

Abstract
Progranulin (GRN) gene mutations are a major cause of frontotemporal dementia (FTD). Mostmutations identified to date are null mutations, which are predicted to cause the pathology via haploinsufficiency. Decreased peripheral progranulin protein (PGRN) levels are associated with the presence of GRN null mutations and are accepted as reliable biomarkers. In this study, our aim was to test whether the presence of specific GRN splice site mutations (c.−8+2T>G and c.708+6_9del), could be predicted by peripheral mRNA or protein GRN levels, by studying affected and asymptomatic individuals from FTD families. We also tested four missense GRN variants to assess if altered GRN levels depended on the type of mutation.Our results confirmed a reduction in both mRNA and protein PGRN levels in the splice site mutation carriers, which is consistent with previous reports for null mutations. Our results also suggested that both decreased peripheral GRN mRNA and serum PGRN levels indicate the presence of pathogenic mutations in affected individuals, and identify the asymptomatic individuals at risk, without previous knowledge of genetic status. Both inferences suggest a potential use of peripheral GRN mRNA or serum PGRN levels as biomarkers for families with FTD.
Keywords: ELISA, frontotemporal dementia, progranulin, serum, splice site mutation
INTRODUCTION
Progranulin protein (PGRN), encoded by the GRN gene, is a secreted growth factor precursor composed of 7.5 tandem repeats of highly conserved 12-cysteine granulin motifs. PGRN can be cleaved by elastase within the linker regions to generate 6 kDa granulin peptides [1]. Both PGRN and granulin peptides are widely expressed and have functions in development, wound repair, inflammation, and tumorigenesis [1]. Mutations in GRN are a significant cause of frontotemporal dementia (FTD), accounting for 5–20% of familial and 1–5% of sporadic FTD patients [2, 3]. Currently in the Human Gene Mutation Database, 180 different GRN mutations have been listed and of these 147 are classified as pathogenic (HGMD; http://www.hgmd.org). Most of these alterations are frameshift, splice site [4] and nonsense mutations [5]; however, there have been reports of complete gene deletions [6]. All pathogenic mutations create null alleles and cause the loss of functional protein, resulting in haploinsufficiency, which can be measured in serum [7–9], plasma [4, 9–12], and CSF [13,14] presenting decreased protein levels. Not only null mutations, but also missense variants, either non-pathogenic or with unclear pathogenicity, have been reported. The clinical findings of FTD associated with GRN mutations are diverse and include behavioral changes, primary progressive aphasia, and movement disorders with extrapyramidal features such as parkinsonism and corticobasal syndrome.
Here, we report molecular data on two Turkish families carrying GRN splice site mutations. One of these families presented a novel splice site mutation (NM_002087.2:c.−8+2T>G), the other family which we have previously reported [15], presented a known deletion also affecting a splice site (NM_002087.2:c.708+6_9del). In order to evaluate the effect of these splice site mutations on the expression of GRN (both at the RNA and protein levels), we have compared the expression of GRN mRNA and serum PGRN between carrier and non-carrier family members and controls. To assess whether any fluctuations in GRN RNA and protein levels were the consequence of the mutation type, we additionally studied four different missense variants.
MATERIALS AND METHODS
Study group and samples
Family ALZ-172 was found to carry the novel c.−8+2T>G mutation with six family members affected with FTD (Supplementary Figure 1). In our study, two asymptomatic carriers and one healthy non-carrier from family ALZ-172 were included in the evaluation of the GRN mRNA and serum PGRN levels (Table 1). We recruited a total of eighteen individuals from Family DEM-35, in which the c.708+6_9del was identified (Supplementary Figure 2). From Family DEM-35, we included for molecular analysis two deletion-carriers affected with FTD, four asymptomatic deletion-carriers and twelve non-carrier first-degree relatives of the index patient (Table 1). The Supplementary Material provides a detailed clinical description of both families.
Table 1.
Descriptive characteristics of patients and families according to GRN variant type
| Patients/Family | Variant | Sex (F/M) | Age | Age of onset | Clinical Status | Diagnosis |
|---|---|---|---|---|---|---|
| Family ALZ-172 (n = 3) | c.−8+2T>G | 2/1 | 55.3 ± 2.1 | - | Asymptomatic carriers (n = 2) | - |
| Non-affected (n = 1) | - | |||||
| Family DEM-35 (n =18) | c.708+6_9del | 10/8 | 37.6 ± 17.4 | 47.5 ± 4.9 | Affected (n = 2) | FTD |
| Asymptomatic carriers (n = 4) | ||||||
| Non-affected (n = 12) | ||||||
| Family FTD-16 (n = 4) | p.Cys139Arg | 2/2 | 44.75 ± 8.7 | - | Asymptomatic carriers (n = 4) | - |
| ALZ-132 | p.Pro209Leu | 0/1 | 51 | 45 | Affected | PCA |
| ALZ-35 | p.Val77Ile | 0/1 | 64 | 50 | Affected | AD |
| ALZ-42 | p.Asp33Glu | 0/1 | 80 | 76 | Affected | AD |
| Controls (n = 20) | 12/8 | 49.9 ± 17.1 | - | Non-affected | - | |
Values are expressed as mean ± SD. AD, Alzheimer’s disease; F, female; FTD, frontotemporal dementia; M, male; PCA, posterior cortical atrophy.
We also included in this study three index patients previously reported as carriers of NM_ 002087.2:c.99C>A p.(Asp33Glu), NM.002087.2: c.229G>Ap.(Val77Ile) andNM_002087.2:c.626C>T p.(Pro209Leu) missense variants [15]. Additionally, we studied four asymptomatic carriers of the missense NM_002087.2:c.415T>C p.(Cys139Arg) variant [15] that we had previously identified in the proband of the respective family.
The control group consisted of 20 non-related individuals with no family history of dementia and without any known GRN mutations. Written informed consent was obtained from all participants and this study was approved by the ethical committee of the Istanbul University.
Peripheral blood samples were drawn into PAXgene tubes, and total RNA was extracted using the PAXgene blood RNA kit (PreAnalytiX, Hombrechtikon, Switzerland) according to the manufacturer’s instructions. Serum samples were isolated according to standard procedures and stored at −80o C.
In silico prediction analysis
PolyPhen-2 [16], MutationTaster [17], and SIFT [18] in silico prediction programs were used to evaluate the effect of missense variants on protein function and structure. MaxEntScan [19] was used to predict the splicing effects. Variants were classified according to the American College of Medical Genetics and Genomics (ACMG) guidelines.
Enzyme linked immunosorbent assay (ELISA)
PGRN levels in the serum samples were determined using the Progranulin (human) ELISA Kit (Adipogen, Incheon, Korea) in duplicate with a 1:200 serum dilution according to manufacturer’s protocol.
Quantitative polymerase chain reaction (qPCR)
GRN gene expression levels were determined using Power SYBR Green PCR Master Mix (Applied Biosystems) on an Agilent Mx3000P qPCR System (Agilent Technologies, Santa Clara, CA, USA). Samples were run in triplicates and normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels. At least two separate qPCR experiments were performed for each sample. The relative expression of GRN was calculated using the ΔΔCt method.
Statistical analysis
The nonparametric Mann-Whitney U test was used to compare PGRN levels between two groups and the Kruskal-Wallis test for comparison of more than two groups. Correlations between age and serum PGRN levels were tested using Spearman’s rho. Results were considered statistically significant for p < 0.05. The receiver operating characteristics (ROC) curve analysis was used to determine the optimal cut-off value of serum PGRN levels in splice site mutation carriers. The diagnostic accuracy of PGRN levels was assessed from the area under the curve (AUC). Optimal cut-off value that yielded the highest Youden index [sensitivity + (specificity −1)] was selected. All analyses were performed using SPSS Statistics 21.0 software (IBM Corp., Armonk, NY, USA).
RESULTS
In silico predictions
Based on MaxEntScan scores, the novel c.−8+2T>G variant was found to likely disturb normal splicing due to loss of a splice donor site. The other splice site mutation (c.708+6_9del) was predicted to decrease the splicing of exon 6 due to decreased 5’ donor site score (Supplementary Table 1). In silico predictions of pathogenicity of the missense variants were presented in Supplementary Table 1.
Serum PGRN levels
Serum samples were available for two asymptomatic individuals carrying the c.−8+2T>G splice site substitution, and one non-carrier first-degree relative. The serum PGRN levels were 64 and 85.1 ng/mL in the asymptomatic carriers and 121 ng/mL in the non-carrier relative (Fig. 1A).
Fig. 1.
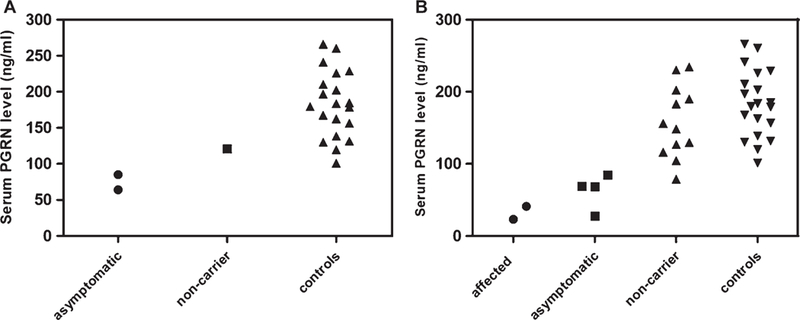
Serum PGRN levels. A) Asymptomatic carriers of c.−8+2T>G, non-carrier and controls. B) Asymptomatic and affected family members with the c.708+6_+9del deletion, non-carrier family members and controls.
Serum PGRN levels were measured in two affected and four asymptomatic family members carrying the c.708+6_9del splice site deletion, and twelve non-carrier first-degree relatives (Supplementary Table 2). The serum PGRN levels in affected patients carrying the deletion were 23 and 41 ng/mL while, the median serum PGRN levels were 68.8 ng/mL (range 27.7–84.7 ng/mL, n: 4) in asymptomatic carriers and 152.3 ng/mL (range 78.8–234.5 ng/mL, n: 12) in non-carrier relatives (Fig. 1B).
Due to the small number of carriers of the splice site mutations in each family, all splice site mutation carriers were combined in a single group in the statistical comparison of serum PGRN levels. In this analysis, the median serum PGRN levels in all splice site mutation carriers (median 66.4 ng/mL, range 23–85.1 ng/mL, n = 8) were significantly (p < 0.001) lower than those of non-related controls (median 181.6 ng/mL, range 101.1–266 ng/mL, n = 20). We then tested whether decreased serum PGRN levels could be used to distinguish, asymptomatic splice site mutation carriers from controls. Asymptomatic carriers (median 68.8 ng/mL, range 27.7–85.1 ng/mL, n = 6) were found to have significantly (p <0.001) lower PGRN levels than controls. ROC curve analysis (Supplementary Figure 3) including all splice site mutation carriers and all non-carriers suggested that a cut-off chosen at 93.1 ng/mL would discriminate null mutation carriers from controls and non-carriers with a sensitivity of 100% and a specificity of 97% (area under the curve, 0.99 p < 0.001).
In addition to the mentioned splice site mutations, the effect on serum PGRN levels of four missense variations was also evaluated in three index patients and four asymptomatic carriers. When considering all variant carriers separately, it was evident that asymptomatic/affected splice site mutation carriers had prominently lower serum PGRN levels (Fig. 2). Strikingly, the PGRN levels in p.Cys139Arg carriers were in the same range as those of individuals carrying splice site mutations (Fig. 2), and the median serum PGRN levels (median, 60.7 ng/mL, range47.8–68.6 ng/mL, n = 4) were significantly lower than those found in controls (p = 0.002). Serum PGRN levels in carriers of p.Val77Ile and p.Pro209Leu missense variants (114.8 ng/mL and 128.9 ng/mL, respectively) were higher than the levels for carriers of splice site mutations and p.Cys139Arg. However, they were still lower than the range observed for controls (Fig. 2). In the p.Asp33Glu carrier the serum PGRN levels (269.8 ng/mL) were prominently higher than the levels for carriers of splice site mutations and even for other missense variants. Interestingly, the serum PGRN levels in the p.Asp33Glu carrier were higher than those observed for controls, as well (Fig. 2). When considering all pathogenic variant carriers (splice site mutations and p.Cys139Arg) together, the PGRN levels in carriers of pathogenic variants were found to be significantly (p = 0.01) decreased compared to those of non-pathogenic variant carriers (p.Asp33Glu, p.Val77Ile, p.Pro209Leu).
Fig. 2.
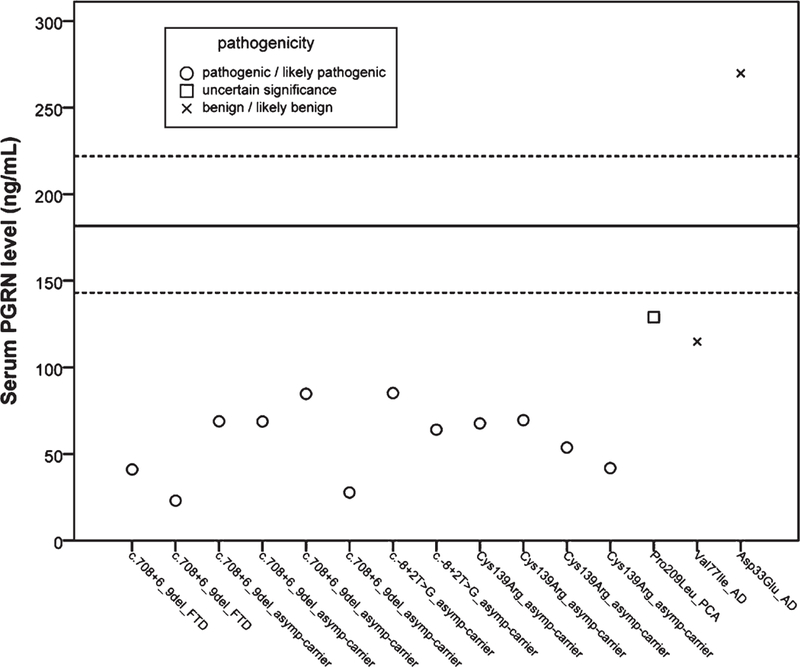
Serum PGRN levels for individual variant carriers relative to median control levels. Circles represent pathogenic and likely pathogenic mutations, squares represent missense variants with uncertain significance and crosses represent benign and likely benign missense variants. The black straight line indicates the median for serum PGRN levels of controls with the 25th and 75th percentiles indicated by the dotted black lines.
GRN mRNA expression levels
To see whether the significant lower serum PGRN protein levels were the consequence of decreased GRN mRNA expression, we analyzed GRN mRNA levels in splice site mutation carriers. We found that mRNA levels were significantly decreased (p = 0.006) in splice site mutation carriers compared to controls (Fig. 3).
Fig. 3.
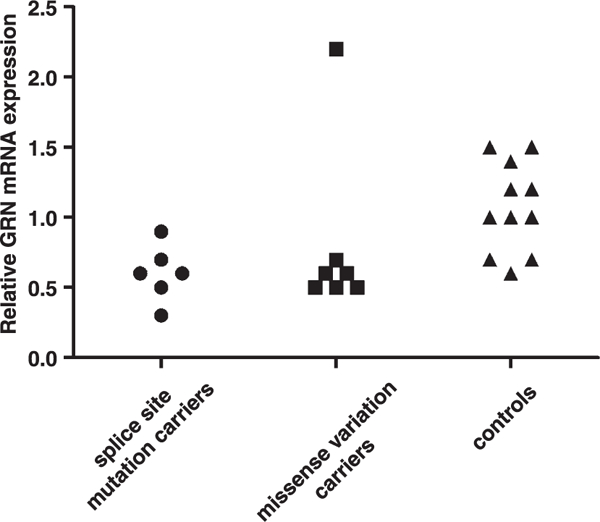
GRN mRNA expression levels in controls, splice site mutation and missense variation carriers. The outlier value in missense variation carriers belongs to the p.Asp33Glu carrier.
When we evaluated GRN mRNA levels in all carriers of missense variants, we found that mRNA levels were significantly decreased (p = 0.025) compared to controls but no significant difference (p = 1) was observed when compared with splice site mutation carriers (Fig. 3). Considering the low serum PGRN levels in p.Cys139Arg carriers, we statistically analyzed p.Cys139Arg carriers separately from other missense carriers (p.Asp33Glu, p.Val77Ile, and p.Pro209Leu) in terms of GRN mRNA levels. Our results showed that mRNA levels in p.Cys139Arg carriers were not significantly different (p = 0.742) from splice site mutation carriers, but were significantly decreased (p = 0.01) compared to controls. When the carrier of p.Asp33Glu was excluded, due to the high serum levels found, and two other missense (p.Val77Ile and p.Pro209Leu) carriers were included in statistical analysis together with carrier of p.Cys139Arg, the difference still remained significant (p = 0.002) when compared to controls. However we did not find any significant difference (p = 0.569) in GRN mRNA levels between pathogenic variant carriers (splice site mutations and p.Cys139Arg) and non-pathogenic variant carriers (p.Asp33Glu, p.Val77Ile, p.Pro209Leu).
DISCUSSION
Most GRN mutations identified to date are predicted to cause the pathology via a haploinsufficiency mechanism by creating premature stop codons, which in turn result in nonsense mediated decay (NMD). In GRN we have previously reported one splice site deletion and 4 missense variants in five different individuals. In this study, we have evaluated the effect of these variants in RNA expression and serum protein levels as well as of the novel c.−8+2T>G splice site substitution by performing mRNA quantification and measuring serum PGRN levels.
The novel c.−8+2T>G variant is located in the splice donor site of intron 0 following non-coding exon 0 of GRN. Near the same region, there are two other mutations (c.−8+3A>T and c.−8+5G>C) that have previously been reported [3, 20–22]. Of these, the c.−8+5G>C mutation was identified in eleven Belgium families mainly diagnosed with FTD [3,21,22] and is known to prevent splicing of intron 0, leading to degradation of the mutant transcript and complete loss of its translation [3]. The novel c.−8+2T>G variant we studied here was found to likely disturb normal splicing due to loss of splice donor site, according to in silico analysis. In our study, we recruited two asymptomatic c.−8+2T>G carriers belonging to the same family with multiple affected family members. We have found that the serum PGRN and mRNA levels in asymptomatic individuals carrying the c.−8+2T>G variant were reduced compared to controls. We were not able to measure protein and mRNA levels in affected individuals, because their serum and RNA samples were not available.
The other splice site mutation (c.708+6_9del) studied here is located within a repeat sequence (TGAGTGAG) next to the splice donor site of GRN intron 6. In silico analysis predicted that this deletion could decrease the splicing of exon 6 due to a decreased 5’ donor site score. Apart from us, two other studies have identified similar deletions in the same repeat sequence in different positions [23–25]. In their study Skoglund and colleagues (2011), reported that the deletion causes alteration in the splicing pattern and generates aberrant transcripts with premature stop codon, which are most probably degraded by NMD. We showed here that serum PGRN levels were reduced in affected patients and asymptomatic individuals carrying the c.708+6_9del. In addition to decreased serum PGRN levels, we have found that GRN mRNA levels were also decreased in deletion carriers when compared with controls. Our findings are consistent with the other two studies that showed reduction in GRN brain mRNA levels in patients carrying the deletion [23, 24]. This decrease was modest both in our and in previous studies. However, this decrease may reflect haploin-sufficiency, even if not at the expected 50% reduction of the normal levels, as seen in most GRN mutations. Interestingly, the decreased CSF amyloid levels found in the index patient of this family suggest the co-occurrence of amyloid-β pathology. A recent study by Tan et al. showed amyloid-β deposition in 43% of FTD cases with GRN mutations but in none of the cases with MAPT mutations [26]. FTD patients may have coexisting amyloid pathology suggesting a potential role of these mutations in the increase of the risk of amyloid accumulation in these cases [27].
All carriers of c.708+6_9del in family DEM-35, are known to also have a novel nonsense variant (NM_001123066.3:c.262C>T p.Gln88*) in the alternatively spliced exon 3 of the Microtubule-associated protein tau (MAPT) gene. In our study, we were not able to determine if the presence of the MAPT p.Gln88* had any effects on serum PGRN levels, because there were no affected individuals carrying only one of the variants in the family. However, MAPT p.Gln88* is not expected to be pathogenic since the vast majority of reported pathogenic mutations in MAPT coding region are missense, deletion, and silent type variants affecting the interaction of tau with microtubules.
More details associated with this variant can be found in the Supplementary Material.
In our study, a cut-off value chosen at 93.1 ng/mL reliably distinguished studied GRN splice site mutation carriers from controls and non-carriers. This threshold value is in line with that previously published by Sleegers et al. [7] establishing a serum PGRN cut-off level of 94 ng/mL to predict GRN null mutations but is higher than more recently proposed plasma or serum cut-off values [4, 8, 11, 12]. The discrepancies between our cut-off value and the values of previous studies may have arisen from inter laboratory variability associated with the ELISA methods. Additionally, cut-off values are expected to show differences depending on the use of different types of biological samples (serum versus plasma). Almeida et al. showed that there was a difference between serum and plasma cut-off values with 8.7% lower values for plasma [9]. Also, these authors determined that mean serum PGRN levels can change depending on the ELISA kit used. It is also important to establish specific cut-offs for specific methodologies and specific laboratories. By using the recently established cut-off of 61ng/mL, our asymptomatic splice site mutation carriers could not be distinguished. Given that there were only eight GRN splice site mutation carriers in our study, more mutation carriers will need to be assessed to more precisely determine the optimal cut-off value for our specific methodology/laboratory.
When considering the two splice site mutations, our results suggest that the measurement of serum PGRN levels may identify asymptomatic individuals at risk for the development of FTD, even without any knowledge of the underlying GRN mutation. As only two affected mutation carriers were available to our study, we were not able to test correlation between serum PGRN levels and age of disease onset.
When we evaluated missense mutation carriers, the serum PGRN levels for the benign p.Asp33Glu carrier were higher than those for splice site mutations and other missense carriers, as expected. Interestingly the serum levels of the p.Asp33Glu carrier were even higher than the levels observed for controls, suggesting that other factors may be affecting serum PGRN levels in this patient. Also, we observed a significant reduction both in protein and RNA levels in p.Cys139Arg carriers. The p.Cys139Arg variant had been previously reported by us and by other studies [10, 28, 29]. Based on in silico predictions, p.Cys139Arg was predicted to be likely pathogenic because it disrupts one of the cysteine disulfide bridges in granulin domain F, responsible for the typical folding of the protein [28]. In our previous study, we identified this variant in a female patient diagnosed with FTD. Unfortunately, serum and mRNA samples of this index patient were not available, therefore it was not possible to include her in this study. We have, however, included four of her asymptomatic first degree relatives carrying this variant. We observed significantly lower serum PGRN levels in asymptomatic p.Cys139Arg carriers when compared to healthy controls. Our findings were consistent with previous studies that reported low serum/plasma PGRN levels in carriers of p.Cys139Arg [10, 30]. This finding can possibly be explained by decreased PGRN activity and/or GRN production due to altered full-length PGRN function and abnormal cleavage of PGRN into granulins [31]. Although mRNA levels are generally not expected to be affected in carriers of missense mutations, in p.Cys139Arg carriers, we observed significantly lower RNA levels. These decreased levels are difficult to explain given the known molecular mechanisms associated with these mutations, but it is possible that decreased GRN mRNA levels resulted independently from the mutation, as reported in a previous study, where patients without any GRN mutation were shown to display decreased peripheral GRN mRNA levels [32]. The levels of serum PGRN observed for carriers of other missense variants (p.Val77Ile and p.Pro209Leu) seem to be in between those observed for carriers of pathogenic variants (splice site mutations and p.Cys139Arg mis-sense variant) and controls. This may suggest that a deregulation of PGRN can occur also in carriers of missense variants and that these variants can play a role in the disease as genetic modifiers or risk factors. Therefore, the significance of lower peripheral GRN mRNA levels in missense mutation carriers needs to be further investigated in larger samples and including different variants. Also, decreased PGRN levels in carriers of pathogenic variants compared to carriers of non-pathogenic variants as shown in our results, may suggest that screening PGRN levels rather than mRNA levels could be more informative to discriminate pathogenic from non-pathogenic GRN variants.
We can summarize our key findings as follows:
-
1)
So far, it was known that GRN mRNA levels of affected c.708+6_9del carriers were decreased in brain tissue. In this study we have shown for the first time that GRN mRNA levels of c.708+6_9del carriers are decreased in peripheral blood, as well.
-
2)
In this study, the c.−8+2T>G and c.708+6_9del splice site mutations were shown for the first time to be associated with lower serum PGRN levels in both asymptomatic and affected mutation carriers.
-
3)
In spite of the limited number of subjects, our results can support the potential use of serum PGRN levels as an easy-to-apply biomarker to predict the presence of GRN mutations in asymptomatic individuals at risk and/or affected individuals. Our results need to be further replicated in larger study groups carrying these same mutations.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all the patients and their families. Ebru Ozer and Meltem Pak helped with obtaining the samples. This work was supported in part by the Research Fund of Istanbul University (Project No: BEK-2017–23839, Project No: 50574) and by Fellowships from the Alzheimer’s Society to Jose Bras and Rita Guerreiro.
Footnotes
Authors’ disclosures available online (www.j-alz.com/manuscript-disclosures/18-0599r1).
SUPPLEMENTARY MATERIAL
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD180599.
REFERENCES
- [1].He Z, Bateman A (2003) Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl) 81, 600–612. [DOI] [PubMed] [Google Scholar]
- [2].Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. [DOI] [PubMed] [Google Scholar]
- [3].Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. [DOI] [PubMed] [Google Scholar]
- [4].Sassi C, Capozzo R, Gibbs R, Crews C, Zecca C, Arcuti S, Copetti M, Barulli MR, Brescia V, Singleton AB, Logroscino G (2016) A novel splice-acceptor site mutation in GRN (c.709–2 A>T) causes frontotemporal dementia spectrum in a large family from Southern Italy. J Alzheimers Dis 53, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mendez MF (2018) Manic behavior and asymmetric right frontotemporal dementia from a novel progranulin mutation. Neuropsychiatr Dis Treat 14, 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Van Langenhove T, van der Zee J, Gijselinck I, Engelborghs S, Vandenberghe R, Vandenbulcke M, De Bleecker J, Sieben A, Versijpt J, Ivanoiu A, Deryck O, Willems C, Dillen L, Philtjens S, Maes G, Baumer V, Van Den Broeck M, Mattheijssens M, Peeters K, Martin JJ, Michotte A, Santens P, De Jonghe P, Cras P, De Deyn PP, Cruts M, Van Broeckhoven C (2013) Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and non-mutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol 70, 365–373. [DOI] [PubMed] [Google Scholar]
- [7].Sleegers K, Brouwers N, Van Damme P, Engelborghs S, Gijselinck I, van der Zee J, Peeters K, Mattheijssens M, Cruts M, Vandenberghe R, De Deyn PP, Robberecht W, Van Broeckhoven C (2009) Serum biomarker for progranulin-associated frontotemporal lobar degeneration. Ann Neurol 65, 603–609. [DOI] [PubMed] [Google Scholar]
- [8].Schofield EC, Halliday GM, Kwok J, Loy C, Double KL, Hodges JR (2010) Low serumprogranulin predicts the presence of mutations: Aprospective study. JAlzheimersDis 22, 981–984. [DOI] [PubMed] [Google Scholar]
- [9].Almeida MR, Baldeiras I, Ribeiro MH, Santiago B, Machado C, Massano J, Guimaraes J, Resende Oliveira C, Santana I (2014) Progranulin peripheral levels as a screening tool for the identification of subjects with progranulin mutations in a Portuguese cohort. Neurodegener Dis 13, 214–223. [DOI] [PubMed] [Google Scholar]
- [10].Finch N, Baker M, Crook R, Swanson K, Kuntz K, Surtees R, Bisceglio G, Rovelet-Lecrux A, Boeve B, Petersen RC, Dickson DW, Younkin SG, Deramecourt V, Crook J, Graff-Radford NR, Rademakers R (2009) Plasma progranulin levels predict progranulin mutation status in frontotemporal dementia patients and asymptomatic family members. Brain 132, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ghidoni R, Stoppani E, Rossi G, Piccoli E, Albertini V, Paterlini A, Glionna M, Pegoiani E, Agnati LF, Fenoglio C, Scarpini E, Galimberti D, Morbin M, Tagliavini F, Binetti G, Benussi L (2012) Optimal plasma progranulin cutoff value for predicting null progranulin mutations in neurodegenerative diseases: A multicenter Italian study. NeurodegenerDis 9, 121–127. [DOI] [PubMed] [Google Scholar]
- [12].Galimberti D, Fumagalli GG, Fenoglio C, Cioffi SMG, Arighi A, Serpente M, Borroni B, Padovani A, Tagliavini F, Masellis M, Tartaglia MC, van Swieten J, Meeter L, Graff C, de Mendonca A, Bocchetta M, Rohrer JD, Scarpini E (2018) Progranulin plasma levels predict the presence of GRN mutations in asymptomatic subjects and do not correlate with brain atrophy: Results from the GENFI study. Neurobiol Aging 62, 245.e9–245.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G (2008) Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology 71, 1235–1239. [DOI] [PubMed] [Google Scholar]
- [14].Meeter LH, Patzke H, Loewen G, Dopper EG, Pijnenburg YA, van Minkelen R, van Swieten JC (2016) Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement Geriatr Cogn Dis Extra 6, 330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guven G, Lohmann E, Bras J, Gibbs JR, Gurvit H, Bilgic B, Hanagasi H, Rizzu P, Heutink P, Emre M, Erginel-Unaltuna N, Just W, Hardy J, Singleton A, Guerreiro R (2016) Mutation frequency of the major frontotemporal dementia genes, MAPT, GRN and C9ORF72 in a Turkish cohort of dementia patients. Plos One 11, e0162592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schwarz JM, Cooper DN, Schuelke M, Seelow D (2014) MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods 11, 361–362. [DOI] [PubMed] [Google Scholar]
- [18].Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. Plos One 7, e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yeo G, Burge CB (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- [20].Le Ber I, Van der Zee J, Hannequin D, Gijselinck I, Campion D, Puel M, Laquerriere A, De Pooter T, Camuzat A, Van den Broeck M, Dubois B, Sellal F, Lacomblez L, Vercelletto M, Thomas-Anterion C, Michel BF, Golfier V, Didic M, Salachas F, Duyckaerts C, Cruts M, Verpillat P, Van Broeckhoven C, Brice A, French Research Network on FTD/FTD-MND (2007) Progranulin null mutations in both sporadic and familial frontotemporal dementia. HumMutat 28, 846–855. [DOI] [PubMed] [Google Scholar]
- [21].Brouwers N, Nuytemans K, van der Zee J, Gijselinck I, Engelborghs S, Theuns J, Kumar-Singh S, Pickut BA, Pals P, Dermaut B, Bogaerts V, De Pooter T, Serneels S, Van den Broeck M, Cuijt I, Mattheijssens M, Peeters K, Sciot R, Martin JJ, Cras P, Santens P, Vandenberghe R, De Deyn PP, Cruts M, Van Broeckhoven C, Sleegers K (2007) Alzheimer and Parkinson diagnoses in progranulin null mutation carriers in an extended founder family. Arch Neurol 64, 1436–1446. [DOI] [PubMed] [Google Scholar]
- [22].Janssens J, Philtjens S, Kleinberger G, Van Mossevelde S, van der Zee J, Cacace R, Engelborghs S, Sieben A, Banzhaf-Strathmann J, Dillen L, Merlin C, Cuijt I, Robberecht C, Schmid B, Santens P, Ivanoiu A, Vandenbulcke M, Van-denberghe R, Cras P, De Deyn PP, Martin JJ, Maudsley S, Haass C, Cruts M, Van Broeckhoven C (2015) Investigating the role of filamin C in Belgian patients with frontotemporal dementia linked to GRN deficiency in FTLD-TDP brains. Acta Neuropathol Commun 3, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Skoglund L, Matsui T, Freeman SH, Wallin A, Blom ES, Frosch MP, Growdon JH, Hyman BT, Lannfelt L, Ingelsson M, Glaser A (2011) Novel progranulin mutation detected in 2 patients with FTLD. Alzheimer Dis Assoc Disord 25, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bit-Ivan EN, Suh E, Shim HS, Weintraub S, Hyman BT, Arnold SE, McCarty-Wood E, Van Deerlin VM, Schneider JA, Trojanowski JQ, Frosch MP, Baker MC, Rademakers R, Mesulam M, Bigio EH (2014)A novel GRN mutation (GRN c.708+6_+9delTGAG) in frontotemporal lobar degeneration with TDP-43-positive inclusions: Clinicopathologic report of 6 cases. J Neuropathol Exp Neurol 73, 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Galimberti D, Cioffi SM, Fenoglio C, Serpente M, Oblak AL, Rodriguez-Porcel F, Oldoni E, Hagen MC, Arcaro M, Scarpini E, Ghetti B, Espay AJ (2017) Rapidly progressive primary progressive aphasia and parkinsonism with novel GRN mutation. Mov Disord 32, 476–478. [DOI] [PubMed] [Google Scholar]
- [26].Tan RH, Kril JJ, Yang Y, Tom N, Hodges JR, Villemagne VL, Rowe CC, Leyton CE, Kwok JBJ, Ittner LM, Halliday GM (2017) Assessment of amyloid beta in pathologically confirmed frontotemporal dementia syndromes. Alzheimers Dement (Amst) 9, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Perry DC, Lehmann M, Yokoyama JS, Karydas A, Lee JJ, Coppola G, Grinberg LT, Geschwind D, Seeley WW, Miller BL, Rosen H, Rabinovici G (2013) Progranulin mutations as risk factors for Alzheimer disease. JAMA Neurol 70,774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I, van der Zee J, Pickut BA, Van den Broeck M, Mattheijssens M, Peeters K, Schymkowitz J, Rousseau F, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C (2008) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71, 656–664. [DOI] [PubMed] [Google Scholar]
- [29].Bernardi L, Tomaino C, Anfossi M, Gallo M, Geracitano S, Costanzo A, Colao R, Puccio G, Frangipane F, Curcio SA, Mirabelli M, Smirne N, Iapaolo D, Maletta RG, Bruni AC (2009) Novel PSEN1 and PGRN mutations in early-onset familial frontotemporal dementia. Neurobiol Aging 30, 1825–1833. [DOI] [PubMed] [Google Scholar]
- [30].Sleegers K, Brouwers N, Maurer-Stroh S, van Es MA, Van Damme P, van Vught PW, van der Zee J, Serneels S, De Pooter T, Van den Broeck M, Cruts M, Schymkowitz J, De Jonghe P, Rousseau F, van den Berg LH, Robberecht W, Van Broeckhoven C (2008) Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71, 253–259. [DOI] [PubMed] [Google Scholar]
- [31].Wang J, Van Damme P, Cruchaga C, Gitcho MA, Vidal JM, Seijo-Martinez M, Wang L, Wu JY, Robberecht W, Goate A (2010) Pathogenic cysteine mutations affect progranulin function and production of mature granulins. J Neurochem 112, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Coppola G, Karydas A, Rademakers R, Wang Q, Baker M, Hutton M, Miller BL, Geschwind DH (2008) Gene expression study on peripheral blood identifies progranulin mutations. Ann Neurol 64, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.