

Abstract
The guanylyl cyclase-activating protein, GCAP1, activates photoreceptor membrane guanylyl cyclase (RetGC) in the light, when free Ca2+ concentrations decline, and decelerates the cyclase in the dark, when Ca2+ concentrations rise. Here, we report a novel mutation, G86R, in the GCAP1 (GUCA1A) gene in a family with a dominant retinopathy. The G86R substitution in a “hinge” region connecting EF-hand domains 2 and 3 in GCAP1 strongly interfered with its Ca2+-dependent activator-to-inhibitor conformational transition. The G86R-GCAP1 variant activated RetGC at low Ca2+ concentrations with higher affinity than did the WT GCAP1, but failed to decelerate the cyclase at the Ca2+ concentrations characteristic of dark-adapted photoreceptors. Ca2+-dependent increase in Trp94 fluorescence, indicative of the GCAP1 transition to its RetGC inhibiting state, was suppressed and shifted to a higher Ca2+ range. Conformational changes in G86R GCAP1 detectable by isothermal titration calorimetry (ITC) also became less sensitive to Ca2+, and the dose dependence of the G86R GCAP1–RetGC1 complex inhibition by retinal degeneration 3 (RD3) protein was shifted toward higher than normal concentrations. Our results indicate that the flexibility of the hinge region between EF-hands 2 and 3 is required for placing GCAP1-regulated Ca2+ sensitivity of the cyclase within the physiological range of intracellular Ca2+ at the expense of reducing GCAP1 affinity for the target enzyme. The disease-linked mutation of the hinge Gly86, leading to abnormally high affinity for the target enzyme and reduced Ca2+ sensitivity of GCAP1, is predicted to abnormally elevate cGMP production and Ca2+ influx in photoreceptors in the dark.
Keywords: calcium-binding protein, eye, cyclic GMP (cGMP), phototransduction, photoreceptor, retinal degeneration, guanylate cyclase (guanylyl cyclase), calcium sensor, GUCA1A, RetGC, retinopathy
Introduction
Guanylyl cyclase-activating proteins (GCAPs),4 N-myristoylated calcium/magnesium-binding proteins of the EF-hand superfamily, are comprised of two pairs of EF-hand domains connected via a “hinge” region (reviewed in Refs. 1 and 2). Among several isoforms of GCAPs expressed in the vertebrate retinas (3–6) two, GCAP1 and GCAP2, regulate visual signaling in all species by properly shaping the sensitivity and kinetics of rod and cone responses (7–10). Vertebrate rods and cones respond to light stimuli by closing cGMP-gated channels in their outer segments via phototransduction cascade-mediated hydrolysis of cGMP (reviewed in Refs. 11 and 12). Following the excitation, cGMP production by retinal membrane guanylyl cyclase (RetGC) (13–15) first becomes accelerated, to speed up the recovery and light adaptation of photoreceptors, and then decelerated again as photoreceptors recover from the excitation back to their dark-adapted state (7, 16). Negative Ca2+ feedback regulates the activity of RetGC via its Ca2+ sensor proteins, GCAPs, such that in the light, when cGMP channels are closed and the influx of Ca2+ through the channels stops, GCAPs release Ca2+ and convert into a Mg2+-liganded state that stimulates RetGC. Once the photoreceptors return to their dark-adapted state, when cGMP channels re-open and the influx of Ca2+ resumes, GCAPs undergo the reverse, activator-to-inhibitor, transition, by replacing Mg2+ in their EF-hands with Ca2+, and decelerate RetGC (reviewed in Refs. 2 and 12). Failure of RetGC to accelerate or decelerate cGMP production within the normal range of the intracellular free Ca2+ alters light sensitivity and kinetics of rod and cone response to light (7–9, 16–18) and has been linked to various forms of retinal blindness in humans, such as Leber congenital amaurosis, dominant cone or cone-rod degenerations (reviewed in Ref. 19–22), and a recessive night blindness (23). Multiple mutations linked to these blinding disorders have been found in the genes coding for RetGC1 isozyme (GUCY2D) (19–27) and GCAP1 (GUCA1A) (28–40). GUCA1A mutations linked to the dominant cone or cone-rod degenerations specifically suppress Ca2+ sensitivity of RetGC1 isozyme in vivo (17, 41, 42), most often by directly altering GCAP1 EF-hand motifs and thus reducing metal binding in EF-hands 3 and 4 (reviewed in Ref. 21), but also indirectly, by affecting EF-hand 4 affinity for Ca2+ via altering a “calcium-myristoyl tug” mechanism connecting the C-terminal EF-hand 4 with the N-terminal myristoyl group buried inside the N-proximal semiglobule of EF-hands 1 and 2 (1, 43, 44). Here we describe a new type of mutation in GCAP1, G86R, leading to a dominant retinopathy in humans. We present evidence that the flexible hinge Gly86 connecting the semiglobules formed by the two pairs of EF-hands reduces GCAP1 affinity for RetGC1 but also adjusts the Ca2+ sensitivity of the activator-to-inhibitor transition to the proper physiological range of RetGC1 regulation by Ca2+. We reason that the G86R mutation in GCAP1 would trigger photoreceptor death by abnormally elevating cGMP production rate in the dark.
Results
G86R GUCA1A causes dominant retinopathy
The patient evaluated at 62 years of age had no history of visual problems (other than myopia) until his 5th decade of life. He was then diagnosed as having a macular dystrophy. Best corrected visual acuities at age 56 were 20/60 and 20/200. Reading and distance vision decreased progressively over many years; and color discrimination became difficult. There was no photosensitivity and no peripheral field or night vision complaints. The patient did not report any general health issues and he was not taking medication with known retinotoxic effects. Visual acuities of the proband were 20/200 (with no significant refractive error) in both eyes. The clinical examination was within normal limits except for the retina. Maculae were granular and thinned in appearance. Peripheral retina, optic nerve, and vessel caliber were normal. Macular functional abnormalities were supported by retinal pigment epithelium (RPE) disease apparent on en face imaging (Results and Fig. S1) and retinal structural changes on cross-sectional imaging. Optical coherence tomography (OCT) (Fig. 1A) showed loss of central retinal lamination suggesting complete degeneration of rod and cone photoreceptors. By 10° from the fovea, however, outer nuclear layer (ONL, where rod and cone photoreceptor nuclei are located, highlighted blue) thickness returned to normal. Beyond the perifoveal region, there was a mild thinning of both cone and rod outer segments extending across the width of the scan (Fig. 1, right panels). Illustrating these photoreceptor abnormalities are magnified images (lower panels). Shortened outer segments where phototransduction occurs is likely one of the contributors to the loss of visual function. Electrophysiological (Fig. 1B) and perceptual (45) (Results and Fig. S1) results suggested mild dysfunction across the retina of both rod and cone photoreceptor-driven function, and a severe loss of macular function.
Figure 1.

Phenotype of GUCA1A G86R patient. A, upper left panels: OCT cross-sectional images along the horizontal meridian of the right eye in a normal subject and the proband. Features are highlighted for visibility: blue, ONL; yellow, near the junction of inner and outer segments (IS/OS); orange, near the cone outer segment tips (COST); light blue, near the rod outer segment tips (ROST); brown, near the interface between RPE and Bruch membrane (BrM/RPE). Lower panels: magnified images of the more peripheral (1) and central (2) retina (indicated by rectangles in the upper panels). Longitudinal reflectivity profiles (LRPs) overlaid onto OCT images are at 20° and 8° temporal to the fovea. ONL, cone outer segment (COS), and rod outer segment (ROS) thickness are bracketed to the left of each magnified image. Upper right panels: ONL, COS, and ROS thicknesses quantified across the horizontal meridian in the proband (black line) and compared with normal limits (gray zones = mean ± 2 S. D.). B, full-field ERG of proband (right) recorded with 4 standard conditions compared with that of an age-related subject with normal vision (left). Rod, dark-adapted ERG with dim blue −1.6 log scot-cd s m−2 flashes; mixed Cone-Rod, dark-adapted ERG with white 1.2 log scot-cd s m−2 flashes; Cone, light-adapted ERG with white 0.8 log phot-cd s m−2 flashes at stimulation rates of 1 and 30 Hz using white adapting backgrounds of 1.5 and 0.8 log phot-cd m−2, respectively. Rod and mixed cone-rod responses are within normal limits, whereas cone responses are reduced in amplitude compared with normal (see “Results” and Fig. S1 for additional details). C, pedigree of family; arrow, proband.
The family history of the proband (Fig. 1C, III, 2) indicated 6 other members with visual symptoms, suggesting an autosomal dominant mode of inheritance, although there was no male-to-male transmission. DNA samples from the proband and two other affected family members (III, 3; and IV, 1) were studied. In the proband, a panel of genes associated with inherited retinal degenerations was screened using next-generation sequencing followed by confirmatory Sanger sequencing. A novel heterozygous missense variant, c.256G → C, in the GUCA1A gene was identified in the proband and in other affected family members by direct testing in the GUCA1A gene. In the disease-causing variant, Gly86, residue that connects the exiting helix of EF-hand 2 and the entering helix of EF-hand 3, thus creating a hinge between the semiglobules of GCAP1 formed by two pairs of EF-hand domains (Fig. 2), was substituted by Arg.
Figure 2.
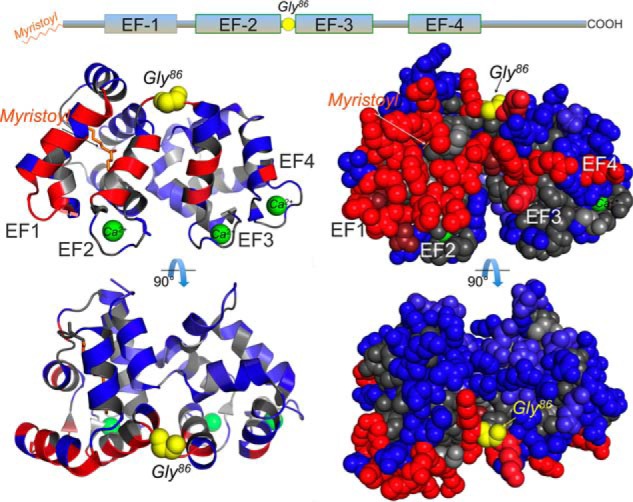
A mutation in the GUCA1A gene of the dominant retinopathy patient replaces the hinge Gly86 of GCAP1. The linear diagram of GCAP1 primary structure (top) indicates the position of four helix-loop-helix EF-hand domains (a nonmetal binding EF1 and the three metal binding EF2, -3, and -4) and the position of the hinge Gly86 residue (marked yellow). A Gly → Cys transversion in codon 256 of a human GUCA1A gene coding for GCAP1 replaces Gly86 with Arg. The hinge Gly86 connects the semiglobules of GCAP1 formed by two pairs of EF-hand domains (EF1 + EF2 and EF3 + EF4), depicted in a ribbon (left) and a space-filled (right) diagram. The N-myristoyl residue, embedded into the EF1/EF2 semiglobule, is marked orange; the side chains required for forming interface with the RetGC1 are shown in red, and the side chains not essential for forming the interface are shown in blue (after Peshenko et al. (46)). The Ca2+ ions coordinated in three EF-hand loops are marked green. The structural model is based on the Stephen et al. (77) GCAP1 crystal structure.
The G86R mutation in GCAP1 reduces Ca2+ sensitivity of guanylyl cyclase regulation
G86R GCAP1 effectively activated human RetGC1 expressed in HEK293 cells at low free Ca2+ concentrations, similarly to the WT GCAP1 (Fig. 3, A and B), but failed to effectively decelerate the cyclase when free Ca2+ concentrations rise. The [Ca]1/2 values became shifted to 5-fold higher free Ca2+ in both bovine and human orthologs of GCAP1. In the case of human GCAP1, the [Ca]1/2 (mean ± S.D.) for G86R increased to 0.94 ± 0.09 μm (n = 4) from 0.14 ± 0.06 μm (n = 3) in WT (p = 0.0004, unpaired/unequal variance Student's t test). As a result, the RetGC1 remained stimulated by the G86R GCAP1 at free Ca2+ concentrations that by far exceeded those found in dark-adapted mammalian photoreceptors (17, 47).
Figure 3.

The G86R mutation in GCAP1 alters RetGC1 regulation by Ca2+. Recombinant RetGC1 was reconstituted with 10 μm bovine (A) or human (B) WT (♢) or G86R (●) GCAP1 and assayed at different free Ca2+ concentrations at 0.9 mm free Mg2+. Data (mean ± S.D.) averaged from 3 to 4 independent experiments in each case were fitted using a Hill function, (Amax − Amin)/(1 + ([Ca]/[Ca]1/2)H) + Amin, where Amax and Amin are the respective maximal and minimal activity, [Ca], free Ca2+ concentration, [Ca]1/2 is the free Ca2+ concentration at half-maximal activity, and H, Hill coefficient. Note that whereas the Amax of the G86R-stimulated cyclase only slightly exceeds the WT levels, the [Ca]1/2 shifts >5-fold toward higher Ca2+ range, and even at saturating Ca2+ the G86R GCAP1 partially activates RetGC1, thus rendering the cyclase constitutively active.
The affinities of GCAP1 to RetGC1 in both the Mg2+ and Ca2+-liganded states are nearly equal (Fig. 4 and Ref. 48). However, replacement of the hinge Gly86 increased the GCAP1 apparent affinity for RetGC1 in the activator state of the mutant, KGCAPMg = 0.34 ± 0.063 μm, n = 4, versus 1.42 ± 0.061 μm, n = 3, in WT (p < 0.0001) (Fig. 4, B and C), whereas the affinity of the Ca2+-liganded G86R was increased to a lesser extent: KGCAPCa = 0.59 ± 0.047 m versus 1.44 ± 0.14 μm in WT, n = 4 (p = 0.001) (Fig. 4C). The G86R GCAP1 gaining higher affinity for the cyclase in Mg2+- than in the Ca2+-liganded form, resulting in change of the KGCAPMg versus KGCAPCa, can account for a ∼2-fold increase in [Ca]1/2 (Fig. 4 and Ref. 48). Nonetheless, this difference alone did not account for the >6-fold [Ca]1/2 increase observed in Fig. 3B, thus suggesting that Gly86, despite its being not a part of the EF-hand motif per se, but a hinge between the two semiglobules of the molecule (Fig. 2) nonetheless affected GCAP1 affinity for Ca2+.
Figure 4.

Change in G86R GCAP1 affinity for the cyclase contributes to its altered Ca2+ sensitivity. A, calcium sensitivity of the RetGC cyclase ([Ca]1/2) depends on three dissociation constants defining the affinity of GCAP for Ca2+ (KCa), the affinity of the Mg2+-liganded GCAP for RetGC (KGCAP*), and the affinity of Ca2+-liganded GCAP for RetGC (KGCAPca) in the equation shown on the right (48). WT GCAP1 has similar affinities for the cyclase regardless of the metal ligand (48), hence the [Ca]1/2 is dominated primarily by KCa. B and C, dose dependence of RetGC1 activation by GCAP1. RetGC1 was reconstituted with increasing concentrations of WT (○) or G86R (●, red) human GCAP1 at saturating 10 mm Mg2+ in the absence of Ca2+ (B) or at 0.9 mm Mg2+ in the presence of 100 μm Ca2+ (C). Note that G86R GCAP1 gains higher affinity for the cyclase in the Mg2+ form (B) than in the Ca2+ form (C), and the respective changes in KGCAP* versus KGCAPCa can account for nearly 2-fold increase in [Ca]1/2, see Equation I. The data (mean ± S.D.) averaged from 3 to 4 independent measurements were fitted using a sigmoidal function, (Amax − Amin)/(1 + ([GCAP]/KGCAPCa)−H) + Amin, where Amax and Amin are the respective maximal and minimal activity, H, Hill coefficient (between 0.9 and 1.3 in all cases). Inset in C, the data for WT GCAP1 at 100 μm Ca2+ shown on an expanded scale.
Activator-to-inhibitor transition in G86R GCAP1 requires higher Ca2+ concentrations
The dependence of the intrinsic GCAP1 Trp fluorescence on Ca2+ (Fig. 5) is biphasic: first reduction and then increase of intensity, reflecting GCAP1 transition from a metal-free to a metal-bound state (49–51). The “phase I” (decrease in fluorescence) reflects GCAP1 conversion to a partially Ca2+- or Mg2+-liganded “cyclase-activator” state, and “phase II” is the increase specifically in Trp94 fluorescence, caused by Ca2+ binding in EF-hand 4, which converts GCAP1 to its “cyclase-inhibitor” state (50, 51). Phase I in the absence of Mg2+ is less pronounced in human GCAP1 compared with the bovine GCAP1 (50, 51), because although both have Trp21 and Trp94, the human ortholog lacks Trp51 contributing to the phase I amplitude. However, the two Ca2+-dependent phases of the fluorescence remained clearly identifiable in the human GCAP1. In G86R GCAP1, Ca2+-sensitivity of the fluorescence spectrum was markedly shifted toward higher range. The Ca2+-dependent phase II can be further isolated by eliminating phase I by first saturating GCAP1 with Mg2+ (50, 51). The resultant phase II (Fig. 5B) in G86R GCAP1 demonstrated a shift toward higher Ca2+ concentrations and drastic reduction of the amplitude at saturating Ca2+ compared with the WT. The Ca2+-specific increase in fluorescence intensity at 200 μm Ca2+ versus 0 μm Ca2+ was lacking, 1.04-fold ± 0.02 (S.D.), compared with the WT (1.51 ± 0.09, n = 3, p = 0.0074, Student's t test), thus indicating that the activator-to-inhibitor conformational transition was restrained even at saturating free Ca2+.
Figure 5.

The G86R substitution affects Ca2+-dependent activator-to-inhibitor transition in GCAP1. A, the Trp fluorescence of purified recombinant human WT (○) or G86R (●) GCAP1 measured in the absence of Mg2+. Inset, SDS-PAGE of purified recombinant WT and G86R human GCAP1 used for the analysis; Coomassie stain. B, Trp fluorescence of GCAP1 measured in the presence of 10 mm free Mg2+ to isolate phase II (51). The data (mean ± S.D., three independent measurements) in each case were normalized per fluorescence intensity at low Ca2+; empirical fit. Note that in G86R GCAP1, the Ca2+ dependence of fluorescence is shifted toward a higher range and the amplitude of the phase II is suppressed.
To further isolate the phase II fluorescence component of the Ca2+-dependent activator-to-inhibitor transition in a human G86R GCAP1, phase I was eliminated using a W21F substitution (51). The remaining single Trp94 fluorescence in human GCAP1 was measured in the absence of Mg2+ (Fig. 6A), in the presence of near-physiological 0.9 mm free Mg2+ (52) (Fig. 6B) or at saturating 9 mm free Mg2+ (Fig. 6C). In all cases, the fold-increase of Ca2+-dependent Trp94 fluorescence of G86R GCAP1, defining its transition to the state decelerating RetGC1 activity, became reduced: 1.50 ± 0.018 in W21F/G86R versus 2.30 ± 0.13 in W21F alone (p = 0.0052, Student's t test), 1.71 ± 0.04 versus 2.68 ± 0.13 (p = 0.01), and 1.85 ± 0.036 versus 3.13 ± 0.055 (p < 0.0001), respectively. It also required higher free Ca2+ concentrations for the transition to occur (Fig. 6, D–F): the respective EC50 values were 0.85 ± 0.065 μm versus 0.050 ± 0.003 μm (p = 0.0023), 0.713 ± 0.03 μm versus 0.133 ± 0.006 μm (p = 0.0009), and 1.53 ± 0.2 μm versus 0.72 ± 0.05 μm (p = 0.011). The difference in the Ca2+-dependent transition to the cyclase-inhibitor state between the W21F mutants in G86R versus WT GCAP1 backbone (Fig. 6, D–F) was also more pronounced in the absence or at low physiological (52) concentrations of Mg2+. The respective [Ca]1/2 ratios (Fig. 6G) at 0, 0.9, and 9 mm Mg2+ were 17.7 ± 1.47, 5.36 ± 0.28, and 2.14 ± 0.27 (p < 0.0001 for all pairs), indicating that the GCAP1 affinity for Mg2+ in the EF-hand 4 also became reduced as a result of the mutation in the hinge region.
Figure 6.

Trp fluorescence of W21F (○) and W21F/G86R (●, red) human GCAP1. A–C, the phase I was eliminated using a W21F substitution (51), and the Ca2+-dependent Trp94 fluorescence was measured in the absence of Mg2+ (A), in the presence of physiological 0.9 mm free Mg2+ (B) or at saturating 9 mm free Mg2+ (C). The fluorescence intensity (mean ± S.D., three independent measurements) was normalized per that at low Ca2+. D-F, the respective fractional changes in fluorescence, (F − Fmin)/(Fmax − Fmin), from panels A–C were fitted using a sigmoidal function, 1/(1 + ([Ca]1/2/[Ca])H, where Fmax and Fmin are the respective maximal and minimal fluorescence, [Ca] 1/2 is a free Ca2+ concentration at half-maximal effect, and H is a Hill coefficient. G, the [Ca]1/2 W21F/G86R: [Ca]1/2 W21F ratio is much larger at the lower concentrations of Mg2+, indicating that the G86R affinity for Mg2+ binding affecting Trp94 exposure is also reduced compared with the WT. Inset in panel D, SDS-PAGE of purified recombinant GCAP1, left to right, W21F GCAP1, W21F/G86R GCAP1, molecular mass markers; Coomassie stain.
G86R mutation alters the thermodynamics of the Ca2+-dependent conformational changes in GCAP1
Changes of the heat release by GCAP1 upon metal binding reflect a superposition of several processes including not only direct interaction of metal and protein, but conformational changes and reorganization of the protein-water shell as well (39, 53, 54) (Fig. 7). Therefore, the parameters for metal binding formally extracted from the ITC measurements describe the apparent metal affinity, but they do not match exactly those revealed by the intrinsic tryptophan fluorescence or RetGC1 regulation sensitivity (53). However, despite this limitation the comparison of the formal parameters extracted from ITC still reflects the change in the GCAP1 thermodynamics imparted by mutations (39, 53–54). In case of G86R in the presence of 1 mm Mg2+ (Fig. 7), the net heat release pattern in response to binding Ca2+ changed from triphasic to biphasic and the endothermic component was more pronounced than in WT (Fig. 7, A and B). However, best fit of the data were obtained with a sequential binding model resulting in two apparent KD values for the WT and the G86R mutant (Fig. 7, A and B, and Table 1). Due to residual free Ca2+ (90–200 nm; see “Experimental procedures”) in the presence of 20–30 μm GCAP1 variants, we assume partial or full saturation of a high affinity Ca2+-binding site in human GCAP1. The apparent affinity of Ca2+-binding was, however, different and became less in case of G86R (see values for KD1 and KD2 in Table 1). Parameters for Ca2+ binding to GCAP1 variants in the absence of Mg2+ exhibited a similar overall pattern (Table 1). In addition, G86R GCAP1 was more sensitive to the metal binding in Mg2+-dependent heat release tested by ITC (Fig. 7, C and D). Enthalpy changes were endothermic in both cases and best fit was obtained with a two-site model indicating conformational changes caused primarily by Mg2+ binding in two EF-hands, occurring in a noncooperative manner.
Figure 7.

ITC measurements of heat release upon metal binding to hGCAP1 and the G86R mutant. A and B, titration with Ca2+ of human WT GCAP1 (A) and G86R mutant (B) in the presence of 1 mm MgCl2 showed large exothermic responses, but in the case of the mutant small endothermic heat pulses were also observed. The upper part shows the heat pulse for every injection, the lower part shows the corresponding normalized integration data in terms of kcal/mol of injectant plotted against the molar ratio. Data analysis by curve fitting to two Ca2+-binding sites yielded apparent dissociations constants with deviation from the best fit of 0.06 ± 0.02 and 1.47 ± 0.25 μm (WT) and 0.5 ± 0.1 and 15.9 ± 2.1 μm (G86R). Titration was with Mg2+– of Ca2+–free WT hGCAP1 (C) and the G86R mutant (D). Fitting results (two-site model) were 7.41 ± 2.48 and 111 ± 2.07 μm for WT and 0.94 ± 0.8 and 14 ± 0.77 μm for the G86R mutant; representative examples of the titration series are shown.
Table 1.
Thermodynamic parameter of Ca2+ and Mg2+ binding to human WT GCAP1 and the mutant G86R derived from ITC measurements
Ca2+ titrations were done in the absence or presence of 1 mm Mg2+ (where indicated in parentheses). Apparent dissociation constants (mean ± S.D., n = 3) were determined from the indicated fitting models.
| GCAP1 form | Apparent dissociation constant, KD |
Enthalpy change, ΔH |
||
|---|---|---|---|---|
| KD1 | KD2 | ΔH1 | ΔH2 | |
| μm | kcal/mol | |||
| Two-site model (Ca2+ titration) | ||||
| WT | 0.16 ± 0.05 | 0.45 ± 0.07 | −17 ± 2 | −2.4 ± 0.8 |
| G86R | 2.5 ± 0.7 | 56 ± 28 | 12.5 ± 3.5 | −6.6 ± 0.4 |
| WT (Mg2+) | 0.07 ± 0.03 | 1.45 ± 0.01 | −12 ± 0.5 | −4 ± 0.05 |
| G86R (Mg2+) | 0.8 ± 0.4 | 19 ± 3.5 | −7.3 ± 0.15 | 1.5 ± 0.9 |
| Two-site model (Mg2+-titration) | ||||
| WT | 120 ± 11.3 | 7.72 ± 1.49 | 0.078 ± 0.006 | 0.43 ± 0.05 |
| G86R | 74.5 ± 0.7 | 1.75 ± 0.77 | 0.1 ± 0.02 | 1.2 ± 0.2 |
Sensitivity of RetGC1–GCAP complex to inhibition by RD3
RD3 (retinal degeneration 3) protein inhibits both RetGCs basal activity and GCAP-stimulated activity (55, 56). The ability of RD3 to suppress activation of RetGC1 is likely required for normal survival of photoreceptors, because rods and cones lacking RD3 degenerate much faster and more severely than those completely lacking both RetGC1 and RetGC2 isozymes, altogether (56). We found that activation of the cyclase by G86R GCAP1 becomes more resistant to inhibition by RD3 than the cyclase activated by WT RD3 (Fig. 8). The EC50 rose to 17.1 ± 2.8 nm, n = 3, versus 1.7 ± 0.4 nm, n = 3, in WT (p = 0.0007).
Figure 8.

Inhibition of the RetGC1–G86R GCAP complex activity by RD3 becomes less efficient. A, dose dependence of RD3-dependent inhibition. The activity of RetGC1 stimulated by 1.5 μm WT (○) or G86R (●) human GCAP1 was assayed in the presence of 2 mm EGTA, 1 mm free Mg2+, and varying concentrations of recombinant human RD3; the data (mean ± S.D., n = 3) were fitted using a Hill function, (Amax − Amin)/(1 + ([RD3]/[RD3]1/2)H), where Amax and Amin are the respective maximal and minimal activity, [RD3], concentration of RD3 in assay, [RD3]1/2, concentration of RD3 causing 50% inhibition, and H, Hill coefficient. B, the fractional inhibition of the RetGC1–GCAP1 complex by RD3. The data from A were normalized per maximal activity of the cyclase in each case. Note a nearly 10-fold, increase in [RD3]1/2 for G86R GCAP1. The overall higher activity of the cyclase in the presence of the G86R GCAP1 at the 1.5 μm GCAP1 is due to the higher than WT affinity of the mutant for the cyclase (see Fig. 4).
Discussion
Clinical features of the disease caused by G86R GCAP1
The dominant retinopathy caused by G86R substitution presents a new example of a photoreceptor dystrophy linked to the mutations in GCAP1 (Fig. 1 and Fig. S1). The clinical disease feature in common with previous studies of families with GUCA1A mutations coding for GCAP1 (28–38) has been maculopathy with central retinal defects documented by functional and structural parameters (reviewed in Refs. 31 and 32). In addition to central cone disease, some patients in the previous studies also had cone photoreceptor dysfunction in extracentral retina and others have had cone as well as rod dysfunction. Of interest, a postmortem donor retina from a 75-year-old patient clinically diagnosed as having a cone degeneration by ERG criteria at age 45 years has been studied by histopathology; and the disease cause was a GUCA1A mutation (36, 57). As in the present study, there was central retinal atrophy, widespread cone disease by cone ERG, and, despite a normal full-field rod ERG, rod inner and outer segment abnormalities beyond the macula. The clinical spectrum due to GUCA1A mutations thus includes macular degeneration, and retina-wide cone and rod dysfunction where cone disease may be equal to or greater than rod disease.
Molecular mechanisms of retinopathies caused by GCAP mutations
The majority of the known examples of retinopathies linked to the GUCA1A gene originate from mutations in GCAP1 EF-hand motifs that disable Ca2+ binding in EF-hands 3 and 4 (28–40, 58, 59). Reduction of Ca2+ affinity in these EF-hands leads to overly active production of cGMP at normal free Ca2+ concentrations in the dark, increasing the fraction of the open cGMP-gated channels and accelerating the influx of Ca2+ into photoreceptor outer segment (17, 18, 41). The overall Ca2+ sensitivity of GCAP1 imparted by the intrinsic ability of their EF-hands to bind Ca2+ or Mg2+ is additionally fine-tuned by a “calcium-myristoyl tug” (1, 43), a structural link between the C-proximal EF-hand 4 and the N-terminal myristoyl residue embedded in the semi-globule formed by the N-proximal EF-hands 1 and 2 (Fig. 2). Leu176 replacement by Phe, through the altered tug action, increases GCAP1 affinity for RetGC1 but also reduces the affinity of the EF-hand 4 for Ca2+ and thus shifts Ca2+ sensitivity of RetGC regulation outside the physiological range (43). The L176F substitution, originally used to change the calcium-myristoyl tug in GCAP1 in vitro (43), was soon after found in patients with a photoreceptor dystrophy (44), likely triggered by elevated cGMP production in the dark.
In contrast to the multiple mutations in GCAP1 affecting RetGC1 sensitivity to Ca2+ causing cone- and cone-rod degenerations, a rare retinopathy accompanied by G157R mutation in GCAP2 (GUCA1B) affects primarily rods (60). The mutant GCAP2 is more prone to accumulating in photoreceptor inner segment (61), but its mechanistic significance in triggering photoreceptor death remains unclear.
Our present results show that not only EF-hands and calcium-myristoyl tug in GCAP1, but the hinge Gly86 located between the two semiglobules formed by EF-hand pairs is critical for defining the normal Ca2+ sensitivity in the whole-molecule structure of GCAP1. A dominant retinopathy (Fig. 1) occurs when the hinge is affected. The two semiglobules in GCAP1 connected by the Gly86 (Fig. 2) can move relative to each other (51, 62). Apparently, this flexibility contributes to interaction with the cyclase and Ca2+ sensitivity of the target enzyme regulation, similarly to the calcium-myristoyl tug (43). Notably, the residues surrounding Gly86 were implicated in forming the complex between GCAP1 and RetGC1 (46). The G86R substitution imparts an abnormally high affinity to the interface for the target enzyme at the expense of making GCAP1 Ca2+ sensitivity abnormally low. The net outcome is a drastic increase of the cyclase activity at the free concentrations of Ca2+ typical for the dark-adapted photoreceptors. Even if the shift in Ca2+ sensitivity of the cyclase caused by the G86R GCAP1 is attenuated by competition with normal GCAP1 and GCAP2 (58), it should inevitably increase the cGMP synthesis rate, open an excessive number of the cGMP-gated channels, and increase the influx of Ca2+ in dark-adapted rods and cones, like other retinal diseases triggered by similar dysregulation of the cyclase (17–18, 41).
Along with the calcium-myristoyl tug in GCAP1 (43), which is functionally and structurally distinct from the calcium-myristoyl switch prototypically observed in recoverin (63, 64), the flexibility of the hinge region in the GCAP1 structure evidently provides the optimal trade-off between the affinity for the target and the sensitivity of its regulation by physiological intracellular Ca2+. EF-hand 1 (Fig. 2) has evolved in GCAPs to become the main part of the target-binding interface, instead of being a metal-binding domain (46, 65). Three other EF-hands contribute differently to the cyclase regulation. EF-hands 2 and 3 bind Mg2+ or Ca2+ to properly present the target-binding interface and thus allow GCAP to dock with the cyclase (66, 67). The pair of the C-proximal EF hands 3 and 4 is the actual Ca2+ sensor part of the molecule that enables the GCAP switch between the Mg2+-bound state in the light to the Ca2+-bound state in the dark and cause the activator-to-inhibitor transition of GCAP1 (51, 53, 66, 67). This transition is affected by the blinding G86R mutation, such that GCAP1's preferential state now gains higher affinity for RetGC1, is more prone to remain in a Mg2+-bound state and less susceptible to undergo the transition to the Ca2+-bound inhibitor state (Figs. 3–7).
Notably, the overall susceptibility of G86R GCAP1 to converting into a Mg2+-liganded state in the ITC experiments increases (Fig. 7), in contrast to the Mg2+ effect on the intrinsic Trp94 fluorescence reflecting GCAP1 transition upon Mg2+ to Ca2+ replacement in EF-hand 4 (Fig. 6), which is more drastically affected at normal physiological Mg2+ than at saturating Mg2+ concentrations. This indicates that in EF-hand 4, responsible for the Ca2+-dependent fluorescence increase in Trp94 in the neighboring EF-hand α-helix (51), the Mg2+ binding affinity is reduced, whereas in the EF-hand 2 and/or -3 it becomes strengthen. The Mg2+ binding in EF-2 and EF-3 is essential for the overall ability of GCAP1 to dock with the cyclase (67) and less critical for the GCAP1 switching between the activator and the inhibitor states, primarily driven by Ca2+ binding in EF-4 (51, 66). The current ITC experiments are unable to identify the EF-hand(s) that stronger binds Mg2+ in G86R GCAP1, but the lack of inhibition of the cyclase at the normal physiological Ca2+ concentrations and the Trp94 fluorescence spectra argue that EF-hand 4 likely has a reduced affinity for both metals, consistent with the ITC titration detecting only two submillimolar-affinity Mg2+-binding sites in the G86R GCAP1. Physiological concentrations of Mg2+ in the photoreceptor outer segment are near 1 mm (52), so the increase of Mg2+ affinity in EF-hands other than EF-4 would unlikely affect RetGC regulation by G86R GCAP1, because their affinities even in WT are already high enough to maintain GCAP1 in the activator state. In contrast, the decrease in metal affinity of EF-4 is critical, preventing the proper activator-to-inhibitor transition in the dark.
Increased cGMP production in the dark is detrimental for photoreceptor viability. A body of evidence from in vivo studies using transgenic animal models also demonstrate that dysregulation of the negative Ca2+ feedback on RetGC, either via altering the Ca2+-sensor properties of GCAPs or via preferentially increasing the affinity of the cyclase for the activator form of GCAP1 lead to severe retinal pathology (17, 18, 41, 80). Our present study argues that triggering a blinding disease by GCAP1 can result by affecting its Ca2+ sensitivity through a mechanistic reason different from directly disabling an EF-hand or calcium-myristoyl tug in GCAP1. This adds a new distinct example to the array of diverse molecular mechanisms through which retinal guanylyl cyclase can become dysregulated by GCAP1 and cause dominant retinopathy.
Competition of the G86R GCAP1 with RD3 becomes more effective
RD3 protein (68, 69) is required for effective accumulation of RetGC in the outer segment (70, 71) and also inhibits RetGC activation by GCAPs (55–56). The inhibitory function of RD3, which likely occurs in the inner segment (71), is essential for the survival of photoreceptors, because rods and cones lacking the RD3-dependent inhibition of RetGC degenerate much faster and more severely than those completely lacking RetGC itself (56). The cyclase activated by G86R GCAP1 resists the inhibition by RD3 more effectively (Fig. 8), because the mutant GCAP1 now has higher affinity for the cyclase (Fig. 3). A similar effect was previously observed in the case of CORD6-linked mutation in Arg838 of RetGC1, which increases RetGC affinity for GCAP1 (18, 80). According to a hypothesis that is currently under investigation, inhibition of RetGC1 by RD3 is required for preventing premature activation of the cyclase by GCAPs, whereas in transit from the inner segment (55, 56, 69). It is conceivable that the G86R GCAP1 outcompetes RD3 and therefore prematurely activates the cyclase while in the photoreceptor inner segment. This, in addition to the abnormal cGMP production in the outer segment, would lead to aberrant cGMP production in the inner segment and aggravate the progression of the G86R GCAP1-produced retinopathy.
Experimental procedures
Clinical studies
Human studies
A family with a multigeneration history of visual loss suggesting an autosomal dominant inheritance was studied. The proband was examined with clinical, electrophysiological, psychophysical, and imaging tests, and records were obtained from other affected members. Informed consent was obtained and procedures followed the Declaration of Helsinki guidelines and were approved by the institutional review board.
Electroretinography (ERG)
Rod, mixed rod-cone, and cone full-field ERGs were performed according to published protocols (72). In brief, bipolar Burian-Allen contact lens electrodes were used with an Espion system (Diagnosys, Lowell, MA). Under dark-adapted conditions, rod photoreceptor driven function was probed with dim blue −1.6 log scot-cd s m−2 flashes, and combined output of rod and cone photoreceptors was probed with white +1.2 log scot-cd s m−2 flashes. Under light-adapted conditions, cone photoreceptor function was isolated with white 0.8 log phot-cd s m−2 flashes at stimulation rates of 1 and 30 Hz using white adapting backgrounds of 1.5 and 0.8 log phot-cd m−2, respectively.
Psychophysical testing
Kinetic visual fields were performed with a Goldmann perimeter using two target sizes (V-4e, 1.72° and I-4e, 0.11°).
Imaging
En face images were obtained using a confocal scanning laser ophthalmoscope (Spectralis HRA, Heidelberg Engineering, Heidelberg, Germany) to determine RPE integrity. Near-IR reduced-illuminance autofluorescence images (NIR-RAFI) and short-wavelength reduced-illuminance autofluorescence imaging (SW-RAFI) were acquired using methods previously reported (73, 74). For NIR-RAFI acquisition, excitation was with 790-nm NIR light (100% laser power) and the detector sensitivity was held invariant at 105%; for SW-RAFI, excitation at 488 nm was used at 25% laser power and 105% detector sensitivity. The automatic normalization feature was turned off and imaging was obtained using the automatic real time feature, which averages multiple frames to improve the signal to noise ratio. Cross-sectional retinal imaging was performed with optical coherence tomography (RTVue-100; Optovue Inc., Fremont, CA, and ultrahigh resolution SDOCT Bi-μ; Kowa Company, Ltd., Tokyo, Japan). Overlapping horizontal line scans were used to create a profile along the horizontal meridian covering eccentricities up to 30° in temporal and nasal directions. Segmentation analysis was performed using custom programs (Matlab 9.1; MathWork, Natick, MA) based on signal feature assignments as previously published (75).
GCAP1 expression and purification
Two orthologs of myristoylated GCAP1 for biochemical and Trp fluorescence analysis, a bovine (D6S variant) and a human (E6S variant), were expressed from pET11d vector (Novagen/Calbiochem) in a BL21(DE3)pLysS Escherichia coli strain (Novagen/Calbiochem) harboring a pBB131 plasmid coding for yeast N-myristoyltransferase and purified using previously published procedures (51) modified as follows. Cells were typically grown in 2.0 liters of a standard LB medium (Thermo Fisher Scientific) containing 50 μg/ml of kanamycin and 100 μg/ml of ampicillin to reach A600 0.6–0.7. Free myristic acid (Sigma) was added from a concentrated ethanol solution to the suspension of bacterial cells to a final concentration of 100 μg/ml, 30 min prior to the induction with 0.5 mm isopropyl β-d-thiogalactopyranoside (Research Products International). Three hours after the induction, the bacterial pellet was harvested by centrifugation at 8,000 × g for 20 min at 4 °C and frozen in −70 °C. The thawed pellet was resuspended in 100 ml of 10 mm Tris-HCl (pH 7.5) containing 2 mm EGTA and 14 mm 2-mercaptoethanol, and the cells were disrupted by ultrasonication. The expressed GCAP1 in the insoluble fraction of the inclusion bodies was collected by centrifugation at 20,000 × g for 20 min, 4 °C, extracted from the pellet by homogenization in 30 mm Tris-HCl (pH 7.5) containing 2 mm EGTA, 14 mm 2-mercaptoethanol, 2 mm MgCl2, and 8 m Sigma Ultra urea for 30 min at 4 °C, and first dialyzed at 4 °C for 3–4 h against 2.0 liters of 10 mm Tris-HCl buffer (pH 7.5) containing 0.5 mm EGTA, 2 mm MgCl2, and 14 mm 2-mercaptoethanol, and then overnight against 2.0 liters of 10 mm Tris-HCl buffer (pH 7.5) containing 0.1 mm EGTA, 2 mm MgCl2, and 14 mm 2-mercaptoethanol. The insoluble material was removed by centrifugation at 20,000 × g for 20 min, 4 °C. The concentration of Tris-HCl buffer (pH 7.5) in the supernatant was adjusted to 50 mm and CaCl2 was added to a final concentration of 10 mm and kept for 20 min at room temperature. The precipitate was removed by centrifugation at 20,000 × g for 20 min, 4 °C. Supernatant was collected and, after adding NaCl to 1 m and DTT to 5 mm, applied on a 1.6 × 5.0-cm butyl-Sepharose Fast Flow column (GE Healthcare) pre-equilibrated with 20 mm Tris-HCl (pH 7.5) containing 1.0 m NaCl. The column was washed with ∼10 volumes of the same buffer and GCAP1 was eluted with 5 mm Tris-HCl (pH 7.5) and concentrated to 5 ml using a Amicon Ultra-15 (10,000 MWCO) centrifugal filter (Thermo Fisher Scientific). Concentrated solution was centrifuged at 200,000 × g for 10 min, 4 °C, in a Beckman Optima TLX centrifuge and chromatographed on a GE Healthcare Sephacryl S-100 column (2.6 × 60 cm) pre-equilibrated with 20 mm Tris-HCl (pH 7.5), 100 mm NaCl. The main peak containing GCAP1 was collected and EDTA was added to 2 mm to remove Ca2+ bound to GCAP1. We observed that using Chelex resin was not sufficient to remove all bound Ca2+ from GCAP1, whereas preincubation with EDTA yielded metal-free GCAP1. The excess EDTA was then removed by 4 cycles of 20-fold concentration/dilution in 10 mm Tris-HCl (pH 7.5) containing 30 μm EDTA using Amicon Ultra-15 (10,000 MWCO) centrifugal filter. We observed that even in solutions prepared using reagents containing <5 ppb Ca2+ and 18 megohms cm water, Ca2+ contaminations could reach as high as 1 μm. Because GCAP1 has high affinity for Ca2+, even these low levels of contamination are sufficient to skew measurement of the Ca2+ binding. Hence, the 30 μm EDTA was used to prevent GCAP1 from rebinding Ca2+ from the solutions during the concentration/dilution cycles. The final concentration of GCAP1 in stock solution was typically 300–350 μm (>10-fold higher than that of EDTA), and the corrections for the presence of the trace amounts of EDTA were made in all subsequent experiments. Concentrated protein was frozen in small aliquots and stored at −70 °C. The purity of GCAP1 preparations estimated by SDS gel electrophoresis was ≥95%.
RetGC1 expression and activity assay
Human recombinant RetGC1 was expressed from a modified Invitrogen pRCCMV vector in HEK293 cells transfected using a calcium-phosphate precipitation method and the membrane fraction containing the expressed cyclase was purified as previously described (48). The guanylyl cyclase activity was assayed as previously described in detail (48, 56). Briefly, the assay mixture (25 μl) containing HEK293 membranes, 30 mm MOPS-KOH (pH 7.2), 60 mm KCl, 4 mm NaCl, 1 mm DTT, 2 mm Ca2+/Mg2+/EGTA buffers, 0.9 mm free Mg2+, 0.3 mm ATP, 4 mm cGMP, 1 mm GTP, and 1 μCi of [α-32P]GTP, 100 μm zaprinast and dipyridamole, and 10 mm creatine phosphate, 0.5 unit of creatine phosphokinase (Sigma) was incubated at 30 °C for 30 min and the reaction was stopped by heat inactivation at 95 °C for 2 min. The resultant [32P]cGMP product was separated by TLC using fluorescently-backed polyethyleneimine-cellulose plates (Merck) developed in 0.2 m LiCl, eluted with 2 m LiCl, and the radioactivity was counted using liquid scintillation. Ca2+/EGTA buffers maintaining variable-free Ca2+ concentrations at 0.9 mm physiological for the photoreceptors (52) free Mg2+ were prepared using Tsien and Pozzan method (76) and verified by fluorescent indicator dyes as previously described in detail (51). Data fit and statistical analysis (Student's t test) was performed using Synergy Kaleidagraph software.
Protein fluorescence spectroscopy
The intrinsic Trp fluorescence of GCAP1 was assayed as previously described in detail (51). In brief, a 332-nm Trp fluorescence emission intensity of GCAP1 in solution containing 100 mm MOPS/KOH (pH 7.2), 40 mm KCl, 1 mm EGTA, and specified concentrations of MgCl2 was recorded at 23 °C (λex = 290 nm). Small aliquots of concentrated CaCl2 were added to obtain the required free Ca2+ concentrations calculated according to the method of Brooks and Stoney (78), utilizing the algorithm of Marks and Maxfield (79). Data fit and statistical analysis (Student's t test) was performed using Synergy Kaleidagraph software.
GCAP1 mutagenesis
The mutations were introduced into GCAP1 cDNA by PCR following conventional “splicing-by-overlap extension” procedure utilizing a high-fidelity Thermo Scientific PhusionFlash polymerase. The mutated cDNA was inserted into the NcoI/BamHI sites of a pET11d vector, downstream from T7 promoter, as described previously (46) and verified by automated Sanger sequencing.
RD3 expression and purification
Recombinant human RD3 was expressed from a pET11d vector in a BL21(DE3)Codon Plus E. coli strain (Stratagene/Agilent Technologies) induced by isopropyl β-d-thiogalactopyranoside, extracted from the inclusion bodies and purified by salt precipitation and dialysis as previously described in detail (55, 56).
ITC experiments
ITC experiments with hGCAP1 and G86R mutant were performed on a VP-ITC from MicroCal (Northhampton, MA). Briefly, purified myristoylated calcium-free (no EGTA) WT GCAP1 and G86R mutant were present in the recording cell in titration buffer (20 mm Hepes, pH 7.4, 60 mm KCl, 4 mm NaCl) at 24 μm and were titrated with 5 μl of 0.5 mm CaCl2 stock solution at T = 25 °C (50 injections, each 5-μl). The titration buffer was decalcified using a self-packed gravity flow Chelex 100 column (Bio-Rad). The remaining Ca2+ concentration was determined by a BAPTA absorbance assay and was found to range between 90 nm in the presence of 24 μm WT and 200 nm in the presence of 30 μm G86R mutant (no EGTA used). All buffers were filtered (0.22 μm) and degassed twice immediately before use. Three independent repetitions were made for each titration set. Protein samples for each repetition were obtained from two to three separate expressions and three separate purification performances. Reference injections of Ca2+ into decalcified buffer were performed without any protein, and the reference was subtracted in each experiment. Each titration was analyzed by applying a model implemented in the Origin software (MicroCal) assuming two Ca2+-binding sites, or two Mg2+-binding sites. The best fitting results were used to obtain dissociation constants KD,app and enthalpy changes (ΔH).
Author contributions
I. V. P., A. V. C., A. Sumaroka, E. V. O., A. Scholten, K.-W. K., and S. G. J. data curation; I. V. P., A. V. C., K.-W. K., S. G. J., and A. M. D. formal analysis; I. V. P., A. V. C., A. Sumaroka, E. V. O., A. Scholten, S. A., K.-W. K., S. G. J., and A. M. D. investigation; I. V. P., A. V. C., and S. G. J. methodology; A. V. C., K.-W. K., S. G. J., and A. M. D. writing-original draft; K.-W. K., S. G. J., and A. M. D. conceptualization; K.-W. K., S. G. J., and A. M. D. funding acquisition; S. G. J. and A. M. D. supervision.
Supplementary Material
This work was supported by National Institutes of Health Grant EY11522 (to A. M. D.), grants from the Macula Vision Research Foundation (to S. G. J., A. V. C.), Pennsylvania Department of Health (to A. M. D.), Deutsche Forschungsgemeinschaft Grant GRK1885 (to S. A., K. W. K., A. S.), and a fellowship from the German Academic Exchange Program (to S. A.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Results and Fig. S1.
- GCAP
- guanylyl cyclase-activating protein
- ERG
- electroretinography
- ITC
- isothermal titration calorimetry
- NIR-RAFI
- near-infrared excited reduced-illuminance autofluorescence imaging
- OCT
- optical coherence tomography
- RD3
- retinal degeneration 3 protein
- RetGC
- retinal membrane guanylyl cyclase
- RPE
- retinal pigment epithelium
- BAPTA
- 1,2-bis(2-aminophenoxyl)ethane-N,N,N′,N′-tetraacetic acid
- ONL
- outer nuclear layer.
References
- 1. Lim S., Dizhoor A. M., and Ames J. B. (2014) Structural diversity of neuronal calcium sensor proteins and insights for activation of retinal guanylyl cyclase by GCAP1. Front. Mol. Neurosci. 7, 19 10.3389/fnmol.2014.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dizhoor A. M., Olshevskaya E. V., and Peshenko I. V. (2010) Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol. Cell. Biochem. 334, 117–124 10.1007/s11010-009-0328-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palczewski K., Subbaraya I., Gorczyca W. A., Helekar B. S., Ruiz C. C., Ohguro H., Huang J., Zhao X., Crabb J. W., Johnson R. S., Walsh K. A., Gray-Keller M. P., Detwiller P. B., and Baehr W. (1994) Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron 13, 395–404 10.1016/0896-6273(94)90355-7 [DOI] [PubMed] [Google Scholar]
- 4. Dizhoor A. M., Olshevskaya E. V., Henzel W. J., Wong S. C., Stults J. T., Ankoudinova I., and Hurley J. B. (1995) Cloning, sequencing, and expression of a 24-kDa Ca2+-binding protein activating photoreceptor guanylyl cyclase. J. Biol. Chem. 270, 25200–25206 10.1074/jbc.270.42.25200 [DOI] [PubMed] [Google Scholar]
- 5. Imanishi Y., Li N., Sokal I., Sowa M. E., Lichtarge O., Wensel T. G., Saperstein D. A., Baehr W., and Palczewski K. (2002) Characterization of retinal guanylate cyclase-activating protein 3 (GCAP3) from zebrafish to man. Eur. J. Neurosci. 15, 63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Imanishi Y., Yang L., Sokal I., Filipek S., Palczewski K., and Baehr W. (2004) Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish, characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1–8) in pufferfish (Fugu rubripes). J. Mol. Evol. 59, 204–217 10.1007/s00239-004-2614-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mendez A., Burns M. E., Sokal I., Dizhoor A. M., Baehr W., Palczewski K., Baylor D. A., and Chen J. (2001) Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 98, 9948–9953 10.1073/pnas.171308998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Makino C. L., Wen X. H., Olshevskaya E. V., Peshenko I. V., Savchenko A. B., and Dizhoor A. M. (2012) Enzymatic relay mechanism stimulates cyclic GMP synthesis in rod photoresponse, biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice. PLoS ONE 7, e47637 10.1371/journal.pone.0047637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sakurai K., Chen J., and Kefalov V. J. (2011) Role of guanylyl cyclase modulation in mouse cone phototransduction. J. Neurosci. 31, 7991–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vinberg F., Peshenko I. V., Chen J., Dizhoor A. M., and Kefalov V. J. (2018) Guanylate cyclase activating protein 2 contributes to phototransduction and light adaptation in mouse cone photoreceptors. J. Biol. Chem. 293, 7457–7465 10.1074/jbc.RA117.001574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Arshavsky V. Y., Lamb T. D., and Pugh E. N. Jr. (2002) G proteins and phototransduction. Annu. Rev. Physiol. 64, 153–187 10.1146/annurev.physiol.64.082701.102229 [DOI] [PubMed] [Google Scholar]
- 12. Koch K.-W., and Dell'Orco D. (2015) Protein and signaling networks in vertebrate photoreceptor cells. Front. Mol. Neurosci. 8, 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dizhoor A. M., Lowe D. G., Olshevskaya E. V., Laura R. P., and Hurley J. B. (1994) The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352 10.1016/0896-6273(94)90449-9 [DOI] [PubMed] [Google Scholar]
- 14. Lowe D. G., Dizhoor A. M., Liu K., Gu Q., Spencer M., Laura R., Lu L., and Hurley J. B. (1995) Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539 10.1073/pnas.92.12.5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang R. B., Foster D. C., Garbers D. L., and Fülle H. J. (1995) Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. U.S.A. 92, 602–606 10.1073/pnas.92.2.602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burns M. E., Mendez A., Chen J., and Baylor D. A. (2002) Dynamics of cyclic GMP synthesis in retinal rods. Neuron 36, 81–91 10.1016/S0896-6273(02)00911-X [DOI] [PubMed] [Google Scholar]
- 17. Woodruff M. L., Olshevskaya E. V., Savchenko A. B., Peshenko I. V., Barrett R., Bush R. A., Sieving P. A., Fain G. L., and Dizhoor A. M. (2007) Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J. Neurosci. 27, 8805–8815 10.1523/JNEUROSCI.2751-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sato S., Peshenko I. V., Olshevskaya E. V., Kefalov V. J., and Dizhoor A. M. (2018) GUCY2D cone-rod dystrophy-6 is a “phototransduction disease” triggered by abnormal calcium feedback on retinal membrane guanylyl cyclase 1. J. Neurosci. 8, 2990–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stone E. M. (2007) Leber congenital amaurosis: a model for efficient genetic testing of heterogeneous disorders, LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 10.1016/j.ajo.2007.08.022 [DOI] [PubMed] [Google Scholar]
- 20. Hunt D. M., Buch P., and Michaelides M. (2010) Guanylate cyclases and associated activator proteins in retinal disease. Mol. Cell. Biochem. 334, 157–168 10.1007/s11010-009-0331-y [DOI] [PubMed] [Google Scholar]
- 21. DelĺOrco D., Behnen P., Linse S., and Koch K.-W. (2010) Calcium binding, structural stability and guanylate cyclase activation in GCAP1 variants associated with human cone dystrophy. Cell Mol. Life Sci. 67, 973–984 10.1007/s00018-009-0243-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharon D., Wimberg H., Kinarty Y., and Koch K.-W. (2018) Genotype-functional-phenotype correlations in photoreceptor guanylate cyclase (GC-E) encoded by GUCY2D. Prog. Retin. Eye Res. 63, 69–91 10.1016/j.preteyeres.2017.10.003 [DOI] [PubMed] [Google Scholar]
- 23. Stunkel M. L., Brodie S. E., Cideciyan A. V., Pfeifer W. L., Kennedy E. L., Stone E. M., Jacobson S. G., and Drack A. V. (2018) Expanded retinal disease spectrum associated with autosomal recessive mutations in GUCY2D. Am. J. Ophthalmol. 190, 58–68 10.1016/j.ajo.2018.03.021 [DOI] [PubMed] [Google Scholar]
- 24. Payne A. M., Morris A. G., Downes S. M., Johnson S., Bird A. C., Moore A. T., Bhattacharya S. S., and Hunt D. M. (2001) Clustering and, frequency of mutations in the retinal guanylate cyclase (GUCY2D) gene in patients with dominant cone-rod dystrophies. J. Med. Genet. 38, 611–614 10.1136/jmg.38.9.611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelsell R. E., Gregory-Evans K., Payne A. M., Perrault I., Kaplan J., Yang R. B., Garbers D. L., Bird A. C., Moore A. T., and Hunt D. M. (1998) Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 7, 1179–1184 10.1093/hmg/7.7.1179 [DOI] [PubMed] [Google Scholar]
- 26. Kitiratschky V. B., Wilke R., Renner A. B., Kellner U., Vadalà M., Birch D. G., Wissinger B., Zrenner E., and Kohl S. (2008) Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest. Ophthalmol. Vis. Sci. 49, 5015–5023 10.1167/iovs.08-1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jacobson S. G., Cideciyan A. V., Peshenko I. V., Sumaroka A., Olshevskaya E. V., Cao L., Schwartz S. B., Roman A. J., Olivares M. B., Sadigh S., Yau K. W., Heon E., Stone E. M., and Dizhoor A. M. (2013) Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum. Mol. Genet. 22, 168–183 10.1093/hmg/dds421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Payne A. M., Downes S. M., Bessant D. A., Taylor R., Holder G. E., Warren M. J., Bird A. C., and Bhattacharya S. S. (1998) A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum. Mol. Genet. 7, 273–277 10.1093/hmg/7.2.273 [DOI] [PubMed] [Google Scholar]
- 29. Wilkie S. E., Li Y., Deery E. C., Newbold R. J., Garibaldi D., Bateman J. B., Zhang H., Lin W., Zack D. J., Bhattacharya S. S., Warren M. J., Hunt D. M., and Zhang K. (2001) Identification and functional consequences of a new mutation (E155G) in the gene for GCAP1 that causes autosomal dominant cone dystrophy. Am. J. Hum. Genet. 69, 471–480 10.1086/323265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jiang L., Katz B. J., Yang Z., Zhao Y., Faulkner N., Hu J., Baird J., Baehr W., Creel D. J., and Zhang K. (2005) Autosomal dominant cone dystrophy caused by a novel mutation in the GCAP1 gene (GUCA1A). Mol. Vis. 20, 143–151 [PubMed] [Google Scholar]
- 31. Jiang L., Wheaton D., Bereta G., Zhang K., Palczewski K., Birch D. G., and Baehr W. A. (2008) A novel GCAP1(N104K) mutation in EF-hand 3 (EF3) linked to autosomal dominant cone dystrophy. Vis. Res. 48, 2425–2432 10.1016/j.visres.2008.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kamenarova K., Corton M., García-Sandoval B., Fernández-San Jose P., Panchev V., Avila-Fernández A., López-Molina M. I., Chakarova C., Ayuso C., and Bhattacharya S. S. (2013) Novel GUCA1A mutations suggesting possible mechanisms of pathogenesis in cone, cone-rod, and macular dystrophy patients. Biomed. Res. Int. 2013, 517570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kitiratschky V. B., Behnen P., Kellner U., Heckenlively J. R., Zrenner E., Jägle H., Kohl S., Wissinger B., and Koch K. W. (2009) Mutations in the GUCA1A gene involved in hereditary cone dystrophies impair calcium-mediated regulation of guanylate cyclase. Hum. Mutat. 30, E782–E796 10.1002/humu.21055 [DOI] [PubMed] [Google Scholar]
- 34. Michaelides M., Wilkie S. E., Jenkins S., Holder G. E., Hunt D. M., Moore A. T., and Webster A. R. (2005) Mutation in the gene GUCA1A, encoding guanylate cyclase-activating protein 1, causes cone, cone-rod, and macular dystrophy. Ophthalmology 112, 1442–1447 10.1016/j.ophtha.2005.02.024 [DOI] [PubMed] [Google Scholar]
- 35. Michaelides M., Hardcastle A. J., Hunt D. M., and Moore A. T. (2006) Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 51, 232–258 10.1016/j.survophthal.2006.02.007 [DOI] [PubMed] [Google Scholar]
- 36. Nishiguchi K. M., Sokal I., Yang L., Roychowdhury N., Palczewski K., Berson E. L., Dryja T. P., and Baehr W. (2004) A novel mutation (I143NT) in guanylate cyclase-activating protein 1 (GCAP1) associated with autosomal dominant cone degeneration. Invest. Ophthalmol. Vis. Sci. 45, 3863–3870 10.1167/iovs.04-0590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nong E., Lee W., Merriam J. E., Allikmets R., and Tsang S. H. (2014) Disease progression in autosomal dominant cone-rod dystrophy caused by a novel mutation (D100G) in the GUCA1A gene. Doc. Ophthalmol. 128, 59–67 10.1007/s10633-013-9420-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sokal I., Dupps W. J., Grassi M. A., Brown J. Jr., Affatigato L. M., Roychowdhury N., Yang L., Filipek S., Palczewski K., Stone E. M., and Baehr W. (2005) A novel GCAP1 missense mutation (L151F) in a large family with autosomal dominant cone-rod dystrophy (adCORD). Invest. Ophthalmol. Vis. Sci. 46, 1124–1132 10.1167/iovs.04-1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sulmann S., DelĺOrco D., Marino V., Behnen P., and Koch K.-W. (2014) Conformational changes in calcium-sensor proteins under molecular crowding conditions. Chemistry 20, 6756–6762 10.1002/chem.201402146 [DOI] [PubMed] [Google Scholar]
- 40. Marino V., Scholten A., Koch K.-W., and Dell'Orco D. (2015) Two retinal dystrophy-associated missense mutations in GUCA1A with distinct molecular properties result in a similar aberrant regulation of the retinal guanylate cyclase. Hum. Mol. Genet. 24, 6653–6666 10.1093/hmg/ddv370 [DOI] [PubMed] [Google Scholar]
- 41. Olshevskaya E. V., Calvert P. D., Woodruff M. L., Peshenko I. V., Savchenko A. B., Makino C. L., Ho Y. S., Fain G. L., and Dizhoor A. M. (2004) The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J. Neurosci. 24, 6078–6085 10.1523/JNEUROSCI.0963-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Olshevskaya E. V., Peshenko I. V., Savchenko A. B., and Dizhoor A. M. (2012) Retinal guanylyl cyclase isozyme 1 is the preferential in vivo target for constitutively active GCAP1 mutants causing congenital degeneration of photoreceptors. J. Neurosci. 32, 7208–7217 10.1523/JNEUROSCI.0976-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peshenko I. V., Olshevskaya E. V., Lim S., Ames J. B., and Dizhoor A. M. (2012) Calcium-myristoyl tug is a new mechanism for intramolecular tuning of calcium sensitivity and target enzyme interaction for guanylyl cyclase-activating protein 1: dynamic connection between N-fatty acyl group and EF-hand controls calcium sensitivity. J. Biol. Chem. 287, 13972–13984 10.1074/jbc.M112.341883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vocke F., Weisschuh N., Marino V., Malfatti S., Jacobson S. G., Reiff C. M., Dell'Orco D., and Koch K.-W. (2017) Dysfunction of cGMP signaling in photoreceptors by a macular dystrophy-related mutation in the calcium sensor GCAP1. Hum. Mol. Genet. 26, 133–144 [DOI] [PubMed] [Google Scholar]
- 45. Jacobson S. G., Voigt W. J., Parel J. M., Apáthy P. P., Nghiem-Phu L., Myers S. W., and Patella V. M. (1986) Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology 93, 1604–16011 10.1016/S0161-6420(86)33522-X [DOI] [PubMed] [Google Scholar]
- 46. Peshenko I. V., Olshevskaya E. V., Lim S., Ames J. B., and Dizhoor A. M. (2014) Identification of target binding site in photoreceptor guanylyl cyclase activating protein 1 (GCAP1). J. Biol. Chem. 289, 10140–10154 10.1074/jbc.M113.540716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Woodruff M. L., Sampath A. P., Matthews H. R., Krasnoperova N. V., Lem J., and Fain G. L. (2002) Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. J. Physiol. 542, 843–854 10.1113/jphysiol.2001.013987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peshenko I. V., Moiseyev G. P., Olshevskaya E. V., and Dizhoor A. M. (2004) Factors that determine Ca2+ sensitivity of photoreceptor guanylyl cyclase: kinetic analysis of the interaction between the Ca2+-bound and the Ca2+-free guanylyl cyclase activating proteins (GCAPs) and recombinant photoreceptor guanylyl cyclase 1 (RetGC-1). Biochemistry 43, 13796–13804 10.1021/bi048943m [DOI] [PubMed] [Google Scholar]
- 49. Sokal I., Otto-Bruc A. E., Surgucheva I., Verlinde C. L., Wang C. K., Baehr W., and Palczewski K. (1999) Conformational changes in guanylyl cyclase-activating protein 1 (GCAP1) and its tryptophan mutants as a function of calcium concentration. J. Biol. Chem. 274, 19829–19837 10.1074/jbc.274.28.19829 [DOI] [PubMed] [Google Scholar]
- 50. Peshenko I. V., and Dizhoor A. M. (2004) Guanylyl cyclase activating proteins (GCAPs) are Ca2+/Mg2+-sensors, implications for photoreceptor guanylyl cyclase (RetGC) regulation in mammalian photoreceptors. J. Biol. Chem. 279, 16903–16906 10.1074/jbc.C400065200 [DOI] [PubMed] [Google Scholar]
- 51. Peshenko I. V., and Dizhoor A. M. (2006) Ca2+ and Mg2+ binding properties of GCAP-1: evidence that Mg2+-bound form is the physiological activator of photoreceptor guanylyl cyclase. J. Biol. Chem. 281, 23830–23841 10.1074/jbc.M600257200 [DOI] [PubMed] [Google Scholar]
- 52. Chen C., Nakatani K., and Koutalos Y. (2003) Free magnesium concentration in salamander photoreceptor outer segments. J. Physiol. 553, 125–135 10.1113/jphysiol.2003.053280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lim S., Peshenko I., Dizhoor A., and Ames J. B. (2009) Effects of Ca2+, Mg2+, and myristoylation on guanylyl cyclase activating protein 1 structure and stability. Biochemistry 48, 850–862 10.1021/bi801897p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Robin J., Brauer J., Sulmann S., Marino V., Dell'Orco D., Lienau C., and Koch K.-W. (2015) Differential nanosecond protein dynamics in homologous calcium sensors. ACS Chem. Biol. 10, 2344–2352 10.1021/acschembio.5b00278 [DOI] [PubMed] [Google Scholar]
- 55. Peshenko I. V., Olshevskaya E. V., Azadi S., Molday L. L., Molday R. S., and Dizhoor A. M. (2011) Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519 10.1021/bi201342b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2016) Functional study and mapping sites for interaction with the target enzyme in retinal degeneration 3 (RD3) protein. J. Biol. Chem. 291, 19713–19723 10.1074/jbc.M116.742288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. To K., Adamian M., Jakobiec F. A., and Berson E. L. (1998) Histopathologic and immunohistochemical study of dominant cone degeneration. Am. J. Ophthalmol. 126, 140–142 10.1016/S0002-9394(98)00085-3 [DOI] [PubMed] [Google Scholar]
- 58. Dizhoor A. M., Boikov S. G., and Olshevskaya E. V. (1998) Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1: possible role in causing human autosomal dominant cone degeneration. J. Biol. Chem. 273, 17311–17314 10.1074/jbc.273.28.17311 [DOI] [PubMed] [Google Scholar]
- 59. Sokal I., Li N., Surgucheva I., Warren M. J., Payne A. M., Bhattacharya S. S., Baehr W., and Palczewski K. (1998) GCAP1 (Y99C) mutant is constitutively active in autosomal dominant cone dystrophy. Mol. Cell 2, 129–133 10.1016/S1097-2765(00)80121-5 [DOI] [PubMed] [Google Scholar]
- 60. Sato M., Nakazawa M., Usui T., Tanimoto N., Abe H., and Ohguro H. (2005) Mutations in the gene coding for guanylate cyclase-activating protein 2 (GUCA1B gene) in patients with autosomal dominant retinal dystrophies. Graefes. Arch. Clin. Exp. Ophthalmol. 243, 235–242 10.1007/s00417-004-1015-7 [DOI] [PubMed] [Google Scholar]
- 61. López-Begines S., Plana-Bonamaisó A., and Méndez A. (2018) Molecular determinants of guanylate cyclase activating protein subcellular distribution in photoreceptor cells of the retina. Sci. Rep. 8, 2903 10.1038/s41598-018-20893-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sokal I., Li N., Klug C. S., Filipek S., Hubbell W. L., Baehr W., and Palczewski K. (2001) Calcium-sensitive regions of GCAP1 as observed by chemical modifications, fluorescence, and EPR spectroscopies. J. Biol. Chem. 276, 43361–43373 10.1074/jbc.M103614200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Burgoyne R. D. (2007) Neuronal calcium sensor proteins, generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193 10.1038/nrn2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ames J. B., Tanaka T., Stryer L., and Ikura M. (1996) Portrait of a myristoyl switch protein. Curr. Opin. Struct. Biol. 6, 432–438 10.1016/S0959-440X(96)80106-0 [DOI] [PubMed] [Google Scholar]
- 65. Ermilov A. N., Olshevskaya E. V., and Dizhoor A. M. (2001) Instead of binding calcium, one of the EF-hand structures in guanylyl cyclase activating protein-2 is required for targeting photoreceptor guanylyl cyclase. J. Biol. Chem. 276, 48143–48148 10.1074/jbc.M107539200 [DOI] [PubMed] [Google Scholar]
- 66. Peshenko I. V., and Dizhoor A. M. (2007) Activation and inhibition of photoreceptor guanylyl cyclase by guanylyl cyclase activating protein 1 (GCAP-1): the functional role of Mg2+/Ca2+ exchange in EF-hand domains. J. Biol. Chem. 282, 21645–21652 10.1074/jbc.M702368200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2008) Binding of guanylyl cyclase activating protein 1 (GCAP1) to retinal guanylyl cyclase (RetGC1): the role of individual EF-hands. J. Biol. Chem. 283, 21747–21757 10.1074/jbc.M801899200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Friedman J. S., Chang B., Kannabiran C., Chakarova C., Singh H. P., Jalali S., Hawes N. L., Branham K., Othman M., Filippova E., Thompson D. A., Webster A. R., Andréasson S., Jacobson S. G., Bhattacharya S. S., Heckenlively J. R., and Swaroop A. (2006) Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070 10.1086/510021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Molday L. L., Jefferies T., and Molday R. S. (2014) Insights into the role of RD3 in guanylate cyclase trafficking, photoreceptor degeneration, and Leber congenital amaurosis. Front. Mol. Neurosci. 7, 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Azadi S., Molday L. L., and Molday R. S. (2010) RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163 10.1073/pnas.1010460107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zulliger R., Naash M. I., Rajala R. V., Molday R. S., and Azadi S. (2015) Impaired association of retinal degeneration-3 with guanylate cyclase-1 and guanylate cyclase-activating protein-1 leads to Leber congenital amaurosis-1. J. Biol. Chem. 290, 3488–3499 10.1074/jbc.M114.616656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jacobson S. G., Yagasaki K., Feuer W. J., and Román A. J. (1989) Interocular asymmetry of visual function in heterozygotes of X-linked retinitis pigmentosa. Exp. Eye Res. 48, 679–691 10.1016/0014-4835(89)90009-2 [DOI] [PubMed] [Google Scholar]
- 73. Cideciyan A. V., Swider M., Aleman T. S., Roman M. I., Sumaroka A., Schwartz S. B., Stone E. M., and Jacobson S. G. (2007) Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 24, 1457–1467 10.1364/JOSAA.24.001457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cideciyan A. V., Swider M., and Jacobson S. G. (2015) Autofluorescence imaging with near-infrared excitation: normalization by reflectance to reduce signal from choroidal fluorophores. Invest. Ophthalmol. Vis. Sci. 56, 3393–3406 10.1167/iovs.15-16726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cideciyan A. V., Hufnagel R. B., Carroll J., Sumaroka A., Luo X., Schwartz S. B., Dubra A., Land M., Michaelides M., Gardner J. C., Hardcastle A. J., Moore A. T., Sisk R. A., Ahmed Z. M., Kohl S., Wissinger B., and Jacobson S. G. (2013) Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum. Gene Ther. 24, 993–1006 10.1089/hum.2013.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tsien R., and Pozzan T. (1989) Measurement of cytosolic free Ca2+ with quin2. Methods Enzymol. 172, 230–262 10.1016/S0076-6879(89)72017-6 [DOI] [PubMed] [Google Scholar]
- 77. Stephen R., Bereta G., Golczak M., Palczewski K., and Sousa M. C. (2007) Stabilizing function for myristoyl group revealed by the crystal structure of a neuronal calcium sensor, guanylate cyclase-activating protein 1. Structure 15, 1392–1402 10.1016/j.str.2007.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Brooks S. P., and Storey K. B. (1992) Bound and determined: a computer program for making buffers of defined ion concentrations. Anal. Biochem. 201, 119–126 10.1016/0003-2697(92)90183-8 [DOI] [PubMed] [Google Scholar]
- 79. Marks P. W., and Maxfield F. R. (1991) Preparation of solutions with free calcium concentration in the nanomolar range using 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid. Anal. Biochem. 193, 61–71 10.1016/0003-2697(91)90044-T [DOI] [PubMed] [Google Scholar]
- 80. Dizhoor A. M., Olshevskaya E. V., and Peshenko I. V. (2016) The R838S mutation in retinal guanylyl cyclase 1 (RetGC1) alters calcium sensitivity of cGMP synthesis in the retina and causes blindness in transgenic mice. J. Biol. Chem. 291, 24504–24516 10.1074/jbc.M116.755553 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.