

Abstract
Plasmodium vivax resistance to chloroquine (CQ) was first reported over 60 years ago. Here we analyzed sequence variations in the multidrug resistance 1 gene (Pvmdr1), a putative molecular marker for P. vivax CQ resistance, in field isolates collected from three sites in Thailand during 2013-2016. Several single nucleotide polymorphisms previously implicated in reduced CQ sensitivity were found. These genetic variations encode amino acids in the two nucleotide-binding domains as well as the transmembrane domains of the protein. The high level of genetic diversity of Pvmdr1 provides insights into the evolutionary history of this gene. Specifically, there was little evidence of positive selection at amino acid F1076L in global isolates to be promoted as a possible marker for CQ resistance. Population genetic analysis clearly divided the parasites into eastern and western populations, which is consistent with their geographical separation by the central malaria-free area of Thailand. With CQ-primaquine remaining as the frontline treatment for vivax malaria in all regions of Thailand, such a population subdivision could be shaped and affected by the current drugs for P. falciparum since mixed P. falciparum/P. vivax infections often occur in this region.
Keywords: Plasmodium vivax, Multidrug resistance 1 gene, Genetic diversity, Phylogenetic relationship, Genetic structure
1. Introduction
Chloroquine (CQ) has been the first-line antimalarial drug to treat Plasmodium vivax infections for more than 70 years. Whereas CQ resistance in Plasmodium falciparum is widespread and appears at high frequencies in most malaria-endemic areas, P. vivax CQ resistance is much less common, despite the first report of treatment failure indicative of CQ resistance in the 1950s (Berliner et al., 1948; Hoekenga, 1952). The slow emergence/spread of CQ resistance in P. vivax may be explained by its ability to form gametocytes before symptoms appear, thus enabling mosquito infection before drug treatment. Nonetheless, ineffective treatments of vivax malaria have been reported in many areas (Price et al., 2014), including, but not limited to, Papua New Guinea (Cooper, 1994; Schuurkamp et al., 1992), Ethiopia (Ketema et al., 2011), Indonesia (Baird et al., 1997; Ratcliff et al., 2007; Tjitra et al., 2008), India (Singh, 2000), and Brazil (de Santana Filho et al., 2007). In the Greater Mekong Subregion (GMS) of Southeast Asia, though CQ is generally effective for treatment of vivax malaria, CQ clinical failure indicative of resistance has been reported sporadically in Myanmar (Htun et al., 2017; Marlar et al., 1995; Myat Phone et al., 1993), Thailand (Congpuon et al., 2011; Rijken et al., 2011), Vietnam (Thanh et al., 2015), and at the China-Myanmar border (Liu et al., 2014; Yuan et al., 2015). Because of high recurrence rates of P. vivax after CQ treatment in some areas, CQ resistance has been considered an important public concern. In Indonesia, for example, CQ as the front-line treatment for P. vivax has been replaced by artemisinin-combination therapy (Baird et al., 1997).
Previous studies have suggested that mutations within the multidrug resistant 1 gene (Pvmdr1) may be used as markers for CQ resistance surveillance (Chung et al., 2015; Huang et al., 2014; Mekonnen et al., 2014). In vitro drug susceptibility assays also revealed association between higher genomic copy number of Pvmdr1 and the increase in CQ IC50 (Marfurt et al., 2007; Suwanarusk et al., 2008). More recently, M908L and T958M mutations were shown to be associated with reduced in vitro CQ sensitivity (Chehuan et al., 2013). In the clinical settings, association was found between the copy number of Pvmdr1 harboring Y976F/F1076L mutations and treatment failure in severe P. vivax malaria cases (Fernandez-Becerra et al., 2009; Melo et al., 2014). However, some studies failed to detect a link between Pvmdr1 mutations and reduced CQ sensitivity (Hamedi et al., 2016; Schousboe et al., 2015; Shalini et al., 2014). Association between Y976F/F1076L mutations and P. vivax CQ resistance was mostly reported based on the cut-off IC50 values of an estimated minimal effective concentration of CQ at 100 ng/ml of the whole blood, a value that is applied for in vitro drug susceptibility in P. falciparum (Baird et al., 1997; Rungsihirunrat et al., 2015). There is not yet a CQ concentration cutoff for defining CQ resistance in P. vivax. Thus, with the unclear link between P. vivax resistance and Pvmdr1 mutations (point mutations and copy number variation) (Suwanarusk et al., 2008), the resistance cut-off value needs future validation.
To better understand the diversity and evolution of Pvmdr1 in P. vivax populations in Thailand, we collected blood samples from P. vivax malaria patients from the western and eastern national borders where malaria remains endemic. In these areas, CQ has been administered with primaquine (PQ) as the standard radical cure for P. vivax malaria since 2007. Through sequencing, we determined the extent of genetic diversity and identified potential mechanisms affecting the evolution of Pvmdr1. We also report haplotype clusters representative of major parasite variations and the population structure of the Thai parasites with respect to the global isolates.
2. Materials and methods
2.1. Study sites and samples collection
Malaria transmission in Thailand is perennial and seasonal, which follows the pattern of rainfall with two transmission peaks: one in July-September and one in November (Cui et al., 2003). Samples were collected in three provinces: Tak and Kanchanaburi in the west bordering Myanmar, Ubon Ratchathani in the northeast bordering Cambodia and Laos (Fig. 1). There are different distribution patterns of malaria vectors between western and eastern regions of Thailand (CDC, 2015; Kittichai et al., 2017). While major vectors in western Thailand are Anopheles dirus, An. minimus and An. maculatus complexes, in eastern Thailand such as Ubon Ratchathani An. barbirostris is widely distributed. The latter species has been shown to be a vector for P. vivax (Sriwichai et al., 2016). Whereas the western border has been the most malaria prevalent, an outbreak occurred in Ubon Ratchathani in 2016, which reported the highest number of malaria cases in Thailand (CDC, 2015). In total, 88 finger-prick blood samples were collected from symptomatic patients in Kanchanaburi (37 during 2013-2014), Tak (22 during 2013-2015), and Ubon Ratchathani (29 during 2015-2016). To reduce the possibility of imported cases, patients were all from local villages who did not have travel histories in the previous month. Genomic DNA was extracted from the bloods using DNeasy Blood & Tissue Kit (Qiagen, Valencia, California, USA). Malaria diagnosis was performed by light microscopy and confirmed by nested PCR targeting the 18S rRNA gene (Snounou et al., 1993). The study was approved by the Ethics Committee of the Faculty of Tropical Medicine, Mahidol University (TMEC11-033).
Fig. 1. Map indicating locations of the sampling sites for parasite isolates from three provinces in Thailand and other regions worldwide.
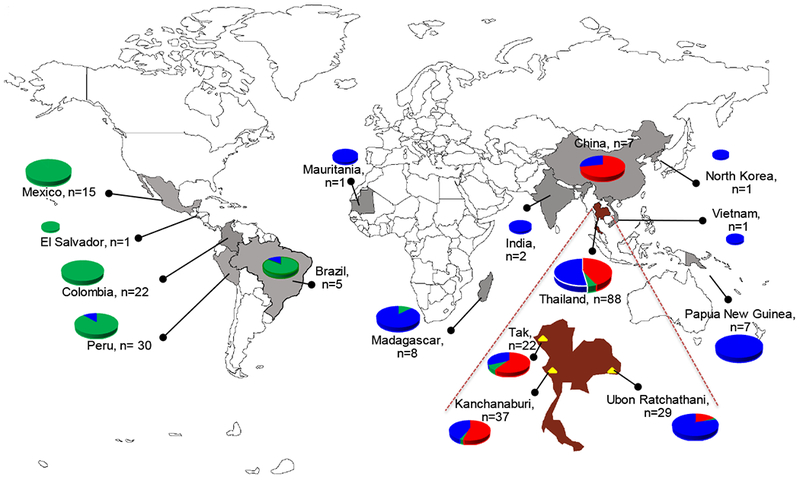
In addition to the origin of the samples, the distribution of the three clusters of Pvmdr1 haplotypes in each country or region based on the STRUCTURE plot (Blue, Green and Red) in Fig. 3 is shown as pie charts. The world map was modified from the one available on: www.freeworldmap.net.
2.2. PCR amplification and sequencing of the Pvmdr1 gene
PCR of the Pvmdr1 gene was performed with Pfu DNA polymerase (Promega) and primer sets MF1/MR5 and MF4/MR7 which generated two amplicons of 2,770 bp and 2,884 bp, respectively (Supplementary Table S1). PCR products were purified using the AccuPrep PCR purification kit (Bioneer Corporation, South Korea), and sequenced in both directions with primers described in the Table S1. All primers were designed based on the reference Pvmdr1 sequence from the Salvador I strain (PVX_080100). Sequences were assembled and edited manually using BioEdit 7.2.5 (Hall, 1999). Sequence alignment was done using the program Muscle 3.8 (Edgar, 2004).
2.3. Domain prediction in Pvmdr1
Nucleotide-binding domains (NBDs) and transmembrane domains (TMs) of the Pvmdr1 protein were identified by mapping the sequence to the P-glycoprotein structure model from PDB: 4Q9H, which shares 31% identity. The suitable protein template was searched via the SWISS-MODELL server (https://swissmodel.expasy.org/interactive).
2.4. Global data collection
A total of 113 full-length or near full-length Pvmdr1 sequences were retrieved from GenBank and PlasmoDB (plasmodb.org) representing parasite isolates from 13 countries: 13 from Thailand, 7 from China, 7 from Papua New Guinea (PNG), 2 from India, 8 from Madagascar, 15 from Mexico, 22 from Columbia, 30 from Peru, 5 from Brazil, and one each from Vietnam, North Korea, Mauritania and El Salvador (Fig. 1). Each sequence was trimmed to remove low-quality segments, yielding 4,257 bp fragments from the 4,395 bp Pvmdr1 open reading frame. Sequences obtained in this study are available in GenBank with the accession numbers KX421155-KX421190 and KX868005-KX868056.
2.5. Sequence polymorphism and diversity
Sequence diversity was estimated using either short sequences through the sliding window technique or the nearly full-length (4,257 bp) sequence. The nucleotide diversity of Pvmdr1 was estimated by DnaSP v5.10 (Librado and Rozas, 2009). Singletons, parsimony, number of segregation sites including both synonymous and nonsynonymous mutations, and haplotypes (H) were determined. For genetic diversity, the average nucleotide diversity (θπ) and haplotype diversity (Hd) were measured across different sites within Thailand as well as across different countries.
2.6. Intragenic Recombination
Detection of recombination events was done using several programs incorporated in DnaSP v5.10 and the RDP4 package (Martin et al., 2010). The minimum numbers of recombination events (Rm) were assessed by the gametic test in DnaSP v5.10. Recombination breakpoints were detected using RDP, MaxChi, GENECONV, BootScan, Chimeara, SIScan, PhylPro, LARD and 3Seq algorithms implemented in the RDP4 package. LDhat in the RDP4 package was used to scan the recombination rate (Rho, ρ) and the mutation rate (Theta, θ). The population-scaled recombination to mutation ratio (ρ/θ) was calculated.
2.7. Tests of departure from neutrality and codon-based tests of selection
Tajima’s D and Fu & Li’s F* and D* tests were used to assess the departure from neutrality using the DnaSP v5.10 software. If the Watterson population nucleotide diversity (θ) and average pairwise nucleotide diversity (π) differ, a significant positive D value is expected, suggesting a balancing selection or a population size shrinking. Likewise, Fu & Li’s F* and D* tests are implemented to evaluate the difference between the number of singletons and the average pairwise nucleotide diversity. If a significant positive value is found, a balancing selection is indicated. However, if the Fu & Li’s statistics are negative, directional selection is implicated. Sliding window analysis also was used to determine nucleotide diversity and to perform all neutrality tests across the loci. Deletions/insertions were excluded from the analyses.
To study the evolutionary force acting on the Pvmdr1 gene, a hypothesis of dN>dS was tested using a bootstrap method with 1,000 replicates. Two-tailed Z-test of selection was used to estimate variance and make a substitution model using the Nei-Gojobori method in MEGA7 (Kumar et al., 2016). While a dN/dS ratio exceeding one is considered as a result of positive selection,a ratio less than one is an indicator of a purifying selection. A P-value of < 0.05 was considered as the level of significance.
A panel of maximum likelihood codons-based tests in the HyPhy package implemented in the online web-server Data Monkey (http://www.datamnkey.org) was conducted in order to determine the existence of specific codons targeted by selection (Kumar et al., 2016).
2.8. Phylogenetic relationship
To determine the genetic interrelationships of all the parasite isolates, phylogenetic analysis was done with aligned Pvmdr1 sequences using PHYLOViZ 2.0 (Francisco et al., 2012). A phylogenetic tree was generated using the neighbor-joining algorithm. The minimum number of substitutions or errors that influence the tree’s branching was measured by the Hamming distance. The Sal I reference strain was represented as the wild type. Haplotype clusters were defined manually using nine global single nucleotide polymorphisms (SNPs) with frequencies of ≥ 5%.
2.9. Population structure and genetic differentiation
Haplotype clustering analysis based on the polymorphic sites in Pvmdr1 was used to analyze the P. vivax populations with reference to the dynamics of the parasite transmission bearing the varied SNP patterns. The pattern was shown by the Thai provinces and countries. STRUCTURE 2.3.2 software was used to assess clustering of haplotypes (Pritchard et al., 2000) based on the admixture model. Twenty iterations were run for each cluster (K= 1-12) with a burn-in of 50,000 steps and then 500,000 Markov Chain Monte Carlo (MCMC) steps. The optimal number of clusters (K) was determined according to a published method based on the change in the posterior probability of the data [Ln P(D)] (Evanno et al., 2005). FSTAT v.2.9.3 (Goudet, 1995) software was used to assess pairwise genetic differentiation (Weir & Cockerham Fst values) using Fst. LIAN 3.7 (Haubold and Hudson, 2000) software was used to calculate the linkage disequilibrium (LD) using 50,000 iterations for burn-in followed by 100,000 MCMC iterations.
3. Results
3.1. Genetic diversity of the Pvmdr1 gene in Thai and global parasite isolates
Based on the alignment with the model structure PDB: 4Q9H, the predicted structure of the Pvmdr1 protein comprises a NBD1, the first six TMs (TM1-6), a NBD2 and the second six TMs (TM7-12). Since the genetic diversity of Pvmdr1 has been evaluated mostly based on partial sequences, we sequenced the near complete Pvmdr1 gene in 88 P. vivax clinical samples from three provinces in Thailand (Table 1). The sequences covering 4,257 bp (1,419 amino acids) contained a total of 34 SNPs, out of which 21 were nonsynonymous and 13 synonymous. The 21 nonsynonymous mutations are located in TM3, NBD1, NBD7-11 and NBD2 (Table 1). 14 out of the 21 nonsynonymous mutations were novel SNPs (K454I, L470H, K672N, N740 D, A763V, L845F, A861E, L936F, E996Q, P1177T, G1232C, G1265W, S1274R, and K1393N). Of the nine most prevalent SNPs, five (L845F, M908L, T958M, Y976F, and F1076L) were found in TM7-12 (Supplementary Fig. S1). In particular, two mutations M908L and T958M previously associated with reduced in vitro CQ sensitivity (Chehuan et al., 2013) reached high frequencies (96.59% and 100%, respectively). Although Ubon Ratchathani has the least number of nonsynonymous mutations of the three Thai sites, it has the highest frequency for the L845F mutant (58.62%). In contrast, the S513R and K1393N combination was the most prevalent in the two western provinces (Table 1). Y976F and F1076L were found in 9-27% and 27-41% of isolates across all three sites, respectively.
Table 1.
SNPs identified in the three P. vivax populations in Thailand and their prevalence (%).
| SNPs | Amino acids* | Tak (N=22) | Kanchanaburi (N=37) | Ubon Rachathani (N=29) | Total (N=88) |
|---|---|---|---|---|---|
| ACC-126-ACT | (T42T) | 3.45 | 1.14 | ||
| AAG-132-AAA | (K44K) | 4.55 | 5.41 | 13.79 | 7.95 |
| GGC-516-GGT | (G172G) | 5.41 | 2.27 | ||
| AAC-936-AAT | (N312N) | 6.90 | 2.27 | ||
| ACG-1065-ACA | (T355T) | 4.55 | 1.14 | ||
| AAA-1367-ACA | (K456I) | 2.70 | 1.14 | ||
| CTT-1409-CAT | (L470H) | 4.55 | 1.14 | ||
| CTA-1477-TTA | (L493L) | 13.64 | 2.70 | 17.24 | 10.23 |
| AGT-1539-AGA | S513R | 63.64 | 37.84 | 17.24 | 37.50 |
| ACA-1587-ACG | T529T | 86.36 | 97.30 | 68.97 | 85.23 |
| AAG-2016-AAC | (K672N) | 2.70 | 1.14 | ||
| AAT-2218-GAT | (N740D) | 2.70 | 1.14 | ||
| CGG-2266-AGG | (R756R) | 4.55 | 8.11 | 4.55 | |
| GCG-2288-GTG | (A763V) | 4.55 | 8.11 | 4.55 | |
| CCA-2424-CCG | (P808P) | 2.70 | 3.45 | 2.27 | |
| CTC-2533-TTC | (L845F) | 2.70 | 58.62 | 20.45 | |
| GCG-2582-GAG | (A861E) | 10.81 | 4.55 | ||
| ATG-2722-CTG | M908L | 95.45 | 97.30 | 96.55 | 96.59 |
| AGC-2739-AGT | S913S | 2.70 | 1.14 | ||
| TTA-2808-TTT | (L936F) | 4.55 | 1.14 | ||
| ACG-2873-ATG | T958M | 100.00 | 100.00 | 100.00 | 100.00 |
| TAC-2927-TTC | Y976F | 9.09 | 27.03 | 20.69 | 20.45 |
| TTT-2936-TCT | F979S | 2.70 | 1.14 | ||
| ATG-2938-GTG | M980V | 2.70 | 1.14 | ||
| GAG-2986-CAG | (E996Q) | 2.70 | 1.14 | ||
| TTT-3226-CTT | F1076L | 31.82 | 40.54 | 27.59 | 34.09 |
| ATC-3414-ATT | (I1138I) | 3.45 | 1.14 | ||
| CCA-3529-ACA | (P1177T) | 4.55 | 1.14 | ||
| GGT-3694-TGT | (G1232C) | 3.45 | 1.14 | ||
| GGG-3793-TGG | (G1265W) | 3.45 | 1.14 | ||
| AGT-3822-AGG | (S1274R) | 2.70 | 1.14 | ||
| TCC-4074-TCT | S1358S | 27.27 | 10.81 | 11.36 | |
| AAG-4179-AAC | (K1393N) | 9.09 | 29.73 | 14.77 | |
| GAG-4188-GAA | (E1396E) | 9.09 | 10.81 | 6.82 |
New mutations identified in this study are in parenthesis, and non-synonymous mutations are in bold.
In total, 15 of the 21 nonsynonymous substitutions were parsimony-informative (9, 12 and 8 were parsimony-informative in Tak, Kanchanaburi and Ubon Ratchathani, respectively) (Table 2). Nineteen singletons were found across the three areas. The nucleotide diversity of the near full-length Pvmdr1 gene was 0.00085 ± 0.00005 (Table 2). Sliding window plot of nucleotide diversity showed two peaks at the nucleotide positions 1,453-1,529 (NBD1) and 3,147-3,222 (TM11), but the two peaks did not reach statistical significance (Fig. 2). Forty eight haplotypes were detected and the haplotype diversity was high (0.961 ± 0.011). Based on the 9 SNPs with ≥ 5% frequency in global isolates (Table 3), nine haplotypes were found among the Thai isolates. Three haplotypes (Hap_10, Hap_29, and Hap_30) were province-specific, while six haplotypes (Hap_2, Hap_5, Hap_11, Hap_17, Hap_20, and Hap_21) with frequencies of 1-11.44% of total isolates were shared among the three provinces (Table 3, Supplementary Table S2). In terms of regions, seven haplotypes were specific to the western border of Thailand (Hap_7, Hap_10, Hap_11, Hap_17, Hap_18, Hap_20, and Hap_29), whereas only one haplotype Hap_30 was unique in the eastern region.
Table 2.
Sequence polymorphisms and summary statistics of the near full-length Pvmdr1 gene in different populations*.
| Population | No. isolates | Polymorphic sites | Singletons | Amino acid changes* | Syn | Non-syn | π ± SD | Haplotypes |
||
|---|---|---|---|---|---|---|---|---|---|---|
| H | Hd ± SD | |||||||||
| Thailand (this study) | 88 | 34 | 19 | 15 | 13 | 21 | 0.00085 ± 0.00005 | 48 | 0.961 ± 0.011 | |
| Tak | 22 | 15 | 7 | 9 | 6 | 9 | 0.00073 ± 0.00008 | 17 | 0.961 ± 0.029 | |
| Kanchanaburi | 37 | 26 | 14 | 12 | 10 | 16 | 0.00090 ± 0.00007 | 27 | 0.961 ± 0.022 | |
| Ubon Ratchathani | 30 | 14 | 6 | 8 | 7 | 7 | 0.00072 ± 0.00083 | 16 | 0.927 ± 0.025 | |
| Thailand (plasmodb) | 13 | 5 | 4 | 1 | 1 | 4 | 0.00025 ± 0.00007 | 5 | 0.692 ± 0.110 | |
| China | 7 | 10 | 8 | 2 | 3 | 7 | 0.00075 ± 0.00018 | 6 | 0.952 ± 0.096 | |
| PNG | 7 | 1 | 0 | 1 | 0 | 1 | 0.00014 ± 0.0003 | 2 | 0.571 ± 0.110 | |
| India | 2 | 4 | 4 | 0 | 3 | 1 | 0.00091 ± 0.00046 | 2 | 1.000 ± 0.500 | |
| Madagascar | 8 | 14 | 10 | 4 | 4 | 10 | 0.00110 ± 0.00023 | 8 | 1.000 ± 0.063 | |
| Mexico | 15 | 7 | 6 | 1 | 3 | 4 | 0.00024 ± 0.00013 | 4 | 0.371 ± 0.153 | |
| Peru | 30 | 8 | 4 | 4 | 2 | 6 | 0.00032 ± 0.00007 | 7 | 0.692 ± 0.057 | |
| Brazil | 5 | 4 | 4 | 0 | 2 | 2 | 0.00038 ± 0.00011 | 4 | 0.900 ± 0.161 | |
| Colombia | 22 | 63 | 59 | 4 | 24 | 39 | 0.00161 ± 0.00108 | 7 | 0.814 ± 0.049 | |
| Other | 4 | 9 | 8 | 1 | 2 | 7 | 0.00111 ± 0.00026 | 4 | 1.000 ± 0.177 | |
| Global | 201 | 46 | 30 | 16 | 14 | 30 | 0.00069 ± 0.00004 | 53 | 0.907± 0.013 | |
For amino acid changes, only the parsimony-informative changes are included here. Syn, synonymous changes; non-syn, nonsynonymous changes; H, number of haplotypes; Hd, haplotype diversity; SD, standard deviation.
Fig. 2. Sliding window plots of nucleotide diversity and results of the neutrality tests from the global (left) and Thai (right) Pvmdr1 sequences.
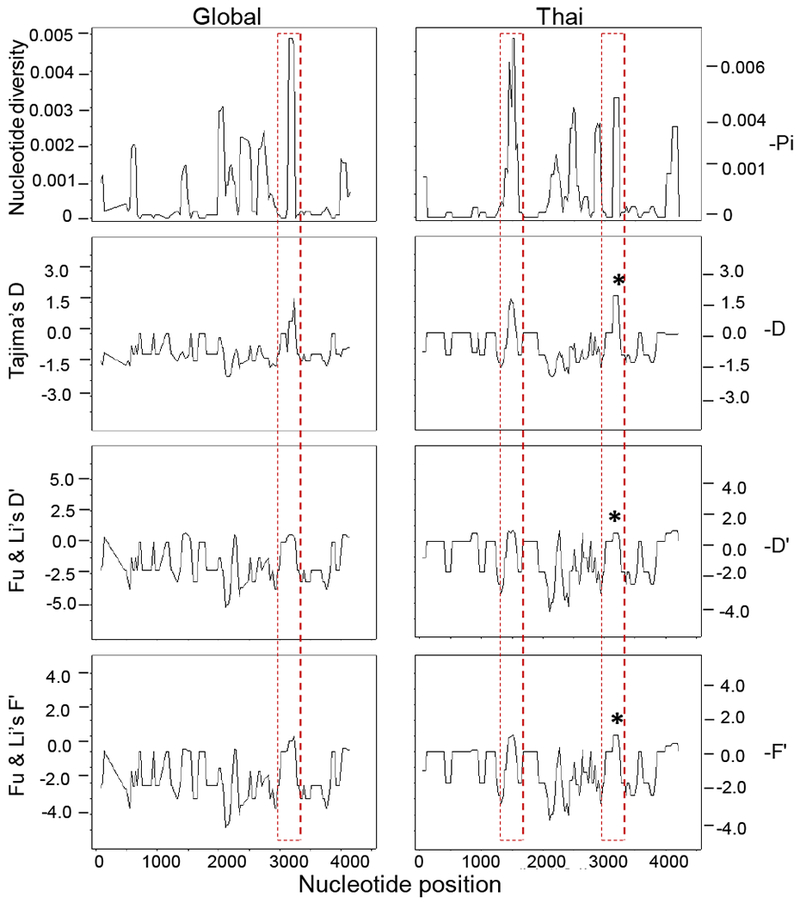
Peaks of diversity consistently shown in all analyses suggestive of balancing selection or population decline, thus, an excess of alleles at intermediate frequencies are marked by red dotted rectangles and correspond to positive values. The star plotted above the peaks depicts the statistical significance of the test at the level 0.05. Sliding window analysis used a window size of 100 bp and a step size of 10 bp. Nucleotide positions correspond to those of Sal I sequence.
Table 3.
Haplotypes of the near full-length Pvmdr1 gene from global P. vivax isolates.
| 2° Structure | TM4 | NBD1 | TM7> | TM8 | TM9 | TM10 | TM11 | NBD2 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | Cluster | V221L | S513R | G698S | L845F | M908L | T958M | Y976F | F1076L | K1393N | N | Frequency (%) |
| Hap_1 | Green | V | S | G | L | L | M | Y | F | K | 37 | 18.41 |
| Hap_2 | Blue | V | R | G | L | L | M | Y | F | K | 23 | 11.44 |
| Hap_3 | Green | L | S | G | L | L | M | Y | F | K | 20 | 9.95 |
| Hap_4 | Blue | V | S | G | L | L | M | F | L | K | 17 | 8.46 |
| Hap_5 | Blue | V | S | G | F | L | M | Y | L | K | 19 | 9.45 |
| Hap_6 | Green | V | S | G | L | M | M | Y | F | K | 15 | 7.46 |
| Hap_7 | Blue | V | S | G | L | L | M | Y | L | K | 7 | 3.48 |
| Hap_8 | Blue | V | S | S | L | L | M | Y | L | K | 9 | 4.48 |
| Hap_9 | Blue | V | S | S | L | L | M | F | L | K | 6 | 2.99 |
| Hap_10 | Red | V | S | G | L | L | M | Y | F | N | 5 | 2.49 |
| Hap_11 | Red | V | R | G | L | L | M | Y | F | N | 5 | 2.49 |
| Hap_12 | Red | V | R | S | L | L | M | Y | F | K | 4 | 1.99 |
| Hap_13 | Blue | V | R | S | L | L | M | F | L | K | 4 | 1.99 |
| Hap_14 | Red | V | S | S | L | L | M | Y | F | N | 3 | 1.49 |
| Hap_15 | Red | V | S | S | L | L | M | Y | F | K | 3 | 1.49 |
| Hap_16 | Green | V | S | G | L | M | T | Y | F | K | 3 | 1.49 |
| Hap_17 | Red | V | R | G | L | M | M | Y | F | K | 3 | 1.49 |
| Hap_18 | Blue | V | R | G | L | L | M | F | L | K | 3 | 1.49 |
| Hap_19 | Red | V | R | S | L | L | M | Y | F | N | 2 | 1.00 |
| Hap_20 | Blue | V | S | G | L | L | M | Y | L | N | 2 | 1.00 |
| Hap_21 | Blue | V | S | G | F | L | M | Y | F | K | 2 | 1.00 |
| Hap_22 | Blue | V | - | S | L | L | M | F | L | K | 1 | 0.50 |
| Hap_23 | Green | V | S | G | L | L | T | Y | F | K | 1 | 0.50 |
| Hap_24 | Blue | V | - | G | L | L | M | Y | L | K | 1 | 0.50 |
| Hap_25 | Blue | V | S | S | F | L | M | Y | L | K | 1 | 0.50 |
| Hap_26 | Green | V | S | G | L | L | M | - | F | K | 1 | 0.50 |
| Hap_27 | Red | V | R | S | L | L | M | Y | F | - | 1 | 0.50 |
| Hap_28 | Red | V | - | G | L | L | M | Y | F | K | 1 | 0.50 |
| Hap_29 | Red | V | R | G | L | L | M | F | F | N | 1 | 0.50 |
| Hap_30 | Blue | V | S | G | L | L | M | F | F | K | 1 | 0.50 |
| Allele frequency (≥ 5% ) | 9.95 | 22.89 | 17.41 | 9.45 | 89.55 | 98.01 | 16.42 | 34.83 | 8.96 | 201 | ||
Hap_16 is Sal I. The assignment of each haplotype to one of the three clusters (colored as blue, green and red) is indicated.
We used 201 Pvmdr1 sequences from 13 countries to assess its global genetic diversity. These sequences contained 46 polymorphic sites, comprising 14 synonymous and 30 nonsynonymous substitutions. All the nonsynonymous mutations were located in TM1, TM3-5, NBD1, TM7-11 and NBD2. Sixteen of those polymorphic sites were parsimony-informative. The nucleotide diversity was 0.00069 ± 0.00004 in global isolates (Table 2). When the analysis was conducted according to the countries, the number of parsimony-informative sites in the Thai population (0.00085) was higher than the rest of countries, and this finding was also consistent with the haplotype number and the degree of haplotype diversity. Sliding window analysis showed that the highest peak was at the nucleotide positions 3,150-3,233 (TM11), coinciding with the second highest peak of the Thai isolates (Fig. 2). Out of the 9 global SNPs with ≥ 5% frequency, 7 were found in the 88 Thai isolates from the current study (Table 1). Based on these 9 SNPs, there were 30 haplotypes distributed across all the continents (Table 3, Supplementary Table S2). Most of them were country-shared haplotypes with a frequency of 1-18% of total isolates/haplotypes. Ten out of the 30 haplotypes (with frequency ≤ 1% of total isolates) were country-specific: Hap_13 for Madagascar; Hap_22-30 for China, Peru, India, North Korea and Thailand, respectively. Four haplotypes were from South America: Hap_3, Hap_16, Hap_23, and Hap_26. Eleven haplotypes were shared by countries or continents; Hap_1 and Hap_4 between Southeast Asia (SEA: Thailand) and the South America, Hap_6 between South America and Africa, Hap_7 and Hap_18 between Thailand and Africa, Hap_12 and Hap_14-15 between Thailand and China. Hap_8 was shared by four subcontinents: SEA (Thailand), South Asia (India), East Asia (China), and the Southwestern Pacific (Papua New Guinea, PNG). Similarly, Hap_9 was shared by three continents: SEA, Africa and the PNG. Seven haplotypes were shared between the western Thailand and the others, but none was shared with the eastern Thai parasite population.
3.2. Departure from neutrality and selection
In order to see whether the Thai Pvmdr1 gene follows the neutral equilibrium model of molecular evolution, Tajima D, as well as Fu & Li’s F* and D* were computed for the full-length Pvmdr1 gene, which all yielded negative values in the Thai populations (Table 4). Only Fu & Li’s F* and D* were statistically deviating from zero (Table 4). Similarly, in the worldwide populations, all the neutrality tests yielded negative values and they were all statistically deviating from zero (Table 4). These findings suggest an excess of low-frequency polymorphisms within the Thai populations as well as the global populations, possibly as a result of directional selection or population expansion. The dN-dS statistic generated by the Z-test was also negative in the total populations, signifying purifying selection on Pvmdr1, albeit the level of significance was not reached (Table 4). To zoom in on particular regions of the gene, sliding window analysis was performed for Tajima’s D (Fig. 2), which showed insignificant positive values at TM11 among the global sequences. When the Thai isolates were analyzed alone, two peaks higher than 1 were observed for both Tajima’s D and Fu & Li’s F*: The first peak was within NBD1, whereas the second peak of significant positive Tajima’s D value was observed at the nucleotide positions 3098-3272 (TM11 encompassing the F1076L mutation). TM11 also matched the highest value of nucleotide and haplotype diversity when partitioning the gene into TM domains (Supplementary Table S3). Interestingly, two codon-based tests of selection revealed that the mutations S513R and F1076L were under positive selection and a few others sites under negative selection (Supplementary Table S4).
Table 4.
Neutrality and recombination tests of the near full-length Pvmdr1 gene in different populations.
| Population | No. isolates | Neutrality tests | Rm rates | Rm events | |||
|---|---|---|---|---|---|---|---|
| Fu & Li’s F | Fu & Li’s D | Tajima’s D | Z test | ρ/θ ratios | |||
| Thailand (this study) | 88 | −3.290** | −3.548** | −1.486 | −1.464 | 3.301 | 6 |
| Tak | 22 | −1-114 | −0.950 | −0.973 | −2.624 | 29.927 | 3 |
| Kanchanaburi | 36 | −2.153 | −2.057 | −1.442 | −0.755 | 3.914 | 3 |
| Ubon Ratchathani | 30 | −0.962 | −1.000 | −0.426 | 0.155* | 1362.087 | 5 |
| Thailand (plasmodb) | 13 | −1.704 | −1.610 | −1.182 | −1610 | 52.094 | 0 |
| China | 7 | −1.168 | −1.053 | −1.109 | −0.653 | 0.080 | 0 |
| PNG | 7 | 1.101 | −0.953 | 1.342 | 1.034 | na | 0 |
| India | 2 | na | na | na | −1.562 | na | 0 |
| Madagascar | 8 | −1.056 | −0.961 | −0.916 | −0.077 | 2.132 | 1 |
| Mexico | 15 | −2.345 | −2.120 | −1.849* | −1.187 | 0.059 | 0 |
| Peru | 30 | −1.381 | −1.211 | −1.118 | −1.565 | 43.900 | 1 |
| Brazil | 5 | −1.113 | −1.094 | −1.094 | −0.542 | 3386.638 | 0 |
| Colombia | 22 | −4.107** | −3.997** | −2.412# | −1.931 | 3.230 | 2 |
| Others | 4 | −0.488 | −0.492 | −0.492 | 0.022* | na | 0 |
| Global | 201 | −5.474** | −6.403** | −2.146** | −1.298 | 1.756 | 6 |
p < 0.01
p < 0.02
p < 0.05
estimated recombination rate; Rm; recombination
estimated mutation rate; na, not analyzed; Other: El Salvador, Mauritania, North Korea, and Vietnam
3.3. Recombination
As recombination can break the linkage, several methods were used to detect the number of recombination events and the locations of breakpoints in the genome. Using seven recombination detection algorithms within the RDP4 software, we only detected one recombination breakpoint by the MaxChi algorithm in the global isolates (Supplementary Fig. S2). For country-wise sequences, DnaSP detected six recombination events in Thailand, two in Colombia and one each in Peru and Madagascar (Table 4), while three methods in the RDP package (BootScan, MaxChi and 3Seq1) only detected one recombination event. The country-wise recombination signal, measured by using the estimated recombination (ρ)/estimated mutation rates (θ) ratio, showed that the ratio in most countries were >1 (Table 4).
3.4. Genetic substructure
The global population genetic structure using the 9 major SNPs (with minor allele frequencies of ≥ 5%) showed optimal clustering at K = 3 (Fig. 3), which are colored as green, blue and red. Whereas all parasite populations had the M908L and T958M mutations at high frequencies, the three clusters also featured discordant, additional mutations (Supplementary Fig. S3). The green cluster represented parasites from South America with prevalent mutations at V221L (Fig. 1). The red cluster containing parasites from China and western Thailand included mutations of S513R, G698S, and K1393N. The blue cluster contained S513R, G698S, L845F, Y976F and F1076L, and it was of multiple origins mostly found in eastern Thailand, Madagascar, and PNG. The expected heterozygosity (HE) corresponding to these clusters was 0.027 (green), 0.048 (red), and 0.063 (blue), respectively (Supplementary Fig. S3). Phylogenetic analysis based on the Neighbor-Joining method among the global parasite isolates also showed a clear lineage separation consistent with the three population clusters (Fig. 4). For the majority of the Thai’s isolates, the red and the blue clusters were also clearly separated.
Fig. 3. Structure analysis of the global Pvmdr1 SNPs by the admixture model.
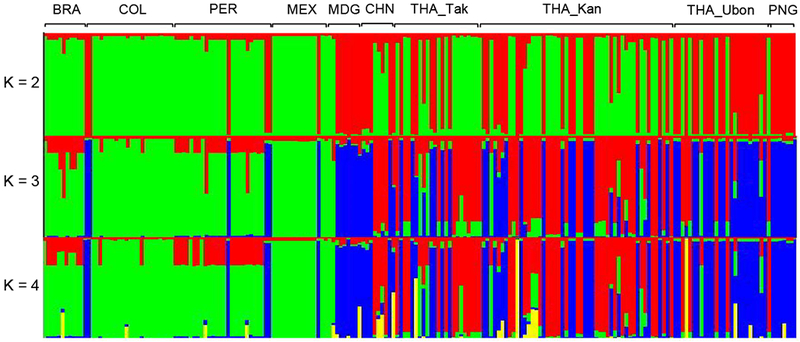
Plots representative of the genetic structure of Pvmdr1 sequences in different countries and regions at K = 2, 3, and 4 respectively. Country codes include of THA_Tak: Tak, THA_Kan: Kanchanaburi, THA_Ubon: Ubon Ratchathani, MEX: Mexico, COL: Colombia, PER: Peru, BRA: Brazil, MDG: Madagascar, CHN: China, and PNG: Papua New Guinea.
Fig. 4. Phylogenetic analysis of Pvmdr1 gene in worldwide isolates.
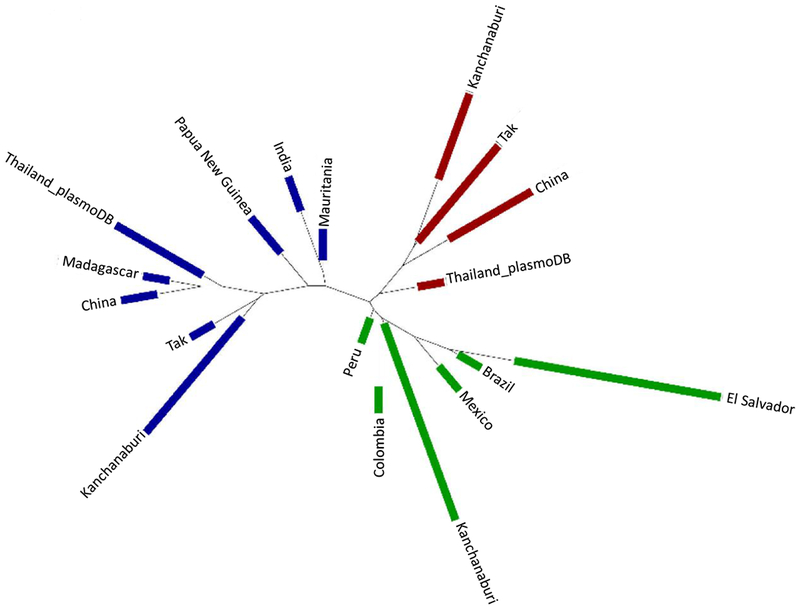
The distribution of the three clusters of Pvmdr1 haplotypes based on the Neighbor-Joining phylogenetic tree is shown. Clusters 1, 2 and 3 are colored in red, green and blue, respectively.
Moderate to high Fst values (0.15-0.76) were observed among the populations from different countries, indicating genetic differentiation of P. vivax populations. Fst showed a high degree of genetic difference among clusters (Fst > 0.25), which was 0.37 between the red and the green clusters, 0.43 between the blue and the green clusters, and 0.41 between the red and the blue clusters. Within Thailand, no significant genetic differentiation was detected between the two western sites, Tak and Kanchanaburi (Fst =0.0283). In contrast, the parasites from eastern Thailand was moderately different from Tak (Fst = 0.2009) and Kanchanaburi (Fst = 0.1505). The result was consistent with the genetic substructure described above.
LD was found in the isolates from Ubon Ratchathani, but not form Tak and Kanchanaburi (Table 6). Likewise, the global isolates displayed limited LD except for those from Brazil and PNG. The low LD might be due to the presence of recombination and could suggest a population expansion/isolation.
Table 6.
Linkage disequilibrium (LD) of the near full-length Pvmdr1 gene in the global isolates.
| Origin | IAS | p value | LD |
|---|---|---|---|
| Thailand (this study) | 0.0043 | 9.16 × 10−03 | Significant |
| Ubon Ratchathani | 0.0072 | 9.32 × 10−03 | Significant |
| Tak | 0.0006 | 3.94 × 10−01 | NS |
| Kanchanaburi | 0.0044 | 5.61 × 10−02 | NS |
| Thailand (plasmodb) | −0.0015 | 6.20 × 10−01 | NS |
| China | 0.0205 | 2.83 × 10−02 | Significant |
| India | NA | 1.00 × 10−05 | Significant |
| Madagascar | 0.0302 | 3.03 × 10−03 | Significant |
| PNG | 0.0008 | 1.00e + 00 | NS |
| Mexico | 0.0443 | 3.00 × 10−05 | Significant |
| Columbia | 0.0128 | 4.20 × 10−04 | Significant |
| Peru | 0.0349 | <1.00 × 10−03 | Significant |
| Brazil | −0.0005 | 8.08 × 10−01 | NS |
Significant LD at p < 0.05 based on 100,000 Monte Carlo Markov chain re-samplings; NA, not analyzed because of the small sample size; NS, not significant.
4. Discussion
Although malaria incidence has significantly declined over the past decade, the problem of drug resistance for both P. falciparum and P. vivax poses a major challenge in malaria control and elimination. The P-glycoprotein encoded by mdr1, an ABC transporter protein, plays an important role in multidrug detoxification in various organisms including the malarial parasites. In P. vivax, though genetic evidence is lacking, SNPs as well as gene duplication of Pvmdr1 are associated with resistance to CQ (Lu et al., 2011; Suwanarusk et al., 2008). This study detected 12 of 34 SNPs in Pvmdr1 occurring at high frequency (≥ 5%) in P. vivax populations in Thailand, among which seven were nonsynonymous. Similar to what has been found in world P. vivax populations, the number of Pvmdr1 SNPs in different Thai parasite populations also varied. Seven SNPs were consistent with those reported from Tak Province (Rungsihirunrat et al., 2015), but different from those reported from Laos (including N133K, S139S and E1261K) (Imwong et al., 2008). Among the SNPs found in this study, one novel SNP (L845F) was found in the eastern province Ubon Ratchathani, whereas another novel SNP (K1393N) was found in Kanchanaburi (Barnadas et al., 2008; Orjuela-Sanchez et al., 2009; Rungsihirunrat et al., 2015). Widespread SNPs such as T958M, Y976F and F1076L, associated with CQR in P. vivax were used in epidemiological surveillance worldwide (Huang et al., 2014; Lu et al., 2011). In previous studies, Pvmdr1 SNPs and gene duplication events (~9%) were reported from countries of the GMS, including Thailand, Myanmar and Laos, albeit the sample sizes are small (Imwong et al., 2008; Lu et al., 2011). High frequency (>62%) F1076L plus gene duplication of the Pvmdr1 was reported in western Thailand, whereas Cambodian parasite isolates near eastern Thailand harbored high frequency Y976F but no gene amplification (Lin et al., 2013). In this study, we did not investigate pvmdr1 gene amplification, and thus some sequences used for analysis could be from different copies of the gene in case of duplicated Pvmdr1. Given that pvmdr1 duplication was mostly associated with mefloquine use and the history of this event was relatively short (Khim et al., 2014), the pvmdr1 gene copies should not have diverged much, which should not have a major effect on our analysis. Findings from in vitro drug susceptibility studies in P. vivax vary (de Santana Filho et al., 2007; Lu et al., 2011; Orjuela-Sanchez et al., 2009; Rungsihirunrat et al., 2015; Shalini et al., 2014), and there is no clear association between any SNP or increased gene copy number and reduced antimalarial drug susceptibility. How P. vivax responds to the antimalarial drug pressure remains controversial (Barnadas et al., 2008; Lu et al., 2011; Orjuela-Sánchez P et al., 2009). This could be studied by performing co-evolution analysis of Pfmdr1 gene in P. vivax/P. falciparum co-endemic regions, given mixed-species infections are frequent and collateral selection can happen.
Our study aimed to analyze the extent of sequence diversity of and potential selection on the Pvmdr1 gene from Thai P. vivax populations. Similar to what was found for the global P. vivax populations, low nucleotide diversity (0.00069 ± 0.00004) was found in Thailand, suggesting the presence of functional/structural constraint on this gene. however, sliding window analysis of Tajima’s D, Fu & Li’s D* and Fu & Li’s F* observed positive values in two domains NBD1 (S513R) and TM11 (F1076L) of Pvmdr1 among the Thai isolates, but only in one domain (TM11) in the global isolates (Fig. 2). This latter result might signify that these two domains of the Pvmdr1 protein might have experienced a balancing selection in Thai populations or a reduction of population size. The multidrug transporter protein is predicted to function by coupling ATP hydrolysis in NBDs to the conformation change in the TM domains leading to extrusion of foreign compounds (Dawson and Locher, 2006). Mutations in both NBDs and TM may therefore negatively impact the function of the transporter protein more than the F1076L alone (Lu et al., 2011; Shalini et al., 2014). Significant signature of selection on Pvmdr1 has not been reported to date, which might be correlated with the unique biology of the parasite such as early gametocyte production before antimalarial drug treatment (Koepfli et al., 2011). However, in the present study, the mutations S513R and F1076L were shown to be under positive selection with two codon-based tests of selection. This observation might be justified since selection often targets specific amino acids instead of the whole protein, which sometimes is masked by purifying selection also acting on the gene (Koepfli et al., 2011).
Two population genomics studies of the global P. vivax populations divided the New and Old World populations as well as detected parasite population substructures according to countries (Hupalo et al., 2016; Pearson et al., 2016). Interestingly, two population clusters in western and eastern Thailand with little gene flow were identified which are separated by the central, malaria-free region. Fst and genetic substructure results both support such a notion. An earlier study identified contrasting genetic structure between Asian and South American populations, and also revealed substantial population differentiation between populations from Thailand and Laos (Imwong et al., 2007). The significant population differentiation between the eastern and western Thai P. vivax populations is similar to this finding. Strong LD within the eastern populations suggested parasite expansion or bottleneck events, which was not found in the two western populations. Furthermore, the eastern parasite population did not share any haplotypes with other parts of the world, further indicating that parasite population from this area might have undergone a unique evolutionary process. The predicted population expansion and bottleneck events are consistent with the malaria outbreaks detected in the eastern province in recent years. Phylogenetic analysis showed three major clusters of haplotypes, where both population-specific and shared haplotypes were observed. It would be interesting to test whether these clusters are correlated with different sensitivities to CQ. It is noteworthy that CQ resistance has only been sporadically reported in the GMS (Myat Phone et al., 1993), where CQ and PQ remain as the standard treatment for uncomplicated P. vivax infections. Thus, the reasons for the differences in the Pvmdr1 gene mutations and their prevalence in the GMS could be related to the epidemiology of sympatric P. falciparum parasites, which are often present in mixed parasite species infections and are treated differently among endemic countries. In addition, the different distribution of major malaria vectors also may contribute to the differentiation of parasites in the eastern and western provinces. Furthermore, different immunity of human hosts may also account for the differences in mutation patterns and their prevalence.
In summary, this study revealed action of directional selection in the evolutionary course of Pvmdr1 gene in Thai P. vivax populations. Particular domains of the gene showing high genetic diversity were found to be under balancing selection or subject to a population size decline. The eastern-western division pattern of the P. vivax parasite populations based on the Pvmdr1 SNPs is consistent with the spread of both Pfmdr1 (Veiga et al., 2016) and K13 propeller mutations in P. falciparum (Menard et al., 2016; Talundzic et al., 2015). Therefore, in addition to the selection from CQ-PQ treatment, we speculate that P. vivax might have been under drug selection pressure from treatment of mixed P. falciparum infections. Obviously, in an area with complex interplays among the multiple vector species, human host and co-endemic P. vivax and P. falciparum, the evolution of drug resistance is also influenced by the multitude of factors. Regardless of the population subdivision between eastern and western Thailand, much higher numbers of recombination events were detected in the Thai P. vivax population, which suggests the presence of a relatively large parasite population despite intensified control efforts in recent years. This is in contrast to the rapidly shrinking P. falciparum population in this region, and highlights that effective targeting the P. vivax reservoir is essential to achieve the goal of malaria elimination in Thailand by 2024.
Supplementary Material
Supplementary Fig. S1. Predicted domain organization of Pvmdr1 and relative positions of the SNPs. Orange bars indicate transmembrane (TM) domains. Red and blue bars indicate loops separating TM domains. SNPs in red (S513R and F1076L) are the positions under positive selection. Model was based on the P-glycoprotein structure PDB:4Q9H.
Supplemental Fig. S2. Recombination events. One significant recombination breakpoint (p<0.05) was detected by the MaxChi algorithm in RDP4 package (Rec). Green bar represents the major parent, blue bars the minor parent, and red bars the recombinant. The breakpoint positions (highlighted by the arrow heads) were compared to that of Sal I reference strain.
Supplementary Fig. S3. Structure plots and SNPs prevalence (A) Genetic structure of global P. vivax populations represented by different numbers of clusters. (B) The prevalence of each of the nine major SNPs in each cluster.
Table 5.
Genetic differentiation of parasite populations based on Pvmdr1 gene in the global isolates.
| Thailand1 | Peru | India | Brazil | China | Colombia | Mexico | PNG | Thailand2 | Madagascar | |
|---|---|---|---|---|---|---|---|---|---|---|
| Thailand1 | - | 0.00002 | NA | 0.00236 | 0.00018 | 0.00002 | 0.00002 | 0.00002 | 0.00002 | NA |
| Peru | 0.1535*** | - | NA | 0.01042 | 0.00002 | 0.00002 | 0.00002 | 0.00002 | 0.00002 | NA |
| India | 0.0982 | 0.4185 | - | NA | NA | NA | NA | NA | NA | NA |
| Brazil | 0.1968NS | 0.2490NS | 0.4683 | - | 0.00220 | 0.00009 | 0.00027 | 0.00116 | 0.00033 | NA |
| China | 0.2018** | 0.4259*** | 0.0906 | 0.3940NS | - | 0.00002 | 0.00004 | 0.00798 | 0.64489 | NA |
| Colombia | 0.2857*** | 0.2544*** | 0.5544 | 0.4291** | 0.5217*** | - | 0.00002 | 0.00002 | 0.00002 | NA |
| Mexico | 0.2568*** | 0.3875*** | 0.6481 | 0.5508* | 0.5514** | 0.5127*** | - | 0.00002 | 0.00002 | NA |
| PNG | 0.2761*** | 0.5934*** | 0.4426 | 0.7385NS | 0.2699NS | 0.6831*** | 0.7564*** | - | 0.01720 | NA |
| Thailand2 | 0.1983*** | 0.3982*** | 0.2923 | 0.4255* | −0.0301NS | 0.5103*** | 0.5376*** | 0.2736NS | - | NA |
| Madagascar | 0.2051 | 0.4976 | 0.0644 | 0.4335 | 0.1568 | 0.5658 | 0.5469 | 0.1662 | 0.2360 | - |
Country-wise comparison included Fst (below diagonal) and p-value (above diagonal). Thailand1 and Thailand2 are from this study and PlasmoDB, respectively. NS, not significant; NA, not analyzed.
Acknowledgement:
We would like to thank all the volunteers who participated in the research project as well as all the surveillance workers. This study was supported by grants from the Fogarty International Center (D43 TW006571) and the National Institute of Allergy and Infectious Diseases (U19 AI089672), National Institutes of Health, USA.
References
- Baird JK, Leksana B, Masbar S, Fryauff DJ, Sutanihardja MA, Suradi, Wignall FS, Hoffman SL, 1997. Diagnosis of resistance to chloroquine by Plasmodium vivax: timing of recurrence and whole blood chloroquine levels. The American journal of tropical medicine and hygiene 56, 621–626. [DOI] [PubMed] [Google Scholar]
- Barnadas C, Ratsimbasoa A, Tichit M, Bouchier C, Jahevitra M, Picot S, Menard D, 2008. Plasmodium vivax resistance to chloroquine in Madagascar: clinical efficacy and polymorphisms in pvmdr1 and pvcrt-o genes. Antimicrobial agents and chemotherapy 52, 4233–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berliner RW, Earle DP, Taggart JV, Zubrod CG, Welch WJ, Conan NJ, Bauman E, Scudder ST, Shannon JA, 1948. Studies on the Chemotherapy of the Human Malarias. Vi. The Physiological Disposition, Antimalarial Activity, and Toxicity of Several Derivatives of 4-Aminoquinoline. The Journal of clinical investigation 27, 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC, 2015. Annual Report, Malaria situation in Thailand Bureau of Vector-Borne Diseases, Department of disease control Nonthaburi, Thailand: ministry of public health., Bangkok, Thailand [Google Scholar]
- Chehuan YF, Costa MR, Costa JS, Alecrim MG, Nogueira F, Silveira H, Brasil LW, Melo GC, Monteiro WM, Lacerda MV, 2013. In vitro chloroquine resistance for Plasmodium vivax isolates from the Western Brazilian Amazon. Malaria journal 12, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DI, Jeong S, Dinzouna-Boutamba SD, Yang HW, Yeo SG, Hong Y, Goo YK, 2015. Evaluation of single nucleotide polymorphisms of pvmdr1 and microsatellite genotype in Plasmodium vivax isolates from Republic of Korea military personnel. Malaria journal 14, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congpuon K, Satimai W, Sujariyakul A, Intanakom S, Harnpitakpong W, Pranuth Y, Cholpol S, Bualombai P, 2011. In vivo sensitivity monitoring of chloroquine for the treatment of uncomplicated vivax malaria in four bordered provinces of Thailand during 2009-2010. Journal of vector borne diseases 48, 190–196. [PubMed] [Google Scholar]
- Cooper RD, 1994. Studies of a chloroquine-resistant strain of Plasmodium vivax from Papua New Guinea in Aotus and Anopheles farauti s.l. The Journal of parasitology 80, 789–795. [PubMed] [Google Scholar]
- Cui L, Mascorro CN, Fan Q, Rzomp KA, Khuntirat B, Zhou G, Chen H, Yan G, Sattabongkot J, 2003. Genetic diversity and multiple infections of Plasmodium vivax malaria in Western Thailand. The American journal of tropical medicine and hygiene 68, 613–619. [DOI] [PubMed] [Google Scholar]
- Dawson RJ, Locher KP, 2006. Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185. [DOI] [PubMed] [Google Scholar]
- de Santana Filho FS, Arcanjo AR, Chehuan YM, Costa MR, Martinez-Espinosa FE, Vieira JL, Barbosa M, Alecrim WD, Alecrim M, 2007. Chloroquine-resistant Plasmodium vivax, Brazilian Amazon. Emerging infectious diseases 13, 1125–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular ecology 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Fernandez-Becerra C, Pinazo MJ, Gonzalez A, Alonso PL, del Portillo HA, Gascon J, 2009. Increased expression levels of the pvcrt-o and pvmdr1 genes in a patient with severe Plasmodium vivax malaria. Malaria journal 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carrico JA, 2012. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC bioinformatics 13, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet J, 1995. FSTAT (Version 1.2): A Computer Program to Calculate F-Statistics. Journal of Heredity 86, 485–486. [Google Scholar]
- Hall TA, 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids. Symp. Ser. 41, 95–98. [Google Scholar]
- Hamedi Y, Sharifi-Sarasiabi K, Dehghan F, Safari R, To S, Handayuni I, Trimarsanto H, Price RN, Auburn S, 2016. Molecular Epidemiology of P. vivax in Iran: High Diversity and Complex Sub-Structure Using Neutral Markers, but No Evidence of Y976F Mutation at pvmdr1. PloS one 11, e0166124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubold B, Hudson RR, 2000. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage Analysis. Bioinformatics 16, 847–848. [DOI] [PubMed] [Google Scholar]
- Hoekenga MT, 1952. Treatment of malaria with a single dose of amodiaquine or chloroquine. Journal of the American Medical Association 149, 1369–1371. [DOI] [PubMed] [Google Scholar]
- Htun MW, Mon NCN, Aye KM, Hlaing CM, Kyaw MP, Handayuni I, Trimarsanto H, Bustos D, Ringwald P, Price RN, Auburn S, Thriemer K, 2017. Chloroquine efficacy for Plasmodium vivax in Myanmar in populations with high genetic diversity and moderate parasite gene flow. Malaria journal 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Huang S, Su XZ, Tong X, Yan J, Li H, Lu F, 2014. Molecular surveillance of pvdhfr, pvdhps, and pvmdr-1 mutations in Plasmodium vivax isolates from Yunnan and Anhui provinces of China. Malaria journal 13, 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, Vallejo AF, Herrera S, Arevalo-Herrera M, Fan Q, Wang Y, Cui L, Lucas CM, Durand S, Sanchez JF, Baldeviano GC, Lescano AG, Laman M, Barnadas C, Barry A, Mueller I, Kazura JW, Eapen A, Kanagaraj D, Valecha N, Ferreira MU, Roobsoong W, Nguitragool W, Sattabonkot J, Gamboa D, Kosek M, Vinetz JM, Gonzalez-Ceron L, Birren BW, Neafsey DE, Carlton JM, 2016. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nature genetics 48, 953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imwong M, Nair S, Pukrittayakamee S, Sudimack D, Williams JT, Mayxay M, Newton PN, Kim JR, Nandy A, Osorio L, Carlton JM, White NJ, Day NP, Anderson TJ, 2007. Contrasting genetic structure in Plasmodium vivax populations from Asia and South America. International journal for parasitology 37, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Imwong M, Pukrittayakamee S, Pongtavornpinyo W, Nakeesathit S, Nair S, Newton P, Nosten F, Anderson TJ, Dondorp A, Day NP, White NJ, 2008. Gene amplification of the multidrug resistance 1 gene of Plasmodium vivax isolates from Thailand, Laos, and Myanmar. Antimicrobial agents and chemotherapy 52, 2657–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketema T, Getahun K, Bacha K, 2011. Therapeutic efficacy of chloroquine for treatment of Plasmodium vivax malaria cases in Halaba district, South Ethiopia. Parasites & vectors 4, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khim N, Andrianaranjaka V, Popovici J, Kim S, Ratsimbasoa A, Benedet C, Barnadas C, Durand R, Thellier M, Legrand E, Musset L, Menegon M, Severini C, Nour BY, Tichit M, Bouchier C, Mercereau-Puijalon O, Menard D, 2014. Effects of mefloquine use on Plasmodium vivax multidrug resistance. Emerging infectious diseases 20, 1637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittichai V, Koepfli C, Nguitragool W, Sattabongkot J, Cui L, 2017. Substantial population structure of Plasmodium vivax in Thailand facilitates identification of the sources of residual transmission. PLoS neglected tropical diseases 11, e0005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepfli C, Ross A, Kiniboro B, Smith TA, Zimmerman PA, Siba P, Mueller I, Felger I, 2011. Multiplicity and diversity of Plasmodium vivax infections in a highly endemic region in Papua New Guinea. PLoS neglected tropical diseases 5, e1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K, 2016. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Molecular biology and evolution 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J, 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. [DOI] [PubMed] [Google Scholar]
- Lin JT, Patel JC, Kharabora O, Sattabongkot J, Muth S, Ubalee R, Schuster AL, Rogers WO, Wongsrichanalai C, Juliano JJ, 2013. Plasmodium vivax isolates from Cambodia and Thailand show high genetic complexity and distinct patterns of P. vivax multidrug resistance gene 1 (pvmdr1) polymorphisms. The American journal of tropical medicine and hygiene 88, 1116–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Yang HL, Tang LH, Li XL, Huang F, Wang JZ, Li CF, Wang HY, Nie RH, Guo XR, Lin YX, Li M, Xu JW, 2014. Monitoring Plasmodium vivax chloroquine sensitivity along China-Myanmar border of Yunnan Province, China during 2008-2013. Malaria journal 13, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Lim CS, Nam DH, Kim K, Lin K, Kim TS, Lee HW, Chen JH, Wang Y, Sattabongkot J, Han ET, 2011. Genetic polymorphism in pvmdr1 and pvcrt-o genes in relation to in vitro drug susceptibility of Plasmodium vivax isolates from malaria-endemic countries. Acta tropica 117, 69–75. [DOI] [PubMed] [Google Scholar]
- Marfurt J, Mueller I, Sie A, Maku P, Goroti M, Reeder JC, Beck HP, Genton B, 2007. Low efficacy of amodiaquine or chloroquine plus sulfadoxine-pyrimethamine against Plasmodium falciparum and P. vivax malaria in Papua New Guinea. The American journal of tropical medicine and hygiene 77, 947–954. [PubMed] [Google Scholar]
- Marlar T, Myat Phone K, Aye Yu S, Khaing Khaing G, Ma S, Myint O, 1995. Development of resistance to chloroquine by Plasmodium vivax in Myanmar. Transactions of the Royal Society of Tropical Medicine and Hygiene 89, 307–308. [DOI] [PubMed] [Google Scholar]
- Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P, 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekonnen SK, Aseffa A, Berhe N, Teklehaymanot T, Clouse RM, Gebru T, Medhin G, Velavan TP, 2014. Return of chloroquine-sensitive Plasmodium falciparum parasites and emergence of chloroquine-resistant Plasmodium vivax in Ethiopia. Malaria journal 13, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo GC, Monteiro WM, Siqueira AM, Silva SR, Magalhaes BM, Alencar AC, Kuehn A, del Portillo HA, Fernandez-Becerra C, Lacerda MV, 2014. Expression levels of pvcrt-o and pvmdr-1 are associated with chloroquine resistance and severe Plasmodium vivax malaria in patients of the Brazilian Amazon. PloS one 9, e105922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, Rahim-Awab G, Barnadas C, Berry A, Boum Y, Bustos MD, Cao J, Chen JH, Collet L, Cui L, Thakur GD, Dieye A, Djalle D, Dorkenoo MA, Eboumbou-Moukoko CE, Espino FE, Fandeur T, Ferreira-da-Cruz MF, Fola AA, Fuehrer HP, Hassan AM, Herrera S, Hongvanthong B, Houze S, Ibrahim ML, Jahirul-Karim M, Jiang L, Kano S, Ali-Khan W, Khanthavong M, Kremsner PG, Lacerda M, Leang R, Leelawong M, Li M, Lin K, Mazarati JB, Menard S, Morlais I, Muhindo-Mavoko H, Musset L, Na-Bangchang K, Nambozi M, Niare K, Noedl H, Ouedraogo JB, Pillai DR, Pradines B, Quang-Phuc B, Ramharter M, Randrianarivelojosia M, Sattabongkot J, Sheikh-Omar A, Silue KD, Sirima SB, Sutherland C, Syafruddin D, Tahar R, Tang LH, Toure OA, Tshibangu-wa-Tshibangu P, Vigan-Womas I, Warsame M, Wini L, Zakeri S, Kim S, Eam R, Berne L, Khean C, Chy S, Ken M, Loch K, Canier L, Duru V, Legrand E, Barale JC, Stokes B, Straimer J, Witkowski B, Fidock DA, Rogier C, Ringwald P, Ariey F, Mercereau-Puijalon O, Consortium K, 2016. A Worldwide Map of Plasmodium falciparum K13-Propeller Polymorphisms. The New England journal of medicine 374, 2453–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myat Phone K, Myint O, Myint L, Thaw Z, Kyin Hla A, Nwe Nwe Y, 1993. Emergence of chloroquine-resistant Plasmodium vivax in Myanmar (Burma). Transactions of the Royal Society of Tropical Medicine and Hygiene 87, 687. [DOI] [PubMed] [Google Scholar]
- Orjuela-Sánchez P, de Santana Filho FS, Machado-Lima A, Chehuan YF, Costa MR, Alecrim Md HA, d.P., 2009. Analysis of single-nucleotide polymorphisms in the crt-o and mdr1 genes of Plasmodium vivax among chloroquine-resistant isolates from the Brazilian Amazon region. Antimicrobial agents and chemotherapy 53, 3561–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjuela-Sanchez P, de Santana Filho FS, Machado-Lima A, Chehuan YF, Costa MR, Alecrim M, del Portillo HA, 2009. Analysis of single-nucleotide polymorphisms in the crt-o and mdr1 genes of Plasmodium vivax among chloroquine-resistant isolates from the Brazilian Amazon region. Antimicrobial agents and chemotherapy 53, 3561–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RD, Amato R, Auburn S, Miotto O, Almagro-Garcia J, Amaratunga C, Suon S, Mao S, Noviyanti R, Trimarsanto H, Marfurt J, Anstey NM, William T, Boni MF, Dolecek C, Tran HT, White NJ, Michon P, Siba P, Tavul L, Harrison G, Barry A, Mueller I, Ferreira MU, Karunaweera N, Randrianarivelojosia M, Gao Q, Hubbart C, Hart L, Jeffery B, Drury E, Mead D, Kekre M, Campino S, Manske M, Cornelius VJ, MacInnis B, Rockett KA, Miles A, Rayner JC, Fairhurst RM, Nosten F, Price RN, Kwiatkowski DP, 2016. Genomic analysis of local variation and recent evolution in Plasmodium vivax. Nature genetics 48, 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RN, von Seidlein L, Valecha N, Nosten F, Baird JK, White NJ, 2014. Global extent of chloroquine-resistant Plasmodium vivax: a systematic review and meta-analysis. The Lancet. Infectious diseases 14, 982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P, 2000. Inference of population structure using multilocus genotype data. Genetics 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff A, Siswantoro H, Kenangalem E, Wuwung M, Brockman A, Edstein MD, Laihad F, Ebsworth EP, Anstey NM, Tjitra E, Price RN, 2007. Therapeutic response of multidrug-resistant Plasmodium falciparum and P. vivax to chloroquine and sulfadoxine-pyrimethamine in southern Papua, Indonesia. Transactions of the Royal Society of Tropical Medicine and Hygiene 101, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijken MJ, Boel ME, Russell B, Imwong M, Leimanis ML, Phyo AP, Muehlenbachs A, Lindegardh N, McGready R, Renia L, Snounou G, Singhasivanon P, Nosten F, 2011. Chloroquine resistant vivax malaria in a pregnant woman on the western border of Thailand. Malaria journal 10, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungsihirunrat K, Muhamad P, Chaijaroenkul W, Kuesap J, Na-Bangchang K, 2015. Plasmodium vivax drug resistance genes; Pvmdr1 and Pvcrt-o polymorphisms in relation to chloroquine sensitivity from a malaria endemic area of Thailand. The Korean journal of parasitology 53, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe ML, Ranjitkar S, Rajakaruna RS, Amerasinghe PH, Morales F, Pearce R, Ord R, Leslie T, Rowland M, Gadalla NB, Konradsen F, Bygbjerg IC, Roper C, Alifrangis M, 2015. Multiple Origins of Mutations in the mdr1 Gene--A Putative Marker of Chloroquine Resistance in P. vivax. PLoS neglected tropical diseases 9, e0004196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurkamp GJ, Spicer PE, Kereu RK, Bulungol PK, Rieckmann KH, 1992. Chloroquine-resistant Plasmodium vivax in Papua New Guinea. Transactions of the Royal Society of Tropical Medicine and Hygiene 86, 121–122. [DOI] [PubMed] [Google Scholar]
- Shalini S, Chaudhuri S, Sutton PL, Mishra N, Srivastava N, David JK, Ravindran KJ, Carlton JM, Eapen A, 2014. Chloroquine efficacy studies confirm drug susceptibility of Plasmodium vivax in Chennai, India. Malaria journal 13, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, 2000. Emergence of chloroquine-resistant vivax malaria in south Bihar (India). Transactions of the Royal Society of Tropical Medicine and Hygiene 94, 327. [PubMed] [Google Scholar]
- Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE, Thaithong S, Brown KN, 1993. High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Molecular and biochemical parasitology 61, 315–320. [DOI] [PubMed] [Google Scholar]
- Sriwichai P, Samung Y, Sumruayphol S, Kiattibutr K, Kumpitak C, Payakkapol A, Kaewkungwal J, Yan G, Cui L, Sattabongkot J, 2016. Natural human Plasmodium infections in major Anopheles mosquitoes in western Thailand. Parasites & vectors 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanarusk R, Chavchich M, Russell B, Jaidee A, Chalfein F, Barends M, Prasetyorini B, Kenangalem E, Piera KA, Lek-Uthai U, Anstey NM, Tjitra E, Nosten F, Cheng Q, Price RN, 2008. Amplification of pvmdr1 associated with multidrug-resistant Plasmodium vivax. The Journal of infectious diseases 198, 1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talundzic E, Okoth SA, Congpuong K, Plucinski MM, Morton L, Goldman IF, Kachur PS, Wongsrichanalai C, Satimai W, Barnwell JW, Udhayakumar V, 2015. Selection and spread of artemisinin-resistant alleles in Thailand prior to the global artemisinin resistance containment campaign. PLoS pathogens 11, e1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanh PV, Hong NV, Van NV, Louisa M, Baird K, Xa NX, Peeters Grietens K, Hung le X, Duong TT, Rosanas-Urgell A, Speybroeck N, D’Alessandro U, Erhart A, 2015. Confirmed Plasmodium vivax Resistance to Chloroquine in Central Vietnam. Antimicrobial agents and chemotherapy 59, 7411–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjitra E, Anstey NM, Sugiarto P, Warikar N, Kenangalem E, Karyana M, Lampah DA, Price RN, 2008. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS medicine 5, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga MI, Dhingra SK, Henrich PP, Straimer J, Gnadig N, Uhlemann AC, Martin RE, Lehane AM, Fidock DA, 2016. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nature communications 7, 11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Wang Y, Parker DM, Gupta B, Yang Z, Liu H, Fan Q, Cao Y, Xiao Y, Lee MC, Zhou G, Yan G, Baird JK, Cui L, 2015. Therapeutic responses of Plasmodium vivax malaria to chloroquine and primaquine treatment in northeastern Myanmar. Antimicrobial agents and chemotherapy 59, 1230–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1. Predicted domain organization of Pvmdr1 and relative positions of the SNPs. Orange bars indicate transmembrane (TM) domains. Red and blue bars indicate loops separating TM domains. SNPs in red (S513R and F1076L) are the positions under positive selection. Model was based on the P-glycoprotein structure PDB:4Q9H.
Supplemental Fig. S2. Recombination events. One significant recombination breakpoint (p<0.05) was detected by the MaxChi algorithm in RDP4 package (Rec). Green bar represents the major parent, blue bars the minor parent, and red bars the recombinant. The breakpoint positions (highlighted by the arrow heads) were compared to that of Sal I reference strain.
Supplementary Fig. S3. Structure plots and SNPs prevalence (A) Genetic structure of global P. vivax populations represented by different numbers of clusters. (B) The prevalence of each of the nine major SNPs in each cluster.