

Abstract
T helper 17 (Th17)‐cell differentiation triggered by interleukin‐6 (IL‐6) via STAT3 activation promotes inflammation in inflammatory bowel disease (IBD) patients. However, leukemia inhibitory factor (LIF), an IL‐6 family cytokine, restricts inflammation by blocking Th17‐cell differentiation via an unknown mechanism. Here, we report that microbiota dysregulation promotes LIF secretion by intestinal epithelial cells (IECs) in a mouse colitis model. LIF greatly activates STAT4 phosphorylation on multiple SPXX elements within the C‐terminal transcription regulation domain. STAT4 and STAT3 act reciprocally on both canonical cis‐inducible elements (SIEs) and noncanonical “AGG” elements at different loci. In lamina propria lymphocytes (LPLs), STAT4 activation by LIF blocks STAT3‐dependent Il17a/Il17f promoter activation, whereas in IECs, LIF bypasses the extraordinarily low level of STAT4 to induce YAP gene expression via STAT3 activation. In addition, we found that the administration of LIF is sufficient to restore microbiome homeostasis. Thus, LIF effectively inhibits Th17 accumulation and promotes repair of damaged intestinal epithelium in inflamed colon, serves as a potential therapy for IBD.
Keywords: intestinal inflammation, intestinal repair, LIF, STAT4, Th17‐cell differentiation
Subject Categories: Immunology, Signal Transduction
Introduction
The intestinal epithelial cells (IECs) and lamina propria lymphocytes (LPLs) of the gastrointestinal tract form the main immunoepithelial frontier against frequent stress from the intestinal microbiota. The intestinal epithelial interface of IECs is populated by numerous commensal microbes that aid in food digestion and markedly influence the functions of both IECs and LPLs (Mowat, 2003). During inflammation, however, microbial invasion activates the innate immune cells located in the lamina propria to initiate early inflammation through pathogen‐recognition receptors (Mankertz & Schulzke, 2007). This chronic and sustained inflammation promotes the activation of the adaptive immune response (Bouma & Strober, 2003; Grivennikov et al, 2012). The robust response of the immunoepithelial frontier system synthesizes and triggers a cytokine storm involving different proinflammatory cytokines and chemokines that contribute to alterations in the intestinal microbiota profile. The altered microbiota initiates and perpetuates intestinal inflammation, leading to inflammatory bowel disease (IBD) (Strober & Fuss, 2011). Among the helper CD4+ T cells involved in IBD induction, T helper 17 (Th17) cells are responsible for inducing both the proinflammatory cytokine storm and colitis (Smith & Garrett, 2011; Belkaid & Hand, 2014). Hence, Th17 cells are responsible for driving inflammatory or autoimmune responses.
Th17‐cell differentiation during inflammation is also cytokine‐dependent. IL‐6 and IL‐23 are associated with many inflammatory disorders, presumably due to their robust effects on the induction of pathogenic Th17‐cell differentiation. Some cytokines promote Th17‐cell differentiation, whereas others inhibit or terminate Th17‐cell differentiation (Park et al, 2005; Yang et al, 2011). Therefore, CD4+ T‐cell differentiation largely depends on the cytokines present in the cellular microenvironment (O'Shea & Paul, 2010). IL‐23 and the IL‐23 receptor are required for the pathology of both IBD and dextran sulfate sodium (DSS)‐induced colitis (Hue et al, 2006; Yen et al, 2006; Elson et al, 2007; Ahern et al, 2010). The secretion of proinflammatory cytokines, including IL‐17A, IL‐17F, and IL‐21, is markedly increased in IBD patients (Monteleone et al, 2005, 2006; Siakavellas & Bamias, 2012). Hence, Th17 cells are the principal effector helper T cell during IBD pathogenesis.
STAT proteins play critical roles in T‐cell differentiation. While Th17 differentiation relies on STAT3 activation by IL‐6, Th1 and Th2 differentiation rely on STAT4 activation by IL‐12 and STAT6 activation by IL‐4, respectively. STAT3 activation by IL‐6 in intestinal inflammation promotes Th17 differentiation and epithelial cell proliferation. Leukemia inhibitory factor (LIF) is a member of the IL‐6 cytokine family and shares specificity with IL‐6 for binding to the signaling receptor gp130 (Garbers et al, 2012). However, neural progenitor cell‐secreted LIF can inhibit Th17‐cell differentiation (Cao et al, 2011). Thus, LIF opposes IL‐6 in the regulation of T‐cell differentiation. The LIF expression level in the colon increases significantly in patients with ulcerative colitis (UC), which belongs to the IBD and is histologically characterized by the infiltration of immune cells, including neutrophils, macrophages, and lymphocytes, into the colonic mucosa (Guimbaud et al, 1998; Podolsky, 2002). However, whether and how LIF affects IBD progression is unclear. We recently reported that LIF can maintain the self‐renewal of mouse embryonic stem cells via activating cytokine storm (Wang et al, 2017b). In a CD4+ T‐cell transfer colitis model, STAT4 deficiency protects mice against colitis pathology (Harbour et al, 2015). However, blocking the IFNγ released from Th1 cells does not noticeably prevent colitis progression (Simpson et al, 1998), suggesting that the function of STAT4 in colitis development does not rely entirely on Th1‐type cytokine production.
To uncover the mechanism by which LIF affects Th17‐cell differentiation, we investigated LIF secretion and LIF‐modulated signal transduction and gene regulation. Lipopolysaccharide (LPS) was previously noted to stimulate IL‐6 and IL‐23 production in dendritic cells (DCs) (Xiao et al, 2008; Chang et al, 2010). In this work, we found that microbial dysregulation and LPS induced LIF expression and secretion in mouse IECs. LIF is a cytokine in the IL‐6 family, which mainly activates the JAK‐STAT3 pathway for signal transduction and transcriptional regulation. Surprisingly, the STAT activation pattern of LIF was quite different in LPLs and IECs. In LPLs, LIF mainly activated STAT4 to downregulate STAT3 activity in Il17 promoter regulation. In IECs, however, LIF mainly activated STAT3 to overcome the effects of STAT4 and induce Yes‐associated protein (YAP) expression, thus promoting epithelial proliferation. Consequently, the repair of damaged intestinal epithelia guarantees intestinal microbiota homeostasis during the amelioration of intestinal injury in response to LIF treatment.
Results
Bacterial endotoxin promotes the secretion of LIF by IECs in the inflamed colon
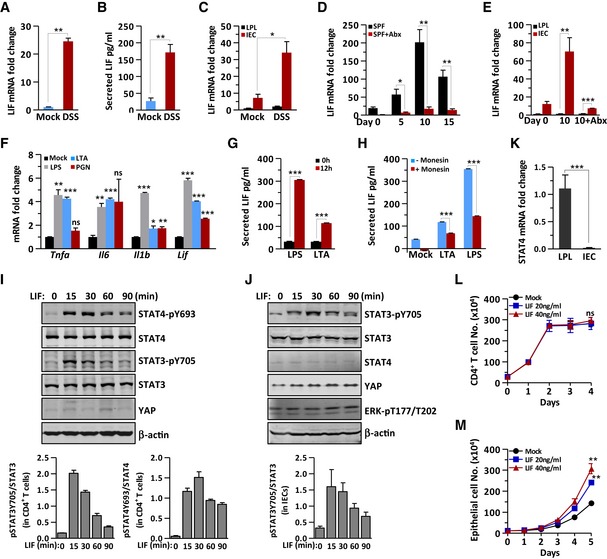
To investigate the potential role of LIF in mucosal immunity, we started by analyzing the LIF expression profile in a mouse colitis model. In mouse colon tissue with DSS‐induced peak disease severity, LIF and LIFR gene expression was markedly elevated (Figs 1A and EV1A), and the LIF protein was detectable in colon explant supernatants (Fig 1B). Notably, gp130 expression showed little increase in the inflamed colon compared to that in the healthy mouse colon (Fig EV1A). According to the RT–PCR analysis of IECs and LPLs isolated from normal mice or DSS‐treated mice, LIF was significantly increased in IECs but not in LPLs (Fig 1C). When antibiotics were used to remove the intestinal microbiota in specific pathogen‐free (SPF) mice, followed by DSS treatment, LIF expression was not induced in the colon of microbiota‐free mice (Fig 1D and E), indicating that microbiota invasion in the inflamed colon is required for LIF induction. Mice with colitis show a dysregulated microbiota composition with invasion into the intestinal mucus layer presumably via Toll‐like receptor (TLR) pathway activation (Mankertz & Schulzke, 2007). Primary mouse IECs were isolated and incubated with bacterial endotoxins such as LPS, peptidoglycan (PGN), or lipoteichoic acid (LTA). In addition to Tnfa, Il6, and Il1b expression, LIF expression was induced by LPS and, to a lesser extent, by LTA or PGN stimulation in IECs (Fig 1F). In addition, LIFR expression increased in LPS‐treated IECs (Fig EV1B). Consequently, LIF secretion was enhanced dramatically in IEC supernatants upon treatment with LPS and, to a lesser extent, LTA (Fig 1G). However, when we used monensin, which is a protein transport inhibitor, to pretreat IECs, the pretreated IECs secreted less LIF than untreated cells under either LPS or LTA stimulation (Fig 1H). Our data suggested that microbiota invasion into the mouse colon is required for IECs to secrete LIF; thus, we next focused on analyzing the physiological function of LIF in regulating colitis progression.
Figure 1. Bacterial dysregulation promotes the secretion of LIF by IECs, which differentially affects immune cells and epithelial cells.
-
AQuantitative expression of Lif mRNA in the colon of DSS‐challenged and control mice (n = 6 per group).
-
BLIF measured by ELISA in mouse colon explant supernatants from DSS‐challenged and control mice (n = 4 per group).
-
CQuantitative expression of Lif mRNA in LPLs and IECs from DSS‐challenged and control mice (n = 4 per group).
-
DQuantitative expression of Lif mRNA in the colon of SPF mice treated with or without antibiotics (Abx) at the indicated time points during colitis induction (n = 3 per group).
-
EQuantitative expression of Lif mRNA in LPLs and IECs from mice treated with or without Abx at the indicated time points during colitis induction (n = 3 per group).
-
FmRNA expression of the indicated genes in IECs stimulated by LPS (2.5 μg/ml), PGN (3 μg/ml), or LTA (2 μg/ml) for 2 h.
-
G, HSerum concentrations of LIF in IEC supernatants untreated (G) or pretreated with monensin (H) followed by LPS (2.5 μg/ml) or LTA (2 μg/ml) for 12 h, as determined by ELISA. The data are representative of three independent experiments.
-
I, JImmunoblot analysis of the expression and modification of the indicated proteins in the total cell lysate of CD4+ T cells (I) and IECs (J) treated with LIF (20 ng/ml) at different time points. The lower panel shows the intensity analysis of the bands from three independent experiments. The data are representative of three independent experiments.
-
KStat4 mRNA expression in LPLs and IECs (n = 6).
-
L, MThe proliferation rate of CD4+ T cells (L) and DLD‐1 cells (M) was determined in the presence or absence of LIF.
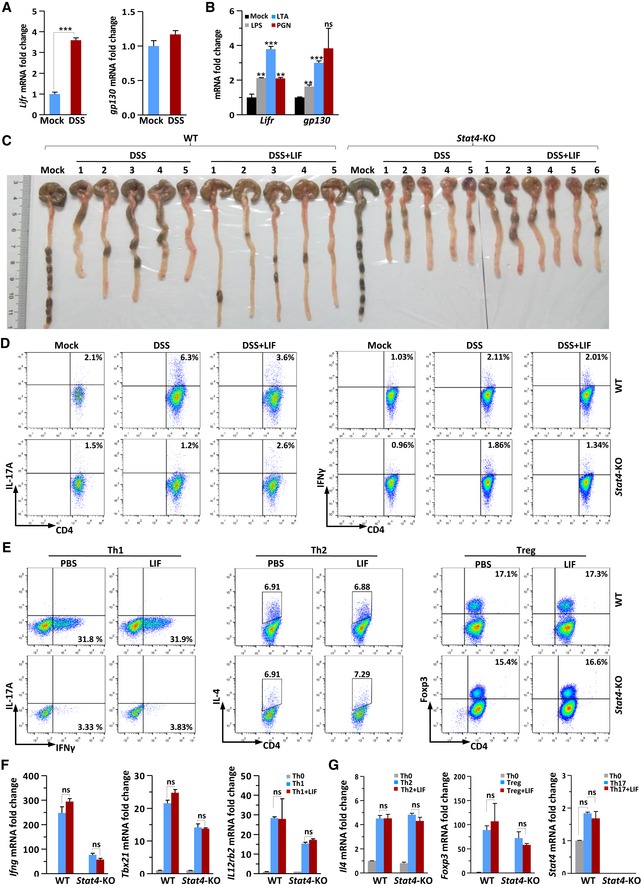
Figure EV1. LIF exhibits therapeutic effects in the wild‐type mouse colitis model and shows minor effect on Th1‐, Th2‐, or Treg‐cell differentiation.
-
A, BQuantitative expression of Lifr and gp130 mRNA in colon tissue from DSS‐challenged or control mice (n = 6 per group) (A) or in IECs following stimulation by LPS (2.5 μg/ml), PGN (3 μg/ml), or LTA (2 μg/ml) for 2 h (B).
-
CMacroscopic changes in the colon of WT and Stat4‐KO mice on day 14 of colitis induction.
-
DFACS staining of LPLs isolated on day 10 from the colon of WT and Stat4‐KO colitis mice receiving PBS or LIF (n = 4 per group). The percentage of IL‐17A+ (left) and IFNγ+CD4+ (right) T cells in vivo was analyzed.
-
EFACS staining of WT and Stat4‐KO naïve CD4+ T cells treated or not treated with LIF on day 4 of induction into Th1‐, Th2‐, or Treg‐cell subsets.
-
F, GqPCR analysis of Th1, Th2, or Treg signature gene expression in WT and Stat4‐KO naïve CD4+ T cells on day 4 of induction into the indicated subsets in the absence or presence of LIF. The data are representative of three independent experiments.
High LIF and LIFR expression in the inflamed colon differentially regulates CD4+ T cells and IECs
The upregulation of LIF in the colon of mice with colitis indicated that LIF might display a biological function in colon tissue. IL‐6 family cytokines bear similar STAT activation patterns (Stahl et al, 1995). Surprisingly, in CD4+ T cells, LIF strongly activated STAT4 in addition to STAT3 (Fig 1I), which has not been reported before, whereas LIF activated only STAT3 in IECs (Fig 1J). This differential STAT4 activation pattern is apparently due to the differential STAT4 expression profile in different types of cells (Fig 1K), in agreement with the idea that STAT4 is mainly expressed in lymphocytes (Frucht et al, 2000). While LIF showed no effect on the CD4+ T‐cell proliferation (Fig 1L), LIF promoted colon epithelial cell proliferation in a dose‐dependent manner (Fig 1M). In contrast, YAP expression, which is induced by IL‐6 and is critical for intestinal epithelial wound healing (Taniguchi et al, 2015), was induced by LIF in epithelial cells but not in immune cells (Fig 1I and J). YAP expression in T cells was quite low (Fig 1I); its paralog, transcriptional coactivator with postsynaptic density 65‐disk large‐zonula occludens 1‐binding (PDZ) motif (TAZ), plays a pivotal role in regulating the differentiation of Treg cells and Th17 cells, but YAP is not involved in this process (Geng et al, 2017). Taken together, these results indicate that LIF activates distinct signaling pathways in different cell types in colon tissue.
Administration of LIF aids mouse colitis remission
Considering that LIF induced STAT4 activation in CD4+ T cells, to verify whether LIF affects intestinal colitis progression via STAT4 involvement, we used DSS to induce colitis in wild‐type (WT) and Stat4 knockout (Stat4 −/−) mice, followed by intraperitoneal injection of LIF or phosphate‐buffered saline (PBS) as a control. Interestingly, we found that although STAT4 deficiency led to enhanced pathology in the colitis model, LIF exhibited a therapeutic effect during colitis pathogenesis only in wild‐type mice (Figs 2A–E and EV1C). LIF‐treated wild‐type colitis mice displayed a less severe decrease in body weight (Fig 2A) and a shorter length of colon blockage than mice treated with DSS only (Fig 2B and C). The disease activity index (DAI), including rectal bleeding and diarrhea, was significantly reduced (Fig 2D), intestinal epithelial damage was decreased, and goblet cells were restored in LIF‐treated wild‐type mice compared to these parameters in DSS‐only‐treated mice (Fig 2E). The apparent amelioration effect of LIF was also achieved in another mouse colitis model, i.e., Rag1 −/− mice with colitis initiated by naïve CD4+ T‐cell transfer (Fig 2F–I). Genetic ablation of STAT4 worsened colitis pathology; however, the disease severity in Stat4 −/− mice showed no apparent remission in response to LIF injection (Fig 2A–E). These results suggested that LIF had a protective role in colitis pathogenesis via STAT4 activation.
Figure 2. LIF administration promotes the amelioration of intestinal colitis.
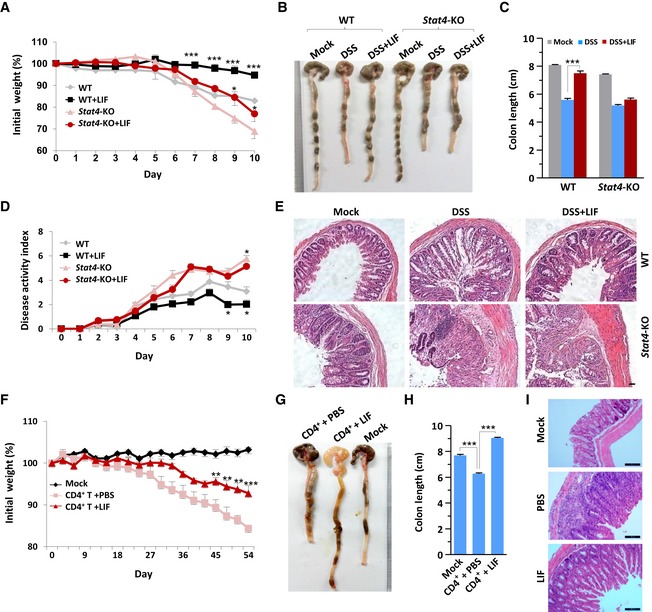
- Weight loss of wild‐type (WT) (n = 6) or Stat4 knockout (Stat4‐KO) (n = 6) mice receiving intraperitoneal injection of PBS or LIF during challenge with 3% DSS.
- Macroscopic changes in the colon of the mice on day 10 of colitis induction as described in (A).
- Comparison of the colon length in the mice on day 10 of colitis induction as described in (A).
- DAI (stool consistency and bleeding score) of colitis mice treated as described in (A).
- H&E histology of representative colons from colitis mice. Scale bar, 20 μm.
- Weight loss of Rag1 −/− (n = 6) mice transferred with CD45RBhi CD4+ T cells and intraperitoneally injected with PBS or LIF for 8 weeks.
- Macroscopic changes in the colon of Rag1 −/− mice treated as described in (F).
- Comparison of colon lengths in the Rag1 −/− mice at week 8 (n = 5).
- H&E histology of representative mouse colons on day 54 of colitis induction as described in (F). Scale bar, 100 μm.
LIF activates STAT4 to block Th17‐cell differentiation in the mouse colitis model and in vitro
Furthermore, cells with activated STAT4 (tyrosine 693‐phosphorylated STAT4) were highly enriched in the colonic lamina propria of wild‐type mice with colitis (Fig 3A). In the spleen extract from mice with colitis, LIF treatment induced a significant increase in STAT4 phosphorylation compared to that in PBS‐treated control mice (Fig 3B). In the inflamed mouse colon, the percentage of proinflammatory Th17 cells was increased. Interestingly, we found that the injection of LIF reduced the accumulation of both IL‐17A single‐positive and IL‐17A and IFNγ double‐positive Th17 cells, which represent nonpathogenic and pathogenic Th17 cells, respectively, in wild‐type mice (Figs 3C and EV1D). In Stat4 −/− mice, however, DSS failed to induce Th17‐cell accumulation, and LIF injection actually slightly increased Th17‐cell induction (Figs 3C and EV1D). LIF showed no effect on Th1‐cell accumulation in either wild‐type or Stat4 −/− mice (Figs 3C and EV1D). Correspondingly, Il17a mRNA level decreased significantly in mesenteric lymph nodes (MLNs) and draining lymph nodes (LNs) of LIF‐treated wild‐type mice, compared with that in wild‐type mice treated with DSS only (Fig 3D). In lymph node tissue from Stat4 −/− mice, LIF treatment moderately enhanced rather than decreasing the percentage of Th17 cells (Fig 3D), suggesting that the inhibitory effect of LIF on Th17 cells involved STAT4.
Figure 3. LIF activates STAT4 to block Th17‐cell differentiation in the mouse colitis model and in vitro .
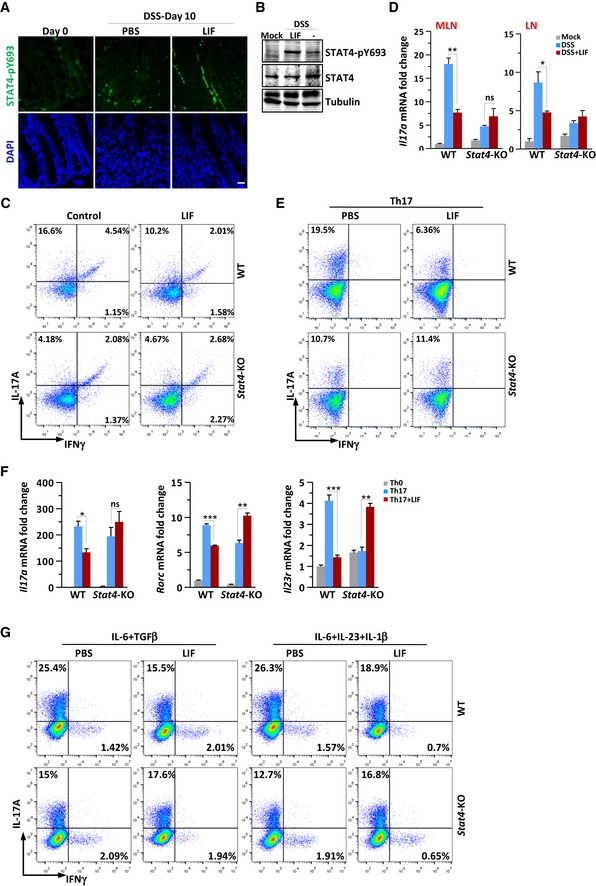
- Colons obtained from WT or Stat4‐KO mice on day 10 of the colitis model induced as in Fig 2A were immunostained with an antibody against phosphorylated STAT4. Scale bar, 50 μm.
- Immunoblot analysis of STAT4 phosphorylation in spleen tissue from colitis mice.
- FACS staining of LPLs isolated on day 10 from the colon of WT or Stat4‐KO colitis mice receiving PBS or LIF (n = 4 per group). The percentage of IL‐17A+ and/or IFNγ+ CD4+ T cells in vivo was analyzed.
- Quantitative expression of Il17a mRNA in MLNs and LNs from colitis mice treated as described in Fig 2A (n = 3 per group).
- FACS staining of WT and Stat4‐KO naïve CD4+ T cells treated with LIF (50 ng/ml) on day 4 of induction into Th17‐cell subsets.
- qPCR analysis of Th17‐related genes in WT or Stat4‐KO naïve CD4+ T cells on day 4 of induction into Th17‐cell subsets in the absence or presence of LIF (n = 3 per group).
- FACS staining of WT or Stat4‐KO naïve CD4+ T cells treated with LIF (50 ng/ml) on day 4 of induction into nonpathogenic or pathogenic Th17 cells. The data are representative of three independent experiments.
To confirm the in vivo phenotype, we isolated naïve CD4+ T cells to induce T helper subset differentiation in vitro in the presence or absence of LIF. STAT4 has previously been noted to be required for Th17‐cell differentiation (Mathur et al, 2007). Indeed, Th17‐cell differentiation was reduced by 50% in Stat4 −/− naïve CD4+ T cells (Fig 3E). However, LIF treatment of naïve CD4+ T cells markedly terminated the differentiation of wild‐type Th17 cells (Fig 3E). This termination effect of LIF on Th17 differentiation was completely abolished in Stat4 −/− T cells. The expression of the Th17‐cell signature genes Il17a, Rorc, and Il23r was significantly inhibited in LIF‐treated Th17 cells (Fig 3F). STAT4 deficiency decreased the expression of these genes, but the expression of these genes increased appreciably in the presence of LIF in Stat4 −/− Th17 cells (Fig 3F). We further tested the inhibitory effect of LIF under two different Th17‐cell induction conditions. IL‐6 and TGFβ primed nonpathogenic Th17‐cell differentiation; IL‐6, IL‐23, and IL‐1β induced pathogenic Th17‐cell development. The results indicated that LIF inhibited the differentiation of both pathogenic and nonpathogenic Th17 cells in wild‐type CD4+ T cells; however, the inhibitory effect of LIF on Th17 cells vanished in Stat4‐deficient CD4+ T cells (Fig 3G). Th1‐cell differentiation relies on STAT4 activation by IL‐12 (Jacobson et al, 1995). As expected, STAT4 deficiency markedly blocked Th1‐cell differentiation, which was not affected by LIF (Fig EV1E). The expression of the Th1‐cell signature genes Ifng, Tbx21, and Il12rb2 was similar in LIF‐treated and LIF‐untreated wild‐type Th1 cells (Fig EV1F). However, neither LIF treatment nor STAT4 deficiency affected Th2‐ or Treg‐cell differentiation (Fig EV1E and G). Together, these in vivo and in vitro results suggested that the inhibitory effect of LIF on Th17‐cell differentiation was largely dependent on STAT4.
“SPXX Repeats” dissociate STAT4 from LIFR for activation
To investigate the means by which LIF activates STAT4 in CD4+ T cells, we pretreated CD4+ T cells with the JAK2 inhibitor LY2784544, which abolished the induction of STAT4 tyrosine phosphorylation by LIF in CD4+ T cells (Fig 4A), indicating that STAT4 activation by LIF occurred via canonical JAK‐STAT activation. Brenner's group recently reported that in human fibroblasts, LIF promoted STAT4 activation via LIFR in an autocrine manner (Nguyen et al, 2017). Furthermore, gp130 is implicated in STAT4 activation during IL‐35 signaling (Collison et al, 2012). We found that the interaction between gp130 and STAT4 displayed a LIF‐independent pattern (Fig EV2A). However, the interaction between LIFR and STAT4 was dynamic and dependent on LIF stimulation in both immune cells (Fig 4B) and HEK293T cells transiently transfected with LIFR and STAT4 (Fig 4C).
Figure 4. LIF‐activated STAT4 is phosphorylated on SPXX motifs within the C‐terminal transcription regulation domain.
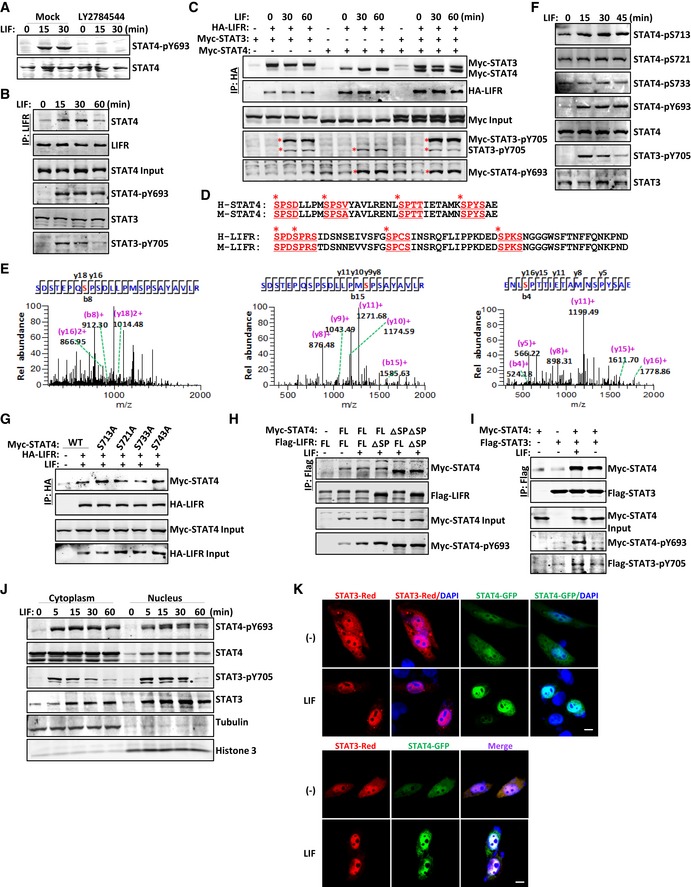
-
AImmunoblot analysis of STAT4 phosphorylation in CD4+ T cells pretreated with PBS or LY2784544 (1 μM) for 12 h followed by LIF for the indicated number of minutes.
-
BImmunoblot analysis of CD4+ T cells treated with LIF for the indicated number of minutes followed by immunoprecipitation with an anti‐LIFR antibody.
-
CCoimmunoprecipitation and immunoblot analysis of LIF‐treated HEK293T cells cotransfected with the indicated constructs. The red asterisks denote phosphorylated STAT bands.
-
DSequence alignment showing that the C‐terminus of STAT4 and LIFR contains four SP repeats in both humans and mice. The SPXX motifs are displayed and underlined in red. The red asterisks point out the potential phosphorylated serine residues.
-
EThe mass spectrometry analysis of purified STAT4 protein revealed the phosphorylation of STAT4 at serine residues in the C‐terminus.
-
FImmunoblot analysis of the indicated protein modifications in the total lysate of CD4+ T cells treated with LIF (20 ng/ml) at different time points.
-
G–ICoimmunoprecipitation and immunoblot analysis of LIF‐treated HEK293T cells cotransfected with the indicated constructs.
-
JImmunoblot analysis of the distribution of the indicated proteins in the CD4+ T‐cell fraction.
-
KCultured HeLa cells transfected with GFP‐tagged STAT constructs were exposed to LIF and imaged 30 min later. Scale bar, 50 μm.
Source data are available online for this figure.
Figure EV2. STAT4 does not alter LIF‐induced STAT3 activation.
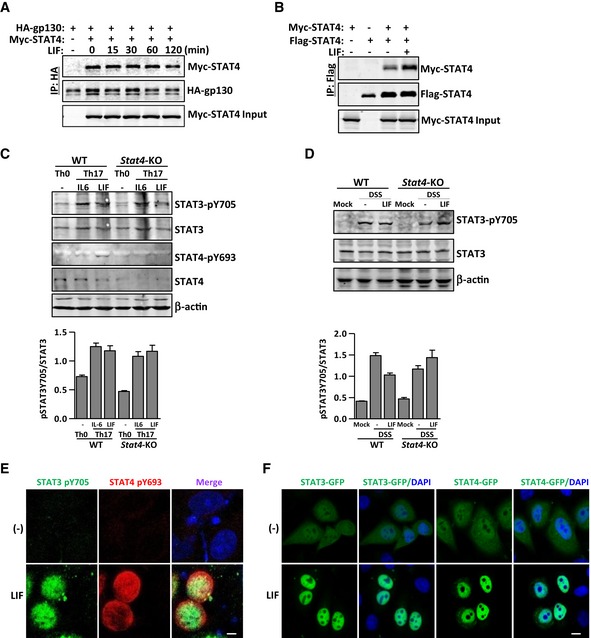
-
A, BCoimmunoprecipitation and immunoblot analysis of LIF‐treated HEK293T cells cotransfected with the indicated constructs.
-
C, DImmunoblot analysis of the expression and modification of the indicated proteins in WT or Stat4‐KO naïve CD4+ T cells on day 4 of induction into Th0 or Th17 cells in the presence or absence of LIF (50 ng/ml) (C) or in spleen extracts from WT or Stat4‐KO colitis mice treated with or without LIF (50 ng/ml) (D). The bottom panel shows the intensity analysis of the bands from three independent experiments. The data are representative of two independent experiments. Error bars represent the SEM.
-
EImmunostaining of cultured HeLa cells transfected with STAT4 and STAT3 after LIF treatment with STAT4 and STAT3 phosphorylation antibody. Scale bar, 20 μm.
-
FCultured HeLa cells transfected with GFP‐tagged STAT4 and RFP‐tagged STAT3 were exposed to LIF and imaged 30 min later. Scale bar, 50 μm.
Source data are available online for this figure.
Interestingly, both STAT4 and LIFR contain four SPXX motifs within their C‐terminal regions (Fig 4D). LIFR is phosphorylated on these four SPXX motifs upon LIF treatment in mouse embryonic stem cells (Wang et al, 2017b). Mass spectrometry analysis revealed that LIF‐activated STAT4 was also phosphorylated on three SPXX motifs: Ser713, Ser721, and Ser733 (Fig 4E). The Pro‐Met‐Ser(721)‐Pro motif is the well‐known motif conserved among all STAT family members (Darnell, 1997). To confirm the mass spectrometry results, we developed a specific antibody to detect phosphorylation on STAT4‐S713 and STAT4‐S733; an anti‐phosphorylated STAT4‐S721 antibody is commercially available. LIF treatment increased phosphorylation on STAT4‐S713 as well as STAT4‐Y693 in immune cells; however, the S721 and S733 residues of STAT4 were constitutively phosphorylated (Fig 4F). We then constructed individual serine‐to‐alanine (S‐to‐A) mutants of STAT4, and STAT4 and LIFR with SPXX motif deletion (ΔSP) vectors. The STAT4‐S713A mutant and, to a lesser extent, the STAT4‐S743A mutant exhibited increased LIFR‐binding activity, whereas the interaction of the STAT4‐S721A and STAT4‐S733A mutants with LIFR was not appreciably affected (Fig 4G). In addition, the interaction between LIFR‐ΔSP and STAT4‐ΔSP in HEK293T cells was noticeably enhanced (Fig 4H). Hence, we hypothesized that phosphorylation on SPXX motifs dissociates LIF‐activated STAT4 from LIFR for nuclear translocation.
STAT proteins undergo dimerization upon activation by cytokines, followed by nuclear translocation and downstream gene promoter activation (Schindler & Darnell, 1995; Heim, 1996). STAT4 homodimers have been linked to the signaling pathways activated by IL‐12, IFNα, and IL‐23 (Jacobson et al, 1995; Nguyen et al, 2002; Parham et al, 2002). Both IL‐12 and IL‐23 can induce STAT3‐STAT4 heterodimer formation in T cells (Jacobson et al, 1995; Parham et al, 2002). We proved that LIF stimulation induced LIFR recruitment of STAT3 and STAT4 (Fig 4C). In addition, LIF stimulation promoted the formation of not only STAT4 homodimers but also STAT3‐STAT4 heterodimers (Figs 4I and EV2B). The presence of STAT4 did not attenuate STAT3 activation either in T cells or in cells ectopically expressing STAT4 (Figs 1I and 4B, and EV2C–E), and STAT4 exhibited only a minor effect on the interaction between LIFR and STAT3 (Fig 4C). STAT3 and STAT4 translocated to and colocalized in the nucleus upon LIF treatment (Figs 4J and K, and EV2F). Therefore, LIF induces canonical tyrosine phosphorylation of STAT4 to activate it and noncanonical serine phosphorylation to dissociate it from LIFR; STAT4 then undergoes homo‐or heterodimerization for nuclear translocation.
STAT4 modulates the gene expression profile during Th17‐cell differentiation
To investigate STAT4 transcriptional activity during Th17‐cell differentiation, we analyzed the gene expression profile in LPLs by performing gene microarray analysis. Unlike IL‐6, which upregulates gene expression mainly via STAT3 activation (Wehinger et al, 1996; Wrighting & Andrews, 2006), LIF both up‐ and downregulated gene expression via STAT4 (Fig EV3A–C). Differential analysis of wild‐type mice versus Stat4 −/− mice revealed that 4,073 genes were upregulated and 4,563 genes were downregulated by LIF‐STAT4 signaling (Fig EV3B). This pattern strongly indicates that STAT4 has more negative than positive regulatory effects of gene expression.
Figure EV3. LIF shows minimal effect on Th17 regulatory genes expression in vitro .
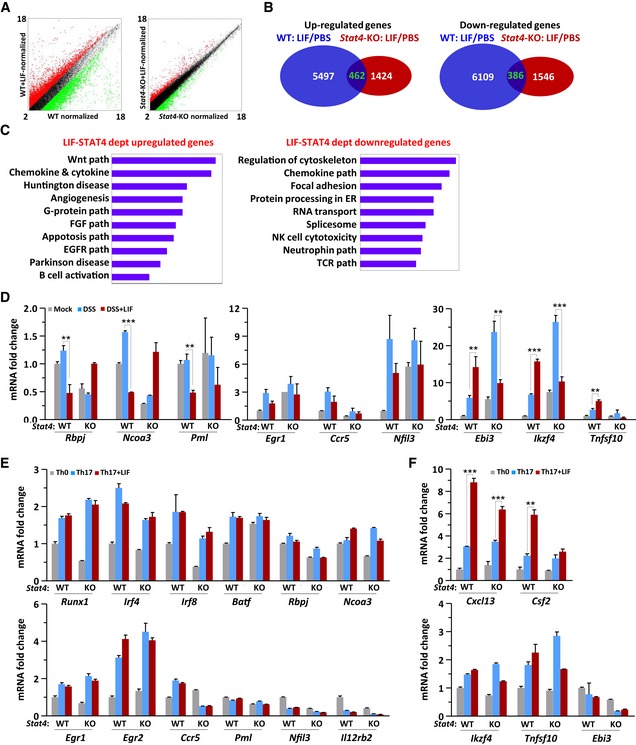
-
A, BGene chip analysis of LPLs purified from colitis mice treated as described in Fig 2A. A scatter plot (A) and the statistical analysis (B) of the average fragments per kilobase of transcript per million mapped reads in WT and Stat4‐KO mice receiving PBS or LIF.
-
CFunctional pathway analysis of LPLs isolated on day 10 from the colon of WT or Stat4‐KO colitis mice receiving injections of PBS or LIF (n = 4 per group).
-
D–FqPCR analysis of the indicated genes in LPLs from colitis mice treated as described in Fig EV3A (D) or in WT or Stat4‐KO naïve CD4+ T cells on day 4 of induction into Th17 subsets in the absence or presence of LIF (E, F). The data are representative of two experiments. **P < 0.01 and ***P < 0.001 (Student's t‐test). Error bars represent the SEM.
In LIF‐treated LPLs, the expression of Th17 signature and positively regulated genes, including Il17f, Il21, Cxcr4, and Ccr5, was inhibited (Yosef et al, 2013), whereas the mRNA expression of signature genes for other T‐cell lineages, including Ifngr1, Il2, and Csf2, was unaffected or even increased (Fig 5A). The gene expression of Th17 master regulators such as Batf, Irf4, and Runx1 was decreased, and the expression of Ikzf4, which is involved in a Th17‐negative regulatory module (Yosef et al, 2013), increased in LIF‐treated LPLs (Figs 5B and C, and EV3D). Here, STAT4 was obviously an essential regulator, because STAT4 deficiency abolished the regulatory effect of LIF on these genes (Fig 5A–C). Since LPLs isolated from the colon were a mixture of various lymphocytes, we harvested Th17 cells differentiated in vitro to further confirm these gene expression profiles. The Il17a gene exhibited a similar expression pattern in both Th17 cells and LPLs (Figs 3F and 5B). The qPCR results showed that inhibitory genes of Th17 differentiation, such as Il2, Ccl3, and Il31ra, exhibited increased expression in response to LIF treatment in wild‐type but not in Stat4 −/− Th17 cells (Fig 5D), consistent with the results of the gene microarray analysis. However, under in vitro differentiation conditions, the expression of Th17‐cell master regulatory genes was not affected by the presence of LIF or by Stat4 deficiency (Fig EV3E and F). The non‐Th17‐cell module genes Cxcl13 and Csf2 were upregulated when Th17 cells were induced in the presence of LIF, but this upregulation was STAT4‐independent (Fig EV3F). These results suggested that we focus on IL‐2, CCL3, and IL‐31Rα in exploring the molecular mechanism; there are few informative studies about the effect of CCL3 or IL‐31Rα on Th17‐cell differentiation, but IL‐2 has been reported to activate STAT5 to inhibit IL‐17 expression (Yang et al, 2011). However, IL‐2 can activate STAT4 only in natural killer cells, not in T cells (Wang et al, 1999). Thus, LIF‐STAT4 signaling inhibits Th17‐cell differentiation by a mechanism independent of Th17‐cell master regulators or via indirect molecules. Furthermore, STAT4 functioned as a negative regulator in immune cells under LIF stimulation to regulate IL‐17 expression.
Figure 5. Reciprocal role of STAT4 and STAT3 on gene regulation in immune cells.
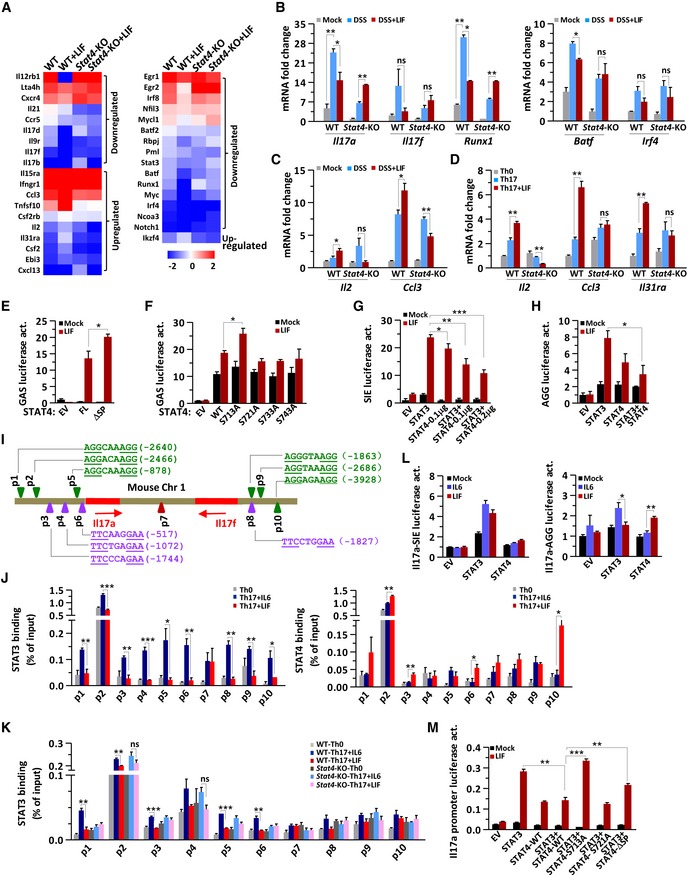
-
AHeatmap showing Th17 signature genes (left) or Th17 regulatory genes (right) up‐ or downregulated in WT or Stat4‐KO LPLs by LIF treatment (n = 2).
-
B, CQuantitative mRNA expression analysis of the indicated genes in LPLs from colitis mice (n = 3 per group).
-
DQuantitative mRNA expression analysis of the indicated genes in WT or Stat4‐KO naïve CD4+ T cells on day 4 of induction into Th17‐cell subsets in the absence or presence of LIF.
-
E, FThe GAS‐luciferase reporter activity assay was performed in Stat3 −/− MEFs transfected with empty vector (EV) or STAT4 constructs and then treated with LIF for 8 h.
-
G, HRelative SIE‐(I) or AGG‐luciferase reporter (J) activities in Stat3 −/− MEFs transfected with EV or the indicated STAT constructs and treated with LIF for 8 h. The data are representative of three independent experiments (D‐H).
-
IThe Il17a/Il17f locus, including qPCR primer sites covering SIEs (purple triangles) or AGG elements (green triangles).
-
JChIP analysis of naïve CD4+ T cells obtained from IL‐17A‐eGFP mice cultured under Th0 or Th17 conditions in the absence or presence of LIF for 3 days. GFP+ cells isolated by flow cytometry were restimulated with IL‐6 in the presence or absence of LIF, crosslinked with formaldehyde and immunoprecipitated with anti‐STAT3 (top) or anti‐STAT4 (bottom) antibodies, followed by the amplification of the immunoprecipitated DNA by quantitative PCR with the p1‐p10 primer pairs. The results are presented relative to the amount of input DNA.
-
KChIP analysis of naïve WT or Stat4‐KO CD4+ T cells cultured under Th0 or Th17 conditions in the absence or presence of LIF for 3 days. The cells were restimulated with IL‐6 or LIF for 30 min, crosslinked with formaldehyde, and immunoprecipitated with the anti‐STAT3 antibody, followed by the amplification of the immunoprecipitated DNA by quantitative PCR with the p1‐p10 primer pairs. The results are presented relative to the amount of input DNA. The data are representative of two independent experiments (J‐K)
-
MRelative Il17a‐SIE‐ (left) or AGG‐luciferase reporter (right) activities in Stat3 −/− MEFs transfected with EV, STAT3, or STAT4 or cotransfected with STAT3 and STAT4 and then treated with IL‐6 or LIF for 8 h.
-
NRelative Il17a promoter‐luciferase reporter activities in Stat3 −/− MEFs transfected with the indicated constructs and then treated with LIF for 8 h. The data are representative of three experiments (L‐M).
Reciprocal regulatory effects of STAT4 and STAT3 on Il17 gene regulation in Th17 cells
We next hypothesized that STAT4 has a direct effect on the Il17 gene locus. STAT3 is a positive transcription factor for both Il17a and Il17f (O'Shea & Murray, 2008; Mukasa et al, 2010). Both STAT3 and STAT4 can bind to similar palindrome DNA elements, cis‐inducible elements (SIE), or IFNγ‐activated sequences (GAS) with different activities (Ehret et al, 2001; Mukasa et al, 2010). In the GAS‐luciferase reporter assay, STAT4 responded to LIF, resulting in GAS‐dependent luciferase reporter activation (Fig 5E). As expected, STAT4‐ΔSP had stronger transcriptional activity than full‐length STAT4 in response to LIF treatment (Fig 5E), suggesting that SPXX phosphorylation plays a negative regulatory role in STAT4 activity. We tested individual S‐to‐A mutants of STAT4 and noted that in HEK293T cells, the S713A mutant exhibited enhanced GAS‐luciferase reporter activity (Fig 5F), in agreement with the findings that S713 phosphorylation dissociates STAT4 from LIFR, as shown in Fig 4G. We previously reported that STAT3 not only binds to the canonical SIE “TTCXXXGAA” but also binds to the AGG element “AGGXXXAGG” (Xu et al, 2015). In STAT3‐null mouse embryonic fibroblasts (MEFs) cotransfected with STAT4 and STAT3, STAT4 further inhibited STAT3 activity during SIE‐driven or AGG element‐driven luciferase reporter activation (Fig 5G and H). STAT4 alone could drive SIE or AGG elements to initiate transcription, but the transcriptional activity of STAT4 was much lower than that of STAT3 (Fig 5G and H). Notably, the SIE had a greater impact than the AGG element on luciferase reporter activation.
The Il17a promoter contains three SIEs; the Il17f promoter, one. However, three AGG elements were identified within the Il17a and Il17f promoters (Fig 5I). To confirm the direct regulation of the Il17a‐Il17f locus by STAT4, we performed a chromatin immunoprecipitation (ChIP) analysis. We obtained naïve CD4+ T cells from IL17A‐eGFP knock‐in mice to selectively study IL‐17A‐producing Th17 cells, and we next screened potential STAT3 and/or STAT4 promoter association at the Il17a‐Il17f locus with designed primers by performing ChIP‐qPCR in Th17 cells. The ChIP‐qPCR results clearly showed that STAT3 binds to both SIE and AGG elements (Fig 5J and K). LIF treatment decreased STAT3 binding but increased STAT4 binding to both SIE and AGG elements. Strikingly, STAT4 bound strongly to the AGG elements p1, p2, and p10 (Fig 5J). We used wild‐type and Stat4‐deficient CD4+ T cells differentiated by IL‐6 and TGFβ for ChIP‐qPCR, and we found that without STAT4, LIF lost the ability to block STAT3 binding to SIEs or AGG elements (Fig 5K). STAT4 binding correlated significantly with the decreased binding of STAT3 to SIE and AGG elements, suggesting that STAT4 directly interfered with the binding ability of STAT3 at these loci. With less STAT3 on SIE and AGG elements and more STAT4 on the inefficient AGG elements, Il17a/f gene transcription decreased. We then constructed luciferase reporter vectors containing Il17a SIE or AGG elements. STAT3 but not STAT4 responded well to either IL‐6 or LIF in inducing Il17a‐SIE‐luciferase reporter activation (Fig 5L, left panel). Whereas STAT3 responded to IL‐6, STAT4 responded to LIF in inducing Il17a‐AGG element luciferase reporter activation (Fig 5L, right panel). Furthermore, we constructed a luciferase reporter vector containing the Il17a promoter sequence with the six AGG elements and SIEs. Wild‐type STAT4 strongly inhibited STAT3 activity in initiating Il17a transcription upon LIF treatment, but the STAT4 S713A mutant and the STAT4 SPXX repeat deletion mutant exhibited lower inhibition of STAT3 function, suggesting that the LIF‐induced noncanonical phosphorylation of STAT4 S713 negatively regulated STAT4 transcriptional activity and was involved in Th17‐cell regulation (Fig 5M). LIF recruited STAT4 to the Il17a‐Il17f locus to restrict STAT3‐dependent Il17 gene expression, in agreement with the previous finding that in IL‐12‐restimulated Th17 cells, STAT4 repressed Il17a and Il17f expression (Ivanov et al, 2008).
Taken together, these results demonstrated that in the presence of LIF, hyperactive SIEs were poorly activated, whereas hypoactive AGG elements became fully occupied by STAT4, leading to less differentiation of Th17 cells.
LIF activates STAT3 and YAP to promote IEC proliferation
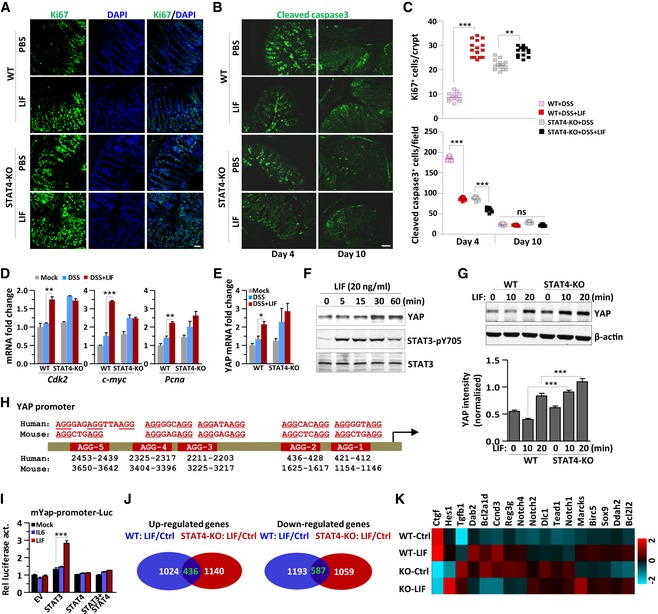
As Fig 1J shows, LIF induced STAT3 activation and YAP expression in IECs expressing extremely low levels of STAT4 (Fig 1J and K). However, LIF stimulated epithelial cell proliferation (Fig 1M). With Th17‐cell inhibition, LIF decreased inflammation in the colon; thus, we further investigated whether LIF can also drive intestinal epithelial wound healing in the damaged colon. We analyzed epithelial cell proliferation via Ki67 and cleaved caspase 3 staining of colon tissues obtained from mice with DSS‐induced colitis. LIF administration dramatically increased the proliferation and inhibited the apoptosis of IECs (Figs 6A–C and EV4A). Significantly enhanced cell proliferation accompanied appreciably reduced apoptosis in Stat4 −/− mice, as revealed by the Ki67 and cleaved caspase 3 staining (Fig 6A–C). Furthermore, genes involved in the cell cycle and cell proliferation, such as Cdk2, c‐Myc, Pcna, and Yap, responded to LIF in wild‐type animals and to DSS challenge in Stat4 −/− mice (Fig 6D and E). However, the expression of other cell cycle promotion genes, such as Ccnd1, e2f3, Mybl1, and the intestinal stem cell marker Cd44, did not change with LIF treatment (Fig EV4B). Therefore, like IL‐6, LIF promoted IEC proliferation and conferred resistance to mucosal erosion, presumably via the activation of STAT3 and/or other pathways (Taniguchi et al, 2015).
Figure 6. STAT3 is responsible for YAP gene expression and cell proliferation in the intestinal.
-
A–CKi67 and cleaved caspase 3 staining of representative colons obtained from mice on day 10 of colitis induction as described in Fig 2A. The number of proliferating cells per crypt in (A) (upper panel) or apoptotic cells per field in (B) was determined (n = 5 per group, 5 crypts were counted per mouse). Scale bar, 50 μm.
-
D, EQuantitative mRNA expression analysis of the indicated genes in colon tissues obtained from WT or Stat4‐KO colitis mice treated as described in Fig 2A (n = 3 per group).
-
FImmunoblot analysis of the expression and modification of the indicated proteins in DLD‐1 cells treated with LIF (20 ng/ml) for different durations.
-
GImmunoblot analysis of YAP expression in WT or Stat4‐KO IECs treated with LIF (20 ng/ml) for different durations; β‐actin was used as a loading control (top). The statistical analysis of YAP protein intensity is shown in the bottom panel. The data are representative of three experiments.
-
HThe human and mouse Yap promoter locus contains multiple AGG elements, as indicated.
-
IRelative YAP promoter‐luciferase reporter activities in HEK293T cells transfected with EV, STAT3, or STAT4 and then treated with IL‐6 or LIF for 8 h. The data are representative of three experiments.
-
J, KRNA sequencing analysis of the gene expression profile in IECs treated with or without LIF (50 ng/ml) for 6 h. Statistical analysis of the average fragments per kilobase of transcript per million mapped reads (J). Heatmap showing the expression profile of cell proliferation‐related genes in LIF‐treated or control IECs (Ctrl) (K).
Figure EV4. STAT4 does not regulate the expression of proliferation‐related genes in IECs.
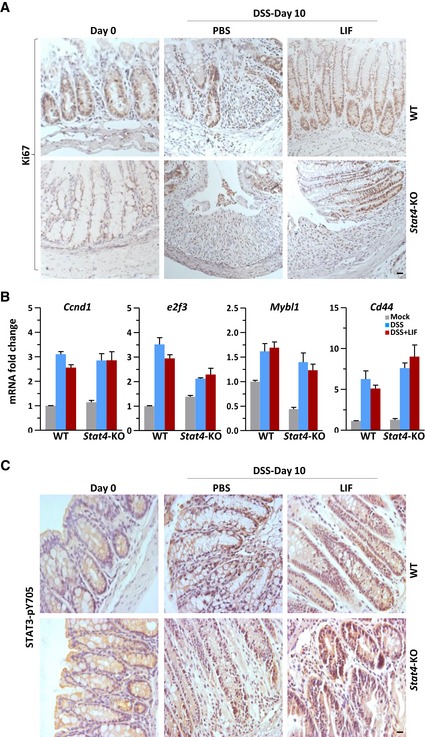
- Ki67 staining of representative colons on day 10 of colitis induction as described in Fig 2A. Scale bar, 20 μm.
- Quantitative mRNA expression analysis of the indicated genes in colon tissue from WT or Stat4‐KO colitis mice treated as described in Fig 2A (n = 3 per group). Error bars represent the SEM.
- Colons obtained from WT or Stat4‐KO mice on day 10 of the colitis model induced as in Fig 2A were immunostained with an antibody against STAT3 phosphorylated on Y705. Scale bar, 20 μm.
As previously reported, YAP protein accumulation in response to gp130‐Src pathway activation is responsible for IEC proliferation (Taniguchi et al, 2015). We found that YAP mRNA and protein level were increased in LIF‐treated epithelial cells (Fig 6E–G), suggesting that transcriptional regulation was involved. Although epithelial cells expressed low levels of STAT4 (Fig 1J), YAP induction by LIF was markedly elevated in Stat4‐deficient IECs (Fig 6G), indicating that STAT4 plays a negative role in gene regulation in epithelial cells. Interestingly, the Yap promoter contains five AGG elements that are largely conserved between humans and mice (Fig 6H). To test whether STAT3 or STAT4 could bind to AGG elements in the Yap promoter, we constructed a luciferase reporter vector containing the mouse Yap promoter, including 2 AGG elements (positions 412–436) (Fig 6H). STAT3 activation by LIF but not IL‐6 induced the activation of this Yap AGG‐luciferase reporter (Fig 6I). In addition, LIF injection resulted in increased nuclear accumulation of activated STAT3 (pY705) in the intestine of mice with DSS‐induced colitis, especially in proliferating crypts of the intestine, compared to that in control mouse intestines (Fig EV4C). In contrast, STAT4 inhibited YAP‐luciferase reporter activation by LIF. However, although exogenously expressed STAT4 blocked STAT3‐induced activation of YAP in vitro, the negative regulation of YAP expression by STAT4 exhibited only a minor effect on IEC proliferation in vivo (Fig 6A).
We applied RNA sequencing to analyze the gene expression profile in IECs. The number of genes upregulated or downregulated by LIF treatment was similar and unaffected by STAT4 deficiency (Fig 6J). We noticed that in primary IECs, LIF stimulation but not STAT4‐deficiency induced YAP mRNA expression; however, the mRNA expression of the YAP target gene connective tissue growth factor (CTGF) was appreciably increased in both Stat4 −/− IECs and LIF‐treated wild‐type IECs (Fig 6K). Neither LIF stimulation nor STAT4 deficiency exhibited a significant effect on other gp130‐activated Notch pathway genes (Taniguchi et al, 2015) (Fig 6K). LIF‐activated STAT4 displays a more negative than positive regulatory effect on gene expression in both LPLs and IECs, but STAT4 exerted a stronger effect in LPLs than in IECs. Therefore, in IECs, LIF bypasses the extraordinarily low level of STAT4 to induce YAP gene expression via STAT3 activation, leading to the repair of damaged colon tissue.
LIF treatment modulates the outgrowth of the proinflammatory microbiota to attenuate colitis pathology
In Fig 1J, we noticed the dynamic expression pattern of LIF in the mouse colon during colitis progression. Bacterial endotoxin was required for IECs to secrete LIF, and LIF expression declined on day 15. Previous studies have fully illustrated that IECs play diverse functions in maintaining intestinal homeostasis, including the physical segregation of commensal bacteria and the integration of microbial signals (Mankertz & Schulzke, 2007; Peterson & Artis, 2014). As Fig 6 shows, we proved that LIF promoted the repair of damaged intestinal epithelia and exhibited a dynamic expression pattern, indicating that repaired IECs might prevent bacterial invasion into the colon. To explore whether LIF‐induced epithelial repair affected the microbiota composition in this DSS‐induced colitis model, we performed 16S rRNA gene sequencing of feces from wild‐type or Stat4 −/− mice treated with or without LIF. The fecal bacteria composition changed in wild‐type and Stat4 −/− mice under DSS challenge alone (Fig 7A). However, LIF treatment kept the bacterial composition of DSS‐challenged wild‐type mice similar to that of control wild‐type mice but displayed only a weak effect on the bacterial community composition in Stat4 −/− mice.
Figure 7. LIF treatment modulates the outgrowth of the proinflammatory microbiota during intestinal inflammation.
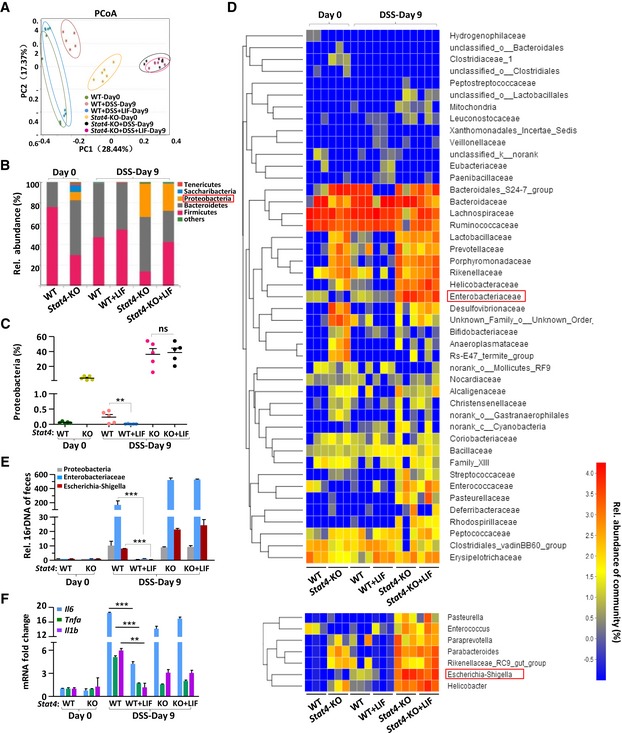
- Principal coordinate analysis (PCoA) of the microbiota composition determined by 16S rRNA gene sequencing of day 0 and day 9 fecal specimens from mice treated as described in Fig 5A (n = 5 or 6 per group). Each symbol represents an individual mouse. PC1 and PC2 represent principal components 1 and 2, respectively.
- The bars depict the average relative abundance of the bacterial phyla of the microbiota in fecal specimens, as determined by taxon‐based analyses as described in (A) (n = 5 per group).
- 16S rRNA sequencing analyses of the Proteobacteria distribution in feces from WT or Stat4‐KO colitis mice. (n = 5 per group).
- Heatmap depicting the relative abundance of microbes at the family level (top) and the genus level (bottom) in the fecal specimens described in (A) (n = 3 per group).
- qPCR of the 16S rRNA genes of microbes in the phylum Proteobacteria, family Enterobacteriaceae, and genera Escherichia‐Shigella in the feces of the indicated mice on day 0 and day 10 (n = 6 per group) of colitis induction.
- Quantitative mRNA expression analysis of proinflammatory genes in the colon of mice (n = 3 per group) treated as described in Fig 2A.
Further analysis showed that STAT4 deficiency greatly impacted the commensal microbiota composition in the colon; the outgrowth of proinflammatory microbes increased dramatically in Stat4 −/− mice (Fig 7B–D). We hypothesized that the low level of effector Th1 and Th17 cells in Stat4‐deficient mice led to inefficient immune defense and bacterial clearance. The abundance of the phylum Proteobacteria was elevated in both wild‐type and Stat4 −/− mice with colitis (Figs 7B and C, and EV5A), consistent with the findings of a previous report (Lupp et al, 2007). However, there was a greater increase in the abundance of phylum Proteobacteria in Stat4 −/− mice under DSS challenge, although more IECs proliferated in Stat4 −/− mice, indicating that without an integrated defense system, Proteobacteria grow much more vigorously. However, the growth of Proteobacteria was inhibited significantly in LIF‐treated wild‐type mice (Fig 7B and C). From the family‐level analysis, we found that although the fecal bacteria composition differed between wild‐type and Stat4 −/− mice, the effect of LIF was obvious only in wild‐type mice. The abundance of the family Enterobacteriaceae in the phylum Proteobacteria decreased in LIF‐treated wild‐type mice (Fig 7D, top). The abundance of Escherichia‐Shigella, genera in the family Enterobacteriaceae, increased more significantly in Stat4 −/− mice than in wild‐type mice with colitis, but LIF treatment effectively inhibited Escherichia‐Shigella growth in wild‐type mice (Fig 7D, bottom and Fig EV5B). Utilizing a qPCR approach, we confirmed the level of fecal Proteobacteria, Enterobacteriaceae, and Escherichia‐Shigella in the feces of wild‐type and Stat4 −/− mice treated or not treated with LIF (Fig 7E). In the damaged colon, dysregulation or altered colonization of commensal microbiota activates innate immune cells to produce more proinflammatory cytokines, such as IL‐6 and TNFα, which are the main cause of overt inflammation (Rakoff‐Nahoum et al, 2004; Grivennikov, 2013). In addition, we found that LIF treatment led to the decreased expression of the proinflammatory cytokines IL‐6, TNFα, and IL‐1β in the colon of wild‐type colitis mice but not Stat4 −/− colitis mice (Fig 7F). In addition to the LIF‐induced transcriptional regulation of the Il17a/f promoter in Th17 cells, the levels of pathogenic and nonpathogenic Th17 cells were unsurprisingly lower in the colon due to the decrease in IL‐6 and IL‐1β (Fig 4C).
Figure EV5. Escherichia‐Shigella but not other bacteria are affected by LIF in the amelioration of colitis.
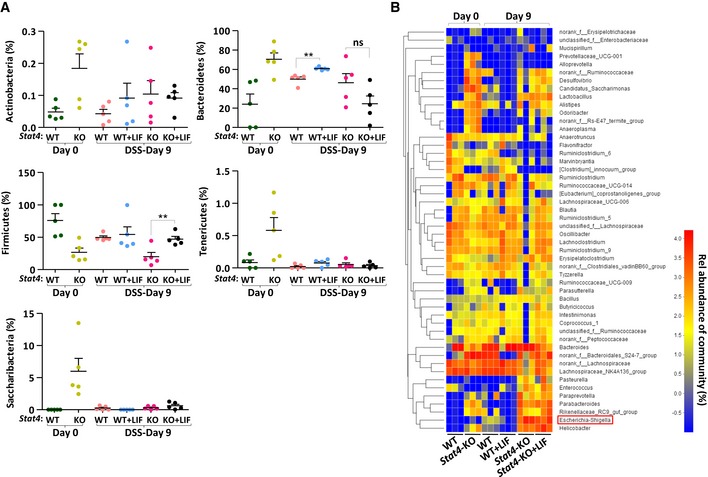
- 16S rRNA sequencing analyses of the microbial distribution at the phylum level in feces from WT and Stat4‐KO colitis mice treated as described in Fig 2A (n = 5 per group). Error bars represent the SEM. **P < 0.01 (Student's t‐test).
- Heatmap depicting the relative microbial abundance at the genus level in the fecal specimens described in (A) (n = 3 per group).
Collectively, our data suggested that bacteria invaded the intestinal epithelium when DSS induced chemical damage to the intestine and that IECs secreted LIF to promote the repair of damaged epithelia as a negative feedback mechanism. Conversely, autocrine LIF helped the intestinal barrier prevent bacterial invasion and stop microbiota dysbiosis in the mouse colon. However, LIF acted directly on inflammatory Th17 cells via STAT4 activation to abrogate the increase in inflammation. Via intestinal repair, microbiota homeostasis, and Th17‐cell inhibition, LIF facilitated the remission of intestinal inflammation (Fig 8).
Figure 8.
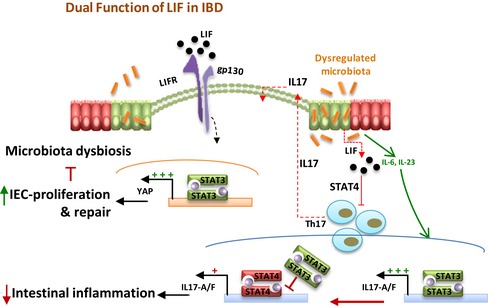
Schematic model of the dual function of LIF in preventing intestinal inflammation progress.
Discussion
The intestine is either the home where host cells and microbiota live and function cooperatively or the arena where these cells wrestle (Thierfelder et al, 1996; Bouma & Strober, 2003). During intestinal inflammation, however, stress on the microbiota triggers cytokine secretion from host cells or even cytokine storm. LIF expression is induced in mice with DSS‐induced colitis, consistent with the finding that LIF expression is increased in UC patients (Guimbaud et al, 1998). LIF secreted from IECs can affect IECs in an autocrine manner or affect LPLs in a paracrine manner to generate quite different outputs that eventually affect intestinal microbiota homeostasis.
Th17 cells play critical roles during the pathogenesis of many autoimmune diseases. For example, LIF suppresses experimental autoimmune encephalomyelitis (EAE) pathology via the inhibition of Th17‐cell differentiation (Cao et al, 2011). Th17 cells also play critical roles in driving intestinal inflammation in IBD. Our findings proved that LIF also decreases the percentage of Th17 cells in the lamina propria in the mouse colitis model, but in a STAT4‐dependent manner. While STAT3 activation by IL‐6 promotes the differentiation of naïve CD4+ T cells into Th17 cells, STAT4 activation by IL‐12 promotes the differentiation of naïve CD4+ T cells into Th1 cells (Kaplan et al, 1996; Thierfelder et al, 1996). However, STAT4 and STAT3 seem to cooperate in Th17‐cell differentiation; naïve CD4+ T cells obtained from Stat4 −/− mice exhibited an apparent defect in Th17‐cell differentiation in response to IL‐6 and IL‐23 (Mathur et al, 2007), suggesting that the positive regulatory effects of STAT4 under Th17‐cell differentiation conditions are involved. This hypothesis is reasonable because IL‐23 receptors are composed of the IL‐23 receptor and the IL‐12 receptor IL‐12Rβ, which can activate STAT4 (Parham et al, 2002). Although the pros and cons of STAT4 activity in Th17 differentiation are rather inexplicable, the different posttranslational patterns of STAT4 under different cytokine induction conditions provide a good explanation.
IL‐6 family cytokines are actively involved in the JAK‐STAT signaling pathway. Although it does not affect STAT3, LIF can activate STAT1 in many different cell types (Durbin et al, 1996; Fujio et al, 1997; Jenab & Morris, 1998). Until recently, Brenner's group reported that LIF expression is elevated in both human and mouse models of arthritis and drives the transcription and activation of STAT4 in fibroblasts, leading to the sustained release of inflammatory mediators, including IL6, IL‐1β, and IL‐11 (Nguyen et al, 2017). This group demonstrated that LIFR and STAT4 form a molecular complex with the JAK1 and TYK2 kinases, controlling STAT4 activation and binding to the Il6 promoter in TNF‐ and IL‐17‐treated fibroblasts, which in turn maintains fibroblast‐mediated inflammation. Fibroblast‐mediated inflammation is distinct from leukocyte‐mediated inflammation. In CD4+ T cells, we noted that LIF can activate STAT4 in addition to STAT3. STAT4 is phosphorylated not only at the canonical Y693 site but also at S713 in the C‐terminal transcriptional regulation domain. Surprisingly, S713 phosphorylation negatively regulated STAT4 transcription and did not improve the ability of STAT4 to compete with STAT3 for promoter binding.
STAT family proteins are structurally related, but they can have opposite regulatory effects on gene expression (Walker et al, 2013). We proved that activated STAT3 and STAT4 can bind to the same DNA sequences, including classical SIEs and newly defined AGG elements on the Il17a/f promoters, but LIF promoted STAT4 competition with STAT3, leading to greater STAT4 occupation of these elements and lower expression of the Il17a/f genes. Therefore, STAT4 becomes an inefficient transcription factor under LIF treatment in Th17 cells. Interestingly, the C‐terminal region of LIFR also contains four SPXX repeats, which are phosphorylated by MAPK to inhibit STAT3 activation by LIFR (Wang et al, 2017b). This mechanism suggests that the SPXX motif plays a negative regulatory role in multiple proteins. Since T cells in the colonic lamina propria are master regulators in maintaining intestinal homeostasis, it is plausible that LIF‐STAT4 signaling regulates colitis pathology. As expected, LIF administration dramatically ameliorated colitis pathology in the mouse model, but only in wild‐type mice, not in Stat4 −/− mice.
Recent studies discovered that innate lymphoid cells (ILCs) control innate immunity at mucosal surfaces and mediate experimental innate immune‐mediated colitis (Buonocore et al, 2010). ILCs are a growing family of immune cells and include IFNγ‐secreting ILC1 cells; IL‐5‐, IL‐9‐, and IL‐13‐secreting ILC2 cells; IL‐17‐, IL‐22‐, and IFNγ‐secreting ILC3 cells; and IL‐10‐ and TGF‐β1‐secreting ILC regulatory (ILCreg) cells (Eberl et al, 2015; Wang et al, 2017a). In humans, IL‐17‐producing ILC3 cells were noted in the inflamed mucosa of patients with Crohn's disease but not in patients with UC (Geremia et al, 2011). Other studies have shown that IL‐17‐producing Th17 cells in the colonic lamina propria were increased in both Crohn's disease and UC (Kobayashi et al, 2008). The characteristics of DSS‐induced colitis in the mouse model resemble those of human UC (Wirtz et al, 2017). Severe combined immunodeficiency (SCID) and Rag −/− mice develop severe intestinal inflammation under DSS challenge, indicating that DSS‐induced acute colitis can progress without the help of adaptive immune cells (Dieleman et al, 1994). However, T cells have been demonstrated to accumulate in inflamed mucosa in DSS‐induced colitis mice (Dieleman et al, 1998). Unsurprisingly, we also found Th17‐cell accumulation in the inflamed colon. However, LIF appreciably inhibited the accumulation of IL‐17A+ Th17 cells, but not IL‐17A+ ILC3 cells (data not shown). IL‐23 and IL‐1β are responsible for the development of IL‐17A‐producing ILC3 cells, and RORγt is the main transcriptional regulator of ILC3 cells; IL‐6 is not required for ILC3 cell development, in contrast to Th17 priming conditions (Buonocore et al, 2010). Our results demonstrated that the inhibitory effect of LIF on Th17 cells functioned through STAT4 and STAT3. We thus inferred that without STAT3 involvement during ILC3‐cell development, LIF could not alter Il17 gene expression, explaining that LIF exhibited a minimal influence on ILC3s accumulation in either wild‐type or Stat4 −/− colitis mice.
These results raise the question of why LIF bestows quite different effects on IECs and LPLs. LIF inhibits Th17‐cell differentiation but stimulates epithelial cell proliferation. We reasoned that these different effects are mainly due to the different expression profile of STAT4 in these two types of cells. STAT4 expression is very high in lymphocytes but quite low in epithelial cells (Jacobson et al, 1995; Frucht et al, 2000; Nguyen et al, 2002; Parham et al, 2002). Both STAT3 and STAT4 can indiscriminately bind to SIEs and AGG elements. However, in T cells, sufficient STAT4 protein is activated by LIF and can form a silent or negative enhanceosome to block the expression of STAT3‐dependent genes. In epithelial cells, though, LIF‐activated STAT4 cannot be visualized due to its extremely low expression level. Thus, STAT4 serves as a good sensor for the expression of STAT‐dependent genes in T cells but not in epithelial cells. LIF‐induced YAP expression also depends on the ability of STAT3 to bind to AGG motifs located in the Yap promoter.
Along with the integrated epithelial barrier, LIF helped prevent the entrance of Proteobacteria into the intestine and maintain microbiota homeostasis in the host mice. Reduced bacterial invasion could also decrease proinflammatory cytokine secretion. Intriguingly, the colonic microbiota is also required for LIF induction during colitis pathology. The means by which LIF is induced by the microbiota remains to be determined.
In summary, we demonstrate that LIF plays central roles in restricting Th17‐cell differentiation by activating STAT4 and in promoting IEC proliferation by activating STAT3. IBD is a clinically challenging illness, often striking at a young age and causing lifelong morbidity. Although cytokines, including IL‐6, are targets in IBD treatment, few have proven to be useful. Our study indicates that LIF is potentially therapeutic in IBD.
Materials and Methods
Mice
Stat4 −/− and Rag1 −/− mice were purchased from the Nanjing Biomedical Research Institute of Nanjing University and back‐crossed to C57BL6 mice. Stat4 −/−, Rag1 −/−, and the corresponding wild‐type control mice were maintained in a SPF facility. All animal experiments were performed in compliance with the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Biomedical Research Ethics Committee of the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences.
Plasmids
A myc‐tagged STAT4 construct was subcloned from a STAT4 construct of mouse origin which is a courtesy from Prof. Mark H. Kaplan. The other tagged wild‐type and mutant STAT4 constructs were subcloned from the above myc‐tagged STAT4 construct. Myc‐ and flag‐tagged STAT3 constructs are of mouse origin. All of the full‐length and deleted constructs of LIFR are of human origin. All of the constructs were transfected into the cell lines we used in our experiments with Lipofectamine 2000 (Invitrogen).
Cell culture and transfection
HEK293T cells were cultured in DMEM (HyClone) supplemented with 10% fetal bovine serum (FBS) (Gemini), 100 units/ml penicillin, and 100 μg/ml streptomycin (Shenggong). SW480 cells were cultured in L‐15 medium (Gibco) supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. DLD‐1 cells were cultured in RPMI‐1640 medium (HyClone) supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cell lines were purchased from the Cell Bank of the Chinese Academy of Sciences.
RNA isolation and quantitative PCR
RNA was extracted from cells and tissues with TRIzol reagent (Invitrogen) and was then transcribed to cDNA with a PrimeScript RT Reagent kit (Takara). Real‐time PCR was performed with SYBR Green Master Mix (YEASEN) on an ABI QuantStudio (Applied Biosystems). The expression of the target genes was normalized to that of β‐actin. “Fold differences” were calculated with the ΔΔC t method.
Immunoprecipitation and Western Blotting
Cells were lysed in RIPA buffer (20‐188; Millipore) containing inhibitors (1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktails, 1 mM Na3VO4, and 1 mM NaF). Cell debris was removed by centrifugation at 4°C and 2,650 g for 15 min, and lysates were incubated overnight with the appropriate antibody and agarose beads. The immunoprecipitates were washed with RIPA buffer three to five times before being boiled and analyzed by Western blotting. The following primary antibodies were commercially obtained: anti‐pSTAT4Y693 (BD Biosciences, 554002); anti‐pSTAT4S721 (Santa Cruz, sc‐28296); anti‐STAT4 (Santa Cruz, sc‐486 and sc‐398228); anti‐pSTAT3Y705 (Santa Cruz, sc‐7993‐R); STAT3 (Santa Cruz, sc‐8019); anti‐YAP (Cell Signaling Technology, 17074); anti‐LIFR (Santa Cruz, sc‐659); anti‐HA (Santa Cruz, sc‐7392); anti‐Myc (Santa Cruz, sc‐40); anti‐Flag (Sigma, F1804); anti‐Histone H3 (Cell Signaling Technology, 4499); anti‐pERK (Cell Signaling Technology, 4370); anti‐tubulin (Sigma, T619); and anti‐β‐actin (Sigma, A1978).
T‐cell isolation and differentiation
Spleens and lymph nodes were collected from wild‐type or Stat4 −/− mice, and single‐cell suspensions were prepared by mechanical disruption in PBS. CD4+ T cells were isolated by magnetic sorting with a Miltenyi Biotec CD4+ CD62L+ T Cell Isolation kit, according to the manufacturer's directions (Miltenyi Biotec, 130‐106‐643). CD4+ T cells were activated with 5 μg/ml precoated anti‐CD3 (eBioscience) and 2 μg/ml anti‐CD28 (eBioscience). Th1 cells were differentiated by the addition of recombinant IL‐12 (1 ng/ml, R&D Systems) and anti‐IL‐4 (10 μg/ml, BD Pharmingen). Th2 cells were differentiated by the addition of IL‐4 (20 ng/ml, R&D Systems) and recombinant IL‐2 (10 ng/ml, PeproTech). Th17 cells were differentiated by the addition of recombinant IL‐6 (20 ng/ml, R&D Systems), recombinant TGF‐β (1 ng/ml, R&D Systems), anti‐IFNγ (10 μg/ml, BD Pharmingen), and anti‐IL‐4 (10 μg/ml, BD Pharmingen), followed by recombinant IL‐23 (30 ng/ml, R&D Systems) on day 3. Treg cells were differentiated by the addition of TGF‐β (5 ng/ml, R&D Systems) and recombinant IL‐2 (40 ng/ml, PeproTech). CD4+ T‐cell cultures were split at ratio of 1:2 on day 3 after activation.
Intracellular cytokine staining
Purified CD4+ T cells were activated for 4–5 h with PMA (50 ng/ml, Sigma) and ionomycin (500 ng/ml, Sigma), and Golgi Stop (BD Pharmingen) was added on day 5. Cells were first stained with Live/Dead‐violet (Life Technologies) followed by a FITC‐conjugated anti‐CD4 (BD Pharmingen) antibody, fixed and permeabilized with Cytofix/Cytoper solution (BD Pharmingen), and intracellularly stained with PerCP‐Cy5.5‐conjugated anti‐IFNγ (BD Pharmingen), PE‐conjugated anti‐IL‐4, PE‐conjugated anti‐IL‐17A, or APC‐conjugated anti‐Foxp3 antibodies. Samples were acquired on a Gallios (Beckman Coulter), and the data were analyzed with FlowJo software.
Chemical induction of colitis
Experimental colitis was induced by treating 8‐week‐old mice with 3% DSS (36,000–50,000 MW, MP Biomedicals) administered via autoclaved drinking water for 7 days. DSS was then replaced by normal autoclaved water for 3–7 days. LIF or PBS was injected intraperitoneally into the mice at a dosage of 10 μg/kg/day. The body weight was monitored daily, and the DAI was determined as previously described (Song et al, 2015), according to the following parameters: rectal bleeding (0 points = negative; 1 point = positive hemoccult test; 2 points = visible blood traces in stool; and 3 points = gross rectal bleeding) and stool consistency (0 points = normal; 1 point = semiformed stool not adhering to the anus; 2 points = semiformed stool adhering to the anus; and 3 points = liquid stool adhering to the anus). The experiments were performed by an independent researcher who was blinded to the group allocation. Mice were sacrificed on days 4, 9, or 10 of different experimental protocols, and colon tissues were obtained for histopathological and RT–PCR analyses.
T‐cell adoptive transfer model of colitis
Wild‐type naïve T cells (CD4+ CD45RBhi) from the spleens of 8‐week‐old wild‐type mice were sorted by flow cytometry (MoFlo Astrios, Beckman Coulter). A total of 5 × 105 cells in 200 μl of sterile PBS were intraperitoneally injected into Rag1 −/− recipient mice. The mice were weighed throughout the colitis model timeline to assess weight loss. Representative colon tissues were harvested for histopathological analyses. LIF or PBS was injected intraperitoneally into the mice at a dosage of 10 μg/kg every 2 days.
Histopathology
Colon tissues were fixed in 4% neutral‐buffered paraformaldehyde, paraffin‐embedded and processed for histological analysis. Colon sections with a 5‐μm thickness were subjected to hematoxylin and eosin (H&E) or immunofluorescence (IF) staining and examined by light microscopy. The following antibodies were used for IF staining: anti‐Ki67 (Cell Signaling Technologies, 9449) and anti‐cleaved caspase 3 (Cell Signaling Technologies, 9664).
Mouse colon explant cultures
Colon explants were cultured as previously described (West et al, 2017). The experimental colitis mouse model was established first, and the mouse proximal colon was then harvested and cut into small segments (0.25 cm2), which were cultured overnight in RPMI medium supplemented with 10% fetal calf serum (FCS) and 10,000 U/ml penicillin/streptomycin. The LIF concentration in the supernatant was quantified by an ELISA kit (R&D Systems, UK) and normalized to the explant weight.
Isolation of IECs
Colon tissues dissected from control or DSS‐challenged mice were washed in PBS buffer and were then cut into pieces and digested at 37°C for 1 h in DMEM supplemented with 1% FBS, penicillin (100 U/ml), streptomycin (100 U/ml), collagenase type XI (0.2 mg/ml, Sigma), and dispase II (200 μg/ml, Roche). After digestion, the crypts containing IECs were isolated from the supernatant of the digestion buffer by centrifugation at 300 × g for 5 min. The isolated crypts were washed with DMEM supplemented with 2.5% FBS, 2% sorbitol, penicillin (100 U/ml), and streptomycin (100 U/ml) and were then centrifuged at 300 × g for 5 min. This step was repeated a minimum of five times. The isolated crypts were then resuspended in F‐12/DMEM culture medium supplemented with 5% FBS, insulin mix (Invitrogen), penicillin (100 U/ml), and streptomycin (100 U/ml) and plated in 12‐well plates coated with Matrigel (BD Pharmingen). The IECs were allowed to attach for 24 h, at which time all unattached material was removed and the cells were supplied with fresh culture medium as described above. The cells were treated with cytokines accordingly and collected in 2 days.
Isolation of LPLs
The dissected colon tissues were washed in PBS, and epithelial cells were removed by shaking at 250 rpm at 37°C for 30 min in PBS buffer containing 30 mM EDTA, 1 mM DTT, and 5% FBS. After sedimentation, the supernatant containing the crypts was discarded, and the remaining colon tissues were further cut into small pieces and digested at 37°C for 1 h with 0.3 mg/ml collagenase VIII (Sigma) and 100 U/ml DNase I (Sigma) in RPMI‐1640 medium supplemented with 5% FBS, penicillin (100 U/ml), and streptomycin (100 U/ml). The cell pellet was collected by centrifugation at 450 × g for 5 min, and the LPLs were further isolated by gradient centrifugation with Percoll (40/80%, GE Healthcare). The isolated LPLs were then subjected to FACS after reactivation and Golgi Stop treatment or to RT–PCR. The following antibodies were used in the analysis of LPLs: anti‐CD4‐FITC (BD Biosciences, 553047); anti‐CD4‐APC (eBioscience, 17‐0041); anti‐IFNγ‐PerCP‐Cy5.5 (BD Biosciences, 560660); anti‐IFNγ‐PE‐Cy7 (BD Biosciences, 561040); anti‐IL17A‐PE (eBioscience, 12‐7177); anti‐Foxp3‐PE (BD Biosciences, 560414); anti‐IL‐4‐PE (BD Biosciences, 554435); anti‐CD45‐PerCP‐Cy5.5 (BioLegend, 103132); anti‐CD3‐APC (BD Biosciences, 561826); and anti‐CD127‐FITC (eBioscience, 11‐1271‐81).
Commensal depletion
The SPF wild‐type mice were treated with a cocktail of antibiotics, as previously described (Song et al, 2015). Briefly, the mice were initially treated with 1 mg/ml neomycin, 0.5 mg/ml vancomycin, 1 mg/ml metronidazole, and 1 mg/ml ampicillin for 4 weeks. Fresh antibiotic solution was supplied every week. Four weeks later, the drinking water was further supplemented with 1 mg/ml streptomycin, 170 μg/ml gentamicin, 125 μg/ml ciprofloxacin, and 1 mg/ml bacitracin for another 5 weeks. With antibiotic treatment, more than 99% of the intestinal microbes were removed. Colitis was induced in the microbe‐free mice 9 weeks later, as described in the section “Chemical Induction of Colitis”. The mice were sacrificed on the indicated days after DSS treatment, and the colon tissues were obtained for further analysis.
Mass spectrometry analysis
Myc‐tagged STAT4 constructs of mouse origin were transfected into HEK293T cells which were cultured in 10‐cm Petri dishes with Lipofectamine 2000. Twenty‐four hours later, the cells from two dishes were treated with LIF for 30 min and collected to immunoprecipitate STAT4 from the cell lysate. The immunoprecipitated STAT4 from the above HEK293T cells was separated via SDS–PAGE. The Coomassie blue‐stained STAT4 band was excised from the gel for protease digestion followed by mass spectrometry analysis with a Thermo LC‐MS/MS system.
ChIP
The ChIP assay was performed as described previously (Nelson et al, 2006; Yang et al, 2011). In brief, Th17 cells were crosslinked for 9 min with 0.9% formaldehyde. The cells were washed with cold PBS twice and collected for sonication. The cell lysates were immunoprecipitated overnight at 4°C with anti‐STAT3 or anti‐STAT4 antibodies. Protein A beads were added to each sample. After washing, crosslinks were reversed with 10% Chelex beads for 10 min at 100°C. The eluted DNA was analyzed by qPCR with custom‐designed primers (Table EV1). Each cycling threshold value was normalized to the corresponding input value.
16S ribosomal RNA sequencing
Fecal samples were collected at the indicated time points, and the microbial DNA was extracted from the feces with a TIAGEN stool DNA isolation kit. The DNA concentration was determined by a NanoDrop (Thermo Scientific), and the DNA quality was assessed by agarose gel electrophoresis. Sequencing was then performed as described previously (Song et al, 2015). The abundances of the indicated intestinal microbial groups were analyzed with previously described real‐time primers (Song et al, 2015).
Accession numbers
The raw data of microarray and RNA sequencing are deposited with the Gene Expression Omnibus (GEO) repository under the accession number GEO: GSE124079
Statistical analysis
Two‐tailed Student's t‐tests were used for all comparisons, including qPCR analysis. The data are presented as the means ± SEMs. Statistical significance is indicated by asterisks (*). A two‐sided P‐value of < 0.05 was considered statistically significant.
Author contributions
YSZ and DEX contributed to the study design and conducted the majority of the experiments; ZW prepared the GFP+ Th17 cells; YS performed the molecular cloning; QCZ, CZ, JMZ, XS, ZL and YJ were involved in the design or execution of several experiments; XZ, TCZ and BS were involved in the analysis of the data and manuscript proof reading. YEC supervised the study and analyzed the data; and YSZ and YEC wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Acknowledgements
This work was supported by National Natural Science Foundation of China Grant 81530083 and Science and Technology Commission of Shanghai Municipality Grant 15JC1403700. We are grateful to M. Cao, J. Wang, and C. Li for technical help in mouse LPL and IEC preparation, L. Zhao for colon dissection and preparation, and H. Liu for the maintenance of the IL17A‐eGFP mice. We are grateful to CS Shao, ZT Wang, and M. Prasad for manuscript review and discussion.
The EMBO Journal (2019) 38:e99595
References
- Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F (2010) Interleukin‐23 drives intestinal inflammation through direct activity on T cells. Immunity 33: 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell 157: 121–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouma G, Strober W (2003) The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol 3: 521–533 [DOI] [PubMed] [Google Scholar]
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, Powrie F (2010) Innate lymphoid cells drive interleukin‐23‐dependent innate intestinal pathology. Nature 464: 1371–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Yang Y, Wang Z, Liu A, Fang L, Wu F, Hong J, Shi Y, Leung S, Dong C, Zhang JZ (2011) Leukemia inhibitory factor inhibits T helper 17 cell differentiation and confers treatment effects of neural progenitor cell therapy in autoimmune disease. Immunity 35: 273–284 [DOI] [PubMed] [Google Scholar]
- Chang J, Voorhees TJ, Liu YS, Zhao YG, Chang CH (2010) Interleukin‐23 production in dendritic cells is negatively regulated by protein phosphatase 2A. Proc Natl Acad Sci USA 107: 8340–8345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, Murray PJ, Vignali DA (2012) The composition and signaling of the IL‐35 receptor are unconventional. Nat Immunol 13: 290–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE Jr (1997) STATs and gene regulation. Science 277: 1630–1635 [DOI] [PubMed] [Google Scholar]
- Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO (1994) Dextran sulfate sodium‐induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107: 1643–1652 [DOI] [PubMed] [Google Scholar]
- Dieleman LA, Palmen MJ, Akol H, Bloemena E, Pena AS, Meuwissen SG, Van Rees EP (1998) Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol 114: 385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE (1996) Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84: 443–450 [DOI] [PubMed] [Google Scholar]
- Eberl G, Colonna M, Di Santo JP, McKenzie AN (2015) Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science 348: aaa6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehret GB, Reichenbach P, Schindler U, Horvath CM, Fritz S, Nabholz M, Bucher P (2001) DNA binding specificity of different STAT proteins. Comparison of in vitro specificity with natural target sites. J Biol Chem 276: 6675–6688 [DOI] [PubMed] [Google Scholar]
- Elson CO, Cong Y, Weaver CT, Schoeb TR, McClanahan TK, Fick RB, Kastelein RA (2007) Monoclonal anti‐interleukin 23 reverses active colitis in a T cell‐mediated model in mice. Gastroenterology 132: 2359–2370 [DOI] [PubMed] [Google Scholar]
- Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S, Centola M, Wu CY, Yamada N, El Gabalawy H, O'Shea JJ (2000) Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1‐mediated inflammation. J Immunol 164: 4659–4664 [DOI] [PubMed] [Google Scholar]
- Fujio Y, Kunisada K, Hirota H, Yamauchi‐Takihara K, Kishimoto T (1997) Signals through gp130 upregulate bcl‐x gene expression via STAT1‐binding cis‐element in cardiac myocytes. J Clin Invest 99: 2898–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbers C, Hermanns HM, Schaper F, Muller‐Newen G, Grotzinger J, Rose‐John S, Scheller J (2012) Plasticity and cross‐talk of interleukin 6‐type cytokines. Cytokine Growth Factor Rev 23: 85–97 [DOI] [PubMed] [Google Scholar]
- Geng J, Yu S, Zhao H, Sun X, Li X, Wang P, Xiong X, Hong L, Xie C, Gao J, Shi Y, Peng J, Johnson RL, Xiao N, Lu L, Han J, Zhou D, Chen L (2017) The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat Immunol 18: 800–812 [DOI] [PubMed] [Google Scholar]
- Geremia A, Arancibia‐Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, Travis SP, Powrie F (2011) IL‐23‐responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med 208: 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G et al (2012) Adenoma‐linked barrier defects and microbial products drive IL‐23/IL‐17‐mediated tumour growth. Nature 491: 254–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI (2013) Inflammation and colorectal cancer: colitis‐associated neoplasia. Semin Immunopathol 35: 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimbaud R, Abitbol V, Bertrand V, Quartier G, Chauvelot‐Moachon L, Giroud J, Couturier D, Chaussade DC (1998) Leukemia inhibitory factor involvement in human ulcerative colitis and its potential role in malignant course. Eur Cytokine Netw 9: 607–612 [PubMed] [Google Scholar]
- Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT (2015) Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci USA 112: 7061–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim MH (1996) The Jak‐STAT pathway: specific signal transduction from the cell membrane to the nucleus. Eur J Clin Invest 26: 1–12 [DOI] [PubMed] [Google Scholar]
- Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, Powrie F, Maloy KJ (2006) Interleukin‐23 drives innate and T cell‐mediated intestinal inflammation. J Exp Med 203: 2473–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR (2008) Specific microbiota direct the differentiation of IL‐17‐producing T‐helper cells in the mucosa of the small intestine. Cell Host Microbe 4: 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson NG, Szabo SJ, Weber‐Nordt RM, Zhong Z, Schreiber RD, Darnell JE Jr, Murphy KM (1995) Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med 181: 1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenab S, Morris PL (1998) Testicular leukemia inhibitory factor (LIF) and LIF receptor mediate phosphorylation of signal transducers and activators of transcription (STAT)‐3 and STAT‐1 and induce c‐fos transcription and activator protein‐1 activation in rat Sertoli but not germ cells. Endocrinology 139: 1883–1890 [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ (1996) Impaired IL‐12 responses and enhanced development of Th2 cells in Stat4‐deficient mice. Nature 382: 174–177 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Okamoto S, Hisamatsu T, Kamada N, Chinen H, Saito R, Kitazume MT, Nakazawa A, Sugita A, Koganei K, Isobe K, Hibi T (2008) IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut 57: 1682–1689 [DOI] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB (2007) Host‐mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2: 119–129 [DOI] [PubMed] [Google Scholar]
- Mankertz J, Schulzke JD (2007) Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr Opin Gastroenterol 23: 379–383 [DOI] [PubMed] [Google Scholar]
- Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O'Malley JT, Kapur R, Levy DE, Kansas GS, Kaplan MH (2007) Stat3 and Stat4 direct development of IL‐17‐secreting Th cells. J Immunol 178: 4901–4907 [DOI] [PubMed] [Google Scholar]
- Monteleone G, Monteleone I, Fina D, Vavassori P, Del Vecchio Blanco G, Caruso R, Tersigni R, Alessandroni L, Biancone L, Naccari GC, MacDonald TT, Pallone F (2005) Interleukin‐21 enhances T‐helper cell type I signaling and interferon‐gamma production in Crohn's disease. Gastroenterology 128: 687–694 [DOI] [PubMed] [Google Scholar]
- Monteleone G, Caruso R, Fina D, Peluso I, Gioia V, Stolfi C, Fantini MC, Caprioli F, Tersigni R, Alessandroni L, MacDonald TT, Pallone F (2006) Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut 55: 1774–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat AM (2003) Anatomical basis of tolerance and immunity to intestinal antigens. Nat Rev Immunol 3: 331–341 [DOI] [PubMed] [Google Scholar]
- Mukasa R, Balasubramani A, Lee YK, Whitley SK, Weaver BT, Shibata Y, Crawford GE, Hatton RD, Weaver CT (2010) Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185 [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O'Shea JJ, Biron CA (2002) Critical role for STAT4 activation by type 1 interferons in the interferon‐gamma response to viral infection. Science 297: 2063–2066 [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Noss EH, Mizoguchi F, Huppertz C, Wei KS, Watts GF, Brenner MB (2017) Autocrine loop involving IL‐6 family member LIF, LIF receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity 46: 220–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea JJ, Murray PJ (2008) Cytokine signaling modules in inflammatory responses. Immunity 28: 477–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea JJ, Paul WE (2010) Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327: 1098–1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, Pflanz S, Zhang R, Singh KP, Vega F, To W, Wagner J, O'Farrell AM, McClanahan T, Zurawski S, Hannum C, Gorman D, Rennick DM, Kastelein RA, de Waal Malefyt R et al (2002) A receptor for the heterodimeric cytokine IL‐23 is composed of IL‐12Rbeta1 and a novel cytokine receptor subunit, IL‐23R. J Immunol 168: 5699–5708 [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D (2014) Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14: 141–153 [DOI] [PubMed] [Google Scholar]
- Podolsky DK (2002) Inflammatory bowel disease. N Engl J Med 347: 417–429 [DOI] [PubMed] [Google Scholar]
- Rakoff‐Nahoum S, Paglino J, Eslami‐Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll‐like receptors is required for intestinal homeostasis. Cell 118: 229–241 [DOI] [PubMed] [Google Scholar]
- Schindler C, Darnell JE Jr (1995) Transcriptional responses to polypeptide ligands: the JAK‐STAT pathway. Annu Rev Biochem 64: 621–651 [DOI] [PubMed] [Google Scholar]
- Siakavellas SI, Bamias G (2012) Role of the IL‐23/IL‐17 axis in Crohn's disease. Discov Med 14: 253–262 [PubMed] [Google Scholar]
- Simpson SJ, Shah S, Comiskey M, de Jong YP, Wang B, Mizoguchi E, Bhan AK, Terhorst C (1998) T cell‐mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)‐4 pathway, but is not conditional on interferon gamma expression by T cells. J Exp Med 187: 1225–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Garrett WS (2011) The gut microbiota and mucosal T cells. Front Microbiol 2: 111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Dai D, He X, Zhu S, Yao Y, Gao H, Wang J, Qu F, Qiu J, Wang H, Li X, Shen N, Qian Y (2015) Growth factor FGF2 cooperates with interleukin‐17 to repair intestinal epithelial damage. Immunity 43: 488–501 [DOI] [PubMed] [Google Scholar]
- Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE Jr, Yancopoulos GD (1995) Choice of STATs and other substrates specified by modular tyrosine‐based motifs in cytokine receptors. Science 267: 1349–1353 [DOI] [PubMed] [Google Scholar]
- Strober W, Fuss IJ (2011) Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 140: 1756–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, Sandborn WJ, Hardiman G, Raz E, Maehara Y, Yoshimura A, Zucman‐Rossi J, Guan KL, Karin M (2015) A gp130‐Src‐YAP module links inflammation to epithelial regeneration. Nature 519: 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN (1996) Requirement for Stat4 in interleukin‐12‐mediated responses of natural killer and T cells. Nature 382: 171–174 [DOI] [PubMed] [Google Scholar]
- Walker SR, Nelson EA, Yeh JE, Pinello L, Yuan GC, Frank DA (2013) STAT5 outcompetes STAT3 to regulate the expression of the oncogenic transcriptional modulator BCL6. Mol Cell Biol 33: 2879–2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KS, Ritz J, Frank DA (1999) IL‐2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J Immunol 162: 299–304 [PubMed] [Google Scholar]
- Wang S, Xia P, Chen Y, Qu Y, Xiong Z, Ye B, Du Y, Tian Y, Yin Z, Xu Z, Fan Z (2017a) Regulatory innate lymphoid cells control innate intestinal inflammation. Cell 171: 201–216.e218 [DOI] [PubMed] [Google Scholar]
- Wang XJ, Qiao Y, Xiao MM, Wang L, Chen J, Lv W, Xu L, Li Y, Wang Y, Tan MD, Huang C, Li J, Zhao TC, Hou Z, Jing N, Chin YE (2017b) Opposing roles of acetylation and phosphorylation in LIFR‐dependent self‐renewal growth signaling in mouse embryonic stem cells. Cell Rep 18: 933–946 [DOI] [PubMed] [Google Scholar]
- Wehinger J, Gouilleux F, Groner B, Finke J, Mertelsmann R, Weber‐Nordt RM (1996) IL‐10 induces DNA binding activity of three STAT proteins (Stat1, Stat3, and Stat5) and their distinct combinatorial assembly in the promoters of selected genes. FEBS Lett 394: 365–370 [DOI] [PubMed] [Google Scholar]
- West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, Coccia M, Gortz D, This S, Stockenhuber K, Pott J, Friedrich M, Ryzhakov G, Baribaud F, Brodmerkel C, Cieluch C, Rahman N, Muller‐Newen G, Owens RJ, Kuhl AA et al (2017) Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor‐neutralizing therapy in patients with inflammatory bowel disease. Nat Med 23: 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz S, Popp V, Kindermann M, Gerlach K, Weigmann B, Fichtner‐Feigl S, Neurath MF (2017) Chemically induced mouse models of acute and chronic intestinal inflammation. Nat Protoc 12: 1295–1309 [DOI] [PubMed] [Google Scholar]
- Wrighting DM, Andrews NC (2006) Interleukin induces hepcidin expression through STAT3. Blood 108: 3204–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK (2008) Retinoic acid increases Foxp3(+) regulatory T cells and inhibits development of Th17 cells by enhancing TGF‐beta‐driven Smad3 signaling and inhibiting IL‐6 and IL‐23 receptor expression. J Immunol 181: 2277–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Ji JJ, Le W, Xu YS, Dou D, Pan J, Jiao Y, Zhong T, Wu D, Wang Y, Wen C, Xie GQ, Yao F, Zhao H, Fan YS, Chin YE (2015) The STAT3 HIES mutation is a gain‐of‐function mutation that activates genes via AGG‐element carrying promoters. Nucleic Acids Res 43: 8898–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XP, Ghoreschi K, Steward‐Tharp SM, Rodriguez‐Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O'Shea JJ, Laurence A (2011) Opposing regulation of the locus encoding IL‐17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol 12: 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, Kleinschek MA, Owyang A, Mattson J, Blumenschein W, Murphy E, Sathe M, Cua DJ, Kastelein RA, Rennick D (2006) IL‐23 is essential for T cell‐mediated colitis and promotes inflammation via IL‐17 and IL‐6. J Clin Invest 116: 1310–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, Wu C, Karwacz K, Xiao S, Jorgolli M, Gennert D, Satija R, Shakya A, Lu DY, Trombetta JJ, Pillai MR, Ratcliffe PJ, Coleman ML, Bix M, Tantin D et al (2013) Dynamic regulatory network controlling TH17 cell differentiation. Nature 496: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6