

Abstract
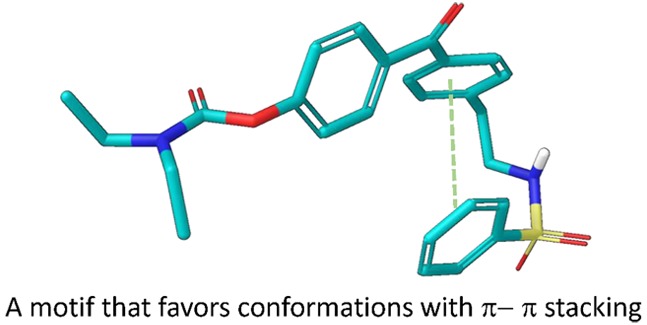
The N-methyl-d-aspartate receptor plays a critical role in central nervous system processes. Its diverse properties, as well as hypothesized role in neurological disease, render NMDA receptors a target of interest for the development of therapeutically relevant modulators. A number of subunit-selective modulators have been reported in the literature, one of which is TCN-201, a GluN2A-selective negative allosteric modulator. Recently, it was determined from a cocrystallization study of TCN-201 with the NMDA receptor that a unique active pose exists in which the sulfonamide group of TCN-201 incorporates a π–π stacking interaction between the two adjacent aryl rings that allows it to make important contacts with the protein. This finding led us to investigate whether this unique structural feature of the diaryl sulfonamide could be incorporated into other modulators that act on distinct pockets. To test whether this idea might have more general utility, we added an aryl ring plus the sulfonamide linker modification to a previously published series of GluN2C- and GluN2D-selective negative allosteric modulators that bind to an entirely different pocket. Herein, we report data suggesting that this structural modification of the NAB-14 series of modulators was tolerated and, in some instances, enhanced potency. These results suggest that this motif may be a reliable means for introducing a π–π stacking element to molecular scaffolds that could improve activity if it allowed access to ligand–protein interactions not accessible from one planar aromatic group.
Keywords: NMDA, glutamate receptor, sulfonamide, GluN2C, GluN2D
The glutamate receptors are ligand-gated, cation-selective channels expressed throughout the central nervous system and comprise three classes: AMPA, kainate, and NMDA receptors (NMDARs).1 The NMDAR plays an important role in nervous system development, synaptic plasticity,2 learning, and memory.3 In addition, NMDARs have been implicated in various neurological disorders such as epilepsy, ischemia, and neurodegenerative disorders.1,4,5 The NMDAR mediates a slow, Ca2+ permeable component of excitatory synaptic transmission, compared to the much faster and briefer synaptic currents mediated by AMPA receptors. NMDARs are unique in that activation requires the binding of glutamate and glycine, which produces an inward current when coincident depolarization of the cell relieves voltage-dependent Mg2+ block of the channel pore. The NMDA receptor is a tetrameric assembly of two different subunits, the glycine-binding GluN1 and glutamate-binding GluN2 subunits. Four different GluN2 subunits exist, referred to as GluN2A-D. These distinct subunits endow the receptor with different response time courses and distinct pharmacological properties and can be targeted by different modulators.1 Like other receptors in its class, each subunit consists of distinct domains, each with its own function: (a) the amino terminal domain (ATD), which controls response properties, such as the open probability for agonist-bound channel and deactivation time course following rapid removal of agonists; (b) the agonist binding domain (ABD), which binds agonists and triggers conformation changes that lead to opening of the ion channel pore; (c) the trans-membrane domain (TMD), which forms the pore of the receptor,6 and (d) the intracellular carboxy terminal domain (CTD), which may direct subcellular localization.
Due to the diverse properties of receptors that contain different GluN2 subunits and the central role that the NMDAR plays in neurological processes, this receptor is an intriguing target for the development of subunit-selective modulators that are therapeutically relevant. There are multiple examples of FDA-approved, nonselective NMDAR inhibitors that block the channel pore of the receptor with similar potencies (see Figure 1). These include memantine (1), dextromethorphan (2), amantadine (3), and ketamine (4), each of which has a different therapeutic use.6,7 Other subunit-specific NMDAR inhibitors have been previously reported, including ifenprodil (5), a noncompetitive selective inhibitor of GluN1/GluN2B. Ifenprodil displays over 100-fold selectivity for GluN2B over GluN2A, GluN2C, and GluN2D.8,9 EVT-101 (10), another example of a GluN1/GluN2B selective antagonist, was claimed by Evotec in a method-of-use patent for cognitive impairment, neurodegenerative diseases, pain, depression, attention deficit hyperactivity disorder, and addiction.16 Additionally, DQP-1105 (6), QNZ-46 (7), and NAB-14 (8) are negative allosteric modulators (NAMs) that display selectivity for GluN2C- and GluN2D-containing NMDARs.
Figure 1.

Structures of known NMDAR channel blockers and negative allosteric modulators; memantine (1), dextromethorphan (2), amantadine (3), ketamine (4) ifenprodil (5),8,9 DQP-110511 (6), QNZ-4610 (7), NAB-1415 (8), TCN-20113 (9, a GluN2A-selective negative allosteric modulator that acts by reducing glycine affinity), and EVT-101 (10).16
TCN-201 (9, Figure 1) is a GluN2A-selective NAM identified by Bettini et al. in 2010.12 A cocrystallization study of TCN-201 described the binding site and active pose at the heterodimer interface between the GluN1 and GluN2A agonist binding domains. The sulfonamide linker endows the structure with a unique pose in which π–π stacking between two adjacent aryl rings is favored in the receptor pocket and allows TCN-201 to make important contacts with amino acid side chains.13,14 The structure and binding pose of TCN-201 (9) are shown in Figure 3A. The unique shape of this biologically active pose led us to hypothesize that it may be helpful to include it in other planar di- and triaromatic compound series that bind at different sites of the receptor and also display relatively flat structure–activity relationships. We reasoned that perhaps these pockets might have space for additional ligand–protein interactions enabled by π–π stacking that could not be realized by substitutions onto planar aromatic systems.
Figure 3.

Conformational searchs results for TCN-201 (9) and the diaryl sulfonamides comparing the shapes adopted when sulfonamide and amide linkers of two different lengths are used. (A) Lowest-energy conformer for each TCN-201 (9) (left) and compound 26 (right), a compound in which the sulfonamide of TCN-201 has been replaced with an amide. (B) Lowest-energy conformer for each 23 (upper) and the corresponding analog 27 (lower) in which the sulfonamide is replaced by an amide. (C) All 50 conformers resulting from the conformational search for compound 18 containing a methylene in the sulfonamide linker (left) and compound 23 containing an ethylene in the sulfonamide linker (right) are superimposed.
Recently, we described a novel N-aryl benzamide analog, represented by NAB-14 (8, Figure 1), a GluN2C/2D-selective NAM. This compound series displayed over 500-fold selectivity for GluN2C/2D over GluN2A/2B, and inhibited GluN1/GluN2C and GluN1/GluN2D with IC50 values of 1–4 μM.15 In addition, this compound series possesses improved drug-like physicochemical properties and achieves modest blood–brain barrier penetration in rodents with minimal off-target effects.15 Structural determinants of action are in the M1 transmembrane helix, suggesting that its binding site is likely distinct from that of TCN-201. The structure–activity relationship for this series was relatively flat,15 leading us to consider whether introduction of the π–π interaction might bring about potency-enhancing ligand–protein interactions within the pocket that we could not access from planar di- and triaryl systems linked by amide bonds.
Part of the SAR to probe the activity of the NAB-14 structure was focused on the indole, where it was first replaced with a phenyl ring (NP11999, Figure 2). This modification showed modest activity in the initial SAR, with an IC50 value of 6 μM for GluN2C/D-containing NMDARs (Table 1). Further expansion of the SAR at this position suggested that the indole side of the NAB series could accommodate larger groups and additional aryl rings without complete loss of activity, depending on the substitution pattern connecting the additional aryl ring (NP12000 and NP12022, Figure 2). We, then, began to investigate whether replacement of the planar amide linker connecting the additional aryl ring with a more flexible sulfonamide linker was tolerated, and, hopefully, improved upon activity. We reasoned that incorporating a sulfonamide linker adjoining the additional phenyl ring should lead to a similar U-shaped π–π stacking motif as in TCN-201, thereby replacing the indole side of the NAB series with a larger π–π stacked aromatic configuration. Figure 3 compares the TCN-201 binding pose (panel A) to one of the modeled low energy candidate binding poses for a proposed compound (panel B), which incorporates a sulfonamide linker. Similar to TCN-201, the proposed sulfonamide moiety endows the scaffold with enough flexibility to obtain a “U-shape.” When the linker contains a single methylene group like TCN-201, all modeled poses contain this orientation of the π–π stacked rings (Figure 3C, left panel); when the linker contains an ethylene group, most but not all poses display π–π stacking (Figure 3C, right panel).
Figure 2.

Structures and activities from first-generation NAB-14 analogs with planar third-ring extensions. Data are for GluN1/GluN2D (n = 6 oocytes).
Table 1. Activity of Previous Compoundsa.
|
I10μM/Icontrol (mean
± SEM, %)b, IC50 (μM)c |
||
|---|---|---|
| cmpd # | GluN2C | GluN2D |
| 8 | 24 ± 2.1 | 16 ± 1.8 |
| 3.7 | 2.2 | |
| 9d | 95 ± 2.1 | 88 ± 1.4 |
| ND | ND | |
| NP11999e | 49 ± 2.1 | 37 ± 4.6 |
| 8.6 | 5.8 | |
Data are from 8 to 9 oocytes from 2 frogs for each compound tested.
The response to drug coapplied with a maximally effective concentration of glutamate (100 μM) and glycine (30 μM) is given as a percent of the control response to glutamate and glycine alone.
IC50 values were determined by fitting the Hill equation to the average composite concentration–response curve and are reported to two significant figures. ND indicates not determined; see ref (15) for methods.
Because TCN-201 is known to be GluN2A-selective, we tested a higher concentration, 30 μM, at GluN2C and GluN2D. There was less than 15% inhibition, which precluded determination of IC50.
Data are from unpublished results.
We chose to extend the SAR of this novel diaryl sulfonamide analog to include meta substitution to the extended aryl ring because of the activity displayed in NP12022 (Figure 2), with both methylene or ethylene units included in the sulfonamide linker. The synthesis of the sulfonamide NAB-modified series is shown in Scheme 1. The first few steps have been previously reported.15 Therefore, commercially available phenol 11 was allowed to react with diethyl carbamoyl chloride to obtain carbamate 12, which was then saponified to produce acid 13. Acid 13 was then coupled with the appropriate aniline intermediate to produce para and meta substituted intermediates 14 (n = 1, 2), which were Boc-deprotected to get the free amines 15. Compounds 15 were coupled with 0.5–1.0 equiv of the appropriate aryl sulfonyl chloride, dependent upon the amount of observed double addition byproduct formed, to yield final compounds 16–25 containing the desired diaryl sulfonamide moiety.
Scheme 1.

The screening results of these compounds on recombinant NMDA receptors expressed in Xenopus oocytes are shown in Table 2. Compounds with a methylene linker (n = 1) showed minimal activity. Compounds with an ethylene linker (n = 2) and para-substitution showed minimal activity, except in the case of the pyridyl sulfonamide (21). The most active compounds in this series are those with an ethylene linker and meta-substitution (compounds 23–25), which were up to 6-fold more potent than the phenyl NAB analogue NP11999, which we used as a reference point. Thus, adding the sulfonamide linker to the scaffold not only retains activity at GluN1/GluN2C and GluN1/GluN2D but it also enhances potency over the phenyl analog NP11999. Interestingly, reducing the length of the diaryl sulfonamide linker by one methylene unit decreases activity despite its ability to better assume the favored π–π stacking motif similar to the TCN series. The differences between the modeled conformations of these two linker geometries are shown Figure 3C, which reveal the expected increased flexibility of the ethylene linker. Given the improved potency, we interpret this increased flexibility as better enabling the stacked rings to fit into the binding pocket for NAB analogues. This emphasizes that there are likely unique requirements for the optimal π–π stacking shape for different pockets, as would be expected.
Table 2. Activity of Bi-aryl Sulfonamidesa.

|
I30μM/Icontrol (mean ± SEM, %) IC50 (μM)b |
|||||||
|---|---|---|---|---|---|---|---|
| Cmpd # | n | R1 | R2 | Y | GluN2C | GluN2D | |
| 16 | para | 1 | H | H | CH | 42 ± 2.5 | 20 ± 2.5 |
| 22 | 10 | ||||||
| 17 | para | 1 | H | H | N | 58 ± 4.6 | 45 ± 3.3 |
| 35 | 24 | ||||||
| 18 | meta | 1 | H | H | CH | 60 ± 2.0 | 46 ± 2.6 |
| 45 | 29 | ||||||
| 19 | meta | 1 | H | H | N | 52 ± 1.4 | 42 ± 1.3 |
| 34 | 23 | ||||||
| 20 | para | 2 | H | H | CH | 77 ± 2.9 | 67 ± 4.4 |
| ND | ND | ||||||
| 21 | para | 2 | H | H | N | 37 ± 2.8 | 26 ± 4.1 |
| 12 | 5.1 | ||||||
| 22 | para | 2 | Cl | F | CH | 79 ± 1.4 | 74 ± 2.0 |
| ND | ND | ||||||
| 23 | meta | 2 | H | H | CH | 9.0 ± 2.8 | 9.0 ± 2.3 |
| 2.4 | 1.7 | ||||||
| 24 | meta | 2 | H | H | N | 8.5 ± 1.3 | 8.6 ± 1.4 |
| 3.2 | 2.7 | ||||||
| 25 | meta | 2 | Cl | F | CH | 41 ± 1.6 | 28 ± 2.6 |
| 2.4 | 1.3 | ||||||
The response to drug coapplied with a maximally effective concentration of glutamate (100 μM) and glycine (30 μM) is given as a percent of the control response to glutamate and glycine alone.
Fitted IC50 values are reported to two significant figures; inhibition in saturating test compound was set to 0 for all except 16, which was 41% for GluN2C and 27% for GluN2D. ND indicates not determined. Data are from 8 to 16 oocytes from 2 to 3 frogs for each compound and receptor tested.
To test whether the enhanced potency of this novel sulfonamide series compared to the phenyl NAB analogue NP11999 is due to the U-shaped motif of adjacent aromatic rings, we reasoned that replacement of the sulfonamide linker with an amide linker would disallow π–π stacking and eliminate activity that is dependent on this specific pose. We first confirmed this premise using modeling as shown in Figure 3A,B, which shows that representative amides cannot achieve the same degree of aryl ring overlap due to the geometry of the amide bond. As a proof of principle, we tested whether this modification altered activity in an analogue of TCN-201 (compound 9, Figure 1) in which the sulfonamide was replaced by the amide (compound 26, Table 3), whose structure is shown in Figure 4. Whereas TCN-201 potently inhibited GluN1/GluN2A, the amide-containing analogue 26 had no detectable activity on GluN1/GluN2A receptors (Table 3), confirming that the π–π stacked pose is essential for activity.
Table 3. Comparison of Sulfonamide to Amide Linkera.
|
Idrug/Icontrol (mean ± SEM, %)b, IC50 (μM)c |
|||||
|---|---|---|---|---|---|
| Cmpd # | cLogPd | Drug μM | GluN2A | GluN2C | GluN2D |
| 9 | 3.55 | 6 | 5.1 ± 1.3 | ND | ND |
| 0.17 | |||||
| 26 | 4.22 | 30 | 102 ± 2.2 | ND | ND |
| ND | |||||
| 23 | 4.34 | 30 | 85 ± 1.2 | 9.0 ± 2.8 | 8.9 ± 2.3 |
| ND | 2.4 | 1.7 | |||
| 27 | 4.59 | 30 | 82 ± 8.6 | 56 ± 1.9 | 49 ± 1.6 |
| ND | 37 | 29 | |||
Data are from 8 to 9 oocytes from 2 frogs for each compound tested.
The response to drug coapplied with a maximally effective concentration of glutamate (100 μM) and glycine (30 μM) is given as a percent of the control response to glutamate and glycine alone.
IC50 values were determined by fitting the Hill equation to the average composite concentration–response curve and are reported to two significant figures. ND indicates not determined.
cLogP calculated using Chemicalize from ChemAxon.
Figure 4.

Structures of compounds 18, 26, 23, and 27.
We subsequently used the same strategy to test the role of π–π stacking in the enhanced potency of compounds 23 and 25. Therefore, we synthesized and tested an analogue of the NAB-modified series of GluN2C/GluN2D-selective inhibitors in which we replaced the sulfonamide group with an amide group (Table 3, compound 27 in Figure 4). In principle, the amide linker should minimize the “U-shape” binding pose due to its linear geometry as shown in the energy-minimized structures in Figure 3A. Indeed, consistent with this idea, amide 27 displayed reduced potency by more than 15-fold as compared to the parent sulfonamide 23. However, an alternative hypothesis could be that the change in potency is due to a change in compound geometry between the amide and the sulfonamide, rather than a requirement of π–π stacking. While this seems unlikely for the one carbon linker in TCN-201, which primarily adopts a single conformation, it is harder to rule out with the increased flexibility of the 2-carbon linker in 23 and 27. Moreover, there is also the possibility that the difference in hydrogen bonding and lipophilicity (Table 3) could alter the way the compound interacts with the receptor. Thus, there remain caveats about our working hypothesis that will require structural data of the ligand-bound receptor to address.
The original NAB series was highly selective for GluN2C- and GluN2D-containing NMDA receptors. We therefore, evaluated whether one of the most active analogues in this diaryl sulfonamide series, compound 23, retained selectivity for GluN1/GluN2C and GluN1/GluN2D over other receptors. Interestingly, while this analogue retained potent activity at GluN2C and GluN2D, it also was active at GluN2B, inhibiting receptors with a fitted IC50 value of 7.6 μM (n = 8 oocytes from 2 frogs). This result raises the possibility that the U-shaped pose of analogue 23 allows it to access a similar pocket on GluN1/GluN2B. Analogue 23 showed minimal activity at AMPA, kainate, and GABA receptors (Table 4), similar to NAB-14. However, while the prototypical compound NAB-14 had no detectable off-target activity at glycine, serotonin, and nicotinic receptors, analogue 23 showed a substantial degree of inhibition at these receptors (Table 4). The reduced selectivity of this compound compared to NAB-14, our starting reference compound, could reflect the increased flexibility of the longer linker and the sulfonamide functionality, or the addition of another aryl group. NAB-14 is a rigid molecule with minimal flexibility, which may contribute to its selectivity, as it fits into the pocket of NMDA receptors with limited ability to fit different geometries required at other receptors. It might be useful in future SAR studies at other targets to test both methylene and ethylene linker that favor the U-shaped pose with different degrees of freedom, which would be predicted to have different off-target profiles.
Table 4. Selectivity of the Sulfonamide Analogue 23a.
| receptor | I30μM/Icontrol (mean ± SEM, %), p, N | |
|---|---|---|
| GluN1/GluN2A | 85 ± 1.2 | p < 0.001 N = 8 |
| GluN1/GluN2B | 24 ± 6.3 | p < 0.001 N = 11 |
| GluA1 | 79 ± 2.0 | p = 0.001 N = 6 |
| GluA2(R607Q) | 95 ± 2.2 | p = 0.083 N = 6 |
| GluA3(L513Y) | 86 ± 2.7 | p = 0.005 N = 6 |
| GluA4(L505Y) | 86 ± 3.1 | p = 0.010 N = 6 |
| GluK2 | 92 ± 3.1 | p = 0.068 N = 6 |
| P2X | 80 ± 2.7 | P < 0.001 N = 8 |
| GABAA α1β2γ2S | 90 ± 4.9 | p = 0.105 N = 10 |
| GABA ρ | 98 ± 3.1 | p = 0.468 N = 5 |
| glycine α1 | 30 ± 2.7 | p < 0.001 N = 8 |
| serotonin 5-HT3Α | 20 ± 2.6 | p < 0.001 N = 6 |
| nicotinic α1βγδ | 27 ± 0.59 | p < 0.001 N = 7 |
The response to test compound coapplied with agonist is given as a percent of the average of control and recovery current responses to agonist alone for receptors expressed in oocytes (VHOLD = −40 mV). Agonist concentrations were 100 μM glutamate plus 30 μM glycine for NMDA receptors, 100 μM glutamate for AMPA and kainate receptors, 100 μM GABA for GABA receptors, 100 μM glycine for glycine receptors, 100 μM serotonin for 5-HT3A receptors, 9 μM ATP for P2X receptors, and 1 μM acetylcholine for nicotinic receptors. cDNAs encoding all receptors were from rat except for GluA2(R607Q), GluA3(L513Y), GluA4(L505Y), and P2X, which were from human. Oocytes expressing GluK2 were incubated for 1–5 min in 1 mg/mL concanavalin A. N indicates the number of oocytes. p values were from a paired t test.
In summary, these results show that the diaryl sulfonamide motif promotes a “U-shape” that can be favorable for some ligands acting at entirely distinct pockets, which for some compounds involves substantial intramolecular π–π interactions. We conclude that the pocket into which the NAB-14 series binds has room to accommodate the bulky structure and/or can make significant favorable interactions with sulfonamide oxygen atoms. This motif, with variable linker lengths, could be used to explore the size of the pocket for other medicinal chemistry campaigns aimed at developing the SAR for other druggable targets. However, longer linkers that allow greater flexibility could bring additional off-target effects.
Acknowledgments
We would like to thank Phuong Le and Jing Zhang for excellent technical assistance. We also acknowledge Kathryn Simon and Francis X. Tavares of ChemoGenics BioPharma for the original synthesis of analogs NP11999, NP12022, and NP12000. The authors thank the Emory Chemical Biology Discovery Center for their assistance.
Glossary
ABBREVIATIONS
- NMDA
N-methyl-d-aspartate
- NMDAR
N-methyl-d-aspartate receptor
- GABA
gamma aminobutyric acid
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ATD
amino terminal domain
- CTD
carboxy terminal domain
- TMD
transmembrane domain
- LBD
ligand binding domain
- NAB
N-aryl benzamide
- NAM
negative allosteric modulator
- IC50
half-maximal inhibitory concentration
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00395.
Chemistry experimental procedures, characterization, and biology experimental procedures (PDF)
Author Contributions
All authors designed experiments. S.L.S., Z.Z., R.L.M., S.A.K., and D.S.M. carried out experiments. S.L.S., Z.Z., R.L.M., S.A.K., D.S.M., and S.F.T. analyzed data. All authors wrote the manuscript, and all authors have given approval for the final version of the manuscript.
The authors acknowledge the use of shared instrumentation provided by grants from NSF (CHE1531620). This project was partially supported by the ELSI Origins Network (EON), which is supported by a grant from the John Templeton Foundation. The study was also supported by the NIH-NINDS (NS065371 S.F.T.) and a research grant from Janssen to Emory University (S.F.T.).
The opinions expressed in this publication are those of the author(s) and do not necessarily reflect the views of the NIH or the John Templeton Foundation.
The authors declare the following competing financial interest(s): D.C.L., S.F.T., S.L.S., and D.S.M. are coinventors on Emory-owned intellectual property associated with allosteric modulators of NMDA receptor function. S.F.T. is a paid consultant for Janssen, the principal investigator on a research grant from Janssen to Emory University, a member of the SAB for Sage Therapeutics, recipient of royalties from licensing software, and a cofounder of NeurOp Inc. D.C.L. is a member of the BOD for NeurOp Inc.
Supplementary Material
References
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496. 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S.; Collin C.; Auerbach J. M.; Meiri N.; Beng-zon J.; Kennedy M. B.; Segal M.; McKay R. D. Hippocampal Synaptic Plasticity in Mice Overexpressing an Embryonic Subunit of the NMDA Receptor. J. Neurosci. 1998, 18, 4177–4188. 10.1523/JNEUROSCI.18-11-04177.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge G. L.; Volianskis A.; Bannister N.; France G.; Hanna L.; Mercier M.; Tidball P.; Fang G.; Irvine M. W.; Costa B. M.; Monaghan D. T.; Bortolotto Z. A.; Molnár E.; Lodge D.; Jane D. E. The NMDA Receptor as a Target for Cognitive Enhancement. Neuropharmacology 2013, 64, 13–26. 10.1016/j.neuropharm.2012.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M. P.; Raymond L. A. Extrasynaptic NMDA Receptor Involvement in Central Nervous System Disorders. Neuron 2014, 82, 279–293. 10.1016/j.neuron.2014.03.030. [DOI] [PubMed] [Google Scholar]
- Gonzalez J.; Jurado-Coronel J. C.; Ávila M. F.; Sabogal A.; Capani F.; Barreto G. E. NMDARs in Neurological Diseases: a Potential Therapeutic Target. Int. J. Neurosci. 2015, 125, 315–327. 10.3109/00207454.2014.940941. [DOI] [PubMed] [Google Scholar]
- Chen H. S. V.; Lipton S. A. The Chemical Biology of Clinically Tolerated NMDA Receptor Antagonists. J. Neurochem. 2006, 97, 1611–1626. 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- Dravid S. M.; Erreger K.; Yuan H.; Nicholson K.; Le P.; Lyuboslavsky P.; Almonte A.; Murray E.; Mosely C.; Barber J.; French A.; Balster R.; Murray T. F.; Traynelis S. F. Subunit-specific Mechanisms and Proton Sensitivity of NMDA Receptor Channel Block. J. Physiol. (Oxford, U. K.) 2007, 581, 107–128. 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. Ifenprodil Discriminates Subtypes of the N-methyl-D-aspartate Receptor: Selectivity and Mechanisms at Recombinant Heteromeric Receptors. Mol. Pharmacol. 1993, 44, 851–859. [PubMed] [Google Scholar]
- Chenard B. L.; Bordner J.; Butler T. W.; Chambers L. K.; Collins M. A.; De Costa D. L.; Ducat M. F.; Dumont M. L.; Fox C. B.; Mena E. E. (1S,2S)-1-(4-hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: a Potent New Neuroprotectant Which Blocks N-methyl-D-aspartate Responses. J. Med. Chem. 1995, 38, 3138–3145. 10.1021/jm00016a017. [DOI] [PubMed] [Google Scholar]
- Mosley C. A.; Acker T. M.; Hansen K. B.; Mullasseril P.; Andersen K. T.; Le P.; Vellano K. M.; Bräuner-Osborne H.; Liotta D. C.; Traynelis S. F. Quinazolin-4-one Derivatives: A Novel Class of Noncompetitive NR2C/D Subunit-selective N-methyl-D-aspartate Receptor Antagonists. J. Med. Chem. 2010, 53, 5476–5490. 10.1021/jm100027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acker T. M.; Yuan H.; Hansen K. B.; Vance K. M.; Ogden K. K.; Jensen H. S.; Burger P. B.; Mullasseril P.; Snyder J. P.; Liotta D. C.; Traynelis S. F. Mechanism for Noncompetitive Inhibition by Novel GluN2C/D N-methyl-D-aspartate Receptor Subunit-selective Modulators. Mol. Pharmacol. 2011, 80, 782–795. 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettini E.; Sava A.; Griffante C.; Carignani C.; Buson A.; Capelli A. M.; Negri M.; Andreetta F.; Senar-Sancho S. A.; Guiral L.; Cardullo F. Identification and Characterization of Novel NMDA Receptor Antagonists Selective for NR2A- over NR2B-containing Receptors. J. Pharmacol. Exp. Ther. 2010, 335, 636–644. 10.1124/jpet.110.172544. [DOI] [PubMed] [Google Scholar]
- Hackos D. H.; Lupardus P. J.; Grand T.; Chen Y.; Wang T.-M.; Reynen P.; Gustafson A.; Wallweber H. J. A.; Volgraf M.; Sellers B. D.; Schwarz J. B.; Paoletti P.; Sheng M.; Zhou Q.; Han-son J. E. Positive Allosteric Modulators of GluN2A-Containing NMDARs with Distinct Modes of Action and Impacts on Circuit Function. Neuron 2016, 89, 983–999. 10.1016/j.neuron.2016.01.016. [DOI] [PubMed] [Google Scholar]
- Yi F.; Mou T.-C.; Dorsett K. N.; Volkmann R. A.; Menniti F. S.; Sprang S. R.; Hansen K. B. Structural Basis for Negative Allosteric Modulation of GluN2A-Containing NMDA Receptors. Neuron 2016, 91, 1316–1329. 10.1016/j.neuron.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanger S. A.; Vance K. M.; Acker T. M.; Zimmerman S. S.; DiRaddo J. O.; Myers S. J.; Bundgaard C.; Mosley C. A.; Summer S. L.; Menaldino D. S.; Jensen H. S.; Liotta D. C.; Traynelis S. F. A Novel Negative Allosteric Modulator Selective for GluN2C/2D-Containing NMDA Receptors Inhibits Synaptic Transmission in Hippocampal Interneurons. ACS Chem. Neurosci. 2018, 9, 306–319. 10.1021/acschemneuro.7b00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D.; Buhl D.; Knafels J.; Chanda P.; Green M.; Sciabola S.; Mony L.; Paoletti P. Pandit. A Novel Binding Mode Reveals Two Distinct Classes of NMDA Receptor GluN2B-selective Antagonists. Mol. Pharmacol. 2016, 89, 541–51. 10.1124/mol.115.103036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.