

Abstract
Peroxisome proliferator–activated receptor α (PPARα) is a transcriptional regulator of lipid metabolism. GW7647 is a potent PPARα agonist that must reach the nucleus to activate this receptor. In cells expressing human fatty acid–binding protein 1 (FABP1), GW7647 treatment increases FABP1's nuclear localization and potentiates GW7647-mediated PPARα activation; GW7647 is less effective in cells that do not express FABP1. To elucidate the underlying mechanism, here we substituted residues in FABP1 known to dictate lipid signaling by other intracellular lipid-binding proteins. Substitutions of Lys-20 and Lys-31 to Ala in the FABP1 helical cap affected neither its nuclear localization nor PPARα activation. In contrast, Ala substitution of Lys-57, Glu-77, and Lys-96, located in the loops adjacent to the ligand-binding portal region, abolished both FABP1 nuclear localization and GW7647-induced PPARα activation but had little effect on GW7647–FABP1 binding affinity. Using solution NMR spectroscopy, we determined the WT FABP1 structure and analyzed the dynamics in the apo and GW7647-bound structures of both the WT and the K57A/E77A/K96A triple mutant. We found that GW7647 binding causes little change in the FABP1 backbone, but solvent exposes several residues in the loops around the portal region, including Lys-57, Glu-77, and Lys-96. These residues also become more solvent-exposed upon binding of FABP1 with the endogenous PPARα agonist oleic acid. Together with previous observations, our findings suggest that GW7647 binding stabilizes a FABP1 conformation that promotes its interaction with PPARα. We conclude that full PPARα agonist activity of GW7647 requires FABP1-dependent transport and nuclear localization processes.
Keywords: protein structure, fatty acid–binding protein, nuclear magnetic resonance (NMR), peroxisome proliferator-activated receptor (PPAR), drug delivery, nuclear receptor, transcription factor, GW7647, PPARα agonist
Introduction
Intracellular lipid-binding proteins (iLBPs)6 are a family of cytosolic proteins that transport poorly the water-soluble ligands, such as long-chain fatty acids (LCFAs) and vitamins, to sites where they exert their functional effects within the cell. Members of the iLBP family include the fatty acid–binding proteins (FABPs) that facilitate the uptake and utilization of LCFAs by acting as molecular transporters to direct the LCFAs to various target organelles (1), as well as the cellular retinoic acid–binding proteins (CRABPs), cellular retinol–binding proteins, and ileal bile acid–binding protein. Although their primary amino acid sequences are not conserved, for example the nine human FABPs show only 20–70% similarity, they retain highly-conserved tertiary structures (2). This common structure includes 10 β-strands that form a ligand-binding cavity and two short α-helices that form a helix-turn-helix motif, which is presumed to act as a portal for ligand entry and exit (3).
ILBPs are expressed in a tissue-specific fashion and have distinct ligand-binding preferences. They are able to bind to a range of LCFAs and other lipophilic molecules, including some drugs (4). In this way, differential iLBP expression and ligand binding preference can precisely influence the distribution and activity of their ligands in various tissues (5).
More recently, it has become clear that in addition to their role in solubilization and transport of poorly water-soluble ligands, iLBPs play important roles in dictating the biological activities of their ligands. For example, adipocyte FABP (FABP4) binds to agonists of peroxisome proliferator–activated receptor γ (PPARγ) to selectively mediate their nuclear transport, thereby allowing enhanced receptor activation (6, 7). The ability of FABPs to regulate biological signaling pathways in this manner is both ligand- and receptor-specific. For example, whereas FABP4 and FABP5 can both bind to a range of agonists of the different PPAR subtypes (PPARα, -γ, and -β/δ), they appear to modulate PPAR activity only for certain ligands (8). Thus, FABP4 appears to selectively potentiate the activity of PPARγ agonists, whereas FABP5 potentiates the activity of PPARβ/δ agonists. Therefore, FABP-ligand binding is not sufficient to dictate PPAR activation.
Some of the mechanisms that underpin these observations have been elucidated. In the case of FABP4 and FABP5, structural studies have revealed a basis for receptor selectivity (6, 9). Binding of PPARγ agonists mediates dimerization of FABP4 that stabilizes a conformational nuclear localization signal (NLS) in the helix-turn-helix motif and thereby enhances importin-meditated nuclear transport (7). Nonactivating ligands can also bind to FABP4 but do not stabilize the NLS, and therefore, they do not cause nuclear localization or receptor activation. FABP5 is also able to form an NLS upon binding of certain ligands, including agonists of PPARβ/δ (9). However, in this case NLS formation does not involve dimerization of FABP5, but rather ligand-binding information is relayed to the NLS residues via an “activation loop” in the portal region, which is located between two of the β-strands (βCD) (9).
The residues that form the conformational NLS in FABP4 and FABP5 are not conserved across the iLBP superfamily, and the importin-mediated nuclear transport mechanism is not common to all iLBPs. Furthermore, for certain iLBPs, where the conformational NLS-forming residues are conserved, translocation to the nucleus does not occur upon ligand binding. For example, CRABP-I and CRABP-II share 74% sequence identity, and each contain the NLS-forming residues in their highly conserved helix-turn-helix motif, but only CRABP-II is translocated to the nucleus upon binding to retinoic acid (RA) (10). However, the mechanism by which CRABP-II enhances the ability of RA to activate the retinoic acid receptor (RAR) appears to involve more than the conformational NLS residues. Thus, CRABP-II, but not CRABP-I, is able to bind directly to RAR, thereby “channeling” RA to its receptor and enhancing transcriptional activity (11). Three residues in the loops surrounding the portal region of CRABP-II are responsible for this channeling activity (12). Substitution of these three residues into the corresponding sequence of CRABP-I transfers the ability to channel RA to RAR and enhances transcriptional activity in cellular assays. Conversely, reverse substitution of the three residues from CRABP-I into CRABP-II renders CRABP-II unable to channel RA to RAR (12).
Other iLBPs also have specific effects on nuclear hormone receptor signaling, although the mechanism by which they do so has not been resolved. For example, FABP1 can bind to the PPARα agonists GW7647 and fenofibrate; however, it only promotes PPARα activation in the presence of GW7647 and not fenofibrate (13). Treatment of cells with GW7647 also results in an increase in nuclear FRET between FABP1 and PPARα, consistent with co-localization of the two proteins in the presence of GW7647 (13). In this case, nuclear localization of FABP1 appears to depend on a decreased rate of nuclear export, rather than the importin-mediated increase in nuclear import observed for FABP4, FABP5, and CRABP-II (9, 10, 13). Consistent with this, the residues that form the conformational NLS in FABP4, FABP5, and CRABP-II are not conserved in FABP1, and importin binding does not play a role in GW7647-mediated nuclear localization of FABP1 (13). This indicates that a different mechanism is responsible for nuclear localization of FABP1 in the presence of GW7647.
Several structures of FABP1 from different species have been reported previously, both in the absence and presence of different LCFA (14–17). These structures reveal that FABP1 possesses a larger cavity than other iLBPs (17), and this has been postulated to explain the ability of FABP1 to bind a diverse range of different ligands. In addition, the backbone dynamics of FABP1 have been evaluated in the absence and presence of oleic acid (OA) (14). These studies reveal pronounced backbone flexibility in the residues around the portal region in both the apo and the ligand-bound protein, and it was hypothesized that these dynamics are essential to enable binding of the bulky ligands in the cavity of FABP1.
Structural and functional data have also been reported for a common variant of FABP1 where Thr-94 is mutated to Ala (18, 19). Thr-94 is located adjacent to one of the loops that form the ligand-binding portal. These data reveal that the T94A mutation does not elicit major changes in the backbone structure, dynamics, and ligand binding preferences of FABP1. However, the T94A mutation was found to diminish the capacity of FABP1 to stimulate PPARα-mediated transcription in cultured hepatocytes (19).
Based on the available structural data for FABP1 and the ligand binding and functional data for PPARα agonists, we hypothesized that PPARα-activating ligands such as GW7647 stabilize a local conformational change in FABP1, which favors its association with PPARα, thereby decreasing export from the nucleus. To evaluate this hypothesis, we made a series of mutations in FABP1. First, we mutated the basic residues (Lys-20 and Lys-31) in the helix-turn-helix motif of FABP1, and we found that this had no effect on either nuclear accumulation of FABP1 or PPARα transactivation in the presence of GW7647. Next, we mutated charged residues in the loops surrounding the portal region of FABP1 (Lys-57, Glu-77, and Lys-96). Mutation of these three residues to Ala had little effect on the affinity or mode of GW7647 binding as determined from isothermal titration microcalorimetry (ITC) and NMR data. Comparison of backbone dynamics in the WT and mutant protein in the absence and presence of GW7647 revealed that mutation to Ala of the three charged residues also had little effect on the backbone dynamics in either the apo or GW7647-bound state. However, the three Ala mutations abolished nuclear accumulation of FABP1 and the associated activation of PPARα in cells treated with GW7647.
Structural analysis of FABP1 in the absence and presence of GW7647 revealed that ligand binding resulted in an opening of the binding cavity through an outward movement of the helix-turn-helix and β-strands βC, βD, βE, and βF. In concert, the side chains of a number of residues that were not involved directly in the interaction with GW7647 in the complex became more solvent-exposed upon ligand binding. These included Lys-57, Glu-77, and Lys-96. These residues are similarly solvent-exposed in the structure of FABP1 in complex with the endogenous PPARα agonist OA.
Thus, the mechanism that underpins the ability of FABP1 to increase activation of PPARα by GW7647 involves a change in conformation of FABP1 on ligand binding, which leads to greater solvent exposure of a number of charged residues that are present in the loops surrounding the portal region. These residues are necessary for both nuclear accumulation of FABP1 upon ligand binding and potentiation of the effects of GW7647 on PPARα signaling.
Results
FABP1 residues Lys-57, Glu-77, and Lys-96 control the activation of PPARα
Two distinct mechanisms have been observed by which iLBPs are able to regulate nuclear hormone receptor signaling. For FABP4 and FABP5, binding to activating ligands results in the stabilization of three positively charged residues in the helical cap into a conformational NLS, which drives nuclear localization of the FABP–ligand complex to promote specific signaling via different PPARs. In CRABP-II, binding of RA promotes nuclear localization through a similar NLS, but also leads to a direct interaction with the ligand-binding domain of the retinoic acid receptor (RAR) that is mediated by residues in the loops around the portal region, which contribute to the electrostatic surface potential. Based on these prior observations, we hypothesized that the ability of FABP1 to promote PPARα activation in the presence of specific PPARα agonists most likely involved structural reorganization of charged residues in one of these regions upon ligand binding.
To test this hypothesis, we first mutated two positively charged residues in the helical cap of FABP1 (Lys-20 and Lys-31) to Ala. We also mutated three charged residues in the loops surrounding the portal region Lys-57, Glu-77, and Lys-96 to Ala and assessed the effect of these mutations on nuclear localization of FABP1 in cells treated with PPARα agonists GW7647 or OA. We also tested PPARα activation following treatment with GW7647 or OA in cells expressing no FABP1, WT FABP1, or one of the FABP1 mutants. Transactivation assays were normalized for transfection efficiency as we have reported previously (13).
These cellular assays revealed that mutations in the helical cap had no effect on nuclear accumulation of FABP1 in response to treatment with either GW7647 or OA (Fig. 1A). Consistent with previously published data, WT FABP1 caused a significant increase in PPARα activation after stimulation with GW7647 or OA (13), and a similar increase in PPARα activation was observed in cells expressing the double mutant K20A/K31A FABP1 (Fig. 1B). In contrast, mutation of the three residues in the loops surrounding the portal K57A/E77A/K96A abolished nuclear accumulation of FABP1 in response to treatment with GW7647 or OA (Fig. 1C), and the triple-mutant FABP1 no longer enhanced PPARα activation in response to treatment with either PPARα agonist (Fig. 1D).
Figure 1.

Nuclear localization and PPAR activation for FABP1 mutants in the presence of GW7647. Each panel depicts the individual data points for the experiment as well as mean and standard deviation. A, quantification of ligand-induced changes in nuclear localization (nuclear to cytosolic ratio) of GFP-FABP1 and the K20A/K31A double mutant using high-content imaging. B, transcriptional activity of PPARα is enhanced by the presence of FABP1 and the K20A/K31A double mutant upon treatment of PPARα-expressing COS-7 cells with either GW7647 or oleic acid. C, quantification of ligand-induced changes in the nuclear localization (nuclear to cytosolic ratio) of GFP-FABP1 and the K57A/E77A/K96A triple mutant using high-content imaging. D, transcriptional activity of PPARα is enhanced by the presence of FABP1 but not the K57A/E77A/K96A triple mutant upon treatment of PPARα-expressing COS-7 cells with either GW7647 or oleic acid. ***, p < 0.001 versus vehicle (B) or vector (C) control, two-way ANOVA with Dunnett's multiple comparisons test.
We have previously reported FRET between FABP1 and PPARα in the presence but not in the absence of GW7647 (20), suggesting that the two proteins are co-localized in the nucleus in the presence of GW7647. Similar experiments with the triple mutant of FABP1 did not show a FRET signal either in the absence or the presence of GW7647 (data not shown), suggesting that even upon binding of GW7647 the triple-mutant FABP1 is not co-localized with PPARα.
Analysis of GW7647 binding to WT FABP1 and triple-mutant FABP1-K57A/E77A/K96A
We have previously shown using ITC that GW7647 binds to WT FABP1 with 1:1 stoichiometry and with an equilibrium dissociation constant KD ≈120 nm (13). Analysis of GW7647 binding to the triple-mutant FABP1 revealed that the mutations had little effect on the ability of FABP1 to bind to GW7647, where the ITC data indicated an equilibrium dissociation constant KD = 250 ± 130 nm. Hence, the three mutations result in only an approximate 2-fold decrease in binding affinity compared with WT FABP1 (Fig. 2A).
Figure 2.

Analysis of GW7647 binding to FABP1. A, ITC titration of K57A/E77A/K96A triple-mutant FABP1 with GW7647. The upper panel shows the raw data from a single ITC experiment, and the lower panel shows the corresponding binding isotherm generated from the data fitted to a one-site binding model. B, 2D 15N HSQC of WT FABP1 in the absence (blue) and presence (red) of a saturating concentration of GW7647. Aliased peaks for Arg-122 that appear in the spectrum upon addition of GW7647 are labeled. C, cross-section of the highlighted peak upon titration of FABP1 with GW7647. The FABP1/GW7647 ratio of each spectrum is shown. The data indicate that the complex is in slow exchange on the NMR timescale where cross-peaks are observed in the titration whose intensity reflects the proportion of apo and holo FABP1. D, chemical structure of GW7647. E, 2D 15N HSQC of K57A/E77A/K96A triple-mutant FABP1 in the absence (blue) and presence (red) of a saturating concentration of GW7647.
To characterize the interaction further using NMR spectroscopy, the binding of GW7647 was first investigated by recording a series of 2D 15N HSQC spectra of WT FABP1 in the presence of increasing concentrations of GW7647 (Fig. 2B). Analysis of the data revealed that the complex was in slow exchange on the NMR timescale as revealed by the observation of separate resonances for apo and holo FABP1, with the intensity of the cross-peaks revealing the relative concentrations for the two species through the titration (Fig. 2C). A notable feature of the spectrum of holo FABP1 was the appearance of a new cross-peak in the HSQC spectrum, which was only detected following addition of GW7647 (Fig. 2B), and this was subsequently assigned as the side-chain guanidinium group of Arg-122. Analysis of HSQC spectra of the triple-mutant FABP1 revealed a similar pattern of chemical shift perturbations upon GW7647 binding (Fig. 2E), and this complex was also in slow exchange on the NMR timescale (Fig. S5), consistent with the KD value measured by ITC. Similar to the WT FABP1, addition of GW7647 resulted in the appearance of a peak in the HSQC spectrum, which was again consistent with the guanidinium side chain of Arg-122.
Resonance assignments and CSP mapping
Because of the complex being in slow exchange and the significant perturbations observed following addition of GW7647, it was necessary to undertake separate 2D and 3D NMR experiments to obtain the sequential assignments of apo and holo FABP1. Full backbone assignments were obtained for the 1H, 15N, and 13C resonances of WT FABP1 (Met-1–Ile-127) in the absence and presence of GW7647. In the case of the triple-mutant FABP1, the protein was observed to have lower solubility, which necessitated acquiring the data at lower protein concentration, and only 98 and 97% of the backbone resonances could be assigned for the apo and holo protein, respectively, due to the lower signal-to-noise arising from the data. The assignments for apo and holo WT and triple-mutant FABP1 in the absence and presence of GW7647 have been deposited into the BMRB database under accession numbers 30477, 30478, 27509, and 27510, respectively. Chemical shift perturbations (CSP) observed upon addition of GW7647 were calculated for each protein from the peak assignments, and these were plotted against amino acid residue number (Fig. 3, A and B) and mapped onto the structure of FABP1 (Fig. 3E). The patterns of CSP observed in the WT and triple-mutant FABP1 upon addition of GW7647 are highly similar. Comparison of the difference in secondary chemical shifts of Cα and Cβ resonances of the WT and triple-mutant FABP1 indicated that the secondary structures of these proteins are similar both in the absence and presence of GW7647 (Fig. 3, C and D).
Figure 3.

Chemical shift perturbations observed upon addition of GW7647 to FABP1 and secondary shift analysis. CSP observed for WT FABP1 (A) and K57A/E77A/K96A triple-mutant FABP1 (B) plotted against amino acid residue number. Missing or ambiguous assignments are highlighted in each panel with an asterisk. In each case, the largest CSP on GW7647 binding was observed for Met-74, which is labeled. Secondary chemical shifts (ΔCα − ΔCβ) were plotted against amino acid residue number for WT (C) and triple-mutant (D) FABP1. ΔCα and ΔCβ represent the difference between observed and random coil Cα and Cβ chemical shifts, respectively. Blue dotted lines indicate the secondary shifts either of +1 or −1 ppm. Regular secondary structure elements observed for WT FABP1 are indicated on the top panels. E, cartoon representation of the structure of FABP1 colored by the extent of CSP. The cartoon is colored from light to dark red on a ramp with dark red representing a perturbation of ≥0.5 ppm. Regular secondary structure elements are labeled.
The CSP observed in the HSQC spectra of the WT and triple-mutant FABP1 along with the appearance of the characteristic Arg-122 guanidinium peak in the HSQC spectra of each protein upon addition of GW7647 suggest that GW7647 binds in a similar manner to both proteins.
Comparison of the dynamics of the WT and triple-mutant FABP1
The backbone dynamics of the WT and triple-mutant FABP1 were assessed by recording 15N-1H heteronuclear NOE data for both proteins in the absence and presence of GW7647. Analysis of these data revealed that the majority of backbone amides displayed 15N-1H heteronuclear NOE values of >0.7, suggesting that overall the FABP1 protein backbone exhibits low flexibility on the nanosecond–picosecond timescale. 15N-1H heteronuclear NOE values indicative of increased internal mobility were detected for residues around the portal region, especially for αII and the linker connecting αI and αII, loops βCD, βEF, and βFG in both apo and holo FABP1 (Fig. 4A). Comparison of the backbone dynamics of apo and holo FABP1 did not reveal any significant difference in protein backbone mobility on the nanosecond–picosecond timescale in the presence of GW7647. These data are largely consistent with the backbone dynamics data reported previously for FABP1 (14). However, the current 15N-1H heteronuclear NOE data did not suggest any loss of backbone mobility in the protein upon binding of GW7647. This is in contrast to previous analysis of dynamics reported for FABP1 in complex with OA, as well as for rat-FABP2 in complex with palmitic acid, where ligand binding was reported to have a stabilizing effect in reducing backbone dynamics (3, 14). Similarly, analysis of the 15N-1H heteronuclear NOE data for the triple mutant revealed that the mutations conferred no profound effects on the backbone dynamics of FABP1 on the nanosecond–picosecond timescale in the absence or presence of GW7647 (Fig. 4B). Minor differences observed in the 15N-1H heteronuclear NOE values between the WT and triple-mutant FABP1 can most likely be attributed to lower signal-to-noise in the spectra for the triple mutant, which were acquired at a lower concentration.
Figure 4.
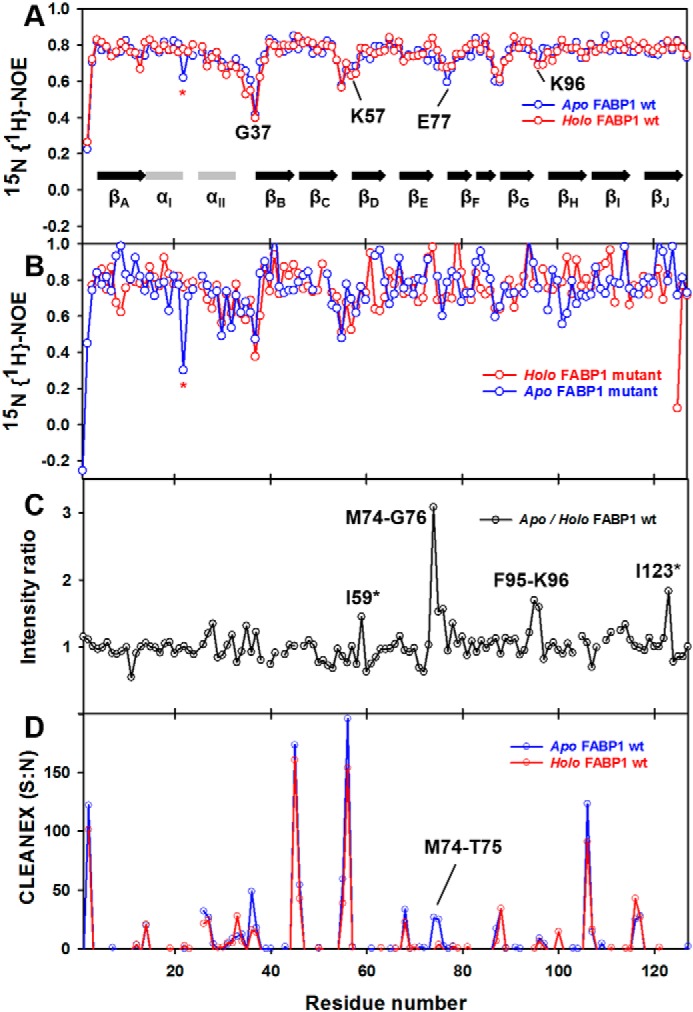
Backbone amide dynamics and solvent-exchange of FABP1 in the absence and presence of GW7647. Comparison of 15N-1H heteronuclear NOE values for apo and holo WT FABP1 (A) and K57A/E77A/K96A triple-mutant FABP1 (B) plotted against amino acid residue number. C, backbone amide resonance intensity ratio between apo and holo WT FABP1 plotted against the amino acid residue number. D, CLEANEX intensity values of apo and holo WT FABP1 plotted against amino acid residue number. Assignments that were missing or ambiguous due to peak overlap are highlighted with an asterisk.
A comparison of the peak intensities in the HSQC spectra of WT FABP1 in the absence and presence of GW7647 indicated some localized line broadening. This was most evident for residues in the βEF (Met-74–Gly-76) and βGH (Phe-95–Lys-96) loops and to a lesser extent for residues in αII, strands βC and βD around the portal region (Fig. 4C). The line broadening may reflect some slower timescale motions in these regions of FABP1 upon binding to GW7647. Conformational flexibility in the loops around the ligand-binding portal is thought to be important for ligand access to the binding cavity of FABP proteins.
Clean chemical exchange phase-modulated (CLEANEX-PM) experiments (21) were performed on both apo and holo WT FABP1 to identify the amide protons in rapid exchange with solvent. Fast-exchanging amide resonances display strong positive intensity in CLEANEX-PM experiments (Fig. 4D). In apo FABP1, fast-exchanging amide protons were predominantly from the residues in the portal region, especially in helix αII and loops αII–βB and βCD. Upon addition of GW7647, a number of residues, mainly in the αII–βB, βCD, and βEF regions, experienced decreased solvent exchange. The differences found between apo and holo FABP1 are minor suggesting that there is no significant change in the secondary structure or solvent exposure of the protein backbone upon ligand binding.
Structural analysis of WT FABP1
To characterize the effects of GW7647 binding on the solution structure of WT FABP1, we determined the structure FABP1 in the absence and presence of GW7647 using NMR spectroscopy under similar solution conditions.
Solution structure of apo FABP1
The solution structure of apo FABP1 was determined using restraints derived from the experimental data (Table 1). The superimposed ensemble of the final 10 conformers of apo FABP1 is depicted in Fig. 5A. The ensemble superimposes with high precision having a root mean square deviation (RMSD) of 0.31 ± 0.03 Å over the backbone atoms (Cα, N, and C′) and RMSD of 0.68 ± 0.04 Å over all heavy atoms (Fig. 5B). A full summary of the structural statistics for the 10 conformers of apo FABP1 (residues 1–127) is presented in Table 1. The structure of apo FABP1 determined at 35 °C is consistent with the previous NMR structure solved at 25 °C (PDB code 2L67) and the crystal structures (PDB code 3STN and 2F73, chain A) with Cα RMSD of 1.36, 1.12, and 1.22 Å, respectively (Tables S1 and S2). All four structures adopt the canonical FABP fold.
Table 1.
Experimental constraints used for structure calculations and structural statistics for the ensemble of 10 best structures for apo and holo FABP1
| Apo FABP1 | Holo FABP1 | |
|---|---|---|
| Chemical shift assignments (2–127)% | ||
| All atoms | 97.2 | 97.3 |
| Backbone atoms (HN, N, Hα1, Hα2, Cα) | 100 | 100 |
| Side-chain protons | 95.2 | 95.2 |
| Chemical shift agreement (%) | ||
| 3D 15N-edited (1H,1H) NOESY | 91.08 | 90.86 |
| 3D 13Cali-edited (1H,1H) NOESY | 90.42 | 86.65 |
| 3D 13Caro-edited (1H,1H) NOESY | 80 | 84.62 |
| NOE upper distance limitsa | ||
| No. of NOEs | 3098 | 2553 |
| Intra-residual (|i − j| = 0) | 762 | 671 |
| Short-range (|i − j| = 1) | 806 | 666 |
| Medium-range (2 ⩽ |i − j| < 6) | 483 | 373 |
| Long-range (|i − j| ≥ 6) | 1047 | 843 |
| Dihedral and hydrogen bond restraints | ||
| Φ and ψ | 208 | 202 |
| Hydrogen bond | 60 | 58 |
| CYANA target function (Å2) | ||
| Cycle 1 | 94.73 ± 1.28 | 130.83 ± 3.81 |
| Cycle 7 | 2.68 ± 0.44 | 3.15 ± 0.52 |
| Residual NOE violations (OPALp) | ||
| Number (≥0.1 Å) | 21 ± 3 | 21 ± 5 |
| Maximum (Å) | 0.02 ± 0.01 | 0.02 ± 0.01 |
| Residual dihedral angle violations (OPALp) | ||
| Number (≥2.5°) | 1 ± 1 | 2 ± 1 |
| Maximum (°) | 3.3 ± 0.9 | 3.5 ± 1.6 |
| RMSD with respect to mean coordinates (Å) | ||
| Backbone atoms (2–127) | 0.31 ± 0.03 | 0.47 ± 0.06 |
| All heavy atoms (2–127) | 0.68 ± 0.04 | 0.81 ± 0.05 |
| Ramachandran statisticsb | ||
| Favored (%) | 94 | 92 |
| Allowed (%) | 6 | 8 |
a NOE upper distances were analyzed by CYANA3.0.
b Data were determined by Molprobity.
Figure 5.

Solution structure of FABP1 in the absence and presence of GW7647. A, ensemble of 10 conformers for apo WT FABP1. B, conformer closest to the mean of the ensemble for apo WT FABP1 in sausage format, with the width of the sausage representing the backbone RMSD. C, ensemble of 10 conformers for holo WT FABP1 bound to GW7647. D, conformer closest to the mean of the ensemble for holo WT FABP1 in sausage format, with the width of the sausage representing the backbone RMSD. E and F, overlay of apo and holo WT FABP1. The conformer closest to the mean of the ensemble is shown in both stick (E) and cartoon (F) format. Mutated residues are labeled. G, number of long-range NOEs assigned in the experimental NMR data presented per residue of apo (cyan) and holo (red) WT FABP1 and plotted against amino acid residue number.
Solution structure of holo FABP1
A similar strategy was used to determine the structure of holo FABP1, where the data were recorded in the presence of a saturating concentration of GW7647. The structure of the protein was first calculated from the experimental data in the absence of GW7647 (Fig. 5C). The experimental constraints used to generate the structure and the structural statistics for the ensemble of 10 conformers chosen to represent the structure of holo FABP1 (residues: Met-1–Ile-127) are shown in Table 1. The structures are of high precision and superimpose with RMSD of 0.46 ± 0.06 Å for the backbone atoms and RMSD of 0.81 ± 0.06 Å for all heavy atoms in FABP1 (Fig. 5D). The lower precision of the structure in complex with GW7647 relative to apo FABP1 structure is most likely due to the lower number of long-range NOE constraints from the holo FABP1 sample, which was recorded at a slightly lower protein concentration (Fig. 5G). Nonetheless, there were a number of residues for which the pattern of intramolecular NOEs observed in the data were different in the absence and presence of GW7647, with some NOEs only being observed in the bound state (Fig. S11), suggesting that there were some local structural changes in FABP1 upon binding.
The high-quality structures of apo and holo FABP1 generated under similar conditions using the same data acquisition and structure calculation protocols enabled us to analyze the structural changes that occur upon binding of GW7647. To do so, the conformers closest to the mean of apo FABP1 and holo FABP1 were compared. The two structures superimpose with Cα RMSD of 0.89 Å over residues Met-1–Ile-127, indicating that only minor differences in backbone conformation were observed between the apo and holo forms of FABP1, which is consistent with the secondary chemical shifts that were observed. The most significant local differences in the backbone structure (Cα, N, and C′ RMSD >1.8 Å) were observed for residues Phe-95–Ile-98 in loop βGH. In addition, smaller but noticeable local backbone differences were observed (Cα, N, and C′ RMSD of 1–1.5 Å) in the portal regions spanning residues Gln-30–Gly-32 in αII, Ala-54–Gly-55 in loop βCD, Glu-77–Lys-80 encompassing loop βEF and strand βF, and Gln-30–Gly-32 in loop βFG and the N- and C-terminal residues. An overlay of the two structures and a comparison of the extent of the structural perturbation on binding are presented in Fig. 5, E and F.
Solution structure of the FABP1–GW7647 complex
The structure of the complex of GW7647 bound to FABP1 was modeled by docking the ligand into the structure of holo FABP1 using unambiguous intermolecular distance constraints derived from NOEs observed between proton resonances of GW7647 and protein side-chain resonances of Ile-52, Met-74, Val-83, and Thr-102, and one hydrogen bond restraint between the protein and ligand (Fig. 6A, Movie S1). The chemical shifts of the aliphatic protons of GW7647 were severely overlapped in the NMR spectrum of the complex, and most could not be assigned unambiguously, resulting in the relatively small number of unambiguous NOE-derived distance constraints. This resulted in some variability in the position of the cyclohexyl rings of GW7647 in the structures (Fig. 6B). Nonetheless, the orientation of the ligand was conserved in the ensemble, with the aromatic ring of GW7647 closer to the helical cap of FABP1 and the aliphatic portion of GW7647 buried more deeply in the cavity. This orientation is consistent with the intermolecular NOEs observed in the complex, where the aromatic protons of GW7647 show NOEs to the methyl groups of Ile-52 and Met-74 (Fig. 5A and Fig. S8), whereas the aliphatic protons of GW7647 show NOEs to the side chains of Phe-50, Phe-63, Thr-81, Val-83, Thr-102, and Asn-111 (Fig. S8), which are located at the bottom of the internal pocket. The H-bonding constraint was supported by a large change in chemical shift observed for the Arg-122–Hϵ proton in the presence of GW7647. The Arg-122–Hϵ proton resonance underwent a downfield shift of 1.66 ppm in the presence of GW7647 (Fig. 2B; δ(GW7647) = 8.74 ppm compared with δ(apo) = 7.08 ppm). The ensemble of 10 conformers chosen to represent the structure of holo FABP1 in the presence of GW7647 superimpose with a RMSD of 0.41 ± 0.05 Å over the backbone atoms and a RMSD of 0.76 ± 0.05 Å over all heavy atoms (Fig. 6C). A representative structure of the GW7647-bound holo FABP1 is shown in Fig. 6D. The structures of holo FABP1 in the absence and presence of GW7647 are similar, and modeling of the ligand into FABP1 did not result in violation of distance constraints derived from the NOE networks in the NMR data that were used to calculate the protein structure. Superposition of the mean coordinates (Ser-2–Ile-127) between the two structures resulted in a RMSD of 0.44 Å over backbone atoms and 0.59 Å over all heavy atoms.
Figure 6.

Solution structure of GW7647 bound to FABP1. A, binding mode of GW7647 represented by the conformer closest to the mean of the ensemble. NOE and H-bond constraints that were used for calculating the structure of the complex are indicated as dotted lines between the side chains of residues in FABP1 and GW7647. B, conformations of GW7647 in the ensemble of 10 conformers when bound to FABP1. C, ensemble of 10 conformers for the complex of GW7647 and FABP1. D, representative structure of the GW7647–FABP1 complex. The protein is shown as a cartoon and colored from white to dark red on a ramp with dark red representing residues having a chemical shift perturbation of ≥0.5 ppm in the 15N HSQC spectrum upon addition of GW7647. GW7647 is shown in stick representation with the carbon atoms colored blue. E, binding site for GW7647 in ensemble of 10 conformers representing the structure of FABP1 is represented with the protein side chains closest to the ligand shown as sticks with the carbon atoms colored white. The structure of GW7647 in the conformer closest to the mean of the ensemble is shown as sticks with carbon atoms colored blue.
Key internal changes in holo FABP1
Large CSPs were observed in 15N/13C-HSQC spectra for the resonances of Ile-52–Hδ1, Met-74–HN, and Arg-122–Hϵ (weighted CSP of 0.52, 0.78, and 1.6 ppm, respectively) upon GW7647 binding (Figs. 2 and 3 and Fig. S13). These residues are in direct contact with GW7647 (Fig. 6E), and unambiguous NOEs were assigned in the NOESY spectrum of the complex (Fig. S8). In addition, conformational changes were observed upon addition of GW7647 to FABP1 for the side-chain atoms of residues Phe-48, Phe-50, Phe-63, Thr-73, Phe-95, and Asn-111 (heavy atom displacement ≥3 Å) inside the β-barrel binding cavity as shown in Fig. 7A. In the holo FABP1 structure, the position of the Arg-122 side chain is somewhat better defined (global side-chain RMSD of 0.88 Å compared with 1.02 Å in apo FABP1). The guanidinium resonances of Arg-122 were only visible in the holo spectra, which provided additional NOE constraints to this residue. Moreover, binding of GW7647 stabilized the conformation of this side chain possibly by making a hydrogen bond between Arg-122–Hϵ and the carboxylate group of GW7647. The appearance of peaks for the guanidinium resonances of Arg-122 in the 2D 15N HSQC spectrum of the complex is consistent with protection from solvent that would result from this interaction with GW7647. Additional residues showing NOEs to GW7647 show line broadening in the 15N/13C HSQC spectra of holo FABP1, specifically the side-chain methyl (Hδ1) resonance of Ile-52 as well as backbone amide resonance of Met-74.
Figure 7.
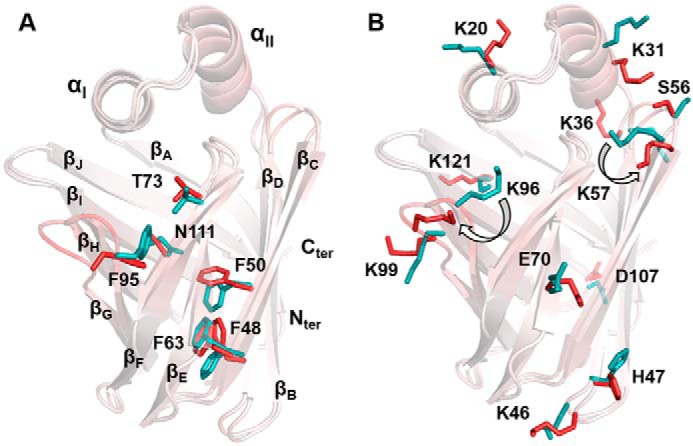
Side chain displacement in the structure of FABP1 upon GW7647 binding. Overlay of the conformer closest to the mean of apo and holo FABP1. The structures are presented as cartoons and colored by atom displacement between the two structures from white (0 Å) to red (5 Å). Side chain atoms of internal residues (within 3 Å of GW7647) showing atom displacement >3 Å (A) and external residues (>3 Å away from GW7647) showing atom displacement >4.5 Å shown as sticks for apo (cyan) and holo (red) FABP1 (B).
External changes in holo FABP1
Binding of GW7647 also altered the conformation of several side chains in holo FABP1 that were not directly involved in contacts with the ligand. When examining the individual structure of apo and holo FABP1 that was closest-to-mean of each ensemble, significant changes in conformation were observed for the side chains of residues Lys-20, Lys-31, Lys-36, Lys-46, His-47, Ser-56, Lys-57, Glu-70, Lys-96, Lys-99, Asp-107, and Lys-121 (RMSD of ≥4.5 Å) upon GW7647 binding (Fig. 7B). In addition, an increase in the solvent accessibility was observed for the residues Lys-57, Glu-77, and Lys-96 upon GW7647 binding (Table 2 and Fig. 8B). These residues are located in the loops βCD, βEF, and βGH, which surround the portal region, and line broadening was observed for the backbone amide residues in each of these loops upon addition of GW7647 (Fig. S12). This change in solvent accessibility also has an effect on the surface electrostatics around the portal region of FABP1 in the complex (Fig. 8 and Figs. S1 and S2).
Table 2.
Solvent accessibility for residues Lys-57, Glu-77, and Lys-96 averaged across each conformer of the NMR ensemble in structures of FABP1 determined in the absence (6DO6) and presence (6DO7) of GW7647, and in the absence (2L67) and presence (2L68) of OA
Solvent accessibility was computed using MOLMOL.
| Amino acids | 6DO6 (apo) | 6DO7 (holo) | 2L67 (apo) | 2L68 (holo) |
|---|---|---|---|---|
| Lys-57 | 22.3 | 28.9 | 10.8 | 39.4 |
| Glu-77 | 29.2 | 37 | 33.2 | 34.3 |
| Lys-96 | 25 | 54.3 | 44 | 49.7 |
Figure 8.

Solvent accessibility, backbone displacement, and electrostatic surface for FABP1 in the absence and presence of GW7647. A, global backbone displacement between apo and holo NMR structures of FABP1 in the absence and presence of GW7647 (6DO7 versus 6DO6) and oleic acid (2L68 versus 2L67) plotted against residue number. B, change in average solvent accessibility per residue for FABP1 in the absence and presence of GW7647 (6DO7 versus 6DO6) and oleic acid (2L68 versus 2L67) plotted against residue number. Solvent accessibility and backbone displacements were calculated in MOLMOL. C, electrostatic surfaces for apo FABP1 (6DO6) and holo FABP1 bound to GW7647 (6DO7). For 6DO6 and 6DO7, the conformer closest to the mean of the ensemble was used to calculate the electrostatic surface. The surface of apo FABP1 (6DO6) in white showing the position of residues Lys-57, Glu-77, and Lys-96. D, electrostatic surfaces for apo FABP1 (2L67) and holo FABP1 bound to oleic acid (2L68). For 2L67 and 2L68, the first conformer of the ensemble was used to calculate the electrostatic surface. Electrostatic surface potentials were calculated using the APBS plug-in within PyMOL.
Discussion
Intracellular lipid–binding proteins, including the FABPs, play an important role in the intracellular transport of poorly water-soluble endogenous ligands such as LCFAs and vitamins (1). More recently, an additional role for some FABPs in binding and intracellular distribution of poorly water-soluble drugs has been uncovered (22, 23). For example, we have previously shown that FABP1 and FABP2 can promote the activity of certain PPAR agonists and that the effect is mediated by co-localization in the nucleus of the FABP and PPAR that follows ligand binding (20). Furthermore, this effect is both FABP- and drug-specific, such that FABP1 can promote the activity of the PPARα agonist GW7647 but has no effect on the activity of the PPARα agonist fenofibrate. Conversely, FABP2 can promote the activity of fenofibrate but not GW7647. Such specificity could be mechanistically explained if ligand binding to the FABP stabilized a conformation that then promoted association with PPARα. Similar mechanisms of ligand-induced conformational change have been reported for other iLBPs, including FABP4, FABP5, and CRABP-II. In the case of FABP4 and FABP5, ligand binding results in the stabilization of a NLS, allowing importin-mediated nuclear import of the FABP–ligand complex and activation of PPARγ in the case of FABP4 (7) or PPARβ/δ in the case of FABP5 (9). Again, this is a drug-specific event; FABP4 can bind agonists of PPARα, PPARβ/δ, and PPARγ, but only the PPARγ agonists appear to stabilize the conformational NLS and promote PPARγ activation. CRABP-II also possesses a conformational NLS that forms upon binding to RA, but in addition utilizes residues in the loops surrounding the ligand portal to mediate a direct interaction with RAR, thereby channeling RA to its receptor. In common with several other iLBPs, FABP1 does not contain the three positively charged residues in the helical cap that assemble to form the NLS in FABP4 and FABP5. We therefore determined the solution structure of FABP1 in the absence and presence of GW7647 in an effort to explain the specific activation of PPARα that is observed with the FABP–GW7647 complex. Binding of GW7647 resulted in small changes in the backbone conformation of FABP1, which were accompanied by larger side-chain movements. Within the binding cavity, a number of residues, including Phe-48, Phe-50, Phe-63, Thr-73, Phe-95, and Asn-111, were observed to undergo conformational changes that were required to accommodate the ligand. In addition, a number of residues on the external surface of FABP1, which did not make direct contact with GW7647 in the complex, were also observed to undergo conformational change and become more surface-exposed upon binding. Some of the largest local conformational changes are observed for residues that are not in direct contact with GW7647 and are located in the loops around the portal regions of FABP1 (Fig. 7). Mutation of three of these residues, Lys-57, Glu-77, and Lys-96 to Ala had little effect on the backbone structure of FABP1, as revealed by analysis of secondary shifts calculated from the NMR assignments for the WT and triple-mutant FABP1 in the absence and presence of GW7647 (Fig. 3). Consistent with this observation, the mutations had only a minor effect on the binding affinity of GW7647 for the triple mutant protein (Fig. 2). Analysis of the CSPs observed in 15N HSQC spectra of WT and triple-mutant FABP1 suggested that the binding mode of GW7647 was similar in both cases (Fig. 2 and Figs. S5, S6, and S7).
The conformational changes on the external surface of FABP1 upon binding of GW7647 result in changes to the surface electrostatics that we hypothesize to be essential for the association of FABP1 with PPARα. The association of FABP1 with PPARα is consistent with both the FRET signal that has been reported between the two in the presence of GW7647 and the decreased rate of nuclear egress that accompanies increased PPARα activation observed in cellular assays (13). A similar co-localization of murine FABP1 and PPARα has previously been observed in mouse primary hepatocytes, and in vitro binding assays revealed a direct interaction between the two proteins (24). In contrast, we observed no FRET signal between the triple-mutant FABP1 and PPARα in either the presence or the absence of GW7647 (data not shown). Mutation of Lys-57, Glu-77, and Lys-96 to Ala also abolished nuclear accumulation of FABP1 and PPARα activation following stimulation with OA (Fig. 1), suggesting that this change in the surface electrostatics may be a common mechanism for PPARα agonists that bind FABP1.
To test this hypothesis, we made a comparison between the structure of FABP1 bound to OA (PDB code 2L68) and the corresponding apo FABP1 structure (PDB code 2L67). These two structures were calculated in solution from NMR data acquired under similar solution conditions. There are several regions where displacement of the backbone atoms is observed between these two structures (Fig. 8A). The largest displacements are observed for residues clustered around the helix-turn-helix motif as well as for the loops containing residues Lys-57, Glu-77, and Lys-96. The patterns of backbone displacements that are observed in the solution structures of FABP1 upon binding of OA and GW7647 also result in an increase in the average solvent-accessible surface area for several residues in these loops (Table 2 and Fig. 8B). With GW7647, the increase in solvent exposure is greatest for residues around Lys-96 and Glu-77, whereas with OA it is residues around Glu-77 and Lys-57 that are most affected (Fig. 8B). However, in both cases the conformational change is accompanied by a change in the surface electrostatics around the ligand portal (Fig. 8, C and D, and Fig. S1–S4), consistent with the suggestion that a common structural change occurs on binding of these ligands that underpins the observed nuclear localization and receptor activation.
Taken together, these data support a mechanism for FABP1-mediated PPARα activation. Ligand binding to FABP1 first provides a means for poorly water-soluble compounds such as LCFA and GW7647 to be transported through the aqueous cytoplasm of the cell. Second, the binding of some ligands is able to stabilize a conformation of FABP1 that promotes co-localization with PPARα. For GW7647 and OA, this mechanism results in a potentiation of their activation of the PPARα receptor. Only ligands that stabilize the appropriate conformation of FABP1 show the characteristic signature of increasing the nuclear localization of FABP1 and increasing the activation of PPARα.
Thus, it appears that iLBPs are able to selectively direct certain ligands to specific nuclear hormone receptors, although they achieve this by using a variety of different mechanisms. In the case of FABP4 and FABP5, ligands are able to stabilize a conformational NLS in the FABP, which delivers the complex to the nucleus. In the case of FABP4, it is PPARγ agonists that stabilize the NLS, which dictates the specificity of FABP4 toward activation of PPARγ (7). For FABP5, it is PPARβ/δ agonists that stabilize the NLS, and in turn FABP5 promotes PPARβ/δ activation (9). With CRABP-II, there appears to be a more complex mechanism, where RA binding first stabilizes the NLS to deliver the complex to the nucleus (10), and then residues in the portal loops of CRABP-II mediate a direct interaction with RAR that results in channeling of the RA to its receptor (11, 12).
The mechanism by which FABP1 promotes activation of PPARα is somewhat different. There is no active transport of the FABP1–GW7647 complex to the nucleus, and nuclear accumulation of the complex is driven instead by a decreased rate of nuclear egress (13). In this case, a change in the conformation of residues on the portal loops upon ligand binding modulates the electrostatic surface to promote association with PPARα.
In the case of each of these nuclear hormone receptor agonists, a concerted system of transport and nuclear localization mediated by a specific FABP protein is required for full activation of the partner receptor. The differential expression of FABP proteins in different tissues and the different mechanisms by which they are able to direct ligands to nuclear hormone receptors may provide a means to regulate the tissue-specific activity of drugs that modulate these receptors.
Experimental procedures
Reagents
The PPAR agonist GW7647 was purchased from Sapphire Bioscience Pty Ltd. (Cayman Chemicals). Ammonium chloride (15NH4Cl), [13C]glucose, and deuterium oxide were purchased from Cambridge Isotopes Laboratories. Competent BL21 Escherichia coli codon plus cells were purchased from Stratagene (Sydney, Australia). X-tremeGENE 9 DNA transfection reagent was from Roche Diagnostics (Castle Hill, Australia). All other reagents were of the highest purity available commercially.
Plasmids
Human FABP1 and human PPARα cDNAs were synthesized by GeneArt (Regensburg, Germany). Recombinant FABP1 for NMR-based structure determination was expressed, purified, and delipidated as described previously (13, 20). The plasmids for FABP1 and GFP–FABP1 used in the reported cell-based assays have been described previously (13). The luciferase reporter construct, PPRE3-TK-LUC, was a kind gift from Dr. Ronald Evans (Salk Institute, La Jolla, CA). The β-gal vector was from Promega (Madison, WI).
ITC data
ITC experiments were undertaken and analyzed as reported previously (13). Data were recorded at 25 °C with stirring at 1000 rpm using an iTC200 microcalorimeter (MicroCal, Malvern, UK). Titrations were performed in ITC buffer (20 mm HEPES, pH 8, 50 mm NaCl, 0.5 mm EDTA). A solution of GW7647 (7 μm) in ITC buffer was prepared by diluting from a concentrated stock in DMSO resulting in a final DMSO concentration of 0.5% v/v in the sample cell. The triple-mutant FABP1 (70 μm) was prepared in an identical buffer and placed into the syringe. 16 serial injections at intervals of 220 s were made with continuous stirring of the solution in the sample cell. The iTC200 control software was used to operate and acquire raw data as power (microcalories/s) versus time (minutes). The data were processed using the Origin7.0 software (MicroCal), and thermodynamic parameters were calculated by fitting the data to a one-site binding model.
NMR sample preparation and processing
All NMR experiments were conducted at 35 °C either on a Bruker 600 or 800 MHz spectrometer equipped with a single axis z-gradient triple resonance CryoProbe.
Both apo and holo samples were prepared in phosphate buffer (20 mm sodium phosphate, 50 mm NaCl, pH 5.5) with 90% H2O and 10% 2H2O. For apo FABP1, uniformly 15N- or 15N,13C-labeled protein samples were concentrated to 1 mm, whereas for holo FABP1, protein samples (0.5–1 mm) were saturated with an equimolar amount of GW7647 (0.5–1 mm), which was added from a concentrated DMSO stock so that the final DMSO concentration was less than 1%. No change in the 15N HSQC spectrum was observed when adding 1% DMSO only. Weighted CSP were calculated using Equation 1,
| (Eq. 1) |
where δHN and δN denote the change in chemical shift between apo FABP1 and holo FABP1 for HN and N resonances, respectively. All spectra were processed using either NMRPipe (25) or Topspin 3.0 (Bruker Biospin) and analyzed with NMRView (26), SPARKY,7 or CARA (40) software. The RMSD values between structures were calculated using MOLMOL (27). CSPs were mapped in PyMOL version 2.0 (Schrödinger, LLC).
NMR assignments
Sequential backbone assignments (1H, 15N, and 13C nuclei) for the proteins were generated from two-dimensional (2D) HSQC and three-dimensional (3D) experiments, including 3D HNCA (28), 3D CBCA(CO)NH (29), and 3D HNCACB (30). 3D experiments for the triple-mutant samples, including HNCA, CBCA(CO)NH, and HNCACB were recorded with nonuniform sampling in the 15N and 13C dimensions. The datasets were processed with the multidimensional decomposition algorithm, MddNMR (31).
Side-chain assignments were obtained from 3D HBHA(CO)NH, 3D 15N-edited (1H,1H)-NOESY, 3D 13Cali-edited (1H,1H)-NOESY, and 3D 13Caro-edited (1H,1H)-NOESY experiments (all recorded with a mixing time of 60 ms) using standard Bruker pulse sequences. For the sample of FABP1 in complex with GW7647, the same three NOESY spectra were collected at a protein concentration of 0.5 mm.
Because of the limited aqueous solubility of GW7647, NMR assignments of GW7647 were first made in 100% DMSO-d6 (DMSO-d6), and these were used to assist in generating the assignments for GW7647 in the complex. Partial assignments for GW7647 in the holo complex containing [U-2H,13C,15N]FABP1 (1 mm) and unlabeled GW7647 (1 mm) were obtained from a 2D ω1,ω2-13C,15N-filtered (1H,1H)-NOESY experiment. The presence of two cyclohexane rings in GW7647 resulted in significant chemical shift degeneracy, such that the resonances for many of the aliphatic protons of GW7647 could not be unambiguously assigned in the bound state.
For the assignment NOEs between GW7647 and FABP1, a series of filtered–edited NOESY experiments were recorded. These included 2D ω1-edited, ω2-13C,15N-filtered (1H,1H)-NOESY, 2D ω1,ω2-13C,15N-filtered (1H,1H)-NOESY, 3D ω1-13C,15N-filtered, ω2-15N-edited (1H,1H)-NOESY (all recorded with a mixing time of 200 ms) using a triple-labeled [U-2H,13C,15N]FABP1 protein sample (1 mm) and 3D ω1-13C,15N-filtered, ω3-13Cali-edited 1H,1H-NOESY using a double-labeled [U-13C,15N]FABP1 sample (∼0.5 mm). In each complex sample, the FABP1/GW7647 ratio was 1:1. Intermolecular distance restraints were derived from the NOE peak intensities (Figs. S9, S10, and S11).
Processed spectra were converted to XEASY format and loaded into CARA. HN, N, Cα, Cα-1, Cβ, and Cβ-1 resonances were manually peak-picked from 3D HNCA, CBCA(CO)NH, and HNCACB spectra for all the observed amide backbone peaks and used as the input for UNIO-MATCH (32) to generate automated backbone assignments. The backbone assignments from UNIO-MATCH were checked manually, verified with the 3D HNCACB, and 3D 15N-edited (1H,1H)-NOESY spectra, and extended where possible. Hβ and Hα resonances were manually assigned in the 3D HBHA(CO)NH spectrum. The three NOESY spectra along with all specific assignments were used as input for UNIO-ATNOS/ASCAN (33) to generate automated side-chain assignments. The output from UNIO-ATNOS/ASCAN was manually checked, and assignments were extended wherever possible.
Structure calculations
Prior to NMR structure determination, backbone torsional angles from HN, N, Cα, Cβ, and Hα chemical shifts were predicted using the default mode on the TALOS+ server (34). The output from TALOS+ for each residue was manually examined against a Ramachandran map for the 10 best database matches. Residues defining consistently “good” predictions based on clustering in the Ramachandran map (8 or 9 out of the 10 best matches) with random coil index predicted order parameter S2 >0.5 were retained and converted to dihedral restraints in CYANA format. Where the TALOS-derived dihedral angles were tightly constrained, the tolerance relaxed to ±15°, otherwise the TALOS values were used. For the assignment list, dihedral restraints from TALOS+ and NOESY data were used as input for UNIO-ATNOS/CANDID (35, 36) using CYANA3.0 (37) for automated structure determination. After the first round of structure calculation, dihedral angle restraints with large violations (>5°) observed in regions of the structure with no well-defined secondary structure were removed, and the structure was recalculated. An ensemble of 20 conformers with lowest target function (no NOE and torsional angle violation >0.2 Å and 5°, respectively) after CYANA was refined using OPALp (38). The structures derived from the NMR data were manually inspected in MOLMOL (27), and hydrogen bonds were calculated using the default settings in MOLMOL and verified using 13Cαβ secondary shifts (39) and dihedral angle restraints from TALOS+. Hydrogen bond constraints were added as input for UNIO-ATNOS/CANDID and CYANA along with dihedral constraints and three NOESY spectra for the final structure calculations. An ensemble of 40 of the CYANA conformers was further energy-refined using OPALp and Maestro's restrained minimization impref utility (Maestro version 11.4.011, Impact version 7.7) with an RMSD convergence of 0.035 Å, before analysis using the PDB validation server. The top 10 conformers without chirality outliers, minimal steric clash, and Ramachandran outliers were selected from the analysis to represent the structure of apo FABP1.
The structure of holo FABP1 was first calculated from the NOESY data without GW7647 in the structure calculation as per the above protocol. Subsequently, the GW7647-bound FABP1 structure was recalculated by CYANA using five unambiguous intermolecular NOEs, one intermolecular H-bond restraint, and all other restraints derived from the NMR data for holo FABP1. The FABP1 structure in the presence of GW7647 (40 CYANA conformers) was refined as above with Maestro's impref utility with the same parameters, and the 10 lowest energy structures were selected for analysis.
Solvent-exchange studies
Phase-modulated CLEAN chemical exchange (CLEANEX-PM) fast-HSQC experiments (21) were recorded for apo and holo WT FABP1 in 20 mm MES buffer, pH 5.5, containing 50 mm NaCl at a sample concentration of 0.5 mm at 35 °C on a Bruker 600 MHz spectrometer equipped with CryoProbe to identify solvent-exposed amide protons. The mixing time for the CLEANEX spin lock was set to 100 ms. Spectra were acquired with 32 scans and 2048 and 128 complex data points for the 1HN and 15N dimensions, respectively. NMR spectra were processed using Topspin and converted to XEASY format for the intensity analysis.
15N-1H heteronuclear NOE studies
Backbone 15N-1H heteronuclear NOE experiments were performed for both apo and holo WT FABP1 and the triple mutant at a sample concentration of 240 and 230 μm, respectively, using the standard Bruker pulse sequence at 35 °C on Bruker 600 MHz spectrometer equipped with CryoProbe. The experiment was acquired in an interleaved mode with a relaxation delay of 5 s, 32 scans, and 2048 and 256 complex data points for the 1HN and 15N dimensions, respectively. NMR data were processed using Topspin and converted to XEASY format for the intensity analysis. Steady-state 15N-1H heteronuclear NOE values were determined from the ratio of peak intensities for spectra collected with and without 5 s of proton presaturation.
Cell culture and transfections
COS-7 cells were kindly provided by Prof. Phillip Nagley (Monash University, Victoria, Australia). The cells were cultured in Dulbecco's modified Eagle's medium with 4 mm glutamine, 100 units/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum in a 95% air, 5% CO2 atmosphere at 37 °C. For transfections, cells were seeded into 6-well plates at a density of 2 × 105 cells/well in culture medium devoid of antibiotics. Transfections were carried out using X-tremeGENE 9 DNA transfection reagent according to the manufacturer's protocol.
Transactivation assays
Transactivation assays were carried out using COS-7 cells in six-well plates (2 × 105 cells/well) according to the procedure published previously (13). Cells were co-transfected with 0.3 μg of the luciferase reporter (PPRE3-TK-LUC), 0.15 μg of β-gal (as a transfection efficiency control), 0.05 μg of either empty vector (pSG5) or PPARα, and 0.3 μg of either empty vector (pSG5), FABP1 WT, FABP1 K20A, K31A, or FABP1 K57A, E77A, K96A. 24 h following transfection, cells were treated with vehicle (0.1% v/v DMSO), oleic acid (10 μm), or GW7647 (600 nm) for 24 h before the cells were lysed and assayed for luciferase activity using the luciferase assay system (Promega). In parallel, lysates were assayed for β-gal activity using the β-gal enzyme assay system (Promega) to correct for differences in transfection efficiency. The data are expressed relative to vehicle-treated control, from three independent experiments conducted in duplicate.
Automated Operetta high-content imaging
The Operetta high-content imager (PerkinElmer Life Sciences) equipped with a live cell chamber was used to obtain unbiased images of the localization of GFP-tagged FABP1 for automated quantification in live cells. COS-7 cells were co-transfected with 0.05 μg of empty vector (pSG5) or 0.05 μg of PPARα plasmid DNA and 0.3 μg of GFP-tagged FABP1 plasmid DNA in six-well plates. Twenty four hours following transfection, cells were subcultured into black-walled, optically clear 96-well plates and treated with vehicle (0.1% v/v DMSO), oleic acid (10 μm), or GW7647 (600 nm) for 24 h. On the day of imaging, cells were incubated with Hoechst stain (2 μg/ml) for 5 min at room temperature (to stain the nuclei), prior to imaging the live cells in Hanks' balanced salt solution at 37 °C. The center of each well of a 96-well plate was imaged with an Olympus LUCPlanFLN ×20 (NA 0.45) objective, and the nuclear redistribution of FABPs was determined by measuring the fluorescence intensity in the nucleus and cytosol of every cell within the field of view using automated protocols within the Harmony High Content Imaging and Analysis software (version 3.5.2). For each experimental repeat, ∼50 cells were analyzed. Experiments were performed on four separate occasions.
Statistical analysis
Data are expressed as the mean ± S.E. from three or more independent replicate experiments. A two-way ANOVA was performed to detect differences among experimental groups with p < 0.05 taken as statistically significant.
Accession numbers
The chemical shift assignments of apo and holo wild type FABP1 have been deposited in BMRB, accession number 30477 and 30478, respectively, and the chemical shifts of triple mutant K57A/E77A/K96A FABP1 in the absence and presence of GW7647 have been deposited in BMRB with accession numbers 27509 and 27510, respectively. Coordinates for the structure of wild type FABP1 calculated from data obtained in the absence and presence of GW7647 have been deposited in the PDB with accession numbers 6DO6 and 6DO7, respectively. Coordinates for the structure of holo FABP1 in complex with GW7647 have been deposited in the PDB with accession number 6DRG.
Author contributions
R. P., P. G., R. J. W., L. V., M. L. H., C. J. P., and M. J. S. conceptualization; R. P., B. M., B. L., I. R. C., S. J. H., M. L. H., C. J. P., and M. J. S. data curation; R. P., B. M., B. L., I. R. C., S. J. H., M. L. W., C. S. C., O. I., B. C. D., P. G., M. L. H., C. J. P., and M. J. S. formal analysis; R. P., M. L. H., C. J. P., and M. J. S. funding acquisition; R. P., B. M., B. L., I. R. C., S. J. H., M. L. W., C. S. C., O. I., B. C. D., M. L. H., and C. J. P. investigation; R. P., B. M., B. L., I. R. C., and M. J. S. writing-original draft; R. P., P. G., R. J. W., L. V., M. L. H., C. J. P., and M. J. S. project administration; R. P., I. R. C., S. J. H., B. C. D., P. G., R. J. W., L. V., M. L. H., C. J. P., and M. J. S. writing-review and editing; M. L. W., C. S. C., O. I., B. C. D., P. G., M. L. H., and C. J. P. methodology; B. C. D. and P. G. validation; P. G., R. J. W., L. V., M. L. H., and C. J. P. resources.
Supplementary Material
This work was supported in part by funding from Les Laboratories Servier and by ARC Grants DP120102930 and DP150102587. P. G., R. J. W., and L. V. were employed by Les Laboratories Servier, a commercial company, at the time this study was conducted.
This article contains Figs. S1–S13, Tables S1–S2, and supporting Movie S1.
The atomic coordinates and structure factors (codes 6DO6, 6DO7, and 6DRG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The chemical shift assignments have been deposited in BMRB with accession numbers 30477, 30478, 27509, and 27510.
T. D. Goddard and D. G. Kneller, unpublished work.
- iLBP
- intracellular lipid-binding protein
- LCFA
- long-chain fatty acid
- FABP
- fatty acid–binding protein
- CRABP
- cellular retinoic acid–binding protein
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- RA
- retinoic acid
- RAR
- retinoic acid receptor
- ANOVA
- analysis of variance
- PPAR
- peroxisome proliferator–activated receptor
- ITC
- isothermal titration microcalorimetry
- NLS
- nuclear localization signal
- OA
- oleic acid
- HSQC
- heteronuclear single quantum coherence
- CSP
- chemical shift perturbation.
References
- 1. Kaikaus R. M., Bass N. M., and Ockner R. K. (1990) Functions of fatty acid–binding proteins. Experientia 46, 617–630 10.1007/BF01939701 [DOI] [PubMed] [Google Scholar]
- 2. Chmurzyńska A. (2006) The multigene family of fatty acid–binding proteins (FABPs): function, structure and polymorphism. J. Appl. Genet. 47, 39–48 10.1007/BF03194597 [DOI] [PubMed] [Google Scholar]
- 3. Hodsdon M. E., and Cistola D. P. (1997) Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid–binding protein: implications for the mechanism of ligand entry. Biochemistry 36, 1450–1460 10.1021/bi961890r [DOI] [PubMed] [Google Scholar]
- 4. Zimmerman A. W., van Moerkerk H. T., and Veerkamp J. H. (2001) Ligand specificity and conformational stability of human fatty acid–binding proteins. Int. J. Biochem. Cell Biol. 33, 865–876 10.1016/S1357-2725(01)00070-X [DOI] [PubMed] [Google Scholar]
- 5. Schug T. T., Berry D. C., Shaw N. S., Travis S. N., and Noy N. (2007) Opposing effects of retinoic acid on cell growth result from alternate activation of two different nuclear receptors. Cell 129, 723–733 10.1016/j.cell.2007.02.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ayers S. D., Nedrow K. L., Gillilan R. E., and Noy N. (2007) Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARγ by FABP4. Biochemistry 46, 6744–6752 10.1021/bi700047a [DOI] [PubMed] [Google Scholar]
- 7. Gillilan R. E., Ayers S. D., and Noy N. (2007) Structural basis for activation of fatty acid–binding protein 4. J. Mol. Biol. 372, 1246–1260 10.1016/j.jmb.2007.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tan N. S., Shaw N. S., Vinckenbosch N., Liu P., Yasmin R., Desvergne B., Wahli W., and Noy N. (2002) Selective cooperation between fatty acid–binding proteins and peroxisome proliferator-activated receptors in regulating transcription. Mol. Cell. Biol. 22, 5114–5127 10.1128/MCB.22.14.5114-5127.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Armstrong E. H., Goswami D., Griffin P. R., Noy N., and Ortlund E. A. (2014) Structural basis for ligand regulation of the fatty acid–binding protein 5, peroxisome proliferator-activated receptor β/δ (FABP5-PPARβ/δ) signaling pathway. J. Biol. Chem. 289, 14941–14954 10.1074/jbc.M113.514646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sessler R. J., and Noy N. (2005) A ligand-activated nuclear localization signal in cellular retinoic acid–binding protein-II. Mol. Cell 18, 343–353 10.1016/j.molcel.2005.03.026 [DOI] [PubMed] [Google Scholar]
- 11. Dong D., Ruuska S. E., Levinthal D. J., and Noy N. (1999) Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 274, 23695–23698 10.1074/jbc.274.34.23695 [DOI] [PubMed] [Google Scholar]
- 12. Budhu A., Gillilan R., and Noy N. (2001) Localization of the RAR interaction domain of cellular retinoic acid–binding protein-II. J. Mol. Biol. 305, 939–949 10.1006/jmbi.2000.4340 [DOI] [PubMed] [Google Scholar]
- 13. Hughes M. L., Liu B., Halls M. L., Wagstaff K. M., Patil R., Velkov T., Jans D. A., Bunnett N. W., Scanlon M. J., and Porter C. J. (2015) Fatty acid–binding proteins 1 and 2 differentially modulate the activation of peroxisome proliferator-activated receptor α in a ligand-selective manner. J. Biol. Chem. 290, 13895–13906 10.1074/jbc.M114.605998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cai J., Lücke C., Chen Z., Qiao Y., Klimtchuk E., and Hamilton J. A. (2012) Solution structure and backbone dynamics of human liver fatty acid–binding protein: fatty acid binding revisited. Biophys. J. 102, 2585–2594 10.1016/j.bpj.2012.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Y., Estephan R., Yang X., Vela A., Wang H., Bernard C., and Stark R. E. (2011) A nuclear magnetic resonance-based structural rationale for contrasting stoichiometry and ligand binding site(s) in fatty acid–binding proteins. Biochemistry 50, 1283–1295 10.1021/bi101307h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma A., and Sharma A. (2011) Fatty acid induced remodeling within the human liver fatty acid–binding protein. J. Biol. Chem. 286, 31924–31928 10.1074/jbc.M111.270165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thompson J., Winter N., Terwey D., Bratt J., and Banaszak L. (1997) The crystal structure of the liver fatty acid–binding protein. A complex with two bound oleates. J. Biol. Chem. 272, 7140–7150 10.1074/jbc.272.11.7140 [DOI] [PubMed] [Google Scholar]
- 18. D'Onofrio M., Barracchia C. G., Bortot A., Munari F., Zanzoni S., and Assfalg M. (2017) Molecular differences between human liver fatty acid–binding protein and its T94A variant in their unbound and lipid-bound states. Biochim. Biophys. Acta 1865, 1152–1159 10.1016/j.bbapap.2017.06.025 [DOI] [PubMed] [Google Scholar]
- 19. Huang H., McIntosh A. L., Martin G. G., Landrock K. K., Landrock D., Gupta S., Atshaves B. P., Kier A. B., and Schroeder F. (2014) Structural and functional interaction of fatty acids with human liver fatty acid–binding protein (L-FABP) T94A variant. FEBS J. 281, 2266–2283 10.1111/febs.12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Patil R., Laguerre A., Wielens J., Headey S. J., Williams M. L., Hughes M. L., Mohanty B., Porter C. J., and Scanlon M. J. (2014) Characterization of two distinct modes of drug binding to human intestinal fatty acid–binding protein. ACS Chem. Biol. 9, 2526–2534 10.1021/cb5005178 [DOI] [PubMed] [Google Scholar]
- 21. Hwang T.-L., van Zijl P. C., and Mori S. (1998) Accurate quantitation of water-amide proton exchange rates using the phase-modulated CLEAN chemical exchange (CLEANEX-PM) approach with a fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR 11, 221–226 10.1023/A:1008276004875 [DOI] [PubMed] [Google Scholar]
- 22. Chuang S., Velkov T., Horne J., Porter C. J., and Scanlon M. J. (2008) Characterization of the drug binding specificity of rat liver fatty acid–binding protein. J. Med. Chem. 51, 3755–3764 10.1021/jm701192w [DOI] [PubMed] [Google Scholar]
- 23. Velkov T., Horne J., Laguerre A., Jones E., Scanlon M. J., and Porter C. J. (2007) Examination of the role of intestinal fatty acid–binding protein in drug absorption using a parallel artificial membrane permeability assay. Chem. Biol. 14, 453–465 10.1016/j.chembiol.2007.03.009 [DOI] [PubMed] [Google Scholar]
- 24. Wolfrum C., Borrmann C. M., Borchers T., and Spener F. (2001) Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors α- and γ-mediated gene expression via liver fatty acid–binding protein: a signaling path to the nucleus. Proc. Natl. Acad. Sci. U.S.A. 98, 2323–2328 10.1073/pnas.051619898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 26. Johnson B. A., and Blevins R. A. (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 27. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 28. Kay L. E., Ikura M., Tschudin R., and Bax A. (2011) Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. 1990. J. Magn. Reson. 213, 423–441 10.1016/j.jmr.2011.09.004 [DOI] [PubMed] [Google Scholar]
- 29. Grzesiek S., and Bax A. (1992) Correlating backbone amide and side-chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 114, 6291–6293 10.1021/ja00042a003 [DOI] [Google Scholar]
- 30. Wittekind M., and Mueller L. (1993) HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the α- and β-carbon resonances in proteins. J. Magn. Reson. 101, 201–205 10.1006/jmrb.1993.1033 [DOI] [Google Scholar]
- 31. Orekhov V. Y., and Jaravine V. A. (2011) Analysis of non-uniformly sampled spectra with multi-dimensional decomposition. Prog. Nucl. Magn. Reson. Spectrosc. 59, 271–292 10.1016/j.pnmrs.2011.02.002 [DOI] [PubMed] [Google Scholar]
- 32. Volk J., Herrmann T., and Wüthrich K. (2008) Automated sequence-specific protein NMR assignment using the memetic algorithm MATCH. J. Biomol. NMR 41, 127–138 10.1007/s10858-008-9243-5 [DOI] [PubMed] [Google Scholar]
- 33. Fiorito F., Herrmann T., Damberger F. F., and Wüthrich K. (2008) Automated amino acid side-chain NMR assignment of proteins using 13C- and 15N-resolved 3D [1H,1H]-NOESY. J. Biomol. NMR 42, 23–33 10.1007/s10858-008-9259-x [DOI] [PubMed] [Google Scholar]
- 34. Shen Y., Delaglio F., Cornilescu G., and Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 10.1007/s10858-009-9333-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herrmann T., Güntert P., and Wüthrich K. (2002) Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J. Mol. Biol. 319, 209–227 10.1016/S0022-2836(02)00241-3 [DOI] [PubMed] [Google Scholar]
- 36. Herrmann T., Güntert P., and Wüthrich K. (2002) Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J. Biomol. NMR 24, 171–189 10.1023/A:1021614115432 [DOI] [PubMed] [Google Scholar]
- 37. Güntert P., Mumenthaler C., and Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 10.1006/jmbi.1997.1284 [DOI] [PubMed] [Google Scholar]
- 38. Koradi R., Billeter M., and Guntert P. (2000) Point-centered domain decomposition for parallel molecular dynamics simulation. Comput. Phys. Commun. 124, 139–147 10.1016/S0010-4655(99)00436-1 [DOI] [Google Scholar]
- 39. Wishart D. S., and Sykes B. D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 4, 171–180 [DOI] [PubMed] [Google Scholar]
- 40. Keller R. (2004) The Computer-aided Resonance Assignment Tutorial. Cantina Verlag, Goldau, Switzerland [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.