

Abstract
Key points
Obesity is associated with disrupted satiety regulation. Mice with diet‐induced obesity have reduced vagal afferent neuronal excitability and a decreased afferent response to satiety signals. A low grade inflammation occurs in obesity with increased expression of inducible nitric oxide synthase (iNOS).
Inhibition of iNOS in diet‐induced obese mice restored vagal afferent neuronal excitability, increased the afferent response to satiety mediators and distention of the gut, and reduced short‐term energy intake.
A prolonged inhibition of iNOS reduced energy intake and body weight gain during the first week, and reduced amounts of epididymal fat after 3 weeks.
We identified a novel pathway underlying disrupted satiety regulation in obesity. Blocking of this pathway might be clinically useful for the management of obesity.
Abstract
Vagal afferents regulate feeding by transmitting satiety signals to the brain. Mice with diet‐induced obesity have reduced vagal afferent sensitivity to satiety signals. We investigated whether inducible nitric oxide synthase (iNOS)‐derived NO contributed to this reduction. C57BL/6J mice were fed a high‐ or low‐fat diet for 6–8 weeks. Nodose ganglia and jejunum were analysed by immunoblotting for iNOS expression; NO production was measured using a fluorometric assay. Nodose neuron excitability and intestinal afferent sensitivity were evaluated by whole‐cell patch clamp and in vitro afferent recording, respectively. Expression of iNOS and production of NO were increased in nodose ganglia and the small intestine in obese mice. Inhibition of iNOS in obese mice by pre‐treatment with an iNOS inhibitor increased nodose neuron excitability via 2‐pore‐domain K+ channel leak currents, restored afferent sensitivity to satiety signals and reduced short‐term energy intake. Obese mice given the iNOS inhibitor daily for 3 weeks had reduced energy intake and decreased body weight gain during the first week, compared to mice given saline, and lower amounts of epididymal fat at the end of 3 weeks. Inhibition of iNOS or blocking the action of iNOS‐derived NO on vagal afferent pathways might comprise therapeutic strategies for hyperphagia and obesity.
Keywords: obesity, vagal afferent, satiety, iNOS, nitric oxide, potassium channel
Key points
Obesity is associated with disrupted satiety regulation. Mice with diet‐induced obesity have reduced vagal afferent neuronal excitability and a decreased afferent response to satiety signals. A low grade inflammation occurs in obesity with increased expression of inducible nitric oxide synthase (iNOS).
Inhibition of iNOS in diet‐induced obese mice restored vagal afferent neuronal excitability, increased the afferent response to satiety mediators and distention of the gut, and reduced short‐term energy intake.
A prolonged inhibition of iNOS reduced energy intake and body weight gain during the first week, and reduced amounts of epididymal fat after 3 weeks.
We identified a novel pathway underlying disrupted satiety regulation in obesity. Blocking of this pathway might be clinically useful for the management of obesity.
Introduction
Obesity and its related morbidity are currently one of the most prevalent public health problems. The prevalence of obesity among US adults was 36.5% during 2011–2014 (Ogden et al. 2015). Globally, the prevalence of being overweight and being obese combined was 36.9% for men and 38.0% for women in 2013 (Ng et al. 2014). Obesity is associated with health risks, such as diabetes, dyslipidaemia, hypertension, cardiovascular disease and cancer, and thus is a primary driver for increasing health costs (Sansbury & Hill, 2014; Shettar et al. 2017). After a protracted debate, many health organizations recently recognized obesity as a pathological state requiring clinical intervention (Bray et al. 2017). Bariatric surgery is effective in weight loss; however, the availability, invasiveness and frequent complications of this procedure prevent its wide application (Kim et al. 2011). Although there is growing evidence that neuromodulation or pharmacotherapy targeting the vagus nerve may be promising treatments (de Lartigue, 2016), our current understanding of the molecular mechanisms underlying the development and maintenance of obesity is still limited.
The vagus nerve innervating the gut plays an important role in regulating feeding behaviour and metabolism by communicating between the gut and brain. A previous study in obese mice by Daly and Park (2011) demonstrated that nodose neurons were less excitable and also that jejunum afferent sensitivity to satiety signals was attenuated. Although our recent work suggested that increased two‐pore domain K+ (K2P) conductance underlies this reduced sensitivity (Park et al. 2018), the causal mechanisms have not been fully clarified.
Some studies suggest that NO may cause a prolonged decrease in nodose neuronal excitability (Bielefeldt et al. 1999) and additionally play an inhibitory neuromodulatory role on a select population of vagal afferents (Page et al. 2009). Both constitutive nitric oxide synthase (NOS) isoforms, neuronal NOS (nNOS) and endothelial NOS (eNOS), are present in the nodose ganglia, whereas inducible nitric oxide synthase (iNOS) is not detectable under normal physiological conditions (Yamamoto et al. 2003). Within the gastrointestinal tract, nNOS and eNOS are found in enteric neurons and epithelial cells (Page et al. 2009; Sansbury & Hill, 2014) and iNOS is expressed in the epithelial cells of inflamed tissue (Kolios et al. 1998). iNOS has the highest capacity to generate NO in response to inflammatory stimuli.
Obesity is associated with chronic low‐grade inflammation. Adipocyte hypertrophy occurs in obesity and results in the increased production of many pro‐inflammatory adipokines and cytokines, such as leptin, resistin, adiponectin, tumor necrosis factor‐α, interleukin (IL)‐6 and IL‐1 (Hosogai et al. 2007; Engin, 2017). Increased leptin, hypoxia and adipocyte cell death are possible contributors to macrophage recruitment in adipose tissue (Surmi & Hasty, 2008). Recruited macrophages during a high‐fat diet display inflammatory properties such as overexpression of iNOS and IL‐6 (Lumeng et al. 2007). Interestingly, iNOS knockout mice fed a high‐fat diet are protected against the development of insulin resistance (Noronha et al. 2005), suggesting a role for iNOS in the regulation of metabolism.
Thus, the present study aimed to test the hypothesis that iNOS‐derived NO is involved in the disruption of vagal afferent signalling in diet‐induced obesity. Furthermore, we hypothesized that iNOS acts via K2P channels because these channels have recently been implicated in the mechanism of reduced sensitivity of vagal afferents after high‐fat feeding.
Methods
Animals and ethical approval
All experiments were performed under an approved protocol from the Queen's University Animal Care Committee (reference number 2014‐1496) in accordance with guidelines from the Canadian Council for Animal Care. The authors understand the ethical principles under which the Journal of Physiology operates and declare that this work complies with those policies and regulations.
Diet‐induced obese and control mice (male C57BL/6J; Stock Numbers: 380050 and 380056; Jackson Laboratory, Bar Harbor, ME, USA) were fed on a high‐fat diet (60 kcal% fat; D12492; Research Diets, Inc., New Brunswick, NJ, USA) or a low‐fat diet (10 kcal% fat; D12450B) for 6–8 weeks (starting from 6 weeks of age). High‐fat fed (HFF) and low‐fat fed (LFF) mice were individually housed under identical conditions with free access to water and food under a standard light/dark cycle (lights on 07.00 h). They were killed by isoflurane inhalation followed by cervical dislocation.
Isolation of single nodose ganglion neurons
The nodose ganglia were isolated as described previously (Daly and Park et al. 2011).
Western blotting
The nodose ganglia and full‐sickness jejunum used for western blotting were conserved in RNALater (AM7020; Invitrogen, Valancia, CA, USA) at −20°C. Samples were lysed in radioimmunoprecipitation assay buffer containing Pierce Protease Inhibitor (catalogue no. 88666; Thermo Scientific, Waltham, MA, USA) cocktail and homogenized using three 10 s bursts of sonication at 1 min intervals. Following a 30 min incubation on ice and a 20 min spin at 16,000 g, supernatants were collected and the protein concentration was determined using a BCA Protein Assay kit (catalogue no. 23225; Thermo Scientific). Next, 50 μg of proteins were separated by SDS‐PAGE (6–15% gradient) at 150 V. Samples were transferred to Immobilon polyvinylidene difluoride membrane (IPFL00010; Millipore, Billerica, MA, USA) at 25 V in transfer buffer (25 mm Tris, 190 mm glycine and 10% methanol) overnight at 4°C. Membrane was blocked with 5% non‐fat milk for 2 h and incubated with primary antibodies (rabbit polyclonal iNOS antibody, dilution 1:500; NB300605; Novus Biologicals, Littleton, CO, USA; mouse monoclonal β‐actin antibody, dilution 1:5000; A5316; Sigma, St Louis, MO, USA) overnight at 4°C, followed by three washes with Tris‐buffered saline with Tween 20 (TBST). HRP‐conjugated secondary antibody (goat anti‐rabbit IgG, dilution 1:5000; catalogue no. 7074; Cell Signaling Technology, Beverly, MA, USA; goat anti‐mouse IgG, dilution 1:5000; catalogue no. 31430; Invitrogen) was added to the membrane, followed by a 1 h incubation at room temperature and three washes with TBST. Immunoreactive bands were visualized by chemiluminescence using SuperSignal West Pico ECL reagents (catalogue no. 34087; Thermo Scientific) or Luminata Forte Western HRP substrate (WBLUF0100; Millipore). Images were acquired and analysed using a ChemiDoc XRS+ System (Bio‐Rad, Hercules, CA, USA) and Image Lab software, version 5.1 (Bio‐Rad). Band boundaries of LFF and HFF samples were adjusted to be identical.
Nitrate/nitrite fluorometric assay
The nitrate and nitrite concentration in the jejunum, serum and culture media of nodose ganglion neuron from both LFF and HFF mice was measured using a fluorometric assay kit (780051; Cayman Chemical Company, Ann Arbor, MI, USA) in accordance with the manufacturer's instructions. Serum and jejunum were collected from mice with/without l‐N 6‐(1‐iminoethyl) lysine dihydrochloride (l‐NIL; i.p. injection, 10 mg kg−1) treatment. Nodose neurons were incubated in media with/without N ω‐propyl‐l‐arginine (N ω, 100 nm) or l‐NIL (10 μm) for 30 min before media collection. About 30 mg of jejunum tissue was collected in PBS, homogenized using a motor‐driven pestle (FS749540‐0000; Fisherbrand, Fisher Scientific Co., Pittsburgh, PA, USA) and ultrafiltered using a 10 kDa ultrafilter (UFC501024; Millipore). The same amount of sample (1 ml) as that used for the serum and nodose ganglion culture media was collected for assay. Total nitrate and nitrite concentration was determined using the nitrate standard curve (ranging from 0.06 to 3.85 μm). For jejunum, the concentration of nitrate/nitrite was normalized to the weight of tissue collected.
Multi‐unit afferent nerve recordings
The nerve activity of afferents innervating mouse small intestine was recorded as described previously (Daly and Park et al. 2011). Most drugs were perfused into the bath, for either a brief 1 min exposure or a 15 min continuous perfusion, whereas only glucagon‐like peptide 1 (GLP‐1) agonist exendin‐4 (ex‐4) was delivered intraluminally for 10 min. Based on work from our group and another group, the GLP‐1 response has a long latency (Kakei et al. 2002) and minimal effect when given by bath perfusion; thus, we applied ex‐4 intraluminally for 10 min to reduce the amount of drug needed (given the much lower intraluminal perfusion rate) and to be able to observe a reliable response that could be compared between LFF and HFF groups.
Single unit analysis was performed using the spike sorting function of Spike2 (Cambridge Electronic Design Ltd, Cambridge, UK) to discriminate the activities of individual afferent units. Based on their sensitivity to ramp distention, afferent units were classified into three subpopulations: low threshold (LT), wide dynamic range (WDR) and high threshold (HT) (Rong et al. 2004). LT afferents responded to low pressure and plateaued after the first few increases in mmHg. WDR afferents also had a low threshold, although their firing rate continued to increase with pressure. HT afferents were defined as units with a threshold over 15 mmHg. Drug responses were assessed by mean change in afferent discharge over a 30 s period at the peak compared to a 30 s baseline period. Drug responses were normalized against the number of responsive units in that nerves. Distention responses were analysed using a customer‐made script and expressed as the increase in firing frequency with increased pressure. To compare distention responses among different preparations, firing frequency was normalized against the number of units. To compare distention responses within the same preparations, firing frequency was normalized against the peak firing rate of the control distention.
Whole‐cell patch clamp recordings
Whole‐cell patch clamp recordings were performed as described previously (Daly and Park et al. 2011). Nodose ganglion neurons were randomly selected for recording, based on our previous work demonstrating that obesity decreases the excitability of both randomly selected neurons and small intestine‐projecting neurons (Daly and Park et al. 2011). Neurons were recorded for 10–15 min and recordings were terminated if membrane potential/access resistance changed during this time. K2P currents were isolated by ion replacement and a pharmacological method as described previously (Park et al. 2018). The K2P channels do not have time‐dependent kinetics (Lesage et al. 1996) and thus a ramp protocol (−90 to 30 mV, 400 ms, 10 mV increment, V h = −60 mV, 2.5 kHz sampling) was applied. Junction potentials (10.1 mV) were corrected after recording. Recordings were analysed using Clampfit, version 10.0 (Axon Instruments Inc., Foster City, CA, USA) and Origin (OriginLab, Northampton, MA, USA).
Food intake measurement
Food intake was monitored using TSE LabMaster Home Cage System (TSE Systems, Inc., Chesterfield, MO, USA). Mice were allowed to acclimate to the metabolic cages for at least 3 days prior to any treatment. Energy intake and meal pattern were analysed using the same software for monitoring. A meal should be separated from other feeding events by more than 10 min and the minimal meal size was defined as 0.03 g. Energy intake was calculated as food intake multiplied by energy density (5.24 and 3.85 kcal g−1 for the high‐ and low‐fat diet, respectively).
To evaluate the influence of iNOS on short‐term energy intake, LFF and HFF mice received an i.p. injection of saline (equal volume as l‐NIL) at 18.00 h followed by an injection of l‐NIL (10 mg kg−1), 24 h later. An i.p. injection was performed using a 1 mL syringe and a 26‐G needle. Feeding patterns during 24 h after treatment were compared with saline injection. To evaluate the long‐term effects of iNOS suppression on energy intake and adiposity, HFF mice were divided into two groups, receiving one daily i.p. injection of l‐NIL or an equivalent volume of saline for 3 weeks, respectively. Injection sites were alternated daily between the left and right lower abdominal quadrants. Epididymal fat pads were weighed after death of the mice.
Drugs
All drugs were prepared as stock solution, kept frozen at −20°C and diluted to their final concentration in either Krebs buffers in afferent recording or Hepes buffer in patch clamp immediately prior to application. Regrading commercial sources: l‐NIL (catalogue no. 80310), 5‐hydroxytryptamine (5‐HT; catalogue no. 14332), exendin‐4 (acetate; catalogue no. 11096), N ω (catalogue no. 80587), N‐([3‐(aminomethyl) phenyl]methyl)ethanimidamide dihydrochloride (1400W; catalogue no. 81520), 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one (ODQ; catalogue no. 81410) and sildenafil citrate (14008) were purchased from Cayman Chemical (Cayman Chemical Company, Ann Arbor, MI, USA); sodium nitroprusside dihydrate (SNP; catalogue no. 71778) was obtained from Sigma; and cholecystokinin octapeptide sulphated (CCK‐8S; catalogue no. ab120209) was obtained from Abcam (Abcam, Cambridge, MA, USA).
Statistical analysis
All data were expressed as the mean ± SD unless otherwise stated. A significant difference was determined using Student's t test (two‐tailed), or a one‐ or two‐way ANOVA with a Bonferroni test as appropriate. N refers to the number of animals, and n indicates the number of cells or samples. P < 0.05 was considered statistically significant. Significance was defined as: P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***). An absence of asterisks indicates no significance or cases where it is not applicable.
Results
Increased iNOS expression and NO production in HFF mice
Immunoblotting with an anti‐iNOS antibody detected protein bands of ∼130 kDa in all nodose ganglion and jejunum samples from HFF mice (Fig. 1 A). Bands were barely visible in all three nodose ganglion pools and two out of eight jejunum samples from LFF mice. Because iNOS protein abundance in the nodose ganglion was much lower than the jejunum, nodose ganglia from four mice were pooled and visualized using a more sensitive HRP substrate. iNOS expression (normalized against β‐actin) was increased in both the nodose ganglia (31.7 ± 5.7% vs. 15.8 ± 3.2%, P < 0.05, unpaired t test, n = 3 pools, N = 12) (Fig. 1 B) and jejunum (85.4 ± 20.5% vs. 48.2 ± 25.9%, P < 0.01, unpaired t test, N = 8) (Fig. 1 C) from HFF mice.
Figure 1.
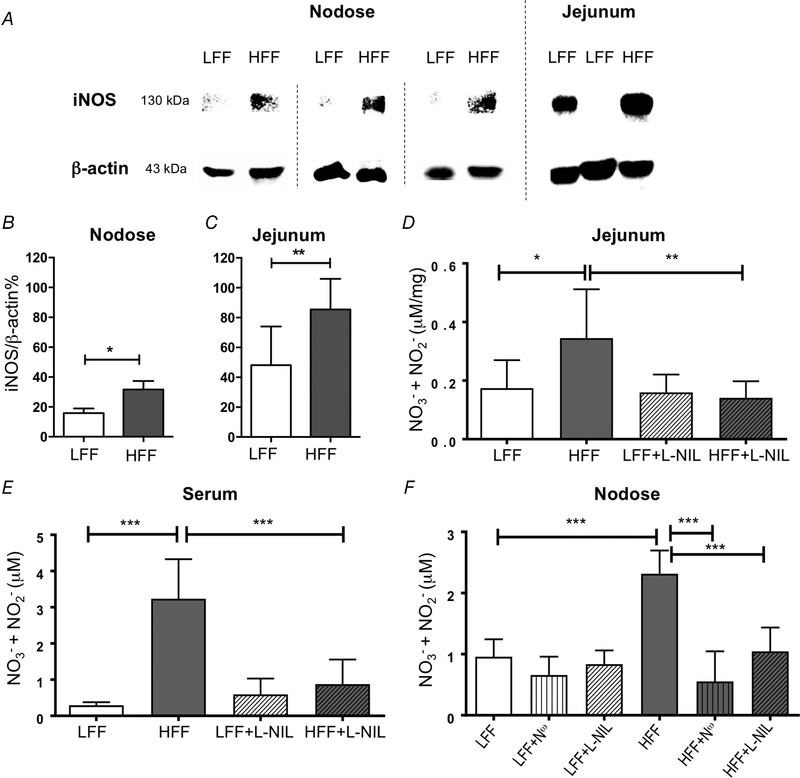
iNOS expression and NO production were increased in HFF mice
A, total iNOS protein in nodose ganglion and jejunum visualized on SDS‐PAGE gel by western blotting. B and C, iNOS expression was increased in HFF mice nodose ganglia (B) (P < 0.05, n = 3 pools, each containing nodose ganglia from four mice, unpaired t test) and jejunum (C) (P < 0.01, N = 8). D and E, total nitrate/nitrite was increased in the jejunum (D) (P < 0.05, N ≥ 8, two‐way ANOVA with Bonferroni test) and serum (E) (P < 0.001, N = 6) from HFF mice. l‐NIL treatment (10 mg/kg, i.p. injection) reversed this change in both jejunum (D) (P < 0.01, N ≥ 7) and serum (E) (P < 0.001, N = 6). F, total nitrate/nitrite was significantly increased in culture media of nodose neurons from HFF mice compared to LFF mice (P < 0.001, N = 6, two‐way ANOVA with Bonferroni test). This augmentation in HFF nodose neurons was significantly attenuated by N ω (100 nm, P < 0.001, N = 6) and l‐NIL (10 μm, P < 0.001, N = 6).
Total nitrate/nitrite was measured as an index for NO production using a fluorometric assay kit. There was a statistically significant interaction between the effects of diet and l‐NIL treatment (a selective iNOS inhibitor, 10 mg kg−1, i.p. injection) on nitrate/nitrite level in both jejunum [F 1,26 = 5.282, P < 0.05, B coefficient = −0.19, 95% confidence interval (CI): −0.36 to −0.02, two‐way ANOVA with Bonferroni test, N ≥ 8] (Fig. 1 D) and serum (F 1,20 = 22.89, P < 0.001, B coefficient = −2.66, 95% CI: −3.82 to −1.50, N = 6) (Fig. 1 E). Simple main effects analysis with Bonferroni post hoc tests revealed that untreated HFF mice had a higher nitrate/nitrite level in the jejunum homogenate (0.35 ± 0.17 vs. 0.17 ± 0.10 μm mg−1, P < 0.05) and serum (3.23 ± 1.09 vs. 0.29 ± 0.08 μm, P < 0.001) compared to untreated LFF mice; in addition, l‐NIL had no significant effect in LFF mice (P > 0.999) but did decrease NO production significantly in HFF mice in both jejunum (0.35 ± 0.17 vs. 0.14 ± 0.06 μm mg−1, P < 0.01) and serum (3.23 ± 1.09 vs. 0.88 ± 0.68 μm, P < 0.001). Similarly, there was a statistically significant interaction between the effects of diet and l‐NIL or N ω (100 nm, nNOS inhibitor) treatment on NO production in the culture media incubated with nodose neurons [F 2,30 = 14.037, P < 0.001, B coefficient (l‐NIL) = −1.146, 95% CI: −1.739 to −0.553, B coefficient (N ω) = −1.463, 95% CI: −2.056 to −0.969, N = 6] (Fig. 1 F). Simple main effects analysis revealed that HFF mice had a higher nitrate/nitrite level in the culture media incubated with nodose neurons (2.32 ± 0.38 vs. 0.96 ± 0.28 μm, P < 0.001) and this was reduced by both l‐NIL (P < 0.001) and N ω (P < 0.001).
Inhibition of NO signalling increased membrane excitability in nodose neurons from HFF mice
High‐fat diet feeding resulted in decreased excitability of nodose ganglion neurons (Daly and Park et al. 2011). Inhibition of NO synthase for 30 min increased the excitability in HFF nodose neurons. N ω (100 nm), l‐NIL (10 μm or i.p. 10 mg kg−1) and 1400W (10 nm, a selective iNOS inhibitor) significantly decreased the rheobase in HFF but not LFF neurons (HFF, 112.1 ± 54.7 pA, n = 14 vs. HFF+N ω, 68.0 ± 13.2, n = 10, P < 0.01; HFF+l‐NIL, 75.5 ± 18.6, n = 11, P < 0.05; HFF+l‐NIL (i.p.), 70.0 ± 17.5, n = 14, P < 0.01; HFF+1400W, 53.3 ± 17.3, n = 9, F 6,73 = 5.894, P < 0.001, one‐way ANOVA with Bonferroni test) (Fig. 2 D and E). The number of action potentials at 2 × rheobase was significantly increased by the iNOS inhibitor, 1400W in HFF but not LFF neurons (HFF, 1.4 ± 0.5, n = 14 vs. HFF+1400W, 2.9 ± 2.3, n = 9, F 6,73 = 2.472, P < 0.05, one‐way ANOVA with Bonferroni test) (Fig. 2 D and E). Input resistance was significantly increased by NO synthase inhibitors in HFF neurons (HFF, 285.3 ± 122.6 MΩ, n = 14 vs. HFF+N ω, 657.6 ± 394.5, n = 10, P < 0.001; HFF+l‐NIL (i.p.), 480.4 ± 202.0, n = 14, P < 0.05; HFF+1400W, 515.7 ± 90.5, n = 9, F 6,73 = 5.429, P < 0.05, one‐way ANOVA with Bonferroni test) (Fig. 2 D and E). One of the targets for NO is soluble guanylate cyclase (sGC), the activation of which results in the accumulation of guanosine 3′,5′‐ cGMP. Inhibitions of sGC by ODQ increased the excitability of HFF neurons. ODQ (10 μm, 30 min incubation) significantly decreased the rheobase in HFF neurons (HFF, 112.1 ± 54.7 pA, n = 14 vs. HFF + ODQ, 54.0 ± 20.7 pA, n = 10, P < 0.001) (Fig. 2 E) and tended to increase input resistance (HFF, 285.3 ± 122.6 MΩ, n = 14 vs. HFF+ODQ, 463.7 ± 90.5 MΩ, n = 10, P = 0.153) and the number of action potentials at 2 × rheobase (HFF, 1.4 ± 0.5, n = 14 vs. HFF+ODQ, 2.4 ± 1.0, n = 10, P = 0.193). Sildenafil (1 μm), an inhibitor of the enzyme that promotes degradation of cGMP, did not significantly affect nodose neuron excitability.
Figure 2.
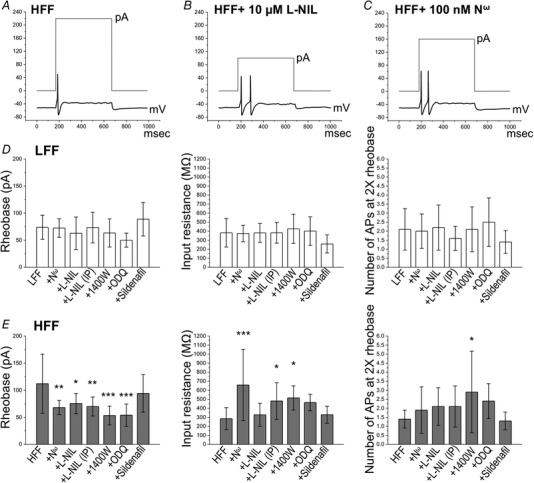
Inhibition of NO signalling pathway increased excitability of nodose neurons from HFF mice
Representative action potential firing elicited by 2 × rheobase current pulse (grey) in HFF nodose neurons (A), HFF+10 μm l‐NIL (B) and HFF+100 nm N ω (C). Histograms illustrating mean rheobase, input resistance and number of action potentials at 2 × rheobase in nodose neurons from LFF (D) and HFF (E) mice. Inhibition of NOS by N ω (100 nm, P < 0.01), l‐NIL (10 μm, P < 0.05), l‐NIL (i.p. 10 mg kg−1, P < 0.01) and 1400W (10 nm, P < 0.001), as well as blockade of sGC by ODQ (10 μm, P < 0.001), significantly decreased rheobase in HFF neurons. N ω (P < 0.001), l‐NIL (i.p., P < 0.05) and 1400W (P < 0.05) significantly increased input resistance in HFF neurons. 1400W (P < 0.05) significantly increased the number of action potential in HFF neurons.
iNOS inhibition decreased K2P conductance in nodose neurons from HFF mice
Figure 3 A shows the representative I–V relationships in the presence of blockers of other ion channels (10 mm TEA, 0.5 mm 4‐AP, 200 μm Ba2+, 200 μM Cd2+ and 25 μm ZD7288), showing the remaining leak K+ (K2P) current on nodose neurons from HFF mice with or without l‐NIL treatment. Leak conductance was determined by a linear fit to the ohmic region (−100 to −50 mV) of the I–V relationship. The current traces in this membrane potential range were well fitted by linear function, indicating leak conductance. There was a statistically significant interaction between the effects of diet and l‐NIL treatment on mean leak conductance (gLeak) [F 2,72 = 3.966, P < 0.05, B coefficient (l‐NIL, i.p.) = −0.257, 95% CI: −0.443 to −0.072, B coefficient (l‐NIL) = −0.138, 95% CI: −0.316 to −0.040, two‐way ANOVA with Bonferroni test, n ≥ 10] (Fig. 3 B). Simple main effects analysis revealed that gLeak was significantly increased in HFF neurons (0.309 ± 0.146 nS, n = 17 vs. 0.466 ± 0.226, n = 18, P < 0.05) and was reversed by inhibition of iNOS [HFF+l‐NIL (i.p.), 0.274 ± 0.095, n = 10, P < 0.05].
Figure 3.
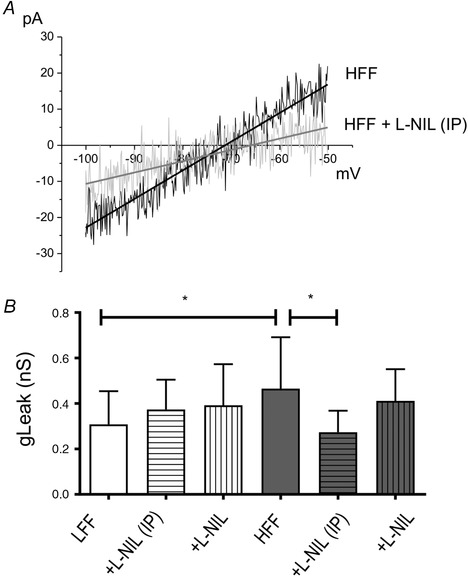
Inhibition of iNOS/NO signalling pathway decreased K2P conductance
A, representative K2P currents were evoked by ramp pulses in the presence of blockers in neurons from HFF (black) and l‐NIL‐pretreated (10 mg kg−1, i.p. injection) HFF mice (grey). I–V relationships showed that l‐NIL elicited a decrease in inward current of the ohmic region (P < 0.05). B, histogram showing mean leak conductance (gLeak) in control and l‐NIL‐pretreated (10 μm, incubation for 30 min. or 10 mg/kg, i.p.) neurons from LFF and HFF mice, respectively.
iNOS inhibition potentiated afferent mechanosensitivity in HFF mouse jejunum
Mechanosensitivity of intestinal afferent nerves was examined by in vitro afferent nerve recording on isolated jejunum segments. Ramp distention of preparations induced a typical biphasic increase in afferent discharge (Fig. 4 A and B), corresponding to the activation of different afferent subpopulations. Mechanosensitive units were classified into LT, WDR and HT subpopulations as described in the Methods. Mechanically insensitive units identified in this set‐up were limited (<2%). It is generally accepted that HT units are exclusively spinal afferents sensing noxious stimuli, whereas LT afferents can be of both vagal and spinal origin. Controversy remains regarding the origin of WDR afferents. HFF mice that received an i.p. injection of l‐NIL (10 mg kg−1) showed an augmented afferent response to distention compared to non‐treated HFF mice (F 1,24 = 4.423, P < 0.05, two‐way ANOVA with Bonferroni test, N = 12) (Fig. 4 C). Single unit analysis revealed that this increase was contributed to by LT (P < 0.05) (Fig. 4 E) and WDR (P = 0.094) afferents. No significant change was observed in LFF mice (F 1,25 = 3.303, P = 0.081, N = 13) (Fig. 4 D).
Figure 4.
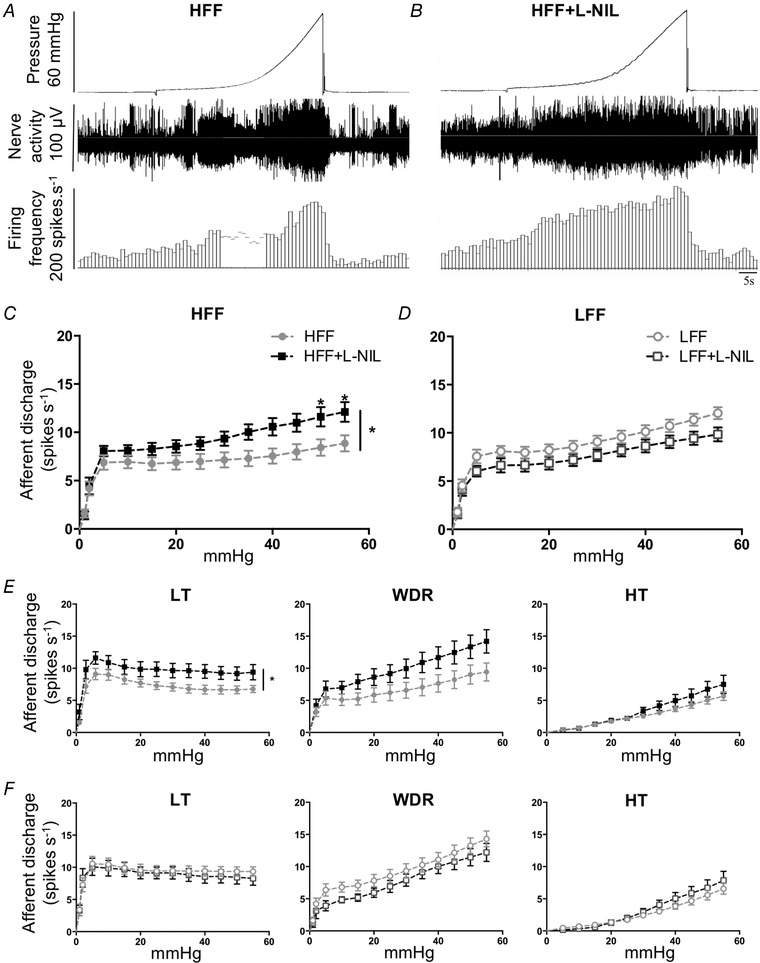
iNOS inhibition potentiated the afferent response to distention in HFF mice jejunum
A and B, representative traces showing multi‐unit afferent response to distention in jejunum segments from HFF mice (A) and l‐NIL‐pretreated (10 mg kg−1, i.p. injection) HFF mice (B). C and E, effects of l‐NIL treatment on mechanosensitivity of total afferents and subpopulations in HFF mouse jejunum. l‐NIL significantly potentiated the total afferent response to increased intraluminal pressure (C) (P < 0.05, two‐way ANOVA with Bonferroni test, N = 12). LT (P < 0.05) and WDR (P = 0.094) instead of HT (P = 0.284) afferents contributed to this potentiation. D and F, no significant change was observed in LFF mice in all units (D) and all subpopulations (F) (N = 13).
iNOS inhibition potentiated satiety signalling in HFF mouse small intestine
The jejunal or ileal afferent response to satiety mediators was investigated using the same recording technique. Only Ex‐4 (GLP‐1 agonist) related experiments were performed on the ileum. In agreement with our previous study, HFF mice had impaired afferent sensitivity to 5‐HT (10 μm), Ex‐4 (100 nm) and CCK (100 nm) (Fig. 5) compared to LFF mice. There was a statistically significant interaction between the effects of diet and l‐NIL treatment (10 mg kg−1, i.p. injection) on the afferent response to 5‐HT (F 1,30 = 6.405, P < 0.05, B coefficient = 2.759, 95% CI: 0.533 to 4.986, two‐way ANOVA with Bonferroni test, N ≥ 7) (Fig. 5 A) and Ex‐4 (F 1,26 = 10.455, P < 0.01, B coefficient = 4.423, 95% CI: 1.611 to 7.235, two‐way ANOVA with Bonferroni test, N ≥ 7) (Fig. 5 B) but not CCK (F 1,28 = 3.254, P = 0.082, B coefficient = 3.221, 95% CI: −0.437 to 6.879, two‐way ANOVA with Bonferroni test, N ≥ 7) (Fig. 5 C). l‐NIL significantly increased the afferent response to 5‐HT in HFF mice (6.4 ± 1.9 vs. 3.5 ± 0.8 spikes s−1, P < 0.01) (Fig. 5 A) by potentiating LT (6.6 ± 2.8 vs. 4.0 ± 1.0 spikes s−1, P < 0.05) and WDR units (5.0 ± 2.9 vs. 2.4 ± 1.5 spikes s−1, P = 0.063). In line with 5‐HT, Ex‐4 sensitivity was enhanced by l‐NIL in HFF mice (5.3 ± 1.6 vs. 2.2 ± 1.1 spikes.s−1, P < 0.05) (Fig. 5 B) and this was also contributed to by LT (5.6 ± 1.8 vs. 2.1 ± 1.3 spikes s−1, P < 0.05). CCK sensitivity in HFF mice was slightly increased by l‐NIL, although it was not statistically significant (8.3 ± 2.5 vs. 6.5 ± 1.6 spikes s−1, P = 0.412) (Fig. 5 C). However, WDR afferents with l‐NIL treatment showed a significant increase in CCK sensitivity (10.4 ± 3.1 vs. 4.1 ± 1.6 spikes s−1, P < 0.01). No significant change was seen in LFF mice for all subpopulations. HT afferents were minimally involved in this satiety signalling (data not shown), in line with evidence that these satiety mediators mainly target vagal afferents (Richards et al. 1996; Hillsley & Grundy, 1998).
Figure 5.
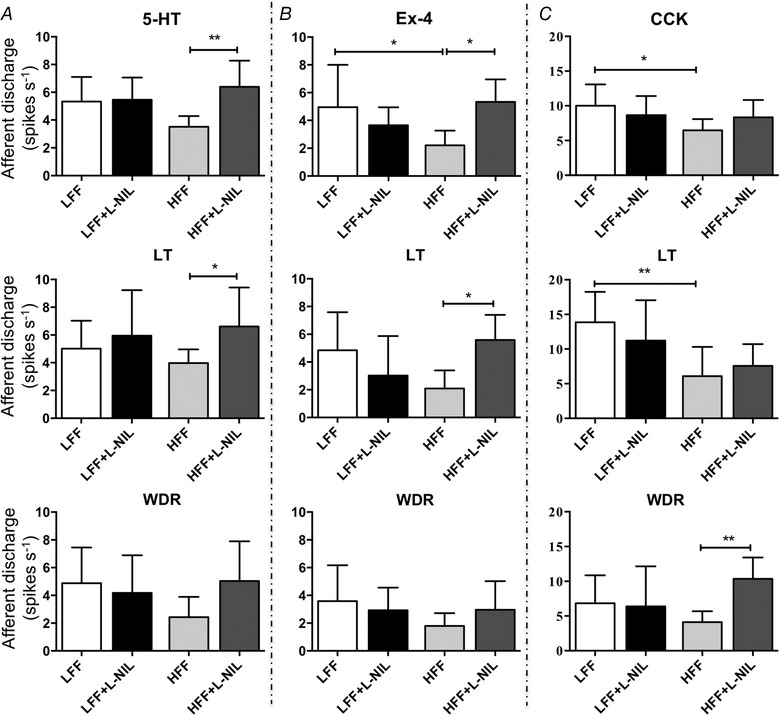
iNOS inhibition potentiated the afferent response to satiety mediators in HFF mice
A, effects of l‐NIL on 5‐HT sensitivity of jejunum afferents and subpopulations. l‐NIL treatment (10 mg kg−1, i.p. injection) resulted in a significant increase in total afferent response to 10 μm 5‐HT (P < 0.01, two‐way ANOVA with Bonferroni test, N = 9), as well as in LT (P < 0.05) and WDR (P = 0.063) afferents, in HFF mice. B, effects of l‐NIL on GLP‐1 signalling in ileum afferents and subpopulations. i.p. injection of l‐NIL in HFF mice augmented the afferent response to 100 nm Ex‐4 in all units (P < 0.05, N = 8) and LT afferents (P < 0.05). C, afferent sensitivity to CCK (100 nm) was not significantly changed by l‐NIL in HFF mice jejunum (P = 0.412, N = 8), whereas only WDR afferents were significantly potentiated (P < 0.01).
Exogenous NO inhibited selected afferent signalling in LFF mouse jejunum
To confirm the inhibitory effects of NO on afferent sensitivity, SNP (10 μm), a NO donor, was perfused into the organ bath for 15 min. Overall, SNP significantly inhibited the afferent response to 5‐HT (F 1,12 = 10.89, P < 0.01, two‐way ANOVA with Bonferroni test, N = 7) (Fig. 6 A), particularly in LFF mice (5.2 ± 2.0 vs. 3.2 ± 2.2 spikes s−1, P < 0.05) by inhibition on LT afferents (7.8 ± 3.1 vs. 4.6 ± 2.0 spikes s−1, P < 0.01). No significant change was observed in WDR and HT afferents (data not shown). Afferent sensitivity to distention was inhibited by SNP in LFF instead of HFF mice (F 1,12 = 32.27, P < 0.001 and F 1,12 = 0.436, P = 0.522, two‐way ANOVA with Bonferroni test, N = 13) (Fig. 6 C and D) and only WDR units in LFF mice showed significant inhibition (P < 0.01) (Fig. 6 E). Conversely, SNP did not change the CCK response in both LFF and HFF mice (F 1,11 = 0.609, P = 0.452, two‐way ANOVA with Bonferroni test, N ≥ 6) (Fig. 6 B). To explain the discrepancy between 5‐HT and CCK data, the effects of SNP on individual unit sensitivity in LFF mice were analysed by single unit analysis (Fig. 6 F) and a noteworthy difference was found in LT afferents. Accordingly, 88.2% of LT units (15 out of 17) in 5‐HT experiments were inhibited by SNP, whereas, in CCK experiments, SNP showed diverse effects on LT afferents (43.5% excitation vs. 26.1% inhibition, total units 23). NO donor SIN‐1 (0.5 mm) also decreased the excitability of cultured nodose neurons from LFF mice, as indicated by increased rheobase (57.9 ± 6.7 pA, n = 14 vs. 79.3 ± 6.6, n = 15, P < 0.05, unpaired t test).
Figure 6.
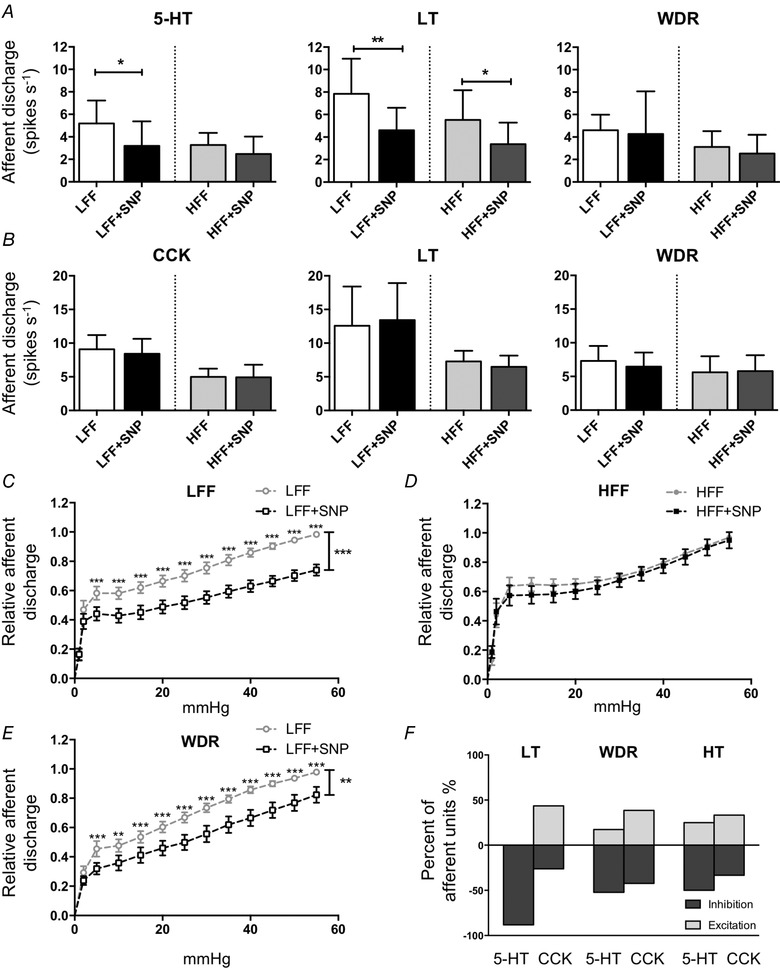
Exogenous NO inhibited the afferent response to 5‐HT and distention instead of CCK in LFF mice
A, effects of NO donor SNP (10 μm) on jejunum afferent sensitivity to 5‐HT (10 μm) in all units and subpopulations. SNP inhibited the afferent response to 5‐HT in LFF mice (P < 0.05, two‐way ANOVA with Bonferroni test, N = 7) instead of HFF mice. Inhibition was observed in LT afferents from both LFF (P < 0.01) and HFF (P < 0.05) mice. No significant change was seen in WDR afferents. B, CCK (100 nm) sensitivity was not affected by SNP in both LFF and HFF mice, as well as in all subpopulations. C and D, the afferent response to distention was inhibited by SNP in LFF mice (C) (P < 0.001, two‐way ANOVA with Bonferroni test, N = 13) but not in HFF mice (D) (P = 0.522, N = 13). E, the only subpopulation showing a significant change in the distention response was the WDR afferents of LFF mice (P < 0.01). F, the effects of SNP on 5‐HT and CCK sensitivity of individual afferent units in LFF mice were diverse. LFF vs. LFF+SNP, HFF vs. HFF+SNP were repeated measurements in the same mice.
iNOS inhibition reduced food intake and adiposity in HFF mice
Feeding patterns during 24 h after i.p. injection of l‐NIL were compared with saline. Saline injection did not change energy intake in both groups of mice (HFF: P = 0.989; LFF: P = 0.451; paired t test, N = 7). There was a statistically significant interaction between the effects of diet and l‐NIL treatment on energy intake (F 1,12 = 12.81, P < 0.01, two‐way ANOVA with Bonferroni test, N = 7) (Fig. 7 A). HFF mice had a higher daily energy intake compared to LFF mice (11.57 ± 1.66 vs. 9.39 ± 1.28 kcal, P < 0.05), indicating the presence of hyperphagia. Energy intake in HFF mice was significantly reduced after l‐NIL injection (11.57 ± 1.66 vs. 9.51 ± 1.75 kcal, P < 0.05), whereas LFF was not significantly changed, despite a tendency to increase (P = 0.168). No difference was observed in average meal size between two groups of mice and between saline and l‐NIL treatment (Fig. 7 B). l‐NIL increased inter‐meal interval in HFF mice (67 ± 7 vs. 85 ± 14 min., P < 0.05, two‐way ANOVA with Bonferroni test) (Fig. 7 C). HFF mice showed higher energy intake than LFF mice in the dark phase (9.33 ± 1.38 vs. 7.18 ± 1.34 kcal, P < 0.05), although without any significant difference in the light phase (2.24 ± 1.29 vs. 2.21 ± 0.62 kcal, P = 0.96). l‐NIL treatment appeared to reduce the energy intake of HFF mice in both the dark phase (9.33 ± 1.38 vs. 7.89 ± 1.83 kcal, P = 0.102) and light phase (2.24 ± 1.29 vs. 1.63 ± 0.76 kcal, P = 0.231), although this was not statistically significant.
Figure 7.
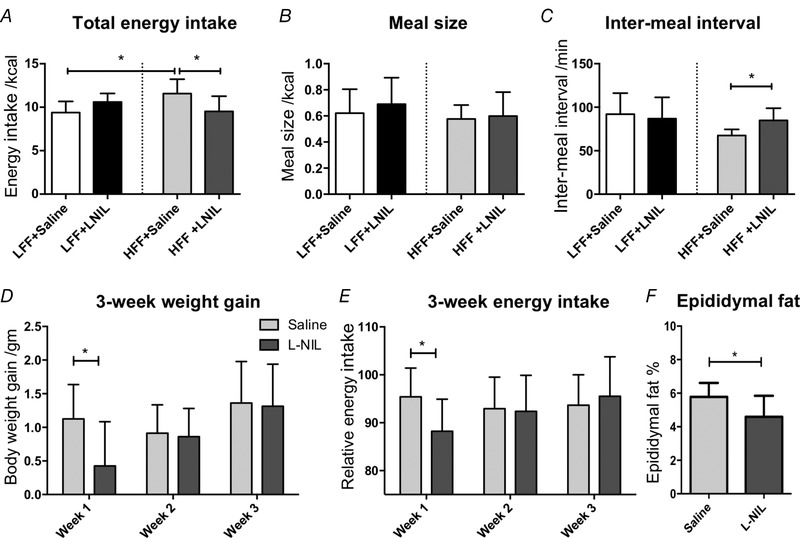
iNOS inhibition reduced food intake and weight gain in HFF mice
A, energy intake during 24 h after l‐NIL injection (10 mg kg−1) was significantly reduced compared to saline treatment in HFF mice (P < 0.05, two‐way ANOVA with Bonferroni test, N = 7). B, no significant alteration in meal size was observed. C, inter‐meal interval was increased by l‐NIL in HFF mice (P < 0.05, two‐way ANOVA with Bonferroni test). D–F, HFF mice were treated with l‐NIL (10 mg kg−1, daily i.p. injection) or saline for 3 weeks. l‐NIL reduced body weight gain (D) (P < 0.05, unpaired t test, N = 8) and energy intake (E) (P < 0.05) in the first week of treatment. Percentage of epididymal fat was lower in l‐NIL treated mice (F) (P < 0.05).
A 3 week treatment of l‐NIL was performed on HFF mice. Differences in weekly body weight gain and food intake between saline and l‐NIL treated HFF mice were compared at three time points using an unpaired t test. Compared to saline‐treated mice, total body weight gain was numerically lower in the l‐NIL treated group (2.6 ± 1.4 vs. 3.4 ± 0.8 g, P = 0.171, unpaired t test, N = 8) and weight gain after the first week was significantly reduced (0.4 ± 0.7 vs. 1.1 ± 0.5 g, P < 0.05) (Fig. 7 D). Energy intake (relative to the week prior to treatment) was significantly reduced in the first week (88.2 ± 6.7% vs. 95.4 ± 6.0%, P < 0.05, N = 8) (Fig. 7 E). No significant difference was observed at weeks 2 and 3. In addition, epididymal fat pad weight, normalized to total body weight, was significantly lower in l‐NIL treated mice at the end of 3 weeks (4.60 ± 1.24% vs. 5.78 ± 0.84%, P < 0.05, unpaired t test, N = 8) (Fig. 7 F).
Discussion
A poor understanding of the molecular mechanisms accounting for the development of obesity and hyperphagia impedes the progress of specific therapy. The present study has provided insight into a mechanism where elevated iNOS‐derived NO impairs vagal afferent sensitivity via increasing K2P channel conductance, disrupts its control of food intake, and drives hyperphagia and obesity. By patch clamping, afferent recording and metabolic monitoring, we have provided evidence at the level of sensory neurons, afferent nerves and feeding behaviour, comprising inhibition of iNOS increased nodose neuron excitability and afferent sensitivity to satiety signals and distention, and reduced acute energy intake in obese mice. In addition, long‐term suppression of iNOS in obese mice reduced visceral fat and decreased body weight gain, although the most prominent effect was in the first week.
iNOS is expressed in multiple cell types, such as macrophages (Lumeng et al. 2007), plasma cells (Fritz et al. 2012), epithelia cells (Kolios et al. 1998) and microglia cells (Sierra et al. 2014), and is probably signalling from multiple sources in the present study. A number of studies have revealed that a high‐fat diet increases NO production or iNOS expression in many different sites, such as plasma (Stanimirovic et al. 2016), liver (Feng et al. 2016), kidney (Sherif, 2014), small intestine (Ou et al. 2012) and adipose tissue (Perreault & Marette, 2001), although not all studies have identified an increase (Perreault & Marette, 2001; Eccleston et al. 2011; Stanimirovic et al. 2016). The present study demonstrated that, after 6–8 weeks of high‐fat diet feeding, the iNOS protein level was increased in nodose ganglia and jejunum from HFF mice, in association with increased NO production in plasma, small intestine and nodose neuron‐incubated culture media. When combined with our electrophysiology results of both nodose ganglion neurons and peripheral intestinal afferents, this indicates that iNOS‐derived NO originates from both peripheral and the nodose ganglion sites. Because iNOS is known to be stimulated by pro‐inflammatory cytokines, our observation is consistent with the proposed low‐grade chronic inflammation associated with obesity. Indeed, other studies have reported that elevated levels of lipopolysaccharides (LPS) are involved in obesity‐induced changes in vagal afferent neurons (de La Serre et al. 2010; de Lartigue et al. 2011). Because LPS is a potent activator of iNOS, the results of the present study may provide a mechanistic explanation.
Our previous studies have demonstrated that obesity decreased the excitability of vagal afferent neurons (Daly and Park et al. 2011) as a result of increased K2P conductance (Park et al. 2018); however, the cellular mechanisms involved were unresolved. In the present study, measurement of electrophysiological membrane properties in nodose neurons revealed that administration of NOS and sGC inhibitors prevented the decreased excitability in HFF neurons. We also demonstrated that the inhibition of iNOS and sGC increased input resistance in HFF neurons, indicating a reduction in K+ conductance. Correspondingly, the increased K2P conductance in HFF neurons was reduced by iNOS inhibitor. Thus, our data suggest that accumulation of iNOS‐derived NO decreases excitability of vagal afferent neurons in obesity, at least in large part, by modulating K2P channels.
A previous study observed impaired afferent sensitivity to 5‐HT, CCK and distention of the jejunum in HFF mice (Daly and Park et al. 2011). The present study proposed an iNOS–NO mediated mechanism to account for this impairment based on inhibition of iNOS potentiating afferent sensitivity to 5‐HT, GLP‐1, CCK and distention in the small intestine of HFF mice. This is consistent with a previous study in which iNOS inhibition increases the afferent response to 5‐HT and distention on a mouse model of intestinal inflammation induced by indomethacin (Xue et al. 2009). Increasing satiety is one of the most important strategies for the pharmacological therapy of obesity. 5‐HT agonists and GLP‐1 agonists are two among five classes of drugs currently approved for obesity treatment in the USA (Shettar et al. 2017). The administration of a CCK‐A agonist was not effective to induce weight loss in clinical trials to date (Kim et al. 2011), although enhanced endogenous CCK signalling might still be beneficial because it is well‐established that CCK activates vagal afferents to inhibit food intake (Dockray, 2014).
Inhibition of iNOS in obesity recovered impaired satiety signalling and hence should reduce food intake. Indeed, we demonstrated that the hyperphagia present in HFF mice was reversed by iNOS suppression. We observed shorter inter‐meal intervals but unaltered meal sizes in HFF mice compared to LFF mice. Changes in meal patterns caused by a high‐fat diet have been variable in previous studies. One study suggested that a high‐fat diet increased meal size and decreased inter‐meal interval (Warwick et al. 2000), whereas another study reported increased meal frequency and reduced meal size on rats, with these changes adapting after long‐term exposure to diet (Paulino et al. 2008). Many factors could influence meal patterns, such as the feeding pattern of mice (i.e. frequent feeding, particularly nocturnally), diet composition and energy density, and exposure time to the diet. Long‐term suppression of iNOS decreased energy intake and body weight gain in the first week of treatment, and these effects diminished afterwards, implying takeover by compensative mechanisms. More importantly, visceral fat was reduced in treated mice at the end of treatment. One week of treatment in mice may be equivalent to a considerable period on humans; thus, the beneficial effects of iNOS suppression on obesity are still relevant. In addition, the observation at the end of the 3 week period of less visceral fat stores, yet a similar weight gain, suggests the possibility of increased lean body mass during treatment with l‐NIL, although we did not measure this directly.
Previous studies addressing the involvement of iNOS in obesity are limited to insulin resistance. iNOS contributes to obesity‐related insulin resistance by reducing insulin receptor substrate‐1 expression (Sugita et al. 2005). Consistently, iNOS knockout mice on a high‐fat diet are protected from insulin resistance (Noronha et al. 2005; Zanotto et al. 2017). The present study clarified the involvement of iNOS in the pathogenesis of obesity in terms of modulation of satiety signalling conveyed by vagal afferents. Interestingly, iNOS knockout did not prevent the excessive weight gain induced by a high‐fat diet (Noronha et al. 2005) because these models may be limited by compensatory mechanisms. However, these observations do not necessarily rule out the potential beneficial effects of iNOS inhibition on existing obesity.
NO has pleiotropic and complex roles in various physiological activities occurring in multiple systems, including the nervous system. Our exogenous NO data confirmed the inhibitory effects of NO on afferent sensitivity to 5‐HT and distention, despite elusive effects on CCK sensitivity. Our single unit analysis revealed that NO had diverse effects on afferent sensitivity depending on the stimuli and target subpopulations. Other studies on non‐obese models have also reported contrasting effects of NO (Kentish et al. 2014; Park et al. 2014). Further investigations are required to determine the complete pathway of the effects of NO on afferent nerve sensitivity.
In summary, obesity is a pandemic health issue with few tangible and safe treatment options. Many ‘distal’ causes of obesity, such as lifestyle, environment, occupation and social influences, are difficult to manipulate. Therefore, molecular pathways more proximal to the origins of obesity may be valuable therapeutic targets. The present study has provided insight into an iNOS–NO–vagal afferent pathway that accounts for the impairment of satiety signalling via vagal afferents in diet‐induced obesity. Therefore, pharmacological manipulation of this pathway is a promising therapy for hyperphagia and obesity.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
All experiments were performed in the Gastrointestinal Disease Research Unit at Queen's University in Kingston, ON, Canada. All authors approved the final version of the manuscript submitted for publication and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. MB supervised the project and obtained grant support. MB, SP and YY designed the research. YY performed the western blotting, afferent recording, feeding monitoring and relevant analyses. SP performed the patch clamp studies, as well as most of the nitrate assays and relevant analyses. YY, SP and MB interpreted the data, and drafted and revised the manuscript.
Funding
The present study was funded by the Canadian Institute of Health Research (MOP 115021).
Translational perspective.
Effective pharmacological treatments for obesity are limited as a result of a poor understanding of the molecular mechanisms underlying the development of obesity. Increasing satiety is one of the most important strategies, and 5‐HT and GLP‐1 agonists (both are satiety mediators) are two among five classes of drugs currently approved for obesity therapy in the USA. However, our previous investigations have demonstrated that obese mice have reduced vagal afferent sensitivity to satiety signals. Thus, the efficacy of exogenous satiety mediators may be limited. A low‐grade inflammation occurs in obesity with increased expression of inducible nitric oxide synthase (iNOS). The present study hypothesized that elevated iNOS‐derived NO impairs vagal afferent sensitivity and disrupts its control of food intake leading to hyperphagia and obesity. We found that inhibition of iNOS in obese mice restored vagal afferent neuronal excitability, increased the afferent response to satiety mediators and reduced short‐term energy intake. A prolonged inhibition of iNOS reduced energy intake and body weight gain during the first week and also reduced amounts of visceral fat after 3 weeks. We identified a novel pathway underlying disrupted satiety regulation in obesity. This finding may drive more studies addressing its upstream and downstream pathways, such as mechanisms modulating iNOS overexpression, involvement of microbiota, possible targets of NO and corresponding subcellular signalling pathways, as well as beneficial effects on relevant metabolic functions. A better understanding of this pathway will promote clinical studies to develop specific treatments for hyperphagia and obesity.
Acknowledgements
We thank Iva Kosatka and Shumei He for their outstanding technical assistance, as well as Dr Stephen Vanner and Dr Alan Lomax for their helpful advice on this manuscript.
Biographies
Yang Yu obtained his master's degree at University College London and his PhD at the University of Sheffield, where he was trained in research about sensory functions of the gastrointestinal tract. He is now a postdoctoral fellow at the Gastrointestinal Diseases Research Unit at Queen's University where he continues his research aiming to understand gut–brain signalling in obesity and other gastrointestinal diseases.

Sung Jin Park received his undergraduate degree in Life Science from Sogang University, and his MSc and PhD degrees in Medicine from Seoul National University. After completing his training in physiology and biophysics, he carried out postdoctoral research training in neurophysiology and gastroenterology at McMaster University. Since 2007, he has been a Research Associate in the Gastrointestinal Disease Research Unit at Queens University where he studies the fundamental cellular mechanisms of ion channels underlying dysfunction of the gastrointestinal system.
Edited by: Kim Barrett and Weifang Rong
This is an Editor's Choice article from the 15 March 2019 issue.
Linked articles: This article is highlighted in a Perspectives article by Page. To read this article, visit https://doi.org/10.1113/JP277490.
References
- Bielefeldt K, Whiteis CA, Chapleau MW & Abboud FM (1999). Nitric oxide enhances slow inactivation of voltage‐dependent sodium currents in rat nodose neurons. Neurosci Lett 271, 159–162. [DOI] [PubMed] [Google Scholar]
- Bray GA, Kim KK & Wilding JPH (2017). Obesity: a chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes Rev 18, 715–723. [DOI] [PubMed] [Google Scholar]
- Daly DM, Park SJ, Valinsky WC & Beyak MJ (2011). Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J Physiol 589, 2857–2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC & Raybould HE (2010). Propensity to high‐fat diet‐induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299, G440–G448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G (2016). Role of the vagus nerve in the development and treatment of diet‐induced obesity. J Physiol 594, 5791–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lartigue G, Barbier de la Serre C, Espero E, Lee J & Raybould HE (2011). Diet‐induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab 301, E187–E195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray GJ (2014). Gastrointestinal hormones and the dialogue between gut and brain. J Physiol 592, 2927–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccleston HB, Andringa KK, Betancourt AM, King AL, Mantena SK, Swain TM, Tinsley HN, Nolte RN, Nagy TR, Abrams GA & Bailey SM (2011). Chronic exposure to a high‐fat diet induces hepatic steatosis, impairs nitric oxide bioavailability, and modifies the mitochondrial proteome in mice. Antioxid Redox Signal 15, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin A (2017). The pathogenesis of obesity‐associated adipose tissue inflammation. Adv Exp Med Biol 960, 221–245. [DOI] [PubMed] [Google Scholar]
- Feng B, Meng R, Huang B, Shen S, Bi Y & Zhu D (2016). Silymarin alleviates hepatic oxidative stress and protects against metabolic disorders in high‐fat diet‐fed mice. Free Radic Res 50, 314–327. [DOI] [PubMed] [Google Scholar]
- Fritz JH, Rojas OL, Simard N, McCarthy DD, Hapfelmeier S, Rubino S, Robertson SJ, Larijani M, Gosselin J, Ivanov II, Martin A, Casellas R, Philpott DJ, Girardin SE, McCoy KD, Macpherson AJ, Paige CJ & Gommerman JL (2012). Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature 481, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillsley K & Grundy D (1998). Serotonin and cholecystokinin activate different populations of rat mesenteric vagal afferents. Neurosci Lett 255, 63–66. [DOI] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M & Shimomura I (2007). Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911. [DOI] [PubMed] [Google Scholar]
- Kakei M, Yada T, Nakagawa A & Nakabayashi H (2002). Glucagon‐like peptide‐1 evokes action potentials and increases cytosolic Ca2+ in rat nodose ganglion neurons. Auton Neurosci 102, 39–44. [DOI] [PubMed] [Google Scholar]
- Kentish SJ, O'Donnell TA, Wittert GA & Page AJ (2014). Diet‐dependent modulation of gastro‐oesphageal vagal afferent mechanosensitivity by endogenous nitric oxide. J Physiol 592, 3287–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GW, Lin JE, Valentino MA, Colon‐Gonzalez F & Waldman SA (2011). Regulation of appetite to treat obesity. Expert Rev Clin Pharmacol 4, 243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolios G, Rooney N, Murphy CT, Robertson DA & Westwick J (1998). Expression of inducible nitric oxide synthase activity in human colon epithelial cells: modulation by T lymphocyte derived cytokines. Gut 43, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage F, Guillemare E, Fink M, Duprat F, Lazdunski M, Romey G & Barhanin J (1996). TWIK‐1, a ubiquitous human weakly inward rectifying K+ channel with a novel structure. EMBO J 15, 1004–1011. [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Deyoung SM, Bodzin JL & Saltiel AR (2007). Increased inflammatory properties of adipose tissue macrophages recruited during diet‐induced obesity. Diabetes 56, 16–23. [DOI] [PubMed] [Google Scholar]
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu‐Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi‐Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez‐Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ & Gakidou E (2014). Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha BT, Li J‐M, Wheatcroft SB, Shah AM & Kearney MT (2005). Inducible nitric oxide synthase has divergent effects on vascular and metabolic function in obesity. Diabetes 54, 1082–1089. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Fryar CD & Flegal KM (2015). Prevalence of obesity among adults and youth: United States, 2011–2014. National Center for Health Statistics, Hyattsville, MD. [PubMed] [Google Scholar]
- Ou Y, Liu R, Wei N, Li X, Qiang O, Huang W & Tang C (2012). Effects of octreotide on nitric oxide synthase expression in the small intestine of high fat diet‐induced obese rats. Obes Res Clin Pract 6, e263–e346. [DOI] [PubMed] [Google Scholar]
- Page AJ, O'Donnell TA, Cooper NJ, Young RL & Blackshaw LA (2009). Nitric oxide as an endogenous peripheral modulator of visceral sensory neuronal function. J Neurosci 29, 7246–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park AR, Lee HI, Semjid D, Kim DK & Chun SW (2014). Dual effect of exogenous nitric oxide on neuronal excitability in rat substantia gelatinosa neurons. Neural Plast 2014, 628531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Yu Y, Wagner B, Valinsky WC, Lomax AE & Beyak MJ (2018). Increased TASK channel mediated currents underlie high fat diet‐induced vagal afferent dysfunction. Am J Physiol Gastrointest Liver Physiol 315, G592–G601. [DOI] [PubMed] [Google Scholar]
- Paulino G, Darcel N, Tome D & Raybould H (2008). Adaptation of lipid‐induced satiatation is not dependent on caloric density in rats. Physiol Behav 93, 930–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault M & Marette A (2001). Targeted disruption of inducible nitric oxide synthase protects against obesity‐linked insulin resistance in muscle. Nat Med 7, 1138–1143. [DOI] [PubMed] [Google Scholar]
- Richards W, Hillsley K, Eastwood C & Grundy D (1996). Sensitivity of vagal mucosal afferents to cholecystokinin and its role in afferent signal transduction in the rat. J Physiol 497, 473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong W, Hillsley K, Davis JB, Hicks G, Winchester WJ & Grundy D (2004). Jejunal afferent nerve sensitivity in wild‐type and TRPV1 knockout mice. J Physiol 560, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansbury BE & Hill BG (2014). Regulation of obesity and insulin resistance by nitric oxide. Free Radic Biol Med 73, 383–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherif IO. (2014). Secoisolariciresinol diglucoside in high‐fat diet and streptozotocin‐induced diabetic nephropathy in rats: a possible renoprotective effect. J Physiol Biochem 70, 961–969. [DOI] [PubMed] [Google Scholar]
- Shettar V, Patel S & Kidambi S (2017). Epidemiology of obesity and pharmacologic treatment options. Nutr Clin Pract 32, 441–462. [DOI] [PubMed] [Google Scholar]
- Sierra A, Navascues J, Cuadros MA, Calvente R, Martin‐Oliva D, Ferrer‐Martin RM, Martin‐Estebane M, Carrasco MC & Marin‐Teva JL (2014). Expression of inducible nitric oxide synthase (iNOS) in microglia of the developing quail retina. PLoS ONE 9, e106048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanimirovic J, Obradovic M, Jovanovic A, Sudar‐Milovanovic E, Zafirovic S, Pitt SJ, Stewart AJ & Isenovic ER (2016). A high fat diet induces sex‐specific differences in hepatic lipid metabolism and nitrite/nitrate in rats. Nitric Oxide 54, 51–59. [DOI] [PubMed] [Google Scholar]
- Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, Martyn JAJ & Kaneki M (2005). Inducible nitric‐oxide synthase and NO donor induce insulin receptor substrate‐1 degradation in skeletal muscle cells. J Biol Chem 280, 14203–14211. [DOI] [PubMed] [Google Scholar]
- Surmi BK & Hasty AH (2008). Macrophage infiltration into adipose tissue: initiation, propagation and remodeling. Future Lipidol 3, 545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warwick ZS, McGuire CM, Bowen KJ & Synowski SJ (2000). Behavioral components of high‐fat diet hyperphagia: meal size and postprandial satiety. Am J Physiol Regul Integr Comp Physiol 278, R196–R200. [DOI] [PubMed] [Google Scholar]
- Xue B, Hausmann M, Müller MH, Pesch T, Karpitschka M, Kasparek MS, Hu WC, Sibaev A, Rogler G & Kreis ME (2009). Afferent nerve sensitivity is decreased by an iNOS‐dependent mechanism during indomethacin‐induced inflammation in the murine jejunum in vitro. J Neurogastroenterol Motil 21, 322–334. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Henrich M, Snipes RL & Kummer W (2003). Altered production of nitric oxide and reactive oxygen species in rat nodose ganglion neurons during acute hypoxia. Brain Res 961, 1–9. [DOI] [PubMed] [Google Scholar]
- Zanotto TM, Quaresma PG, Guadagnini D, Weissmann L, Santos AC, Vecina JF, Calisto K, Santos A, Prada PO & Saad MJ (2017). Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol Metabol 6, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]