

Abstract
Cellular retinol-binding proteins (CRBPs) facilitate the uptake and intracellular transport of vitamin A. They integrate retinoid metabolism, playing an important role in regulating the synthesis of bioactive vitamin A metabolites. Thus, CRBPs constitute potential pharmacological targets to modulate cellular retinoid status that in turn may have applications in the treatment of certain immunological, metabolic, and ocular disorders. Here we identify abnormal cannabidiol (abn-CBD) as a nonretinoid inhibitor of cellular retinol-binding protein 1 (CRBP1). X-ray crystal structures of CRBP1 in complex with abn-CBD and its derivatives revealed a distinctive mode of protein–ligand interaction and provided a molecular basis for the high affinity and selectivity of this compound. We demonstrated that abn-CBD modulates the flux of retinoids via the retinoid cycle in vivo. Furthermore, the biological activity of abn-CBD was evidenced by its ability to protect against light-induced retinal damage in Balb/cJ mice. Altogether, our findings indicate that targeting selected CRBPs with a small-molecule inhibitor can potentially lead to the development of new therapeutic agents to counteract diseases with etiologies involving imbalance in retinoid metabolism or signaling.
Graphical Abstract
INTRODUCTION
Vitamin A (all-trans-retinol, atROL) is an essential micronutrient that exerts biological activities via its metabolites. The oxidized form of atROL, all-trans-retinoic acid, is a ligand for three nuclear receptors (RARα, β, and γ), which bind to retinoic acid response elements on the DNA of target genes regulating their transcription.1–4 Vitamin A is also a precursor for 11-cis-retinal.5,6 This chromophore binds to rod and cone opsins to form the photosensitive visual pigments necessary for vision.7–9 Thus, understanding of the enzymatic pathways that lead to the formation of vitamin A metabolites is crucial for comprehending the physiological functions of retinoids. It also opens exciting avenues for pharmacological intervention in the processes governed by these bioactive compounds.
Decades of intensive studies led to the identification of the key protein components involved in the production of all-trans-retinoic acid and the regeneration of visual chromophore via the retinoid (visual) cycle. Nevertheless, this knowledge has not been fully explored for potential therapeutic interventions. The main limitation in targeting enzymes involved in retinoid metabolism was the inability to find potent and selective small-molecule modulators of their enzymatic activities. The examples of such modulators are competitive inhibitors of the retinoid isomerase RPE65 (retinylamine, emixustat, and their derivatives10–12), which protect against light-induced retinal degeneration and arrest the accumulation of retinaldehyde condensation products by slowing the regeneration of visual chromophore.13–15 However, retinoid metabolism does not depend solely on retinoid processing enzymes; it also involves several classes of retinoid-binding proteins represented by serum retinol-binding protein (RBP4), cellular retinol-binding proteins (CRBPs), cellular retinaldehyde-binding protein (CRALBP), and cellular retinoic acid-binding proteins (CRABPs).16,17 They play important roles in integrating vitamin A metabolism by sequestering hydrophobic retinoids, facilitating their transport across the aqueous milieu, and directing them to dedicated enzymes and receptors.18,19 Nevertheless, the pharmacological potential of these classes of carrier proteins in controlling the metabolic and physiological functions of retinoids has not been fully explored. The only known compounds that target retinoid-binding proteins are competitive inbibitors of RBP4, which includes 4-hydroxy-(phenyl)retinamide (fenretinide) and two nonretinoid-based experimental drugs A1120 and BPN-14136.20–22 They show efficacy in the treatment of certain types of cancer as well as slowing retinal degeneration in a mouse model of Stargardt disease.22–26 However, their cell and tissue-specific effects are achieved by significantly depleting the systemic level of circulating atROL, which leads to vitamin A deficiency in the peripheral tissues.20,22,27 Thus, the long-term administration of these drugs may put patients at risk for adverse health effects that are exemplified by pathogenic mutations in RBP4, including acne, night blindness, retinal dystrophies, or ocular developmental abnormalities in offspring.28–31
In our attempt to influence vitamin A metabolism for therapeutic benefit via pharmacologic methods, we hypothesized that targeting retinol carriers inside cells might represent a safer and more specific alternative than utilizing RBP4 inhibitors. There are four representatives of the CRBP protein family in the human genome (CRBP1–4).16 Although they are very similar to each other structurally, their affinities for atROL range between less than 1 nM for CRBP1 to 200 nM for CRBP4.32–34 CRBPs also differ in their relative abundance and tissue distribution, which may suggest diverse and cell-specific functions for these proteins.16 The most ubiquitous is CRBP1 (encoded by the RBP1 gene). Although present in numerous tissues, it is particularly abundant in hepatocytes and the retinal pigment epithelium (RPE), where it enhances intracellular vitamin A uptake.35–39 The role of CRBP1 in RPE cells is principally important. CRBP1 is directly involved in the regeneration of visual chromophore via the retinoid cycle by facilitating both the recycling of vitamin A from photoreceptor cells and its efficient esterification. Studies on CRBP1-deficient mice (Rbp1−/−) revealed a diminished amount of all-trans-retinyl esters in the RPE and the transient accumulation of atROL upon recovery from exposure to bright light.38 This physiological function of CRBP1 preordains this protein to become a potential pharmaceutical target for modulating the flux of retinoids via the visual cycle and, thus, offers an opportunity to manage the pathological processes associated with an imbalance in ocular retinoid homeostasis. Importantly, the deactivation of the Rbp1 gene does not cause spontaneous pathological changes in the murine retina.
To test this hypothesis, we developed a high-throughput screening (HTS) methodology that led to the identification of abnormal cannabidiol (abn-CBD) as a potent nonretinoid ligand of CRBP1. We delineated the mode of protein–ligand interaction at the atomic level by solving high-resolution X-ray structures of CRBP1 in complex with abn-CBD and its derivatives. We also examined the effects of abn-CBD on ocular vitamin A homeostasis in vivo and provided evidence that the systemic administration of this compound protects mouse retinas from light-induced damage. Thus, we discovered a first-in-class drug candidate that affects retinoid metabolism by targeting CRBPs
RESULTS
The Identification of abn-CBD as a Ligand for CRBP1.
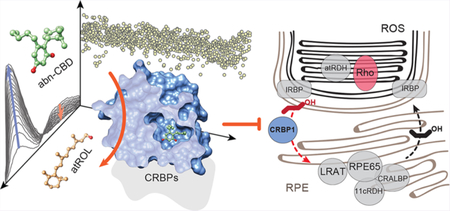
To search for compounds interacting with CRBP1, we developed an HTS strategy that minimalized experimental bias associated with investigating the binding of hydrophobic ligands to a protein specialized for interaction with small lipophilic molecules. It is based on the fluorescence properties of CRBP1 in complex with its natural ligand, atROL. The excitation of the protein scaffold by 285 nm light leads to a robust fluorescence resonance energy transfer (FRET) between the tryptophan residues and the retinoid moiety. Consequently, two maxima at 350 and 480 nm are observed in the CRBP1 fluorescence emission spectrum corresponding fluorescence emission from tryptophan residues and atROL, respectively. Upon the liberation of atROL or its replacement from the binding site by an alternative molecule, the efficiency of FRET decreases, leading to a dramatic increase in the fluorescence signal at 350 nm accompanied by a proportional drop in the emission at 480 nm (Figure 1a,b). The signal intensities for apo-CRBP1 and CRBP1/atROL complex were used as the 0% and +100% change controls, respectively. The final assay quality was characterized by a Z′-factor of 0.61, a signal-to-background ratio of 14.3, and a coefficient of variation of 10.5%. The primary HTS was performed using 986 lipid compounds from the Bioactive Lipid I Screening (Cayman Chemicals) and Screen-Well Bioactive Lipid (Enzo Life Science) libraries. After the elimination of duplicated molecules and compounds with spectral properties interfering with the fluorescence assay, the HTS revealed a unique hit that corresponded to abnormal cannabidiol (abn-CBD; Figure 1c). This synthetic derivative of plant cannabidiol does not interact with cannabinoid receptor 1 or 2 and, thus, does not cause psychoactive effects.40
Figure 1. Biophysical principles and the results of the HTS for CRBP1 ligands.
(a) A schematic representation of the vitamin A-displacement assay. Replacement or liberation of atROL from holo-CRBP1 by an alternative nonretinoid ligand results in diminishing of FRET between the retinoid moiety and the protein scaffold. (b) Differences in the fluorescence emission spectra between CRBP1 in complex with atROL and the apo form of the protein were used as a readout in a high-throughput assay. (c) The primary screening of a chemical library composed of bioactive lipids revealed a single hit that corresponded to a synthetic derivative of cannabidiol, abn-CBD.
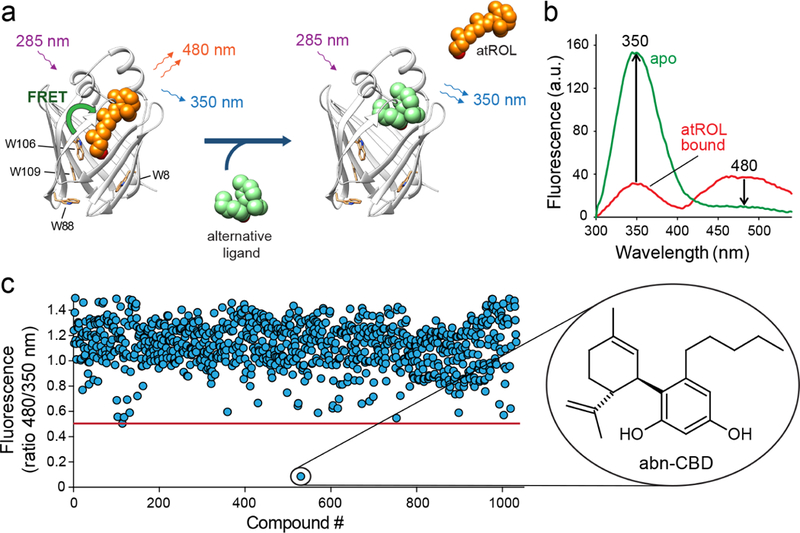
To determine the potency of abn-CBD interaction with CRBP1, we titrated CRBP1/atROL complex with increasing concentrations of the ligand (Figure 2a). Changes in the fluorescence of the protein or the retinoid moiety plotted as a function of abn-CBD concentration were best fitted with a one-site saturation-binding model. The value of the inhibition constant (Ki) was calculated to be ~67 nM. (Figure 2b, Supporting Information Table 1). In the reverse experiment, CRBP1 loaded with abn-CBD was titrated with atROL. In this setting, the Ki value for replacing abn-CBD by vitamin A was ~34 nM (Supporting Information Figure 1, Supporting Information Table 1), indicating that the affinity of abn-CBD for CRBP1 is comparable to the endogenous ligand.
Figure 2. Determination of the Ki values for abn-CBD.
(a) Fluorescence spectra of holo-CRBP1 upon titration with abn-CBD. (b) Changes in the emission maxima at 350 and 480 nm (inset) plotted vs concentration of abn-CBD were fitted to the one-site saturation ligand-binding model (Rsqr = 0.997 and 0.990 for the fluorescence signal at 350 and 480 nm, respectively) and used to calculate Ki values. The experiments were repeated three separate times, each time in duplicate. Data are presented as mean values ± sd.
Although suggestive, changes in the fluorescence of CRBP1 upon titration with a nonretinoid ligand do not provide information on whether the retinoid liberation is caused by a direct competition of abn-CBD for the vitamin A binding site. To validate the fluorescence data and further investigate the in teraction of abn-CBD with CRBP1, the protein prebound to atROL was incubated with the cannabinoid in a 1:2 molar ratio and repurified on an ion exchange column. The lipid composition carried by the protein at the end of the experiment was analyzed by liquid chromatography/mass spectrometry (LC/MS). Consistent with the fluorescence data, the incubation of holo-CRBP1 with abn-CBD resulted in diminished absorbance at 325 nm as compared to the control sample, indicating the loss of the retinoid (Figure 3a). To attest whether the removal of atROL was accompanied by the binding of abn-CBD, we extracted lipids associated with the repurified protein with hexane and examined their composition. As shown in Figure 3b, abn-CBD was readily detectable in the samples obtained from the incubation of apo- or holo-CRBP1 with this compound. Notably, the quantification of atROL and abn-CBD indicated the proportional replacement of the retinoid by the cannabinoid (Figure 3c). These biochemical data suggest that abn-CBD interacts tightly with CRBP1, causing the replacement of vitamin A from the protein’s binding site and, thus, acting as a competitive inhibitor of this carrier protein.
Figure 3. Biochemical evaluation of the interaction of CRBP1 with abn-CBD.
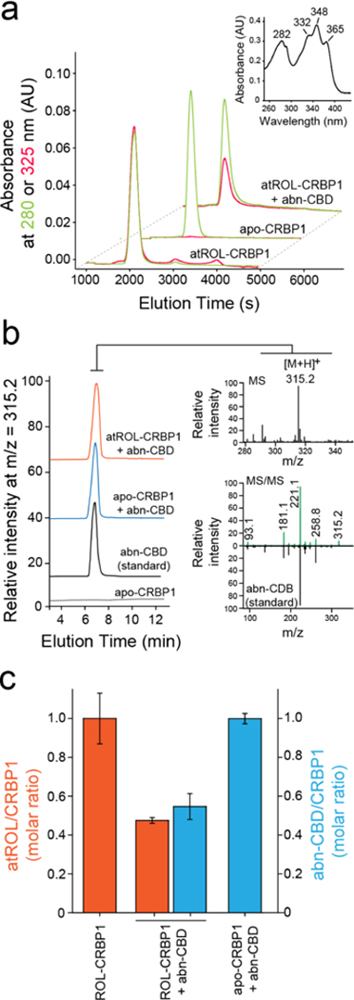
(a) Incubation of vitamin A-bound CRBP1 with abn-CBD led to depletion of atROL as evident by decreased absorption at 325 nm (red trace) in relation to the protein absorbance at 280 nm (green trace) in the repurified sample. (inset) UV/vis absorbance spectrum of atROL-bound CRBP1. (b) MS-based detection of abn-CBD in the CRBP1 fractions. The extracts of the protein samples preincubated abn-CBD and repurified revealed presence of intense ion peak at m/z = 315.2 [M + H]+. The molecular identification of this parent ion as corresponding to abn-CBD was achieved by comparing the MS/MS fragmentation pattern with a synthetic standard of the ligand. (c) Quantification of the ligand/protein ratios after incubation of vitamin A-bound CRBP1 with 2 molar excess of abn-CBD. The degree of atROL elimination from the protein is proportional to the amount of abn-CBD bound suggesting that these two ligands compete for the same binding site.
The Structure of CRBP1 in Complex with abn-CBD.
To obtain insight into the molecular aspect of the interaction between CRBP1 and its nonretinoid ligand, we crystallized this protein in the presence of abn-CBD and solved the complex structure at 1.17 Å resolution (PDB No. 6E5L; Supporting Information Table 2). The electron density of the ligand was readily observed and well-defined, allowing for the unambiguous modeling of abn-CBD molecules inside the binding pocket of the protein (Figure 4a,b). As predicted by the biochemical data, abn-CBD binds in the same binding cavity as atROL. Remarkably, the interaction of CRBP1 with the cannabinoid dramatically decreased the flexibility of the protein’s portal region (residues 24–36, 53–60, and 73–81) by inducing identical conformational changes as observed upon binding of vitamin A.33 Also, the averaged B-factor calculated for the main chain of the portal region dropped from 30.6 Å2 for apo-CRBP1 (PDB No. 5H9A) to 9.8 Å2 upon abn-CBD binding. This value is comparable to that observed for the protein in complex with atROL (10.5 Å2, PDB No. 5H8T).33 However, despite promoting identical conformational changes upon binding, the mode of interaction with the protein differs considerably for atROL and abn-CBD (Figure 4c,d). Although the comparison of the spatial position of these two ligands revealed overlap between the orientation of the β-ionone and the cyclohexene rings, the substituted aromatic ring of abn-CBD is positioned in a part of the binding cavity that is normally not occupied by the retinoid moiety. Importantly, this part of abn-CBD makes specific contacts by forming hydrogen bonds with the main and side chains of residues within the binding pocket (Figure 4b). The hydroxyl group in the ortho position (with respect to the cyclohexene ring) forms a single hydrogen bond with the carboxyl oxygen of the Ala33, whereas the para-hydroxyl is part of an extensive network of hydrogen bonds that involve the side chains of Asn13, Lys40, and Gln128 as well as at least four ordered water molecules. Lastly, the aliphatic chain of abn-CBD occupies a hydrophobic cavity enclosed by the side chain of Phe16, Tyr19, Leu20, Ile77, and Met119 that is common with the binding site of the polyene chain of vitamin A.
Figure 4. Crystal structure of CRBP1 in complex with abn-CBD.
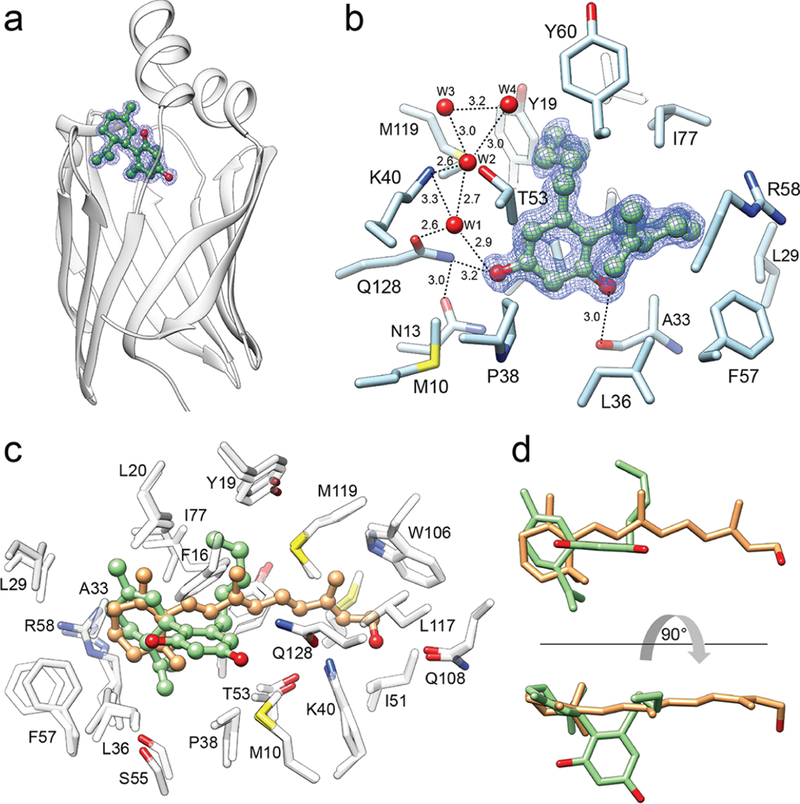
(a) Ribbon representation of the overlay structures of CRBP1 (PDB No. 6E5L). Position of abn-CBD is indicated by ball-and-stick model of the ligand. The blue mesh corresponds to the 2Fo − Fc electron density map contoured at 1.2σ. (b) Molecular organization of the abn-CBD binding pocket with selected residues present in the vicinity or interacting with the ligand. Ordered water molecules (W) are shown as red spheres; dashed lines indicate hydrogen bonds. Distances are shown in angstroms. (c) Superimposed structures of CRBP1 in complex with atROL (PDB No. 5HBS) or abn-CBD (PDB No. 6E5L). Spatial positions of amino acids within the binding site are nearly identical in both structures, suggesting that interaction with abn-CBD (colored green) provokes similar conformational changes in CRBP1 as observed for atROL (colored orange). (d) Comparison of the ligands’ positions within the binding pocket of CRBP1. The hydroxylated aromatic ring of abn-CBD (green) utilizes a part of the binding cavity that is not occupied by the retinoid moiety (orange).
The Correlation between the Ligands’ Structures and Their Binding Affinities.
The structural data identified two types of interactions contributing to abn-CBD binding: hydrophobic that involve the cyclohexene ring and the pentyl aliphatic chain (analogous to the β-ionone ring and the polyene chain of vitamin A) and the hydrogen bond formation by the hydroxyl groups of the ligand. To dissect the relative contributions of these two modes of interaction to the Ki value of this ligand, we examined three derivatives of abn-CBD: abn-CBDO (5-methyl-4-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene-1,3-diol), in which the pentyl chain was shortened to a methyl group; CBDO (5-methyl-2-[(6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene-1,3-diol), in which positions of hydroxyl and methyl groups were swapped as compared to abn-CBDO; and O-1918 (1,3-dimethoxy-5-methyl-2-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcy-clohex-2-en-1-yl]benzene), which cannot form hydrogen bonds due to the methylation of its hydroxyl groups (Figure 5a). The fluorescence atROL-replacing assay revealed major differences in the Ki values for these compounds. Compared with abn-CBD, shortening the aliphatic chain (represented by abn-CBDO) resulted in the apparent affinity being lowered by 4.3 times (Figure 5b, Supporting Information Table 1). Even more profound change was observed for CBDO. Switching positions of the para-hydroxyl and ortho-methyl groups caused a further 6.3-fold increase of the Ki value to ~1.7 μM (Figure 5c). Finally, the inability of O-1918 to form hydrogen bonds abolished the meaningful binding of this derivative in the 0–10 μM range (Supporting Information Figure 2a). Similarly, we did not observe limonene interacting (representing the cyclohexene portion of the ligand) with CRBP1 (Supporting Information Figure 2b). These data strongly suggest that, although abn-CBD competes for the same site with atROL, its high affinity is largely determined by the unique interactions inside the binding pocket that differ from those observed with vitamin A.
Figure 5. A relationship between structure of cannabinoid ligands and their affinity.
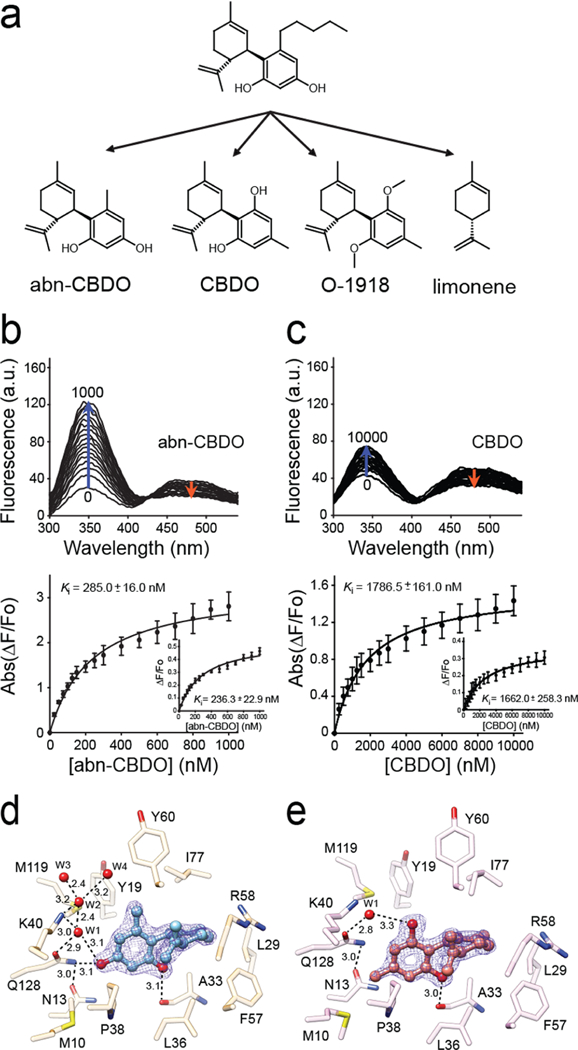
(a) Chemical structures of abn-CBD derivatives used in the experiments. (b, c) Changes in the fluorescence emission spectra upon titration with abn-CBDO and CBDO, respectively. Ki values were calculated by fitting the experimental data to the one-site saturation ligand-binding model. (d) Interactions of abn-CBDO inside the binding cavity of CRBP1 as revealed by the X-ray crystallography (PDB No. 6E5T). (e) Orientation of CBDO inside of the binding pocket (PDB No. 6E6M). The absence of a hydroxyl group in the para position in CBDO determines weaker interaction of this ligand with CRBP1. The 2Fo − Fc electron density maps were contoured at 1.4σ. Ordered water molecules (W) are shown as red spheres; dashed lines indicate hydrogen bonds. Distances are shown in angstroms.
To better understand the correlation between the affinity of the abn-CBD derivatives and their binding modes, we cocrystallized CRBP1 with abn-CBDO or CBDO and determined structures of these complexes at 1.55 Å resolution (Supporting Information Table 2, PDB No. 6E5T and PDB No. 6E6M, respectively). In both structures, the ligands were clearly identified based on their distinct residual electron densities (Figure 5d,e). The cyclohexene and aromatic moieties common to these compounds were found in essentially identical configurations inside the binding pocket. The only difference in binding modes between these two compounds was the extent to which they interact with the protein via hydrogen bonds. The absence of the para hydroxyl effectively eliminates the interaction of CBDO with the side chain of Gln128 and the ordered water molecules that were part of a larger network of hydrogen bonding seen in the abn-CBD and abn-CBDO complexes (Figure 4b and Figure 5d). This deficiency is neither compensated by the hydrogen bonds between the ortho hydroxyl and the main chain of Ala33 nor substituted by the involvement of the swapped hydroxyl group in a hydrogen bond with an isolated water molecule (Figure 5e). Thus, the affinity and specificity of the interaction between abn-CBD and CRBP1 is not determined by the similarities to the retinoid moiety but rather by the interactions of the hydroxyl substituents on the aromatic ring that are distinct from those observed for atROL.
The Interaction of abn-CBD with Other Members of the CRBP Protein Family.
High structural similarity between CRBPs raises the question whether abn-CBD interacts specifically with CRBP1 or if it also binds to other members of this protein family. Although the structures of human CRBPs revealed highly similar binding cavities, there are several amino acid substitutions that might contribute to the selectivity of this ligand. Comparing the binding sites of CRBP1 and CRBP2 revealed four amino acid substitutions in the vicinity of abn-CBD: Leu20/Met, Pro38/Gln, Gly76/Ser, and Ile77/Leu. They do not affect the architecture of the binding cavity with the exception of Pro38/Gln. The much larger side chain of glutamine present in CRBP2 protrudes into the binding pocket and occupies part of the space, so that it cannot accommodate abn-CBD, and thus effectively disables binding of this compound (Supporting Information Figure 3a). The absence of interaction between abn-CBD and CRBP2 was also evident in the fluorescence-binding assay (Supporting Information Figure 3b). Consequently, we were not able to obtain crystals of CRBP2 in complex with this ligand.
The differences in the binding pocket of CRBP3 and CRBP4 as compared to CRBP1 are less profound. They include substitutions of Phe16/Met and Ile77/Leu in CRBP3 and Leu20/Met, Phe57/Leu, and Ile77/Leu in CRBP4. As a result, these two proteins bind abn-CBD similarly to CRBP1. The crystal structures of CRBP3 and CRBP4 in complex with this ligand revealed that the orientation of abn-CBD within the binding pocket was nearly identical to that observed in CRBP1 (Supporting Information Figure 3b–e, Supporting Information Table 2, PDB No. 6E5W and PDB No. 6E6K, respectively). A subtle shift in abn-CBD’s position was seen while in complex with CRBP3, attributable to the smaller size of the Leu57 side chain, which eliminates some of the steric restraints that define the spatial position of the ligand’s aromatic ring.
The Effect of abn-CBD on Ocular Vitamin A Metabolism.
The physiological significance of CRBP1 stems from its role in the intracellular transport of vitamin A. CRBP1 is particularly important for the regeneration of visual chromophore. Highly expressed in the RPE cells, this carrier protein facilitates the reuptake of atROL from photoreceptor cells, enabling the efficient formation of all-trans-retinyl ester and its subsequent enzymatic conversion into 11-cis-retinol by RPE65. Consequently, studies on Rbp1−/− mice revealed a transient accumulation of atROL during the regeneration of visual chromophore after exposure to bright light.38 This well-documented phenotype can serve as a reference for the investigation of the effect of CRBP1 inhibitors on retinoid metabolism in vivo.
Influencing retinoid metabolism in the eye requires the efficient delivery of an active compound to retina tissue. Thus, we first investigated whether abn-CBD crosses the retina/blood or RPE/blood barrier by directly measuring the quantity of this compound in the eye after intraperitoneal (IP) injection. Mouse eyes perfused with phosphate-buffered saline (PBS) were collected between 1 and 15 h after administration of a single dose of 15 mg kg−1. Abn-CBD was readily detectable in the eye extracts by LC/MS (Supporting Information Figure 5). Using deuterated abn-CBD (d3-abn-CBD) as an internal standard, we determined that the ocular level of this compound reached the maximum value of 58 pmol/eye 2 h after the injection (Supporting Information Figure 5). Importantly, abn-CBD persisted in the ocular tissues for several hours post-treatment with a half-life of ~7 h. These results indicate that abn-CBD efficiently distributes into the eye targeting CRBPs expressed in the RPE and the retina.
Next, we examined the impact of abn-CBD on vitamin A metabolism in vivo. For this purpose, we analyzed the flow of retinoids via the visual cycle during the regeneration of visual chromophore after exposure to bright light. Dark-adapted mice were treated with 0.25 mg of the compound 1.5 h prior to exposure to a flash of light. The light intensity was set up to cause photoisomerization in 75% of rhodopsin as indicated by the decrease in the amount of 11-cis-retinal measured immediately after the light exposure (Figure 6). As anticipated, the levels of all-trans-retinal increased transiently in the first minutes of the recovery and faded away, as the bolus of retinoid released by photoactivated rhodopsin was first reduced to atROL, which was subsequently transported to RPE cells, esterified by lecithin:retinol acyltransferase (LRAT), converted to 11-cis-retinol, and oxidized to the corresponding aldehyde.41
Figure 6. The effect of abn-CBD on the temporal retinoid composition upon regeneration of the visual chromophore.
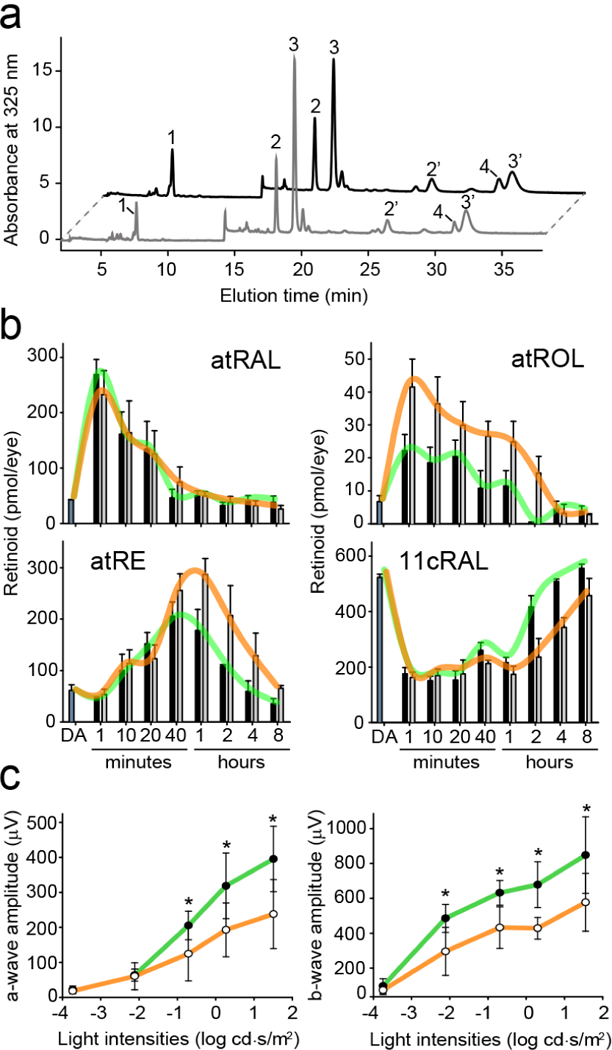
(a) HPLC separation of retinoids extracted from a mouse eye during recovery from a flash. Peak 1–all-trans-retinyl esters (atRE), 2,2′−11-cis-retinal oxime (syn and anti, respectively) (11cRAL), 3,3′−all-trans-retinal oxime (syn, anti, respectively) (atRAL), 4–all-trans-retinol (atROL). (b) Quantification of visual cycle retinoid during recovery from the light stimulus. Black bars correspond to data obtained for control (DMSO-treated) mice, where gray bars represent samples from animals administrated with abn-CBD. The trend lines for both groups are colored in green and orange, respectively. The bars represent the mean values ± sd (n = 4). (c) Single-flash ERG responses of increasing light intensity for abn-CBD-treated mice. ERG responses were recorded in mice 4 h after a flash of light that bleached ~50% of their visual pigment. ERG data represent the means and s.d. of both a-wave and b-wave amplitudes (n = 8). Mice were administrated with a single dose of abn-CBD (30 mg kg−1) 1 h before exposure to light. Systemic administration of abn-CBD (○) caused slight delay in the dark adaptation as compared to the vehicle-treated animal (●). Two-factor ANOVA test for treated and control groups of mice revealed p-values of 0.002 and 0.035 for a- and b-waves amplitudes, respectively. The asterisks depict significance: *p < 0.02 as determined by equal variance t test between groups with the same light stimulus.
Remarkably, the comparison of the time-resolved retinoid composition between the treated and the control groups of mice indicated no differences in the rate of all-trans-retinal clearance. However, transient accumulation of atROL became apparent in the presence of abn-CBD (Figure 6a,b). This prolonged persistence of atROL had a trickledown effect on the accumulation of all-trans-retinyl esters in the RPE cells and their delayed enzymatic isomerization. Consequently, the rate of 11-cis-retinal regeneration was noticeably slower in the abn-CBD treated mice. Notably, the alteration in atROL clearance upon pharmacological treatment was comparable to what has been described for Rbp1−/− mice, suggesting that abn-CBD exerts its biological activity by targeting CRBP1 in vivo.38
Abn-CBD Does Not Inhibit the Activities of Key Enzymes in the Visual Cycle.
The transient accumulation of atROL upon the regeneration of visual chromophore might result from the off-target inhibition of LRAT, RPE65, or retinol dehydrogenases activities by abn-CBD. Thus, to further probe the biological effect of this compound, we tested its effect on the key enzymes of the visual cycle. For this purpose, microsomes isolated from bovine RPE cells were preincubated with 10–50 μM of abn-CBD (1:1 to 1:5 molar excess of cannabinoid) prior to the initiation of the esterification, isomerization, or oxidation reactions by addition of an adequate retinoid substrate. The initial rates of the reactions were calculated by quantifying products of the enzymatic assays by high-performance liquid chromatography (HPLC). As shown in Supporting Information Figure 6, abn-CBD did not cause a measurable decline in the enzymatic activities of LRAT, RPE65 or RDH5, further indicating that the delay in the regeneration of the visual chromophore in vivo results from blocking CRBP function, rather than interfering with the enzyme machinery of visual cycle.
The Impact of the CRBP1 Inhibitor on the Mouse ERG.
To quantify the effect of the CRBP1 inhibitor on the recovery of visual sensitivity after a light stimulus, we measured full-field electroretinography (ERG) responses in mice. Prior to the illumination, the animals were either treated with DMSO (vehicle) or abn-CBD. Both drugs were administrated in a dose of 30 mg/kg. Scotopic ERG responses at varying light intensities were recorded 4 h after the exposure to a flash of light that photobleached ~50% of the visual pigment. As illustrated in Figure 6c, the recovery of both a-wave and b-wave amplitudes after light illumination in mice treated with abn-CBD is slower as compared to the untreated group of animals. However, even in a relatively high dose used in this experiment, the inhibitor of CRBP1 had much smaller impact on the visual cycle without causing its blockage, as it was reported for retinylamine and emixustat.10,13,42
The Protective Effect of abn-CBD against Light-Induced Retinal Degeneration.
Retinaldehyde (retinal) reactivity and the accumulation of its cytotoxic condensation products is an etiologic factor in retinal and macular degenerations of multiple causes.41,43 Thus, the regulation of the ocular metabolism of vitamin A was shown to be an effective strategy for control progress of a subset of ocular diseases resulting from a combination of certain environmental insults, such as prolonged exposure to intense light.10,13,44,45 The ability of CRBP1 inhibitors to modulate visual cycle retinoid flow might represent an alternative strategy to protect the retina from the cytotoxicity of retinal and its metabolites. Thus, to examine the effectiveness of abn-CBD in protection against retinal phototoxicity, we administered this compound to Balb/cJ mice by IP injection 1.5 h prior to exposure to white light of sufficient intensity to cause retinal damage in the absence of the treatment. A week later, the integrity of the retinas was examined in vivo by optical coherence tomography (OCT) followed by a morphological analysis of the isolated mouse eyes. As shown in Figure 7a, exposure to bright light caused severe retinal degeneration in the untreated mice as exemplified by the diminished thickness of the outer nuclear layer (ONL) and the deterioration of the cellular organization of the retinas. Notably, the animals pretreated with abn-CBD were largely protected against the light-induced retinal damage, as their retinal morphologies closely resembled those of mice not exposed to light. The protective effect of abn-CBD was dose-dependent with the estimated ED50 at 15 mg kg−1 (Figure 7b,c). To confirm the results obtained from the in vivo analyses, we performed a histological examination of retinal sections after Hemotoxylin and Eosin (H&E) staining. The diminished ONL thickness in DMSO-treated mice exposed to bright light as well as the protective effect of treatment with abn-CBD agreed with the findings from the OCT imaging (Figure 7d). Altogether, these data indicate that the modulation of ocular retinoid metabolism by the CRBP1 inhibitor is effective in alleviating pathophysiological changes resulting from the overstimulation of the photoreceptors and retinaldehyde toxicity.
Figure 7. Abn-CBD protects against acute light-induced retinal degeneration in Balb/cJ mice.
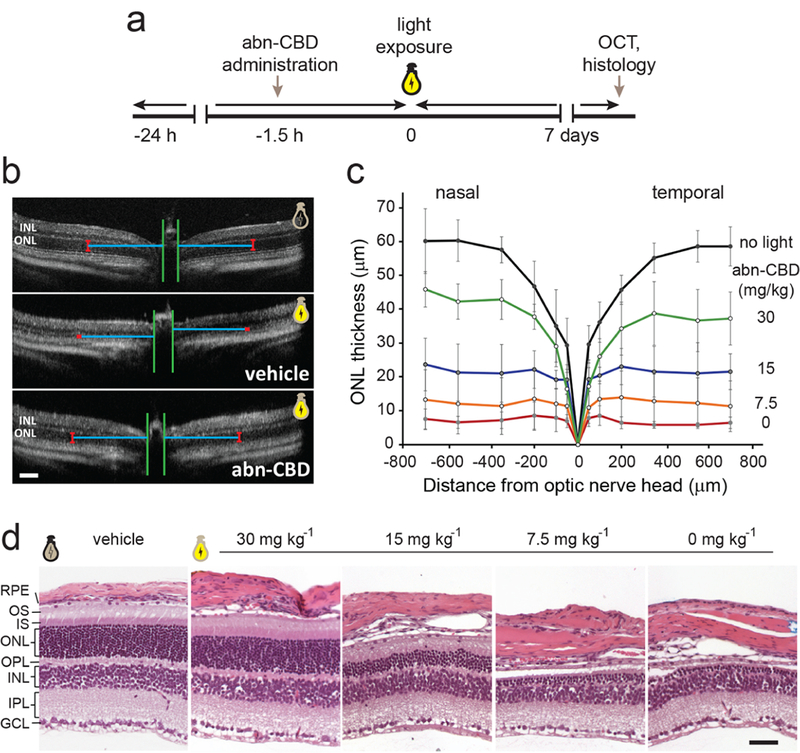
(a) Schematic representation of the experimental design. (b) Representative OCT images of the retinas for Balb/cJ mice not exposed to light (top) and animals pretreated with DMSO (vehicle) or abn-CBD (30 mg kg−1) and subjected to strong illumination (middle and bottom, respectively). The images indicate the protective effect of abn-CBD on photoreceptor cells. ONL, outer nuclear layer; INL, inner nuclear layer. Bars indicate 100 μm. (c) Quantification of the changes in the retinal morphology based on the thickness of the ONL of the OCT images (n = 6). The retinal morphology was preserved by the CRBP1 inhibitor in a dose-dependent manner. (d) Representative retinal images of the inferior retina of Balb/cJ mice after light exposure. Severe retinal degeneration is observed 7 d post illumination in untreated mice, whereas systemic administration of abn-CBD resulted in partial preservation of the photoreceptor cells as compared to the eyes not exposed to light. OS, photoreceptor outer segments, IS, photoreceptor inner segments, OPL, outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Bars indicate 50 μm.
DISCUSSION
Here we report the discovery of abn-CBD as a nonretinoid ligand of selected CRBPs. This non-psychoactive derivative of cannabidiol exerts its biological activity by competing with atROL for the binding site of CRBPs at pharmacologically relevant concentrations (Figure 2). The high affinity of abn-CBD stems from the unique chemical structure of this compound that perfectly accommodates the architecture of CRBP binding sites. Thus, despite belonging to a large and diverse family of cannabinoids, abn-CBD emerged as a specific ligand for CRBPs. Atomic-resolution crystal structures of protein–ligand complexes revealed the principles of this high specificity. Comparisons between the structures of complexes with ligand derivatives clearly emphasized the significance of the hydrogen bonding interactions between the protein and the examined cannabidiols, as well as the importance of the relative position of the hydroxyl groups within these compounds. Notably, the interactions between this ligand and the protein scaffold are distinct from those observed for the endogenous ligand, atROL (Figure 4). Detailed structural information can also be used to make predictions about the interactions of CRBPs with other cannabinoids, particularly tetrahydrocannabinol and cannabidiol, that could not be examined directly in this study due to restricted access to these compounds. However, on the basis of the position of CBDO inside the binding cavity of CRBP1, it becomes apparent that neither of these abundant natural cannabinoids can bind to CRBPs. They not only lack a key hydroxyl group in the preferable position but also their pentyl aliphatic chains cannot be accommodated inside the binding pocket without causing interference with the protein backbone (Supporting Information Figure 7). Conversely, the structure of CRBP1 in complex with abn-CBD provides suggestions for how to improve the pharmacodynamic properties of this ligand. Several suitable polar modifications could be proposed to further strengthen the ligand’s affinity by increasing the number of hydrogen bonding interactions. However, any potential gains in binding affinity will need to be balanced with appropriate pharmacokinetic characteristics.
To our knowledge, abn-CBD is the first inhibitor of CRBPs that can potentially serve as a pharmacological tool to influence vitamin A metabolism in vivo. Thus, as a proof of concept, we investigated the effect of abn-CBD on the flux of retinoids via the visual cycle during the regeneration of visual chromophore after light exposure. Congruently with the physiological role of CRBP1 inferred from the studies on Rbp1−/− mice, the administration of the inhibitor of this carrier protein affected the transport of atROL between the photoreceptor and RPE cells and its efficient esterification, which in turn resulted in the moderately delayed regeneration of visual chromophore. Importantly, the impaired flow of retinoids between these two cell types did not result in impaired clearance of retinaldehyde (Figure 6). Although the conversion of retinol to retinal is at equilibrium in the two-cell system, atROL seems to be rapidly removed from the photoreceptor outer segments by partitioning into the inter-photoreceptor matrix, where it is sequestered by inter-photoreceptor retinoid-binding protein.46,47 Thus, atROL does not persist in the photoreceptor cells, preventing its mass action-driven oxidation back to retinaldehyde.
The significance of the identification of abn-CBD as a competitive inhibitor of CRBP1 stems from the potential clinical application of vitamin A metabolism modulators. As shown by genetic and pharmacological studies, limiting the rate of visual chromophore regeneration could be therapeutically advantageous by averting light-induced retinal degeneration and lessening the accumulation of cytotoxic retinaldehyde metabolites.48–51 These effects are largely attributed to the averted overstimulation of photoreceptor cells and decreased steady-state concentration of retinaldehyde. Among strategies to modulate ocular vitamin A metabolism, inhibition of the visual cycle by RPE65 inhibitors such as retinylamine was one of the first to advance to clinical testing.10 Subsequently, a derivative of 11-cis-retinylamine, emixustat, entered clinical trials for the treatment of dry age-related macular degeneration (AMD).11,50,52 However, in a phase IIb/IIIa clinical trial, emixustat failed to protect against the progression of geographic atrophy in patients with AMD (NCT01802866). The main factor that contributed to this result was the inability of this drug to achieve a balance of vision-related adverse reactions, including night blindness and dyschromatopsia, with the desired therapeutic effects.52 In contrast to directly targeting visual cycle enzymes, controlling retinoid metabolism with fenretinide was partially effective in slowing the rate of progression of geographic retinal atrophy in AMD patients enrolled in phase II clinical trials (NCT02141958).53 Fenretinide binds to RBP4, preventing its interaction with transthyretin, resulting in a loss of RBP4 to glomerular filtration.54 The treatment of Abca4−/− mice with fenretinide arrested the accumulation of retinal conjugates.55 Despite the effects of this drug on bisretinoid accumulation, it is a retinoid-based compound with a broad and still-unresolved therapeutic specificity.56,57 To alleviate some of the side effects, nonretinoid inhibitors of RBP4, A1120, and BPN-14136 were introduced.22,26 Nevertheless, they still induce systemic vitamin A deficiency, which is a concerning therapeutic disadvantage. Thus, inhibitors of CRBP1 emerge as an alternative, potentially more efficient and safer approach to achieve similar therapeutic effects. The potency of abn-CBD in preventing acute light-induced retinal damage in Balb/cJ mice demonstrates the potential of this strategy (Figure 7). In contrast to the RBP4 inhibitors, a single dose of abn-CBD was sufficient to alter retinoid metabolism for long enough to provide protection against a light stimulus that in untreated animals caused irreversible retinal degeneration. This favorable effect relates to the fact that during light illumination the vast majority of visual chromophore is produced from recycled all-trans-retinal within the photoreceptor and RPE cells rather than synthesized from vitamin A newly acquired from bloodstream.58
Although information about the biological activity of abn-CBD is limited, this compound has been shown to modulate inflammatory response in a mouse model of colitis, presumably by acting as an antagonist of GPR18 receptor.59,60 It is unknown whether the anti-inflammatory properties of abn-CBD extend beyond gastrointestinal system. Nevertheless, it is tempting to speculate that this off-site activity could have a beneficial synergistic effect on the treatment of retinopathies.
The question remains whether long-term treatment with a CRBP1 inhibitor is safe. Studies on Rbp1−/− mice indicated that, in addition to a relatively mild ocular phenotype, the deficiency in CRBP1 causes a lower storage capacity and higher turnover of vitamin A in the liver.39 Importantly, vitamin A-sufficient animals were asymptomatic with the absence of CRBP1, but mice kept on a vitamin A-deficient diet had faster depletion of retinoid storage. Thus, although the presence of functional CRBP1 is evolutionarily beneficial in organisms frequently experiencing nutrient insufficiencies, its inhibition should be tolerated in individuals having access to a balanced diet. Importantly, previous studies that investigated the biological activity of abn-CBD in the context of wound healing, vasodilation, or diabetes did not report toxicity of this compound in rodents.59,61,62 Additionally, abn-CBD does not interact with CRBP2. This protein is exclusively expressed in enterocytes in the small intestine, where it mediates the uptake of dietary vitamin A. The inhibition of CRBP2 could impair the efficient absorption of vitamin A as evidenced by the phenotype of Rbp2−/− mice.63 Thus, the lack of toxicity and inability to interfere with CRBP2 activity make abn-CBD suitable for the long-term treatment of progressive retinal degenerative diseases.
Although the modulation of ocular vitamin A metabolism by abn-CBD can be mainly attributed to it binding to CRBP1, the interaction of this compound with CRBP3 and 4 may contribute to certain off-target effects. Unfortunately, there is not much information about the physiological function of these proteins, mostly because these two macromolecules are represented by just one homologous protein in mice. Expressed in heart, muscle, adipose, and mammary tissue, mouse CRBP3 plays a role in the regulation of lipid metabolism.64 The genetic disruption of CRBP3 protected mice kept on a high-fat diet from hepatic steatosis, contributed to decreased free fatty acid output from adipose tissue, and increased mitochondrial respiratory rate.64 If the phenotype of CRBP3-deficent mice holds true in humans, inhibitors of this protein, including abn-CBD, might find an alternative application in the treatment of metabolic disorders.
In summary, we have identified a first-in-class drug candidate that targets selected CRBPs and, thus, influences retinoid metabolism in vivo. Abn-CBD, a competitive inhibitor of these vitamin A chaperones, showed efficacy in protecting mouse retinas from light-induced retinal degeneration. The biological activity and pharmacokinetic properties of abn-CBD meet criteria for further development as a compound for the treatment of retinal pathologies with etiologies involving imbalances in ocular retinoid homeostasis.
METHODS
Chemicals and Reagents.
4-[(1R,6R)-3-Methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]-5-pentylbenzene-1,3-diol (abn-CBD) was purchased form Cayman Chemical Company and Toronto Research Chemicals. Derivatives of abn-CBD including 4-[(6R)-3-methyl-6-(1-methylethenyl)-2-cyclohexen-1-yl]-5-pentyl-1,3-benzenediol-5,5,5-d3 (3d-abn-CBD), 5-methyl-4-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene-1,3-diol (abn-CBDO), 5-methyl-2-[(6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene-1,3-diol (CBDO), and 1,3-dimethoxy-5-methyl-2-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene (O-1918) were obtained from Cayman Chemical Company. The racemic mixture of limonene was obtained from Sigma-Aldrich. atROL was purchased from Toronto Research Chemicals, whereas 11-cis-retinol was produced as described in Arne et al.65 HPLC-grade organic solvents used in this study were purchased from Thermo Fisher Scientific.
Animals and Animal Care.
Balb/cJ mice at five to eight weeks of age were purchased from The Jackson Laboratory. All mice were housed in the Animal Resource Center at the School of Medicine, Case Western Reserve University (CWRU), and fed with a standard laboratory mouse diet supplemented with 10–12 IU g−1 of vitamin A. Mice were dark adapted for at least 24 h prior to experiments. Manipulations in the dark were performed under dim red light. All animal procedures and experimental protocols were approved by the Institutional Animal Care and Use Committee at CWRU and conformed to recommendations of both the American Veterinary Medical Association Panel on Euthanasia as well as the Association for Research in Vision and Ophthalmology.
Expression and Purification of CRBP1.
Human apo-CRBP1 was expressed and purified as described by Silvaroli et al.33 without any further modification to the established protocol.
Expression and Purification of CRBP2.
The cDNA of human CRBP2 was purchased from Origene Technologies and amplified by polymerase chain reaction (PCR) using the following primers: forward –GCAGATCATATGACAAGGGACCAGAATGGAACC and reverse –CGTCTACTCGAGTCCCGGGGATCCACGCGGAACCAGTTTCTTTTTGAACACTTGACGG to subclone into pET30b expression vector (Millipore Sigma), thereby modifying the C-terminal sequence to introduce thrombin cut site that allowed for removal of a 6x histidine tag from the purified protein. The recombinant protein was expressed in the BL21 (DE3) E. coli strain (New England Biolabs). The bacteria were grown at 37 °C in the presence of 50 μM kanamycin to OD600 = 0.6–0.8 prior to addition of isopropyl β-D-1 thiogalactopyranoside (Roche) to a final concentration of 0.5 mM. After 4 h of incubation, the cells were harvested by centrifugation (6000g, 15 min, 4 °C). Bacteria were disrupted by osmotic shock, and the lysate was centrifuged at 36 000g for 30 min at 4 °C. The supernatant was collected, and its buffer composition was adjusted to match the loading buffer (50 mM Tris-HCl, pH 8.0, 250 mM NaCl). Next, the protein was loaded onto a 5 mL HisTrap column (GE Healthcare). The column was washed with 100 mL of the loading buffer containing 5 mM imidazole prior to the elution of bound proteins with 250 mM imidazole. Eluted fractions were examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), combined, and concentrated to 5 mL in an Amicon Ultra-4 centrifugal filter, cutoff 10 000 Da (Millipore). The protein was loaded onto a gel filtration column Superdex 200 (GE Healthcare) equilibrated with 10 mM Tris/HCl buffer, pH 8.0. Fractions containing CRBP2 were pooled together and incubated with thrombin (USB Affymetrix) for 16 h at 4 °C to remove the histidine tag. After the ion strength of the protein solution was adjusted by adding NaCl to the final concentration of 300 mM, digested CRBP2 was reloaded onto a HisTrap column. Unbound fraction that contained digested protein was collected, diluted 20 times with 10 mM Tris/HCl buffer, pH 8.0 buffer, and loaded onto HiTrap Q HP ion exchanger column (GE Healthcare) for the final step of purification. CRBP2 was eluted with a gradient of NaCl (0–0.5 M) over 30 min at a flow rate 1.0 mL min−1. Purity of the collected protein fractions were checked by SDS-PAGE. The mass of an intact protein was verified by mass spectrometry. Purified CRBP2 was concentrated to ~9 mg mL−1, and 0.2 mL aliquots were stored at −80 °C.
Expression and Purification of CRBP3.
The cDNA of human CRBP3 was purchased from Origene Technologies. To introduce a thrombin cut site at the C-terminus of the protein, the cDNA was amplified by PCR using the following primers: forward –GCAGATCATATGCCTCCCAACCTCACTGGCTACTACC and reverse –CGTCTACTCGAGTCCCGGGGATCCACGCGGAACCAGTCTGACCTTCCTGAAGACCTGC and subcloned into pET30b vector. The next steps of the expression and purification of CRBP3 were identical to the procedure described above for CRBP2.
Expression and Purification of CRBP4.
The cDNA of human CRBP4 was purchased from Origene Technologies. The introduction of a thrombin cut site at the C-terminus was achieved by PCR amplification of the cDNA using the following primers: forward –GCAGATCATATGCCCGCCGACCTCAGCGGTACTTGG and reverse –GTCTACTCGAGTCCCGGGGATCCACGCGGAACCAGGGCTCTCTGGAATGTCTGTTTGC. The PCR product was subcloned into pET30b vector. For the most part, CRBP4 was expressed and purified as described for CRBP2. However, after digestion with thrombin and reloading onto HisTrap column, the unbound fraction of the protein was further purified on a HiTrap SP HP ion exchanger column (GE Healthcare). CRBP4 was loaded in 10 mM Tris/HCl buffer, pH 8.0 buffer, and subsequently eluted with a gradient of NaCl (0–0.5 M) over 30 min at a flow rate 1.0 mL min−1. Purified CRBP4 was concentrated to 6 mg mL−1 and liquated at −80 °C.
Obtaining CRBP1 in Complex with atROL.
To prepare holo-CRBP1, 3 mg of purified apoprotein was incubated for 15 min on ice with 2 molar excess of atROL (Toronto Research Chemicals) in 10 mM Tris-HCl, pH 8.0, 5% glycerol (v/v). The retinoid was delivered in acetonitrile (<1%, v/v). To remove excess of atROL, the protein solution was diluted 10× with 10 mM Tris-HCl, pH 8.0, centrifuged (36 000g, 20 min, 4 °C) and loaded onto a 5 mL HiTrap Q HP column. Holo-CRBP1 was eluted from the column in conditions described in CRBP1 purification method. The efficiency of formation of holo-CRBP1 was examined by recording UV/vis spectrum of the repurified protein. The complex of CRBP1 with atROL revealed characteristic absorbance spectrum with maxima at 282 nm, corresponding to the protein scaffold and triple maxima at 327, 350, and 370 nm, indicating bound retinoid.
Fluorescence Binding Assays.
The apparent affinity of non-retinoid ligands was evaluated by monitoring changes in the fluorescence of holo-CRBP1. In the atROL bound form, the protein excited at 285 nm emitted fluorescence at 350 and 480 nm due to FRET between the protein scaffold and the retinoid moiety (Figure 1A). Replacement of atROL in the binding site by tested ligands leads to disruption of FRET and consequently an increase in the fluorescence intensity at 350 nm with concomitant diminishing signal at 480 nm. All fluorescence measurements were performed using a PerkinElmer Life Sciences LS55 spectrofluorometer. The titrations were performed at 25 °C in 67 mM phosphate-buffered saline buffer, pH 7.4, containing 5% glycerol (v/v) by adding an increasing amount of an examined compound delivered in acetonitrile. The final concentrations of the organic solvent did not exceed 0.4% of the sample’s total volume. All fluorescence spectra were corrected for the inner filter effect. The Ki values were calculated by fitting intensities of the protein fluorescence at the maximum emission (350 nm) or the intensity of retinoid fluorescence at 480 nm to the saturation single ligand-binding model. The data fitting and calculations of the Ki values were performed using SigmaPlot 11 software package (Systat Software).
High-Throughput Screening for CRBP1 Ligands.
The principle of the fluorescence binding assays described above was adapted for the HTS in a 96-well plate format. Each well contained 0.2 mL of 20 mM Tris/HCl, pH 7.4 and 1 μM of holo-CRBP1. Tested compounds from two independent libraries (Bioactive Lipid I Screening Library form Cayman Chemicals and Screen-Well Bioactive Lipid Library from Enzo Life Science, total of 986 chemicals) were delivered in dimethyl sulfoxide (DMSO) to the final concentration of 5 μM. After 15 min of incubation at room temperature (RT), the samples were excited at 285 nm, and the fluorescence signals from CRBP1 (at 350 nm) and atROL (at 480 nm) were recorded in a Flexstation3 microplate reader (Molecular Devices). Changes in the fluorescence emissions were evaluated by calculating ratio between signals at 480 and 350 nm. To eliminate false positives, only those wells in which the increase in protein fluorescence was accompanied by proportional decline in the fluorescence of atROL were considered as potential hits.
Retinol Replacement Assay.
holo-CRBP1 (500 μg) in 0.5 mL (67 μM) was incubated with tested compound (1:2 molar ratio) delivered in ethanol in 20 mM Tris/HCl, pH 8.0, 200 mM NaCl, 5% glycerol (v/v) at RT for 15 min. The sample was then diluted with 10 788 mM Tris/HCl, pH 8.0 to the final volume of 5 mL, loaded onto HiTrap Q HP column, and eluted as described above. The elution profiles were monitored at both 280 and 325 nm to concurrently record signals for the protein and the retinoid moieties.
Mass Spectrometry Analysis.
After incubation of apo- or holo-CRBP1 with abn-CBD, the protein was repurified on HiTrap Q HP column as described above. The protein fractions were collected, pooled together, and concentrated to 0.25 mL in an Amicon Ultra-4 centrifugal filter, cutoff 10 000 Da. For quantification purpose, 100 pmol of 3d-abn-CBD (internal standard) were added to the protein samples. To extract organic compounds associated with the repurified CRBP1, 0.25 mL of ethanol was added followed by 4 mL of hexane. The sample were shaken vigorously and centrifuged at 26 000g for 5 min. The resulting supernatant was dried under a stream of nitrogen, and the residual compounds were resuspended in 0.3 mL of ethanol. The samples were injected onto an X-Bridge C18 column, 3.5 μm, 2.1 × 100 mm (Waters) equilibrated with 30% water in methanol (v/v). Abn-CBD was eluted in a gradient of methanol in water (70% to 100%) developed over 10 min at a flow rate 0.5 mL min−1. The eluent was directed into an LTQ ion trap mass spectrometer (Thermo Scientific) via an electrospray ionization source working in the positive mode. The mass spectrometer operation parameters were optimized using synthetic abn-CBD standard. Abn-CBD and its deuterated derivative were detected in the selected reaction monitoring mode using m/z 315.2 → 221.2 and 318.2 → 224.1 transitions, respectively. Calibration curves were calculated based on the linear relationship between areas under SRM ion intensity peaks corresponding to abn-CBD and the internal standard versus molar ratios of these compounds in a range from 20 to 500 pmol.
Detection and Quantification of abn-CBD in the Ocular Tissues.
Balb/cJ mice were i.p. injected with 15 mg kg−1 dose of abn-CBD. At the time points between 1 and 15 h, they were deeply anesthetized by ketamine injection and perfused with PBS. Perfused eyes were collected and homogenized in 1 mL of PBS/methanol (v/v). During the homogenization, 100 pmol of an internal standard (3d-abn-CBD) was added. The samples were extracted with 4 mL of hexane and centrifuged at 15 000g for 5 min at 4 °C. The organic solvent was transferred to glass test tubes and dried in a Savant speedvac concentrator (Thermo Fisher Scientific). The extracted compounds were dissolved in 0.3 mL of ethanol and subjected to LC/MS analysis and quantification as described above.
Crystallization of CRBPs.
Crystals of CRBP1 in complex abn-CBD and its derivatives (abn-CBDO or CBDO) were grown essentially as described by Silvaroli et al.33 The apo protein at 3 mg mL−1 was preincubated with 300 μM of a ligand in 10 mM Tris-HCl buffer, pH 8.0, 150 mM NaCl for 15 min on ice. This sample (1 μL) was mixed in 1:1 ratio with a crystallization solution composed of 0.1 M BisTris, pH 5.5, and 25% poly(ethylene glycol) (PEG) 3350 (w/v). Crystals of CRBP3 with abn-CBD were obtained by preincubating apoprotein at 8 mg mL−1 with 300 μM of the ligand for 15 min and setting up crystallization drops by mixing 1 μL of the protein sample with 1 μL of 0.2 M sodium malonate, pH 5.0 and 20% PEG 3350 (w/v) solution. For CRBP4, the crystallization conditions included 0.2 M NaCl, 0.1 M BisTris, pH 6.5, and 25% PEG 3350 (w/v). Similar to the other CRBPs, CRBP4 (6 mg mL−1) was preincubated with abn-CBD prior to the crystallization. All of the protein crystals were obtained by the sitting drop vapor diffusion method at RT. Mature crystals were collected and flash-cooled in liquid nitrogen in preparation for X-ray diffraction experiments.
X-ray Data Collection, Processing, and Model Building.
X-ray diffraction data were collected at the Advanced Photon Source beamlines NE-CAT 24-ID-C and 24-ID-E or Stanford Synchrotron Radiation Lightsource beamline 9–2. Data from single crystals were integrated and scaled with MOSFLM.66 The structures of CRBPs in complexes with its ligands were solved by molecular replacement with PHASER_MR 67 using the following structural templates: PDB No. 5HBS, 1GGL, and 1LPJ for CRBP1, 3, and 4, respectively.33,34,68 Initial models were manually adjusted in COOT69 and refined with PHENIX70 using a riding hydrogen model, individual anisotropic temperature factors, and occupancy refinement for alternative conformers and waters. Geometry of the refined model was verified with the MolProbity server.71 The accession codes as well as the data collection and refinement statistics are summarized in Supporting Information Table 2. Visualization of the macromolecules were performed in the CHIMERA software package version 1.12.72
Photobleaching and Visual Chromophore Regeneration Protocols.
Balb/cJ mice were dark adapted for at least 24 h before the experiment. Mouse pupils were dilated with 1% tropicamide, and the animals were subjected to a single light flash from a photographic flash unit TT560 (Neewer) at the maximum setting that resulted in photobleach of ~70% of visual pigment. After the light exposure, the mice were kept in dark and euthanized at time intervals between 1 min to 8 h. The eyes were removed, flash frozen in liquid nitrogen, and stored in −80 °C until retinoid analysis.
Detection and Quantification of Ocular Retinoids.
All procedures related to the retinoid analysis were done under dim red light. To extract retinoids, enucleated eyes were transferred to a glass/glass homogenizer and submerged in 1 mL of 40 mM hydroxylamine in a 1:1 solution of PBS/methanol (v/v). The eyes were thoroughly homogenized and incubated at RT for 20 min to allow derivatization of retinylaldehydes to retinyl oximes. The retinoids were extracted with 4 mL of hexane. To facilitate the fraction separation, the samples were centrifuged at 5000g for 10 min, after which the upper organic phase was collected in a glass test tube. The organic solvent was dried in a rotary SpeedVac, and the residual retinoids were redissolved in 250 mL of hexanes, and subjected to HPLC analysis. Separation of retinoids was achieved on a normal phase column (Agilent Zorbax Sil 5 μm 4.6 × 250 mm) in a step gradient of ethyl acetate in hexane (1% for 10 min followed by 10% for 40 min at a constant flow rate of 1.5 mL min−1).73 Elution of retinoids was monitored at 325 and 360 nm wavelengths. All-trans-retinyl esters, all-trans- and 11-cis-retinal oximes as well as atROL were identified based on their elution times and characteristic UV/vis spectra. Retinoids were quantified based on linear correlations between the amount of injected synthetic standards and the area under chromatographic peaks.
Retinol Esterification Assay.
The enzymatic activity of LRAT was assayed using isolated UV-treated bovine RPE microsomes as described previously.74 The reaction mixture included 20 mM Tris/HCl buffer, pH 7.4, 1 mM DTT, 1% bovine serum albumin (BSA; w/v), and RPE microsomes (~100 μg of total protein) in the total volume of 0.1 mL. To test the influence of abn-CBD on the LRAT activity, 10, 20, or 50 μM of this compound was added prior to the addition of atROL (final concentration of 10 μM). To record the initial rate of the esterification, the mixture was incubated at RT for 5 min. The reaction was quenched with 0.2 mL of methanol, and the retinoids were extracted with 0.3 mL of hexane. The extracted retinoids were separated on a normal phase column (Agilent Zorbax Sil 5 μm 4.6 × 250 mm) in an isocratic flow of 10% ethyl acetate in hexane (flow rate of 1.5 mL min−1). Elution of retinoids was monitored at 325 nm. All-trans-retinyl esters were quantified based on a correlation between the amount of injected synthetic standards and the area under chromatographic peaks.
11-cis-Retinol Oxidation Assay.
Dehydrogenase activity was assayed by monitoring oxidation of 11-cis-retinol to a corresponding aldehyde in bovine RPE microsomes. The reaction mixture containing 20 mM Tris/HCl buffer, pH 8.0, 1% BSA (w/v), 20 μM NAD+, and RPE microsomes (~100 μg of total protein) in the total volume of 0.1 mL. 11-cis-Retinol (10 μM) was incubated at 30 °C for 10 min. The reaction was stopped by 0.2 mL of methanol that contained 40 mM hydroxylamine. After subsequent 15 min of incubation at RT, the retinoids were extracted with 0.3 mL of hexane and analyzed by normal-phase HPLC as described for the retinol esterification assay.
RPE65 Isomerization Activity Assay.
For the retinoid isomerization assay, UV-bleached RPE microsomes isolated from bovine eyes served as a source of robust RPE65 enzymatic activity.74 The isomerization reaction was performed in the presence of cellular retinaldehyde-binding protein (CRALBP) according to an established methodology published previously.10,73,75 The enzymatic reaction was initiated by addition of atROL the final concentration of 10 μM. After incubation at 30 °C for 30 min, the reaction products were extracted with hexane and analyzed by HPLC as described above.
Bright Light-Induced Retinal Degeneration.
Retinal degeneration was induced by exposing dark-adapted Balb/cJ mice to white light with an intensity of ~50 000 lx, delivered from a 100 W AC90–145 V light-emitting diode (LED) lamp (Home Depot) for 1 h. Abn-CBD in doses of 30, 15, or 7.5 mg/kg was administered by i.p. injection in sterilized 80% DMSO/water (v/v) 1 h before exposure to bright light. The injection volume did not exceed 25 μL. Retinal morphology was analyzed in vivo by OCT 7 d after the light exposure. Then the mice were euthanized, and their eyes were subjected to staining with H&E, immuno-histochemistry, and imaging.
Optical Coherence Tomography (OCT).
To assess the effect of abn-CBD on light-induced retinal degeneration in Balb/cJ mice, the retinas were examined by ultrahigh resolution OCT (Bioptigen). Pupils of mice were first dilated with 1% tropicamide, and the animals were anesthetized with ketamine/xylazine cocktail. Twenty-five frames of OCT images scanned at 0° and 90° were acquired in the B-scan and averaged.
Electroretinography (ERG) Analyses.
Before the ERG recording, dark-adapted Balb/cJ mice were anesthetized with 20 mg mL−1 ketamine and 1.75 mg mL−1 xylazine in PBS at a dose of 0.1–0.13 mL per 25 g of body weight, and pupils were dilated with 1% tropicamide. Contact lens electrodes were placed on the eyes, and a reference electrode was positioned between two ears, while ground electrode was placed on the tail. Scotopic ERGs were recorded for both eyes of each mouse using a UTAS E-3000 universal testing and ERG system (LKC Technologies). To evaluate an effect of abn-CBD on retinal function, ERGs were performed on the control and abn-CBD-treated group of mice 4 h after exposure to a light illumination that resulted in photobleach 40% of visual pigment.
Retinal Histology.
The structural morphology of mouse retinas challenged with bright light were assessed using H&E staining of paraffin sections. Mouse eyes were fixed in 4% paraformaldehyde and 1% glutaraldehyde followed by paraffin sectioning. Paraffin sections (5 μm thick) were stained with H&E and imaged by light microscopy (Leica).
Statistical Analysis.
Four to six mice per treatment group were used. The data are expressed as mean ± standard deviation (sd). For two-group comparisons, student’s t test was performed, whereas for multiple groups comparisons, one-way ANOVA was used. Differences were considered statistically significant at p-value of less than 0.05.
Supplementary Material
ACKNOWLEDGMENTS
We thank H. Hill (Case Western Reserve Univ.) for her contribution to editing of the text. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on 24-ID-C beamline is funded by an NIH-ORIP HEI grant (No. S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. DOE, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Funding
This work was supported by Grant No. EY023948 from the National Eye Institute of the National Institutes of Health (NIH) (M.G.) and by Grant No. IK2BX002683 from the Department of Veterans Affairs (P.D.K.).
ABBREVIATIONS:
- ABCA4
ATP-binding cassette transporter 4
- abn-CBD
abnormal cannabidiol (4-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]-5-pentylbenzene-1,3-diol)
- abn-CBDO
(5-methyl-4-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene-1,3-diol)
- atROL
all-trans-retinol
- AMD
age-related macular degeneration
- BSA
bovine serum albumin
- CBDO
(5-methyl-2-[(6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene-1,3-diol)
- CRALBP
retinaldehyde-binding protein
- CRBPs
cellular retinol-binding proteins
- DMSO
dimethyl sulfoxide
- ERG
electroretinography
- FRET
fluorescence resonance energy transfer
- H&E
hematoxylin and eosin stain
- HPLC
high-performance liquid chromatography
- HTS
high-throughput screening
- i.p.
intraperitoneal
- Ki
inhibition constant
- LC/MS
liquid chromatography–mass spectrometry
- LRAT
lecithin:retinol acyltransferase
- MS
mass spectrometry
- O-1918
(1,3-dimethoxy-5-methyl-2-[(1R,6R)-3-methyl-6-prop-1-en-2-ylcyclohex-2-en-1-yl]benzene)
- OCT
optical coherence tomography
- ONL
outer nuclear layer
- RBP4
serum retinol-binding protein
- RPE
retinal pigment epithelium
- RPE65
retinoid isomerase (retinal pigment epithelium-specific 65 kDa protein)
- sd
standard deviation
Footnotes
ASSOCIATED CONTENT
Supporting Information
A table that summarizes the Ki values for abn-CBD and its derivatives; X-ray crystallographic table; titrations of CRBP1 with atROL, O-1918, and limonene; comparison of the binding sites architecture of CRBP1 and CRBP2; structures of CRBP3 and CRBP4 in complex with abn-CBD; detection of abn-CBD in the ocular tisssues; evaluation of the effect of abn-CBD on the activity of key enzymes of the visual cycle; and structural base for the inability of cannabidiol or tetrahydrocannabinol to interact with CRBPs (PDF)
Accession Codes
The coordinates and structure factor amplitudes were deposited in the Protein Data Bank under Accession Nos. 6E5L, 6E5T, 6E6M, 6E5W, and 6E6K.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Duester G (2008) Retinoic acid synthesis and signaling during early organogenesis. Cell 134, 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stephensen CB (2001) Vitamin A, infection, and immune function. Annu. Rev. Nutr 21, 167–192. [DOI] [PubMed] [Google Scholar]
- (3).Maden M (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci 8, 755–765. [DOI] [PubMed] [Google Scholar]
- (4).Niederreither K, and Dolle P (2008) Retinoic acid in development: towards an integrated view. Nat. Rev. Genet 9, 541–553. [DOI] [PubMed] [Google Scholar]
- (5).Wald G (1968) The molecular basis of visual excitation. Nature 219, 800–807. [DOI] [PubMed] [Google Scholar]
- (6).Park PS, Lodowski DT, and Palczewski K (2008) Activation of G protein-coupled receptors: beyond two-state models and tertiary conformational changes. Annu. Rev. Pharmacol. Toxicol 48, 107–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, and Miyano M (2000) Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- (8).Nathans J (1990) Determinants of visual pigment absorbance: identification of the retinylidene Schiff’s base counterion in bovine rhodopsin. Biochemistry 29, 9746–9752. [DOI] [PubMed] [Google Scholar]
- (9).Nathans J, Merbs SL, Sung CH, Weitz CJ, and Wang Y (1992) Molecular genetics of human visual pigments. Annu. Rev. Genet 26, 403–424. [DOI] [PubMed] [Google Scholar]
- (10).Golczak M, Kuksa V, Maeda T, Moise AR, and Palczewski K (2005) Positively charged retinoids are potent and selective inhibitors of the trans-cis isomerization in the retinoid (visual) cycle. Proc. Natl. Acad. Sci. U. S. A 102, 8162–8167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kubota R, Boman NL, David R, Mallikaarjun S, Patil S, and Birch D (2012) Safety and effect on rod function of ACU-4429, a novel small-molecule visual cycle modulator. Retina 32, 183–188. [DOI] [PubMed] [Google Scholar]
- (12).Kiser PD, Zhang J, Badiee M, Kinoshita J, Peachey NS, Tochtrop GP, and Palczewski K (2017) Rational Tuning of Visual Cycle Modulator Pharmacodynamics. J. Pharmacol. Exp. Ther 362, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Maeda A, Maeda T, Golczak M, Imanishi Y, Leahy P, Kubota R, and Palczewski K (2006) Effects of potent inhibitors of the retinoid cycle on visual function and photoreceptor protection from light damage in mice. Mol. Pharmacol 70, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Golczak M, Maeda A, Bereta G, Maeda T, Kiser PD, Hunzelmann S, von Lintig J, Blaner WS, and Palczewski K (2008) Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J. Biol. Chem 283, 9543–9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Maeda A, Maeda T, Golczak M, Chou S, Desai A, Hoppel CL, Matsuyama S, and Palczewski K (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem 284, 15173–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Napoli JL (2017) Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther 173, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang YR, Zhao YQ, and Huang JF (2012) Retinoid- binding proteins: similar protein architectures bind similar ligands via completely different ways. PLoS One 7, e36772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ross AC (1993) Cellular metabolism and activation of retinoids: roles of cellular retinoid-binding proteins. FASEB J 7, 317–327. [DOI] [PubMed] [Google Scholar]
- (19).Ong DE (1994) Cellular transport and metabolism of vitamin A: roles of the cellular retinoid-binding proteins. Nutr. Rev 52, S24–31. [DOI] [PubMed] [Google Scholar]
- (20).Motani A, Wang Z, Conn M, Siegler K, Zhang Y, Liu Q, Johnstone S, Xu H, Thibault S, Wang Y, Fan P, Connors R, Le H, Xu G, Walker N, Shan B, and Coward P (2009) Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem 284, 7673–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Peng YM, Dalton WS, Alberts DS, Xu MJ, Lim H, and Meyskens FL Jr. (1989) Pharmacokinetics of N-4-hydroxyphenyl-retinamide and the effect of its oral administration on plasma retinol concentrations in cancer patients. Int. J. Cancer 43, 22–26. [DOI] [PubMed] [Google Scholar]
- (22).Racz B, Varadi A, Kong J, Allikmets R, Pearson PG, Johnson G, Cioffi CL, and Petrukhin K (2018) A non-retinoid antagonist of retinol-binding protein 4 rescues phenotype in a model of Stargardt disease without inhibiting the visual cycle. J. Biol. Chem 293, 11574–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lois N, Halfyard AS, Bird AC, Holder GE, and Fitzke FW (2004) Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am. J. Ophthalmol 138, 55–63. [DOI] [PubMed] [Google Scholar]
- (24).Mata NL, Weng J, and Travis GH (2000) Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. U. S. A 97, 7154–7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mata NL, Tzekov RT, Liu X, Weng J, Birch DG, and Travis GH (2001) Delayed dark-adaptation and lipofuscin accumulation in abcr± mice: implications for involvement of ABCR in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci 42, 1685–1690. [PubMed] [Google Scholar]
- (26).Dobri N, Qin Q, Kong J, Yamamoto K, Liu Z, Moiseyev G, Ma JX, Allikmets R, Sparrow JR, and Petrukhin K (2013) A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscino-genesis. Invest. Ophthalmol. Visual Sci 54, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Berni R, and Formelli F (1992) In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett 308, 43–45. [DOI] [PubMed] [Google Scholar]
- (28).Cukras C, Gaasterland T, Lee P, Gudiseva HV, Chavali VR, Pullakhandam R, Maranhao B, Edsall L, Soares S, Reddy GB, Sieving PA, and Ayyagari R (2012) Exome analysis identified a novel mutation in the RBP4 gene in a consanguineous pedigree with retinal dystrophy and developmental abnormalities. PLoS One 7, e50205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Seeliger MW, Biesalski HK, Wissinger B, Gollnick H, Gielen S, Frank J, Beck S, and Zrenner E (1999) Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest. Ophthalmol. Vis. Sci 40, 3–11. [PubMed] [Google Scholar]
- (30).Biesalski HK, Frank J, Beck SC, Heinrich F, Illek B, Reifen R, Gollnick H, Seeliger MW, Wissinger B, and Zrenner E (1999) Biochemical but not clinical vitamin A deficiency results from mutations in the gene for retinol binding protein. Am. J. Clin. Nutr 69, 931–936. [DOI] [PubMed] [Google Scholar]
- (31).Chou CM, Nelson C, Tarle SA, Pribila JT, Bardakjian T, Woods S, Schneider A, and Glaser T (2015) Biochemical Basis for Dominant Inheritance, Variable Penetrance, and Maternal Effects in RBP4 Congenital Eye Disease. Cell 161, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li E, Qian SJ, Winter NS, d’Avignon A, Levin MS, and Gordon JI (1991) Fluorine nuclear magnetic resonance analysis of the ligand binding properties of two homologous rat cellular retinol-binding proteins expressed in Escherichia coli. J. Biol. Chem 266, 3622–3629. [PubMed] [Google Scholar]
- (33).Silvaroli JA, Arne JM, Chelstowska S, Kiser PD, Banerjee S, and Golczak M (2016) Ligand Binding Induces Conformational Changes in Human Cellular Retinol-binding Protein 1 (CRBP1) Revealed by Atomic Resolution Crystal Structures. J. Biol. Chem 291, 8528–8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Folli C, Calderone V, Ottonello S, Bolchi A, Zanotti G, Stoppini M, and Berni R (2001) Identification, retinoid binding, and x-ray analysis of a human retinol-binding protein. Proc. Natl. Acad.Sci. U. S. A 98, 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Herr FM, and Ong DE (1992) Differential interaction of lecithin-retinol acyltransferase with cellular retinol binding proteins. Biochemistry 31, 6748–6755. [DOI] [PubMed] [Google Scholar]
- (36).Farias EF, Ong DE, Ghyselinck NB, Nakajo S, Kuppumbatti YS, and Mira y Lopez R (2005) Cellular retinol-binding protein I, a regulator of breast epithelial retinoic acid receptor activity, cell differentiation, and tumorigenicity. J. Natl. Cancer Inst 97, 21–29. [DOI] [PubMed] [Google Scholar]
- (37).Pierzchalski K, Yu J, Norman V, and Kane MA (2013) CrbpI regulates mammary retinoic acid homeostasis and the mammary microenvironment. FASEB J 27, 1904–1916. [DOI] [PubMed] [Google Scholar]
- (38).Saari JC, Nawrot M, Garwin GG, Kennedy MJ, Hurley JB, Ghyselinck NB, and Chambon P (2002) Analysis of the visual cycle in cellular retinol-binding protein type I (CRBPI) knockout mice. Invest. Ophthalmol. Vis. Sci 43, 1730–1735. [PubMed] [Google Scholar]
- (39).Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, Dierich A, Nilsson CB, Hakansson H, Sauvant P, Azais-Braesco V, Frasson M, Picaud S, and Chambon P (1999) Cellular retinol-binding protein I is essential for vitamin A homeostasis. EMBO J 18, 4903–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Pertwee RG (2005) Pharmacological actions of cannabinoids. Handb. Exp. Pharmacol 168, 1–51. [DOI] [PubMed] [Google Scholar]
- (41).Kiser PD, Golczak M, and Palczewski K (2014) Chemistry of the retinoid (visual) cycle. Chem. Rev 114, 194–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang J, Kiser PD, Badiee M, Palczewska G, Dong Z, Golczak M, Tochtrop GP, and Palczewski K (2015) Molecular pharmacodynamics of emixustat in protection against retinal degeneration. J. Clin. Invest 125, 2781–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Maeda T, Golczak M, and Maeda A (2012) Retinal photodamage mediated by all-trans-retinal. Photochem. Photobiol 88, 1309–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Petrukhin K (2013) Pharmacological inhibition of lipofuscin accumulation in the retina as a therapeutic strategy for dry AMD treatment, Drug Discovery Today: Ther. Drug Discovery Today: Ther. Strategies 10, e11–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sparrow JR (2016) Vitamin A-aldehyde adducts: AMD risk and targeted therapeutics. Proc. Natl. Acad. Sci. U. S. A 113, 4564–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Jin M, Li S, Nusinowitz S, Lloyd M, Hu J, Radu RA, Bok D, and Travis GH (2009) The role of interphotoreceptor retinoid-binding protein on the translocation of visual retinoids and function of cone photoreceptors. J. Neurosci 29, 1486–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Palczewski K, Van Hooser JP, Garwin GG, Chen J, Liou GI, and Saari JC (1999) Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry 38, 12012–12019. [DOI] [PubMed] [Google Scholar]
- (48).Chen Y, Okano K, Maeda T, Chauhan V, Golczak M, Maeda A, and Palczewski K (2012) Mechanism of all-trans-retinal toxicity with implications for stargardt disease and age-related macular degeneration. J. Biol. Chem 287, 5059–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Maeda A, Maeda T, Golczak M, and Palczewski K (2008) Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem 283, 26684–26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Bavik C, Henry SH, Zhang Y, Mitts K, McGinn T, Budzynski E, Pashko A, Lieu KL, Zhong S, Blumberg B, Kuksa V, Orme M, Scott I, Fawzi A, and Kubota R (2015) Visual Cycle Modulation as an Approach toward Preservation of Retinal Integrity. PLoS One 10, e0124940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Maeda A, Golczak M, Chen Y, Okano K, Kohno H, Shiose S, Ishikawa K, Harte W, Palczewska G, Maeda T, and Palczewski K (2012) Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat. Chem. Biol 8, 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kubota R, Al-Fayoumi S, Mallikaarjun S, Patil S, Bavik C, and Chandler JW (2014) Phase 1, dose-ranging study of emixustat hydrochloride (ACU-4429), a novel visual cycle modulator, in healthy volunteers. Retina 34, 603–609. [DOI] [PubMed] [Google Scholar]
- (53).Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, and Singerman LJ (2013) Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina 33, 498–507. [DOI] [PubMed] [Google Scholar]
- (54).Noy N, and Xu ZJ (1990) Interactions of retinol with binding proteins: implications for the mechanism of uptake by cells. Biochemistry 29, 3878–3883. [DOI] [PubMed] [Google Scholar]
- (55).Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, Widder K, Travis GH, and Mata NL (2005) Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest. Ophthalmol. Visual Sci 46, 4393–4401. [DOI] [PubMed] [Google Scholar]
- (56).Sabichi AL, Hendricks DT, Bober MA, and Birrer MJ (1998) Retinoic acid receptor beta expression and growth inhibition of gynecologic cancer cells by the synthetic retinoid N-(4-hydroxyphenyl) retinamide. J. Natl. Cancer Inst 90, 597–605. [DOI] [PubMed] [Google Scholar]
- (57).Poliakov E, Samuel W, Duncan T, Gutierrez DB, Mata NL, and Redmond TM (2017) Inhibitory effects of fenretinide metabolites N-[4-methoxyphenyl]retinamide (MPR) and 4-oxo-N-(4-hydroxyphenyl)retinamide (3-keto-HPR) on fenretinide molecular targets beta-carotene oxygenase 1, stearoyl-CoA desaturase 1 and dihydroceramide Delta4-desaturase 1. PLoS One 12, e0176487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wald G (1968) Molecular basis of visual excitation. Science 162, 230–239. [DOI] [PubMed] [Google Scholar]
- (59).Krohn RM, Parsons SA, Fichna J, Patel KD, Yates RM, Sharkey KA, and Storr MA (2016) Abnormal cannabidiol attenuates experimental colitis in mice, promotes wound healing and inhibits neutrophil recruitment. J. Inflammation (London, U. K.) 13, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, and Bradshaw HB (2010) N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci 11, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Jarai Z, Wagner JA, Varga K, Lake KD, Compton DR, Martin BR, Zimmer AM, Bonner TI, Buckley NE, Mezey E, Razdan RK, Zimmer A, and Kunos G (1999) Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc. Natl. Acad. Sci. U. S. A 96, 14136–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Matouk AI, Taye A, El-Moselhy MA, Heeba GH, and Abdel-Rahman AA (2018) Abnormal cannabidiol confers cardioprotection in diabetic rats independent of glycemic control. Eur. J. Pharmacol 820, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).E, X., Zhang L, Lu J, Tso P, Blaner WS, Levin MS, and Li E (2002) Increased neonatal mortality in mice lacking cellular retinol-binding protein II. J. Biol. Chem 277, 36617–36623. [DOI] [PubMed] [Google Scholar]
- (64).Zizola CF, Schwartz GJ, and Vogel S (2008) Cellular retinol-binding protein type III is a PPARgamma target gene and plays a role in lipid metabolism. Am. J. Physiol. Endocrinol. Metab 295, E1358–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Arne JM, Widjaja-Adhi MA, Hughes T, Huynh KW, Silvaroli JA, Chelstowska S, Moiseenkova-Bell VY, and Golczak M (2017) Allosteric modulation of the substrate specificity of acyl-CoA wax alcohol acyltransferase 2. J. Lipid Res 58, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr., Sect. D: Biol. Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Folli C, Calderone V, Ramazzina I, Zanotti G, and Berni R (2002) Ligand binding and structural analysis of a human putative cellular retinol-binding protein. J. Biol. Chem 277, 41970–41977. [DOI] [PubMed] [Google Scholar]
- (69).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (70).Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (73).Golczak M, Bereta G, Maeda A, and Palczewski K (2010) Molecular biology and analytical chemistry methods used to probe the retinoid cycle. Methods Mol. Biol 652, 229–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Stecher H, Gelb MH, Saari JC, and Palczewski K (1999) Preferential release of 11-cis-retinol from retinal pigment epithelial cells in the presence of cellular retinaldehyde-binding protein. J. Biol. Chem 274, 8577–8585. [DOI] [PubMed] [Google Scholar]
- (75).Crabb JW, Chen Y, Goldflam S, West K, and Kapron J (1998) Methods for producing recombinant human cellular retinaldehyde-binding protein. Methods Mol. Biol 89, 91–104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.