

Summary
Growing evidences are suggesting that extra-long genes in mammals are vulnerable for full-gene length transcription and dysregulation of long genes is a mechanism underlying human genetic disorders. How long-distance transcription is achieved is a fundamental question to be elucidated. In previous study, we had discovered that RNA-binding protein SFPQ preferentially binds to long pre-mRNAs and specifically regulates the cluster of neuronal genes >100 kbp. Here we investigated the roles of SFPQ for long gene expression, target specificities, and also physiological functions in skeletal muscle. Loss of Sfpq selectively downregulated genes >100 kbp including Dystrophin, which is 2.26 Mbp in length. Sfpq knockout (KO) mice showed progressive muscle mass reduction and metabolic myopathy characterized by glycogen accumulation and decreased abundance of mitochondrial oxidative phosphorylation complexes. Functional clustering analysis identified energy metabolism pathway genes as SFPQ's targets. These findings indicate target gene specificities and tissue-specific physiological functions of SFPQ in skeletal muscle.
Subject Areas: Molecular Biology, Molecular Mechanism of Gene Regulation, Pathophysiology
Graphical Abstract
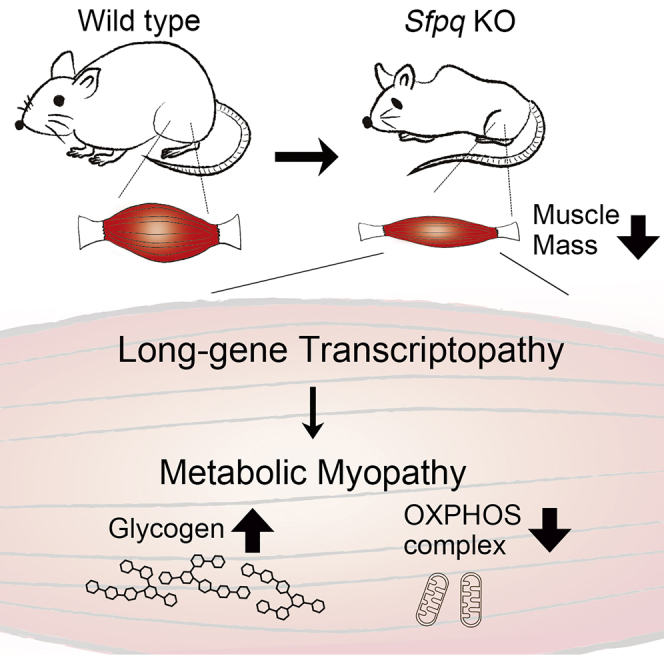
Highlights
-
•
SFPQ is essential for long gene expression, including Dystrophin, in skeletal muscle
-
•
Disruption of Sfpq caused severe muscle mass reduction and premature death
-
•
SFPQ is required for metabolic pathway gene expression in skeletal muscle
-
•
Loss of Sfpq decreased OXPHOS complexes and caused glycogen accumulation
Molecular Biology; Molecular Mechanism of Gene Regulation; Pathophysiology
Introduction
Evolutionarily, genome size and gene length of vertebrates are quite expanded and pre-mRNA transcripts expressed in mammals, especially in brains, are significantly longer (Gabel et al., 2015, Polymenidou et al., 2011). It has been suggested that extra-long genes are subjects of specific regulatory machineries that sustain their long-distance transcription. For long gene transcription, RNA-binding proteins (RBPs) or splicing factors have been demonstrated to play central roles as shown in SFPQ, FUS, TDP-43 (Lagier-Tourenne et al., 2012, Polymenidou et al., 2011, Takeuchi et al., 2018), or U1 snRNP (Oh et al., 2017). In addition, dysregulations of long gene transcription have been revealed as the causes of several genetic diseases in human, such as neurodegenerative and psychiatric diseases (Cortese et al., 2014, Gabel et al., 2015, King et al., 2013, Lagier-Tourenne et al., 2012, Polymenidou et al., 2011, Rogelj et al., 2012, Sugino et al., 2014).
In our previous study, we found that SFPQ is a key regulator of long genes in brain that sustains the full-gene length transcription through serially activating RNA polymerase II during transcriptional elongation. SFPQ preferentially binds to genes harboring GA- and CA-repeats in their long introns. Neuron-specific disruption of Sfpq resulted in the downregulation of long genes essential for brain formation and caused neural apoptosis. We have termed this selected disruption of long-gene expression as “long-gene transcriptopathy” (Takeuchi et al., 2018). Skeletal muscle is known to express several extra-long genes longer than 100 kbp, such as Dystrophin (Dmd, 2.26 Mbp), Titin (Ttn, 279 kbp), Dysferlin (Dysf, 202 kbp), and Sarcoglycan delta (Sgcd, 1.09 Mbp), which are essential for structures or motor functions of skeletal muscle cells, and mutations of those genes are known to cause muscular dystrophies (Savarese et al., 2016, Vainzof et al., 2008). In this study, we investigated whether SFPQ-dependent machinery for long genes expression is also essential for muscle cells, what is the regulatory target genes and physiological functions in skeletal muscle, and also whether dysregulations of long genes could cause disorders in muscle functions or diseases. Here we show that loss of Sfpq disturbed extra-long gene expression, indicating the significance of SFPQ for the full-length transcription of extra-long genes not only in the nervous system but also in muscle cells. Phenotypically, skeletal-muscle-specific disruption of Sfpq caused progressive muscle mass reduction. Mechanistically, SFPQ regulates the expression of multiple metabolic pathway genes that are identified by functional clustering analysis. Our results show that SFPQ-dependent transcriptional regulation of extra-long genes is required in muscle tissue and SFPQ plays crucial roles to regulate muscle-specific target genes essential for the energy metabolism and to maintain muscle mass.
Results
Loss of Sfpq Caused Severe Growth Defect
To elucidate whether SFPQ regulates muscle long genes and what the physiological role(s) of SFPQ is in skeletal muscle in vivo, we developed conditional gene-knockout (KO) mice for Sfpq. As Sfpq-null mutant mice in whole body caused early embryonic lethality (Takeuchi et al., 2018), we selectively deleted Sfpq gene in skeletal muscle by crossing Sfpq-floxed mice (Sfpqf/f) with Mlc1f-Cre transgenic mice. Mlc1f-Cre mice express Cre recombinase under the control of the myosin light chain (Mlc)1f promoter, which selectively expresses Cre in skeletal muscle except heart and thus enables us to analyze the physiological functions of SFPQ in skeletal muscle eliminating heart-related symptoms (Bothe et al., 2000). Homozygous KO mice (Sfpqf/f;Mlc1f-Cre) (KO mice) were born at the expected Mendelian ratios (Figure S1A) and were indistinguishable from littermates control mice, indicating that loss of Sfpq did not cause detectable abnormalities during the embryonic period. We next monitored the growth of KO mice during the postnatal period. Total body weight and appearance were indistinguishable from those of control mice until postnatal day 14 (P14). However, KO mice started to show a marked and progressing reduction in body weight around P16 relative to their littermate controls, whereas the body weight of heterozygous (Sfpqf/+;Mlc1f-Cre) mice did not differ from that of littermate controls (Figure 1A). KO mice caused sudden death starting from P33, and all of them had died by P50 (Figure 1B). KO mice showed smaller body size and pronounced kyphosis, which became evident around P30, the typical phenotypes frequently observed in association with muscle weakness (Figure 1C). Given that loss of Sfpq caused quite severe growth defects throughout the body with premature death, we examined whether KO mice selectively disrupted Sfpq gene only in skeletal muscle tissue using genomic PCR targeting the floxed allele of Sfpq. In KO mice, deletion of Sfpq gene was detected in all skeletal muscles examined, including the diaphragm (DIA), gastrocnemius (GC), tibialis anterior (TA), and soleus (SOL), but not in the heart, brain, liver, kidney, or spleen, confirming that observed systemic phenotype is originated from the skeletal muscle-specific disruption of the Sfpq gene (Figure S1B; KO allele). As we found remaining weak band from floxed alleles in skeletal muscle tissues (Figure S1B; floxed allele), we histologically accessed muscle-specific disruption of Sfpq in KO mice. SFPQ expression in myofibers was detected in nuclei that attach to the inside of the sarcolemma (Laminin2 positive plasma membrane of myofiber) as observed in control GC, TA, and SOL muscles (Figures S1C and S1D, open arrow heads). We also detected accumulation of nuclei in the fibroblasts or vascular cells in connective tissue between myofibers that are negative for laminin (Figure S1C, Control-GC). In KO mice, we found selective loss of SFPQ staining in the nuclei of myofibers of GC muscles with remaining expression of SFPQ in the connective tissues (Figure S1C, KO-GC), suggesting that Mlc1f-Cre mice efficiently disrupted SFPQ expression selectively in skeletal muscle cells by P30. From these observations, we confirmed that severe growth abnormalities and premature death were caused by the selective disruption of Sfpq in skeletal muscle tissues. Since Cre expression under the control of Mlc1f promoter is restricted to fast fibers (Mourkioti et al., 2008, Schiaffino and Reggiani, 1994), we examined whether efficiency of Cre excision is different between slow-twitch (SOL) and fast-twitch (TA) muscles. We observed SFPQ-positive nuclei in SOL in KO mice by immunostaining (Figure S1D, upper panels, open arrow heads in KO-SOL), indicating remaining expression of SFPQ. Furthermore, expression level of SFPQ protein was confirmed by western blotting (WB) (Figure S1D, lower panel, fold change [FC] for KO/Ct: 0.46 [TA] and 0.66 [SOL]), indicating the difference of SFPQ disruption between slow-twitch and fast-twitch muscles. These results are consistent with the previous report generating and characterizing Mlc1f-Cre mice (Bothe et al., 2000, Mourkioti et al., 2008, Patel et al., 2008), indicating that Cre excision is rather weak in slow-twitch muscle and intact in non-myogenic cells such as fibroblasts, Schwann cells, and satellite cells contained in skeletal muscle tissues, thus caused remaining weak band from floxed alleles in entire skeletal muscle tissues in genotype PCR.
Figure 1.

Sfpq Mutants Exhibit Skeletal Muscle Growth Defects with Kyphosis and Premature Death
(A) Growth charts of male skeletal muscle-specific Sfpq homozygous knockout mice (KO-homo) (n = 4), heterozygous knockout mice (KO-hetero) (n = 7), and littermate control mice (n = 16). Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 versus control (Student's or Welch's t test).
(B) Kaplan-Meier survival curves for male and female KO-homo (n = 14) and control (KO-hetero and others) (n = 27) mice.
(C) Kyphosis phenotype observed in P35 male KO mice.
(D) Body weight and weight of various skeletal muscles in male KO and control mice (n = 5). Representative skeletal muscle samples from KO and control mice (right). DIA, diaphragm; GC, gastrocnemius; SOL, soleus; TA, tibialis anterior. Data are presented as mean ± SD. ***p < 0.001 (Student's test).
(E) Grip strength test performance normalized to body weight of male KO (n = 19) and control (n = 20) mice from P26 to P30. Data are presented as mean ± SEM. ***p < 0.001 (Student's t test).
(F) Cross-section of laminin-stained TA muscle from 1-month-old male KO and control mice (left). Scale bars: 100 μm. Histogram of cross-sectional area (μm2) of laminin-stained myofibers from KO and control mice (right) (n = 3). Data are presented as mean ± SD. The difference of the distribution of cross-sectional area between KO and control mice was statistically significant (p < 0.001) (totally nine sections from three mice in each genotypes) using Mann-Whitney U test.
(G) Number of myofibers in TA muscle from 1-month-old male KO and control mice (n = 3, totally three sections from three mice in each genotype). Data are presented as mean ± SEM. There was no significant difference with Student's t test.
See also Figure S1.
Sfpq KO Mice Exhibited Reduced Muscle Mass
To further characterize skeletal muscle abnormalities in KO mice, muscle tissues were morphologically analyzed. At 1 month old (P30), the average body weight in KO mice was significantly reduced to 55% of that in littermate controls (Figure 1D, left bar graph). Accordingly, the mass of various types of skeletal muscles, including fast-twitch glycolytic muscles such as TA and GC, slow-twitch oxidative muscles such as SOL, and also mixed-fiber muscle of DIA, was about 45% lower in KO mice than in controls (Figure 1D, right bar graphs). As KO mice showed smaller body size than controls (Figure S1E), we examined whether loss of Sfpq caused systemic growth arrest by analyzing morphology of bone and brain. The length of long bones, such as the tibia, and the size and shape of the skull bone and brain was indistinguishable between KO mice and their littermate controls (Figure S1F), indicating that growth-hormone-dependent skeletal development was intact in KO mice. Instead, all KO mice exhibited growing pronounced kyphosis during development, which made the mice look smaller (Figure 1C).
Next, we assessed the muscle function in KO mice by several behavioral examinations. The grip-strength test was conducted to examine the muscle strength of forelegs. As the body weight in KO mice was relatively smaller than in control mice (Figure 1A), we evaluated the muscle strength by calculating force per body weight (Connolly et al., 2001). The grip strength per body weight of KO mice was significantly lower than that of controls (FC = 0.75; Figure 1E). We further examined the muscle force with endurance and motor skill by the hanging wire and the rotarod tests. Muscle strength and motor function in KO mice were significantly decreased with these tests (Figures S1G and S1H), indicating that loss of Sfpq caused muscle mass reduction with significant impairment of motor function.
To further investigate the cause of severe muscle mass reduction and impairment of motor function, we examined the number and size of myofiber using quantitative analysis of the cross-sectional area in TA muscle. The size of each myofiber was significantly reduced in KO compared with that in control mice (Figure 1F). When we counted the number of total myofibers in TA muscle samples, there was no difference with the myofiber number itself between KO and control mice (Figure 1G). Since the total number of myofibers is defined during the embryonic period and myofiber size is developed during the postnatal periods, it is indicated that loss of Sfpq does not affect myofiber generation but reduces the size of each myofiber, resulting in an overall decrease in body weight after P14.
Loss of Sfpq in Skeletal Muscle Causes Metabolic Myopathy but Not Typical Dystrophic Phenotypes
To elucidate the pathogenesis of Sfpq disruption to cause severe muscle mass reduction, histological analyses were conducted using standard H&E staining. Given that SFPQ is essential for regulating the transcription of extra-long genes in neural tissue (Takeuchi et al., 2018), it could be expected that loss of Sfpq disrupts the expression of long genes including Dmd in KO mice, which causes muscular dystrophy. Then we examined sections of TA and DIA muscles from KO mice from P14 to P30 (from starting point of body weight loss and before sudden death); however, H&E sections of TA and DIA muscles from P30 KO were indistinguishable from those in control mice except for the smaller fiber size observed in KO mice samples (Figure 2A). We could not detect any symptoms of myofiber degeneration such as inflammation and neutrophil infiltration, regeneration with centrally located myonuclei, fiber splitting, or replacement of myofibers with connective tissues. This result indicated that severe muscle mass reduction in KO mice was not caused as the symptoms of muscular dystrophy. A series of histopathological analyses was further conducted to characterize the observed myopathy in KO mice. In nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR) and modified Gomori trichrome staining, no obvious irregular intermyofibrillar network and abnormal aggregates, such as “raggedred” and “moth-eaten” fibers, were detected in the TA muscle sections from 1-month-old KO mice. However, myofibers that are rather densely stained with modified Gomori trichrome staining in TA muscle of KO mice were increased in number, indicating the increment in the proportion of oxidative fibers (Figure 2B, Gomori). The intensity with NADH-TR staining became weaker in TA muscle of KO mice, indicating the decrease of NADH dehydrogenase activity in mitochondria complex I (Figure 2B, NADH-TR). The result of Gomori trichrome staining suggests the possibility that loss of Sfpq caused fiber type change. Thus, we analyzed and quantified fiber type distribution using immunohistochemistry for myosin heavy chain (MyHC) expression. The number of MyHC I and IIA-positive fibers in the TA muscle was 4% and 26% higher, respectively, whereas the number of MyHC IIB-positive fibers was 10% lower in 1-month-old KO mice compared with those in control littermates (Figure 2C). Given that fibers expressing MyHC I and IIA have greater oxidative capacity than those expressing MyHC IIB, these findings indicate that loss of Sfpq induced a shift toward more oxidative fiber types, which is a well-observed phenomenon in myopathies. We further examined the amount of mitochondrial oxidative phosphorylation (OXPHOS) complexes from the observation of lower NADH dehydrogenase activity in mitochondria complex I of TA muscles in KO mice (Figure 2B). Then we found significant decreases in the abundance of OXPHOS complex I (35%), complex II (18%), and complex IV (35%) in TA muscle in KO relative to those in control mice (Figure 2D). We next examined the glycogen accumulation using periodic acid Schiff staining. Surprisingly, glycogen in entire GC muscle in KO was more intensively stained than that in control mice, indicating that loss of Sfpq caused excess glycogen accumulation (Figure 2E). We quantified the amount of glycogen content in GC muscle and found a 2.5-fold increase in KO compared with that in control mice (Figure 2F). Together, loss of Sfpq caused decreased OXPHOS complexes in mitochondria; in the meantime, excess glycogen was accumulated in skeletal muscles, indicating features of metabolic myopathy.
Figure 2.

Loss of Sfpq in Skeletal Muscle Causes Metabolic Myopathy but Not Typical Dystrophic Phenotypes
(A and B) (A) Representative cross-section of H&E-stained TA muscle from 1-month-old male KO and control mice. Scale bars: 100 μm. (B) Representative cross-section of modified Gomori trichrome stained TA muscle from 1-month-old male KO and control mice. Scale bars: 100 μm (left). Representative cross-section of NADH-TR stained TA muscle from 1-month-old male KO and control mice. Scale bars: 100 μm (right).
(C) Percentage distribution of MyHC isoforms I, IIA, and IIB in TA muscle of male KO and control mice (n = 3, totally three sections from three mice in each genotype). Data are presented as mean ± SD. *p < 0.05, **p < 0.01 (Student's t test).
(D) Representative results of WB using an OXPHOS antibody cocktail on TA muscle samples from male KO and control mice. Lamin B1 was used as a loading control. Signal intensities were quantified by densitometry (n = 3). CI, complex I (Ndufb8); CII, complex II (Sdhb); CIII, complex III (Uqcrc2); CIV, complex IV (mt-Co1); and CV, complex V (Atp5a1). Data are presented as mean ± SD. *p < 0.05, **p < 0.01 (Student's or Welch's t test).
(E) Representative periodic acid Schiff staining of GC sections from 1-month-old male KO and control mice. Scale bars: 100 μm.
(F) Glycogen content in GC muscles from 1-month-old male KO and control mice (n = 3). Data are presented as mean ± SD. **p < 0.01 (Student's t test).
Loss of Sfpq Specifically Downregulated Long Genes in Muscle Cells
We next tried to investigate whether SFPQ regulates specific target long genes in muscles using transcriptome study. Histological analyses revealed that the size of each myofiber was significantly reduced in KO mice and the relative amount of connective tissue and non-myogenic cells contained in muscle was much higher in KO than in control mice (Figure 1F). To eliminate the contamination of non-muscle cells in the transcriptome study, we decided to use primary myotubes isolated from KO and control mice for RNA sequencing (RNA-seq). We raised primary myoblasts from 1-month-old KO and control mice, induced differentiation into myotubes, and extracted total RNA for RNA-seq. SFPQ protein levels in Sfpq−/− myotubes (KO myotubes) were confirmed as ∼35% compared with control myotubes by WB, indicating that differentiation-induced Cre expression successfully disrupted Sfpq gene and decreased its protein level in KO myotubes (Figure S2A). Similarly, in vivo, we histologically examined SFPQ disruption in myotube and confirmed that SFPQ expression is selectively downregulated in the nuclei of MyHC-positive differentiated myotubes from Sfpq-KO mice (Figure S2B). In RNA-seq analysis using polyA-selected mRNAs for mature mRNA expression, observed gene expression changes between KO and control myotubes were quite small among 10,810 expressed genes (transcripts per million [TPM] ≥ 2.0 in either KO or control myotubes) (Figure S2C), which is consistent with our previous observation that SFPQ regulates limited numbers of target extra-long genes in embryonic mouse brains (Takeuchi et al., 2018). To see whether SFPQ specifically regulates long genes similarly in muscle, we examined the relationship between FC with pre-mRNA length of expressed genes. As gene length increased from 100 kbp and beyond, the FC in KO myotubes tended to be more negative (Figure 3A; median FC = 0.89 for genes 100–500 kbp, 0.79 for genes 500–1,000 kbp, and 0.59 for genes >1,000 kbp), indicating extra-long gene-specific downregulation. In brain, SFPQ-regulated genes caused gradual downregulation of pre-mRNAs when Sfpq is disrupted owing to the impairment of transcriptional elongation (Takeuchi et al., 2018). We next examined whether downregulated long genes in KO myotubes show this typical gradual downregulation by RNA-seq using rRNA depleted total RNAs for pre-mRNAs [RNA-Seq Ribo(−)]. For Dmd genes, pre-mRNA level was not downregulated or rather upregulated in the 5′ region (ratio >1.0) but decreased beyond the middle of the transcript close to 5′ region (ratio <1.0) and plateaued out to the 3′ end (Figure 3B, left panel). We further confirmed that the pre-mRNA ratio (KO/Control) of the 3′ site was significantly lower than that of the 5′ site using reverse transcriptase-quantitative PCR (RT-qPCR) (hereinafter called “3′-site downregulation”), indicating the impairment of transcriptional elongation of Dmd gene (Figure 3B, right panel). These results indicate that Sfpq is an essential regulator of transcriptional elongation of extra-long genes such as Dmd in skeletal muscle.
Figure 3.

Sfpq Deficiency Inhibits the Expression of Extra-Long Genes and the Metabolism-Related Gene Expression
(A) Box plots showing changes in gene expression in KO myotubes relative to control myotubes. Bins were defined according to gene length. *p < 0.01, **p < 0.001 for changes between two bins (Mann-Whitney U test).
(B) Pre-mRNA ratio of Dmd calculated by pre-mRNA levels in KO vs. control myotubes using Ribo(−) RNA-seq data (left) and the result of RT-qPCR for Dmd pre-mRNA between KO and control myotubes at the indicated positions (n = 3) (right). Data are presented as mean ± SD. **p < 0.01, ***p < 0.001 (Student's t test).
(C and D) List of significantly downregulated gene sets identified by GSEA analysis using KEGG (C) and Reactome (D) gene sets in KO as compared with control myotubes. Rank is based on the normalized enrichment score (NES) of each pathway. Gene sets (q < 0.01) are listed. FDR, false discovery rate; NOM, nominal. **p, q < 0.01, ***p, q < 0.001 (permutation test).
See also Figures S2–S4.
Sfpq Regulates Energy Metabolism Pathway Genes in Muscle Cells
In our histological analyses, KO mice did not show typical dystrophic phenotypes (Figure 2A). We examined the mature mRNA level of Dmd gene in GC muscles (P35) using RT-PCR and found that the Dmd mRNA level in KO was about 25% of that in control mice (FC = 0.25 vs. control mice, data not shown). As 25% of expression is still not critically low for causing muscular dystrophy, this can be the explanation why KO mice did not exhibit phenotypes that are typically observed in Dmd mutant mice (Vainzof et al., 2008, van Putten et al., 2012). Recent studies and our own have indicated that RBPs regulate functional clusters of genes known as regulons (Cosker et al., 2016, Keene, 2007, Takeuchi et al., 2018). To identify SFPQ regulons in skeletal muscle, gene set enrichment analysis (GSEA) was performed (Subramanian et al., 2005) using gene sets from the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome platforms (Kanehisa and Goto, 2000, Matthews et al., 2009). Gene sets significantly up- or downregulated in KO myotubes were identified using FDR (false discovery rate), or q values <0.01. In significantly downregulated genes, 15 and 9 pathways were identified using the KEGG and Reactome platforms, respectively. Surprisingly, 13 of the 15 downregulated gene sets in KEGG exclusively comprised metabolism-related gene clusters encompassing the tricarboxylic acid (TCA) cycle, pyruvate metabolism, OXPHOS, gluconeogenesis, and fatty acid metabolism, with the other two sets relating to Parkinson and Alzheimer diseases, which include mitochondria-related genes (Figures 3C and S2D). The nine downregulated gene sets enriched in Reactome comprised metabolism-related gene clusters relating to gluconeogenesis, glucose metabolism, TCA cycle, or respiratory electron transport (Figures 3D and S2D). Significantly upregulated gene sets were also identified using KEGG or Reactome, but none of them has a clear relation with muscle mass reduction (data not shown). Together, it is suggested that SFPQ regulates a wide range of energy metabolism pathway gene expression, and loss of Sfpq disturbed metabolic regulation leading to severe muscle mass reduction in KO mice.
Next, we focused individual downregulated genes in energy metabolism pathways. Using the threshold of FC < 0.769 (1/1.3) and q value <0.1 in all expressed genes (TPM ≥2), totally 32 genes were identified as significantly downregulated genes in GSEA, and 16 (50%) among them are longer than 100 kbp (Table S1). On the other hand, 1,027 genes were included in entire GSEA among expressed genes, however only 84 of them were longer than 100 kbp (8.2%) (Figure 4A), indicating that downregulated genes in GSEA are significantly enriched long gene >100 kbp as SFPQ target genes. We also found that 11 long genes were involved in pathways related to ATP production, i.e., glycogen, pyruvate, fatty acid, amino acid, and mitochondrial metabolism (Figure 4B and Table S1). Of the downregulated long genes longer than 100 kbp but not included in the GSEA-identified gene sets, three genes related to ATP production were significantly downregulated in KO relative to control myotubes; these encoded transcriptional regulators of mitochondria and oxidative metabolism (Ppargc1b [peroxisome proliferative activated receptor, gamma, coactivator 1 beta] [PGC1β]: 102 kbp, FC = 0.73, p < 0.01; Esrrb [estrogen-related receptor β]: 161 kbp, FC = 0.39, p < 0.001; and Esrrg [estrogen-related receptor γ]: 217 kbp, FC = 0.52, p < 0.001). Loss of PGC-1α/β and estrogen-related receptors (Esrrs) decreased mitochondrial OXPHOS complexes or changed the slow- or fast-myofiber type distribution (Gan et al., 2013, Gudiksen and Pilegaard, 2017, Zechner et al., 2010) as similarly observed in Sfpq-KO mice. To identify SFPQ target genes, we examined the pre-mRNA level of the downregulated metabolic pathway relating long genes (genes in Figure 4B, upper panel) in a similar way with Dmd gene using RT-qPCR (Figure 3B). Thirteen genes showing 3′-site downregulation were identified as SFPQ target genes (Figure 4B, lower panel) and highlighted with red characters (Figure 4B, upper panel). These results indicated that SFPQ is broadly involved in metabolic pathway regulations of skeletal muscle. Of 32 downregulated genes in GSEA in upper panel of Figure 4B, 7 were associated with inherited metabolic diseases according to the Human Gene Mutation Database (HGMD), and Phkb (phosphorylase kinase beta) and Suclg2 (succinate-Coenzyme A ligase, GDP-forming, beta subunit) are identified as SFPQ target genes (Figure 4B, lower panel, Figure S2E). Phkb is known as the causative gene for glycogen storage disease type IX (Beauchamp et al., 2007, Burwinkel et al., 1997) that is consistent with our observations of increased glycogen accumulation of GC muscle in KO mice (Figures 2E and 2F).
Figure 4.

Loss of Sfpq Downregulates Energy Metabolism Pathway Gene Expression in Muscle Cells
(A) Venn diagram of downregulated genes, genes≥100 kbp, and genes on GSEA identified pathways.
(B) Upper panel: Schematic diagram of downregulated genes in KO myotubes, which are GSEA-identified metabolic-pathway-related genes and also transcriptional regulators for mitochondrial activity. Asterisk indicates HGMD-identified inherited metabolic-diseases-related genes in Figure S2E. Lower panel: RT-qPCR for 5′ and 3′ sites of pre-mRNAs (n = 3). Pre-mRNA ratio (fold change) was calculated between KO and control myotubes for each position (Control = 1). Idh1 gene was used as negative control (the gene length <100 kbp). Genes showing 3′-site downregulation are highlighted with red characters in upper panel. Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 (Student's or Welch's t test).
See also Figures S2–S4.
Next, we confirmed downregulation of long genes and metabolic pathway genes in vivo using RNA-seq analysis in GC muscles from P35 male KO and control mice. First, the relationship between FC with pre-mRNA length of expressed genes were similarly analyzed in vitro. As gene length increased from 100 kbp and beyond, the FC tended to be more negative in KO GC muscle (Figure S3A), indicating long-gene transcriptopathy. We further examined mRNA expression changes of long genes relating to energy metabolism pathways that were identified in vitro and confirmed well-overlapped downregulated genes in vivo with in vitro (Figure S3B). Using RT-qPCR and WB, we validated the downregulation of mRNA and proteins of SFPQ target genes in vivo for the representative genes (Phkb, Me3 [malic enzyme 3, NADP(+)-dependent, mitochondrial], and Gphn [gephyrin]) (Figures S3C and S3D). These results indicated that metabolic-pathway-related genes were downregulated in vivo. We further assessed systemic energy metabolism by measuring blood glucose concentration in KO mice. In fasted condition, the blood glucose level was significantly lower in KO mice than in control mice (Figure S3E), indicating that loss of Sfpq impaired energy metabolism regulation in skeletal muscle, presumably increased glucose uptake, and further affected systemic energy regulation. This lower glucose level in KO mice potentially worsens muscle mass reduction and could be the cause of premature death.
We next tried to access why loss of Sfpq caused severe muscle mass reduction in KO mice. First, we examined the possibility of catabolic pathway activation (protein degradation pathway). We checked the mRNA expression of 18 genes relating to Ubiqutin-Proteasome, Calpine-Calpastatin, and Lysosome-Autophagy system (Carmignac et al., 2011, Stefanetti et al., 2015, Tipton et al., 2018, Yamamoto et al., 2015). There was limited upregulation only in Ctsl (Cathepsin L) (FC = 1.44), but the expression of all other genes was comparable between KO and control muscle (Figure S4A). In addition, we considered the possibility of dysfunction in anabolic pathway (protein synthesis). We examined protein synthesis in KO muscle by the surface sensing of translation (SUnSET) technique (Goodman et al., 2011). There was no significant reduction of puromycin-labeled peptides in KO muscle compared with controls, indicating that protein synthesis was not significantly impaired in KO muscles (Figure S4B). Together, it is indicated that both catabolic and anabolic pathways were not disturbed as the cause of severe muscle mass reduction in KO mice.
For further evaluation of muscle growth defect in KO mice, we determined the correlation between Sfpq and its target metabolic pathway relating genes during postnatal muscle development (P2, P7, P14, P21, and P28) in wild-type mice. Only modest change in the level of Sfpq mRNA was detected during postnatal muscle development, whereas that of all target genes except Plce1 and Hs6st2 were comprehensively upregulated during P14 to P28 (Figure S4C), consistent with our observation that body weight reduction in KO mice became evident in this stage. These results suggest that SFPQ is critically required for the upregulation of metabolic pathway genes and the muscle growth from P14.
Discussion
In this study, we found that skeletal muscle-specific disruption of Sfpq inhibited the expression of extra-long genes, indicating that SFPQ-dependent machinery is generally crucial for the full-length transcription of extra-long genes in muscle cells as we observed in the nervous system. We initially expected that extra-long genes such as Ttn, nebulin, Dysf, laminin α2, and Sgcd that are related to skeletal muscle structure or muscular dystrophy would be downregulated in KO mice, but this was not the case. Thus, Sfpq regulation does not extend to all long genes. In the developing brain, SFPQ regulates fewer than 10% of expressed long genes >100 kbp in length; a Gene Ontology analysis showed that target genes are related to neuronal development and maturation (Takeuchi et al., 2018). In skeletal muscle, we found that downregulated genes in KO myotube were highly enriched in metabolism such as glucose metabolism, TCA cycle, and respiratory electron transport indicating energy metabolism pathways as regulons of SFPQ. These results indicate that SFPQ specifically regulates tissue-specific target genes and is essential for regulating energy metabolism in skeletal muscle, which is closely related to the maintenance of muscle mass and motor function (Cunningham et al., 2007, Laplante and Sabatini, 2012, Mouisel et al., 2014). In addition, a recent study demonstrated that disruption of splicing factors Rbfox1 and Rbfox2 caused severe loss of muscle mass (Singh et al., 2018), indicating that RBPs are essential for the maintaining muscle mass as we observed in Sfpq-KO mice.
Energy metabolism in muscle is classified into two distinct energy-supplying systems: anaerobic metabolism, which provides energy from glycogenolysis/glycolysis for rapid movements, and aerobic metabolism, regulated by mitochondria for sustained exercise (Zierath and Hawley, 2004). The dependency on two energy-supplying systems are flexibly changed and compensated with each other for adapting to the energy requirement status such as exercises, whereas dysregulation of energy metabolism is associated with muscle mass reduction and dysfunction in pathophysiological conditions such as aging (sarcopenia), chronic diseases (cachexia), and atrophy of the other origin (Brocca et al., 2012, Carson et al., 2016, Dirks et al., 2006). Actn3 (Actinin alpha 3) is essential for glycogenolysis by activating glycogen phosphorylase. In Actn3-KO mice, glycogen conversion to glucose is impaired and glycogen accumulates in muscle, resulting in a slight reduction in muscle mass and decreased body weight (MacArthur et al., 2008, Quinlan et al., 2010), similar to what is observed in Sfpq-KO mice.
Regulation of energy metabolism is closely related to the maintenance of muscle mass and motor function and involves factors such as myostatin as well as IGF1-AKT-mTOR (insulin-like growth factor 1-AKT-mechanistic target of rapamycin) signaling pathway and its downstream transcriptional regulators PGC-1α and -1β (Cunningham et al., 2007, Laplante and Sabatini, 2012, Mouisel et al., 2014). For aerobic ATP production in mitochondria, PGC-1α and -1β play essential roles in coordinately activating nuclear and mitochondrial genes. Yy1 (Yin yang 1) is a transcription factor modulated by PGC-1α that regulates mitochondrial biogenesis. Skeletal-muscle-specific Yy1-knockout mice show decreases in mitochondrial OXPHOS complexes and have a dwarf-like appearance. Surprisingly, identified pathways downregulated in the skeletal muscles of Sfpq-KO mice well overlapped to those in Yy1-KO mice, indicating that genes regulating mitochondrial metabolism are synergistically regulated by transcription factor and RBP. Major differences in these two KO mice is that glycogenolysis was unaffected in Yy1-KO but was impaired in Sfpq-KO mice, and muscle mass and body weight in Yy1-KO mice are normal until 2 months of age (Blattler et al., 2012). This implies that diminished oxidative capacity can be compensated by glycogenolysis to meet energy requirements, resulting in milder phenotypes related to muscle mass and body weight in Yy1-KO as compared with Sfpq-KO mice. This is supported by a previous report that double knockout of PGC1α/β in skeletal muscle caused dysregulation of mitochondrial biogenesis but was indistinguishable from control mice in normal condition; however, these mice exhibited dramatic reductions in exercise tolerance owing to rapid depletion of muscle glycogen stores (Zechner et al., 2010).
Muscle mass is regulated by the signaling pathway of IGF1-AKT-mTOR (Schiaffino and Mammucari, 2011), and mTOR is a primary metabolic regulator of both glycogen and mitochondrial metabolism, indicating that maintenance of muscle mass is closely related to metabolism regulation. Mice with skeletal-muscle-specific mTOR-KO showed glycogen accumulation and impaired mitochondrial OXPHOS, which was accompanied by reduced muscle mass and body weight and premature death (Risson et al., 2009). Observed phenotypes in Sfpq-KO mice well overlapped with those in mTOR-KO mice, and these were much severe than those in Actn3-, Yy1-, and Pgc1α/β-KO mice. In Pgc1α/β- and Actn3-KO mice, either glycogenolysis or mitochondrial metabolism is increased, indicating that these metabolic pathways can mutually compensate each other and that disruption of both can potentially lead to more severe phenotypes as observed in Sfpq- or mTOR-KO mice. Additionally, patients with metabolic myopathies such as mitochondrial myopathies and glycogen storage diseases often present with growth failure (Blattler et al., 2012, Slonim et al., 1984, Wolny et al., 2009). These findings support our conjecture that dysregulated energy metabolism in skeletal muscle is an underlying cause of impaired postnatal skeletal muscle development and whole-body growth.
Loss of Sfpq downregulated genes the lengths of which are not extra-long. We had found that SFPQ co-transcriptionally activates the transcriptional elongation of pre-mRNAs possessing relatively longer introns (Takeuchi et al., 2018) and affects the expression of genes <100 kbp. Moreover, SFPQ has multifunction such as mRNA processing, transcriptional regulation, and DNA repair (Dong et al., 2007, Ha et al., 2011, Kaneko et al., 2007, Patton et al., 1993, Yarosh et al., 2015). It is possible that loss of Sfpq disrupted shorter gene expression and mRNA regulation and subsequently affected the phenotype. As we are analyzing the binding rules of SFPQ to pre-mRNAs and relationship between SFPQ bindings and mRNA regulations, further studies would decipher the roles of SFPQ more precisely in muscle energy metabolism.
SFPQ has been implicated in various diseases including familial amyotrophic lateral sclerosis (ALS) (Thomas-Jinu et al., 2017), frontotemporal lobar degeneration (Ishigaki et al., 2017), and autism spectrum disorder (Chang et al., 2015, O'Roak et al., 2012). About 40% of myopathies have yet to be genetically characterized (Risson et al., 2009). The disease-causing mechanism and pathogenesis of aging-related diseases are still largely unknown. Mitochondrial dysfunction is a cornerstone of aging and loss of muscle mass (Dirks et al., 2006, Konopka and Sreekumaran Nair, 2013). Thus, mutations in SFPQ or dysregulation of SFPQ-dependent energy metabolism could underlie idiopathic myopathies or sarcopenia (Risson et al., 2009, Thomas-Jinu et al., 2017). It remains unclear whether exercise tolerance and metabolic changes are reduced in KO mice, whether SFPQ actively regulates energy production in skeletal muscle, and whether therapeutic strategies targeting SFPQ are a viable treatment for metabolic myopathies that should be addressed with further studies. The finding of metabolic pathway gene expression maintained by SFPQ provides an insight into the synergistic network of energy metabolism by RBP with signal transduction and transcriptional regulations in skeletal muscle associating with muscle growth and maintenance and also pathogenesis of muscular dystrophies or metabolic myopathies.
Limitations of the Study
We demonstrated that loss of Sfpq in skeletal muscle caused long-gene transcriptopathy and downregulated metabolic pathway-relating genes. RBPs regulate broader genes and add variety and complexity. Thus, it is expected that RBPs plays multiple and essential roles; however, analyses of RBPs tend to be difficult to simply show their functions. We observed severe muscle mass reduction in KO mice but could not eliminate the possibility that effects other than long-gene transcriptopathy affect the observed phenotype. We are challenging to decipher the relationship between binding rules, target specificities, and functions of RBPs that would overcome these technical limits in the near future. In addition, loss of Sfpq caused premature death in KO mice with unknown reasons. Severe growth defect with reduced muscle mass, kyphosis, and dysregulation of systemic metabolism affect each other and might contribute to the observed phenotypes. Further study using a different Cre-line may help understand the cause of death and the network regulating muscle mass, muscle energy metabolism, and systemic homeostasis.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the Medical Research Support Center for performing high-throughput sequencing analysis and mouse behavioral tests and the Institute of Laboratory Animals, Graduate School of Medicine, Kyoto University, for providing animal care; Dr. Steven J. Burden of Skirball Institute for Molecular Medicine for providing Mlc1f-Cre transgenic mice; Dr. Satoru Noguchi and Dr. Miho Miyakawa of Department of Neuromuscular Research, National Institute of Neurosciences, National Center of Neurology and Psychiatry (NCNP) for technical advice in histological analysis; A. Hagiwara, K. Wanezaki, and Y. Takahashi for technical assistance; Dr. T. Tsubota, Dr. Y. Okuno, M. Denawa, Y. Sako, Y. Qi, and members of the M.H. laboratory at Kyoto University for their helpful comments and technical advice; and Editage (www.editage.jp) for English language editing. This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT; Japan Society for the Promotion of Science KAKENHI 19500269, 25500288, 21249013, 15H05721) (to M. Hagaiwara and A.T.); Innovative Cell Biology by Innovating Technology (Cell Innovation) (to M. Hagaiwara, K.O., and A.T.); a Core Research for Evolutional Science and Technology grant from the Japan Science and Technology Agency (to M. Hagaiwara); Intramural Research Grant 25-5 and 28-6 for Neurological and Psychiatric Disorders of NCNP (to M. Hagaiwara); a grant from the Japan Agency for Medical Research and Development (AMED) (to M. Hagaiwara); Platform for Dynamic Approaches to Living System from the MEXT and AMED (to M.H.); iCeMS Cross-Disciplinary Research Promotion Project of Kyoto University (to A.T.); and the Fujiwara Memorial Foundation (to A.T.).
Author Contributions
M. Hosokawa conducted most of the experiments, contributed to data analysis, and wrote the manuscript. A.T. conceived and designed the project, conducted animal experiments including the mouse survival assay, supervised all experiments, and prepared the manuscript. J.T. and S.T. provided technical supervision and assisted with muscle phenotype analyses in animal experiments. K.I. analyzed the high-throughput sequencing data. M. Hagiwara conceived the project and prepared the manuscript. All authors contributed to manuscript preparation.
Declaration of Interests
The authors declare no conflicts of interest.
Published: March 29, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.02.023.
Contributor Information
Akihide Takeuchi, Email: takeuchi.akihide.8r@kyoto-u.ac.jp.
Masatoshi Hagiwara, Email: hagiwara.masatoshi.8c@kyoto-u.ac.jp.
Supplemental Information
References
- Beauchamp N.J., Dalton A., Ramaswami U., Niinikoski H., Mention K., Kenny P., Kolho K.L., Raiman J., Walter J., Treacy E. Glycogen storage disease type IX: high variability in clinical phenotype. Mol. Genet. Metab. 2007;92:88–99. doi: 10.1016/j.ymgme.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Blattler S.M., Verdeguer F., Liesa M., Cunningham J.T., Vogel R.O., Chim H., Liu H., Romanino K., Shirihai O.S., Vazquez F. Defective mitochondrial morphology and bioenergetic function in mice lacking the transcription factor Yin Yang 1 in skeletal muscle. Mol. Cell. Biol. 2012;32:3333–3346. doi: 10.1128/MCB.00337-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothe G.W., Haspel J.A., Smith C.L., Wiener H.H., Burden S.J. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26:165–166. [PubMed] [Google Scholar]
- Brocca L., Cannavino J., Coletto L., Biolo G., Sandri M., Bottinelli R., Pellegrino M.A. The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J. Physiol. 2012;590:5211–5230. doi: 10.1113/jphysiol.2012.240267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burwinkel B., Maichele A.J., Aagenaes O., Bakker H.D., Lerner A., Shin Y.S., Strachan J.A., Kilimann M.W. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB) Hum. Mol. Genet. 1997;6:1109–1115. doi: 10.1093/hmg/6.7.1109. [DOI] [PubMed] [Google Scholar]
- Carmignac V., Svensson M., Korner Z., Elowsson L., Matsumura C., Gawlik K.I., Allamand V., Durbeej M. Autophagy is increased in laminin alpha2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 2011;20:4891–4902. doi: 10.1093/hmg/ddr427. [DOI] [PubMed] [Google Scholar]
- Carson J.A., Hardee J.P., VanderVeen B.N. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin. Cell Dev. Biol. 2016;54:53–67. doi: 10.1016/j.semcdb.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Gilman S.R., Chiang A.H., Sanders S.J., Vitkup D. Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci. 2015;18:191–198. doi: 10.1038/nn.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly A.M., Keeling R.M., Mehta S., Pestronk A., Sanes J.R. Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha2-deficient mice. Neuromuscul. Disord. 2001;11:703–712. doi: 10.1016/s0960-8966(01)00232-2. [DOI] [PubMed] [Google Scholar]
- Cortese A., Plagnol V., Brady S., Simone R., Lashley T., Acevedo-Arozena A., de Silva R., Greensmith L., Holton J., Hanna M.G. Widespread RNA metabolism impairment in sporadic inclusion body myositis TDP43-proteinopathy. Neurobiol. Aging. 2014;35:1491–1498. doi: 10.1016/j.neurobiolaging.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosker K.E., Fenstermacher S.J., Pazyra-Murphy M.F., Elliott H.L., Segal R.A. The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat. Neurosci. 2016;19:690–696. doi: 10.1038/nn.4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham J.T., Rodgers J.T., Arlow D.H., Vazquez F., Mootha V.K., Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature. 2007;450:736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- Dirks A.J., Hofer T., Marzetti E., Pahor M., Leeuwenburgh C. Mitochondrial DNA mutations, energy metabolism and apoptosis in aging muscle. Ageing Res. Rev. 2006;5:179–195. doi: 10.1016/j.arr.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Dong X., Sweet J., Challis J.R., Brown T., Lye S.J. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell. Biol. 2007;27:4863–4875. doi: 10.1128/MCB.02144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel H.W., Kinde B., Stroud H., Gilbert C.S., Harmin D.A., Kastan N.R., Hemberg M., Ebert D.H., Greenberg M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522:89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z., Rumsey J., Hazen B.C., Lai L., Leone T.C., Vega R.B., Xie H., Conley K.E., Auwerx J., Smith S.R. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J. Clin. Invest. 2013;123:2564–2575. doi: 10.1172/JCI67652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman C.A., Mabrey D.M., Frey J.W., Miu M.H., Schmidt E.K., Pierre P., Hornberger T.A. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J. 2011;25:1028–1039. doi: 10.1096/fj.10-168799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudiksen A., Pilegaard H. PGC-1alpha and fasting-induced PDH regulation in mouse skeletal muscle. Physiol. Rep. 2017;5:e13222. doi: 10.14814/phy2.13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K., Takeda Y., Dynan W.S. Sequences in PSF/SFPQ mediate radioresistance and recruitment of PSF/SFPQ-containing complexes to DNA damage sites in human cells. DNA Repair (Amst.) 2011;10:252–259. doi: 10.1016/j.dnarep.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki S., Fujioka Y., Okada Y., Riku Y., Udagawa T., Honda D., Yokoi S., Endo K., Ikenaka K., Takagi S. Altered Tau Isoform ratio caused by loss of FUS and SFPQ function leads to FTLD-like phenotypes. Cell Rep. 2017;18:1118–1131. doi: 10.1016/j.celrep.2017.01.013. [DOI] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S., Rozenblatt-Rosen O., Meyerson M., Manley J.L. The multifunctional protein p54nrb/PSF recruits the exonuclease XRN2 to facilitate pre-mRNA 3' processing and transcription termination. Genes Dev. 2007;21:1779–1789. doi: 10.1101/gad.1565207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene J.D. RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 2007;8:533–543. doi: 10.1038/nrg2111. [DOI] [PubMed] [Google Scholar]
- King I.F., Yandava C.N., Mabb A.M., Hsiao J.S., Huang H.S., Pearson B.L., Calabrese J.M., Starmer J., Parker J.S., Magnuson T. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501:58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka A.R., Sreekumaran Nair K. Mitochondrial and skeletal muscle health with advancing age. Mol. Cell. Endocrinol. 2013;379:19–29. doi: 10.1016/j.mce.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C., Polymenidou M., Hutt K.R., Vu A.Q., Baughn M., Huelga S.C., Clutario K.M., Ling S.C., Liang T.Y., Mazur C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012;15:1488–1497. doi: 10.1038/nn.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M., Sabatini D.M. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur D.G., Seto J.T., Chan S., Quinlan K.G., Raftery J.M., Turner N., Nicholson M.D., Kee A.J., Hardeman E.C., Gunning P.W. An Actn3 knockout mouse provides mechanistic insights into the association between alpha-actinin-3 deficiency and human athletic performance. Hum. Mol. Genet. 2008;17:1076–1086. doi: 10.1093/hmg/ddm380. [DOI] [PubMed] [Google Scholar]
- Matthews L., Gopinath G., Gillespie M., Caudy M., Croft D., de Bono B., Garapati P., Hemish J., Hermjakob H., Jassal B. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009;37:D619–D622. doi: 10.1093/nar/gkn863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouisel E., Relizani K., Mille-Hamard L., Denis R., Hourde C., Agbulut O., Patel K., Arandel L., Morales-Gonzalez S., Vignaud A. Myostatin is a key mediator between energy metabolism and endurance capacity of skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014;307:R444–R454. doi: 10.1152/ajpregu.00377.2013. [DOI] [PubMed] [Google Scholar]
- Mourkioti F., Slonimsky E., Huth M., Berno V., Rosenthal N. Analysis of CRE-mediated recombination driven by myosin light chain 1/3 regulatory elements in embryonic and adult skeletal muscle: a tool to study fiber specification. Genesis. 2008;46:424–430. doi: 10.1002/dvg.20419. [DOI] [PubMed] [Google Scholar]
- O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.M., Di C., Venters C.C., Guo J., Arai C., So B.R., Pinto A.M., Zhang Z., Wan L., Younis I. U1 snRNP telescripting regulates a size-function-stratified human genome. Nat. Struct. Mol. Biol. 2017;24:993–999. doi: 10.1038/nsmb.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S., Doble B.W., MacAulay K., Sinclair E.M., Drucker D.J., Woodgett J.R. Tissue-specific role of glycogen synthase kinase 3beta in glucose homeostasis and insulin action. Mol. Cell. Biol. 2008;28:6314–6328. doi: 10.1128/MCB.00763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J.G., Porro E.B., Galceran J., Tempst P., Nadal-Ginard B. Cloning and characterization of PSF, a novel pre-mRNA splicing factor. Genes Dev. 1993;7:393–406. doi: 10.1101/gad.7.3.393. [DOI] [PubMed] [Google Scholar]
- Polymenidou M., Lagier-Tourenne C., Hutt K.R., Huelga S.C., Moran J., Liang T.Y., Ling S.C., Sun E., Wancewicz E., Mazur C. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011;14:459–468. doi: 10.1038/nn.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan K.G., Seto J.T., Turner N., Vandebrouck A., Floetenmeyer M., Macarthur D.G., Raftery J.M., Lek M., Yang N., Parton R.G. Alpha-actinin-3 deficiency results in reduced glycogen phosphorylase activity and altered calcium handling in skeletal muscle. Hum. Mol. Genet. 2010;19:1335–1346. doi: 10.1093/hmg/ddq010. [DOI] [PubMed] [Google Scholar]
- Risson V., Mazelin L., Roceri M., Sanchez H., Moncollin V., Corneloup C., Richard-Bulteau H., Vignaud A., Baas D., Defour A. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009;187:859–874. doi: 10.1083/jcb.200903131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogelj B., Easton L.E., Bogu G.K., Stanton L.W., Rot G., Curk T., Zupan B., Sugimoto Y., Modic M., Haberman N. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci. Rep. 2012;2:603. doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese M., Sarparanta J., Vihola A., Udd B., Hackman P. Increasing role of titin mutations in neuromuscular disorders. J. Neuromuscul. Dis. 2016;3:293–308. doi: 10.3233/JND-160158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S., Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet. Muscle. 2011;1:4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S., Reggiani C. Myosin isoforms in mammalian skeletal muscle. J. Appl. Physiol. (1985) 1994;77:493–501. doi: 10.1152/jappl.1994.77.2.493. [DOI] [PubMed] [Google Scholar]
- Singh R.K., Kolonin A.M., Fiorotto M.L., Cooper T.A. Rbfox-splicing factors maintain skeletal muscle mass by regulating calpain3 and proteostasis. Cell Rep. 2018;24:197–208. doi: 10.1016/j.celrep.2018.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonim A.E., Coleman R.A., Moses W.S. Myopathy and growth failure in debrancher enzyme deficiency: improvement with high-protein nocturnal enteral therapy. J. Pediatr. 1984;105:906–911. doi: 10.1016/s0022-3476(84)80075-x. [DOI] [PubMed] [Google Scholar]
- Stefanetti R.J., Lamon S., Wallace M., Vendelbo M.H., Russell A.P., Vissing K. Regulation of ubiquitin proteasome pathway molecular markers in response to endurance and resistance exercise and training. Pflugers Arch. 2015;467:1523–1537. doi: 10.1007/s00424-014-1587-y. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K., Hempel C.M., Okaty B.W., Arnson H.A., Kato S., Dani V.S., Nelson S.B. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci. 2014;34:12877–12883. doi: 10.1523/JNEUROSCI.2674-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A., Iida K., Tsubota T., Hosokawa M., Denawa M., Brown J.B., Ninomiya K., Ito M., Kimura H., Abe T. Loss of Sfpq causes long-gene transcriptopathy in the brain. Cell Rep. 2018;23:1326–1341. doi: 10.1016/j.celrep.2018.03.141. [DOI] [PubMed] [Google Scholar]
- Thomas-Jinu S., Gordon P.M., Fielding T., Taylor R., Smith B.N., Snowden V., Blanc E., Vance C., Topp S., Wong C.H. Non-nuclear pool of splicing factor SFPQ regulates axonal transcripts required for normal motor development. Neuron. 2017;94:322–336.e5. doi: 10.1016/j.neuron.2017.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton K.D., Hamilton D.L., Gallagher I.J. Assessing the role of muscle protein breakdown in response to nutrition and exercise in humans. Sports Med. 2018;48(Suppl 1):53–64. doi: 10.1007/s40279-017-0845-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainzof M., Ayub-Guerrieri D., Onofre P.C., Martins P.C., Lopes V.F., Zilberztajn D., Maia L.S., Sell K., Yamamoto L.U. Animal models for genetic neuromuscular diseases. J. Mol. Neurosci. 2008;34:241–248. doi: 10.1007/s12031-007-9023-9. [DOI] [PubMed] [Google Scholar]
- van Putten M., Hulsker M., Nadarajah V.D., van Heiningen S.H., van Huizen E., van Iterson M., Admiraal P., Messemaker T., den Dunnen J.T., t Hoen P.A. The effects of low levels of dystrophin on mouse muscle function and pathology. PLoS One. 2012;7:e31937. doi: 10.1371/journal.pone.0031937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolny S., McFarland R., Chinnery P., Cheetham T. Abnormal growth in mitochondrial disease. Acta Paediatr. 2009;98:553–554. doi: 10.1111/j.1651-2227.2008.01148.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto J., Kamata S., Miura A., Nagata T., Kainuma R., Ishii I. Differential adaptive responses to 1- or 2-day fasting in various mouse tissues revealed by quantitative PCR analysis. FEBS Open Bio. 2015;5:357–368. doi: 10.1016/j.fob.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarosh C.A., Iacona J.R., Lutz C.S., Lynch K.W. PSF: nuclear busy-body or nuclear facilitator? Wiley Interdiscip. Rev. RNA. 2015;6:351–367. doi: 10.1002/wrna.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner C., Lai L., Zechner J.F., Geng T., Yan Z., Rumsey J.W., Collia D., Chen Z., Wozniak D.F., Leone T.C. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 2010;12:633–642. doi: 10.1016/j.cmet.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierath J.R., Hawley J.A. Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS Biol. 2004;2:e348. doi: 10.1371/journal.pbio.0020348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.