

Abstract
Human cells express the UDG superfamily of glycosylases, which excise uracil (U) from the genome. The three members of this structural superfamily are uracil DNA glycosylase (UNG/UDG), single-strand selective monofunctional uracil DNA glycosylase (SMUG1), and thymine DNA glycosylase (TDG). We previously reported that UDG is efficient at removing U from DNA packaged into nucleosome core particles (NCP) and is minimally affected by the histone proteins when acting on an outward-facing U in the dyad region. In an effort to determine whether this high activity is a general property of the UDG superfamily of glycosylases, we compare the activity of UDG, SMUG1, and TDG on a U:G wobble base pair using NCP assembled from Xenopus laevis histones and the Widom 601 positioning sequence. We found that while UDG is highly active, SMUG1 is severely inhibited on NCP and this inhibition is independent of sequence context. Here we also provide the first report of TDG activity on an NCP, and found that TDG has an intermediate level of activity in excision of U and is severely inhibited in its excision of T. These results are discussed in the context of cellular roles for each of these enzymes.
Keywords: nucleosome core particle, base excision repair, uracil DNA glycosylase, single-strand selective monofunctional uracil DNA glycosylase, thymine DNA glycosylase
1. Introduction
Cellular DNA is exposed to a wide variety of both exogenous and endogenous damaging agents, and the resulting lesions may carry mutagenic, apoptotic, or transformative potential for the cell. Nucleobase lesions, for example the deamination of C to U, are repaired via the base excision repair (BER) pathway (1, 2). A DNA glycosylase, such as a uracil DNA glycosylase, recognizes and cleaves the glycosidic bond of the U, creating an abasic site that is subsequently incised by apurinic/apyrimidinic endonuclease 1 (APE1). Polymerase β (polβ) then removes the deoxyribosephosphate (dRP) and inserts a C opposite the unpaired G, and ligase seals the nick (1). Humans have a total of four glycosylases responsible for removing U lesions: uracil DNA glycosylase (UNG/UDG), single-strand selective monofunctional uracil DNA glycosylase (SMUG1), thymine DNA glycosylase (TDG), and methyl binding domain 4 (MBD4) (2). With the exception of MBD4, the human uracil glycosylases belong to the structural UDG superfamily of glycosylases (2, 3). This structural superfamily is defined by a common central α/β fold (highlighted in royal blue in Fig. 1A-C) (3).
Fig. 1.

Graphical representation of co-crystal structures of uracil DNA glycosylases bound to duplex DNA, structure of an NCP, and U:G base pair. (A) UNG2 (PDB 1emh), (B) SMUG1 (PDB 1oe4), and (C) TDG (PDB 5hf7) bound to DNA with the central fold highlighted in royal blue and oriented using the β sheet motif. The helical wedge unique to SMUG1 is highlighted in orange. (D) Merged crystal structure of an NCP containing Widom 601 DNA (PDB 3lz0) and a histone octamer including histone N-terminal tails (PDB 1kx5). The U lesion is highlighted in red using PyMol and the dyad axis is indicated by an arrow. The DNA strand containing the U is numbered from the 5'-end (Widom 601 “i” strand), with the lesion located at position 75. (E) U:G wobble base pair used in this study as a mimic of C deamination.
Though all three members of the UDG superfamily remove U, their substrate specificities and roles within the cell differ dramatically. UDG has the narrowest range of substrates and is able to excise U from single-stranded (ss) DNA, U opposite A or G from duplex DNA, and 5-fluorouracil (5FU) opposite A (4-6). UDG is expressed at high levels and is upregulated during S-phase to remove U misincorporated from the dNTP pool (6). It is noteworthy that UDG is the only member of the superfamily to have a mitochondrial isoform (UNG1), which alone is responsible for the removal of U from the mitochondrial genome (3, 7). SMUG1 has increased substrate tolerance relative to UDG, with the ability to excise U from ssDNA, U opposite A or G, and halogenated and oxidized uracil derivatives (6, 8-11). Unlike the other two members of the superfamily, SMUG1 does not undergo cell cycle regulation, but rather is expressed at constant, low levels (6). Interestingly, SMUG1 tends to accumulate in nucleoli (6), which contain both regions of active transcription and condensed chromatin (12). Like SMUG1, TDG has a wider substrate range than UDG, able to excise U or T opposite G, and a variety of U and C derivatives, including the oxidation products of the epigenetic silencer 5-methylcytosine (5mC), from duplex DNA with greater efficiency in CpG sequence contexts (13-22). TDG, furthermore, interacts with a diverse array of proteins, including chromatin remodelers, transcription factors, and other DNA repair proteins (23-29). The regulation of TDG is maintained through post-translational modifications (PTM) (23, 28, 30-33) and degradation of the protein at the G1/S transition (34, 35).
The biochemical activities of these glycosylases have been characterized in duplex DNA, however there have been fewer examinations of UDG and SMUG1 activity on chromatin or packaged DNA (36-40). The studies on packaged DNA have revealed that UDG and SMUG1 activity is generally lower than on duplex, and that local dynamics of the DNA and associated proteins can modulate activity. There are not any reports of TDG activity on packaged DNA. The nucleosome core particle (NCP) is the most basic unit of DNA packaging (41). It consists of 145-147 bp wrapped ~1.7 times around a core of histone proteins and has a two-fold rotational axis of pseudosymmetry called the dyad axis (Fig. 1D). We recently reported that UDG is unique among a group of glycosylases representing four structural superfamilies in its ability to excise an outward-facing and highly solution-accessible lesion at the often-studied dyad axis region of an NCP with activity comparable to duplex (38). These results led us to question whether the high activity is a property of the entire UDG superfamily or solely of UDG. Here we report that, unlike UDG, SMUG1 exhibits sharply reduced activity for excising an outward-facing U at the dyad relative to U in duplex DNA, while TDG displays intermediate levels of activity. Implications of these observations for the potential biological roles of these glycosylases is discussed.
2. Materials and Methods
2.1. Oligonucleotide synthesis and purification
Oligonucleotides were synthesized using a MerMade 4 (BioAutomation) DNA synthesizer using standard phosphoramidite chemistry. The reagents used for synthesis were from Glen Research. The final dimethoxytrityl (DMT) group was retained for HPLC purification at 90 °C (Agilent PLRP-S column, 250 mm × 4.6 mm; A= 100 mM triethylammonium acetate [TEAA] in 5% [v/v] aqueous acetonitrile [MeCN], B= 100 mM TEAA in MeCN; 5:95 to 30:70 A:B over 30 min at 1 mL/min). Oligonucleotides were subject to detritylation through incubation for 60 min at room temperature in 20% (v/v) aqueous glacial acetic acid. The reaction was quenched by ethanol precipitation. A second round of HPLC was then performed at 90 °C (Agilent PLRP-S column, 250 mm × 4.6 mm; A= 100 mM triethylammonium acetate [TEAA] in 5% [v/v] aqueous acetonitrile [MeCN], B= 100 mM TEAA in MeCN; 0:100 to 25:75 A:B over 40 min at 1 mL/min).
2.2. Preparation of 145mer oligonucleotides
145mer oligonucleotides were assembled by ligation of shorter strands (Scheme S1). The component oligonucleotides were 5'-phosphorylated in 2 mM ATP by T4 polynucleotide kinase (New England BioLabs). Phosphorylated oligonucleotides were annealed in a 1:1 ratio with scaffolds by heating to 95 °C for 5 min and cooling at a rate of 1 °C/min to 25 °C. The annealed oligonucleotides were ligated overnight at room temperature by T4 DNA ligase (New England BioLabs). The full-length 145mer oligonucleotides were then purified by 8% denaturing PAGE (0.8 mm thickness). The concentrations of oligonucleotides were determined by absorbance at 260 nm using molar extinction coefficients calculated by OligoAnalyzer 3.1 (www.idtdna.com).
2.3. Glycosylase expression and purification
Recombinant human TDGFL (full length) and TDG82-308 (residues 82-308 of 410 total) were expressed in Escherichia coli and purified essentially as previously described (19), except that the final chromatographic step for purification of TDGFL employed a Mono Q anion exchange column (GE Healthcare). E. coli UDG and human SMUG1 were purchased from New England BioLabs. E. coli UDG is over 55% identical and 73% similar to human UNG, with the active site completely conserved (42). Furthermore, E. coli UDG and human UNG are highly similar in overall conformation, with root mean square deviation of <1 Å with Cα alignment (43). We note that several studies have examined the activity of both E. coli UDG and human UNG and both enzymes generally have activity in excising U from NCP (36-39, 44). The concentrations of UDG and SMUG1 were determined by Bradford assay against γ-globulin standards (Bio-Rad Laboratories), while the concentrations of TDGFL and TDG82-308 were determined by absorbance at 280 nm (19).
2.4. Reconstitution of nucleosome core particles
The expression and purification of recombinant histone proteins from X. laevis and histone octamer assembly were performed according to the published method of Luger and colleagues (45, 46). Reconstitution of NCP were carried out via the salt dialysis method as reported previously (37-39). More specifically, a Slide-a-Lyzer MINI dialysis device (0.1 mL capacity, 3.5 kDa MWCO; Thermo Fisher Scientific) was equilibrated for 30 min at 4 °C in deionized water followed by 10 min in buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1 mM dithiothreitol [DTT], 2 M NaCl). 5'-32P-labeled 145mer U- or T-containing lesion strand was annealed to its complement (1:1.05 lesion strand:complement strand) by heating to 95 °C for 5 min and cooling at a rate of 1 °C/min to 25 °C in 10 mM Tris-HCl (pH 8.0), 30 mM NaCl. The annealed lesion-containing duplexes were used in the NCP reconstitution as well as for the analogous duplex control experiments. 20 μL (5'-UpG/CpG-3' or 5'-TpG/CpG-3' duplex) or 50 μL (5'-UpT/ApG-3' duplex) of 1 μM duplex was added to the dialysis device and equilibrated for 30 min prior to the addition of histone octamer which was in 10 mM Tris-HCl (pH 7.5), 1.8 M NaCl, 42% (v/v) glycerol (1:1.05 duplex:octamer). The NaCl concentration in the dialysis buffer was lowered stepwise every hour (1.2 M, 1.0 M, 0.6 M, 0 M). The final dialysis in 0 M NaCl was performed for 3 h. The samples were then filtered by centrifugation at 14,000 rpm for 5 min in a Spin-X Centrifuge Tube Filter (0.22 μm, Corning Incorporated) to remove precipitated products. The formation of NCP was confirmed by 7% non-denaturing PAGE (60:1 acrylamide:bisacrylamide, 0.25X TBE; 3 h at 150 V, 4 °C). A representative non-denaturing gel is shown in Fig. S1. Only NCP preparations for which there was < 5% duplex (as determined by densitometry) were used for kinetic experiments.
2.5. Glycosylase kinetics experiments
Samples of 5'-32P-labeled U- or T-containing substrate and glycosylase were prepared. The substrate samples contained 40 nM DNA (duplex or NCP) in Buffer TNK [20 mM Tris-HCl (pH 8.0), 25 mM NaCl, 75 mM KCl, 1 mM EDTA, 1 mM DTT, 200 μg/mL BSA]. SMUG1 and UDG (1.28 μM) solutions were prepared in Buffer TNK. TDG (800 nM), both TDGFL and TDG82-308, was prepared in Buffer TNK plus 8% (v/v) glycerol. The substrate and glycosylase samples were pre-incubated at 37 °C for 2 min, followed by mixing of equal volumes (8 μL each) of DNA substrate and glycosylase for a final reaction condition of 20 nM DNA substrate, 640 nM UDG or SMUG1 in Buffer TNK; or 20 nM DNA substrate (duplex or NCP), 400 nM TDGFL or TDG82-308 in Buffer TNK plus 4% (v/v) glycerol. Enzyme concentrations were selected to ensure single-turnover (STO) conditions and the same kobs was observed in experiments performed at 2-fold higher concentration of glycosylase. Reactions were incubated at 37 °C and quenched by the addition of 16 μL 1 M NaOH (final concentration 500 mM). For each time course two negative controls were included. The no enzyme sample (−E) was prepared by mixing 8 μL DNA substrate with 8 μL Buffer TNK, and incubation at 37 °C for the longest time point followed by addition of 16 μL 1 M NaOH quench. The quench control (QC) sample was prepared by mixing 8 μL DNA substrate with 16 μL 1 M NaOH quench, followed immediately by mixing with 8 μL glycosylase and incubation at 37 °C for the longest time point. This control serves not only to identify any baseline levels of damage in the strands, but also to confirm that the quench is immediate. To evaluate whether low product yields were the result of loss of enzyme activity, a separate control was used (marked “120*” in Fig. S3 and “60*” in Fig. S5). For these two reactions, the reactions were initiated as described above. At either 60 min (Fig. S3) or 30 min (Fig. S5), an additional 8 μL 1.28 μM SMUG1 (Fig. S3) or 800 nM TDGFL or TDG82-308 (Fig. S5) were added. The reactions were incubated for another 60 min (SMUG1, Fig. S3) or 30 min (TDGFL or TDG82-308, Fig. S5) followed by addition of 24 μL 1 M NaOH quench. All samples were then heated at 90 °C for 2 min to produce strand breaks at abasic sites. DNA from NCP substrates was isolated from the histone proteins via 25:24:1 phenol:choloroform:isoamyl alcohol extraction. All samples (NCP and duplex) were desalted via ethanol precipitation. DNA substrate and product were resolved by 8% denaturing PAGE, imaged by phosphorimagery, and quantitated by densitometry.
The fraction product, Fp, at each time point t was determined as follows:
in which δs(t) and δp(t) represent the densities of substrate and product bands, respectively, at time t. The product yield at each time point, P(t), was corrected for baseline levels of damage as follows:
in which Fp(0) is the fraction product in the QC sample. The product yield for each time course was plotted as a function of time and fit using nonlinear least-squares regression to the modified first-order integrated rate law:
or
where P(t) is the product yield at time t, P(∞) is the maximum product yield (Kaleidagraph). Double exponential fits were used when single exponential fits yielded R2 values <0.95. Reaction rates, kobs, and product yield were reported from the fits. The reported rates represent mean ± standard error. All time courses with NCP were performed on at least two separate NCP reconstitutions.
Rates on duplex substrates were compared by analysis of variance (ANOVA) followed by a post hoc Tukey test (α=0.01). Rates on NCP substrates were compared separately by ANOVA followed by a post hoc Tukey test (α=0.01). All rates and product yields were compared to the rate of UDG on duplex using ANOVA followed by a post hoc Tukey test (α=0.01). Within individual glycosylases, rates and yields on duplex and NCP were compared by two-tailed Welch’s t test (α=0.01). ANOVA, specifically, was used for multiple comparisons while Welch’s t test was used only for single pairwise comparisons. All statistical tests were performed using R. Here we consider p <0.01 to be significant.
3. Results
3.1. Rationale and experimental design
In a previous survey of five glycosylases, we observed that UDG is exceptional in its rapid excision of an outward-facing lesion from the dyad region of an NCP (38). In light of this observation, we sought to examine whether this result is unique to UDG, or is a property of the UDG superfamily of glycosylases. We designed a substrate based on the 145 bp Widom 601 DNA sequence and containing a U:G wobble bp at position 75, the same location as our previous work (Fig. 1D and E; Scheme S1) (38). The Widom 601 sequence binds the histone octamer core in a single orientation, producing a homogenous population of NCP (47). We chose a U:G bp mimicking the deamination of a C:G bp as recent reports have shown that mice lacking both UNG and SMUG1 have higher levels of U resulting from C deamination (48). The U:G bp was positioned within a CpG dinucleotide (5'-UpG/CpG-3') as TDG shows the highest activity in this sequence context (17). Neither UDG nor SMUG1 are biased for or against this sequence context in duplex (6). Furthermore, the tumors of mice lacking both UNG and SMUG1 have increased incidence of C to T transition mutations specifically in CpG dinucleotides, demonstrating a role for these two glycosylases in the maintenance of CpG sites (48).
Given the severe product inhibition of both SMUG1 and TDG (6, 49), and to a lesser extent UDG (50), we performed all experiments under single-turnover (STO) conditions, with an excess of glycosylase over substrate. Under these conditions, the observed rate (kobs) is reflective of the slowest kinetic step up to and including chemistry, where chemistry is cleavage of the glycosidic bond.
3.2. Rapid excision of uracil from duplex and NCP by UDG
When UDG is incubated with the U:G duplex control, product rapidly accumulates with a kobs of 24.9 ± 1.2 min−1, which is comparable to rates previously reported for UDG acting on 145 bp duplex (Fig. 2A, Table 1) (38, 39). Though this kobs is significantly slower than excision of U from a 19 bp duplex (50), similar discrepancies between STO values on longer duplexes have been observed for other glycosylases (38) and are likely due to increased nonspecific binding (51). We also observed rapid product formation using ssDNA, consistent with the known activity of UDG on such substrates (Fig. S2). Since kobs for UDG excision of U from duplex is the fastest observed in this work, we used it as a benchmark for comparison of all other glycosylases and substrates.
Fig. 2.

Single-turnover kinetics of UDG superfamily of glycosylases. Kinetic time courses were used to evaluate the excision of U from a U:G bp in (A) duplex DNA and (B) NCP. Reactions consisted of 20 nM substrate (duplex or NCP), 640 nM SMUG1 or UDG in Buffer TNK and 400 nM TDGFL or TDG82-308 in Buffer TNK plus 4% (v/v) glycerol. Data were fit to a single or double exponential equation (see Materials and Methods). Error bars represent standard error (n=3 for all duplex, TDG82-308 on the NCP; n=4 for UDG, SMUG1, and TDGFL on the NCP). Black circles( ), UDG; red squares (
), UDG; red squares ( ), SMUG1; blue diamonds (
), SMUG1; blue diamonds ( ), TDGFL; green triangles (
), TDGFL; green triangles ( ), TDG82-308.
), TDG82-308.
Table 1.
kobs Values and Product Yield for U:G in Duplex DNA and NCP.
| Enzyme | Substrate | kobs min−1 (Fraction product)a | |
|---|---|---|---|
| UDG | Duplex | 24.9 ± 1.2 (0.98) | |
| NCP | 5.7 ± 0.4 * (0.98) | ||
| SMUG1 | Duplex | 2.1 ± 0.1 * (0.99) | |
| NCP | 1.1 ± 0.6 * (0.04) | (4.9 ± 4.2)×10−3 * (0.21) | |
| TDGFL | Duplex | 6.2 ± 0.4 * (0.98) | |
| NCP | 3.4 ± 0.9 * (0.09) | 0.03 ± 0.01 * (0.62) | |
| TDG82-308 | Duplex | 7.2 ± 1.7 * (0.98) | |
| NCP | 1.2 ± 0.2 * (0.18) | 0.04 ± 0.01 * (0.65) | |
Rates reported as mean ± standard error (n=3 for duplex substrates, TDG82-308 NCP; n=4 for UDG, SMUG1, TDGFL NCP substrates). Asterisk (*) denotes p<0.01 compared to UDG duplex.
In accordance with our previous observation (38), UDG rapidly excises an outward-facing U from the dyad region of an NCP with kobs of 5.7 ± 0.4 min−1 (Fig. 2B, Table 1), a rate that is only ~4-times slower than that observed for duplex. On both NCP and duplex, UDG exhibits monophasic kinetics.
3.3. SMUG1 exhibits differential activity on duplex and NCP substrates
SMUG1 rapidly excises U from the duplex control and complete conversion to product was observed as previously reported (36), with kobs of 2.1 ± 0.1 min−1 (Fig. 2A, Table 1).
When SMUG1 is incubated with the NCP, striking differences are observed when compared to UDG. SMUG1 does not fully convert the NCP substrate to product, consistent with previous reports (36), and in fact, exhibits very little product formation (Fig. 2B, Table 1). Supplementing the reaction with more SMUG1 after the longest reaction time does not result in a substantial increase in product, indicating that the low level of product formation is not the result of SMUG1 losing activity over the course of the reaction (Fig. S3). Also in contrast to UDG, SMUG1 exhibits biphasic kinetics on the NCP and the rates are significantly different from UDG on both duplex and NCP.
Though SMUG1 does not exhibit strong sequence bias in duplex DNA, it does exhibit bias against U in a 5'-UpG-3' ssDNA context, as would be formed by deamination of C in a CpG dinucleotide (11). Given the sequence preferences of SMUG1 on ssDNA, we questioned whether the low SMUG1 activity on the NCP reflects a sequence preference. We performed experiments on ssDNA, duplex, and NCP substrates in the 5'-UpT/ApG-3' sequence context, as SMUG1 displays a preference for this sequence context in ssDNA (11) (Fig. 3, Table S1). In accordance with a previous report (11), we observed a bias against 5'-UpG/CpG-3' in ssDNA but not duplex substrates. In the NCP, we again observed biphasic kinetics; however, there was no significant difference in either product accumulation or between the fast rates or the slow rates in the two sequence contexts. Overall, we observed similar results for both sequence contexts in both duplex and NCP.
Fig. 3.

Single-turnover kinetics of SMUG1 activity in different sequence contexts. (A) Kinetic time courses were used to evaluate the excision of U in ssDNA substrates in 5'-UpT-3' (dashed line) and 5'-UpG -3' (solid line) sequence contexts. Reactions consisted of 20 nM DNA substrate, 640 nM SMUG1 in Buffer TNK. Data were fit to a single exponential equation. Error bars represent standard error (n=2 for 5'-UpT-3'; n=3 for 5'-UpG-3'). The observed rates and fraction product are kobs=0.14 ± 0.01 min−1, 0.92 (5'-UpT-3') and kobs=0.02 ± 0.01 min−1, 1.0 (5'-UpG-3'). (B) Kinetic time courses were used to evaluate the excision of U in duplex and NCP in a 5'-UpT/ApG-3' sequence context. Reactions consisted of 20 nM DNA substrate, 640 nM SMUG1 in buffer described in Materials and Methods. Data were fit to a single exponential equation (duplex), or double exponential equation (NCP). Error bars represent standard error (n=2 for duplex; n=6 NCP). Open squares ( ), duplex; filled squares (
), duplex; filled squares ( ), NCP. The observed rates (fraction product) are kobs=4.4 ±0.1 min−1 (0.99) (duplex) and kobs-fast=4.0 ± 0.9 min−1 (0.06), kobs-slow = 0.05 ± 0.02 min−11 (0.14) (NCP).
), NCP. The observed rates (fraction product) are kobs=4.4 ±0.1 min−1 (0.99) (duplex) and kobs-fast=4.0 ± 0.9 min−1 (0.06), kobs-slow = 0.05 ± 0.02 min−11 (0.14) (NCP).
3.4. TDG is capable of excising U from NCP substrates
For our experiments with TDG, we used two different versions of the enzyme: (1) the full-length enzyme (TDGFL) and (2) a truncated enzyme (residues 82-308, TDG82-308) (19) that is missing portions of the largely unstructured N- and C-termini. We tested the truncated version of TDG to investigate the influence, if any, of the unstructured portions of the protein on enzyme activity, specifically on the NCP substrate.
When TDGFL and TDG82-308 are incubated with duplex, monophasic kinetics and full conversion to product are observed, with kobs of 6.2 ± 0.4 min−1 and 7.2 ± 1.7 min−1 for TDGFL and TDG82-308, respectively (Fig. 2A, Table 1). These rates for excision of U are comparable to each other, consistent with findings for excision of T from a T:G mismatch in duplex for these two versions of TDG (16, 19) (Fig. S4). Additionally, the rate of excision of U from the U:G bp for TDGFL is comparable to that reported previously for a 19 bp duplex (52).
Similar to SMUG1, TDG processes the NCP differently than duplex. Both TDGFL and TDG82-308 exhibit biphasic kinetics, with fast and slow phases of 3.4 ± 0.9 min−1 and 0.03 ± 0.01 min−1 for TDGFL and 1.2 ± 0.2 min−1 and 0.04 ± 0.01 min−1 for TDG82-308 (Fig. 2B, Table 1). Though for each individual TDG construct the fast and slow rates differ dramatically (kobs-fast >> kobs-slow), the fast and slow rates of TDGFL are not statistically different from those of TDG82-308. Additionally, the fast rates observed for the NCP are not significantly different from duplex for either version of TDG. Although all the rates on the NCP are significantly different from UDG, they are not different from SMUG1.
In accordance with SMUG1, neither TDGFL nor TDG82-308 fully converts the NCP to product over the time course, though both versions of TDG accumulate significantly more product over the time course as compared to SMUG1 and significantly less than UDG. As with SMUG1, addition of more TDG at the end of the time course did not yield a significant increase in product (Fig. S5).
Since we observed the capability of TDG to excise U from an NCP, we sought to determine whether T could be excised similarly from a T:G mismatch. In this case, T:G in 5'-TpG/CpG-3' mimics the deamination of 5mC:G. In contrast to excision of U from an NCP, we observed minimal excision of T (≤10%) for both TDGFL and TDG82-308, with kobs of 0.3 ± 0.1 min−1 and 0.5 ± 0.1 min−1, respectively. These kobs values are consistent with observed rates for excision of T from 145 bp duplex (Fig. S4) as well as 19 bp duplex (16, 19).
4. Discussion
All the UDG superfamily glycosylases examined here, UDG, SMUG1, TDGFL, and TDG82-308, are capable of completely converting U-containing duplex substrates to product, though at different rates. Under STO conditions, kobs reflects the slowest kinetic step up to and chemistry. These steps include DNA binding, distortion of the DNA helix for base flipping into the active site, intercalation of an amino acid residue into the DNA helix to plug the resulting hole, and chemistry. In comparing the three UDG superfamily glycosylases excising U from U:G in duplex, it is clear that UDG is the fastest, while SMUG1, TDGFL, and TDG82-308 all have comparable kobs that are ~3-12 times slower than UDG. That UDG is fastest then begs the question as to whether SMUG1 and TDG serve functions only redundant to UDG for excision of U in duplex-like DNA, that is, DNA that is not wrapped around histone proteins. Duplex-like DNA may be found in the cell in several situations: between adjacent nucleosomes, during chromatin remodeling, or transiently unwrapped from nucleosomes during transcription and replication.
The cell cycle regulation of each of the UDG superfamily glycosylases may suggest which of these types of duplex-like DNA, if any, are substrates for the enzymes. UDG is upregulated during S-phase and tends to accumulate specifically at replication foci to remove U misincorporated from the dNTP pool (6). UDG may exploit the existence of unwrapped DNA at this time to maximize excision of U from duplex-like or even ssDNA. SMUG1, on the other hand, does not undergo appreciable variation of expression throughout the cell cycle, and instead is expressed continuously at low levels, in both actively replicating and quiescent cells (6). It is possible that SMUG1, with observed higher activity on duplex substrates, may take advantage of transient duplex-like DNA throughout the cell cycle. Like UDG, TDG levels are regulated, but TDG is actively degraded at the G1/S transition (34, 35). Therefore, unlike UDG and SMUG1, TDG is not available during replication. Should TDG require duplex-like DNA for efficiency, it would need to rely on other processes, such as transcription or chromatin remodelers, to access its substrate.
Though it is clear that UDG superfamily glycosylases are capable of excising U in duplex DNA, their activities on packaged DNA have not been as well characterized. Here we have shown that for an outward-facing U at the dyad axis of an NCP compared to the analogous duplex, UDG is minimally inhibited, SMUG1 is the most inhibited, and TDG exhibits intermediate levels of inhibition. UDG is unique among these three UDG superfamily glycosylases because of its monophasic kinetics and full conversion to product for the NCP substrate. These results are especially interesting given that the UDG enzyme used in this study is the E. coli ortholog, which was not influenced by the presence of nucleosomes through its evolution. We note, however, that the E. coli and human orthologues of UDG are highly similar in both sequence (42) and overall conformation. Comparison of the crystal structures of E. coli UDG and human UNG show root mean square deviation of <1 Å when Cα are aligned (43). The single-phase kinetics reveal that the entire population of NCP is in a form that is readily accessible to UDG for glycosidic bond cleavage, with a kobs that is only ~4-times slower than duplex. This observation is consistent with what we and others have observed previously, a slower kobs for UDG excision at the dyad region versus duplex (38, 53). We note, however, that Cole et al. observed a more dramatic decrease in kobs at the dyad than what we describe here (44). We attribute this discrepancy to a difference in the DNA positioning sequence, namely, the 5S rDNA versus Widom 601 positioning sequences. For the NCP substrates, kobs could represent the same kinetic step as kobs on duplex. However, it is also possible that kobs on the NCP represents a rate-limiting step prior to chemistry.
In contrast to UDG, SMUG1 exhibits dramatically lower product yield in its excision of U, and this is not dependent on sequence context. Though we cannot rule out that a small amount of the product is derived from a duplex contaminant, our extensive purification techniques limit the amount of contaminating duplex in the NCP sample. The limited amount of duplex (<5%) combined with the observation that SMUG1 exhibits biphasic kinetics on this substrate lead us to conclude that some product results from SMUG1 excising U from the NCP. Due to the low product yield associated with kobs-fast, we cannot rule out that this fast phase represents SMUG1 acting on a small amount of duplex. In this case, kobs-slow represents SMUG1 acting on NCP. The product yield reveals that only −20% of the NCP population is accessible to SMUG1. On the other hand, both phases may reflect SMUG1 acting on the NCP. The fast phase would therefore correspond to SMUG1 excising U on a readily accessible population, while the slow phase reflects a population that requires, for example, a conformational change prior to SMUG1 binding and/or cleavage. Nevertheless, a substantial proportion of the NCP substrate is inaccessible to glycosidic bond cleavage by SMUG1.
The significant inhibition of SMUG1 on the NCP substrate may be due, at least in part, to the presence of an additional α-helix directly C-terminal to its intercalation loop (9). Though UDG, SMUG1, and TDG all possess a similar intercalation loop, SMUG1 is unique in its possession of this additional α-helix, which forms a “helical wedge” (highlighted orange in Fig 1B) and is believed to promote DNA distortion and U extrusion (9). In contrast to UDG and TDG, which disrupt only the base pair containing the scissile U, SMUG1 also disrupts the base pair on the 5′-side of the U, leading to a greater distortion of the DNA helix (9). In the context of an outward-facing lesion at the dyad, the torsional flexibility of the DNA is significantly decreased relative to duplex DNA in solution (47, 54), and may provide a barrier to DNA distortion necessary for glycosidic bond cleavage. This barrier to DNA distortion may be responsible for the low product yields observed for the excision of U from NCP. In this case, the requirement for DNA distortion may also prevent efficient repair of sites that are less solution accessible. In contrast, lesions displaced from the dyad, especially at the DNA ends where there are increased dynamics (55-58), may be more easily repaired by SMUG1. The results then bring into question whether an outward-facing U at the dyad is a substrate for SMUG1 in a cellular context. It may be the case that SMUG1 relies on chromatin remodelers, transcriptional machinery, or the degree of DNA packaging variable with the cell cycle to make lesions, both at the dyad and throughout the NCP, more accessible. Conversely, these lesions at the dyad may be removed primarily by UDG, and thus may seldom be a substrate for SMUG1. While SMUG1 may defer to UDG for lesion removal in some contexts, it tends to accumulate in nucleoli, which lack UDG (6). Therefore, there may be factors specific to nucleoli that increase SMUG1 activity on packaged DNA. Conversely, SMUG1 may take advantage of the high transcription of ribosomal genes in the nucleoli and its more duplex-like character (12) for its excision of U.
TDGFL and TDG82-308 exhibit biphasic kinetics on the NCP, suggesting that SMUG1 and TDG are more similar to each other than to UDG. For both TDGFL and TDG82-308, kobs-fast does not differ significantly from kobs for duplex, suggesting that these rates may correspond to the same kinetic step. Additionally, the rates obtained for TDGFL and for TDG82-308 are not statistically different. Again, we cannot rule out that a small amount of contaminating duplex contributes to the product yield for the fast phase, or, in contrast, this phase corresponds to an NCP population readily accessible to TDG. Recent molecular dynamic simulations examining the steps after binding and up to but excluding bond cleavage have indicated that the rate-limiting step for TDG on duplex substrates is the intercalation of R275 to plug the hole left by the extruded nucleobase (59, 60). Due to the decreased dynamics of DNA in the dyad region (47, 54), there may be a subset of TDG-NCP complexes for which intercalation of R275 is slowed, and thus requires a conformational change in the NCP in order to proceed.
TDG, furthermore, is inhibited in its excision of U on the NCP relative to duplex, though not to the same extent as SMUG1. There is no significant difference between TDGFL and TDG82-308, consistent with a previous report that the N- and C-termini missing from TDG82-308 do not affect the excision of U or T from duplex (19). Nevertheless, there is a proportion of the NCP substrate inaccessible to glycosidic bond cleavage by TDG. This inaccessible substrate may be a population of TDG-NCP complexes for which U extrusion and/or intercalation of R275 is inhibited. On the other hand, this proportion of the substrate may be inaccessible to initial binding of TDG, due to steric clash of the enzyme with the histone core. Excision of T from a T:G wobble bp in the dyad region is significantly inhibited, with product yields less than 10% for both TDGFL and TDG82-308. Due to the increased steric hindrance of extruding T versus U, T is excised at a slower rate than U (61). Furthermore, it is known that a specific conformation of the N-terminus is required for the recognition and removal of T and deletion of the entire N-terminus results in loss of the ability to excise T (15, 62). Indeed, it has been demonstrated that more stable binding of TDG to the DNA is required for the excision of T versus U (63). Steric clash of the enzyme with the histone core may prevent productive binding to the NCP, thus resulting in low product yields.
Since TDG is degraded prior to S-phase and cannot take advantage of the more duplex-like character of DNA during this phase of the cell cycle (34, 35), the question remains as to which factors, if any, may increase the efficiency of removal of U and T. Possible mechanisms may include PTM of TDG or the histone proteins, interaction with and/or the presence of chromatin remodelers, and interactions with other proteins. TDG has been demonstrated to be acetylated (23, 28, 29, 32) and phosphorylated (32) on its N-terminus, and SUMOylated (30, 31, 33, 64, 65) on its C-terminus. Phosphorylation has been demonstrated to increase activity, SUMOylation severely inhibits excision while augmenting APE1-induced product release, and acetylation has either stimulatory or inhibitory effects, based on the lesion in question (28, 32, 33, 65). TDG also interacts with a wide variety of chromatin remodelers, including the methyltransferases DMT3A (26), DMT3B (27), the lysine acetyltransferase CBP/p300 (23, 29), and the NAD+-dependent deacetylase SIRT1 (28). TDG has been demonstrated to interact with several other proteins, including the nucleotide excision repair protein XPC (24) and the transcription factor estrogen receptor α (66). The presence and/or absence of these factors separately or in combination may modulate the efficiency of TDG, resulting in precise spatiotemporal activity.
In light of our results, it is curious that UDG is unique among the UDG superfamily glycosylases in its rapid and complete excision of an outward-facing U at the dyad. SMUG1 and TDG, on the other hand, do not convert the substrate fully to product. It is noteworthy that UDG differs from TDG and SMUG1 in its intercalating residue, which stabilizes the duplex when the U is extruded from the helix. While the intercalating residue of UDG is a leucine (67), both SMUG1 and TDG use an arginine (9, 18, 19, 59, 60). The positive charge on the arginine may cause electrostatic repulsion from the overall positive charge of the histone core, thus limiting the intercalation step and preventing glycosidic bond cleavage. Indeed, a mutation of the intercalating leucine in UNG to arginine (L272R) modestly decreases steady-state kinetic parameters kcat, Km, and kcat/Km on duplex substrates (68). The helical wedge of SMUG1 presents another barrier to intercalation, as the rather constrained DNA at the dyad axis must be disrupted further for catalysis to occur. Given these results, UDG may be the main glycosylase for excision of outward-facing U near the dyad axis, with roles for SMUG1 and TDG in specific spatiotemporal and/or sequence contexts, perhaps aided by the presence of other factors and/or PTM.
Conclusion
In summary, we show that UDG is unique among the UDG superfamily glycosylases in its rapid excision of an outward-facing U from the dyad of an NCP. Though both SMUG1 and TDG demonstrate inhibition in excision of U from the NCP compared to duplex, SMUG1 displays the most drastic inhibition. These results bring into question whether other cellular factors, including PTM, chromatin remodelers, or phases of the cell cycle may influence the efficiency of U excision. Further studies combining in vitro and in vivo approaches will help to elucidate the roles of these uracil glycosylases within the cell.
Supplementary Material
Fig. 4.
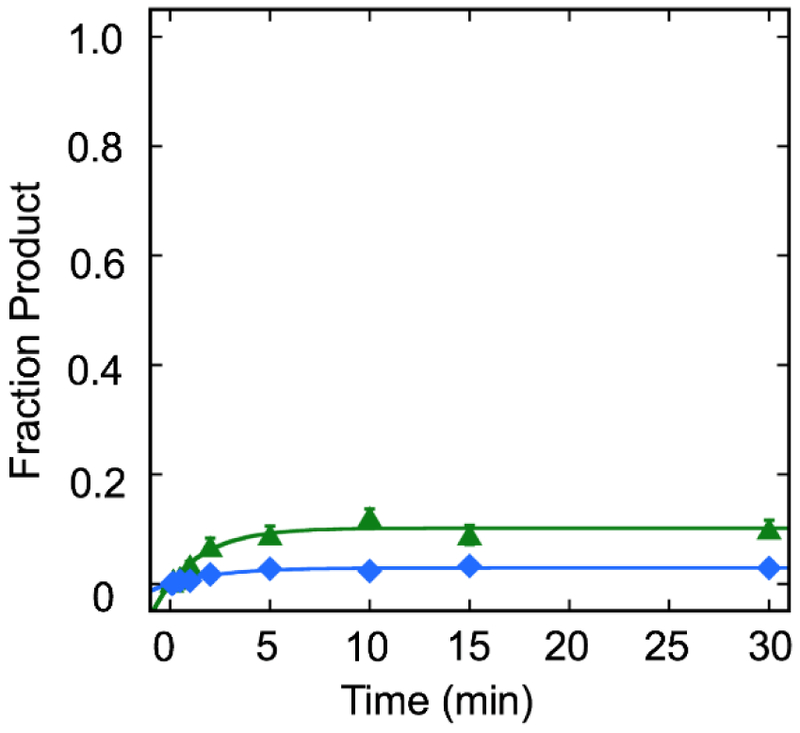
Single-turnover kinetics of TDGFL and TDG82-308 excising T from a T:G mismatch in an NCP. Kinetic time courses were used to evaluate the excision of T in NCP substrates in a 5'-TpG/CpG-3' sequence context. Reactions consisted of 20 nM DNA substrate, 400 nM TDGFL and TDG82-308 in Buffer TNK plus 4% (v/v) glycerol. Data were fit to a single exponential equation. Error bars represent standard error (n=3 for TDGFL; n=4 for TDG82-308). Blue diamonds ( ), TDGFL; green triangles (
), TDGFL; green triangles ( ), TDG82-308. The observed rates (fraction product) are kobs=0.3 ± 0.1 min−1 (0.03) (TDGFL) and kobs=0.5 ± 0.1 min−1 (0.10) (TDG82-308).
), TDG82-308. The observed rates (fraction product) are kobs=0.3 ± 0.1 min−1 (0.03) (TDGFL) and kobs=0.5 ± 0.1 min−1 (0.10) (TDG82-308).
Acknowledgements
This work was supported by the National Science Foundation (MCB-1817417 to S.D.) and National Institutes of Health (R01-GM72711 to A.C.D.). We thank members of the Delaney laboratory for careful reading of the manuscript and helpful discussion.
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Appendix A: Supplementary Data
Supplementary data for this article can be found in the online version.
References
- 1.Schermerhorn KM, Delaney S, A Chemical and Kinetic Perspective on Base Excision Repair of DNA. Acc. Chem. Res 47, 1238–1246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks SC, Adhikary S, Rubinson EH, Eichman BF, Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta, Proteins Proteomics 1834, 247–271 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aravind L, Koonin EV, The α/β fold uracil DNA glycosylases: a common origin with diverse fates. Genome Biol. 1, research0007.0001 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B DNA N-Glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem 252, 3286–3294 (1977). [PubMed] [Google Scholar]
- 5.Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE, Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry 34, 128–138 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, Slupphaug G, hUNG2 is the major repair enzyme for removal of uracil from U: A matches, U: G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J. Biol. Chem 277, 39926–39936 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Prakash A, Doublié S, Base excision repair in the mitochondria. J. Cell. Biochem 116, 1490–1499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nilsen H, Haushalter KA, Robins P, Barnes DE, Verdine GL, Lindahl T, Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil–DNA glycosylase. EMBO J. 20, 4278–4286 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH, Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol. Cell 11, 1647–1659 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Matsubara M, Tanaka T, Terato H, Ohmae E, Izumi S, Katayanagi K, Ide H, Mutational analysis of the damage-recognition and catalytic mechanism of human SMUG1 DNA glycosylase. Nucleic Acids Res. 32, 5291–5302 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doseth B, Ekre C, Slupphaug G, Krokan HE, Kavli B, Strikingly different properties of uracil-DNA glycosylases UNG2 and SMUG1 may explain divergent roles in processing of genomic uracil. DNA Repair 11, 587–593 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Németh A, Längst G, Genome organization in and around the nucleolus. Trends Genet. 27, 149–156 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Wiebauer K, Jiricny J, In vitro correction of G to T mispairs to G o C pairs in nuclear extracts from human cells. Nature 339, 234 (1989). [DOI] [PubMed] [Google Scholar]
- 14.Neddermann P, Jiricny J, Efficient removal of uracil from GU mispairs by the mismatch-specific thymine DNA glycosylase from HeLa cells. Proc. Natl. Acad. Sci. U.S.A 91, 1642–1646 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallinari P, Jiricny J, A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature 383, 735 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Bennett MT, Rodgers MT, Hebert AS, Ruslander LE, Eisele L, Drohat AC, Specificity of human thymine DNA glycosylase depends on N-glycosidic bond stability. J. Am. Chem. Soc 128, 12510–12519 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan MT, Bennett MT, Drohat AC, Excision of 5-Halogenated Uracils by Human Thymine DNA Glycosylase: Robust activity for DNA contexts other than CpG. J. Biol. Chem 282, 27578–27586 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maiti A, Morgan MT, Pozharski E, Drohat AC, Crystal structure of human thymine DNA glycosylase bound to DNA elucidates sequence-specific mismatch recognition. Proc. Natl. Acad. Sci. U.S.A 105, 8890–8895 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coey CT, Malik SS, Pidugu LS, Varney KM, Pozharski E, Drohat AC, Structural basis of damage recognition by thymine DNA glycosylase: Key roles for N-terminal residues. Nucleic Acids Res. 44, 10248–10258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohli RM, Zhang Y, TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Y-F, Li B-Z, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu G-L, Luo C, Jiang H, He C, Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol 8, 328–330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tini M, Benecke A, Um S-J, Torchia J, Evans RM, Chambon P, Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell 9, 265–277 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Shimizu Y, Iwai S, Hanaoka F, Sugasawa K, Xeroderma pigmentosum group C protein interacts physically and functionally with thymine DNA glycosylase. EMBO J. 22, 164–173 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lucey MJ, Chen D, Lopez-Garcia J, Hart SM, Pheonix F, Al-Jehani F, Alao JP, White R, Kindle KB, Losson R, T: G mismatch-specific thymine-DNA glycosylase (TDG) as a coregulator of transcription interacts with SRC1 family members through a novel tyrosine repeat motif. Nucleic Acids Res. 33, 6393–6404 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y-Q, Zhou P-Z, Zheng X-D, Walsh CP, Xu G-L, Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 35, 390–400 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boland MJ, Christman JK, Characterization of Dnmt3b: thymine-DNA glycosylase interaction and stimulation of thymine glycosylase-mediated repair by DNA methyltransferase (s) and RNA. J. Mol. Biol 379, 492–504 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madabushi A, Hwang B-J, Jin J, Lu A-L, Histone deacetylase SIRT1 modulates and deacetylates DNA base excision repair enzyme thymine DNA glycosylase. Biochem. J 456, 89–98 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henry RA, Mancuso P, Kuo Y-M, Tricarico R, Tini M, Cole PA, Bellacosa A, Andrews AJ, Interaction with the DNA Repair Protein Thymine DNA Glycosylase Regulates Histone Acetylation by p300. Biochemistry 55, 6766–6775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinacher R, Schär P, Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation. Curr. Biol 15, 616–623 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Mohan RD, Rao A, Gagliardi J, Tini M, SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Mol. Cell. Biol 27, 229–243 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohan RD, Litchfield DW, Torchia J, Tini M, Opposing regulatory roles of phosphorylation and acetylation in DNA mispair processing by thymine DNA glycosylase. Nucleic Acids Res. 38, 1135–1148 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLaughlin D, Coey CT, Yang W-C, Drohat AC, Matunis MJ, Characterizing requirements for small ubiquitin-like modifier (SUMO) modification and binding on base excision repair activity of thymine-DNA glycosylase in vivo. J. Biol. Chem 291, 9014–9024 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibata E, Dar A, Dutta A, CRL4Cdt2 E3 ubiquitin ligase and proliferating cell nuclear antigen (PCNA) cooperate to degrade thymine DNA glycosylase in S phase. J. Biol. Chem 289, 23056–23064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slenn TJ, Morris B, Havens CG, Freeman RM, Takahashi TS, Walter JC, Thymine DNA glycosylase is a CRL4Cdt2 substrate. J. Biol. Chem 289, 23043–23055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nilsen H, Lindahl T, Verreault A, DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 21, 5943–5952 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye Y, Stahley MR, Xu J, Friedman JI, Sun Y, McKnight JN, Gray JJ, Bowman GD, Stivers JT, Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry 51, 6028–6038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olmon ED, Delaney S, Differential Ability of Five DNA Glycosylases to Recognize and Repair Damage on Nucleosomal DNA. ACS Chem. Biol 12, 692–701 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bilotti K, Kennedy EE, Li C, Delaney S, Human OGG1 activity in nucleosomes is facilitated by transient unwrapping of DNA and is influenced by the local histone environment. DNA Repair 59, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee DR, Deckard CE, Elinski MB, Buzbee ML, Wang WW, Batteas JD, Sczepanski JT, Plug-and-Play Approach for Preparing Chromatin Containing Site-Specific DNA Modifications: The Influence of Chromatin Structure on Base Excision Repair. J. Am. Chem. Soc 140, 8260–8267 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Odell ID, Wallace SS, Pederson DS, Rules of engagement for base excision repair in chromatin. J. Cell. Physiol 228, 258–266 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsen LC, Aasland R, Wittwer CU, Krokan HE, Helland D, Molecular cloning of human uracil-DNA glycosylase, a highly conserved DNA repair enzyme. EMBO J. 8, 3121–3125 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao G, Tordova M, Jagadeesh J, Drohat AC, Stivers JT, Gilliland GL, Crystal structure of Escherichia coli uracil DNA glycosylase and its complexes with uracil and glycerol: Structure and glycosylase mechanism revisited. Proteins: Struct., Funct., Bioinf 35, 13–24 (1999). [PubMed] [Google Scholar]
- 44.Cole HA, Tabor-Godwin JM, Hayes JJ, Uracil DNA Glycosylase Activity on Nucleosomal DNA Depends on Rotational Orientation of Targets. J. of Biol. Chem 285, 2876–2885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luger K, Rechsteiner TJ, Richmond TJ, Expression and purification of recombinant histones and nucleosome reconstitution. Chromatin Protocols, 1–16 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Luger K, Rechsteiner TJ, Richmond TJ, Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 304, 3–19 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Chua EY, Vasudevan D, Davey GE, Wu B, Davey CA, The mechanics behind DNA sequence-dependent properties of the nucleosome. Nucleic Acids Res. 40, 6338–6352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alsøe L, Sarno A, Carracedo S, Domanska D, Dingier F, Lirussi L, SenGupta T, Tekin NB, Jobert L, Alexandrov LB, Galashevskaya A, Rada C, Sandve GK, Rognes T, Krokan HE, Nilsen H, Uracil Accumulation and Mutagenesis Dominated by Cytosine Deamination in CpG Dinucleotides in Mice Lacking UNG and SMUG1. Sci. Rep 7, 7199 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waters TR, Gallinari P, Jiricny J, Swann PF, Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem 274, 67–74 (1999). [DOI] [PubMed] [Google Scholar]
- 50.Drohat AC, Jagadeesh J, Ferguson E, Stivers JT, Role of Electrophilic and General Base Catalysis in the Mechanism of Escherichia coli Uracil DNA Glycosylase. Biochemistry 38, 11866–11875 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Stivers JT, Jiang YL, A Mechanistic Perspective on the Chemistry of DNA Repair Glycosylases. Chem. Rev 103, 2729–2760 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Maiti A, Drohat AC, Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and processing of G·T mispairs. DNA Repair 10, 545–553 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez Y, Smerdon MJ, The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem 288, 13863–13875 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vasudevan D, Chua EY, Davey CA, Crystal structures of nucleosome core particles containing the ‘601’strong positioning sequence. J. Mol. Biol 403, 1–10 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Li G, Widom J, Nucleosomes facilitate their own invasion. Nat. Struc. Mol. Biol 11, 763–769 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Li G, Levitus M, Bustamante C, Widom J, Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol 12, 46 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Tomschik M, Zheng H, van Holde K, Zlatanova J, Leuba SH, Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. U.S.A 102, 3278–3283 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei S, Falk SJ, Black BE, Lee T-H, A novel hybrid single molecule approach reveals spontaneous DNA motion in the nucleosome. Nucleic Acids Res. 43, e111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Da L-T, Yu J, Base-flipping dynamics from an intrahelical to an extrahelical state exerted by thymine DNA glycosylase during DNA repair process. Nucleic Acids Res. 46, 5410–5425 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dodd T, Yan C, Kossmann BR, Martin K, Ivanov I, Uncovering universal rules governing the selectivity of the archetypal DNA glycosylase TDG. Proc. Natl. Acad. Sci. U.S.A 115, 5974–5979 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maiti A, Noon MS, MacKerell AD, Pozharski E, Drohat AC, Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A 109, 8091 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smet-Nocca C, Wieruszeski J-M, Chaar V, Leroy A, Benecke A, The Thymine–DNA Glycosylase Regulatory Domain: Residual Structure and DNA Binding. Biochemistry 47, 6519–6530 (2008). [DOI] [PubMed] [Google Scholar]
- 63.Hardeland U, Bentele M, Jiricny J, Schär P, Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J. Biol. Chem 275, 33449–33456 (2000). [DOI] [PubMed] [Google Scholar]
- 64.Hardeland U, Steinacher R, Jiricny J, Schär P, Modification of the human thymine-DNA glycosylase by ubiquitin-like proteins facilitates enzymatic turnover. EMBO J. 21, 1456–1464 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coey CT, Drohat AC, Defining the impact of sumoylation on substrate binding and catalysis by thymine DNA glycosylase. Nucleic Acids Res. 46, 5159–5170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen D, Lucey MJ, Phoenix F, Lopez-Garcia J, Hart SM, Losson R, Buluwela L, Coombes RC, Chambon P, Schar P, T: G mismatch-specific thymine-DNA glycosylase potentiates transcription of estrogen-regulated genes through direct interaction with estrogen receptor α. J. Biol. Chem 278, 38586–38592 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA, Uracil-DNA glycosylase–DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl. Acad. Sci. U.S.A 97, 5083–5088 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA, A nucleotide-flipping mechanism from the structure of human uracil–DNA glycosylase bound to DNA. Nature 384, 87–92 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.