

Abstract
Pathways linking activation of the insulin receptor to downstream targets of insulin have traditionally been studied using a candidate gene approach. To elucidate additional pathways regulating insulin activity, we performed a forward chemical–genetics screen based on translocation of a glucose transporter 4 (Glut4) reporter expressed in murine 3T3-L1 adipocytes. To identify compounds with known targets, we screened drug-repurposing and natural product libraries. We identified, confirmed, and validated 64 activators and 65 inhibitors that acutely increase or rapidly decrease cell-surface Glut4 in adipocytes stimulated with submaximal insulin concentrations. These agents were grouped by target, chemical class, and mechanism of action. All groups contained multiple hits from a single drug class, and several comprised multiple structurally unrelated hits for a single target. Targets include the β-adrenergic and adenosine receptors. Agonists of these receptors increased and inverse agonists/antagonists decreased cell-surface Glut4 independently of insulin. Additional activators include insulin sensitizers (thiazolidinediones), insulin mimetics, dis-inhibitors (the mTORC1 inhibitor rapamycin), cardiotonic steroids (the Na+/K+-ATPase inhibitor ouabain), and corticosteroids (dexamethasone). Inhibitors include heterocyclic amines (tricyclic antidepressants) and 21 natural product supplements and herbal extracts. Mechanisms of action include effects on Glut4 trafficking, signal transduction, inhibition of protein synthesis, and dissipation of proton gradients. Two pathways that acutely regulate Glut4 translocation were discovered: de novo protein synthesis and endocytic acidification. The mechanism of action of additional classes of activators (tanshinones, dalbergiones, and coumarins) and inhibitors (flavonoids and resveratrol) remains to be determined. These tools are among the most sensitive, responsive, and reproducible insulin-activity assays described to date.
Keywords: adipocyte, insulin, membrane trafficking, glucose transporter type 4 (GLUT4), drug discovery, chemical genetics, drug repurposing, library screening, small molecule
Introduction
Insulin increases the rate of glucose uptake into adipocytes 20–50-fold. Glucose transport into adipocytes plays a key and essential role in the clearance of lipid from the blood after a meal. Disruption of this pathway leads to systemic metabolic derangement. Restoration of insulin-sensitive lipid metabolism is an important target in the treatment of obesity and diabetes (1).
The primary purposes of adipose tissue are synthesis/storage of triacylglycerol (TAG)4 in periods of energy excess (after a meal) and hydrolysis of TAG to generate fatty acids (FAs) for use by other organs during energy deprivation (fasting and intense exercise) (2, 3). Although it is traditionally thought that excess carbohydrates are converted into lipids for storage by de novo lipogenesis, most FAs in adipose tissue come from dietary fat (4). After a meal, the chylomicron-associated TAG in the blood is hydrolyzed by endothelial lipoprotein lipase, and the released FAs cross cell membranes and are then re-esterified into TAGs. In adipocytes, this re-esterification requires glucose. Adipocytes do not express glycerol kinase; the glycerol phosphate required for TAG synthesis comes from glycolysis (5). Thus, the glucose disposed of in fat after a meal becomes the glycerol backbone of the re-esterified TAGs (6). Disruption of insulin-induced glucose transport in fat (e.g. in obesity) elevates serum-free FAs and TAGs, eliciting insulin resistance in muscle and liver (lipotoxicity) (7, 8). Drugs that restore insulin-sensitive glucose uptake into adipocytes improve overall insulin sensitivity (9, 10).
Glucose uptake into adipocytes is rate-limited by the number of Glut4 facilitative glucose transport proteins inserted into the plasma membrane (PM) (11–13). Insulin increases glucose transport by recruiting Glut4 from intracellular compartments to the PM (14–17). In basal adipocytes, only 1–2% of the total cellular Glut4 is on the cell surface. After insulin stimulation, 30–50% is inserted into the PM. Thus, the large increase in cell-surface Glut4 is the product of both the very efficient sequestration of Glut4 inside of the cell under fasting conditions (when adipocytes need very little glucose) and the rapid redistribution of Glut4 to the PM after insulin stimulation.
A signal transduction cascade linking activation of the insulin receptor to regulated secretory vesicles containing Glut4 has been described (18–20). It includes five proteins as follows: PI 3-kinase; Akt; Rab-GAP AS160; and two GTP-binding proteins that regulate vesicle trafficking, Rab10 and Rab 14 (21–29). PI 3-kinase is activated by the insulin receptor through its substrate IRS-1. The resultant PI 3-phosphate in cell membranes recruits kinases that activate Akt. Akt phosphorylates and inhibits AS160, which is co-localized with Glut4 in intracellular compartments. Inhibition of the GAP activity of AS160 increases GTP loading and activation of its substrates Rab10 and Rab14, which are also resident on Glut4 vesicles. This increases Glut4 exocytosis.
Previous work by our lab and others relied on a candidate gene approach; reverse genetics (shRNA knockdown) and reverse chemical genetics (small molecule inhibitors) were used to perturb the proteins known to affect cell-surface Glut4, and the effects of these perturbations on trafficking were measured (17, 29–34). To elucidate novel protein machineries and pathways regulating Glut4 trafficking, a forward chemical genetics screen was done. Classical forward genetics involves random DNA mutation and selection of cells/organisms with a specific phenotype (e.g. changes in cell-surface Glut4 levels). In chemical genetics, compounds that produce a specific phenotype are discovered and then the targets of “hits” identified. When libraries of approved, well-characterized drugs are used, the targets of hits are known. The power of forward genetics is the ability to develop a comprehensive, systems-level map of all proteins/pathways affecting a selected phenotype. This unbiased approach allows identification of unexpected relationships.
This paper describes assays to identify and validate small molecules that enhance or inhibit insulin action in adipocytes using changes in cell-surface Glut4 as a marker for insulin activity. These high-throughput assays are among the most sensitive, responsive, and reproducible insulin-activity assays that have been reported. It also describes assays to distinguish the mechanisms of action of these compounds. Using these assays, small molecule screens of four drug-repurposing and natural products libraries (3858 compounds) were performed. Confirming the power of the chemical genetics approach, both novel activators and inhibitors of insulin action were discovered. These hits revealed previously unidentified regulatory pathways for insulin signaling as well as for Glut4 trafficking. FDA-approved drugs and commonly consumed supplements/“nutraceuticals” that inhibit Glut4 translocation were also identified (e.g. quinines/antimalarials, cyclic antidepressants, resveratrol, and flavonoids). These may worsen insulin resistance in patients with diabetes. Consistent with this, several of the inhibitors identified are known to cause acute insulin resistance (hyperinsulinemia and normoglycemia) in humans through unknown mechanisms. The potential role of inhibition of Glut4 translocation in these adverse drug affects is discussed.
Results
Assay
An assay to screen for the effects of small molecules on acute insulin action was developed that is highly sensitive, responsive, specific, and reproducible. It is among the most sensitive and reproducible assays for insulin activity described to date. It is a high-throughput, content-rich assay that allows rapid collection of data from thousands of individual cells/samples, and hundreds of samples/experiment. The screening assay has two key features: the insulin sensitivity/responsiveness of the cells (cultured adipocytes) and the sensitivity, speed, and accuracy of the detection system (flow cytometry). Importantly, these screens identify compounds that have acute effects in adipocytes pretreated with insulin (increase or decrease cell-surface Glut4). Although compounds may reverse or increase insulin signaling, this protocol eliminates confounders that prevent activation of the insulin receptor or severely impair membrane trafficking.
Using this system, a 2–3-fold increase over basal surface Glut4 levels can be reproducibly detected with 0.05 nm insulin, with a maximal increase of 20–30-fold at 100 nm insulin (EC50 = 1.4 nm), and a sample-to-sample standard deviation of <15% (Fig. 1A). Insulin rapidly increases cell-surface Glut4 (t½ = 4 min), and this remains elevated for several hours (Fig. 1B) (17, 30, 32). The PI 3-kinase inhibitor LY294002 (LYi) inhibits Glut4 exocytosis, causing a rapid decrease in cell-surface Glut4 (up to 90%, t½ = 5 min, EC50 = 10 μm). For screening, cells were preincubated with insulin for 45 min, followed by a 1-h treatment with compounds. Cells were then placed on ice; surface Glut4 was labeled with Alexafluor647-conjugated antibody against the HA epitope (AF647–α-HA), and the cells were detached from the culture dish using collagenase. The total HA/Glut4-GFP and cell-surface AF647–labeled Glut4 were measured in the disassociated cells using flow cytometry. Adipocytes were distinguished from fibroblasts and necrotic cells in the cell cultures based on light scatter (from the lipid droplets) and cellular autofluorescence (17).
Figure 1.

Assay. 3T3-L1 fibroblasts stably expressing HA–Glut4/GFP were differentiated into adipocytes in 96-well plates. The cells develop a three-dimensional co-culture of adipocytes embedded in a collagen matrix, with a monolayer of fibroblasts on the plastic. The cultures were incubated in serum-free media for 2 h (basal) at 37 °C. Insulin was added, and cells were incubated for 45 min. Compound was added, and cells were incubated for 1 h. Cells were placed on ice, and surface Glut4 was labeled with AF647-conjugated anti-HA antibody. Cells were detached from the plate with collagenase and analyzed by flow cytometry. A, insulin dose response. B, time course. C, pilot screen. D, identification of hits. For activators (green), relative geometric mean fluorescence (rGMF) was ≥1.5, and for inhibitors (red), rGMF was ≤0.62. 64 activators and 65 inhibitors were verified by rescreening, and representative compounds were confirmed with pure compound and validated by concentration response, SAR, and time course (Table S1).
The distribution of total fluorescence per cell in a single sample (∼5000 cells) is non-Gaussian (roughly log normal; Fig. S1A). This is an inherent property of protein expression in cells (35). Therefore, the geometric mean fluorescence (GMF) is computed for each sample (Fig. S1B; the mean of the log10 fluorescence per cell is determined and then the antilog10 of this mean is calculated). These values are standardized relative to the average GMF of the DMSO samples from the same 96-well assay plate. In our system, the distribution of the relative GMF (rGMF) values across a large population of control DMSO samples is Gaussian (Fig. S1C; solid line, n = 1005). Thus, the probability that a sample treated with compound is the same as treatment with DMSO alone (lies within the same population as the control samples) or is representative of a treatment that is affecting cell-surface Glut4 can be estimated using the mean and standard deviation of the rGMF values (mean of means) of all control samples from each screen.
Pilot screen
The Prestwick Chemical Library of FDA-approved drugs (1120 compounds) was screened at a single concentration as a single sample under two conditions. For activators, cells were pretreated for 45 min with 0.05 nm insulin and then incubated with compounds for 1 h. The positive reference for activators (REFactivator, 100 nm insulin) increased cell-surface Glut4 an additional 26-fold under these conditions. For inhibitors, cells were pretreated with 1 nm insulin and then incubated with compounds for 1 h. The positive reference for inhibitors (LYi) decreased cell-surface Glut4 by 75% in these samples. Insulin (100 nm) increased Glut4 3-fold in cells pretreated with 1 nm insulin; thus activators could also be detected in the “inhibitor” screen.
Of the 1120 compounds screened, 25 were “shifters” that directly affected either autofluorescence or scatter of the cells (e.g. Chicago sky blue, propidium iodide, and sanguinarine). This could be detected in the uninfected cells present in each sample (17, 30–34). These compounds were not further analyzed.
Compounds were selected for verification by retesting if the sample had a relative geometric mean fluorescence (rGMF) >3 S.D. from the average rGMF of all control DMSO samples from the screen. Using these criteria, hits lie outside the range of 99.7% of the control DMSO samples (∼3 compounds per 1000 are expected to randomly screen as false positives). rGMFDMSO ± S.D. = 1 ± 0.13 (n = 224 samples; eight samples/plate, 28 plates). Therefore, compounds with an rGMF ≥1.4 were selected as potential “activators,” whereas compounds with an rGMF ≤ 0.6 were selected as potential “inhibitors.”
Using these criteria, 24 of the 1120 compounds screened positive as activators and none as inhibitors on cells pretreated with 0.05 nm insulin. Two of the control DMSO samples (n = 112) falsely screened positive as activators under these conditions. In cells pretreated with 1 nm insulin, 32 screened positive as activators and 31 screened positive as inhibitors. None of the control DMSO samples (n = 112) screened positive under these conditions. Three of the activators were identified as positive hits on both plates. Hits were subjected to verification by repeat testing in triplicate using compounds from the library. Of the 42 activators that were rescreened, 10 were verified. However, it was much more cost-effective to verify hits by rescreening in triplicate than it would have been to screen all compounds in triplicate to reduce false positives. Using two plates increased sensitivity (increased true positives) but decreased specificity (increased the number of false positives) for activators. Of the 24 inhibitors that were rescreened, 23 were verified.
Z-factors
Z-factors are statistical parameters used to evaluate assay robustness in high-throughput screens (36). They are dependent on both the signal dynamic range (the difference between known positive reference samples and untreated control samples) and the sample-to-sample variability (standard deviation) using Equation 1,
| (Eq. 1) |
In an excellent assay, 0.5 ≤ Z′ ≤ 1; if Z′ = 0–0.5, the assay is less robust but is still acceptable. In the pilot screen, Z′ = 0.82 or 0.62 for activators when screened in cells pretreated with either 0.05 or 1 nm insulin, respectively (REF: 100 nm insulin). For inhibitors, Z′ = 0.35 in cells pretreated with 1 nm insulin but was below 0 in cells treated with 0.05 nm insulin, making this condition unsuitable for discovery of inhibitors (REF: 50 μm LYi). However, despite the lower Z′ score, the specificity was higher for the inhibitor screen than for the activator screen (there was a significantly lower false-positive rate).
Library screens
Based on the results of the pilot screen, compounds were assayed as single samples on a single plate at an intermediate insulin concentration (0.3 nm) in subsequent screens of the Spectrum Collection of bioactive chemicals and natural products, the National Institutes of Health Clinical Collection, and the Selleck natural product library (2000, 707, and 131 compounds, respectively). Under these conditions, maximal insulin (100 nm) increased cell-surface Glut4 an additional 3–5-fold, whereas LYi decreased cell-surface Glut4 by 80–90% (Fig. 1, A and B). The Z′-factors calculated from a test experiment were 0.59 and 0.13 (n = 80 control DMSO and n = 8 each reference). The average Z′ scores from the completed screens were 0.52 and 0.17 (n = 224 DMSO and 68 of each reference sample from 34 plates). To reduce the number of false-positive activators and false-negative inhibitors, the criteria for selection of hits were made more stringent (activators, rGMF ≥1.5, and inhibitors, rGMF ≤0.65).
The distribution of rGMF values for all unknown samples in the screens (2769 total after elimination of shifters) was approximately Gaussian (Fig. 1, C and D). 85 activators and 99 inhibitors screened positive and were verified. A number of these were redundant, found in multiple libraries.
Using a lower concentration of insulin for pretreatment (0.3 nm) significantly increased the assay sensitivity (decreased false negatives) for activators. Nine compounds were identified in the subsequent screens that were present in the Prestwick library, but not hits in the initial screen in cells pretreated with higher insulin (1 nm). However, these screening conditions significantly increased the false-negative rate for inhibitors: seven compounds identified as hits in the initial screen at 1 nm insulin did not screen positive as hits in subsequent screens at 0.3 nm insulin. Although all significantly decreased cell-surface Glut4 when assayed as pure compounds, their effects were too small at the lower insulin concentration to screen positive. Thus, screening in cells pretreated with 0.3 nm insulin is optimized for finding compounds that increase but not that decrease cell-surface Glut4.
Structure/activity relationships (SAR)
Overall, 64 activators and 65 inhibitors were identified in the combined screens and were verified in replicate assays using compounds from the libraries (Table S1). The hits can be sorted into drug classes based on structure, targets, and pathways. Multiple hits from a single drug class and multiple structurally unrelated hits for a single target provide additional verification for compounds. Eight classes of activators and seven classes of inhibitors were identified (Table 1). Representative compounds from each class were selected for further analysis. Thirty two activators and nine inhibitors were validated using pure compound. Based on the drug classes, five additional activators and three additional inhibitors were identified by testing related compounds. Hits from each class were verified by determining their concentration response (EC50) and time course.
Table 1.
Summary of hits: mechanisms of action, drug classes, and representative compounds
| Activators | Inhibitors | |
|---|---|---|
| Insulin sensitizers | Thiazolidinediones: pioglitazone, ciglitazone, and rosiglitazone | |
| Disulfiram (thiuram) | ||
| Dis-inhibitors | Inhibitors of mechanistic target of rapamycin complex 1 (mTORC1): rapamycin | |
| β-Adrenergic | Agonists: isoproterenol, metaproteranol, terbutiline, and epinephrine | Inverse agonists/antagonists: propranolol |
| Adenylate cyclase activator: forskolin | ||
| Adenosine receptor | Agonists: 2-chloroadenosine | Inverse agonists/antagonists: CGS 15943, IBMX |
| Steroids | Corticosteroids: flumethasone, halcinonide, prednicarbate, and dexamethasone | |
| Androgenic: flutamide | Androgenic: testosterone | |
| Na+/K+-ATPase inhibitors | Cardiotonic steroids: proscillaridin A, medrysone, strophanthidin, and ouabain | |
| Antibiotics | Proguanil, niclosamide, amsacrin | |
| Protein synthesis inhibitors | Puromycin, cycloheximide, anisomycin, cephaeline, ementine, lycorine | |
| Heterocyclic amines | Antihistamines: homochlorcyclizine, azelastine | |
| Antipsychotics: haloperidol, bromperidol | ||
| Phenothiazine anti-psychotics: promazine, promethazine, thioridazine | ||
| Dibenzazepines: tricyclic antidepressants; imipramine, desipramine, protriptyline | ||
| Tetracyclic anti-depressants: maprotiline | ||
| Lysosomotropic amines | Chloroquine, ammonium chloride | |
| Phytochemicals: supplements and medicinal herbal extracts | Tanshinone I, dihydrotanshinone I, cryptotanshinone, GSPE | Parthenolide, piperlongumine, resveratrol, LY294002/LYi, Akti |
| Flavonoids | Icariin, myricetin, and dihydromyricetin | Kaempferol, quercitin, geraldol |
Insulin sensitizers
All of the thiazolidinediones present in the libraries (pioglitazone, ciglitazone, and rosiglitazone) screened positive and were verified as activators. Thiazolidinediones improve insulin sensitivity in cultured adipocytes, as well as in humans, through activation of the transcription factor PPARγ (37). The concentration response of pioglitazone is consistent with an effect through PPARγ (Fig. 2A; EC50 = 0.6 μm for increased cell-surface Glut4 and EC50 = 1 μm for increased differentiation in 3T3-L1 cells) (38). As expected for an insulin sensitizer, addition of a saturating concentration of pioglitazone (10 μm) to cells stimulated with submaximal insulin caused a left shift in the insulin dose response (Fig. 3A). The cells responded as if they had been treated with a 4-fold higher concentration of insulin (EC50 insulin = 0.8 nm with DMSO, 0.2 nm with pioglitazone). Thiazolidinediones had little effect in quiescent cells or in cells treated with saturating insulin (0.003 or 100 nm; Fig. 3B). Thiazolidinediones act as insulin sensitizers in cells and animals through changes in adipocyte differentiation (chronic long-term exposure). An acute effect of these drugs, within minutes of treatment, on insulin sensitivity and/or glucose transport has not previously been reported. However, rapid (within minutes), non-genomic effects of PPARγ activators have previously been reported in cells and in animals (39, 40). These affects were blocked by GW9662, a PPARγ antagonist. Interestingly, the acetaldehyde dehydrogenase inhibitor disulfiram also acts like an acute insulin sensitizer in cultured adipocytes (Fig. 3A).
Figure 2.
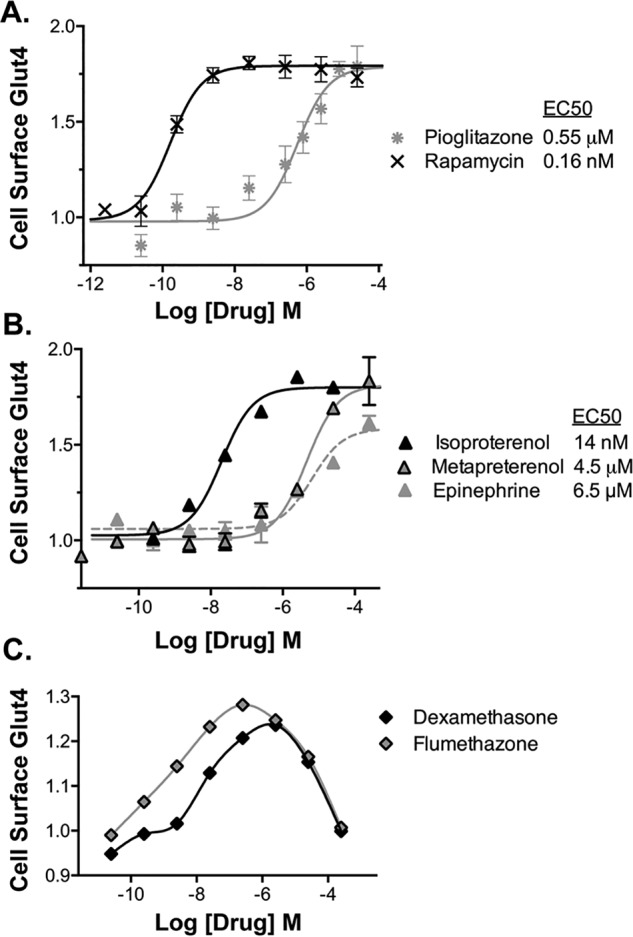
Validation of activators, concentration response. Verified hits were assigned into drug classes based on shared structures and targets (Table S1). Representative compounds from each group were chosen for further analysis. For confirmation, representative hits were confirmed by ordering pure compound and rescreening. For concentration response, cells were pretreated with insulin (0.3 nm), and compounds were added at increasing concentrations, and cells were incubated for 1 h. Cells were placed on ice, and cell-surface Glut4 was determined. A, insulin sensitizers and dis-inhibitors. B, β-adrenergic receptor agonists. C, glucocorticoids. To verify targets, the measured EC50 values (Table S1) were compared with published values.
Figure 3.

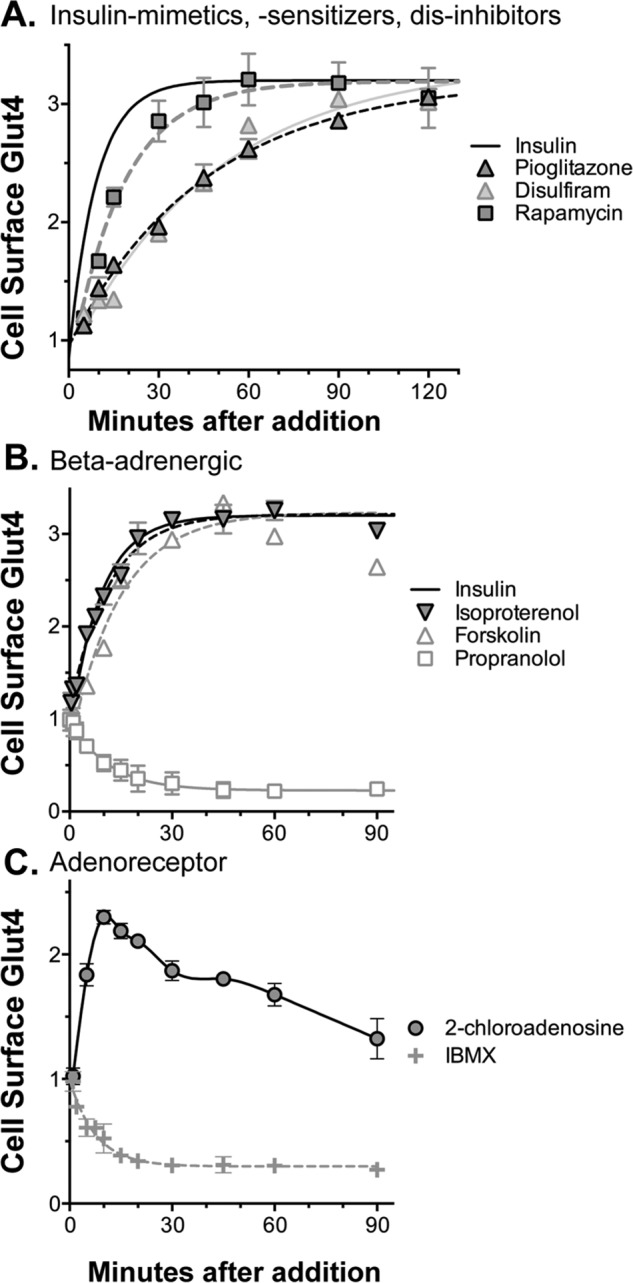
Mechanism of action of activators, insulin concentration response. Cells were pretreated with increasing concentrations of insulin; compounds were added at saturating concentrations, and cells were incubated for 1 h. Cells were placed on ice, and cell-surface Glut4 was determined. The shapes of the curves distinguish insulin mimetics, insulin-sensitizers, dis-inhibitors, and drugs that work through parallel (insulin-independent) pathways. A, all compounds increase cell-surface Glut4 ∼2-fold at sub-maximal (0.3 nm) insulin (red oval). B, drug classes are distinguished by their effects on cells preincubated at low (0.003 nm) and saturating (100 nm) insulin (summarized in Table 2).
Insulin mimetics
Thiazolidinediones are not acting as insulin mimetics. Their effect on the shape of the insulin concentration–response curve is different. Insulin mimetics elevate surface Glut4 in cells treated with low doses of insulin, to the same extent at all concentrations below the “effective” insulin concentration (submaximal insulin, 0.6 nm, is used as an example). An insulin mimetic will shift the dose response to the right (Fig. 3A; in this example from 1 to 2 nm) and have no effect in cells treated with saturating insulin (Fig. 3B).
To further distinguish the mechanism of action of insulin-sensitizers versus insulin mimetics, time-course experiments were performed (Fig. 4A). In cells pretreated with submaximal insulin (0.3 nm), 0.6 nm insulin increased cell-surface Glut4 significantly more rapidly than pioglitazone (t½ = 4 min insulin and 35 min pioglitazone), consistent with different mechanisms of action. Disulfiram showed kinetics very similar to pioglitazone. Although we did not identify any insulin mimetics in our screen, small molecule insulin mimetics were discovered in targeted screens for activators of the tyrosine kinase activity of the isolated insulin receptor (41, 42), providing proof of concept that they exist in small-molecule libraries.
Figure 4.
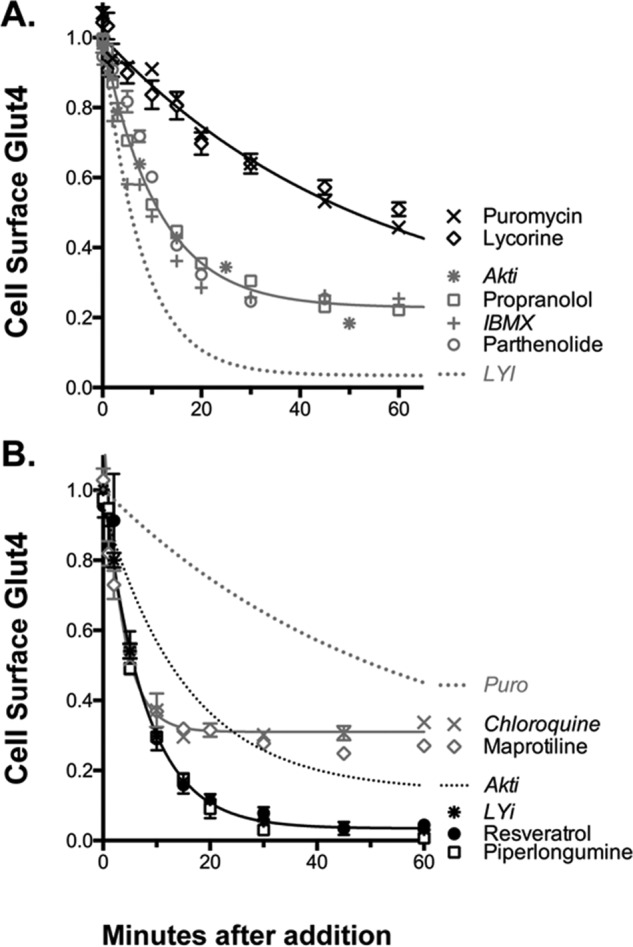
Mechanism of action of activators, time course. Cells were pretreated with insulin (0.3 nm); compounds were added at saturating concentrations, and cells were incubated for increasing times. Cells were placed on ice, and cell-surface Glut4 was determined. At saturating concentrations, compounds with the same mechanism of action will have similar time courses. Differences in the shape of the time courses indicate a different mechanism of action. A, insulin mimetics (t½ = 4 min), dis-inhibitors (t½ = 12 min), and insulin sensitizers (t½ = 35 min) increase cell-surface Glut4 at different rates. B, β-adrenergic receptor agonists and forskolin show kinetics similar to insulin (t½ = 5–10 min), consistent with effects on a parallel signal transduction pathways. C, adenosine receptor agonists transiently increase cell-surface Glut4 with a maximal effect at 10 min that slowly decreases. B and C, propranolol and IBMX act as reverse agonists, rapidly decreasing cell-surface Glut4 (t½ = 8–10 min) in the absence of agonists.
Dis-inhibitors
The mechanistic target of rapamycin complex 1 (mTORC1) inhibitor rapamycin also screened positive and was verified as an activator. The concentration response of rapamycin is consistent with a direct effect on mTORC1 (Fig. 2A; EC50 for increased cell-surface Glut4 = 0.16 nm, IC50 for mTORC1 inhibition in tissue culture cells = 0.1 nm) (43). Rapamycin acutely enhances Glut4 translocation in 3T3-L1 adipocytes through inhibition of an mTORC1-catalyzed feedback inhibition loop for insulin signaling via S6 kinase (44). Consistent with this mechanism, rapamycin had little effect in quiescent cells (0.003 nm) and was synergistic with insulin at saturating concentrations (Fig. 3, A and B). In cells pretreated with submaximal insulin (0.3 nm), rapamycin increased cell-surface Glut4 more slowly than insulin (Fig. 4A; t½ = 12 min). Thus, reversal of inhibition of signaling is slower than direct stimulation of the insulin receptor. However, rapamycin acted significantly faster than pioglitazone and disulfiram, again consistent with their different mechanisms of action. Rapamycin acutely enhances insulin sensitivity and effectiveness in humans, making it an attractive potential therapeutic (45). However, chronic rapamycin treatment inhibits insulin activity and causes hyperglycemia due to indirect effects on mTOR complex 2 (46–48).
β-Adrenergic receptor agonists and antagonists/inverse agonists
Activators also include 10 β-adrenergic agonists (e.g. isoproterenol, metaproterenol, and terbutaline). The β-agonist epinephrine and the adenylate cyclase activator forskolin also increase cell-surface Glut4. The concentration responses for the β-agonists (isoproterenol ≫ metaproterenol and epinephrine, EC50 = 14 nm versus 4.5 and 6.5 μm; Fig. 2B) are consistent with an effect via β3-adrenergic receptors (49). In contrast to the insulin sensitizers, β-agonists had their maximal effect in quiescent cells, with a synergistic effect that was additive to insulin at all insulin concentrations (Fig. 3, A and B). Thus, the β-agonists have an effect on Glut4 trafficking that is independent of signal transduction through the insulin receptor. Like insulin, the effect of the β-agonists is very fast (t½ = 5 min), consistent with an effect via signal transduction (Fig. 4B). Forskolin also rapidly increased cell-surface Glut4 (t½ = 10 min), although it is slower than isoproterenol.
The β-adrenergic antagonist propranolol screened positive as an inhibitor in insulin-stimulated cells. Propranolol is acting as an inverse agonist, rapidly decreasing cell-surface Glut4 even in the absence of added β-agonist (t½ = 8 min; Figs. 4B and 7A). Propranolol had little effect on cell-surface Glut4 in quiescent cells (data not shown). Activation of β-adrenergic receptors acutely increases glucose transport in isolated adipocytes, cardiomyocytes, and skeletal muscle (50–55). In muscle, this increase is through insulin-independent activation of the mTORC2 pathway and translocation of Glut4 (56). β-Agonists and antagonists affect glucose transport in primary adipocytes through effects on Vmax and not transporter affinity (57). They also induce translocation of glucose transport activity from microsomes to the plasma membrane fraction in adipocytes. We have verified that these compounds directly effect Glut4 translocation in adipocytes.
Figure 7.
Mechanism of action of inhibitors, time course. Cells were treated as described in Fig. 4. The shapes of the curves distinguish four different mechanisms (summarized in Table 3). A, protein synthesis inhibitors (slow, 80%; e.g. puromycin) and inhibitors of signal transduction (intermediate, 80%; e.g. Akti). B, inhibitors of Glut4 exocytosis (fast, 80–90%; e.g. LYi) and inhibitors of endosomal acidification (very fast, 50%; e.g. chloroquine).
Adenosine receptor agonists and antagonists/inverse agonists
Activators also included the adenoreceptor agonist 2-chloroadenosine (Fig. 4C). The nonxanthine α1/α2 adenosine receptor antagonist CGS 15943 screened positive as an inhibitor, and the adenosine receptor antagonist isobutylmethylxanthine (IBMX) is an inhibitor when assayed as a pure compound. The dose response observed for IBMX (EC50 = 0.5 mm; Fig. 6A) indicates that its effects are due to adenoreceptor inverse agonism and not to effects on cAMP as a phosphodiesterase inhibitor (IC50 PDE = 2–50 μm) (58). Because of this low affinity, IBMX would not have screened positive as an inhibitor if it had been in the libraries (Fig. 6A). This is a limitation of screening compounds at a single concentration (25 μm). This can contribute to false negatives.
Figure 6.
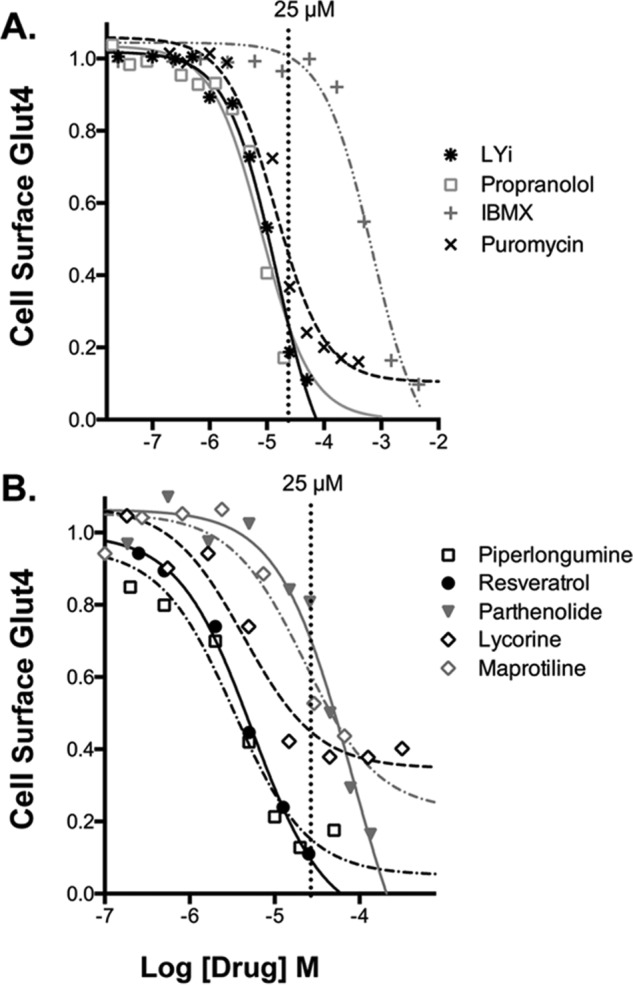
Validation of inhibitors, concentration response. Verified hits were assigned into drug classes based on shared structures and targets (Table S1). Representative hits were confirmed using pure compound and validated by dose response. A, drugs with known targets/mechanisms of action. Note: IBMX (EC50 = 0.5 mm) is acting via adenoreceptor inverse agonism and not through effects on cAMP as a phosphodiesterase inhibitor (IC50 PDE = 2–50 μm). Because of its low affinity, IBMX would not have screened positive as an inhibitor (25 μm). B, inhibitors identified in the screen with unknown mechanism of action.
Like propranolol, IBMX also rapidly decreased cell-surface Glut4 even in the absence of added adenosine receptor agonist (t½ = 10 min; Figs. 4C and 7A). Interestingly, adenosine receptor stimulation transiently increased cell-surface Glut4 (maximal increase at 10 min), while β-adrenergic agonist stimulation was sustained (Fig. 4, B and C). Xanthines, including caffeine, induce acute insulin resistance in humans, decreasing whole-body glucose disposal ∼30% when measured in either an oral glucose tolerance test or insulin clamp (59). Acute effects of IBMX on glucose transport in isolated human and rat adipocytes have also been reported (57, 60). Consistent with our results, IBMX inhibited glucose transport in adipocytes at concentrations inconsistent with effects on cAMP. However, this may be due to direct effects of IBMX on the activity of the glucose transporters. Effects of IBMX on Glut4 translocation have not previously been reported.
Corticosteroids
13 steroids screened positive as activators (e.g. halcinonide, flumethasone, and prednicarbate). The corticosteroid dexamethasone was also an acute activator when assayed as a pure compound. Interestingly, the androgen receptor agonist testosterone screened positive as an inhibitor, whereas the nonsteroidal androgen receptor antagonist/reverse agonist flutamide screened positive as an activator. The steroids showed a U-shaped concentration-response curve with maximal effects at concentrations lower than the screening concentration (<25 μm), and decreasing effects at higher concentrations (Fig. 2C). Acute effects of dexamethasone on Glut4 translocation have not previously been reported. Chronic treatment with dexamethasone and other corticosteroids inhibits insulin signaling in primary adipocytes and 3T3-L1 adipocytes and causes hyperglycemia in humans (61).
Cardiotonic steroids
Three cardioglycoside inhibitors of the Na+/K+-ATPase screened positive as activators (proscillaridin A, medrysone, and strophanthidin). Ouabain also increased Glut4 translocation when assayed as a pure compound. The ouabain dose response (EC50 = 13 μm; Fig. 5A) is consistent with an effect via the ubiquitously expressed α1 isozyme (IC50 = 40–50 μm) rather than the tissue-specific α2 isozyme (IC50 = 50 nm), although both are expressed in adipocytes and muscle (α1 in rodents is very low affinity relative to other species (62)). As observed for insulin sensitizers, ouabain has no effect in quiescent cells (0.05 nm insulin; Fig. 5A).
Figure 5.
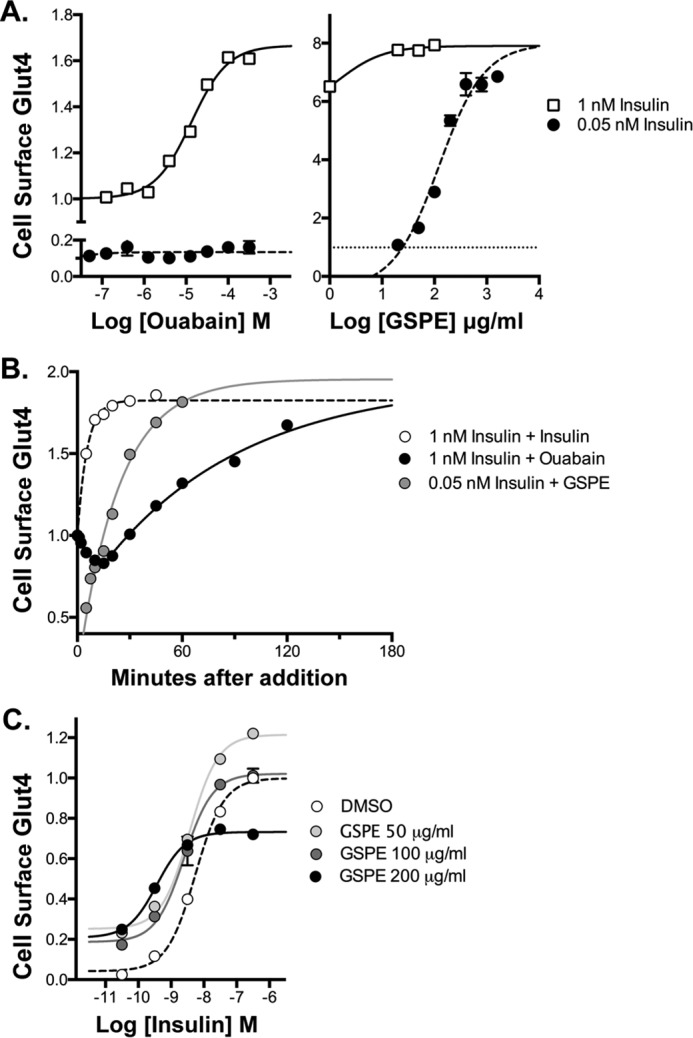
Novel mechanisms of action, Na+/K+-ATPase inhibitors (cardiotonic steroids) and GSPE. Cells were treated as described in Figs. 2–4. A, concentration response. The increase in cell-surface Glut4 was dose-dependent and saturable for both ouabain and GSPE. Unlike other activators, GSPE had its largest effect in quiescent cells (filled symbols), increasing cell-surface Glut4 to the same extent as 1 nm insulin (open symbols). Ouabain had no effect in quiescent cells. B, time course. The effect of GSPE was slow relative to insulin (t½ = 20 min) but much faster than ouabain (t½ = 60 min). Ouabain had a complex kinetics profile, with an initial rapid decrease in cell-surface Glut4, followed by a slow increase (black filled symbols). C, insulin concentration response. GSPE increased cell-surface Glut4 at sub-maximal insulin (<3 nm) but inhibited translocation at saturating insulin (black filled symbols).
The kinetics of the ouabain-induced increase in insulin-stimulated cells were significantly slower than insulin, insulin sensitizers, the dis-inhibitor rapamycin, and the β-adrenergic agonists, indicating a novel mechanism of action (t½ = 60 min; Fig. 5B). Ouabain increased cell-surface Glut4 in cells pretreated with 1 nm insulin ∼80–90% under screening conditions (1 h treatment) and nearly 3-fold with longer incubation times. Consistent with a unique mechanism of action, the kinetics profile of ouabain shows a very distinctive shape. Initially, there is a rapid decrease in cell-surface Glut4, which is followed by a slow rise. The kinetics of the slow rise is comparable with the rate constant for Glut4 exocytosis at 1 nm insulin (30, 31), suggesting that ouabain may be effecting Glut4 trafficking directly rather than through signaling. In support of this idea, ouabain has acute effects on endocytic trafficking through the effects on endosomal pH (63, 64). Glut4 traffics through insulin-sensitive acidic compartments in adipocytes (26, 65–68).
Cardiotonic steroids were originally isolated from plant extracts, but highly related compounds are endogenously expressed in experimental animals and humans (69). These endogenous ligands have been implicated in both normal and disease processes. Interestingly, insulin stimulates the translocation of the Na+/K+-ATPase from intracellular compartments to the cell surface in a number of cell types, including muscle (70–72).
Protein synthesis inhibitors
Inhibitors included 12 antibiotics/anti-neoplastics (e.g. proguanil, niclosamide, and amsacrin). Five of these affect the same pathway, protein synthesis, via distinct mechanisms (puromycin, cycloheximide, anisomycin, cephaeline, and ementine). Loss of Glut4 from the cell surface using a saturating concentration of puromycin (Fig. 6A) was slow compared with inhibition of proteins known to be important for insulin signaling (t½ = 35 min for puromycin versus 5 min for LYi and 10 min for the Akt inhibitor Akti; Fig. 7A). The protein synthesis inhibitors did not decrease cell-surface Glut4 in quiescent cells (data not shown). The plant phytochemical lycorine shows nearly identical kinetics to puromycin (slow, t½ = 30–40 min). Lycorine is a protein synthesis inhibitor (73). Lycorine has also been reported to inhibit Akt. However, the kinetics of inhibition of glucose transporter translocation by lycorine is inconsistent with Akt inhibition as its major mechanism of action.
A requirement for continuous protein synthesis to maintain Glut4 translocation has not been previously reported; however, a requirement for continuous protein synthesis for insulin receptor trafficking has been described (74). Interestingly, Glut4 trafficking utilizes a protein component that is consumed during the process, TUG (Tether containing UBX domain, for Glut4). Both expression of full-length TUG and proteolytic cleavage of TUG is required for proper Glut4 targeting in adipocytes (75, 76). Insulin-induced proteolytic cleavage of TUG (TUG consumption) occurs with kinetics similar to those observed for inhibition of Glut4 trafficking after addition of protein synthesis inhibitors.
Heterocyclic amines
Inhibitors also include two antihistamines (homochlorcyclizine and azelastine), two “typical” antipsychotics (haloperidol and bromperidol), three phenothiazine antipsychotics (promazine, promethazine, and thioridazine), five dibenzazepines (tricyclic antidepressants; e.g. imipramine, desipramine, and protriptyline), and the tetracyclic antidepressant maprotiline. The antihistamines, antipsychotics, phenothiazines, dibenzazepines, and heterocyclic antidepressants act as antagonists or inverse agonists for multiple overlapping receptors (e.g. histamine, dopamine, and muscarinic acetylcholine), with differences in relative potencies. However, the relative concentration responses of these compounds on Glut4 translocation are not consistent with effects on any of these receptors (Table S1). Compounds with hundreds of fold differences in affinity for particular receptors have similar effects on Glut4 translocation, and compounds with similar affinities for specific receptors have very different effects on Glut4 translocation.
Although their targets are diverse, the heterocyclic amines are structurally similar. Their planar, hydrophobic ring structures allow them to cross lipid bilayers. Their amines bind protons (pKa values are listed in Table S1). Thus, all would be expected to act as lysosomotropic amines and dissipate pH gradients in acidic endosomal compartments (63, 77, 78). Consistent with this, the kinetics of the effects of the heterocyclic amines on Glut4 translocation are very similar to those of the vacuologenic amine chloroquine, and different from other classes of inhibitors (Fig. 7B; maprotiline). There is a very rapid but only partial decrease in cell-surface Glut4 at saturating drug concentrations (t½ = 3 min; maximal inhibition 50%). The vacuologenic amine NH4Cl also inhibits Glut4 trafficking with very similar kinetics and to a similar extent (data not shown). All three of these compounds decrease cell-surface Glut4 in quiescent as well as insulin-stimulated cells (0.05 nm insulin; data not shown). Consistent with the idea that the heterocyclic amines affect surface Glut4 through effects on endosomal pH, toxicology studies have shown that many therapeutic drugs in this class, including several of the hits in this screen, are lysosomogenic in humans and mice (79). They accumulate in acidic compartments in cells, particularly in adipose tissue. Phenothiazines and dibenzazepines cause acute insulin resistance (hyperinsulinemia and normoglycemia) in humans, through unknown mechanisms (80–82). Because of this side effect, tricyclic and tetracyclic antidepressants are no longer first line therapeutics for treatment of individuals with depression, although they are still used in patients that fail to respond to other drug classes. It is possible that alternative dosing regimens can be identified that ameliorate the adverse effects of this class of drugs on glucose homeostasis.
Supplements and medicinal herbs
All four of the libraries include natural products/phytochemicals in addition to FDA-approved drugs. 42 of these compounds screened positive, 16 as activators and 26 as inhibitors. Of the 16 activators identified, eight are available as highly concentrated nutraceuticals and eight are active ingredients isolated from medicinal herbal extracts. Activators include three compounds related to tanshinone I (tanshinone I, dihydrotanshinone I, and cryptotanshinone). Tanishinone IIA, which is an additional bioactive compound found in the same extracts, did not screen positive. Dihydrotanshinone I decreases blood glucose in diabetic rodents, via an unknown mechanism (83–86).
The nutritional supplement grape seed proanthocyanidin extract (GSPE) was also identified as an activator. GSPE induced a saturable, dose-dependent increase in cell-surface Glut4. However, unlike other activators such as ouabain, GSPE had its largest effect at low insulin (0.05 nm), increasing cell-surface Glut4 to the same extent as 1 nm insulin (Fig. 5A). GSPE had a smaller effect at submaximal insulin (1 nm). The time course of the effect of GSPE on quiescent cells (0.05 nm insulin; t½ = 20 min) was slow relative to insulin but much faster than ouabain. At higher concentrations (200 μg/ml), GSPE elevated basal cell-surface Glut4 but inhibited Glut4 translocation at saturating insulin (Fig. 5C), indicating a complex mechanism. GSPE has previously been shown to acutely increase 2-deoxyglucose transport and Glut4 translocation in quiescent 3T3-L1 adipocytes at comparable concentrations (140 μg/ml) and to improve glucose homeostasis in animal models of metabolic disease (87–89).
Of the 26 phytochemicals that were identified as inhibitors in this screen, 17 are available in highly concentrated form as nutraceuticals, and five others are active ingredients isolated from medicinal herbal extracts. Inhibitors include 13 flavonoids (e.g. kaempferol, quercitin, and geraldol). Interestingly, three structurally related flavonoids screened positive as activators (icariin, myricetin, and dihydromyricetin). There were also flavonoids that had no effect on cell-surface Glut4. These compounds are often found together in plants and herbal extracts, and differences in their relative abundance could potentially cause significant variability in the biological responses seen with these extracts/supplements. Interestingly, two phytochemical flavonoids structurally very similar to icariin promote adipogenesis (increase lipid droplet and triglyceride content) in 3T3-L1 cells. They also up-regulate expression of aP2 and Glut4 (both mRNA and protein) to the same extent as rosiglitazone (used as a positive control in these experiments) (90). A third related flavonoid isolated from the same medicinal herbal extract had little to no effect on these parameters.
The biological targets of many of these inhibitory supplements remain unclear. To explore the mechanisms of action of a selection of these compounds, time-course studies were done. As noted above, the transition kinetics of the inhibitors showed several distinct phenotypes (Fig. 7). These can be used to tentatively assign the novel phytochemical inhibitors of Glut4 translocation to specific mechanistic groups. For example, known inhibitors of signal transduction, including propranolol and IBMX, show kinetics and extent of inhibition very similar to Akti (t½ = 8–12 min). The kinetics and extent of inhibition by parthenolide are consistent with an effect on signal transduction (Fig. 7A). Parthenolide has been reported to have antitumor and anti-inflammatory activities through inhibition of signal transduction pathways, including HDAC1, NF-κB, and Akt (91).
In contrast to inhibitors of signal transduction, the PI 3-kinase inhibitor LYi very rapidly clears 90% of Glut4 from the cell surface in insulin-stimulated cells (fast, t½ = 5 min; Fig. 6B). PI 3-kinase inhibition causes rapid loss of cell-surface Glut4 through effects on membrane trafficking. LYi rapidly and nearly completely blocks Glut4 exocytosis without effecting endocytosis (30, 31). The nutritional supplements piperlongumine and resveratrol show kinetics and extent nearly identical to LYi. Consistent with this, these supplements acutely inhibit PI 3-kinase both in cultured cells and in vitro at micromolar concentrations (IC50 Glut4 translocation = 3–5 μm and IC50 PI 3-kinase = 10–25 μm; Fig. 6B) (92, 93). Thus, piperlongumine and resveratrol likely decrease cell-surface Glut4 through inhibition of endosomal membrane trafficking. LYi, piperlongumine, and resveratrol all decrease cell-surface Glut4 in both quiescent and insulin-stimulated cells, indicating an effect on constitutive and well as insulin-stimulated trafficking pathways. Resveratrol has previously been shown to inhibit insulin action in isolated human adipocytes at micromolar concentrations (94).
Discussion
Previous work in our lab and others utilized reverse genetics to identify proteins and pathways that affect cell-surface Glut4 levels and insulin action. In reverse genetics, the phenotype of an organism or cell is determined following the disruption of a known candidate gene. The candidate genes tested were found either biochemically (e.g. by co-localization or co-immunoprecipitation) or by analogy to other trafficking processes. The role of these candidate genes in Glut4 trafficking and insulin signaling was tested using a combination of chemical reagents (small molecule inhibitors and activators) and DNA-based reagents (shRNA/siRNA-induced knockdown and expression of exogenous proteins). This work yielded a framework outlining both the trafficking itinerary of Glut4 and signal transduction pathways linking activation of the insulin receptor to regulation of this trafficking. However, significant gaps in our understanding of these processes remain.
The goal of this small molecule screen was to discover novel probes and new proteins/pathways that affect Glut4 trafficking and insulin action in highly insulin-responsive cells (adipocytes) using forward chemical genetics. Forward genetics is an approach used to identify genes/groups of genes responsible for a particular phenotype in cells/organisms. Classically, forward genetics involves selection of cells/organisms exhibiting a specific phenotype after random DNA mutagenesis (spontaneous or induced) and identification of the genes by mapping. As an alternative to random mutagenesis, shRNA, siRNA, and CRISPR–Cas9 libraries have been used. In these experiments, the targets are identified by the sequence of the probes and verified using multiple sequences from the same protein. In chemical forward genetics, compounds that produce a specific phenotype are discovered and then the targets of hits identified. When libraries of approved drugs are used, the targets of the compounds are known. Targets are verified by their pharmacology and by using chemically unrelated compounds affecting the same protein/pathway. The power of forward genetics is the ability to develop a comprehensive, systems-level map of all proteins affecting a selected phenotype, in an unbiased way. This facilitates the discovery of novel, unpredicted relationships.
We identified, confirmed, and validated 64 compounds that acutely increase cell-surface Glut4 and 65 compounds that rapidly decrease cell-surface Glut4 in adipocytes stimulated with sub-maximal concentrations of insulin. These hits could be organized into 11 groups based on their targets and chemical classes (Tables 1 and Table S1). Targets were verified by their pharmacology (EC50, time course; Figs. 2–7), and by SAR identification of multiple hits of the same chemical class and chemically unrelated compounds affecting the same protein/pathway. With the exception of the dis-inhibitor rapamycin, all groups contained multiple hits identified in more than one of the independent screens of the four libraries. Several of the groups include both agonists and antagonists of specific targets (e.g. the β-adrenergic and adenosine receptors) with opposite effects on cell-surface Glut4. Based on these groupings, we discovered an additional eight compounds that affect cell-surface Glut4 that were not identified in the screens. These include the four components of the standard cell culture adipocyte differentiation mixture: insulin, IBMX, dexamethasone, and thiazolidinediones. All four showed acute effects on cell-surface Glut4 at concentrations equivalent to those used during adipogenesis.
As a further verification of the screen, the hits included a number of compounds known to affect glucose transport or insulin action in cells, animals, and/or humans. However, with the exception of rapamycin, the details of how these compounds exert their effects on glucose uptake/homeostasis were incompletely understood. None had previously been reported to directly and acutely effect Glut4 translocation in adipocytes. This is due in part to the technical difficulty of screening Glut4 translocation in adipocytes. Previous Glut4 translocation screens used nonphysiological, insulin-insensitive fibroblasts (HEK293 and CHO) expressing Glut4 reporter constructs as their cell culture model (95–97). The pathways identified in our screen would not be expected to affect Glut4 trafficking in fibroblasts. Adipocytes have been used previously in screens for compounds that affect differentiation (long-term treatment) (98–101), but not for acute effects on insulin signaling. We overcame these difficulties by carefully optimizing our cell culture conditions and by utilizing a novel detection system, flow cytometry. This technique allows the rapid collection of five (or more) individual attributes per cell, 5000 cells per sample, and hundreds of samples per experiment (Fig. 1). The sensitivity and reproducibility of our screening assay allowed the identification of compounds that affect cell-surface Glut4 under suboptimal conditions for many of the compounds (e.g. at a single incubation time and a single concentration of drug and insulin).
In addition to identifying distinct chemical classes and targets that affect cell-surface Glut4, we were able to determine the mechanisms underlying a number of these groups based on their distinctive phenotypes (Tables 2 and 3). Compounds can increase cell-surface Glut4 by acting as insulin mimetics (0.6 nm insulin), insulin sensitizers (thiazolidinediones, disulfiram), and dis-inhibitors of feedback regulatory loops of insulin signaling (rapamycin) (Figs. 3 and 4). We also identified signal transduction pathways that stimulate Glut4 translocation that are independent of insulin signaling (β-adrenergic), as well as compounds that affect Glut4 trafficking directly (ouabain) (Fig. 5). Compounds that affect trafficking can increase cell-surface Glut4 through either increasing exocytosis or decreasing endocytosis. Furthermore, compounds can effect Glut4 trafficking either through insulin-dependent processes or via constitutive pathways. Consistent with the mechanisms identified for activators, inhibitors were identified that decreased cell-surface Glut4 through effects on signal transduction (propranolol, IBMX, and parthenolide) and effects on Glut4 trafficking (PI 3-kinase inhibition, LYi, resveratrol, and piperlongumine) (Fig. 7). Two novel pathways regulating Glut4 trafficking were identified: protein synthesis (puromycin and lycorine) and endosomal pH (lysosomogenic heterocyclic amines: chloroquine and maprotiline; Na+/K+-ATPase inhibitors/cardiotonic steroids, e.g. ouabain).
Table 2.
Phenotypes of activators: effects on cell-surface Glut4
| 0.003 nm, quiescent | 0.3 nm, 1/3 maximal | 100 nm, maximal | |
|---|---|---|---|
| Mechanism | |||
| Insulin mimetic | ⇑ | ⇑ | ⇒ |
| Insulin sensitizer | ⇒ | ⇑ | ⇒ |
| Dis-inhibition | ⇒ | ⇑ | ⇑ |
| Insulin-independent | ⇑ | ⇑ | ⇑ |
| Trafficking | |||
| Regulated | ⇒ | ⇑ | ⇒ |
| Constitutive | ⇑ | ⇑ | ⇑ |
Table 3.
Phenotypes of inhibitors: effects on cell-surface Glut4
| Time course (t½) | Extent | |
|---|---|---|
| Mechanism | ||
| Signal transduction | Intermediate (8–12 min) | 80% |
| Protein synthesis | Slow (30–40 min) | 80% |
| Trafficking | ||
| Endosomal pH ⇑ | Very fast (3 min) | 50% |
| Exocytosis ⇓ | Fast (5 min) | 80–90% |
| Endocytosis ⇑ | Fast (5 min) | Variable (>40%) |
The mechanisms of action of the groups of compounds were determined based on the effects of representative compounds on insulin concentration-response curves (Fig. 3), their time course of action (Figs. 4, 5, and 7), known biochemical targets (Table 1), and effects on Glut4 trafficking kinetics (data not shown). For several of these mechanisms, compounds with independent targets that affect the same processes were identified, including signal transduction, protein synthesis, and endosomal pH. Importantly, these assays defined specific phenotypes that allowed tentative assignment of novel compounds whose targets are less well established to mechanistic categories. For the novel compounds tested in this study (e.g. disulfiram, lycorine, parthenolide, resveratrol, piperlongumine, and heterocyclic amines), published biochemical analysis of their effects on specific targets support their assignment to specific mechanistic categories.
Two preclinical strategies are used to find new drugs: 1) modification of natural substances/biologics, and 2) screening, either target-based or phenotypic. Modified biologics and natural products are central to the treatment of diabetes (e.g. insulin, incretins, amylin, phlorizin/SGLT2 inhibitors, and DPP4 inhibitors). In target-based screening, potential drugs are identified based on effects on isolated proteins, and then their effects in cells/animals/humans are examined (their phenotype is determined; e.g. α-glucosidase inhibitors and small molecule activators of the insulin receptor tyrosine kinase). In phenotypic screens, compounds that cause a desired outcome in cells or organisms are discovered, and then their targets are identified. For example, sulfonylureas were discovered during World War II when typhoid fever patients treated with sulfonamides developed hypoglycemia (102). Metformin was discovered as the active ingredient in a plant extract used since the Middle Ages to relieve frequent urination (in what we now know was diabetes) (103). A study of new drugs approved by the FDA over a 10-year period showed that although less common than target-based screening, phenotypic screening contributed twice as many first-in-class small-molecule drugs with new molecular mechanisms of action (104). The first thiazolidinediones were discovered by screening for hypoglycemic action in ob/ob mice (105). The first cystic fibrosis drugs that specifically target genetic defects in the cystic fibrosis transmembrane regulator (CFTR) were discovered by screening for translocation of the ΔF508-CFTR expressed in NIH3T3 cells (106).
Our screen for compounds that target acute insulin action is phenotypic and not target based. Insulin-stimulated Glut4 translocation was used as a surrogate readout for activation of the insulin receptor, as well as effects on downstream pathways in a physiologically relevant target cell (adipocytes). This ensured that the compounds identified were active in a complex cellular signaling network on native receptors in their natural environment (membranes). Our chemical genetics screen showed that Glut4 translocation was the mechanistic target for a number of drug classes known to affect glucose homeostasis in humans through incompletely understood pathways. We also identified two novel, previously unknown pathways affecting cell-surface Glut4: protein synthesis and endosomal pH. Future work with chemically diverse libraries containing compounds not currently used as drugs will identify additional unknown regulatory pathways that are potential future therapeutic targets. It will also identify inhibitory drug classes and targets that may exacerbate insulin resistance and should be avoided in patients with diabetes.
Experimental procedures
Libraries
We used the following libraries: Prestwick Chemical Library of off-patent, approved drugs and natural products (1120 compounds); Spectrum Chemical collection of bioactive chemicals and natural products (2000 compounds); National Institutes of Health Clinical Collection (707 compounds); and Selleck Chemical natural products library (131 compounds).
Cells
3T3-L1 adipocytes have been used extensively to investigate Glut4 trafficking and acute insulin action. This model was optimized for maximal insulin sensitivity and responsiveness (Fig. 1). Low-passage 3T3-L1 fibroblasts stably expressing a Glut4 reporter at low levels were used (17, 30–34). Low-level expression is required for proper trafficking. The Glut4 reporter is a C-terminal GFP fusion protein with an exofacial HA epitope (HA–Glut4/GFP). HA–Glut4/GFP has been well characterized and traffics with endogenous Glut4 when expressed at the low levels used in our assay (17). The cells were differentiated into adipocytes on standard tissue culture 96-well plates. Differentiation produces a collagen based three-dimensional (3D) co-culture that contains adipocytes (suspended in the collagen gel), fibroblasts (attached to the tissue culture plate), and highly fluorescent necrotic cells (17). All experimental treatments and labeling were done with cells maintained in the 3D co-culture developed during differentiation. This is critical: Glut4 traffics differently when the adipocytes are detached from the plates or are replated (17).
Screening
Cells were incubated for 2 h at 37 °C in serum-free (sf) DMEM (with 1% BSA). Insulin (0.05, 0.3, or 1 nm, as indicated) was added for the final 45 min of the preincubation. Compounds in DMSO were diluted into warm sfDMEM with insulin and were added as a 2× stock (final concentration 25 μm, 0.25% DMSO). Incubation was continued for 1 h at 37 °C. After treatment with insulin and compounds, the culture plates were placed on ice, and cell-surface Glut4 labeled using Alexafluor647-conjugated antibody against the HA epitope (AF647–α-HA), as described previously (17, 30–34). After labeling, cells were treated with collagenase to dissociate the cells from the plates, and samples were analyzed by flow cytometry.
Reference samples
For control, DMSO (0.25%) was used; for activators, insulin (100 nm) was used; and for inhibitors, the PI 3-kinase inhibitor LYi (50 μm) was used. Reference samples were included on each plate. All samples were standardized to the control DMSO samples from the same plate (4–8 samples; values are reported as relative mean fluorescence, see below).
Flow cytometry
The presence of multiple cell types in the 3T3-L1 adipocyte cultures precludes their use in “whole plate” assays (17). To distinguish among the three cell types, single cell analysis was done using flow cytometry. After treatment and antibody labeling, cells were detached using collagenase and resuspended in buffer. Data were collected from all cells in the culture. Insulin-sensitive, lipid droplet-filled adipocytes are distinguished from the fibroblasts, and necrotic cells present in the co-culture based on light scatter (forward and side scatter) and cellular autofluorescence (488 excitation/>670 emission). Two parameters are used to identify hits, AF647–α-HA (cell-surface Glut4) and GFP (total Glut4). In true positive hits, there will be a correlation between the cell-surface Glut4 and GFP for all cells within a treated sample (both fluorescence values are dependent on the levels of expression of HA–Glut4/GFP in each cell).
The distribution of fluorescence values per cell in a population of cells is non-Gaussian (approximately log normal; Fig. S1A) (35). A Gaussian fit of data from a population of cells plotted on a logarithmic scale yields the sample geometric mean fluorescence or GMF (Fig. S1B). To compare samples run on different plates, sample GMF values (∼5000 cells per sample) were standardized to the average GMF of control DMSO samples run on the same plate (4–8 samples/96-well plate), and data for each sample were analyzed as relative GMF or rGMF as shown in Equation 2.
| (Eq. 2) |
Hits
Compounds that increased or decreased cell-surface Glut4 by more than three standard deviations from the average of all control (DMSO) samples across the screen were selected as hits as shown in Equation 3.
| (Eq. 3) |
Validation (Table S1)
Verification
Hits were rescreened in triplicate using compounds from the libraries.
SAR
Hits were assigned into drug classes based on shared structures and targets. Representative hits from each group were chosen for further analysis.
Confirmation
Representative hits were confirmed by ordering pure compound and rescreening in triplicate.
Concentration response
Cells in triplicate were pretreated with insulin as indicated; compounds were added at increasing concentrations, and cells were further incubated for 1 h. Cells were placed on ice, and cell-surface Glut4 was determined. To verify targets, the measured EC50 value was compared with published EC50 values for cell-based assays in adipocytes and other cell types (Figs. 2, 5, and 6).
Mechanism of action
Insulin dose response
Cells in triplicate were pretreated with increasing concentrations of insulin for 45 min; the compounds were added at saturating concentrations, and the cells were incubated for an additional 1 h. The shapes of the curves distinguish insulin mimetics, insulin sensitizers, dis-inhibitors, and drugs that work through parallel (insulin-independent) pathways (Figs. 3 and 5C).
Time course
Cells in triplicate were pretreated with insulin as described, and then compounds were added, and cells were further incubated for increasing times. Cells were placed on ice, and cell-surface Glut4 was labeled and analyzed. Hits were sorted into classes based on the kinetics of response to compounds and the shape of the transition curves (Figs. 4 and 7). At saturating concentrations, compounds that work through the same mechanism will show similar responses.
Author contributions
P. D. B. and C. C. M. conceptualization; P. D. B., I. R., and C. C. M. investigation; P. D. B., I. R., and C. C. M. methodology; P. D. B., I. R., and C. C. M. writing-review and editing; I. R. and C. C. M. data curation; C. C. M. formal analysis; C. C. M. funding acquisition; C. C. M. writing-original draft.
Supplementary Material
This work was supported by National Institutes of Health Grant P20 GM103440 from NIGMS and American Diabetes Association Grant 1-12-BS-132. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1 and Table S1.
- TAG
- triacylglycerol
- FA
- fatty acid
- PM
- plasma membrane
- LYi
- PI 3-kinase inhibitor LY294002
- GMF
- geometric mean fluorescence
- rGMF
- relative geometric mean fluorescence
- mTORC1
- mechanistic target of rapamycin complex 1
- IBMX
- isobutylmethylxanthine
- Akti
- Akt ½ inhibitor
- GSPE
- grape seed proanthocyanodin extract
- HA-Glut4/GFP
- Glut4 reported construct with exofacial HA-epitope and C-terminal GFP fusion
- AF647
- AlexaFluor647
- α-HA
- anti-HA antibody
- sf
- serum-free
- DMEM
- Dulbecco's modified Eagle's medium
- PI
- phosphatidylinositol
- FDA
- Food and Drug Administration
- PPAR
- peroxisome proliferator-activated receptor
- SAR
- structure/activity relationship
- PDE
- phosphodiesterase
- REF
- reference
- CFTR
- cystic fibrosis transmembrane regulator.
References
- 1. Vazirani R. P., Verma A., Sadacca L. A., Buckman M. S., Picatoste B., Beg M., Torsitano C., Bruno J. H., Patel R. T., Simonyte K., Camporez J. P., Moreira G., Falcone D. J., Accili D., Elemento O., et al. (2016) Disruption of adipose Rab10-dependent insulin signaling causes hepatic insulin resistance. Diabetes 65, 1577–1589 10.2337/db15-1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahmadian M., Duncan R. E., Jaworski K., Sarkadi-Nagy E., and Sul H. S. (2007) Triacylglycerol metabolism in adipose tissue. Future Lipidol, 2, 229–237 10.2217/17460875.2.2.229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rutkowski J. M., Stern J. H., and Scherer P. E. (2015) The cell biology of fat expansion. J. Cell Biol. 208, 501–512 10.1083/jcb.201409063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herman M., and Kanh B. (2012) Adipose tissue de novo lipogenesis: unanticipated benefits in health and disease. ASBMB Today 2012 02, 30–32 [Google Scholar]
- 5. Lebeck J. (2014) Metabolic impact of the glycerol channels AQP7 and AQP9 in adipose tissue and liver. J. Mol. Endocrinol. 52, R165–R178 10.1530/JME-13-0268 [DOI] [PubMed] [Google Scholar]
- 6. Salans L. B., Bray G. A., Cushman S. W., Danforth E. Jr, Glennon J. A., Horton E. S., and Sims E. A. (1974) Glucose metabolism and the response to insulin by human adipose tissue in spontaneous and experimental obesity. Effects of dietary composition and adipose cell size. J. Clin. Invest. 53, 848–856 10.1172/JCI107625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qatanani M., and Lazar M. A. (2007) Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 21, 1443–1455 10.1101/gad.1550907 [DOI] [PubMed] [Google Scholar]
- 8. Chaurasia B., and Summers S. A. (2015) Ceramides–lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 26, 538–550 10.1016/j.tem.2015.07.006 [DOI] [PubMed] [Google Scholar]
- 9. Leroyer S. N., Tordjman J., Chauvet G., Quette J., Chapron C., Forest C., and Antoine B. (2006) Rosiglitazone controls fatty acid cycling in human adipose tissue by means of glyceroneogenesis and glycerol phosphorylation. J. Biol. Chem. 281, 13141–13149 10.1074/jbc.M512943200 [DOI] [PubMed] [Google Scholar]
- 10. Tordjman J., Chauvet G., Quette J., Beale E. G., Forest C., and Antoine B. (2003) Thiazolidinediones block fatty acid release by inducing glyceroneogenesis in fat cells. J. Biol. Chem. 278, 18785–18790 10.1074/jbc.M206999200 [DOI] [PubMed] [Google Scholar]
- 11. Kandror K. V., and Pilch P. F. (2011) The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic 12, 665–671 10.1111/j.1600-0854.2011.01175.x [DOI] [PubMed] [Google Scholar]
- 12. Foley K., Boguslavsky S., and Klip A. (2011) Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 50, 3048–3061 10.1021/bi2000356 [DOI] [PubMed] [Google Scholar]
- 13. Stöckli J., Fazakerley D. J., and James D. E. (2011) GLUT4 exocytosis. J. Cell Sci. 124, 4147–4159 10.1242/jcs.097063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. James D. E., Brown R., Navarro J., and Pilch P. F. (1988) Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature 333, 183–185 10.1038/333183a0 [DOI] [PubMed] [Google Scholar]
- 15. Slot J. W., Geuze H. J., Gigengack S., Lienhard G. E., and James D. E. (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. J. Cell Biol. 113, 123–135 10.1083/jcb.113.1.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malide D., Ramm G., Cushman S. W., and Slot J. W. (2000) Immunoelectron microscopic evidence that GLUT4 translocation explains the stimulation of glucose transport in isolated rat white adipose cells. J. Cell Sci. 113, 4203–4210 [DOI] [PubMed] [Google Scholar]
- 17. Muretta J. M., Romenskaia I., and Mastick C. C. (2008) Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J. Biol. Chem. 283, 311–323 10.1074/jbc.M705756200 [DOI] [PubMed] [Google Scholar]
- 18. Saltiel A. R., and Kahn C. R. (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 10.1038/414799a [DOI] [PubMed] [Google Scholar]
- 19. Klip A., Sun Y., Chiu T. T., and Foley K. P. (2014) Signal transduction meets vesicle traffic: the software and hardware of GLUT4 translocation. Am. J. Physiol. Cell Physiol. 306, C879–C886 10.1152/ajpcell.00069.2014 [DOI] [PubMed] [Google Scholar]
- 20. Sakamoto K., and Holman G. D. (2008) Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. Am. J. Physiol. Endocrinol. Metab. 295, E29–E37 10.1152/ajpendo.90331.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sano H., Kane S., Sano E., Mîinea C. P., Asara J. M., Lane W. S., Garner C. W., and Lienhard G. E. (2003) Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J. Biol. Chem. 278, 14599–14602 10.1074/jbc.C300063200 [DOI] [PubMed] [Google Scholar]
- 22. Mîinea C. P., Sano H., Kane S., Sano E., Fukuda M., Peränen J., Lane W. S., and Lienhard G. E. (2005) AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem. J. 391, 87–93 10.1042/BJ20050887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larance M., Ramm G., Stöckli J., van Dam E. M., Winata S., Wasinger V., Simpson F., Graham M., Junutula J. R., Guilhaus M., and James D. E. (2005) Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J. Biol. Chem. 280, 37803–37813 10.1074/jbc.M503897200 [DOI] [PubMed] [Google Scholar]
- 24. Sano H., Eguez L., Teruel M. N., Fukuda M., Chuang T. D., Chavez J. A., Lienhard G. E., and McGraw T. E. (2007) Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 5, 293–303 10.1016/j.cmet.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 25. Sano H., Roach W. G., Peck G. R., Fukuda M., and Lienhard G. E. (2008) Rab10 in insulin-stimulated GLUT4 translocation. Biochem. J. 411, 89–95 10.1042/BJ20071318 [DOI] [PubMed] [Google Scholar]
- 26. Jiang L., Fan J., Bai L., Wang Y., Chen Y., Yang L., Chen L., and Xu T. (2008) Direct quantification of fusion rate reveals a distal role for AS160 in insulin-stimulated fusion of GLUT4 storage vesicles. J. Biol. Chem. 283, 8508–8516 10.1074/jbc.M708688200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishikura S., Bilan P. J., and Klip A. (2007) Rabs 8A and 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem. Biophys. Res. Commun. 353, 1074–1079 10.1016/j.bbrc.2006.12.140 [DOI] [PubMed] [Google Scholar]
- 28. Sadacca L. A., Bruno J., Wen J., Xiong W., and McGraw T. E. (2013) Specialized sorting of GLUT4 and its recruitment to the cell surface are independently regulated by distinct Rabs. Mol. Biol. Cell 24, 2544–2557 10.1091/mbc.e13-02-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reed S. E., Hodgson L. R., Song S., May M. T., Kelly E. E., McCaffrey M. W., Mastick C. C., Verkade P., and Tavaré J. M. (2013) A role for Rab14 in the endocytic trafficking of GLUT4 in 3T3-L1 adipocytes. J. Cell Sci. 126, 1931–1941 10.1242/jcs.104307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Habtemichael E. N., Brewer P. D., Romenskaia I., and Mastick C. C. (2011) Kinetic evidence that Glut4 follows different endocytic pathways than the receptors for transferrin and α2-macroglobulin. J. Biol. Chem. 286, 10115–10125 10.1074/jbc.M111.217935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brewer P. D., Romenskaia I., Kanow M. A., and Mastick C. C. (2011) Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J. Biol. Chem. 286, 26287–26297 10.1074/jbc.M111.253880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brewer P. D., Habtemichael E. N., Romenskaia I., Mastick C. C., and Coster A. C. (2014) Insulin-regulated Glut4 translocation: membrane protein trafficking with six distinctive steps. J. Biol. Chem. 289, 17280–17298 10.1074/jbc.M114.555714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brewer P. D., Habtemichael E. N., Romenskaia I., Mastick C. C., and Coster A. C. (2016) Glut4 is sorted from a Rab10 GTPase-independent constitutive recycling pathway into a highly insulin-responsive Rab10 GTPase-dependent sequestration pathway after adipocyte differentiation. J. Biol. Chem. 291, 773–789 10.1074/jbc.M115.694919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brewer P. D., Habtemichael E. N., Romenskaia I., Coster A. C., and Mastick C. C. (2016) Rab14 limits the sorting of Glut4 from endosomes into insulin-sensitive regulated secretory compartments in adipocytes. Biochem. J. 473, 1315–1327 10.1042/BCJ20160020 [DOI] [PubMed] [Google Scholar]
- 35. Furusawa C., Suzuki T., Kashiwagi A., Yomo T., and Kaneko K. (2005) Ubiquity of log-normal distributions in intra-cellular reaction dynamics. Biophysics 1, 25–31 10.2142/biophysics.1.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang J. H., Chung T. D., and Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 37. Berger J., and Moller D. E. (2002) The mechanisms of action of PPARs. Annu. Rev. Med. 53, 409–435 10.1146/annurev.med.53.082901.104018 [DOI] [PubMed] [Google Scholar]
- 38. Kletzien R. F., Clarke S. D., and Ulrich R. G. (1992) Enhancement of adipocyte differentiation by an insulin-sensitizing agent. Mol. Pharmacol. 41, 393–398 [PubMed] [Google Scholar]
- 39. Moraes L. A., Spyridon M., Kaiser W. J., Jones C. I., Sage T., Atherton R. E., and Gibbins J. M. (2010) Non-genomic effects of PPARγ ligands: inhibition of GPVI-stimulated platelet activation. J. Thromb. Haemost. 8, 577–587 10.1111/j.1538-7836.2009.03732.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Griggs R. B., Donahue R. R., Morgenweck J., Grace P. M., Sutton A., Watkins L. R., and Taylor B. K. (2015) Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPARγ mechanisms. Pain 156, 469–482 10.1097/01.j.pain.0000460333.79127.be [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang B., Salituro G., Szalkowski D., Li Z., Zhang Y., Royo I., Vilella D., Díez M. T., Pelaez F., Ruby C., Kendall R. L., Mao X., Griffin P., Calaycay J., Zierath J. R., Heck J. V., et al. (1999) Discovery of a small molecule insulin mimetic with antidiabetic activity in mice. Science 284, 974–977 10.1126/science.284.5416.974 [DOI] [PubMed] [Google Scholar]
- 42. Qiang G., Xue S., Yang J. J., Du G., Pang X., Li X., Goswami D., Griffin P. R., Ortlund E. A., Chan C. B., and Ye K. (2014) Identification of a small molecular insulin receptor agonist with potent antidiabetes activity. Diabetes 63, 1394–1409 10.2337/db13-0334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edwards S. R., and Wandless T. J. (2007) The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J. Biol. Chem. 282, 13395–13401 10.1074/jbc.M700498200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tremblay F., Gagnon A., Veilleux A., Sorisky A., and Marette A. (2005) Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3-L1 and human adipocytes. Endocrinology 146, 1328–1337 10.1210/en.2004-0777 [DOI] [PubMed] [Google Scholar]
- 45. Krebs M., Brunmair B., Brehm A., Artwohl M., Szendroedi J., Nowotny P., Roth E., Fürnsinn C., Promintzer M., Anderwald C., Bischof M., and Roden M. (2007) The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 56, 1600–1607 10.2337/db06-1016 [DOI] [PubMed] [Google Scholar]
- 46. Bogan J. S., McKee A. E., and Lodish H. F. (2001) Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol. Cell. Biol. 21, 4785–4806 10.1128/MCB.21.14.4785-4806.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pereira M. J., Palming J., Rizell M., Aureliano M., Carvalho E., Svensson M. K., and Eriksson J. W. (2012) mTOR inhibition with rapamycin causes impaired insulin signalling and glucose uptake in human subcutaneous and omental adipocytes. Mol. Cell. Endocrinol. 355, 96–105 10.1016/j.mce.2012.01.024 [DOI] [PubMed] [Google Scholar]
- 48. Lamming D. W., Ye L., Katajisto P., Goncalves M. D., Saitoh M., Stevens D. M., Davis J. G., Salmon A. B., Richardson A., Ahima R. S., Guertin D. A., Sabatini D. M., and Baur J. A. (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335, 1638–1643 10.1126/science.1215135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baker J. G. (2010) The selectivity of β-adrenoceptor agonists at human β1-, β2- and β3-adrenoceptors. Br. J. Pharmacol. 160, 1048–1061 10.1111/j.1476-5381.2010.00754.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Joost H. G., Göke R., and Steinfelder H. J. (1985) Dual effect of isoprenaline on glucose transport and response to insulin in isolated adipocytes. Biochem. Pharmacol. 34, 649–653 10.1016/0006-2952(85)90259-X [DOI] [PubMed] [Google Scholar]
- 51. Eckel J., Gerlach-Eskuchen E., and Reinauer H. (1990) G-protein-mediated regulation of the insulin-responsive glucose transporter in isolated cardiac myocytes. Biochem. J. 272, 691–696 10.1042/bj2720691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Omatsu-Kanbe M., and Kitasato H. (1992) Insulin and noradrenaline independently stimulate the translocation of glucose transporters from intracellular stores to the plasma membrane in mouse brown adipocytes. FEBS Lett. 314, 246–250 10.1016/0014-5793(92)81481-Z [DOI] [PubMed] [Google Scholar]
- 53. Shimizu M., Blaak E. E., Lonnqvist F., Gafvels M. E., and Arner P. (1996) Agonist and antagonist properties of β3-adrenoceptors in human omental and mouse 3T3-L1 adipocytes. Pharmacol. Toxicol. 78, 254–263 10.1111/j.1600-0773.1996.tb00214.x [DOI] [PubMed] [Google Scholar]
- 54. Chernogubova E., Cannon B., and Bengtsson T. (2004) Norepinephrine increases glucose transport in brown adipocytes via β3-adrenoceptors through a cAMP, PKA, and PI 3-kinase-dependent pathway stimulating conventional and novel PKCs. Endocrinology 145, 269–280 10.1210/en.2003-0857 [DOI] [PubMed] [Google Scholar]
- 55. Merlin J., Sato M., Nowell C., Pakzad M., Fahey R., Gao J., Dehvari N., Summers R. J., Bengtsson T., Evans B. A., and Hutchinson D. S. (2018) The PPARγ agonist rosiglitazone promotes the induction of brite adipocytes, increasing β-adrenoceptor-mediated mitochondrial function and glucose uptake. Cell. Signal. 42, 54–66 10.1016/j.cellsig.2017.09.023 [DOI] [PubMed] [Google Scholar]
- 56. Mukaida S., Evans B. A., Bengtsson T., Hutchinson D. S., and Sato M. (2017) Adrenoceptors promote glucose uptake into adipocytes and muscle by an insulin-independent signaling pathway involving mechanistic target of rapamycin complex 2. Pharmacol. Res. 116, 87–92 10.1016/j.phrs.2016.12.022 [DOI] [PubMed] [Google Scholar]
- 57. Kashiwagi A., and Foley J. E. (1982) Opposite effects of a β-adrenergic agonist and a phosphodiesterase inhibitor on glucose transport in isolated human adipocytes: isoproterenol increases Vmax and IBMX increases Ks. Biochem. Biophys. Res. Commun. 107, 1151–1157 10.1016/0006-291X(82)90642-8 [DOI] [PubMed] [Google Scholar]
- 58. Sacramento J. F., Ribeiro M. J., Yubero S., Melo B. F., Obeso A., Guarino M. P., Gonzalez C., and Conde S. V. (2015) Disclosing caffeine action on insulin sensitivity: effects on rat skeletal muscle. Eur. J. Pharm. Sci. 70, 107–116 10.1016/j.ejps.2015.01.011 [DOI] [PubMed] [Google Scholar]
- 59. Shearer J., and Graham T. E. (2014) Performance effects and metabolic consequences of caffeine and caffeinated energy drink consumption on glucose disposal. Nutr. Rev. 72, Suppl. 1, 121–136 10.1111/nure.12124 [DOI] [PubMed] [Google Scholar]
- 60. Kashiwagi A., Huecksteadt T. P., and Foley J. E. (1983) The regulation of glucose transport by cAMP stimulators via three different mechanisms in rat and human adipocytes. J. Biol. Chem. 258, 13685–13692 [PubMed] [Google Scholar]
- 61. Kuo T., McQueen A., Chen T. C., and Wang J. C. (2015) Regulation of glucose homeostasis by glucocorticoids. Adv. Exp. Med. Biol. 872, 99–126 10.1007/978-1-4939-2895-8_5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jewell E. A., and Lingrel J. B. (1991) Comparison of the substrate dependence properties of the rat Na,K-ATPase α1, α2, and α3 isoforms expressed in HeLa cells. J. Biol. Chem. 266, 16925–16930 [PubMed] [Google Scholar]
- 63. Cain C. C., Sipe D. M., and Murphy R. F. (1989) Regulation of endocytic pH by the Na+,K+-ATPase in living cells. Proc. Natl. Acad. Sci. U.S.A. 86, 544–548 10.1073/pnas.86.2.544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fuchs R., Schmid S., and Mellman I. (1989) A possible role for Na+,K+-ATPase in regulating ATP-dependent endosome acidification. Proc. Natl. Acad. Sci. U.S.A. 86, 539–543 10.1073/pnas.86.2.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lopez J. A., Burchfield J. G., Blair D. H., Mele K., Ng Y., Vallotton P., James D. E., and Hughes W. E. (2009) Identification of a distal GLUT4 trafficking event controlled by actin polymerization. Mol. Biol. Cell 20, 3918–3929 10.1091/mbc.e09-03-0187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Y., Rubin B. R., Orme C. M., Karpikov A., Yu C., Bogan J. S., and Toomre D. K. (2011) Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J. Cell Biol. 193, 643–653 10.1083/jcb.201008135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen Y., Wang Y., Zhang J., Deng Y., Jiang L., Song E., Wu X. S., Hammer J. A., Xu T., and Lippincott-Schwartz J. (2012) Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J. Cell Biol. 198, 545–560 10.1083/jcb.201111091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Xu Y., Nan D., Fan J., Bogan J. S., and Toomre D. (2016) Optogenetic activation reveals distinct roles of PIP3 and Akt in adipocyte insulin action. J. Cell Sci. 129, 2085–2095 10.1242/jcs.174805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lingrel J. B. (2010) The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 72, 395–412 10.1146/annurev-physiol-021909-135725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hundal H. S., Marette A., Mitsumoto Y., Ramlal T., Blostein R., and Klip A. (1992) Insulin induces translocation of the α2 and β1 subunits of the Na+/K+-ATPase from intracellular compartments to the plasma membrane in mammalian skeletal muscle. J. Biol. Chem. 267, 5040–5043 [PubMed] [Google Scholar]
- 71. Sennoune S., Gerbi A., Duran M. J., Grillasca J. P., Compe E., Pierre S., Planells R., Bourdeaux M., Vague P., Pieroni G., and Maixent J. M. (2000) Effect of streptozotocin-induced diabetes on rat liver Na+/K+-ATPase. Eur. J. Biochem. 267, 2071–2078 10.1046/j.1432-1327.2000.01211.x [DOI] [PubMed] [Google Scholar]
- 72. Alves D. S., Thulin G., Loffing J., Kashgarian M., and Caplan M. J. (2015) Akt substrate of 160 kD regulates Na+,K+-ATPase trafficking in response to energy depletion and renal ischemia. J. Am. Soc. Nephrol. 26, 2765–2776 10.1681/ASN.2013101040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Vrijsen R., Vanden Berghe D. A., Vlietinck A. J., and Boeyé A. (1986) Lycorine: a eukaryotic termination inhibitor? J. Biol. Chem. 261, 505–507 [PubMed] [Google Scholar]
- 74. Reed B. C., Ronnett G. V., and Lane M. D. (1981) Role of glycosylation and protein synthesis in insulin receptor metabolism by 3T3-L1 mouse adipocytes. Proc. Natl. Acad. Sci. U.S.A. 78, 2908–2912 10.1073/pnas.78.5.2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bogan J. S., and Kandror K. V. (2010) Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr. Opin. Cell Biol. 22, 506–512 10.1016/j.ceb.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bogan J. S., Rubin B. R., Yu C., Löffler M. G., Orme C. M., Belman J. P., McNally L. J., Hao M., and Cresswell J. A. (2012) Endoproteolytic cleavage of TUG protein regulates GLUT4 glucose transporter translocation. J. Biol. Chem. 287, 23932–23947 10.1074/jbc.M112.339457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cain C. C., and Murphy R. F. (1986) Growth inhibition of 3T3 fibroblasts by lysosomotropic amines: correlation with effects on intravesicular pH but not vacuolation. J. Cell. Physiol. 129, 65–70 10.1002/jcp.1041290110 [DOI] [PubMed] [Google Scholar]
- 78. Cain C. C., and Murphy R. F. (1988) A chloroquine-resistant Swiss 3T3 cell line with a defect in late endocytic acidification. J. Cell Biol. 106, 269–277 10.1083/jcb.106.2.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Goldman S. D., Funk R. S., Rajewski R. A., and Krise J. P. (2009) Mechanisms of amine accumulation in, and egress from, lysosomes. Bioanalysis 1, 1445–1459 10.4155/bio.09.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen Y. C., Shen Y. C., Hung Y. J., Chou C. H., Yeh C. B., and Perng C. H. (2007) Comparisons of glucose-insulin homeostasis following maprotiline and fluoxetine treatment in depressed males. J. Affect. Disord. 103, 257–261 10.1016/j.jad.2007.01.023 [DOI] [PubMed] [Google Scholar]
- 81. Liao T. V., and Phan S. V. (2014) Acute hyperglycemia associated with short-term use of atypical antipsychotic medications. Drugs 74, 183–194 10.1007/s40265-013-0171-7 [DOI] [PubMed] [Google Scholar]
- 82. Ghaeli P., Shahsavand E., Mesbahi M., Kamkar M. Z., Sadeghi M., and Dashti-Khavidaki S. (2004) Comparing the effects of 8-week treatment with fluoxetine and imipramine on fasting blood glucose of patients with major depressive disorder. J. Clin. Psychopharmacol. 24, 386–388 10.1097/01.jcp.0000132441.27854.0d [DOI] [PubMed] [Google Scholar]
- 83. Müller G., Satoh Y., and Geisen K. (1995) Extrapancreatic effects of sulfonylureas–a comparison between glimepiride and conventional sulfonylureas. Diabetes Res. Clin. Pract. 28, S115–S137 10.1016/0168-8227(95)01089-V [DOI] [PubMed] [Google Scholar]
- 84. Basit A., Riaz M., and Fawwad A. (2012) Glimepiride: evidence-based facts, trends, and observations (GIFTS). (corrected). Vasc. Health Risk Manag. 8, 463–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Syamsudin Ia W. (2008) The effect of Inai (Lawsonia inermis Linn) leaves extract on blood sugar level: an experimental study. Res. J. Pharmacol. 2, 20–23 [Google Scholar]
- 86. Liu Q., Zhang Y., Lin Z., Shen H., Chen L., Hu L., Jiang H., and Shen X. (2010) Danshen extract 15,16-dihydrotanshinone I functions as a potential modulator against metabolic syndrome through multi-target pathways. J. Steroid Biochem. Mol. Biol. 120, 155–163 10.1016/j.jsbmb.2010.03.090 [DOI] [PubMed] [Google Scholar]
- 87. Pinent M., Blay M., Bladé M. C., Salvadó M. J., Arola L., and Ardóvol A. (2004) Grape seed-derived procyanidins have an antihyperglycemic effect in streptozotocin-induced diabetic rats and insulinomimetic activity in insulin-sensitive cell lines. Endocrinology 145, 4985–4990 10.1210/en.2004-0764 [DOI] [PubMed] [Google Scholar]
- 88. Pinent M., Bladé M. C., Salvadó M. J., Arola L., and Ardévol A. (2005) Metabolic fate of glucose on 3T3-L1 adipocytes treated with grape seed-derived procyanidin extract (GSPE). Comparison with the effects of insulin. J. Agric. Food Chem. 53, 5932–5935 10.1021/jf050601f [DOI] [PubMed] [Google Scholar]
- 89. Montagut G., Bladé C., Blay M., Fernández-Larrea J., Pujadas G., Salvadó M. J., Arola L., Pinent M., and Ardévol A. (2010) Effects of a grapeseed procyanidin extract (GSPE) on insulin resistance. J. Nutr. Biochem. 21, 961–967 10.1016/j.jnutbio.2009.08.001 [DOI] [PubMed] [Google Scholar]
- 90. Hu X., Ji J., Wang M., Wu J. W., Zhao Q. S., Wang H. Y., and Hou A. J. (2011) New isoprenylated flavonoids and adipogenesis-promoting constituents from Morus notabilis. Bioorg. Med. Chem. Lett. 21, 4441–4446 10.1016/j.bmcl.2011.06.020 [DOI] [PubMed] [Google Scholar]
- 91. Nakabayashi H., and Shimizu K. (2012) Involvement of Akt/NF-κB pathway in antitumor effects of parthenolide on glioblastoma cells in vitro and in vivo. BMC Cancer 12, 453 10.1186/1471-2407-12-453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fröjdö S., Cozzone D., Vidal H., and Pirola L. (2007) Resveratrol is a class IA phosphoinositide 3-kinase inhibitor. Biochem. J. 406, 511–518 10.1042/BJ20070236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wang F., Mao Y., You Q., Hua D., and Cai D. (2015) Piperlongumine induces apoptosis and autophagy in human lung cancer cells through inhibition of PI3K/Akt/mTOR pathway. Int. J. Immunopathol. Pharmacol. 28, 362–373 10.1177/0394632015598849 [DOI] [PubMed] [Google Scholar]
- 94. Gomez-Zorita S., Tréguer K., Mercader J., and Carpéné C. (2013) Resveratrol directly affects in vitro lipolysis and glucose transport in human fat cells. J. Physiol. Biochem. 69, 585–593 10.1007/s13105-012-0229-0 [DOI] [PubMed] [Google Scholar]
- 95. Liu F., Dallas-Yang Q., Castriota G., Fischer P., Santini F., Ferrer M., Li J., Akiyama T. E., Berger J. P., Zhang B. B., and Jiang G. (2009) Development of a novel GLUT4 translocation assay for identifying potential novel therapeutic targets for insulin sensitization. Biochem. J. 418, 413–420 10.1042/BJ20082051 [DOI] [PubMed] [Google Scholar]
- 96. Vijayakumar M. V., Ajay A. K., and Bhat M. K. (2010) Demonstration of a visual cell-based assay for screening glucose transporter 4 translocation modulators in real time. J. Biosci. 35, 525–531 10.1007/s12038-010-0060-0 [DOI] [PubMed] [Google Scholar]
- 97. Lanzerstorfer P., Stadlbauer V., Chtcheglova L. A., Haselgrübler R., Borgmann D., Wruss J., Hinterdorfer P., Schröder K., Winkler S. M., Höglinger O., and Weghuber J. (2014) Identification of novel insulin mimetic drugs by quantitative total internal reflection fluorescence (TIRF) microscopy. Br. J. Pharmacol. 171, 5237–5251 10.1111/bph.12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kawazoe Y., Tanaka S., and Uesugi M. (2004) Chemical genetic identification of the histamine H1 receptor as a stimulator of insulin-induced adipogenesis. Chem. Biol. 11, 907–913 10.1016/j.chembiol.2004.04.017 [DOI] [PubMed] [Google Scholar]
- 99. Dragunow M., Cameron R., Narayan P., and O'Carroll S. (2007) Image-based high-throughput quantification of cellular fat accumulation. J. Biomol. Screen. 12, 999–1005 10.1177/1087057107306502 [DOI] [PubMed] [Google Scholar]
- 100. Hino K., Nagata H., Shimonishi M., and Ido M. (2011) High-throughput screening for small-molecule adiponectin secretion modulators. J. Biomol. Screen. 16, 628–636 10.1177/1087057111403474 [DOI] [PubMed] [Google Scholar]
- 101. Lahrita L., Kato E., and Kawabata J. (2015) Uncovering potential of Indonesian medicinal plants on glucose uptake enhancement and lipid suppression in 3T3-L1 adipocytes. J. Ethnopharmacol. 168, 229–236 10.1016/j.jep.2015.03.082 [DOI] [PubMed] [Google Scholar]
- 102. Loubatières-Mariani M. M. (2007) The discovery of hypoglycemic sulfonamides. J. Soc. Biol. 201, 121–125 10.1051/jbio:2007014 [DOI] [PubMed] [Google Scholar]
- 103. Thomas I., and Gregg B. (2017) Metformin; a review of its history and future: from lilac to longevity. Pediatr. Diabetes 18, 10–16 10.1111/pedi.12473 [DOI] [PubMed] [Google Scholar]
- 104. Swinney D. C., and Anthony J. (2011) How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519 10.1038/nrd3480 [DOI] [PubMed] [Google Scholar]
- 105. Fujiwara T., Yoshioka S., Yoshioka T., Ushiyama I., and Horikoshi H. (1988) Characterization of new oral antidiabetic agent CS-045. Studies in KK and ob/ob mice and Zucker fatty rats. Diabetes 37, 1549–1558 10.2337/diab.37.11.1549 [DOI] [PubMed] [Google Scholar]
- 106. Van Goor F., Straley K. S., Cao D., González J., Hadida S., Hazlewood A., Joubran J., Knapp T., Makings L. R., Miller M., Neuberger T., Olson E., Panchenko V., Rader J., Singh A., et al. (2006) Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L1117–L1130 10.1152/ajplung.00169.2005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.