 25% shorter lifespan than wild-type mice, but the difference is not comparable to human DMD patients, whose lifespan is reduced by 75% (47). Disease progression of the mdx mouse is also relatively mild and does not mimic the human course of the disease. Following birth, the muscles of mdx mice are histologically normal. Extensive necrosis, myofiber regeneration with centrally located nuclei, and elevated levels of serum creatine kinase are evident during the crisis period at 4–6 weeks of age (48, 49). Then disease progression slows, and the mice display a moderate myopathic disease state until 12–15 months of age, when muscle wasting, scoliosis, and cardiomyopathy develop (50–53). Although mdx mice develop a milder form of fibrosis and inflammatory cell infiltration in cardiac and skeletal muscle, their diaphragm presents severe pathological changes comparable to those in human DMD patients (54).
Four chemical variants of the mdx mouse have been developed, known as mdx2cv, mdx3cv, mdx4cv, and mdx5cv. Each of these mouse strains carries a point mutation that leads to loss of full-length dystrophin and results in expression of different dystrophin isoforms, which make these mutant mice useful for deciphering the role of these isoforms in DMD (55). Several other dystrophin-deficient mouse lines were established using different genetic engineering techniques, including Dup2 for exon 2 duplication (56) and MD-null for deletion of the entire dystrophin gene (57). Genome editing technology has played a major role in expanding DMD mouse models. For example, CRISPR/Cas9 was used to create a mouse model with deletion of exon 50, the most common mutational hotspot in DMD patients (22). Two other mouse models, hDMDdel45/mdx and hDMDdel52/mdx, were also generated using CRISPR/Cas9 to remove a specific exon in the knocked-in humanized dystrophin gene in the mdx mouse background (58, 59).
The relatively mild dystrophin-deficient phenotype of mice compared with DMD patients is thought to be due to the upregulation of compensatory mechanisms, such as increased utrophin expression, as well as robust skeletal muscle regenerative capacity in mice, plus differences in body size, lifespan and form of locomotion. To mimic the dystrophic phenotype in human DMD, several double-knockout mouse models have been generated with genetic elimination of the dystrophin gene and deletion of an additional gene that contributes to the compensatory mechanism, such as the genes for utrophin or α7β1-integrin (60–63). Another DMD mouse line that has features more representative of human DMD was generated by deleting telomerase RNA in mdx mice (64, 65). Mice deficient in dystrophin and telomerase RNA have increased telomere shortening, which is thought to disrupt muscle stem cell maintenance.
Clinical diagnosis of muscular dystrophy has been reported in dogs (66, 67). Unlike mdx mice, affected dogs share a clinical resemblance to human DMD patients, exhibiting limb muscle atrophy, fibrosis, joint contracture, and hypersalivation (44). Several dog models of DMD with spontaneous mutations that cause dystrophinopathy have been identified (68, 69). The age of onset of limb weakness and cardiac defects and the expected lifespan are comparable to those of human DMD patients (70, 71). These clinically similar pathologies make DMD dogs an excellent model for preclinical gene therapy studies.
CRISPR-Mediated Gene Editing Strategies
CRISPR/Cas-mediated genome editing has been shown to permanently correct DMD mutations and restore dystrophin function in mouse models. Germline editing by injecting zygotes with CRISPR/Cas9 editing components was first accomplished in mdx mice by correcting the mutated exon 23 using either HDR or NHEJ (25). Postnatal editing of mdx mice was then achieved using recombinant adeno-associated virus to deliver CRISPR/Cas9 genome editing components and correct the dystrophin gene by skipping or deleting the mutated exon 23 in vivo (72–75). Germline and postnatal CRISPR/Cas9 genome editing approaches both successfully restored dystrophin expression and improved muscle function in mdx mice. Other CRISPR systems, including CRISPR/Cpf1, have been used to correct DMD mutations in both mdx mice and human-derived iPSCs by exon skipping or HDR (18). Additionally, CRISPR/Cas9 adenine base editors have been deployed to repair a DMD mutant mouse with an exon 20 nonsense mutation by adenine-to-guanine single-nucleotide substitutions (76). These encouraging results suggest that CRISPR technology offers therapeutic potential to correct various DMD mutations. Different strategies for correcting DMD mutations with the CRISPR system are discussed in detail in the following sections and are outlined in Figure 2.
Figure 2.
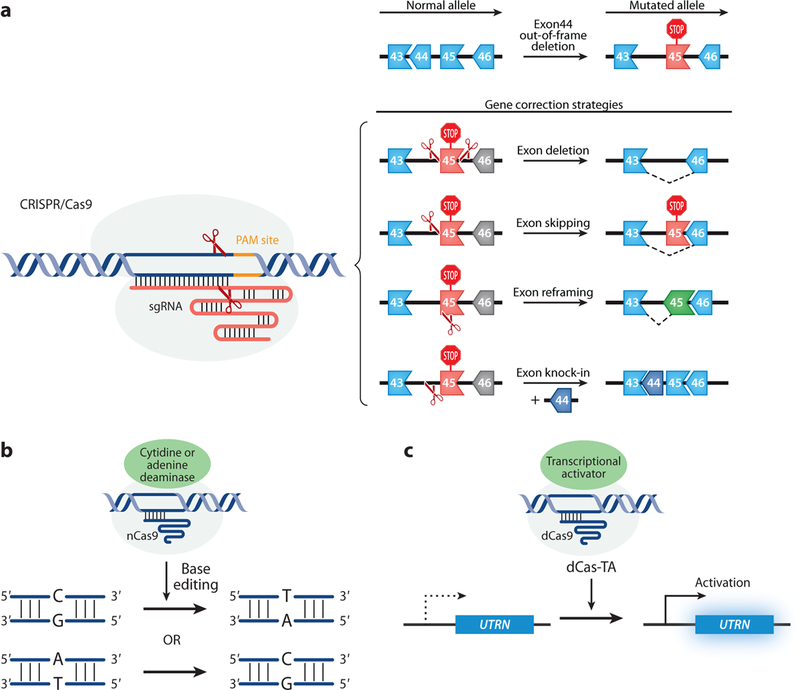
CRISPR-mediated editing strategies to correct DMD. (a) CRISPR/Cas9-mediated strategies that require double-strand breaks, including exon deletion, exon skipping, exon reframing, and exon knock-in. (b) CRISPR/nCas9 attached with a cytidine or adenine deaminase carry out a C:G to T:A or A:T to C:G pair base substitution. (c) CRISPR/dCas9-mediated gene regulation. UTRN, a gene that codes for utrophin, a compensatory protein for dystrophin, can be upregulated by CRISPR/dCas9 fused with a transcriptional activator. Abbreviations: CRISPR, clustered regularly interspaced short palindromic repeats; dCas9, catalytically deficient Cas9; DMD, Duchenne muscular dystrophy; nCas9, Cas9 nickase.
Exon deletion.
Approximately 65–72% of all DMD patients carry a deletion of one or more exons. Deletions tend to cluster in a hotspot region between exons 45 and 55 of the dystrophin gene (77, 78). A common strategy for correcting single or multiple exon deletions is to delete the out-of-frame exon and restore the ORF. This can be achieved by deleting one exon or the entire hotspot region. Specifically, two sgRNAs flanking the targeted exon(s) can be delivered with Cas9 to excise the single or multiple mutated exons, resulting in restoration of the ORF by splicing adjacent in-frame exons (Figure 2a) (30, 40, 79). Exon deletion strategies may also be used to correct exon duplication mutations, which occur in∼5%ofDMDpatients. A single sgRNA can be designed to target the intron region adjacent to the duplicated exon, and in the presence of Cas9, the single sgRNA will generate two cuts and delete one of the duplicated exons. Removal of one of the duplicated exons can renew the dystrophin gene ORF and produce full-length dystrophin protein, indistinguishable from normal dystrophin (27).
Exon skipping.
Skipping mutant exons leads to a shortened but semifunctional dystrophin protein, thereby transforming a severe DMD phenotype into the milder symptoms of BMD. In ∼83% of DMD patients, the deletions in the dystrophin gene can be targeted by exon skipping strategies, permitting one or more exons to be excluded to restore the dystrophin ORF (80). Anti-sense oligonucleotide (AON)-based exon skipping therapy for exon 51 has been approved by the US Food and Drug Administration (FDA) since 2016. However, AON-based exon skipping only modifies dystrophin mRNA, leaving the underlying mutation still present in the dystrophin gene. Therefore, for AON-based therapy to be effective, the DMD patient must undergo lifelong biweekly administration of this drug. In contrast, CRISPR gene editing treatment would potentially be “one and done,” as this technology corrects the underlying mutation in the genome.
Approaches used to correct DMD by CRISPR gene editing can be designed to minimize the loss of genomic DNA. Instead of using two sgRNAs flanking a mutant exon, one can design a single sgRNA to abolish either the splice acceptor site or splice donor site of the out-of-frame exon (Figure 2a). Once the sgRNA has guided Cas9 to cut near the exon junction, the sequence encoding the exon splice acceptor or donor site will be destroyed by NHEJ, resulting in splicing to the next available exon and skipping the out-of-frame exon. For example, CRISPR-mediated exon skipping was reported to correct a DMD mouse model with a deletion of exon 50. Splicing of exon 49 into exon 51 in these mice disrupts the dystrophin ORF. A single sgRNA can be used to correct this out-of-frame mutation. Targeting the splice acceptor site of exon 51 results in skipping exon 51 and restoration of the dystrophin ORF, as well as rescue of dystrophin expression and muscle function (22, 81). Furthermore, a single-sgRNA approach for exon skipping also improves the editing efficiency in comparison to using two sgRNAs to flank the exon for removal.
Exon reframing.
Another strategy to restore the dystrophin ORF is NHEJ-based reframing. When using a sgRNA to induce NHEJ in an out-of-frame exon, the generated indels lead to a targeted frameshift with a one-in-three possibility of putting the dystrophin gene back in frame (Figure 2a). Several groups have demonstrated successful restoration of the dystrophin ORF through exon reframing (18, 38, 40, 73, 79). Since exon reframing generates small indels during the repair, this strategy is an effective approach to preserve maximal dystrophin genomic sequence while circumventing the DMD mutation.
Like exon skipping, exon reframing has the advantage of requiring only a single sgRNA. When using two sgRNAs, successful exon excision requires two DNA DSBs around the targeted exon(s), and these cuts need to occur simultaneously. In contrast, single-sgRNA-mediated NHEJ targets one intron–exon junction and requires only a single cut to restore the dystrophin ORF by either exon skipping or exon reframing.
Exon knock-in.
All three strategies described above, using CRISPR gene editing to excise exons, result in a truncated form of dystrophin. In contrast, the exon knock-in strategy uses CRISPR-induced HDR, which incorporates a DNA donor template, resulting in restoration of full-length dystrophin protein (Figure 2a). However, there is a limit to the length of the DNA donor template allowed by certain delivery approaches, making it problematic to apply this strategy to large deletion mutations of dystrophin. In addition, the efficiency of HDR is low in postmitotic cells. The HDR machinery is not readily available in G1-arrested cells, such as mature myofibers. To overcome this problem, a recently developed CRISPR/Cas9-based technology named homology-independent targeted integration (HITI) supports targeted gene insertion in nondividing cells (82). The HITI method precisely knocks in a missing exon(s) at a specific locus using NHEJ and bypasses the requirement of HDR.
Base editing.
It is estimated that 25–35% of DMD patients have point mutations (77, 78). A newly developed strategy called base editing has been added to the CRISPR tool box to tackle DMD point mutations. These base editing tools can be divided into two categories: cytosine base editors that convert a C:G base pair to a T:A pair (83, 84) and adenine base editors that convert an A:T pair to a G:C pair (Figure 2b) (85). These RNA-guided nucleotide-specific base editors, which consist of a cytidine deaminase or an engineered adenine deaminase fused with a Cas9 nickase (nCas9) or catalytically deficient Cas9 (dCas9), do not produce DNA DSBs like Cas9 and do not rely on the NHEJ repair pathway. A donor DNA template for HDR is not required, and small indels through error-prone NHEJ at the target site are not produced. Most recently, CRISPR/Cas9 adenine base editors were used to substitute a single adenine to guanine in a DMD mouse model that harbors an exon 20 nonsense mutation (76). This strategy has been used to disrupt splicing acceptor sites (86) and can be used to disrupt premature stop codons for inducing exon skipping. This opens new therapeutic opportunities for DMD.
The alternatives.
The CRISPR technology has evolved not only for gene editing but also for gene regulation. By using dCas9, a deactivated form of Cas9, fused to a transcriptional activator or a repressor, CRISPR technology can be applied to transcriptional upregulation or downregulation (87, 88). For example, dCas9 fused with VP160 was shown to boost utrophin expression as a compensatory therapy in DMD (Figure 2c) (89). An advantage of this strategy is that it does not require a DNA DSB to regulate the target gene. However, prior to clinical use of this approach, maintenance and monitoring of the epigenetic state of the target must be ensured.
Delivery of CRISPR In Vivo
Effective in vivo postnatal genome editing requires an efficient delivery system. Genome editing components, Cas9 and sgRNA, can be delivered to target organs in various forms and by different systems. The forms of Cas9 and sgRNA can be DNA/DNA, mRNA/sgRNA, or protein/sgRNA, respectively. Viral and nonviral delivery systems are the CRISPR delivery systems commonly used in DMD.
Viral delivery.
Lentivirus, adenovirus, and adeno-associated virus (AAV) have been used for delivery of CRISPR/Cas9 components. Among these viruses, AAV has the advantage of low immunogenicity, minimal integration risk, tissue tropism, little toxicity, and long-term transgene expression from the episomal viral genome, making it a suitable viral vector for the delivery of gene editing components in patients with DMD (90). Indeed, the FDA has approved AAV for gene replacement therapy in spinal muscular atrophy, and clinical trials are in progress (91).
AAV has a relatively small cargo capacity (<4.7 kb) compared to other viral vectors. The SpCas9 ORF is ∼4.2 kb in length, which nears the maximum capacity of AAV cargo. This necessitates including an additional AAV vector harboring the sgRNA or the donor template. For example, a dual-AAV system has been shown to successfully edit hepatocytes and muscle cells in vivo (92). To circumvent the need for a dual-vector system, a smaller Cas9 protein such as SaCas9, which is encoded by a 3.2-kb cDNA, has been used for gene editing in mdx mouse strains (73–75).
AAV serotypes 1, 6, 8, 9, rh10, and rh74 have tropism for skeletal muscle and heart. Several studies have demonstrated successful delivery of CRISPR gene editing components using these AAV serotypes for postnatal genome editing (90, 93). This approach can be modified to correct other monogenic neuromuscular disorders.
Nonviral delivery.
Cas9 and sgRNAs, in various forms such as DNA, mRNA, or ribonucleoprotein (RNP), can be delivered in vivo using nonviral delivery systems. Electroporation delivers editing components by pulsing cells with high-voltage currents and creating nanometer-sized pores in the cell membrane. This allows negatively charged DNA or mRNA to enter the cells. This method has been used to deliver Cas9 and sgRNA constructs directly into skeletal muscle in mdx mice, resulting in restoration of dystrophin expression (94). Lipid-mediated nanoparticle delivery is another delivery option to carry CRISPR components in the forms of RNP or mRNA/sgRNA into the cells (95, 96). The cationic Cas9 protein is mixed with the highly anionic sgRNA to create a RNP complex that is highly anionic. This complex can be encapsulated by the cationic lipid nanoparticles and delivered into cells through endocytosis and macropinocytosis. Cationic lipid-based delivery is a relatively easy, low-cost process to deliver CRISPR components into cells. Furthermore, using RNPs as CRISPR components has been shown to reduce possible off-target mutations relative to nucleic acids (97). In addition to lipid-based nanoparticles, gold nanoparticles conjugated to DNA and complexed with cationic endosomal disruptive polymers have been reported to successfully deliver CRISPR RNP to correct the mutation in mdx mice (98). Therefore, CRISPR/Cas9 RNP and mRNA/sgRNA delivery could be a desirable method for further CRISPR/Cas9 applications in DMD therapy. However, given the requirement for body-wide delivery to muscles and the heart, major impracticalities remain to be overcome.
Potential Advantages Over Other Duchenne Muscular Dystrophy Therapies
How does gene editing differ from other therapeutic approaches that have been attempted for DMD? First, by eliminating the genetic mutations responsible for the disease, gene editing has the potential in principle to prevent further progression following a single treatment (caveats are discussed in the next section). Second, gene editing can allow the production of normal or nearly normal forms of dystrophin, which likely will maintain maximal function. This depends, of course, on the type of mutation being corrected. Nevertheless, optimized truncated forms of dystrophin, referred to as microdystrophins, which are currently being tested for gene therapy, are ∼116 kDa (1,001 amino acids) and contain the minimal functional domains; in contrast, the form of dystrophin generated by editing of the most commonly deleted exon, exon 50, is 3,606 amino acids in length if it is generated by skipping exon 51 and can be even larger (3,684 amino acids) if corrected by reframing. Finally, because expression of dystrophin following gene editing originates from the endogenous dystrophin gene, which contains the normal cis-regulatory elements within the native genomic location, the temporospatial expression and the level of expression are normal, whereas with microdystrophin gene replacement therapy, expression depends on the cis-regulatory sequences within the AAV vector.
FUTURE CHALLENGES
Despite the potential advantages of gene editing as a therapeutic strategy for DMD, this remains an experimental approach in the early stages of development, and there are many unknowns and potential challenges. Perhaps the biggest challenge is efficient body-wide delivery of gene editing components to all affected muscles and the heart. At present, AAV has provided promising results in mice and dogs, but cost-effective large-scale production of the quantities of AAV that will be needed for treatment of large cohorts of patients will be a challenge. It also remains to be determined whether gene editing will allow long-term maintenance of dystrophin expression over months, years, and even decades. Because cardiomyocytes are thought to be extremely long lived with an estimated turnover rate of only ∼1% per year in humans, it seems likely that dystrophin correction will be durable in the heart. Less clear is the turnover rate of adult skeletal muscle fibers, particularly in the setting of DMD. If we assume that gene editing will not fully rescue all muscle fibers, then there will likely be gradual muscle turnover and regeneration via satellite cell recruitment. As new satellite cells fuse with damaged myofibers, perhaps their nuclei will be exposed to the gene editing components within the injured myofibers and will be subsequently edited to allow dystrophin expression, thereby sustaining therapeutic rescue. However, it remains unknown whether satellite cell nuclei will be localized appropriately within a damaged myofiber so as to be exposed to gene editing components throughout life. Gene editing within satellite cells represents a means of maintaining a reservoir of myonuclei with the potential to restore dystrophin expression in DMD. However, with the exception of one report (75), efficient infection of satellite cells with AAV has not been observed (99). Whether the resistance of satellite cells to AAV-mediated editing reflects the lack of tropism of currently employed serotypes for satellite cells or the inaccessibility of satellite cells, which reside below the basal lamina, to AAV infection is uncertain.
The longevity of expression of Cas9 and sgRNAs from AAV is also unknown, although estimates of AAV half-life indicate sustained expression over many years (100). Because exposure to AAV elicits an immune response, at present only a single dose can be delivered. Thus, if expression is not sustained, subsequent injections with the same AAV serotype seem unlikely to be effective. It may be possible to use plasmapheresis to remove AAV antibodies to allow subsequent injections, or to use different AAV serotypes for subsequent injections, but such efforts remain in early development.
The risk of off-target mutations in the genome has been raised as a safety concern for gene editing in vivo, although there have been no documented examples of such mutations having deleterious effects in mice. Another theoretical concern for gene editing is the consequence of long-term expression of Cas9 throughout the body. Might this increase the likelihood of off-target effects or other pathological sequelae? To date, there has been no evidence for off-target mutagenesis in animals treated with AAV9-Cas9 and sgRNAs. Nevertheless, the possibility of a deleterious off-target mutation within a rogue cell somewhere in the body sometime in the lifespan cannot easily be ruled out. To minimize possible off-target effects in nonmuscle tissues, we and others have used muscle-specific cis-regulatory elements to drive expression of Cas9, thus restricting expression to muscle and preventing expression in the liver or kidney, which are known to accumulate AAV (22, 73, 93). Moreover, because muscle tissues are permanently postmitotic, possible oncogenic off-target effects in these tissues are likely to be insignificant.
There are several immunological considerations with respect to restoration of dystrophin expression by gene editing. As a foreign bacterial protein, Cas9 is likely to evoke an immune response, although studies to date have not observed this to be significant. As in any gene replacement therapy, the possibility of immune rejection of the protein being replaced is a concern. In the case of DMD, patients typically contain a small number of dystrophin-positive revertant fibers that arise because inaccurate splicing allows exon skipping and dystrophin expression. Because of these fibers, the immune system of DMD patients has been exposed to dystrophin, which may mitigate immune rejection of dystrophin-expressing cells following gene editing. In addition, DMD patients are typically treated with corticosteroids, which may also diminish possible adverse immune reactions.
CONCLUSIONS AND FUTURE PERSPECTIVES
While we have focused here on the potential application of gene editing to DMD, the lessons learned from these studies will undoubtedly be applicable to other monogenic disorders of muscle. Indeed, a study on facioscapulohumeral muscular dystrophy showed that CRISPR/dCas9 fused with a transcriptional repressor represses the expression of Double homeobox 4 (DUX4), a potent transcription factor that activates various genes that lead to oxidative stress and muscle atrophy (101). CRISPR/Cas9-mediated genome editing has also been shown to successfully correct genomic mutations by HDR in iPSCs derived from limb-girdle muscular dystrophy patients (33). Another report used CRISPR/Cas9-mediated NHEJ DNA repair to excise the entire region of expanded CTG/CAG repeats, thereby removing the pathogenic hallmarks of myotonic dystrophy (102).
It is remarkable to realize that CRISPR was only described about five years ago as a possible gene editing tool for mammalian cells and has already advanced into initial clinical studies. Despite the challenges and uncertainties of the CRISPR/Cas9 system, this technology has opened up new opportunities for correcting monogenic neuromuscular diseases, and there will undoubtedly be many unforeseen advances and applications of this technology in the years to come.
ACKNOWLEDGMENTS
We thank J. Cabrera for graphics. This work was supported by grants from the National Institutes of Health (AR-067294, HL-130253, HL-138426, and HD-087351) and the Robert A. Welch Foundation (grant 1–0025 to E.N.O.).
Footnotes
DISCLOSURE STATEMENT
Y.-L. Min has patent applications relevant to this review. R. Bassel-Duby is a consultant for Exonics Therapeutics and has patent applications relevant to this review. E. Olson is the founder of and a consultant for Exonics Therapeutics and has patent applications relevant to this review.
LITERATURE CITED
- 1.Bonne G, Rivier F, Hamroun D. 2017. The 2018 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul. Disord 27:1152–83 [DOI] [PubMed] [Google Scholar]
- 2.Hoffman EP, Brown RH Jr., Kunkel LM. 1987. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–28 [DOI] [PubMed] [Google Scholar]
- 3.Lee T, Takeshima Y, Kusunoki N, et al. 2014. Differences in carrier frequency between mothers of Duchenne and Becker muscular dystrophy patients. J. Hum. Genet 59:46–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darras BT, Urion DK, Ghosh PS. 1993–2018. Dystrophinopathies Seattle: Univ. Washington Press; [PubMed] [Google Scholar]
- 5.Andrews JG, Wahl RA. 2018. Duchenne and Becker muscular dystrophy in adolescents: current perspectives. Adolesc. Health Med. Ther 9:53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salmaninejad A, Valilou SF, Bayat H, et al. 2018. Duchenne muscular dystrophy: an updated review of common available therapies. Int. J. Neurosci 128:854–64 [DOI] [PubMed] [Google Scholar]
- 7.Ahn AH, Kunkel LM. 1993. The structural and functional diversity of dystrophin. Nat. Genet 3:283–91 [DOI] [PubMed] [Google Scholar]
- 8.Gao QQ, McNally EM. 2015. The dystrophin complex: structure, function, and implications for therapy. Compr Physiol 5:1223–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang NC, Chevalier FP, Rudnicki MA. 2016. Satellite cells in muscular dystrophy 2013; lost in polarity. Trends Mol. Med 22:479–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fayssoil A, Nardi O, Orlikowski D, et al. 2009. Cardiomyopathy in Duchenne muscular dystrophy: pathogenesis and therapeutics. Heart Fail. Rev 15:103. [DOI] [PubMed] [Google Scholar]
- 11.Ginjaar IB, Kneppers AL, v d Meulen J-DM, et al. 2000. Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family. Eur. J. Hum. Genet 8:793–96 [DOI] [PubMed] [Google Scholar]
- 12.Duan D. 2015. Duchenne muscular dystrophy gene therapy in the canine model. Hum. Gene Ther. Clin. Dev 26:57–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodino-Klapac LR, Montgomery CL, Bremer WG, et al. 2010. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol. Ther 18:109–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M, Chylinski K, Fonfara I, et al. 2012. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cong L, Ran FA, Cox D, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mali P, Yang L, Esvelt KM, et al. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ran FA, Cong L, Yan WX, et al. 2015. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520:186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Long C, Li H, et al. 2017. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv 3:e1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Müller M, Lee CM, Gasiunas G, et al. 2016. Streptococcus thermophilus CRISPR-Cas9 systems enable specific editing of the human genome. Mol. Ther 24:636–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hou Z, Zhang Y, Propson NE, et al. 2013. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. PNAS 110:15644–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Long C, Bassel-Duby R, et al. 2018. Myoediting: toward prevention of muscular dystrophy by therapeutic genome editing. Physiol. Rev 98:1205–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amoasii L, Long C, Li H, et al. 2017. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med 9:eaan8081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemos BR, Kaplan AC, Bae JE, et al. 2018. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. PNAS 115:E2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reinig AM, Mirzaei S, Berlau DJ. 2017. Advances in the treatment of Duchenne muscular dystrophy: new and emerging pharmacotherapies. Pharmacother. J. Hum. Pharmacol. Drug Ther 37:492–99 [DOI] [PubMed] [Google Scholar]
- 25.Long C, McAnally JR, Shelton JM, et al. 2014. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 345:1184–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim EY, Page P, Dellefave-Castillo LM, et al. 2016. Direct reprogramming of urine-derived cells with inducible MyoD for modeling human muscle disease. Skeletal Muscle 6:32–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long C, Li H, Tiburcy M, et al. 2018. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv 4:eaap9004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai W-F, Huang W, Wang L, et al. 2016. Induced pluripotent stem cells derived muscle progenitors effectively mitigate muscular dystrophy through restoring the dystrophin distribution. J. Stem Cell Res. Ther 6:1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi IY, Lim H, Estrellas K, et al. 2016. Concordant but varied phenotypes among Duchenne muscular dystrophy patient-specific myoblasts derived using a human iPSC-based model. Cell Rep 15:2301–12 [DOI] [PubMed] [Google Scholar]
- 30.Young CS, Hicks MR, Ermolova NV, et al. 2016. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 18:533–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snider L, Geng LN, Lemmers RJLF, et al. 2010. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLOS Genet 6:e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caron L, Kher D, Lee KL, et al. 2016. A human pluripotent stem cell model of facioscapulohumeral muscular dystrophy-affected skeletal muscles. Stem Cells Transl. Med 5:1145–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turan S, Farruggio AP, Srifa W, et al. 2016. Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy. Mol. Ther 24:685–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu J, Hunt SD, Matthias N, et al. 2017. Generation of an induced pluripotent stem cell line (CSCRMi001-A) from a patient with a new type of limb-girdle muscular dystrophy (LGMD) due to a missense mutation in POGLUT1 (Rumi). Stem Cell Res 24:102–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueki J, Nakamori M, Nakamura M, et al. 2017. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep 7:42522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Cao J, Xiong M, et al. 2015. Engineering human stem cell lines with inducible gene knockout using CRISPR/Cas9. Cell Stem Cell 17:233–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adkar SS, Willard VP, Brunger JM, et al. 2016. Targeted genome editing of human induced pluripotent stem cells using CRISPR/CAS9 to generate a knock-in type II collagen reporter for the purification of chondrogenic cells. Mol. Ther 24:S128 [Google Scholar]
- 38.Li Hongmei L, Fujimoto N, Sasakawa N, et al. 2015. Precise correction of the dystrophin gene in Duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep 4:143–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kyrychenko V, Kyrychenko S, Tiburcy M, et al. 2017. Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight 2:e95918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ousterout DG, Kabadi AM, Thakore PI, et al. 2015. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun 6:6244–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bock C, Kiskinis E, Verstappen G, et al. 2011. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144:439–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boulting GL, Kiskinis E, Croft GF, et al. 2011. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol 29:279–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandai M, Watanabe A, Kurimoto Y, et al. 2017. Autologous induced stem-cell–derived retinal cells for macular degeneration. N. Engl. J. Med 376:1038–46 [DOI] [PubMed] [Google Scholar]
- 44.McGreevy JW, Hakim CH, McIntosh MA, Duan D. 2015. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Models Mech 8:195–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulfield G, Siller WG, Wight PA, et al. 1984. X chromosome-linked muscular dystrophy (mdx) in the mouse. PNAS 81:1189–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sicinski P, Geng Y, Ryder-Cook AS, et al. 1989. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244:1578–80 [DOI] [PubMed] [Google Scholar]
- 47.Chamberlain JS, Metzger J, Reyes M, et al. 2007. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 21:2195–204 [DOI] [PubMed] [Google Scholar]
- 48.Carnwath JW, Shotton DM. 1987. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci 80:39–54 [DOI] [PubMed] [Google Scholar]
- 49.Dangain J, Vrbova G. 1984. Muscle development in mdx mutant mice. Muscle Nerve 7:700–4 [DOI] [PubMed] [Google Scholar]
- 50.Pastoret C, Sebille A. 1995. mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci 129:97–105 [DOI] [PubMed] [Google Scholar]
- 51.Bostick B, Yue Y, Long C, et al. 2008. Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ. Res 102:121–30 [DOI] [PubMed] [Google Scholar]
- 52.Lefaucheur Jean P, Pastoret C, Sebille A. 1995. Phenotype of dystrophinopathy in old MDX mice. Anat. Rec 242:70–76 [DOI] [PubMed] [Google Scholar]
- 53.Hakim CH, Grange RW, Duan D. 2011. The passive mechanical properties of the extensor digitorum longus muscle are compromised in 2- to 20-mo-old mdx mice. J. Appl. Physiol 110:1656–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stedman HH, Sweeney HL, Shrager JB, et al. 1991. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352:536–39 [DOI] [PubMed] [Google Scholar]
- 55.Chapman VM, Miller DR, Armstrong D, et al. 1989. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. PNAS 86:1292–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vulin A, Wein N, Simmons TR, et al. 2015. The first exon duplication mouse model of Duchenne muscular dystrophy: a tool for therapeutic development. Neuromuscul. Disord 25:827–34 [DOI] [PubMed] [Google Scholar]
- 57.Kudoh H, Ikeda H, Kakitani M, et al. 2005. A new model mouse for Duchenne muscular dystrophy produced by 2.4 Mb deletion of dystrophin gene using Cre-loxP recombination system. Biochem. Biophys. Res. Commun 328:507–16 [DOI] [PubMed] [Google Scholar]
- 58.Young CS, Mokhonova E, Quinonez M, et al. 2017. Creation of a novel humanized dystrophic mouse model of Duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J. Neuromuscul. Dis 4:139–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Veltrop M, van Vliet L, Hulsker M, et al. 2018. A dystrophic Duchenne mouse model for testing human antisense oligonucleotides. PLOS ONE 13:e0193289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deconinck AE, Rafael JA, Skinner JA, et al. 1997. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90:717–27 [DOI] [PubMed] [Google Scholar]
- 61.Rooney JE, Welser JV, Dechert MA, et al. 2006. Severe muscular dystrophy in mice that lack dystrophin and α7 integrin. J. Cell Sci 119:2185–95 [DOI] [PubMed] [Google Scholar]
- 62.Guo C, Willem M, Werner A, et al. 2006. Absence of α7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet 15:989–98 [DOI] [PubMed] [Google Scholar]
- 63.Grady RM, Teng H, Nichol MC, et al. 1997. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell 90:729–38 [DOI] [PubMed] [Google Scholar]
- 64.Sacco A, Mourkioti F, Tran R, et al. 2010. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 143:1059–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mourkioti F, Kustan J, Kraft P, et al. 2013. Role of telomere dysfunction in cardiac failure in Duchenne muscular dystrophy. Nat. Cell Biol 15:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Valentine BA, Cooper BJ, Cummings JF, et al. 1986. Progressive muscular dystrophy in a golden retriever dog: light microscope and ultrastructural features at 4 and 8 months. Acta Neuropathol 71:301–10 [DOI] [PubMed] [Google Scholar]
- 67.Funkquist B, Haraldsson I, Stahre L. 1980. Primary progressive muscular dystrophy in the dog. Vet Rec 106:341–43 [DOI] [PubMed] [Google Scholar]
- 68.Sharp NJH, Kornegay J, Van Camp SD, et al. 1992. An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics 13:115–21 [DOI] [PubMed] [Google Scholar]
- 69.Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, et al. 2010. A Duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier King Charles spaniels is amenable to exon 51 skipping. PLOS ONE 5:e8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Smith BF, Yue Y, Woods PR, et al. 2011. An intronic LINE-1 element insertion in the dystrophin gene aborts dystrophin expression and results in Duchenne-like muscular dystrophy in the corgi breed. Lab. Investig. J. Tech. Methods Pathol 91:216–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Valentine BA, Cooper BJ, de Lahunta A, et al. 1988. Canine X-linked muscular dystrophy: an animal model of Duchenne muscular dystrophy: clinical studies. J. Neurol. Sci 88:69–81 [DOI] [PubMed] [Google Scholar]
- 72.Long C, Amoasii L, Mireault AA, et al. 2016. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351:400–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bengtsson NE, Hall JK, Odom GL, et al. 2017. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun 8:14454–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nelson CE, Hakim CH, Ousterout DG, et al. 2016. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351:403–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tabebordbar M, Zhu K, Cheng JKW, et al. 2016. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351:407–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ryu S-M, Koo T, Kim K, et al. 2018. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol 36:536–39 [DOI] [PubMed] [Google Scholar]
- 77.Bladen CL, Salgado D, Monges S, et al. 2015. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat 36:395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aartsma-Rus A, Van Deutekom JCT, Fokkema Ivo F, et al. 2006. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 34:135–44 [DOI] [PubMed] [Google Scholar]
- 79.Maggio I, Liu J, Janssen JM, et al. 2016. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep 6:37051–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kole R, Krieg AM. 2015. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Del. Rev 87:104–7 [DOI] [PubMed] [Google Scholar]
- 81.Amoasii L, Hildyard JCW, Li H, et al. 2018. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362:86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Suzuki K, Tsunekawa Y, Hernandez-Benitez R, et al. 2016. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540:144–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Komor AC, Kim YB, Packer MS, et al. 2016. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishida K, Arazoe T, Yachie N, et al. 2016. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353:aaf8729 [DOI] [PubMed] [Google Scholar]
- 85.Gaudelli NM, Komor AC, Rees HA, et al. 2017. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551:464–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gapinske M, Luu A, Winter J, et al. 2018. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol 19:107–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chavez A, Scheiman J, Vora S, et al. 2015. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12:326–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Perez-Pinera P, Kocak DD, Vockley CM, et al. 2013. RNA-guided gene activation by CRISPR-Cas9– based transcription factors. Nat. Methods 10:973–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wojtal D, Kemaladewi Dwi U, Malam Z, et al. 2016. Spell checking nature: versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet 98:90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang J-Z, Wu P, Shi Z-M, et al. 2017. The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev 39:547–56 [DOI] [PubMed] [Google Scholar]
- 91.Mendell JR, Al-Zaidy S, Shell R, et al. 2017. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med 377:1713–22 [DOI] [PubMed] [Google Scholar]
- 92.Yang Y, Wang L, Bell P, et al. 2016. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol 34:334–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lau C-H, Suh Y. 2017. In vivo genome editing in animals using AAV-CRISPR system: applications to translational research of human disease. F1000Research 6:2153–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xu L, Park KH, Zhao L, et al. 2016. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther 24:564–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miller JB, Zhang S, Kos P, et al. 2016. Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle co-delivery of Cas9 mRNA and sgRNA. Angew. Chemie Int. Ed 56:1059–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zuris JA, Thompson DB, Shu Y, et al. 2014. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol 33:73–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim S, Kim D, Cho SW, et al. 2014. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee K, Conboy M, Park HM, et al. 2017. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng 1:889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arnett ALH, Konieczny P, Ramos JN, et al. 2014. Adeno-associated viral vectors do not efficiently target muscle satellite cells. Mol. Ther. Methods Clin. Dev 1:14038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kessler PD, Podsakoff GM, Chen X, et al. 1996. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. PNAS 93:14082–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Himeda CL, Jones TI, Jones PL. 2016. CRISPR/dCas9-mediated transcriptional inhibition ameliorates the epigenetic dysregulation at D4Z4 and represses DUX4-fl in FSH muscular dystrophy. Mol. Ther 24:527–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van Agtmaal EL, Andre´ LM, Willemse M, et al. 2017. CRISPR/Cas9-induced (CTGcCAG)n˙ repeat instability in the myotonic dystrophy type 1 locus: implications for therapeutic genome editing. Mol. Ther 25:24–43 [DOI] [PMC free article] [PubMed] [Google Scholar]