

Abstract
Bile acids are critical metabolites in the gastrointestinal tract and contribute to maintaining intestinal immune homeostasis through cross-talk with the gut microbiota. The conversion of bile acids by the gut microbiome is now recognized as a factor affecting both host metabolism and immune responses, but its physiological roles remain unclear. We conducted a screen for microbiome metabolites that would function as inflammasome activators and herein report the identification of 12-oxo-lithocholic acid (BAA485), a potential microbiome-derived bile acid metabolite. We demonstrate that the more potent analogue 11-oxo-12S-hydroxylithocholic acid methyl ester (BAA473) can induce secretion of interleukin-18 (IL-18) through activation of the inflammasome in both myeloid and intestinal epithelial cells. Using a genome-wide CRISPR screen with compound induced pyroptosis in THP-1 cells, we identified that inflammasome activation by BAA473 is pyrin-dependent (MEFV). To our knowledge, the bile acid analogues BAA485 and BAA473 are the first small molecule activators of the pyrin inflammasome. We surmise that pyrin inflammasome activation through microbiota-modified bile acid metabolites such as BAA473 and BAA485 plays a role in gut microbiota regulated intestinal immune response. The discovery of these two bioactive compounds may help to further unveil the importance of pyrin in gut homeostasis and autoimmune diseases.
Keywords: inflammasome, CRISPR/Cas, microbiome, mucosal immunology, bile acid, pyrin, familial Mediterranean fever, CRISPR screen, microbiota, biotransformation, immune response, gut homeostasis, autoimmune disease
Introduction
The stimulation of microbe-associated molecular patterns or damage-associated molecular patterns triggers the assembly of the cytosolic inflammasome, a multiprotein complex that leads to the release of interleukin 1β (IL-1β)3 and interleukin 18 (IL-18) and induction of pyroptosis, a type of inflammasome-associated cell death (1). Accumulating evidence suggests a critical role of the inflammasome in coordinating the immune response against pathogens (2), and excessive inflammasome signaling is associated with autoinflammatory diseases such as Muckle–Wells syndrome (3). Proper modulation of the inflammasome activity is therefore necessary to maintain immune system homeostasis.
The inflammasome complex components are highly expressed in both the epithelial and immune cells of the gut. Recent studies have indicated that perturbations of the inflammasome pathway may be an axis of dysbiosis in human inflammatory bowel disease. For example, mice deficient in the inflammasome sensors NLRP3, NLRP6, and pyrin all display impaired immune responses, exacerbation of chemically induced colitis, and dysbiosis of the gut microbiota (4–6). Interestingly, human genetics studies have identified inflammatory bowel disease–associated SNPs in inflammasome-related genes such as ILRAP (7). Whereas numerous studies have focused on the physiological functions of intestinal inflammasomes, specific ligands that elicit inflammasome activation and their underlying mechanisms are poorly understood.
Bile acids are a family of steroid acids produced in the liver from cholesterol. In a process called enterohepatic circulation, the primary bile acids, such as chenodeoxycholic acid and cholic acid, are synthesized in the liver, secreted into the duodenum via the bile, reabsorbed in the ileum, and circulated back to the liver via the portal vein (8). During enterohepatic circulation, about 3% of the total bile acids remain in the gut, where they exist at high concentrations (about 200–1000 μm) (9). Bile acids can be further modified by the gut microbiota. For instance, cholic acid can be converted into the secondary bile acid deoxycholic acid (DCA), which can compose up to 20–30% of the human bile acid pool in the gut (10). Although there is emerging appreciation of the bioconversion of bile acids by the gut microbiota in impacting host metabolism and immune responses (8, 11), the exact physiological roles of gut microbiota–mediated biotransformation of bile acids are still largely unknown.
In this study, we screened a focused set of predicted microbiome-derived metabolites and identified potential bacteria-metabolized bile acid analogues as inflammasome activators. Further, we used pooled CRISPR screen technology to identify pyrin as the inflammasome sensor of these bile acid analogues. Our results suggest that intestinal microbiota-mediated bile acid conversion may regulate intestinal immune homeostasis by modulating pyrin inflammasome signaling.
Results
Bile acid analogues, BAA485 and BAA473, induce IL-18
Accumulating evidence suggests that proper modulation of the intestinal inflammasome is required for maintenance of immune homeostasis in the gut; however, the specific ligands that regulate the inflammasome response are not fully deciphered. We hypothesized that the intestinal inflammasome may sense the presence of gut bacteria by recognizing secondary metabolites derived from commensal bacteria. Therefore, we screened a small collection of known and predicted microbiome-derived metabolites for induction of IL-18 in human peripheral blood mononuclear cells (PBMCs) primed with LPS. Whereas none of the principal bile acids, including DCA, induced IL-18 production (Fig. S1 and see Fig. 6, B and C), 12-oxo-lithocholic acid (BAA485), a putative intestinal bacteria-produced derivative of DCA (12), induced secretion of IL-18 in LPS-primed PBMCs (Fig. 1, A and B). These results suggest that BAA485 could be an inflammasome activator and that C12 oxidation of DCA by the gut microbiota may potentially play a role in the immunogenicity of BAA485 (13). We performed structure–activity relationship studies and identified a more potent analog, BAA473, as a robust activator of IL-18 secretion in PBMCs (Fig. 1, A and B). The enhanced potency of BAA473 led us to focus on this compound for further mechanistic experiments.
Figure 6.
A, BAA473 induces inflammasome activation in U937 cells overexpressing pyrin. Control and pyrin overexpression lines were established via lentiviral transduction in U937 cells lacking endogenous pyrin expression. Cells were primed with 1 μg/ml LPS and treated with increasing concentrations of BAA473. IL-1β secretion was measured by HTRF. B and C, BAA473 is a stronger pyrin inflammasome activator than BAA485. U937-pyrin cells and PBMCs were primed overnight with LPS (1 μg/ml and 0.1 ng/ml, respectively) and treated with BAA473, BAA485, and DCA for 16 h followed by measurement of IL-18 secretion. Data presented are mean ± S.E. (error bars) (n = 3). *, p < 0.001; **, p < 0.01 versus vehicle (paired t test).
Figure 1.
A, structures of inflammasome inducer BAA485 and BAA473. Structure–activity relationship studies revealed that moving the ketone from the 12-oxo position of the weak inflammasome inducer BAA485 to the 11-oxo position of BAA473 as well as adding the methyl pentanoate group results in a more potent inflammasome activity. B, BAA485 induces secretion of IL-18 in PBMCs. PBMCs were primed with 0.1 ng/ml LPS overnight. Following treatment with BAA485 (4-((3R,5R,10S,13R,17R)-3-hydroxy-10,13-dimethyl-12-oxohexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoic acid) or BAA473 (methyl 4-((3R,5R,10S,12S,13R,17R)-3,12-dihydroxy-10,13-dimethyl-11-oxohexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)pentanoate) for 16 h, IL-18 secretion was measured by AlphaLISA. Data presented are mean ± S.E. (error bars) (n = 3). *, p value <0.004 versus vehicle (paired t test).
BAA473 activates inflammasome in both myeloid and intestinal epithelial cells
Several approaches were used to demonstrate that BAA473 activates the inflammasome pathway. BAA473 selectively induced secretion of the inflammasome-regulated cytokines, IL-1β and IL-18, but not IL-6 and IL-8, in LPS-primed PBMCs (Figs. 1B and 2A). Likewise, in THP-1 cells, BAA473 induced the secretion of IL-1β and IL-18 (Fig. 2B), activated caspase-1 (CASP1) and stimulated release of lactate dehydrogenase (LDH) (Fig. 2, C and D). Moreover, the CASP1 inhibitor VX-765 blocked BAA473-induced IL-1β production in THP-1 cells (Fig. 2E). Together, these data suggest that BAA473 functions as an inflammasome activator.
Figure 2.
A, BAA473 selectively induces secretion of IL-1β but not IL-6 and IL-8. PBMCs were primed with 0.1 ng/ml LPS overnight followed by BAA473 treatment for 16 h and HTRF detection of secreted levels of IL-1β, IL-6, and IL-8. Data presented are mean ± S.E. (error bars) (n = 3). *, p < 0.005 versus vehicle (paired t test). B–D, BAA473 induces secretion of cytokines as well as caspase-1 and LDH activity in THP-1 cells. THP-1 cells were primed with 1 μg/ml LPS overnight followed by compound treatment for 24 h. B, cytokine secretion in supernatant was measured by HTRF (*, p < 0.0007). C, caspase-1 activity was assessed using the Caspase-Glo® 1 assay. Nigericin was used as an NALP3 activator. D, LDH activity (*, p < 0.003) was measured using a colorimetric LDH assay kit. E, BAA473 induces cytokine production via ASC/CASP1. THP-1 cells primed with 1 μg/ml LPS were pretreated with 10 μm BAA473 or 5 mm ATP for 1 h, followed by overnight treatment with caspase-1 inhibitor (VX-765). The level of IL-1β secretion was measured by an HTRF assay. Data presented are mean ± S.E. (n = 3).
In immune cells, the functional role of the inflammasome is well-appreciated; however, the role in epithelial cells remains understudied. Many inflammasome components are highly expressed by intestinal epithelial cells (IECs), which act as a barrier between the host and the intestinal microbiota. Recent reports have identified functional roles of the IEC inflammasomes in mucosal immune defense, inflammation, and tumorigenesis (14). To understand the potential connection of the bile acid analogues' role in gut homeostasis, we evaluated BAA473 in IECs. In addition to its effects on immune cells, BAA473 induced secretion of IL-18 but not IL-1β in the T84 human colonic adenocarcinoma cell line (Fig. 3A). To mimic a differentiated and polarized gut epithelium, we used an epithelial monolayer derived from primary human intestinal organoids as an in vitro system (15, 16). The addition of BAA473 on the luminal side of the epithelial monolayer resulted in apical secretion of IL-18, whereas no secretion of IL-18 was detected on the basolateral side (Fig. 3B). Together, these results demonstrate that BAA473 is an inflammasome activator in both myeloid and intestinal epithelial cells, two key cell types involved in immune homeostasis in the gut.
Figure 3.
A, BAA473 induces secretion of IL-18 in T84 cells as well as in the intestinal organoid-derived epithelial monolayer. TLR-unprimed T84 cells constitutively expressing pro-IL-18 were treated with BAA485 and BAA473 overnight. IL-1β cytokine secretion was measured by HTRF, and IL-18 cytokine secretion was measured by AlphaLISA. Data presented are mean ± S.E. (error bars) (n = 3). *, p < 0.05 versus vehicle (paired t test). B, TLR-unprimed human intestinal organoid-derived epithelial cells were plated in a monolayer in 96-well transwell membrane cell culture inserts and treated with BAA473 overnight followed by measurement of IL-18 secretion by AlphaLISA. Data presented are mean ± S.E. (n = 3). *, p < 0.04 versus vehicle (paired t test).
BAA473 activates pyrin inflammasome
To understand mechanistically how BAA473 activates the inflammasome pathway, we first evaluated whether BAA473 was NLRP3-dependent. Interestingly, BAA473-induced IL-1β and IL-18 secretion was blocked by CASP1 inhibition, but cytokine secretion was not blocked by the NLRP3 inhibitor, MCC950 (17). This suggests that BAA473-induced inflammasome activation is independent of the NLRP3 pathway (Figs. 2E and 4A). To define the inflammasome pathway modulated by BAA473, we took an unbiased approach and performed a whole-genome pooled CRISPR screen using BAA473-mediated pyroptosis as a readout (Fig. 4C). A similar approach recently identified NEK7 as an essential component of the inflammasome (18). To conduct our genome-wide screen, THP-1-Cas9 cells were infected with a whole-genome gRNA library and treated with BAA473 to induce cell death (Fig. 4B). The gRNAs enriched in “nonresponding” cells, unaffected by BAA473-mediated pyroptosis, were analyzed by next-generation sequencing. We identified ASC, a common adaptor for multiple inflammasome pathways, and the inflammasome sensor pyrin (MEFV) (19) as the two strongest hits in the screen (Fig. 4D). To validate ASC and pyrin, each gene was knocked out with two independent gRNAs in THP-1-Cas9 cells (Fig. 5A). Validating the screening results, ASC and pyrin were both required for BAA473-induced cell death as well as IL-1β and IL-18 secretion (Fig. 5, B–D). These data suggest that pyrin is likely the responding inflammasome sensor for BAA473 and mediates the classical inflammasome pathway activation involving ASC.
Figure 4.

A, BAA473 induces cytokine production via ASC/CASP1 but not NLRP3. LPS-primed (0.1 ng/ml) PBMCs were pretreated with 5 μm BAA473 or 5 mm ATP for 1 h followed by overnight treatment with increasing concentrations of the NLRP3 inhibitor MCC950. IL-1β secretion was measured by HTRF. B–D, genome-wide CRISPR screen to determine the mechanism of action of BAA473. B, screening workflow. C, LPS-primed (1 μg/ml) THP-1 cells were treated with BAA473 overnight, after which cell death was assessed by CTG, whereas IL-1β secretion was measured by HTRF. Induced cell death observed in LPS-primed (1 μg/ml) THP-1 cells upon treatment with BAA473 was used as a readout in the genome-wide CRISPR screen. D, THP-1-Cas9 cells were transduced with a genome-wide lentiviral CRISPR library. Cells were primed with LPS and after 24 h were treated with 75 μm BAA473. The next day, live cells were FACS-sorted using fluorescent live stain. The screen was run in duplicate. Gene-centric visualization is shown as the average log2 -fold change versus significance score in live cells. Error bars, S.E.
Figure 5.
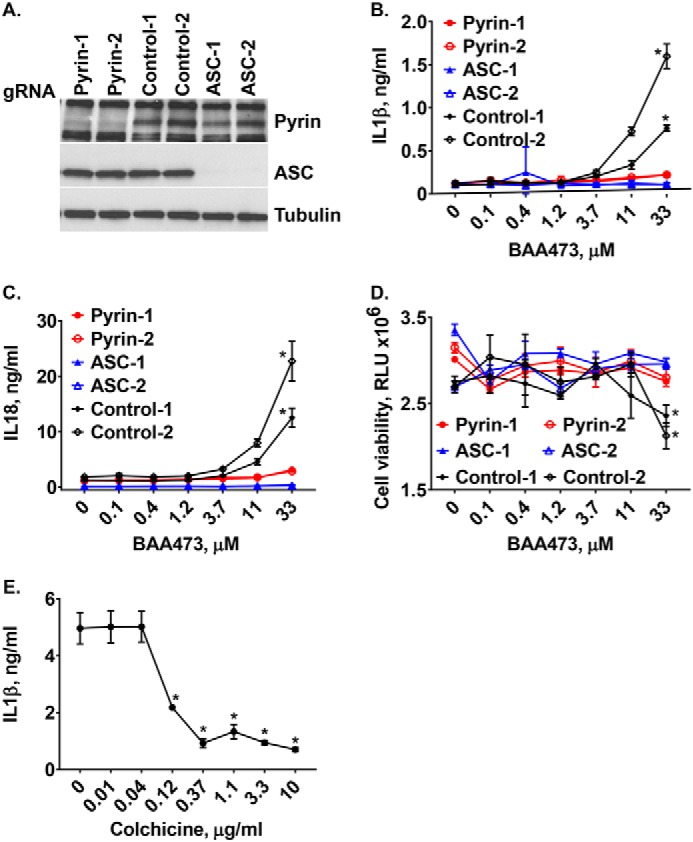
A, pyrin hit validation. Pyrin and ASC knockout cells were validated by Western blotting using anti-pyrin and anti-ASC antibodies. B–D, BAA473-induced production of IL-1β and IL-18 as well as pyroptosis were blocked in TLR-primed THP-1-Cas9 cells with knockout of pyrin and ASC. Two knockout cell lines each for pyrin and ASC were generated by transducing THP-1-Cas9 cells with lentiviral vectors expressing the corresponding gRNAs. The knockout cells were primed with 1 μg/ml LPS overnight and then treated with increasing concentrations of BAA473. Data presented are mean ± S.E. (n = 3). *, p < 0.002 (IL-1β); *, p < 0.01 (IL-18); *, p < 0.03 (CTG) versus control (paired t test). E, colchicine blocks cytokine production but not cell death. TLR-primed (1 μg/ml LPS) THP-1 cells were pretreated with 10 μm BAA473 for 1 h followed by overnight treatment with colchicine. IL-1β secretion was measured by HTRF, and cell death was assessed by CTG. Data presented are mean ± S.E. (error bars) (n = 3). *, p < 0.02 versus vehicle (paired t test).
Pyrin is genetically associated with familial Mediterranean fever (FMF), a monogenic autoinflammatory disease. More than 80 mutations in pyrin associated with FMF have been identified (https://infevers.umai-montpellier.fr/web/)4 (40). The first-line treatment for FMF is the drug colchicine, which inhibits activation of pyrin by disrupting the microtubule network (20). Intriguingly, colchicine blocked inflammasome activation by BAA473 (Fig. 5E), which supports pyrin as the sensor for BAA473. To confirm that inflammasome activity of BAA473 is pyrin-dependent, we generated a U937-pyrin stable cell line and observed that BAA473 enhanced secretion of IL-18 and cell death in cells stably expressing pyrin, but not in cells expressing the empty vector (Fig. 6A). Whereas BAA485 was too weak to induce inflammasome activation in THP-1 cells (data not shown), it could induce IL-18 production in the U937-pyrin reporter cell line as well as in PBMCs (Figs. 1B and 6 (B and C)), which suggests that both BAA473 and BAA485 activate the pyrin inflammasome with a similar mechanism.
The BAA473-sensitized inflammasome CRISPR screen also identified other hits; some of these may help define potential pathways that converge on regulation of bile acid metabolite-mediated pyrin inflammasome activation (Fig. S3). It is worth noting that none of these hits scored in the reported NLRP3 CRISPR screen (18). Molecular signatures database (MSigDB) analysis identified a few enriched gene sets (Table S1), including the inositol phosphate signaling regulators (IPMK, IPPK, and ITPK1) and components of the transcription factor complex (IRF8 and SPI1) that controls interferon-induced gene expression (21, 22). Pyrin has been reported to be up-regulated following interferon stimulation, it is possible that IRF8 and SPI1 regulate BAA473-mediated inflammasome activation by modulation of pyrin expression. Further studies are needed to explore how these signaling pathways might influence pyrin inflammasome activation.
Discussion
In an unbiased approach, we developed a short path from focused screen to identification of the ligand–target component of the inflammasome. Our focused screen concept could be readily expandable to identify other potential endogenous ligand–inflammasome component pairs, with rapid target validation by genome-wide CRISPR screening. The combination of stable pyrin-expressing U937 cells with the control compound BAA473 should enable a larger screening effort to identify other potential metabolites or drugs that would modulate the pyrin inflammasome. Modulation of the pyrin inflammasome has been limited to the use of colchicine and the bacterial toxin TcdB, which as indirect modulators have limitations. The pyrin inflammasome activators, BAA485 and BAA473, will hopefully enable further insights and better mechanistic understandings around pyrin in the context of gut homeostasis and autoimmune diseases.
Accumulating evidence supports that the inflammasome has a key role in shaping epithelial responses at the host–lumen interface. Previously, microbiota metabolites such as taurine have been identified to have a role in regulating NLRP6 inflammasome signaling (23). We show that BAA485 and BAA473 activate the pyrin inflammasome pathway in cell types relevant to maintaining intestinal immune homeostasis. Intriguingly, pyrin inflammasome activation in the gut can have multiple consequences. Minimal activation of pyrin in the resting state may promote intestinal barrier integrity and prevent inflammation and tumorigenesis in the colon (4); however, acute or excessive activation (e.g. by the toxin TcdB produced by Clostridium difficile) can lead to pathogenic inflammation (19). One can speculate that the level of pyrin activation in the gut might be controlled by gut microbiota-modified bile acids such as BAA485, which are likely to be present at significantly lower concentrations than the principal bile acids, thereby preventing full activation of pyrin inflammasome.
FMF patients display a dramatic range of disease severity, which is attributable to mutations in pyrin (24, 25); however, the range in symptom intensity is also attributed to environmental factors (26). Previous work has established that the gut microbial diversity is specifically restructured in FMF. In a study done in Armenian patients with FMF, those who settled in the United States were shown to develop secondary amyloidosis with a much lower incidence than those settled in Armenia (27). A similar effect was observed in Turkish patients with FMF who had spent their childhood years in Turkey compared with Germany or the United States (28, 29). Together, this suggests the possibility that diet and diversity of the regional bacterial milieu may impact pyrin function and gut homeostasis. Our data also prompt us to suggest that bile acid metabolites produced by gut microbiota may mediate some of these effects (30–32).
Both the bile acid analogues BAA485 and BAA473 utilized in this study were synthetically derived molecules (33–35). However, it is not unreasonable that gut microbiota could be envisaged to generate such bile acid species via a two-step transformation starting with DCA (Fig. S2) (12). Generation of our initial screening hit BAA485 could be envisioned through enzymatic conversion of DCA by 12α-hydroxysteroid dehydrogenase produced by gut bacteria and a second transformation step of BAA485 methylation yielding the more potent analogue BAA473 (36–39). Unfortunately, to date we have been unable to identify intestinal bacteria that can convert DCA to BAA485 or BAA473.
In conclusion, we have identified bile acid analogues that activate the pyrin inflammasome pathway in immune and intestinal epithelial cells. Because bile acid analogues, but not the principal bile acids, induce pyrin inflammasome, we hypothesize that the gut microbiota metabolizes endogenous bile acids into metabolites such as BAA485 to regulate pyrin inflammasome activation and intestinal immune homeostasis. The identification of BAA485 suggests a novel interplay between gut microbiota and host immune system.
Experimental procedures
Antibodies and reagents
Anti-pyrin (AL196) and anti-ASC (AL177) antibodies were obtained from Adipogen, and anti-tubulin antibody was from Sigma (T6199). Lipopolysaccharide from Escherichia coli O26:B6 was from Sigma-Aldrich (L2654). ATP (tlrl-atpl), Nigericin (tlrl-nig), VX-765 (inh-vx765i), and MCC950 (inh-mcc) were purchased from Invivogen.
Protein extraction and Western blot analysis
THP-1 Cas9 cell pellets were lysed in 100 μl of radioimmune precipitation assay buffer (Cell Signaling Technology, catalog no. 9806) in the presence of Halt Protease Inhibitor Mixture (Thermo Fisher Scientific, catalog no. 78430). Cell lysis was performed with an ultrasonic bath. Lysates containing proteins were cleared by centrifugation at 13,000 rpm at 4 °C. Total protein concentration was determined using the BCA protein assay kit (Pierce, catalog no. 23225). 30 μg of lysate was loaded per lane, and the proteins were separated using a CriterionTM TGXTM 4–15% gel system (Bio-Rad, catalog no. 5671084); transferred to Trans-Blot Turbo, 0.2-μm nitrocellulose membrane (Bio-Rad, catalog no. 1704159); and subjected to immunoblotting.
Cell culture
Blood was collected from healthy human donors after obtaining their informed consent from an internal Novartis Cambridge Research Donor Program. Blood-related research at Novartis is being overseen by an institutional review board, a group that performs independent review of research studies (New England IRB, Newton, MA). This study was conducted in accordance with the ethical principles of the Declaration of Helsinki. Human PBMCs were isolated from peripheral human blood in the presence of heparin as an anticoagulant using Ficoll–Paque (GE Healthcare, catalog no. 17-1440-03). The cells were cultured in RPMI 1640 medium (Gibco, catalog no. 11875-093) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, catalog no. 16140), 1% penicillin/streptomycin (P/S; Gibco, catalog no. 15140-122), 0.1 mm minimal essential medium (MEM) non-essential amino acids solution (Gibco, catalog no. 11140-050), 1 mm sodium pyruvate (Gibco, catalog no. 11360-070), 2 mm l-glutamine (Gibco, catalog no. 25030-081), and 10 mm HEPES (Gibco, catalog no. 15630-070). THP-1 (ATCC, TIB-202) and U937 (ATCC, CRL-1593.2) cell lines were cultured in RPMI 1640 medium (ATCC, catalog no. 30-2001) containing 10% FBS and 1% P/S, whereas T84 (ATCC, CCL-248) cells were grown in a 1:1 mixture of complete Ham's F-12 medium and Dulbecco's modified Eagle's medium (ATCC, catalog no. 30-2006) containing 10% FBS and 1% P/S. The cells were either not primed or primed with LPS before plating and were incubated in a humidified chamber at 37 °C and 5% CO2. Primary human colon cell culture was established according to Moon (15) and VanDussen (16). Cells in monolayer were cultured in a 96-well insert plate (Falcon, catalog no. 351130), and BAA473 was applied to the top chamber.
Assays
For the CellTiter-Glo assay, CTG reagent (Promega, G7570) was mixed at a 1:1 ratio with supernatant from the treatment plate. The mix was incubated for 10 min at room temperature on the shaker, followed by luminescence measurement. The HTRF IL-1β immunoassay (Cisbio, 62HIL1BPEH) was carried out according to the manufacturer's instructions for a 384-well plate. The human IL-18 AlphaLISA detection kit (PerkinElmer Life Sciences, AL241C) was used to assess IL-18 secretion according to the manufacturer's protocol. Caspase-1 activity was determined using the Caspase-Glo 1 inflammasome assay from Promega (G9951). LDH released into the media was measured using the LDH colorimetric assay kit from Abcam (ab102526).
CRISPR screen and validation
THP-1 cells were engineered to stably express Cas9 by lentiviral delivery of the pNGx-LV-c004 Cas9 construct followed by blasticidin selection. A genome-wide gRNA library targeting 18,360 genes covering most protein-coding genes was constructed, and the pooled CRISPR screen was performed as described previously (41). Packaging of the lentiviral gRNA library was done according to Hoffman et al. (42). THP-1-Cas9 cells were transduced with the lentiviral gRNA library, and 7 days post-transduction, the cells were primed with 1 μg/ml LPS followed by treatment with 75 μm BAA473 after 24 h. The next day, cells were stained using the LIVE/DEAD® Fixable Far Red Dead Cell Stain kit (Thermo Scientific, L10120), and live cells were sorted by FACS.
For validation experiments, individual single guide RNAs were selected according to the screening results and cloned into the BbsI restriction site of pNGx-LV-g003 lentiviral backbone. gRNA sequences are listed in Table S2. The g003 vector expresses the sgRNA of interest along with an RFP marker. 293T cells were transfected with 0.28 μg of lentiviral packaging mix (4:1 mix of Gag/Pol and VSV-G) along with 0.23 μg of the lentiviral vector using the transfection reagent TransIT-293 (Mirus, MIR 2700). Supernatant was collected 48 h post-transfection and cleared by centrifugation at 2000 rpm for 5 min followed by syringe filtering using a 0.45-μm filter (Millipore, HAWP04700). THP-1 cells were transduced in the presence of 5 μg/ml Polybrene via spin infection at 2100 rpm for 90 min. Cells were rested for 24 h before being selected with 1.5 μg/ml puromycin for 72 h.
Generation of U937 pyrin reporter cell line
The human MEFV ORF (isoform NP_000234) was subcloned into the pXP1510 lentiviral vector, allowing for expression driven by the EF1-α promoter utilizing NotI and AscI restriction sites. To establish a stable cell line, the lentiviral vector was packaged, and viral supernatant was produced as described above. U937 cells were transduced in the presence of 5 μg/ml Polybrene via spin infection at 2100 rpm for 90 min. Cells were allowed to recover for 24 h before being selected with 0.4 mg/ml neomycin (G418:Geneticin, Gibco, catalog no. 10131027) for 72 h.
Author contributions
I. A., N. C., and A. L. data curation; I. A., N. C., R. M., Z. Y., G. H., S. M. C., and X. C. formal analysis; I. A., N. C., R. M., Q. W., J. A., E. H., L. L., A. L., and X. C. investigation; I. A., N. C., J. B. C., J. R.-H., and X. C. methodology; I. A., S. M., S. M. C., and X. C. writing-original draft; S. M., N. C., J. B. C., and G. H. resources; S. M., G. H., C. R., J. R.-H., S. M. C., and X. C. supervision; S. M., G. H., C. R., S. M. C., and X. C. project administration; S. M., S. M. C., and X. C. writing-review and editing; N. C. validation; Z. Y. visualization; T. S., S. M. C., and X. C. conceptualization.
Supplementary Material
Acknowledgments
We thank Xiaowei Xiong for help with the gut organoid culture; Christopher Ng Thow Hing, Rajiv Chopra, and Michael Schaefer for biophysics and structural help; and Christopher Faraday, Philip Smith, Henry Haiser, and Bindu Sukumaran for reviewing the manuscript. We also thank Leon Murphy, Aron Jaffe, Rishi Jain, John Tallarico, and Jeff Porter for support and input through the course of this work.
I. A., S. M., E. H., L. L, N. C., R. M., Q. W., J. A., J. B. C., Z. Y., A. L., G. H., T. S., C. R. J. R.-H., S. M. C. are full-time employees of Novartis Institutes for BioMedical Research (NIBR). X. C. is a full-time employee of Sanofi US.
This article was selected as one of our Editors' Picks.
This article contains Tables S1 and S2 and Figs. S1–S3.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- IL
- interleukin
- DCA
- deoxycholic acid
- PBMC
- peripheral blood mononuclear cell
- CASP1
- caspase-1
- LDH
- lactate dehydrogenase
- IEC
- intestinal epithelial cell
- gRNA
- guide RNA
- FMF
- familial Mediterranean fever
- FBS
- fetal bovine serum
- P/S
- penicillin/streptomycin
- HTRF
- homogeneous time-resolved fluorescence
- TLR
- Toll-like receptor.
References
- 1. Broz P., and Dixit V. M. (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunology 16, 407–420 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- 2. Guo H., Callaway J. B., and Ting J. P. (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687 10.1038/nm.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hoffman H. M., Mueller J. L., Broide D. H., Wanderer A. A., and Kolodner R. D. (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305 10.1038/ng756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharma D., Malik A., Guy C. S., Karki R., Vogel P., and Kanneganti T. D. (2018) Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology 154, 948–964.e8 10.1053/j.gastro.2017.11.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elinav E., Strowig T., Kau A. L., Henao-Mejia J., Thaiss C. A., Booth C. J., Peaper D. R., Bertin J., Eisenbarth S. C., Gordon J. I., and Flavell R. A. (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 10.1016/j.cell.2011.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Allen I. C., TeKippe E. M., Woodford R. M., Uronis J. M., Holl E. K., Rogers A. B., Herfarth H. H., Jobin C., and Ting J. P. (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 10.1084/jem.20100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhernakova A., Festen E. M., Franke L., Trynka G., van Diemen C. C., Monsuur A. J., Bevova M., Nijmeijer R. M., van 't Slot R., Heijmans R., Boezen H. M., van Heel D. A., van Bodegraven A. A., Stokkers P. C., Wijmenga C., et al. (2008) Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 82, 1202–1210 10.1016/j.ajhg.2008.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wahlström A., Sayin S. I., Marschall H. U., and Bäckhed F. (2016) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 10.1016/j.cmet.2016.05.005 [DOI] [PubMed] [Google Scholar]
- 9. Hamilton J. P., Xie G., Raufman J. P., Hogan S., Griffin T. L., Packard C. A., Chatfield D. A., Hagey L. R., Steinbach J. H., and Hofmann A. F. (2007) Human cecal bile acids: concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G256–G263 10.1152/ajpgi.00027.2007 [DOI] [PubMed] [Google Scholar]
- 10. Macdonald I. A., Bokkenheuser V. D., Winter J., McLernon A. M., and Mosbach E. H. (1983) Degradation of steroids in the human gut. J. Lipid Res. 24, 675–700 [PubMed] [Google Scholar]
- 11. Yao L., Seaton S. C., Ndousse-Fetter S., Adhikari A. A., DiBenedetto N., Mina A. I., Banks A. S., Bry L., and Devlin A. S. (2018) A selective gut bacterial bile salt hydrolase alters host metabolism. eLife 7, e37182 10.7554/eLife.37182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Groh H., Schade K., and Hörhold-Schubert C. (1993) Steroid metabolism with intestinal microorganisms. J. Basic Microbiol. 33, 59–72 10.1002/jobm.3620330115 [DOI] [PubMed] [Google Scholar]
- 13. Devlin A. S., and Fischbach M. A. (2015) A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat. Chem. Biol. 11, 685–690 10.1038/nchembio.1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lei-Leston A. C., Murphy A. G., and Maloy K. J. (2017) Epithelial cell inflammasomes in intestinal immunity and inflammation. Front. Immunol. 8, 1168 10.3389/fimmu.2017.01168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moon C., VanDussen K. L., Miyoshi H., and Stappenbeck T. S. (2014) Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol. 7, 818–828 10.1038/mi.2013.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. VanDussen K. L., Marinshaw J. M., Shaikh N., Miyoshi H., Moon C., Tarr P. I., Ciorba M. A., and Stappenbeck T. S. (2015) Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64, 911–920 10.1136/gutjnl-2013-306651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coll R. C., Robertson A. A., Chae J. J., Higgins S. C., Muñoz-Planillo R., Inserra M. C., Vetter I., Dungan L. S., Monks B. G., Stutz A., Croker D. E., Butler M. S., Haneklaus M., Sutton C. E., Núñez G., et al. (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255 10.1038/nm.3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schmid-Burgk J. L., Chauhan D., Schmidt T., Ebert T. S., Reinhardt J., Endl E., and Hornung V. (2016) A genome-wide CRISPR (clustered regularly interspaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem. 291, 103–109 10.1074/jbc.C115.700492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu H., Yang J., Gao W., Li L., Li P., Zhang L., Gong Y. N., Peng X., Xi J. J., Chen S., Wang F., and Shao F. (2014) Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 10.1038/nature13449 [DOI] [PubMed] [Google Scholar]
- 20. Gao W., Yang J., Liu W., Wang Y., and Shao F. (2016) Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl. Acad. Sci. U.S.A. 113, E4857–E4866 10.1073/pnas.1601700113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kubosaki A., Lindgren G., Tagami M., Simon C., Tomaru Y., Miura H., Suzuki T., Arner E., Forrest A. R., Irvine K. M., Schroder K., Hasegawa Y., Kanamori-Katayama M., Rehli M., Hume D. A., et al. (2010) The combination of gene perturbation assay and ChIP-chip reveals functional direct target genes for IRF8 in THP-1 cells. Mol. Immunol. 47, 2295–2302 10.1016/j.molimm.2010.05.289 [DOI] [PubMed] [Google Scholar]
- 22. Liberzon A. (2014) A description of the Molecular Signatures database (MSigDB) Web site. Methods Mol. Biol. 1150, 153–160 10.1007/978-1-4939-0512-6_9 [DOI] [PubMed] [Google Scholar]
- 23. Levy M., Thaiss C. A., Zeevi D., Dohnalová L., Zilberman-Schapira G., Mahdi J. A., David E., Savidor A., Korem T., Herzig Y., Pevsner-Fischer M., Shapiro H., Christ A., Harmelin A., Halpern Z., et al. (2015) Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 10.1016/j.cell.2015.10.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fidder H., Chowers Y., Ackerman Z., Pollak R. D., Crusius J. B., Livneh A., Bar-Meir S., Avidan B., and Shinhar Y. (2005) The familial Mediterranean fever (MEVF) gene as a modifier of Crohn's disease. Am. J. Gastroenterol. 100, 338–343 10.1111/j.1572-0241.2005.40810.x [DOI] [PubMed] [Google Scholar]
- 25. Villani A. C., Lemire M., Louis E., Silverberg M. S., Collette C., Fortin G., Nimmo E. R., Renaud Y., Brunet S., Libioulle C., Belaiche J., Bitton A., Gaudet D., Cohen A., Langelier D., et al. (2009) Genetic variation in the familial Mediterranean fever gene (MEFV) and risk for Crohn's disease and ulcerative colitis. PLoS One 4, e7154 10.1371/journal.pone.0007154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yepiskoposyan L., and Harutyunyan A. (2007) Population genetics of familial Mediterranean fever: a review. Eur. J. Hum. Genet. 15, 911–916 10.1038/sj.ejhg.5201869 [DOI] [PubMed] [Google Scholar]
- 27. Schwabe A. D., and Peters R. S. (1974) Familial Mediterranean fever in Armenians: analysis of 100 cases. Medicine 53, 453–462 10.1097/00005792-197411000-00005 [DOI] [PubMed] [Google Scholar]
- 28. Ozen S., Aktay N., Lainka E., Duzova A., Bakkaloglu A., and Kallinich T. (2009) Disease severity in children and adolescents with familial Mediterranean fever: a comparative study to explore environmental effects on a monogenic disease. Ann. Rheum. Dis. 68, 246–248 10.1136/ard.2008.092031 [DOI] [PubMed] [Google Scholar]
- 29. Batu E. D., Kara Eroglu F., Tsoukas P., Hausmann J. S., Bilginer Y., Kenna M. A., Licameli G. R., Fuhlbrigge R. C., Ozen S., and Dedeoglu F. (2016) Periodic fever, aphthosis, pharyngitis, and adenitis syndrome: analysis of patients from two geographic areas. Arthritis Care Res. (Hoboken) 68, 1859–1865 10.1002/acr.22901 [DOI] [PubMed] [Google Scholar]
- 30. Khachatryan Z. A., Ktsoyan Z. A., Manukyan G. P., Kelly D., Ghazaryan K. A., and Aminov R. I. (2008) Predominant role of host genetics in controlling the composition of gut microbiota. PLoS One 3, e3064 10.1371/journal.pone.0003064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ktsoyan Z. A., Beloborodova N. V., Sedrakyan A. M., Osipov G. A., Khachatryan Z. A., Manukyan G. P., Arakelova K. A., Hovhannisyan A. I., Arakelyan A. A., Ghazaryan K. A., Zakaryan M. K., and Aminov R. I. (2013) Management of familial Mediterranean fever by colchicine does not normalize the altered profile of microbial long chain fatty acids in the human metabolome. Front. Cell. Infect. Microbiol. 3, 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ktsoyan Z. A., Mkrtchyan M. S., Zakharyan M. K., Mnatsakanyan A. A., Arakelova K. A., Gevorgyan Z. U., Sedrakyan A. M., Hovhannisyan A. I., Arakelyan A. A., and Aminov R. I. (2016) Systemic concentrations of short chain fatty acids are elevated in salmonellosis and exacerbation of familial Mediterranean fever. Front. Microbiol. 7, 776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marker R. E., and Lawson E. J. (1938) Sterols. XXXIII. 3,11-Dihydroxy-12-ketocholanic acid and derivatives. J. Am. Chem. Soc. 60, 1334–1337 10.1021/ja01273a017 [DOI] [Google Scholar]
- 34. Gallagher T. F. (1946) Partial synthesis of compounds related to adrenal cortical hormones: VI. The structure of the “3(α),11-dihydroxy-12-ketocholanic acid” of Marker and Lawson and of the products obtained by “Wolff-Kishner reduction”. J. Biol. Chem. 162, 539–548 [PubMed] [Google Scholar]
- 35. Hershberg E. B., Herzog H. L., Coan S. B., Weber L., and Jevnik M. (1952) Cortisone intermediates. I. A new preparation of 12α-bromo-24,24-diphenylchol-23-en-3α-ol-11-one acetate. J. Am. Chem. Soc. 74, 2585–2589 10.1021/ja01130a036 [DOI] [Google Scholar]
- 36. Edenharder R., and Mielek K. (1984) Epimerization, oxidation and reduction of bile acids by Eubacterium lentum. Syst. Appl. Microbiol. 5, 287–298 10.1016/S0723-2020(84)80031-4 [DOI] [Google Scholar]
- 37. Edenharder R., and Schneider J. (1985) 12β-dehydrogenation of bile acids by Clostridium paraputrificum, C. tertium, and C. difficile and epimerization at carbon-12 of deoxycholic acid by cocultivation with 12α-dehydrogenating Eubacterium lentum. Appl. Environ. Microbiol. 49, 964–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Edenharder R., and Hammann R. (1985) Deoxycholic acid methyl ester—a novel bacterial metabolite of cholic acid. Syst. Appl. Microbiol. 6, 18–22 10.1016/S0723-2020(85)80005-9 [DOI] [Google Scholar]
- 39. Kelsey M. I., and Sexton S. A. (1976) The biosynthesis of ethyl esters of lithocholic acid and isolithocholic acid by rat intestinal microflora. J. Steroid Biochem. 7, 641–647 10.1016/0022-4731(76)90059-5 [DOI] [PubMed] [Google Scholar]
- 40. Van Gijn M. E., Ceccherini I., Shinar Y., Carbo E. C., Slofstra M., Arostegui J. I., Sarrabay G., Rowczenio D., Omoyımnı E., Balci-Peynircioglu B., Hoffman H. M., Milhavet F., Swertz M. A., and Touitou I. J. (2018) New workflow for classification of genetic variants' pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). Med. Genet. 55, 530–537 10.1136/jmedgenet-2017-105216 [DOI] [PubMed] [Google Scholar]
- 41. DeJesus R., Moretti F., McAllister G., Wang Z., Bergman P., Liu S., Frias E., Alford J., Reece-Hoyes J. S., Lindeman A., Kelliher J., Russ C., Knehr J., Carbone W., Beibel M., et al. (2016) Functional CRISPR screening identifies the ufmylation pathway as a regulator of SQSTM1/p62. eLife 5, e17290 10.7554/eLife.17290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoffman G. R., Rahal R., Buxton F., Xiang K., McAllister G., Frias E., Bagdasarian L., Huber J., Lindeman A., Chen D., Romero R., Ramadan N., Phadke T., Haas K., Jaskelioff M., et al. (2014) Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. U.S.A. 111, 3128–3133 10.1073/pnas.1316793111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.