

Abstract
Quantifying health-related biological effects, like genotoxicity, could provide a way of distinguishing between tobacco products. In order to develop tools for using genotoxicty data to quantitatively evaluate the risk of tobacco products, we tested five carcinogens found in cigarette smoke, 4-aminobiphenyl (4- ABP), benzo[a]pyrene (BaP), cadmium (in the form of CdCl2), 2-amino-3,4-dimethyl-3H-imidazo[4,5-f] quinoline (MelQ) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), in the mouse lymphoma assay (MLA). The resulting mutagenicity dose responses were analyzed by various quantitative approaches and their strengths and weaknesses for distinguishing responses in the MLA were evaluated. L5178Y/Tk+/− 3.7.2C mouse lymphoma cells were treated with four to seven concentrations of each chemical for 4 h. Only CdCl2 produced a positive response without metabolic activation (S9); all five chemicals produced dose-dependent increases in cytotoxicity and mutagenicity with S9. The lowest dose exceeding the global evaluation factor, the benchmark dose producing a 10%, 50%, 100% or 200% increase in the background frequency (BMD10, BMD50, BMD100 and BMD200), the no observed genotoxic effect level (NOGEL), the lowest observed genotoxic effect level (LOGEL) and the mutagenic potency expressed as a mutant frequency per micromole of chemical, were calculated for all the positive responses. All the quantitative metrics had similar rank orders for the agents’ ability to induce mutation, from the most to least potent as CdCl2(-S9) > BaP(+S9) > CdCl2(+S9) > MeIQ(+S9) > 4-ABP(+S9) > NNK(+S9). However, the metric values for the different chemical responses (i.e. the ratio of the greatest value to the least value) for the different chemicals ranged from 16-fold (BMD10) to 572-fold (mutagenic potency). These results suggest that data from the MLA are capable of discriminating the mutagenicity of various constituents of cigarette smoke, and that quantitative analyses are available that can be useful in distinguishing between the exposure responses.
Introduction
Smoking is the leading preventable cause of death in the USA (1). Worldwide, smoking results in hundreds of billions of dollars of economic damage each year and is ultimately the cause of death for up to half of the world’s 1 billion smokers (2).Epidemiological evidence indicates that cigarette smoking harms nearly every organ in the body and is causally related to a number of human cancers, including acute myeloid leukemia, colorectal cancer and cancer of the bladder, cervix, esophagus, kidney and ureter, larynx, liver, oropharynx (includes parts of the throat, tongue, soft palate and the tonsils), pancreas, stomach, trachea, bronchus, and lung (1).
The Family Smoking Prevention and Tobacco Control Act of 2009 gave the U.S. Food and Drug Administration authority to regulate tobacco products. Cigarette smoke is a complex mixture of >7000 gaseous and particulate compounds (3), over 70 of which have been classified as carcinogens by the International Agency for Research on Cancer (IARC) (4). In addition, these carcinogens belong to different chemical classes, including polycyclic aromatic hydrocarbons (PAHs), tobacco-specific N-nitrosamines (TSNAs), aromatic amines, aldehydes, metals and volatile hydrocarbons; and all of these classes of chemicals likely contribute to the carcinogenic activity of tobacco smoke (4,5). A challenge is in defining which changes to these >7000 constituents impact the overall toxicity and carcinogenicity of cigarette smoke.
Because genotoxicity is considered an important mechanism for the carcinogenicity of chemical substances (6), genotoxicity measurements have the advantage of measuring a property related to health risk that can be assessed using a test article relevant to human exposure. Various preparations of cigarette smoke (e.g. cigarette mainstream smoke, whole smoke, total particulate matter or cigarette smoke condensate and the gas/vapour phase from different cigarette types) have been tested in several in vitro genotoxicity assays, including the bacterial Ames mutagenicity test and a number of mammalian cell assays including the Comet assay, the mouse lymphoma gene mutation assay (MLA), the micronucleus assay, and the chromosome aberration assay (7–13). Although different genotoxicity assays have different sensitivities and specificities, and measure different genotoxicity endpoints, cigarette smoke is positive in nearly all assays. Therefore, if genotoxicity data were used to assess different cigarette products, quantitative assessments of data from several types of genotoxicity assays may convey useful information for assisting the decision making.
Genotoxicity evaluation has historically resulted in a qualitative determination and test materials are categorized as negative or positive, although a positive dose-response is one of the criteria for a positive determination. However, several quantitative approaches have been utilised for in vitro genetic toxicology studies. The mutagenic potency, calculated using the slope of the liner regression of dose-response curves, has been used to describe the genotoxicity of cigarette smoke condensates tested in several in vitro genotoxicity assays, for example the Ames test, the micronucleus assay, the Comet assay, the chromosome aberration assay and the MLA (8,10). In addition, quantitative assessment of the dose-response relationships and point-of-departure (PoD) metrics have been reported for in vivo and in vitro genetic toxicology data bases developed for several model mutagens (14,15). Most recently, the Working Group on Quantitative Approaches to Genetic Toxicology Risk Assessment (QWG) of the International Workshops on Genotoxicity testing (IWGT) has reported on the need for quantitative dose-response analysis of genetic toxicology data, the methods to derive PoDs, and the approaches to define exposure-related risks. This group also indicates that PoD metrics calculated from in vitro mammalian genotoxicity data can be used for potency ranking among related agents with similar modes of action (16,17).
The goal of the current study was to utilise a number of different metrics that might be useful to evaluate the relative mutagenicity of several chemicals, in this case different constituents of tobacco products. The metrics used for these analyses include the benchmark dose (BMD), the no observed genotoxic effect level (NOGEL), and the nonlinear slope-transition dose. The NOGEL and the lowest observed genotoxic effect level (LOGEL) are strongly affected by the experimental design (i.e. dose range and dose selection). BMD is calculated from mathematically modeled dose-response relationships (i.e. the shapes of the curves). Determination of the lower and upper limits of the BMD (BMDL and BMDU) can be used in comparisons of chemical-induced toxicity across multiple genotoxicity endpoints (14). Recently, these approaches have been used to analyse data generated in the gpt-delta transgenic mouse (18), the in vitro micronucleus test (19,20), and the Ames mutagenicity rest (21).
Quantitative dose-response modeling approaches for the derivation of PoD metrics have been adopted for safety and risk assessments of potential toxic materials by some US regulatory and homeland security agencies and the European Food Safety Authority (17,22,23). The BMDL has been used for non-cancer and cancer risk assessments by the US Environmental Protection Agency (24) in the context of assigning a level of risk to particular exposure levels, generally with the goal of determining some type of acceptable exposure limit or to assess the margin of exposure. The dose-response metrics calculated in our study, however, are not used for defining an acceptable human exposure level for a particular chemical but for comparing genotoxic potencies. We anticipate that these quantitative approaches may be useful for assessing genotoxicty data to distinguish the mutagenic responses between tobacco products.
Here, we describe results from our pilot study to assess the ways in which genotoxicity data associated with tobacco smoke might be evaluated quantitatively to provide insights to inform regulatory decision making. The five chemicals studied are all found in cigarette smoke, represent different classes of chemicals, and are all human carcinogens or suspected human carcinogens: 4-amino- biphenyl (4-ABP), an aromatic amine; benzo[a]pyrene (BaP), a PAH; cadmium chloride (CdCl2), a metal; 2-amino-3,4-dimethyl- 3H-imidazo[4,5-f]quinoline (MeIQ), a heterocyclic amine (HCA); and 4 - (me thy lnitro samino) −1 - (3 -py ridyl) −1 -butanone (NNK), a TSNA. Table 1 provides information on the chemical classes and carcinogenicity of these test agents, and representative amounts of these chemicals in cigarette mainstream smoke and sidestream smoke. It should be noted that the amount of these and other chemical constituents may vary with tobacco type, agricultural conditions, curing methods, manufacturing processes and smoking patterns (33).
Table 1.
Chemical classes and typical amounts of five chemical constituents of tobacco smoke
| Chemical abbreviation | Chemical name | CAS | Class | Carcinogenicitya,b | Mainstream smoke (ng/cigarette) | Sidestream smoke (ng/cigarette) |
|---|---|---|---|---|---|---|
| 4-ABP | 4-Aminobiphenyl | 92–67-1 | Aromatic amine | Group 1a | 0.2–23c | Up to 140c |
| BaP | Benzo[a]pyrene | 50–32-8 | PAH | Group 1d | 8.5–11.6e | 52–95d |
| CdCl2 | Cadmium chloride | 10108–64-2 | Inorganic compound | Group 1a | 0–6670a | 122–265f |
| MeIQ | 2-Amino-3,4-dimethylimidazo[4,5-f]quinoline | 77094–11-2 | HCA | Group 2Bb | 0.28–0.75b | 0.25–0.45g |
| NNK | 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone | 64091–91-4 | TSNA | Group 1h | 3–1749h, 20–4200c | 200–15700c |
For this study, we used the MLA to investigate the cytotoxicity and mutagenicity of five chemicals. The MLA detects the widest range of genotoxic events of any in vitro genotoxicity test, detecting both gene mutations and chromosomal events (34,35). Mutagenicity data generated from testing these agents in the MLA were used to evaluate the features of the different metrics for distinguishing between test articles.
Materials and methods
Materials
CdCl2 (CAS# 10108–64-2), 4-ABP (CAS# 92–67-1), BaP (CAS# 50–32-8), trifluorothymidine (TFT, CAS# 70–00-8), and dimethyl sulfoxide (DMSO, CAS# 67–68-5) were purchased from Sigma-Aldrich (St. Louis, MO). NNK (CAS# 64091–91-4) and MeIQ (CAS# 77094–11-2) were obtained from Toronto Research Chemicals (North York, ON, Canada). Aroclor 1254 induced male Sprague-Dawley rat liver post-mitochondrial fraction (S9) was purchased from Moltox (Boone, NC). Fischer’s medium was obtained from Quality Biological (Gaithersburg, MD), and all other cell culture supplies were acquired from Invitrogen Life Technologies (Carlsbad, CA).
Cells and culture conditions
The MLA forward mutation assay was performed using the L5178Y/Tk+/− 3.7.2C mouse lymphoma cell line. The methods for cell growth and cleansing to purge spontaneous Tk−/− mutants were described previously (36). Cell cultures were maintained in Fischer’s Medium for Leukemic Cells of Mice with l-glutamine, supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 0.1% pluronic F68, 1 mM sodium pyruvate, and 10% heat-inactivated horse serum (F10p). Fischer’s medium containing 5% (F5p) or 20% (F20p) heat-inactivated horse serum was used for treatment and cloning, respectively. The cultures were gassed with 5% (v/v) CO2 in air and maintained in a shaker-incubator at 37°C.
S9-mix preparation
The S9 liver fraction was mixed with a reduced nicotinamide adenine dinucleotide phosphate (NADPH)-generating system (37). The standard S9 mix contained 50 mM sodium phosphate buffer (pH 8.0), 30 mM KCl, 10 mM MgCl2, 4 mM NADP, 5 mM glucose-6-phosphate, 10 mM CaCl2 and 2 mg/ml of S9 protein. Two millilitres of freshly prepared S9 mix were added to 8 ml of medium for the treatment. The final S9 concentration was 0.4 mg protein/ml in the treatment medium (about 1% v/v).
Cell treatments with five chemicals
Dose range finding tests were performed to establish an appropriate concentration range for each chemical. To initiate the main mutation assay, 6 × 106 cells were suspended in 10 ml (in the absence of S9) or 8 ml (in the presence of S9) of F5p medium, and treated with 4–7 concentrations of each chemical, with the concentrations based on the results of dose range finding tests. The working solutions (100×) for each test agent concentration were prepared fresh from stocks using a series of dilutions in DMSO or distilled water, and 100 μl of the working solutions were added to the cell cultures. For treatment with metabolic activation, 2 ml of S9 mix were added to the cell cultures just prior to the chemical treatment. The cells then were gassed with 5% (v/v) CO2 in air and placed on a roller drum (15 rpm) in a 37°C incubator for the 4-h treatment. In all cases, the final concentration of DMSO in the medium was 1%, including in the DMSO solvent controls and the positive controls [0.3 μg/ml BaP with S9 and 0.1 μg/ml 4-nitroquinoline-1-oxide (4-NQO) without S9]. After treatment, the cells were washed twice with basic medium by centrifugation and resuspended in 20 ml of F10p. The culture tubes were placed on a roller drum at 37°C for the 2-day expression of Tk-deficient phenotype mutations. Cell cultures were counted ~24 h after treatment and adjusted with fresh F10p medium to give a density of 2 × 105 cells/ml.
The Tk microwell mutant assay
Tk mutants were selected as described previously (38). Briefly, following the 2-day expression period, the cells were resuspended in F20P at a density of 1 × 104 cells/ml. Three μg/ml of TFT selective agent were added to the cells, and 200 μl of the cell culture were seeded into each well of four 96-well flat-bottom microtitre plates at a final density of 2000 cells/well. Plating efficiency was determined by plating 200 μl F20p without TFT containing 8 cells/ml into the wells of two 96-well flat-bottom microtitre plates. After 11 days of incubation, mutant colonies were counted visually and categorized as small colony or large colony mutants (37). Relative total growth (RTG) was used to measure chemical-induced cytotoxicity. RTG includes a relative (compared to the untreated/solvent control) measurement of cell growth for the treated cultures covering the 4-h treatment, 2-day expression and 11-day cloning phases. Mutant frequencies (MFs) were calculated using the Poisson distribution (37).
Data analysis
All of the MF data are expressed as the mean ± 1 SD from 3 to 5 independent experiments. The data evaluation criteria developed by the MLA Expert Workgroup of the IWGT were used for determining positive and negative MF responses (39). Positive responses were defined as those where the induced MF in one or more treated cultures exceeded the global evaluation factor (GEF, 126 mutants per 106 cells for the microwell version of the MLA) and where there was also a dose-related increase in MF. We designated the lowest dose exceeding the GEF as one of the metrics used for quantitatively differentiating between mutational responses and for comparing with other metrics.
The mutagenic potencies of the five chemicals were calculated from the slope of linear regression lines fit to the increasing portions of the dose-response curves as previously described (8). The potency values were expressed as MF per micromole of chemical. The NOGEL (defined as the highest tested dose that did not induce a significant increase in MF) and the lowest observed genotoxic effect level (LOGEL, defined as the lowest tested dose that induced a significant increase in MF) were determined by one way analysis of variance (ANOVA) followed by Dunnett’s test using SigmaPlot 11.0 (Systat Software, San Jose, CA).
BMD analysis was conducted using the EPA’s Benchmark Dose Software (version 2.4.0, released in 2013; http://www.epa.gov/ncea/bmds/index.html) according to the BMD technical guidance. The BMD is a dose that produces a predetermined change in the response of an adverse effect over the control (e.g. a 10% increase over the background MF or BMD10); and the BMDU and BMDL refer to the upper and lower bounds of the 95% confidence interval of the BMD. For example, the BMDL10 is the lower one-sided confidence limit of the BMD for a 10% change in response (24). For this study, a continuous model was used since the data sets displayed dose-dependent trends. The Benchmark Dose software contains five models for continuous data, that is exponential, Hill, linear, polynomial and power models. First, the BMD10 for each data set was calculated using all five models. The models with absolute values of scaled residuals >2 were excluded, because scaled residuals of this magnitude indicate a disagreement between the predicted and observed means in the region of particular interest. Then, the best fitting model was selected based on Akaike’s Information Criterion (24), and the BMDs, BMDLs and BMDUs were derived from the selected model for 10%, 50%, 100%, 200% and 300% changes in response above the control.
Results
Dose-response relationships for five chemicals in the MLA: comparisons of responses using IWGT criteria
Based on the dose range finding tests, 4–7 concentrations from 0.2 μM to 2 mM were used for testing the five chemicals in the main experiments. Data were analyzed and positive responses were determined based on IWGT MLA workgroup recommendations (39). In the absence of metabolic activation, only CdCl2 produced doserelated cytotoxicity and mutagenicity following the 4-h treatment; the lowest positive concentration (based on the GEF) was 0.5 μM. The other four agents showed no mutagenic response in the absence of S9. However, in the presence of S9, all five chemicals produced dose-dependent decreases in RTG (Figure 1A) and increases in MF (Figure 1B). Based on the GEF, the lowest concentrations giving a positive response in the presence of S9 were 2, 3, and 10 μM for BaP, CdCl2 and MelQ, respectively, and 50 μM for 4-ABP and NNK(Table 2). Three chemicals (BaP, NNK and MelQ) produced positive mutagenic responses at RTGs ≥82% (Figure 1), indicating that they are mutagenic even at slightly cytotoxic concentrations. For cytotoxic compounds that are not limited by solubility, the highest recommended concentration for the MLA is based on an RTG between 20% and 10% (39). That concentration was 0.6 μM, 4 μM, 7 μM, 100 μM, 125 μM and 2 mM for CdCl2(-S9), CdCl2(+S9), BaP(+S9), 4-ABP(+S9), MeIQ(+S9) and NNK(+S9), respectively.
Figure 1.
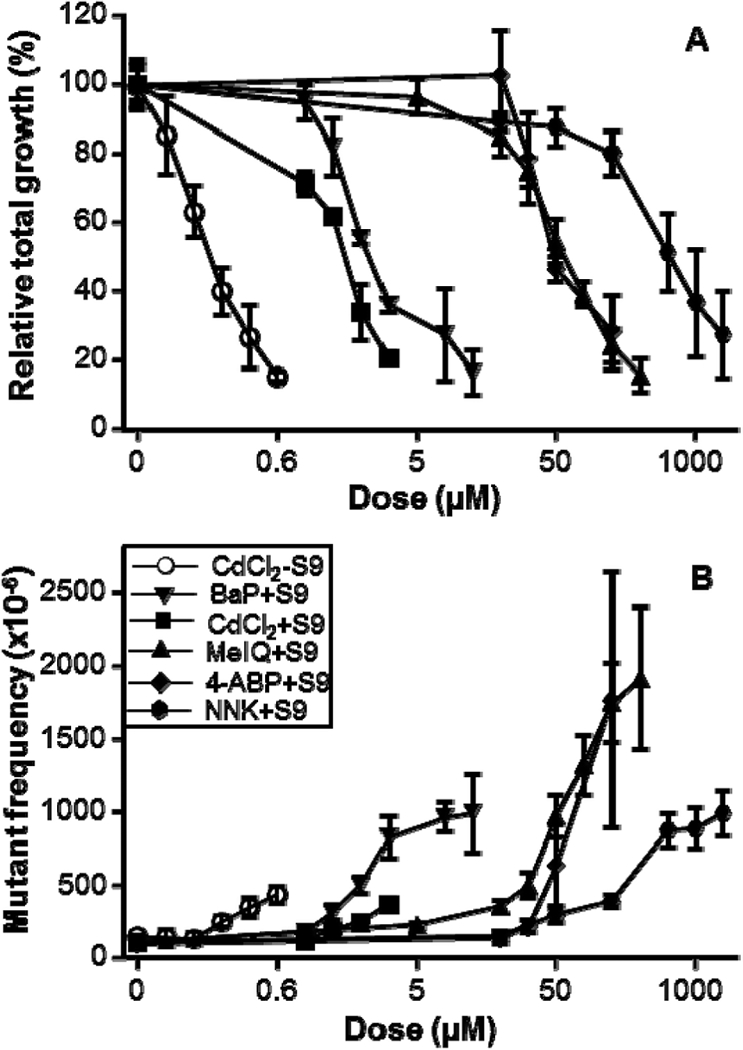
Cytotoxic and mutagenic effects in mouse lymphoma cells treated by five chemicals for 4 h. (A) Cytotoxicity is presented as RTG (%); (B) Mutagenicity is presented as mutant frequencies (MF) per 106 cells; Open circles, treatment in the absence of S9; Closed symbols, treatment in the presence of S9. All data are expressed as the mean ± 1 SD from 3 to 5 independent experiments.
Table 2.
Relative mutagenicity of five tobacco-related chemicals in the mouse lymphoma assay
| Chemical | S9 | PoD metrics |
LOGELd (μM) | Lowest positive response based on GEFe (μM) |
Mutagenic potencyf (MF × 10−6 per μM) |
||
|---|---|---|---|---|---|---|---|
| BMDL10a(μM) | BMD10b (μm) | NOGELc (μM) | |||||
| 4-ABP | + | 2.25 | 2.65 | 25 | 50 | 50 | 17.1 |
| BaP | + | 0.17 | 0.19 | 1 | 2 | 2 | 141.5 |
| CdCl2 | − | 0.12 | 0.27 | 0.5 | 0.6 | 0.5 | 800.9 |
| CdCl2 | + | 0.26 | 0.29 | 1 | 2 | 3 | 64.9 |
| MeIQ | + | 0.48 | 0.84 | 5 | 10 | 10 | 14.9 |
| NNK | + | 2.01 | 2.98 | 10 | 50 | 50 | 1.4 |
BMDL10, calculated using the EPA’s Benchmark Dose Software, is the lower limit of the 10% Benchmark Dose.
BMD10, calculated using the EPA’s Benchmark Dose Software, is a dose that produces a predetermined change in the response of an adverse effect over control [in this case, 10% increase in mutant frequency (MF)].
NOGEL, determined by one way ANOVA followed by Dunnett’s test, is the highest dose that did not induce a significant increase in MF.
LOGEL, determined by one way ANOVA followed by Dunnett’s test, is the lowest dose that induced a significant increase in MF.
Lowest positive response based on the GEF, as defined by the MLA Expert Workgroup of the IWGT (39), is the lowest dose which induced a MF that exceeded the global evaluation factor (GEF) of 126 mutants per 106 cells for the microwell version of the MLA.
Mutagenic potency is calculated from the slope of the linear regression of the dose-response curve (8).
Mutagenic potency
The mutagenic potencies of the five chemicals were calculated using the slope of the linear regression fit to the most rapidly increasing portion of the dose-response curves (R2 = 0.937–0.994), and are expressed in Table 2 as MF per μM of chemical. Among the five chemicals, CdCl2(-S9) was the most potent, exhibiting a mutagenic potency of 801 × 10−6 MF per μM. The mutagenic potency of CdCl2(- S9) was 12-fold higher than CdCl2(+S9) and more than 572-fold higher than NNK(+S9), the least mutagenic chemical in this study. MeIQ(+S9) and 4-ABP(+S9) had similar potencies of 14.9 × 10−6 and 17.1 × 10−6 MF per μM, respectively.
Calculation of NOGELs, LOGELs and BMDs for five chemicals
Quantitative analysis of in vitro dose-response data for the induction of Tk gene mutations by the five chemicals representative of cigarette smoke constituents also was performed as recommended by Gollapudi et al. (14). ANOVA followed by Dunnett’s test was employed to determine the NOGEL and LOGEL (Table 2). EPA’s BMD Software fitted three or four models to the MLA data for each chemical and Akaike’s Information Criterion was used to select the model with the overall best fit for the data. As suggested by Gollapudi et al. (14), initially only the BMD10 and BMDL10 metrics were calculated. Following the EPA’s BMD technical guidance, some of the responses for the top dose(s) were removed for better modeling. For example, the variance in NNK treatment appeared to increase as the means increased and the higher doses of NNK had responses on a plateau. Accordingly, the three top doses of NNK were eliminated during the quantitative analysis for an adequate model fit. The exponential model provided the best fit to calculate the BMD10 and BMDL10 for 4-ABP(+S9), BaP(+S9) and CdCl2(+S9), the polynomial model was used to calculate the BMD10 and BMDL10 for CdCl2(- S9) and NNK(+S9), and the linear model was used for MeIQ(+S9). Figure 2 shows the dose-response BMD modeling results for Tk gene MFs following the chemical treatments.
Figure 2.
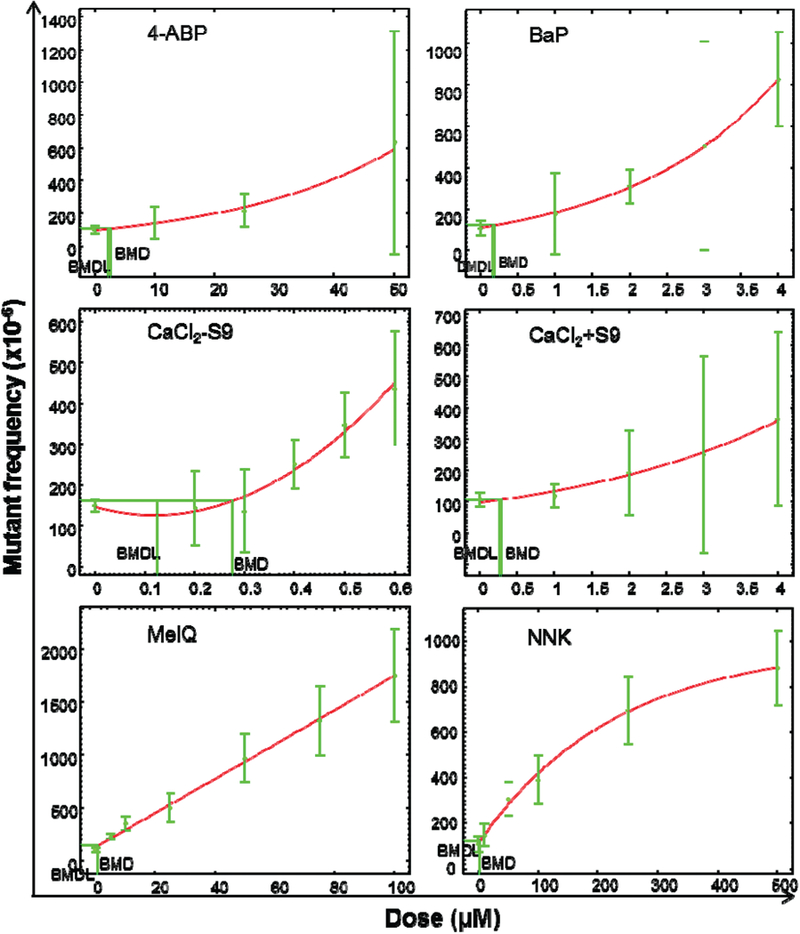
Quantitative analysis of the dose-response induced by five chemicals in the mouse lymphoma cells. BMD10 and BMDL10, calculated by EPA’s BMDS software, are shown in each graph. The data points represent the mean mutant frequencies (MF) from Figure 1 and the bars represent the calculated 95% confidence interval of each MF
Comparison of MLA mutagenicity of five chemicals using different quantification metrics
As shown in Table 2, the lowest dose giving a positive mutagenic response for each chemical was calculated based on two criteria, the application of the GEF developed by the IWGT and the LOGEL calculated by ANOVA followed by Dunnett’s test. The values were quite comparable. Not surprisingly, the NOGEL values for each agent were somewhat lower than the lowest positive concentrations. The ordering of agents from the most to least potent [CdCl2(-S9) < BaP(+S9) ≤ CdCl2(+S9) < MeIQ(+S9) < NNK(+S9) ≤ 4-ABP(+S9)] and the ranges in mutagenic effect (50- to 100-fold differences between the least to most potent) were similar for all three metrics (GEF, LOGEL and NOGEL).
The mutagenic potency expressed as MF per μM of chemical produced the highest numerical values of all the metrics evaluated (Table 2). When nonlinear responses were observed, such as a plateau (e.g. NNK), only the linear portion of the dose range was used to calculate the mutagenic potency. The calculated mutagenic potencies displayed the widest range of values of all the quantitative analyses (the most potent vs. the least potent = 572-fold). The order of potency (from greatest to least) was: CdCl2(-S9) > BaP(+S9) > CdCl2(+S9) > 4-ABP(+S9) > MeIQ(+S9) > NNK(+S9).
The BMDL10s and the BMD1Qs were the lowest numerical values of all metrics evaluated (Table 2). A 16-fold range in BMD10s was calculated for the test agent dose responses, while the BMDL10s (the lower the more toxic) for the in vitro MLA increased in the order of CdCl2(-S9) < BaP(+S9) < CdCl2(+S9) < MeIQ(+S9) < NNK(+S9) < 4-ABP(+S9), with a range in values of 19-fold. Because the BMD metrics in this study were being used for distinguishing between dose responses, rather than setting a safe dose (as commonly is done using the BMD10), we also calculated BMDs for 50%, 100%, 200%, and 300% increases in the background response (Table 3). The fold- change in BMDs for the different agents increased from 16-fold for the BMD10, to 40-, 69-, 116- and 163-fold for BMD50, BMD100, BMD200 and BMD300. Setting the BMD at increasing levels also had a similar effect on increasing the fold-changes associated with the BMDL. Because the BMD300s for the CdCl2 responses were higher than the highest tested doses for this agent (Table 3), the BMD300 was not considered further. Using the BMDL and BMDU values calculated for the BMD10s, it was possible to divide the six dose responses into three groups whose upper and lower 95% confidence intervals did not overlap (Figure 3A). Similarly, it was possible to divide the BMD50 (Figure 3B) and BMD100 metrics (Figure 3C) into five different groups, while the BMD200s for each of the six dose responses all had non-overlapping 95% confidence limits (Figure 3D).
Table 3.
The benchmark doses (BMD) producing a 10%, 50%, 100%, 200% and 300% increase in the background frequency (BMD10–300) for the six dose responses and their lower limits (BMDLs)
| Chemical | S9 | Model | 10% |
50% |
100% |
200% |
300% |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BMDL10 | BMD10 | BMDL50 | BMD50 | BMDL100 | BMD100 | BMDL200 | BMD200 | BMDL300 | BMD300 | |||
| 4-ABP | + | exponential | 2.25 | 2.65 | 9.60 | 11.30 | 16.30 | 19.30 | 25.89 | 30.53 | 32.67 | 38.52 |
| BaP | + | exponential | 0.17 | 0.19 | 0.72 | 0.81 | 1.22 | 1.38 | 1.94 | 2.19 | 2.45 | 2.77 |
| CdCl2 | − | polynomial | 0.12 | 0.27 | 0.25 | 0.38 | 0.35 | 0.46 | 0.47 | 0.59 | 0.57 | 0.69a |
| CdCl2 | + | exponential | 0.26 | 0.29 | 1.11 | 1.23 | 1.89 | 2.11 | 3.00 | 3.34 | 3.79 | 4.22a |
| MeIQ | + | liner | 0.48 | 0.84 | 2.40 | 4.20 | 4.81 | 8.40 | 9.60 | 16.81 | 14.41 | 25.22 |
| NNK | + | exponential | 2.01 | 2.98 | 10.26 | 15.28 | 21.09 | 31.66 | 44.75 | 68.38 | 71.68 | 112.12 |
| Fold changeb | 19 | 16 | 41 | 40 | 60 | 69 | 95 | 116 | 126 | 163 | ||
For CdCl2 both with and without S9, the BMD300 is beyond the range of the actual data (4 and 0.6 μM, respectively).
The ratio of the greatest value to the least value in each column.
Figure 3.
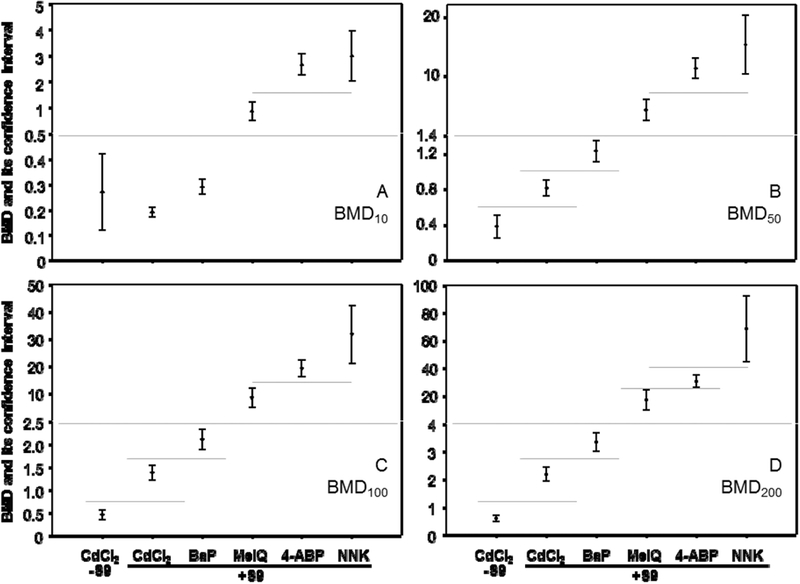
Comparison of BMD values for five chemicals in the mouse lymphoma assay. The model was selected in Figure 2 and the BMD estimates producing a 10%, 50%, 100% and 200% increase in the background frequency calculated (A, BMD10; B, BMD50; C, BMD100; D, BMD200).The bars represent the calculated 95% confidence interval of each value. The lower and upper limits derived from the BMD estimates were used to differentiate between responses based on non-overlapping confidence intervals.
Discussion
Genotoxicity data have been used extensively in chemical safety assessments and historically have been considered as a qualitative determination of potential hazard. Recently there has been an increased focus on providing more quantitative assessments of the dose response data from genetic toxicology studies (14,15), with an ultimate goal of applying PoDs to genotoxicity data to estimate human risk (16,17). We are using these metrics here to address a more limited problem, that is whether or not genotoxicity models can be used to compare the toxic properties of tobacco products and how to use genotoxicity data to differentiate between the genotoxic properties of tobacco products.
In this pilot study, MLA data for five chemicals representative of classes of chemicals found in cigarette smoke were evaluated using several different quantitative methods. This included using the more ‘traditional’ GEF criteria developed by the IWGT and a mutagenic potency calculation, as well as several metrics recently considered by the Health and Environmental Science Institute of the International Life Sciences Institute (ILSI-HESI) (14,15), that is BMD and NOGEL/LOGEL. Note that slope transition dose models, which also were evaluated by the ILSI-HESI group, were not used in our analyses because of an insufficient number of values at low response levels for some of the data sets.
As expected, all five chemicals resulted in dose-related cytotoxic and mutagenic effects in the presence of S9 (Figure 1). Only CdCl2 produced a positive mutagenic response without S9, indicating that metabolic activation was required for the other four chemicals’ mutagenic activity. This observation is in agreement with previous reports indicating that the majority of human carcinogens, including PAHs, HCAs, aromatic amines and N-nitrosamines, require metabolic activation to elicit their carcinogenic effects (40–42). S9-activation decreased the mutagenic activity of CdCl2 [e.g. decreasing the mutagenic potency by 12-fold and increasing the BMDL10 more than 2-fold (Table 2)]. Likely this is due to the formation of a Cd-metallothionein complex in the presence of S9 (43).
Overall, BMDL10, BMD10, NOGEL, LOGEL and the lowest positive response by GEF (whose values focus on the lower end of the dose-response curve) and the mutagenic potency (whose values are calculated over a broader portion of the dose-response curve), ordered the relative mutagenicity of the five test articles similarly (Table 2). The quantitative analysis indicated that CdCl2(-S9) and BaP(+S9) were the most potent mutagens, while NNK(+S9) and 4-ABP(+S9) generally were the weakest mutagens. Exceptions to the ordering included NNK, which while consistently among the weakest mutagens, was ranked higher than 4-ABP in the BMD and NOGEL calculations, while NNK tied for least potent using the LOGEL and GEF criteria. A possible reason for this is both LOGEL and GEF use the same type of endpoint (the lowest positive dose, albeit determined differently) for estimating the value used for comparison, and these types of estimates will benefit from data from additional treatment doses in the region where that value lies.
Our expectation was that metrics producing the greatest range in values for our five test agents would be better able to distinguish between their genotoxicities. The data in Table 2 indicate that there were relatively large differences in the ranges of metric values for the five agents determined by the different methods. MF data in this study resulted in a 572-fold range (the most potent vs. the least potent) of calculated mutagenic potency values, while there were much smaller ranges in values for BMDL10 (19-fold), BMD10 (16-fold), NOGEL (50-fold), LOGEL (83-fold) and the lowest positive response based on the GEF (100-fold), suggesting that mutagenic potency allows the best separation to distinguish between the chemicals selected for this study. As indicted above, the accuracy of NOGEL, LOGEL and GEF estimates are greatly influenced by the number of concentrations tested and the scatter in the data (44). The BMD and mutagenic potency estimates are also affected by data scatter, but both use the entire (or a large portion of the) mutagenicity dose-response. There are however differences in how the BMD and mutagenic potency model dose responses. The BMD approach explicitly takes the shape of the dose-response into account and selects one of five models based on how well each model describes the shape of the dose-response relationship, thus always yielding a good fit to the data (44). It was the preferred approach for dose-response analysis and PoD derivation for both quantal and continuous genotoxicity data by the QWG group because of the following advantages (17).
The BMD analysis can be performed on minimum datasets with three doses plus control; the BMD values are minimally affected by experimental design and dose selection; confidence intervals can be derived; and, most importantly, the genotoxic potency can be monitored because any fold-change (i.e. 10–200%) in response can be calculated from the entire dose-response curve, such as the determination of BMD10, BMD50, BMD100 and BMD200 in the present study. It should be noted, that while the BMD10 level has historically been used for quantitative risk assessment, particularly for in vivo data, an increase of this magnitude falls well within the normal range of the background MF of the MLA. Such a small increase would not be seen as biologically relevant and BMD values obtained with higher fold-increases are much more likely to give values that are biologically relevant.
By contrast, the mutagenic potency estimate relies on ‘expert judgment’ to fit a linear regression to the increasing portion of the dose-response (8). Although this is common practice, it is possible that statistical tests could be applied to make estimating a mutagenic potency a more rigorous procedure. It should be noted that one of the five models available in the BMD software was a linear model and indeed, the BMD procedure for MeIQ(+S9) chose the linear model (Figure 2) as the one best fitting the data.
The differences in the ranges of metric values might be explained by the fact that the different approaches focus on different parts of the mutagenicity dose responses. The BMDL10 describes the lower limit of a dose that produces a 10% increase over the control, and the control MF for the microwell version of MLA is about 50–170 × 10−6 (39). A MF of 10% above control is well below the minimum increase considered to be positive in the assay (126 × 10−6) and thus is considered to be on the extreme lower end of the dose-response relationship. The slope of a linear regression fit to a dose-response, however, will be greatly influenced by the higher values at the upper end of the dose-response. The values for NOGEL, LOGEL and the lowest positive response by GEF, on the other hand, will identify doses at the point of the dose-response where the response is turning from negative to positive. This area of the dose-response will be greater than the BMDL10 dose, and very likely less than the doses influencing the mutagenic potency estimate. Because BMDs can be calculated for any increase relative to the control, we calculated the BMD50, BMD100 and BMD200 values for the dose responses (Table 3), which are the doses that produce a 50%, 2-fold, and 3-fold increase in the background MF, respectively. While the BMD10s for the five agents had a range of 16, the ranges for BMD50, BMD100 and BMD200 were 40, 69 and 116, respectively, close to the GEF, NOGEL and LOGEL ranges, which we expect to occur at a point in the dose- response curve higher than the BMD10, likely in the mid-portion of our dose-response curves.
A final consideration in evaluating quantitative methods for genetic toxicity data, is distinguishing between the values derived for different test agents in a scientifically rigorous manner. GEF, NOGEL and LOGEL estimates do not lend themselves to deriving confidence intervals and hypothesis testing of differences. Establishing rules by which mutagenic potency estimates are derived might lend themselves to multiple regression analysis of the dose responses produced by different test agents. The number of data points required to make suitably powered estimates, however, would be an issue that must be addressed, and as indicated by our findings, fitting a linear regression to data might not be the best way of describing most mutagenicity dose responses. On the other hand, BMD estimates generate upper and lower confidence intervals, which can be used to differentiate between responses. For instance, the upper and lower confidence bounds derived from the BMD10 estimates of the mutagenicity dose responses produced in this study, divided the five test agents (six dose responses) into three groups having non-overlapping upper and lower 95% confidence intervals (Figure 3A). Moving further up on the dose responses to a BMD200, distinguished all six dose responses (Figure 3D).
The BMDL is often used as the starting point for evaluating allowable exposure levels in animal studies and comparisons with other toxicological endpoints (45). Accordingly, quantitative BMD approaches have been recommended for future applications using genotoxicity data (16,17,19). The results of our study indicate that the BMD approach has advantages for our application in terms of scientific rigor. It uses all of the dose response data, performs curve fitting to find the best way of describing the dose-response relationship, and provides confidence intervals that may be used to distinguish between the dose responses produced by different test agents. The BMDL10 has been proposed as a reasonable metric for calculating margin of exposure (MOE) from genotoxicity data for deriving estimates of human risk (15). Estimating MOE, however, is not the goal for our use of these quantitative approaches. Although the doses of test agents used at the lower end of our MLA dose-response curves may be more similar to what humans are exposed (Table 1) (25–32), the nature of in vitro assays (e.g. short treatment, artificial system of metabolic activation, transformed and rapidly dividing cell line) make any kind of direct estimate of human risk from in vitro genetic toxicology data problematic. What can be estimated is the intrinsic mutagenicity of the test article. Our observations with these model compounds indicate that greater discrimination between responses are made using BMDs of 50%, 100% or higher (e.g. 200%) over the control so that these higher response BMDs are likely the most useful, and they are certainly more biologically relevant than the BMD10. We intend to test this hypothesis using responses generated by cigarette smoke extracts and whole smoke generated by a smoking machine.
In summary, we have employed several metrics to evaluate the relative dose-response mutagenicity of five chemicals representative of tobacco smoke constituents. Mutagenicity was measured in the MLA using the standard 4-h treatment, with and without S9 activation. Application of quantitative analysis methods to these data revealed some strengths and weakness in the abilities of the methods to discriminate between the potency of mutagens. All the methods we employed ordered the potency of the five test agents similarly. The NOGEL, LOGEL and BMD10 metrics produced the smallest range in potency values while the mutagenic potency expressed as a mutant frequency per micromole of chemical produced the highest range. Arguably, the BMD approach produced the most useful comparisons and the ranges could be increased (likely making the results more biologically relevant) by sampling different portions of the dose-response curves and utilizing the BMD50, BMD100 or perhaps even higher responses. Overall, these results demonstrate that the MLA is capable of quantitatively discriminating the relative MF induced by a set of structurally diverse chemicals.
Acknowledgments
This research was supported by an inter-center agreement within US Food and Drug Administration (FDA) between the Center for Tobacco Products (CTP) and National Center for Toxicological Research (NCTR). This manuscript has been reviewed by the NCTR and the CTP. The information in these materials is not a formal dissemination of information by the US FDA and does not represent agency position or policy.
Footnotes
Conflict of interest statement: None declared.
References
- 1.CDC (2014) Health Effects of Cigarette Smoking. Smoking and Tobacco Use. http://www.cdc.gov/tobacco/data_statistics/fact_sheets/health_effects/effects_cig_smoking/index.htm#overview (accessed May 7, 2015).
- 2.WHO (2011) WHO Report on the Global Tobacco Epidemic. World Health Organization; http://whqlibdoc.who.int/publications/2009/9789241563918_eng_full.pdf (accessed May 7, 2015). [Google Scholar]
- 3.CDC (2014) Mechanisms of Cancer Induction by Tobacco Smoke In U.S. Department of Health and Human Services; (ed.), 2014 Surgeon General’s Report: The Health Consequences of Smoking—50 Years of Progress. pp. 148–151. [Google Scholar]
- 4.IARC (2012) Tobacco smoking, a review of human carcinogens: personal habits and indoor combustions. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 100E, 43–212. [PMC free article] [PubMed] [Google Scholar]
- 5.Hecht SS (2003) Tobacco carcinogens, their biomarkers and tobacco- induced cancer. Nat. Rev. Cancer, 3, 733–744. [DOI] [PubMed] [Google Scholar]
- 6.EPA (2005) Guildelines for carcinogen risk assessment. U.S. Environmental Protection Agency; http://www2.epa.gov/sites/production/files/2013-09/documentsZcancer_guidelines_final_3-25-05.pdf (accessed May 7, 2015). [Google Scholar]
- 7.DeMarini DM (2004) Genotoxicity of tobacco smoke and tobacco smoke condensate: a review. Mutat. Res, 567, 447–474. [DOI] [PubMed] [Google Scholar]
- 8.DeMarini DM, Gudi R, Szkudlinska A, Rao M, Recio L, Kehl M, Kirby PE, Polzin G and Richter PA (2008) Genotoxicity of 10 cigarette smoke condensates in four test systems: comparisons between assays and condensates. Mutat. Res, 650, 15–29. [DOI] [PubMed] [Google Scholar]
- 9.Foy JW, Bombick BR, Bombick DW, Doolittle DJ, Mosberg AT and Swauger JE (2004) A comparison of in vitro toxicities of cigarette smoke condensate from Eclipse cigarettes and four commercially available ultra low-”tar” cigarettes. Food Chem. Toxicol, 42, 237–243. [DOI] [PubMed] [Google Scholar]
- 10.Guo X, Verkler TL, Chen Y, Richter PA, Polzin GM, Moore MM and Mei N (2011) Mutagenicity of 11 cigarette smoke condensates in two versions of the mouse lymphoma assay. Mutagenesis, 26, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lou J, Zhou G, Chu G, Jiang J, Huang F, Zheng S, Lu Y, Li X and He J (2010) Studying the cyto-genotoxic effects of 12 cigarette smoke condensates on human lymphoblastoid cell line in vitro. Mutat. Res, 696, 48–54. [DOI] [PubMed] [Google Scholar]
- 12.Roemer E, Stabbert R, Rustemeier K, Veltel DJ, Meisgen TJ, Reininghaus W, Carchman RA, Gaworski CL and Podraza KF (2004) Chemical composition, cytotoxicity and mutagenicity of smoke from US commercial and reference cigarettes smoked under two sets of machine smoking conditions. Toxicology, 195, 31–52. [DOI] [PubMed] [Google Scholar]
- 13.Schramke H, Meisgen TJ, Tewes FJ, Gomm W and Roemer E (2006) The mouse lymphoma thymidine kinase assay for the assessment and comparison of the mutagenic activity of cigarette mainstream smoke particulate phase. Toxicology, 227, 193–210. [DOI] [PubMed] [Google Scholar]
- 14.Gollapudi BB, Johnson GE, Hernandez LG et al. (2013) Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ. Mol. Mutagen, 54, 8–18. [DOI] [PubMed] [Google Scholar]
- 15.Johnson GE, Soeteman-Hernández LG, Gollapudi BB et al. (2014) Derivation of point of departure (PoD) estimates in genetic toxicology studies and their potential applications in risk assessment. Environ. Mol. Mutagen, 55, 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacGregor JT, Frötschl R, White PA et al. (2015) IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutat Res, 783, 66–78. [DOI] [PubMed] [Google Scholar]
- 17.MacGregor JT, Frötschl R, White PA, et al. (2015) IWGT report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure-response relationships and points of departure (PoDs). Mutat Res, 783, 55–65. [DOI] [PubMed] [Google Scholar]
- 18.Cao X, Mittelstaedt RA, Pearce MG, Allen BC, Soeteman-Hernán- dez LG, Johnson GE, Bigger CA and Heflich RH (2014) Quantitative dose-response analysis of ethyl methanesulfonate genotoxicity in adult gpt-delta transgenic mice. Environ. Mol. Mutagen, 55, 385–399. [DOI] [PubMed] [Google Scholar]
- 19.Hernández LG, van Benthem J and Johnson GE (2013) A mode- of-action approach for the identification of genotoxic carcinogens. PLoS One, 8, e64532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Recio L, Shepard KG, Hernández LG and Kedderis GL (2012) Dose-response assessment of naphthalene-induced genotoxicity and glutathione detoxication in human TK6 lymphoblasts. Toxicol. Sci, 126, 405–412. [DOI] [PubMed] [Google Scholar]
- 21.Tang L, Guérard M and Zeller A (2014) Quantitative assessment of the dose-response of alkylating agents in DNA repair proficient and deficient ames tester strains. Environ. Mol. Mutagen, 55, 15–23. [DOI] [PubMed] [Google Scholar]
- 22.EFSA (2009) Guidance of the Scientific Committee on a request from EFSA on the use of the benchmark dose approach in risk assessment. EFSA J, 1150, 1–72. [Google Scholar]
- 23.Burgoon LD and Zacharewski TR (2008) Automated quantitative dose-response modeling and point of departure determination for large toxicogenomic and high-throughput screening data sets. Toxicol. Sci, 104, 412–418. [DOI] [PubMed] [Google Scholar]
- 24.Davis JA, Gift JS and Zhao QJ (2011) Introduction to benchmark dose methods and U.S. EPA’s benchmark dose software (BMDS) version 2.1.1. Toxicol. Appl. Pharmacol, 254, 181–191. [DOI] [PubMed] [Google Scholar]
- 25.Smith CJ, Livingston SD and Doolittle DJ (1997) An international literature survey of “IARC Group I carcinogens” reported in mainstream cigarette smoke. Food Chem. Toxicol, 35, 1107–1130. [DOI] [PubMed] [Google Scholar]
- 26.Smith CJ, Perfetti TA, Rumple MA, Rodgman A and Doolittle DJ (2001) “IARC Group 2B carcinogens” reported in cigarette mainstream smoke. Food Chem. Toxicol, 39, 183–205. [DOI] [PubMed] [Google Scholar]
- 27.National Toxicology Program (2011) NTP 12th Report on Carcinogens. Rep Carcinog, 12, iii–499. [PubMed] [Google Scholar]
- 28.IARC (2012) Second-hand tobacco smoke, a review of human carcinogens: personal habits and indoor combustions. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 100E, 213–264. [PMC free article] [PubMed] [Google Scholar]
- 29.Ding YS, Ashley DL and Watson CH (2007) Determination of 10 carcinogenic polycyclic aromatic hydrocarbons in mainstream cigarette smoke. J. Agric. Food Chem, 55, 5966–5973. [DOI] [PubMed] [Google Scholar]
- 30.IARC (2004) Involuntary smoking, tobacco smoke and involuntary smoking. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, 83, 1189–1414. [PMC free article] [PubMed] [Google Scholar]
- 31.National Toxicology Program (2002) Selected Heterocyclic Amines: PhIP, MeIQ, and MeIQx. Final Report on Carcinogens Background Document. http://ntp.niehs.nih.gov/ntp/newhomeroc/roc11/hcaspub.pdf (accessed May 7, 2015).
- 32.IARC (2012) Benzo[a]pyrene, a review of human carcinogens: chemical agents and related occupations. IARC Monographs on the E-valuation of Carcinogenic Risks to Humans, 100F, 111–144. [Google Scholar]
- 33.Hammond D and O’Connor RJ (2008) Constituents in tobacco and smoke emissions from Canadian cigarettes. Tob. Control, 17(Suppl 1), i24–i31. [DOI] [PubMed] [Google Scholar]
- 34.Applegate ML, Moore MM, Broder CB, Burrell A, Juhn G, Kas- weck KL, Lin PF, Wadhams A and Hozier JC (1990) Molecular dissection of mutations at the heterozygous thymidine kinase locus in mouse lymphoma cells. Proc. Natl Acad. Sci. U. S. A, 87, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Sawyer JR, Chen L, Chen T, Honma M, Mei N and Moore MM (2009) The mouse lymphoma assay detects recombination, deletion, and aneuploidy. Toxicol. Sci, 109, 96–105. [DOI] [PubMed] [Google Scholar]
- 36.Mei N, Xia Q, Chen L, Moore MM, Chen T and Fu PP (2006) Photomutagenicity of anhydroretinol and 5,6-epoxyretinyl palmitate in mouse lymphoma cells. Chem. Res. Toxicol, 19, 1435–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mei N, Guo X and Moore MM (2014) Methods for using the mouse lymphoma assay to screen for chemical mutagenicity and photo-mutagenicity In Caldwell GW and Yan Z (eds.), Optimization in Drug Discovery: In-vitro Methods. Humana Press, New York, pp. 561–592. [Google Scholar]
- 38.Mei N, Zhang Y, Chen Y et al. (2012) Silver nanoparticle-induced mutations and oxidative stress in mouse lymphoma cells. Environ. Mol. Mutagen, 53, 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moore MM, Honma M, Clements J et al. (2006) Mouse lymphoma thymidine kinase gene mutation assay: follow-up meeting of the International Workshop on Genotoxicity Testing-Aberdeen, Scotland, 2003-Assay acceptance criteria, positive controls, and data evaluation. Environ. Mol. Mutagen, 47, 1–5. [DOI] [PubMed] [Google Scholar]
- 40.Arlt VM, Stiborová M, Henderson CJ et al. (2008) Metabolic activation of benzo[a]pyrene in vitro by hepatic cytochrome P450 contrasts with detoxification in vivo: experiments with hepatic cytochrome P450 reductase null mice. Carcinogenesis, 29, 656–665. [DOI] [PubMed] [Google Scholar]
- 41.Schrader E, Hirsch-Ernst KI, Scholz E, Kahl GF and Foth H (2000) Metabolism of 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in primary cultures of rat alveolar type II cells. Drug Metab. Dispos, 28, 180–185. [PubMed] [Google Scholar]
- 42.Luch A (2005) Nature and nurture—lessons from chemical carcinogenesis. Nat. Rev. Cancer, 5, 113–125. [DOI] [PubMed] [Google Scholar]
- 43.Klaassen CD, Liu J and Diwan BA (2009) Metallothionein protection of cadmium toxicity. Toxicol. Appl. Pharmacol, 238, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slob W and Setzer RW (2014) Shape and steepness of toxicological dose-response relationships of continuous endpoints. Crit. Rev. Toxicol, 44,270–297. [DOI] [PubMed] [Google Scholar]
- 45.Travis KZ, Pate I and Welsh ZK (2005) The role of the benchmark dose in a regulatory context. Regul. Toxicol. Pharmacol, 43, 280–291. [DOI] [PubMed] [Google Scholar]