

All cells are required to resolve the different types of RNA-DNA hybrids that form in vivo. When RNA-DNA hybrids persist, cells experience an increase in mutation rate and problems with DNA replication. Okazaki fragment synthesis on the lagging strand requires an RNA primer to begin synthesis of each fragment. The mechanism of RNA removal from Okazaki fragments remains unknown in bacteria that lack RNase HI. We examined Okazaki fragment processing in vitro and found that RNase HIII in conjunction with DNA polymerase I represent the most efficient repair pathway. We also assessed the contribution of YpcP and found that YpcP is a 5′ to 3′ exonuclease that prefers RNA substrates with activity on Okazaki and flap substrates in vitro.
KEYWORDS: Bacillus subtilis, DNA polymerase I, DNA repair, DNA replication, Okazaki fragment, RNA-DNA hybrid, RNase H, YpcP
ABSTRACT
RNA-DNA hybrids are common in chromosomal DNA. Persistent RNA-DNA hybrids result in replication fork stress, DNA breaks, and neurological disorders in humans. During replication, Okazaki fragment synthesis relies on frequent RNA primer placement, providing one of the most prominent forms of covalent RNA-DNA strands in vivo. The mechanism of Okazaki fragment maturation, which involves RNA removal and subsequent DNA replacement, in bacteria lacking RNase HI remains unclear. In this work, we reconstituted repair of a linear model Okazaki fragment in vitro using purified recombinant enzymes from Bacillus subtilis. We showed that RNase HII and HIII are capable of incision on Okazaki fragments in vitro and that both enzymes show mild stimulation by single-stranded DNA binding protein (SSB). We also showed that RNase HIII and DNA polymerase I provide the primary pathway for Okazaki fragment maturation in vitro. Furthermore, we found that YpcP is a 5′ to 3′ nuclease that can act on a wide variety of RNA- and DNA-containing substrates and exhibits preference for degrading RNA in model Okazaki fragments. Together, our data showed that RNase HIII and DNA polymerase I provide the primary pathway for Okazaki fragment maturation, whereas YpcP also contributes to the removal of RNA from an Okazaki fragment in vitro.
IMPORTANCE All cells are required to resolve the different types of RNA-DNA hybrids that form in vivo. When RNA-DNA hybrids persist, cells experience an increase in mutation rate and problems with DNA replication. Okazaki fragment synthesis on the lagging strand requires an RNA primer to begin synthesis of each fragment. The mechanism of RNA removal from Okazaki fragments remains unknown in bacteria that lack RNase HI. We examined Okazaki fragment processing in vitro and found that RNase HIII in conjunction with DNA polymerase I represent the most efficient repair pathway. We also assessed the contribution of YpcP and found that YpcP is a 5′ to 3′ exonuclease that prefers RNA substrates with activity on Okazaki and flap substrates in vitro.
INTRODUCTION
RNA-DNA hybrids are nucleic acid structures that form when RNA is included in DNA. Throughout the cell cycle, RNA is routinely included in DNA as covalent RNA-DNA chimeras or by base pairing with complementary DNA (for review, see references 1 to 3). In all organisms, RNA-DNA hybrids can exist in at least three prominent forms. The first is an R-loop, which occurs when single-stranded RNA pairs with DNA, displacing the nontemplate strand (4). The second occurs when single ribonucleotides and short polymers of RNA become covalently embedded in DNA during replication by DNA polymerases, which results in an RNA-DNA junction on both sides of the embedded ribonucleotide(s) (5, 6). The third occurs when RNA is used to prime DNA synthesis of Okazaki fragments during lagging-strand replication (7, 8). Okazaki fragment synthesis can be distinguished from embedded ribonucleotides because Okazaki fragments have an RNA-DNA junction on only the 3′ end of the ribonucleotide covalently bonded to DNA (9). It is well established that RNA is far less stable than DNA, leading to a potent source of genome instability when RNA is not removed (5, 10, 11). Moreover, when RNA-DNA hybrids persist, compromised genome integrity can lead to disease in humans and an increase in mutation rate in bacteria and yeast (5, 10, 12–18).
The primary enzymes responsible for recognizing and cleaving RNA-DNA hybrids are of the RNase H family. RNase H enzymes are highly conserved among all three domains of life (for review, see references 19 and 20). Two classes have been described within the RNase H family. Class one is represented by RNase HI and the archaeal hybrid binding domain (21–23). Both hydrolyze RNA-DNA hybrids containing polymers of four or more ribonucleotides hybridized to DNA or as covalent chimeras nested in duplex DNA. Class two consists of RNase HII and prokaryote-specific RNase HIII (20, 24). RNase HII hydrolyzes 5′ to a single ribonucleotide and 5′ to the ribonucleoside monophosphate located at the RNA-DNA junction in polymers of four or more ribonucleotides nested in DNA (25–27). RNase HIII has a biochemical function similar to that of RNase HI (28). It has been suggested that these enzymes are redundant because functional RNase HI and HIII are often mutually exclusive (29, 30). Therefore, bacteria generally have two functional RNase H enzymes, RNase HI and HII or RNase HII and HIII (29).
Single ribonucleotides incorporated in genomic DNA by DNA polymerases are corrected through a process known as ribonucleotide excision repair (RER) (17, 31, 32). Bacterial RER has been reconstituted with enzymes from the Gram-positive bacterium Bacillus subtilis on a linear substrate (11). This process involves hydrolysis 5′ to the ribonucleotide by RNase HII, resulting in a nick that serves as a point for entry and resynthesis by DNA polymerase I (Pol I) (11). Pol I then removes the single ribonucleotide using its 5′ to 3′ exonuclease activity and replaces it with the corresponding deoxyribonucleotide (11). Loss of RNase HII in B. subtilis results in a sequence context-dependent 2-fold increase of G→A transitions specific to the lagging strand (11).
The process of RER shares similarities with Okazaki fragment metabolism, wherein the RNA primers used for replication must be removed and replaced with DNA. Discontinuous lagging-strand replication is initiated by DnaG (primase), a DNA-dependent RNA polymerase that synthesizes 10- to 15-bp RNA primers approximately every 1.5 kbp (7, 8, 33). RNA primers account for the vast majority of RNA covalently bound to DNA and must be subsequently removed and replaced with DNA to complete replication (2). It has been estimated that ∼23,000 ribonucleotides are placed into the Escherichia coli chromosome during each round of replication (1). The human genome is estimated to contain ∼150 million ribonucleotides after one round of replication, most of which originate from synthesis of Okazaki fragments (2). Therefore, Okazaki fragment synthesis is a major source of RNA-DNA hybrids that form during each round of DNA replication.
Eukaryotic Okazaki fragment maturation can be performed by several different proteins in conjunction with FEN1 (for review, see reference 34). A prominent pathway begins when PCNA-bound DNA Pol δ displaces the RNA primer, resulting in a single-stranded flap (9, 31). The flap is then processed by FEN1, and the remaining nick is sealed by DNA ligase I in a process reminiscent of eukaryotic RER (9, 31). There is some evidence that RNase H2 may play a role in this process by digesting the primer before displacement (9, 35).
The maturation of Okazaki fragments in bacteria is not as well understood as Okazaki fragment processing in eukaryotes. Studies of the Gram-negative bacterium E. coli suggest that this process is likely performed by two enzymes, DNA polymerase I and RNase HI (36). Mutants compromised for Pol I contained short nascent fragments with RNA linked to DNA, as determined by alkaline sucrose density sedimentation experiments and by 5′-end labeling of nascent fragments (37). The appearance of shorter nascent DNA fragments with longer primer RNA was exacerbated when mutations in rnhA (RNase HI) and polA (Pol I) were combined (37). Therefore, these data indicate that RNase HI and DNA polymerase I are important for Okazaki fragment maturation in vivo, with Pol I providing the major contribution for RNA primer removal (37).
Many Gram-positive bacteria do not contain RNase HI. Further, how Okazaki fragments are matured in these organisms remains unclear (28). It is also unclear whether RNase HII, RNase HIII, or both contribute to Okazaki fragment maturation in vitro or in vivo. We examined Okazaki fragment maturation using the B. subtilis RNase H enzymes and provide evidence for two separate mechanisms of RNA primer removal and repair. We found that Pol I and RNase HIII function in the repair pathway and that the 5′ to 3′ exonuclease YpcP can serve to remove RNA during Okazaki fragment maturation. Furthermore, our experiments showed that YpcP is most active on RNA in duplex DNA. We also identified a striking cold-sensitive phenotype for cells deficient in RNase HIII and Pol I-dependent or YpcP-dependent 5′ to 3′ exonuclease activity, suggesting that the phenotype is exacerbated by failures in Okazaki fragment maturation.
RESULTS
RNase HII and HIII hydrolyze Okazaki fragments.
To begin to understand the contribution of RNase HII and HIII to the initiation of RNA removal from Okazaki fragments, we tested the ability of RNase HII and RNase HIII to hydrolyze an RNA-DNA hybrid using an RNA-DNA chimera with a 3′ overhang to model a single Okazaki fragment in vitro. Isolated RNase HII and HIII were purified as described previously (11, 30), with purity of the preparations shown via SDS-PAGE (Fig. 1A). To create the RNA-DNA chimera 3′-overhang substrate, a 3′-labeled 20-bp RNA-DNA covalent chimera oligonucleotide (oJR339, 12 bp RNA followed by 8 bp DNA) was annealed to a 90-bp DNA oligonucleotide (oJR343) to create a 70-base single-stranded 3′ DNA overhang (Fig. 1B and C). This substrate was hydrolyzed with NaOH to generate a ladder representing the location of ribonucleotides in the RNA-containing strand. The substrate was incubated with RNase HII and HIII during a time course followed by resolution of the 3′-labeled RNA-DNA chimera products using denaturing urea-PAGE (Fig. 1B).
FIG 1.

RNase HII and HIII process Okazaki fragments in vitro. (A) SDS-PAGE of 2 μg purified SSB, RNase HIII, and RNase HII stained with Coomassie brilliant blue. (B) Denaturing urea-PAGE of a 3′ overhang containing a single Okazaki fragment substrate (oJR339, oJR343) treated with NaOH, RNase HII, and RNase HIII over 0.1 to 10 min with 1 mM MgCl2 and 10 μM MnCl2. (C) Schematic of the model 3′-overhang substrate used in panel B with the cleavage sites for RNase HII and HIII indicated and the predicted melting temperature of each fragment after cleavage shown. Melting temperature was predicted using a tool found at Integrated DNA Technologies (https://www.idtdna.com/site/order/oligoentry/index/rna). (D) Native PAGE of the 3′-overhang single Okazaki fragment substrate from panel C incubated with BSA or SSB. (E) Fold stimulation of RNase HII and RNase HIII cleavage of the substrate shown in panel C on addition of SSB relative to addition of BSA at 4 nM. The squiggly lines indicate the position of RNA, and the smooth lines indicate the position of DNA.
We monitored RNase HII and HIII activities on the Okazaki fragment substrate (described above) in a time course assay at 25°C and found that, although RNase HII was slightly more active during the 10-minute reaction time under the conditions tested here, both HII and HIII were similarly capable of incising a model Okazaki fragment in vitro (Fig. 1B). Furthermore, both enzymes showed activity on the substrate near the RNA-DNA junction consistent with their enzymatic activities (Fig. 1B) (11, 28, 30). The locations of hydrolysis varied between the two enzymes, with RNase HII cleaving exclusively one nucleotide 5′ to the RNA-DNA junction and RNase HIII cleaving in multiple locations two, three, and four nucleotides 5′ to the junction. The melting temperatures of the remaining RNA fragments after RNase H cleavage are indicated in Fig. 1C. The predicted melting temperature of fragments resulting from RNase HIII incision are lower, and differences in cleavage location result in different lengths of RNA fragments. These differences may affect fragment removal by a 5′ to 3′ exonuclease, such as Pol I or YpcP, during the next step of maturation (38).
Previous studies showed that E. coli RNase HI is stimulated by the presence of single-stranded DNA binding protein (SSB) (39). We therefore asked whether SSB stimulated the activity of either B. subtilis RNase H enzyme at 25°C. We used an electrophoretic mobility shift assay to test the ability of purified SSB (Fig. 1A) to bind and shift the modeled Okazaki substrate (Fig. 1D). RNase HII and HIII were then assayed for cleavage of the single Okazaki fragment substrate with bovine serum albumin (BSA) or SSB added to the reaction. The results were quantified, and the fold stimulation of SSB for each enzyme over the addition of equimolar BSA was calculated. We used BSA as a control for comparison because BSA addition alone showed stimulation of RNase HII and HIII incision (data not shown). We found that SSB showed modest stimulation of both RNase H enzymes over the BSA control. RNase HII was stimulated by 1.5-fold, whereas RNase HIII showed 2-fold stimulation (Fig. 1E). Kinetic analysis of SSB stimulation on E. coli RNase HI showed a 4-fold increase in catalytic efficiency and ∼2.5-fold increase in activity (39). We now show that SSB causes a mild stimulation of incision by RNase HII and HIII, with HIII showing the greatest increase in activity after addition of SSB.
RNase HIII increases DNA polymerase I processing of Okazaki fragments.
These results indicate that both RNase HII and RNase HIII act on an RNA-DNA hybrid with a 3′ overhang modeling a single Okazaki fragment in vitro. An important difference between RNase HII and HIII is their different locations of incision in the RNA primer. The various sites of incision may have downstream effects on the efficiency of RNA removal and replacement with DNA by a DNA polymerase. We therefore explored whether RNase HII or HIII worked more efficiently with a DNA polymerase in vitro. To begin, we asked if either of the B. subtilis replicative DNA polymerases, PolC or DnaE, was capable of strand displacement synthesis of an Okazaki fragment. In addition, we tested DNA Pol I for the same displacement synthesis activity (Fig. 2A). All three DNA polymerases were able to extend the primed substrate (oJR361 annealed to oJR362), but only Pol I was able to extend the same primed substrate flanked by an RNA-DNA chimera (oJR361, oJR362, and oJR367) (Fig. 2A), which supports our prior results for Pol I action during ribonucleotide excision repair (11). The 5′ to 3′ exonuclease domain of Pol I likely allows for degradation of the RNA while polymerizing deoxyribonucleotides to fill in the gap.
FIG 2.

RNase HIII stimulates Pol I during Okazaki fragment maturation in vitro. (A) Denaturing urea-PAGE of the products following primer extension with or without the presence of an annealed RNA-DNA chimera fragment downstream for PolC, DnaE, and Pol I. Primer set oJR361 and oJR362 and primer set oJR361, oJR362, and oJR367 were used for “primed” and “blocked” substrates, respectively. For blocked substrates, only the green 5′ label from oJR362 was imaged. (B) Denaturing urea-PAGE showing products of Pol I extension (red 5′ label) and 5′ to 3′ exonuclease activity (green) through an RNA-DNA chimera or DNA substrates labeled on 3′ ends. Primer set oJR338, oJR339 and oJR340 and primer set oJR338, oJR348, and oJR340 were used for RNA and DNA substrates, respectively. (C) Denaturing urea-PAGE showing products of Pol I extension through an Okazaki fragment labeled on the 3′ end (red) with or without the addition RNase HII or RNase HIII at the indicated time points. Pol I extension was monitored by a 5′ label (green) on the extended strand. The primer set oJR361, oJR362, and oJR367 was used for assembly of the Okazaki fragment substrate. (D) Quantification of three reactions from panel C showing the percentage of substrates extended past the RNA portion of the Okazaki fragment over time for each reaction. Error bars indicate standard deviation between experiments. Primers oJR364 and oJR365 were used as size standards.
We assayed the exonuclease and polymerase activities of Pol I by monitoring 3′- and 5′-fluorescent-end-labeled the substrates. Substrates consisted of a 5′-labeled DNA primer (oJR338) and an unlabeled complementary strand (oJR340), as well as a downstream 3′-labeled RNA-DNA chimera (oJR339) or a 3′-labeled DNA strand (oJR348). We monitored the 3′ green fluorescent label on the excised strand for both RNA-DNA chimera and DNA to measure the 5′ to 3′ exonuclease activity and found that Pol I was active on both substrates but more efficient on DNA than on RNA (Fig. 2B, green bands). Pol I extension was monitored by the red fluorescent label on the 5′ end of the extended strand, which demonstrated that Pol I was also more efficient at extending a primer with DNA immediately downstream than an RNA-DNA hybrid (Fig. 2B, red bands).
With Pol I extension reactions on a model Okazaki fragment established, we asked whether the efficiency of Pol I-dependent extension increased in the presence of RNase HII or HIII at 25°C. Using a substrate with the primer (oJR362) labeled on the 5′ end to monitor extension (green bands) and the RNA-DNA chimera (oJR367) labeled on the 3′ end to monitor degradation (red bands), we added Pol I alone, Pol I with RNase HII (Pol I + HII), or Pol I with RNase HIII (Pol I + HIII) to measure the percentage of primer extension past the RNA portion of the excised oligonucleotide (indicated as percentage repaired) over time. We visualized the products by denaturing urea-PAGE (Fig. 2C and D). We found that Pol I was far more efficient at removing and replacing RNA when supplemented with RNase HIII than Pol I alone at 25°C. To this end, we found that removal of RNA by Pol I alone showed little activity compared with near-complete excision on addition of RNase HIII (Fig. 2C, compare red bands in lanes 2 to 4 and 8 to 10). Repair was complete at 2 min in the reaction with Pol I and RNase HIII, whereas Pol I alone only repaired 71% after 5 min. Addition of RNase HII had a modest increase in efficiency compared to Pol I alone, with 79% repaired after 5 min. We conclude that the activity of RNase HIII in conjunction with DNA polymerase I provides the most efficient pathway for Okazaki fragment repair in vitro.
YpcP processes Okazaki fragments in vitro.
Prior results have shown that single deletions of polA (Pol I) or ypcP, the gene coding for a putative 5′ to 3′ exonuclease, are viable in B. subtilis. YpcP is paralogous to the N-terminal 5′ to 3′ exonuclease domain of polA (38, 40); however, deletion of both genes is lethal, which suggests similar functions between Pol I and YpcP in vivo (38, 40). Given the putative 5′ to 3′ exonuclease activity of YpcP in addition to the lethality of the ΔpolA ΔypcP mutant (38, 40, 41), we reasoned that YpcP might have an overlapping function in Okazaki fragment maturation.
Because YpcP activity has not yet been tested, we began by purifying recombinant YpcP and a variant we predicted would be catalytically inactive based on the conservation of YpcP D192N with Mycobacterium smegmatis FenA (42) (Fig. 3A; see Fig. S1 in the supplemental material). We cloned several predicted catalytic mutants based on alignment with the structure of M. smegmatis FenA (42). YpcP D192N was the only soluble variant we constructed (data not shown). On purification, we noticed a minor proteolytic fragment in our purification of YpcP D192N (Fig. S1), which we confirmed was indeed YpcP by tandem mass spectrometry (data not shown). We suspect that the YpcP D192N has a slight conformational change that causes it to be more susceptible to proteolysis during cell lysis. With that noted, YpcP D192N was purified using the same procedure as wild-type YpcP, making it an appropriate negative control for wild-type YpcP activity in our assays (see Materials and Methods).
FIG 3.

YpcP is an exonuclease showing preference for RNA-DNA hybrids. (A) SDS-PAGE of 2 μg of purified YpcP. (B) Metal-dependent activity of YpcP on a 3′-overhang single Okazaki fragment substrate (oJR339, oJR340) after 10 min incubation at 25°C using denaturing urea-PAGE to separate the products. Metals were used at 1 mM, except for the final lane, which used 1 mM MgCl2 and 10 μM MnCl2. (C) Activity of YpcP and YpcP D192N on the substrate used in (B) with 1 mM MgCl2 and 10 μM MnCl2 after 15 min incubation at 25°C. (D) Activity of YpcP on several different RNA and DNA substrates visualized after electrophoresis in denaturing urea-PAGE. YpcP was used at a final concentration of 4 nM using the Okazaki repair assay buffer (see Materials and Methods). Oligonucleotide sets used are as follows: oJR339 and oJR340, 3′ overhang with an RNA-DNA chimera; oJR340 and oJR348, 3′ overhang DNA only; oJR339, ssRNA-DNA chimera; oJR348, ssDNA; oJR338, oJR339, and oJR340, RNA-DNA chimera nicked; oJR338, oJR340, and oJR348, DNA nicked; oJR339 and oJR365, RNA-DNA chimera blunt; oJR348 and oJR365, DNA blunt. The squiggly line indicates the position of RNA, and the smooth line indicates the location of DNA for the substrate schematics. See “Exonuclease assays” in Materials and Methods for substrate formation. Time points assayed were 10 s, 1 min, and 10 min, with 1 mM MgCl2 and 10 μM MnCl2 at 25°C. (E) Assay of YpcP on flapped substrates. The oligonucleotides used are as follows: oJR339, oJR366, and oJR368, RNA-DNA chimera flap; oJR348, oJR366, and oJR368, DNA flap. The incubation time was 15 min at 25°C.
To understand the substrate specificity of YpcP, activity was assayed on a 3′-overhang substrate with an RNA-DNA chimera modeling a single Okazaki fragment. The 3′-overhang substrate was created by annealing a 20-bp RNA-DNA chimera (oJR339) hybridized to a 32-bp DNA oligonucleotide (oJR340) to create a 12-bp 3′ overhang. This substrate was incubated with purified YpcP in the presence of various metals, and the products were visualized using denaturing urea-PAGE. YpcP showed 5′ to 3′ nucleolytic activity on the RNA-containing strand of the 3′-overhang substrate and favored Mn2+ over Mg2+ in the reaction at 25°C (Fig. 3B). We then assayed YpcP and YpcP D192N on the same substrate and found that YpcP cleaved the single Okazaki-like substrate and D192N failed to show activity (Fig. 3C). With these controls established, we assayed YpcP activity on several different substrates. We assayed double-stranded substrates with a 3′-overhang, single-stranded-only, double-stranded with a nick, blunt-ended, and flapped substrates using a physiologically relevant mixture of Mn2+ and Mg2+ as described previously (30) (Fig. 3D and E). In each case, the 5′ to 3′ activity was monitored on an oligonucleotide that was either DNA or an RNA-DNA chimera to compare activity. We found that YpcP degraded RNA-containing substrates more efficiently than the DNA substrates (Fig. 3D), including the flapped and nicked substrates (Fig. 3D, E). Thus, we showed that YpcP has activity on every substrate tested, with a clear preference for RNA (Fig. 3D). We suggest that the activity of YpcP on Okazaki fragments in addition to the preference of YpcP for RNA substrates supports a role for YpcP in Okazaki fragment maturation (38). Moreover, our biochemical results suggest that deletion of both polA and ypcP causes synthetic lethality in B. subtilis due to a lack of 5′ to 3′ exonuclease activity that, in part, contributes to decreased Okazaki fragment maturation (38).
RNase HIII-deficient cells are cold sensitive.
To understand the contribution of RNase HII, RNase HIII, Pol I, and YpcP to Okazaki fragment maturation in vivo, we sought to develop a genetic approach. We reasoned that growth at lower temperatures may stabilize Okazaki fragments, potentially revealing genetic requirements for their removal in the form of a cold-sensitive phenotype. We serially diluted and assayed the growth of wild-type RNase H-deficient strains and polA and ypcP deletion strains on LB agar at 37°C, 30°C, and 25°C (Fig. 4A, top five rows). Growth of all deletion strains was nearly identical to that of wild-type strains at 30°C and 37°C, with the exception of smaller colony sizes in the ΔrnhC (RNase HIII) background at 30°C. In contrast, the ΔrnhC mutant showed a more severe decrease in growth at 25°C than the other strains tested (Fig. 4A). Furthermore, we performed growth curves on wild-type, ΔrnhB (RNase HII), and ΔrnhC strains and found that the growth rate was decreased in the ΔrnhC but not the ΔrnhB strain, relative to wild-type strains (see Fig. S2 in the supplemental material).
FIG 4.

RNase H mutants show cold-temperature growth interference. (A) Spot titer assays for each strain grown on LB agar at the indicated temperatures. (B) Spot titer assays with the indicated strains plated on mitomycin C (MMC) or methyl methanesulfonate (MMS) using the concentrations shown.
To test whether the cold sensitivity was exacerbated on deletion of RNase HII and RNase HIII, we built a ΔrnhC rnhB::erm double-mutant strain as described (11, 30, 38). We noticed that the phenotype diminished over time and required that the strain be rebuilt. This observation motivated us to perform whole-genome resequencing of five isolates, revealing several mutations in the five ΔrnhC rnhB::erm isolates relative to the wild-type reference genome (see Table S1 in the supplemental material). We speculate that prolonged use of the strain leads to mutations that may be important for long-term survival of the double mutant. Given what appeared to be the accumulation of mutations in the ΔrnhC rnhB::erm double mutant, we chose not to pursue the characterization of this strain further and instead focused our experiments on ΔrnhC.
Deletion of polA or ypcP has a synergistic effect on cold sensitivity of RNase HIII-deficient cells.
Given the cold-sensitive phenotype of RNase HIII-deficient cells in vivo, we asked whether deletion of polA or ypcP further sensitized ΔrnhC cells to growth at 25°C. To test this, we performed serial dilutions of ΔrnhC ΔpolA and ΔrnhC ΔypcP strains plated on LB agar and incubated the plates at 37°C, 30°C, and 25°C (Fig. 4A). Although we found no difference in growth between ΔrnhC cells and the double deletions grown at 37°C, we observed a synergistic decrease in growth of ΔrnhC ΔpolA and ΔrnhC ΔypcP cells relative to ΔrnhC cells at 30°C and an even more striking decrease in growth at 25°C. At 30°C, we observed an ∼100-fold decrease in growth in the ΔrnhC ΔpolA and ΔrnhC ΔypcP cells with virtually no growth of the double-deletion strains detected at 25°C.
The growth interference of the ΔrnhC ΔpolA and ΔrnhC ΔypcP strains suggests that the primary pathways for Okazaki fragment maturation involves the activity of RNase HIII and the 5′ to 3′ exonuclease activities of Pol I and/or YpcP in vivo. Deletion of both RNase HII and Pol I (ΔrnhB ΔpolA) does not show a cold-sensitive phenotype, further supporting our in vitro data that RNase HII is probably not involved in Okazaki fragment maturation. With this genetic data, we suggest that RNase HIII and Pol I function together in Okazaki fragment maturation and that YpcP contributes to Okazaki repair.
Although YpcP was not well characterized before this study, it was implicated in repair of photodimers (41). To further address this observation, we asked if a strain bearing a ΔypcP deletion was sensitive to DNA damage. We tested ΔypcP and ΔpolA strains as controls on LB plates with mitomycin C or methyl methanesulfonate. We found that the ΔypcP strain was not sensitive to either DNA-damaging agent, whereas the ΔpolA strain was very sensitive when challenged with both exogenous DNA-damaging agents (Fig. 4B). Based on these results, it seems unlikely that YpcP makes an important contribution to genome maintenance outside of Okazaki fragment and other RNA-DNA hybrid repair.
Here, we showed that RNase HIII functions with Pol I or YpcP in Okazaki fragment maturation in B. subtilis. RNase HIII has also been shown to affect R-loop stability in B. subtilis (12). We certainly cannot exclude the possibility that the cold-sensitive phenotype we observed was due to a failure in R-loop resolution. However, if the cold-sensitive phenotype was strictly due to a failure in R-loop resolution, we would not have expected to find an exacerbation of the ΔrnhC phenotype with deletion of polA because Pol I has not been shown to resolve R-loop structures. Therefore, we suggest that the cold-sensitive phenotype is due at least in part to a failure in Okazaki fragment maturation in vivo.
DISCUSSION
Here we show that RNase HIII and DNA polymerase I reconstitute an efficient pathway for the maturation of Okazaki fragments for B. subtilis. B. subtilis RNase HII and HIII both cleave an Okazaki fragment in vitro near the RNA-DNA junction, leaving short spans of RNA annealed to DNA. As demonstrated with E. coli RNase HI (39), SSB modestly stimulates the activity of RNase HII and HIII, possibly acting to recruit these enzymes to the replisome in vivo. After cleavage, lower temperatures have the potential to stabilize the short remaining RNA-DNA hybrids hindering Pol I removal. We noticed that the 5ʹ to 3ʹ exonuclease activity of Pol I appears to slow during excision at 25°C (Fig. 2C, lanes 2 to 4). The addition of RNase HIII to these reactions allows for rapid Pol I extension. RNase HII has a much more modest effect at lower temperatures, possibly due to its differing cleavage site, which would leave behind a longer RNA-DNA hybrid that would be more stable at 25°C. Therefore, we suggest that the maturation of Okazaki fragments is primarily performed by DNA Pol I and RNase HIII.
B. subtilis contains another putative 5ʹ to 3ʹ exonuclease, YpcP, which has rescued an RNase H-deficient cell elongation phenotype when overexpressed (38). Furthermore, deletion of the gene for DNA polymerase I (polA) and ypcP was shown to be synthetically lethal, suggesting that YpcP serves a similar function to Pol I in vivo (40). In addition, this enzyme was recently implicated in the processing of DNA photodimers in vitro (41). Our results show that recombinant YpcP has 5ʹ to 3ʹ exonuclease activity on both DNA and RNA (Fig. 3). YpcP appears to be significantly more effective on RNA hybridized to DNA than on double-stranded DNA, suggesting that it also provides the nuclease activity needed for RNA removal from Okazaki fragments in conjunction with or as a backup to DNA polymerase I.
B. subtilis ΔrnhC cells grow poorly in the presence of hydroxyurea, high salt, lysozyme, and paraquat stresses and in engineered strains where genes are oriented in the head-on direction from a strong inducible promoter (12, 30). We report here that ΔrnhC cells show growth interference at 25°C. Thus, ΔrnhC cells have severe growth defects in the presence of a wide variety of stressors. Prior work showed that ΔrnhC cells have an increase in RNA-DNA hybrid formation in vivo, and it was proposed that the phenotypes of ΔrnhC cells are caused by persistent R-loop formation (12). Our work shows that RNase HIII is important for the efficient removal of RNA from Okazaki fragments during their maturation process in vitro. To study the process in vivo requires a phenotype that might separate the contribution of RNase HIII in Okazaki fragment maturation from the role of RNase HIII in R-loop resolution. We hypothesize that the cold sensitivity may be caused in part by the stabilization of Okazaki fragments during removal. If the cold-sensitive phenotype were mediated by persistent R-loop formation, we would not expect deletions of ypcP or polA (Pol I) to exacerbate the phenotype. Instead, we found that ΔypcP or ΔpolA alleles did indeed exacerbate the phenotype of the ΔrnhC strain at 25°C. When the temperature was increased, the Okazaki fragments were processed more readily in the absence of RNase HIII, and the phenotype was rescued. We suggest that the lower temperature causes defects in Okazaki fragment maturation that are observed as growth interference at 25°C; however, this does not preclude the possibility of persistent R-loops contributing to the observed phenotype.
The above data suggest a primary model for Okazaki fragment removal in B. subtilis involving RNase HIII cleavage upstream of the RNA-DNA junction and removal and replacement of the remaining RNA with DNA by DNA Pol I. In addition, YpcP displays the enzymatic activity necessary to remove the RNA primer in the absence of or in conjunction with DNA Pol I but not the polymerase activity necessary to replace it. We suggest that once the RNA primer is removed by YpcP, either replicative polymerase PolC or DnaE could fill the gap when Pol I is not available (Fig. 5). RNase HIII is unique to many infectious bacteria and has been identified as important for listeria pathogenesis (12). RNase HIII has also been suggested as important for R-loop removal (12). Because we found that RNase HIII plays an important role in aiding Pol I in Okazaki fragment maturation, we suggest that many of the phenotypes observed for rnhC-deleted cells may be a result of problems in Okazaki fragment maturation in addition to failures in R-loop removal.
FIG 5.
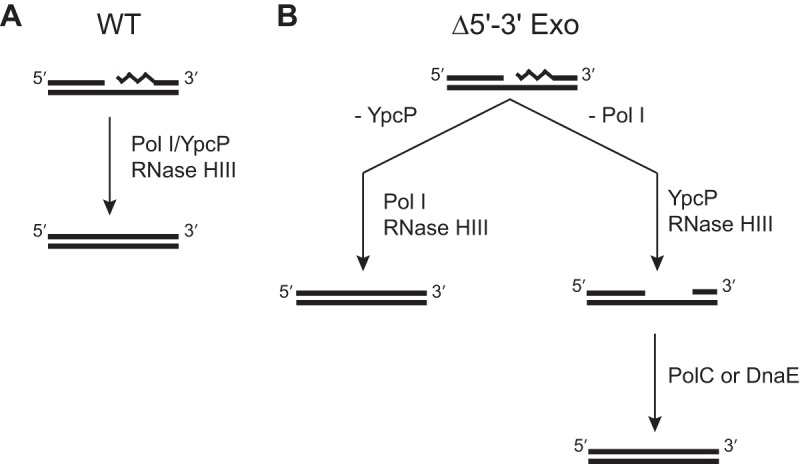
A model for Okazaki fragment maturation in B. subtilis. (A) In wild-type cells, DNA Pol I and RNase HIII serve as the primary pathway. YpcP may help facilitate primer removal, and RNase HII may provide a less-efficient backup for RNase HIII activity. (B) In the absence of YpcP, the pathway remains the same. However, when DNA Pol I is absent, YpcP can excise the RNA primer possibly with help from the RNase H enzymes. After removal, the remaining gap would need to be filled by another DNA polymerase, likely DnaE, because it has an established role in lagging strand synthesis (43). The squiggly line denotes the position of RNA.
MATERIALS AND METHODS
Strain construction.
All strains used in this work are described in Table S2 in the supplemental material. All primers and plasmids used in this work are described in Tables S3 and S4, respectively, in the supplemental material. Strains were built by obtaining B. subtilis 168 cells with an erythromycin resistance cassette replacing the desired gene open reading frame flanked by loxP sites from the Bacillus Genetic Stock Center (http://www.bgsc.org). Genomic DNA from these strains was isolated and used to transform competent wild-type JR27 (PY79) or JR48 (ΔrnhC) cells. The erythromycin resistance cassette of each strain was then removed by transformation of each recipient strain with pDR244 expressing Cre recombinase. Plasmid pDR244 confers spectinomycin resistance and contains a temperature-sensitive origin of replication. The plasmid was then cured by growth at 45°C, and the resulting strains were screened for loss of erm and spec resistance. Finally, each strain was characterized by PCR genotyping for loss of the corresponding coding region.
Protein purification.
Proteins were purified by transforming chemically competent BL21DE3 (JRR1) cells and plating on LB agar supplemented with 25 μg/ml kanamycin. After growing a 25-ml subculture, 3 liters of culture was grown to an optical density at 600 nm (OD600) of 0.7 at 37°C, and then IPTG (isopropyl-β-d-thiogalactopyranoside) was added to 200 μM. Cultures were allowed to grow for an additional 4 h and then pelleted using centrifugation. Cells were resuspended in lysis buffer (50 mM Tris HCl, pH 8, 10% wt/vol sucrose, 300 mM NaCl, 10 mM imidazole) and lysed via sonication. Cell debris was removed by centrifugation, and the lysate was passed over a Ni2+-agarose (Qiagen) gravity column. The column was washed with 50 mM Tris HCl, pH 8, 2 M NaCl, and 20 mM imidazole and eluted with 5 ml of 50 mM Tris HCl, pH 8, 50 mM NaCl, and 400 mM imidazole. The eluted fractions were dialyzed with SUMO protease in protease buffer (50 mM Tris HCl, pH 8, 50 mM NaCl, 2 mM β-mercaptoethanol) at 4°C overnight and passed back over a Ni2+-agarose gravity column. The flowthrough was collected and loaded onto a Q anion-exchange column (GE no. 17115301) and eluted with a gradient of 50 to 1,000 mM NaCl in buffer (50 mM Tris HCl pH 8, 1 mM dithiothreitol [DTT]). Fractions were analyzed by SDS-PAGE, pooled, exchanged into storage buffer (50 mM Tris HCl, pH 8, 100 mM NaCl, 25% glycerol), and frozen in liquid nitrogen. Purified PolC, DnaE, and SSB were a generous gift from Charles McHenry (43).
Plasmid cloning.
Plasmid cloning was performed by amplifying each insert (ypcP) from PY79 genomic DNA with primers containing 5′-overlapping pE-SUMO complementary DNA (oJR153 and oJR154; oJR378 and oJR379). The ypcP D192N mutant primers contained the point mutation necessary for the change (see Table S4 in the supplemental material). Inserts were then joined with pE-SUMO vector via Gibson assembly (44) and used to transform chemically competent JRR2 cells, followed by plating on LB agar supplemented with 25 μg/ml kanamycin. Plates were grown at 37°C overnight, and colonies from overnight growth were screened by PCR for the ypcP insert. The plasmid DNA was isolated, and the ypcP sequence was confirmed by Sanger sequencing (University of Michigan Sequencing Core).
Growth curves.
Growth curves were generated by streaking each strain on LB agar and growing at 37°C overnight. Each strain was then plate washed and diluted to an OD600 of 0.05, and 250 μl was added to each of three wells of a 96-well plate per strain. The 96-well plate was incubated with shaking at either 25°C or 37°C in a FLUOstar Omega plate reader for 10 h, with reads taken at OD600 every 30 min. Reads were normalized to LB alone, averaged, and plotted using R. Growth rate estimates and corresponding confidence intervals were obtained by fitting a modified Gompertz growth model, (45), where the parameters A, μm, and λ represent time (t) when the growth rate equals zero (asymptote), maximum growth rate, and lag time, respectively, to the replicates for each strain. Doubling-time estimates were calculated as ln(2)/μm, where μm indicates estimated growth rate from the modified Gompertz growth model.
Spot titer.
Strains were struck out on LB agar and grown overnight at 37°C. A single colony was then picked and grown in 2 ml of LB at 37°C to an OD600 between 0.5 and 1. Cultures were normalized to an OD600 of 0.5 and serially diluted 10−5 in 0.85% saline. A volume of 5 μl of each dilution was spotted onto LB agar and grown at the indicated temperatures.
Genome resequencing.
Genomic DNA was purified from five isolates of the ΔrnhC rnhB::erm strain (JRR33-3) and submitted to the University of Michigan Sequencing Core for library preparation and sequencing. A total of 76-bp paired-end reads were sequenced on the MiSeq platform, 150 cycles, v3. Sequence alignment to the Bacillus subtilis PY79 reference genome (46) and subsequent variant calling were performed using breseq v0.33 (47). The variants and the frequency in which they appeared in the isolates sequenced here are indicated. The rnhC deletion was detected in all strains, and the rnhB region was indicated as unassigned missing coverage in all strains due to replacement with the erm cassette.
RNase H and YpcP assays.
Assays were performed at 25°C in a buffer containing 10 mM Tris HCl, pH 8, 50 mM NaCl, 1 mM MgCl2, and 10 μM MnCl2, representing the in vivo ratios of these two metals that support incision (30). For YpcP, 1 mM DTT was added to the reaction. First, the substrate was generated by annealing oJR339 at 1 μM and oJR343 at 2 μM in the same buffer by heating to 100°C for 30 s and then allowing the solution to cool back to room temperature. For SSB stimulation, 4 μM SSB was added after annealing and allowed to sit at room temperature for 30 min. We then added 4 nM RNase HII, RNase HIII, or YpcP to the reactions with 100 nM substrate, and the reactions were stopped with an equal volume of stop buffer (95% formamide, 20 mM EDTA) at the indicated time points. Unless otherwise stated in the figure legends, reaction mixtures were incubated for 10 min. The reaction mixtures were then boiled for 5 min and immediately snap cooled in an ice-water bath. Reaction products were resolved using denaturing urea-PAGE and imaged with a LI-COR Odyssey infrared imager.
Extension reaction.
Extension reactions were performed at 25°C in a buffer containing 40 mM Tris-acetate, pH 7.8, 12 mM magnesium acetate, 300 mM potassium glutamate, 3 μM ZnSO4, 2% (wt/vol) polyethylene glycol, 0.02% pluronic F68, and 1 mM DTT by procedures similar to those described previously (11, 30, 43). Figure 1A shows substrates that were generated by annealing 1 μM oJR361 and 2 μM oJR362 in the same buffer, heating to 100°C, and allowing the reaction to cool back to room temperature. For the RNA-DNA chimera-blocked substrate, 2 μM oJR367 was added. Each DNA polymerase was added to 100 nM in the presence of 100 nM of the substrate with 50 μM deoxynucleoside triphosphates (dNTP). Each reaction was stopped with stop buffer (95% formamide, 20 mM EDTA), the reaction mixture was boiled and snap cooled, and the reaction products were resolved via denaturing urea-PAGE and imaged with a LI-COR Odyssey infrared imager. For imaging of the blocked substrate, only the 5′ green label from oJR362 was imaged to assay extension.
Okazaki repair assay.
Okazaki fragment repair assays were performed at 25°C in a buffer containing 10 mM Tris-HCl, pH 8, 50 mM NaCl, 1 mM MgCl2, 10 μM MnCl2, 1 mM DTT, and 50 μM dNTP as described previously (11, 30). The substrate was generated by annealing oJR361, oJR362, and oJR367 at 1, 1.5, and 1.75 μM, respectively. The oligonucleotides were heated to 100°C and allowed to cool back to room temperature. Each reaction mixture contained 100 nM substrate and 100 nM DNA Pol I, and the reaction was stopped with stop buffer (95% formamide, 20 mM EDTA) at the indicated time points. RNase H-containing reaction mixtures had either RNase HII or RNase HIII added to 50 nM. Reaction products were then resolved by denaturing urea-PAGE with oJR363 and oJR364 added to separate lanes to distinguish repair efficiency. Each gel was visualized with a LI-COR Odyssey infrared imager. Percent repaired was calculated by dividing the portion of the lane with length greater than the RNA portion (repaired) by the total lane signal. Each lane was normalized to a reaction mixture containing no DNA polymerase and plotted against time.
Exonuclease assays.
Assays were performed at 25°C as described above in the RNase H assay section with addition of 1 mM DTT to the buffer. All substrates were annealed in the same buffer by heating to 100°C for 30 s and allowing them to cool at room temperature. 3′-overhang substrates annealed 1 μM oJR339 (RNA) or oJR348 (DNA) to 2 μM oJR340 (Fig. 3). Nicked substrates included the addition of 2 μM oJR338 to the 3′-overhang substrate single Okazaki fragment substrate (Fig. 2B and 3). Blunt substrates annealed 1 μM oJR339 (RNA) or oJR348 (DNA) to 2 μM oJR365 (Fig. 3). Flap substrates annealed 1 μM oJR339 (RNA) or oJR348 (DNA) to 2 μM oJR366 and oJR368 (Fig. 3E). Exonuclease reaction mixtures contained 100 nM substrate with either 4 nM YpcP or 100 nM DNA Pol I, and reactions were stopped with stop buffer (95% formamide, 20 mM EDTA) at the indicated time points. Reaction mixtures were then boiled at 100°C for 5 min and snap cooled in an ice-water bath. Reaction products were resolved via 20% denaturing urea-PAGE and visualized with a LI-COR Odyssey infrared imager.
Supplementary Material
ACKNOWLEDGMENTS
We are indebted to Charles McHenry (University of Colorado, Boulder) for providing purified DnaE, PolC, and SSB. We thank Peter Freddolino and Rebecca Hurto for help with analysis of the mutations in the rnhC rnhB double mutant.
J.R.R. was supported in part by an NIH Cellular Biotechnology Training grant (T32 GM008353) and a predoctoral fellowship from the Rackham Graduate School at the University of Michigan. T.M.N. was supported by a predoctoral fellowship from the National Science Foundation (DEG 1256260). K.J.W. was supported by an NIH Cellular Biotechnology Training grant (T32 GM008353) and a predoctoral fellowship from the National Science Foundation (DEG 1256260). This research was further supported by NIH grant GM107312 to L.A.S.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00686-18.
REFERENCES
- 1.Schroeder JW, Randall JR, Matthews LA, Simmons LA. 2015. Ribonucleotides in bacterial DNA. Crit Rev Biochem Mol Biol 50:181–193. doi: 10.3109/10409238.2014.981647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams JS, Kunkel TA. 2014. Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst) 19:27–37. doi: 10.1016/j.dnarep.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vaisman A, Woodgate R. 2018. Ribonucleotide discrimination by translesion synthesis DNA polymerases. Crit Rev Biochem Mol Biol 53:382–402. doi: 10.1080/10409238.2018.1483889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas M, White RL, Davis RW. 1976. Hybridization of RNA to double-stranded DNA: formation of R-loops. Proc Natl Acad Sci U S A 73:2294–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom EB, Burgers PM, Johansson E, Chabes A, Kunkel TA. 2010. Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci U S A 107:4949–4954. doi: 10.1073/pnas.0914857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao NY, Schroeder JW, Yurieva O, Simmons LA, O'Donnell ME. 2013. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc Natl Acad Sci U S A 110:12942–12947. doi: 10.1073/pnas.1309506110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowen L, Kornberg A. 1978. A ribo-deoxyribonucleotide primer synthesized by primase. J Biol Chem 253:770–774. [PubMed] [Google Scholar]
- 8.Rowen L, Kornberg A. 1978. Primase, the dnaG protein of Escherichia coli. An enzyme which starts DNA chains. J Biol Chem 253:758–764. [PubMed] [Google Scholar]
- 9.Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA. 1994. Enzymatic completion of mammalian lagging-strand DNA replication. Proc Natl Acad Sci U S A 91:9803–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Breaker RR. 1999. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2' hydroxyl group. J Am Chem Soc 121:5364–5372. doi: 10.1021/ja990592p. [DOI] [Google Scholar]
- 11.Schroeder JW, Randall JR, Hirst WG, O'Donnell ME, Simmons LA. 2017. Mutagenic cost of ribonucleotides in bacterial DNA. Proc Natl Acad Sci U S A 114:11733–11738. doi: 10.1073/pnas.1710995114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, Pollock AJ, Woodward JJ, Dreifus JE, Merrikh H. 2017. Replication-transcription conflicts generate R-loops that orchestrate bacterial stress survival and pathogenesis. Cell 170:787–799.e18. doi: 10.1016/j.cell.2017.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundstrom EB, Johansson E, Chabes A, Kunkel TA. 2010. Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6:774–781. doi: 10.1038/nchembio.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pizzi S, Sertic S, Orcesi S, Cereda C, Bianchi M, Jackson AP, Lazzaro F, Plevani P, Muzi-Falconi M. 2015. Reduction of hRNase H2 activity in Aicardi-Goutieres syndrome cells leads to replication stress and genome instability. Hum Mol Genet 24:649–658. doi: 10.1093/hmg/ddu485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reijns MA, Jackson AP. 2014. Ribonuclease H2 in health and disease. Biochem Soc Trans 42:717–725. doi: 10.1042/BST20140079. [DOI] [PubMed] [Google Scholar]
- 16.Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vaisman A, McDonald JP, Huston D, Kuban W, Liu L, Van Houten B, Woodgate R. 2013. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet 9:e1003878. doi: 10.1371/journal.pgen.1003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouzminova EA, Kadyrov FF, Kuzminov A. 2017. RNase HII saves rnhA mutant Escherichia coli from R-loop-associated chromosomal fragmentation. J Mol Biol 429:2873–2894. doi: 10.1016/j.jmb.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerritelli SM, Crouch RJ. 2009. Ribonuclease H: the enzymes in eukaryotes. FEBS J 276:1494–1505. doi: 10.1111/j.1742-4658.2009.06908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohtani N, Haruki M, Morikawa M, Kanaya S. 1999. Molecular diversities of RNases H. J Biosci Bioeng 88:12–19. [DOI] [PubMed] [Google Scholar]
- 21.Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. 2005. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell 121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 22.Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. 2007. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol Cell 28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 23.Ohtani N, Yanagawa H, Tomita M, Itaya M. 2004. Identification of the first archaeal type 1 RNase H gene from Halobacterium sp. NRC-1: archaeal RNase HI can cleave an RNA-DNA junction. Biochem J 381:795–802. doi: 10.1042/BJ20040153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohtani N, Haruki M, Muroya A, Morikawa M, Kanaya S. 2000. Characterization of ribonuclease HII from Escherichia coli overproduced in a soluble form. J Biochem 127:895–899. [DOI] [PubMed] [Google Scholar]
- 25.Itaya M. 1990. Isolation and characterization of a second RNase H (RNase H II) encoded by the rnhB gene. Proc Natl Acad Sci U S A 87:8587–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohtani N, Tomita M, Itaya M. 2008. Junction ribonuclease: a ribonuclease HII orthologue from Thermus thermophilus HB8 prefers the RNA-DNA junction to the RNA/DNA heteroduplex. Biochem J 412:517–526. doi: 10.1042/BJ20080140. [DOI] [PubMed] [Google Scholar]
- 27.Ohtani N, Tomita M, Itaya M. 2008. Junction ribonuclease activity specified in RNases HII/2. FEBS J 275:5444–5455. doi: 10.1111/j.1742-4658.2008.06673.x. [DOI] [PubMed] [Google Scholar]
- 28.Ohtani N, Haruki M, Morikawa M, Crouch RJ, Itaya M, Kanaya S. 1999. Identification of the genes encoding Mn2+-dependent RNase HII and Mg2+-dependent RNase HIII from Bacillus subtilis: classification of RNases H into three families. Biochemistry 38:605–618. doi: 10.1021/bi982207z. [DOI] [PubMed] [Google Scholar]
- 29.Kochiwa H, Tomita M, Kanai A. 2007. Evolution of ribonuclease H genes in prokaryotes to avoid inheritance of redundant genes. BMC Evol Biol 7:128. doi: 10.1186/1471-2148-7-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randall JR, Hirst WG, Simmons LA. 2017. Substrate specificity for bacterial RNase HII and HIII is influenced by metal availability. J Bacteriol:JB.00401-17. doi: 10.1128/JB.00401-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM. 2012. RNase H2-initiated ribonucleotide excision repair. Mol Cell 47:980–986. doi: 10.1016/j.molcel.2012.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaisman A, McDonald JP, Noll S, Huston D, Loeb G, Goodman MF, Woodgate R. 2014. Investigating the mechanisms of ribonucleotide excision repair in Escherichia coli. Mutat Res 761:21–33. doi: 10.1016/j.mrfmmm.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corn JE, Berger JM. 2006. Regulation of bacterial priming and daughter strand synthesis through helicase-primase interactions. Nucleic Acids Res 34:4082–4088. doi: 10.1093/nar/gkl363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kao HI, Bambara RA. 2003. The protein components and mechanism of eukaryotic Okazaki fragment maturation. Crit Rev Biochem Mol Biol 38:433–452. doi: 10.1080/10409230390259382. [DOI] [PubMed] [Google Scholar]
- 35.Murante RS, Henricksen LA, Bambara RA. 1998. Junction ribonuclease: an activity in Okazaki fragment processing. Proc Natl Acad Sci U S A 95:2244–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kogoma T. 1986. RNase H-defective mutants of Escherichia coli. J Bacteriol 166:361–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogawa T, Okazaki T. 1984. Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli. Mol Gen Genet 193:231–237. [DOI] [PubMed] [Google Scholar]
- 38.Fukushima S, Itaya M, Kato H, Ogasawara N, Yoshikawa H. 2007. Reassessment of the in vivo functions of DNA polymerase I and RNase H in bacterial cell growth. J Bacteriol 189:8575–8583. doi: 10.1128/JB.00653-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petzold C, Marceau AH, Miller KH, Marqusee S, Keck JL. 2015. Interaction with single-stranded DNA-binding protein stimulates Escherichia coli ribonuclease HI enzymatic activity. J Biol Chem 290:14626–14636. doi: 10.1074/jbc.M115.655134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomaides HB, Davison EJ, Burston L, Johnson H, Brown DR, Hunt AC, Errington J, Czaplewski L. 2007. Essential bacterial functions encoded by gene pairs. J Bacteriol 189:591–602. doi: 10.1128/JB.01381-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patlán AG, Corona SU, Ayala-García VM, Pedraza-Reyes M. 2018. Non-canonical processing of DNA photodimers with Bacillus subtilis UV-endonuclease YwjD, 5'→3' exonuclease YpcP and low-fidelity DNA polymerases YqjH and YqjW. DNA Repair (Amst) 70:1–9. doi: 10.1016/j.dnarep.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Uson ML, Carl A, Goldgur Y, Shuman S. 2018. Crystal structure and mutational analysis of Mycobacterium smegmatis FenA highlight active site amino acids and three metal ions essential for flap endonuclease and 5' exonuclease activities. Nucleic Acids Res 46:4164–4175. doi: 10.1093/nar/gky238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanders GM, Dallmann HG, McHenry CS. 2010. Reconstitution of the B. subtilis replisome with 13 proteins including two distinct replicases. Mol Cell 37:273–281. doi: 10.1016/j.molcel.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 44.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 45.Zwietering MH, Jongenburger I, Rombouts FM, van 't Riet K. 1990. Modeling of the bacterial growth curve. Appl Environ Microbiol 56:1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schroeder JW, Simmons LA. 2013. Complete genome sequence of Bacillus subtilis strain PY79. Genome Announc 1:e01085-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.