

ABSTRACT
There is now a consensus that efficient peptide vaccination against cancer requires that peptides should (i) be exclusively presented by professional APC and (ii) stimulate both CD4 and CD8-specific T cell responses. To this aim, in recent trials, patients were vaccinated with pools of synthetic long peptides (SLP) (15–30 aa long) composed of a potential class I epitope(s) elongated at both ends with native antigen sequences to also provide a potential class II epitope(s). Using MELOE-1 as a model antigen, we present an alternative strategy consisting in linking selected class I and class II epitopes with an artificial cathepsin-sensitive linker to improve epitope processing and presentation by DC. We provide evidence that some linker sequences used in our artificial SLPs (aSLPs) could increase up to 100-fold the cross-presentation of class I epitopes to CD8-specific T cell clones when compared to cross-presentation of the corresponding native long peptide. Presentation of class II epitopes were only slightly increased. We confirmed this increased cross-presentation after in vitro stimulation of PBMC from healthy donors with aSLP and assessment of CD8-specific responses and also in vivo following aSLP vaccination of HLA*A0201/HLA-DRB0101 transgenic mice. Finally, we provide some evidence that vaccination with aSLP could inhibit the growth of transplanted tumors in mice. Our data thus support the use of such aSLPs in future cancer vaccination trials to improve anti-tumor CD8 T cell responses and therapeutic efficacy.
KEYWORDS: Cancer vaccines, melanoma, synthetic long peptides
Introduction
It is now established that cancer vaccines triggering T cell activation against tumor antigens can be beneficial to cancer patients1 although many improvements in the vaccination strategies are still necessary to achieve long-term survival of patients.2 Various immunogens have been tested, ranging from minimal CTL epitope to the full-length recombinant protein. Minimal CTL epitope, usually 9–10 amino acids in length led to limited induction of effector T cells associated with disappointing clinical efficacy. This was likely due to the induction of anergic CD8 T cells. This anergy probably resulted from exogenous loading of the short epitope and direct presentation to CD8 T cells, thus bypassing intracellular processing of the antigen by DC and co-signaling by matured DC.3 On the other hand, vaccination with full-length recombinant proteins does not seem to be the best alternative. Indeed, in vivo mouse studies showed that intracellular routes of cross-presentation were more efficient with synthetic long peptide (SLP) than with full-length antigen.4,5 Therefore nowadays, SLP, usually defined as 25–35 amino-acid (aa)-long peptides encompassing a well-defined CD8 epitope extended to include putative CD4 epitopes are regarded as the most efficient immunogens. They are usually administered as a mix of up to a dozen units to cover either a wide range of HLA haplotype and/or a wide range of epitopes.6 Notably, synthetic long peptides have shown clinical efficiency against HPV induced cervical and vulvar neoplasia7 and recently they have been used in melanoma patients to vaccinate them against tumor neoantigens.8 In most cases reported, the choice of the SLP relied primarily on a defined CD8 epitope and assumed the presence of a CD4 helper epitope in the vicinity. An alternative strategy for designing SLP vaccines relies on a careful selection of well-defined CD8 and CD4 epitopes, for which a wide repertoire exists and/or elicits strong immune responses.9,10 Selection of both CD4 and CD8 epitopes offers a wide range of opportunities: separation of naturally overlapping epitopes, linking epitopes that are otherwise far apart on the natural antigen or creation of chimeric epitopes containing, for example, a CD4 epitope from one antigen coupled to a CD8 epitope from another tumor antigen.11 In line with this, universal CD4 helper epitopes, capable of binding to a broad range of HLA haplotypes, and thus eliciting responses in a large population of patients have been described.12,13 We suggest to coin these modified immunogenic peptides, artificial SLP (aSLP) to distinguish them from native SLP.
We have previously characterized MELOE-1 antigen as an IRES-dependent, melanoma-specific translation product from a lncRNA mainly transcribed in the melanocytic lineage.14–16 MELOE-1 contains numerous class II epitopes17,18 and one HLA-A*0201-restricted CD8 epitope (Figure 1b) eliciting a frequent repertoire of high avidity T cells.19 Our previous studies allowed us to produce CD4 and CD8 T cells clones against these various epitopes which constitute valuable tools to study SLP processing and presentation by DC in vitro. Therefore, as a first step, using the MELOE-1 antigen as a model, we designed various SLPs comprising a CD4 epitope coupled to the CD8 epitope by a series of linkers of 4 to 6 aa and studied the efficacy of T cell clone activation by SLP-loaded DC in vitro. This allowed us to select the most efficient linker sequences for processing and presentation of the MELOE-1 epitopes. We then replace the HLA*A0201-restricted MELOE-1 epitope by an HLA*A0201 Melan-A/MART-1 epitope with the same linkers and re-assessed cross-presentation by DC. We next evaluated the ability of a few selected SLPs to stimulate specific T cells proliferation of healthy donors’ PBMC in vitro. Finally, we explored the immunogenicity and anti-tumor potential of our best SLP candidate in vivo in an HLA-A*0201/HLA-DRB1*0101 transgenic mouse.20
Figure 1.
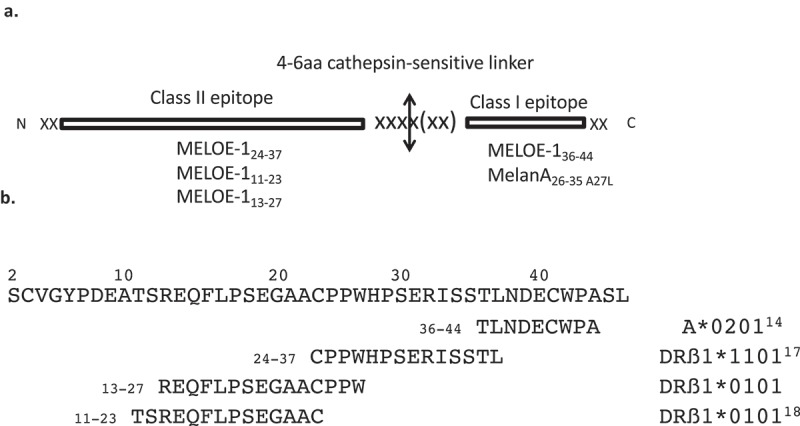
Panel A. Schematic representation of an artificial Synthetic Long Peptide (aSLP). The designed SLP comprises a class II epitope in N-ter fused to a class I epitope via a cathepsin-sensitive linker. SLP length ranges from 30 to 35 aa. Panel B. Sequence of MELOE-1 antigen. MELOE-1 is a 46 aa antigen containing multiple class II epitopes presented in various HLA context and a HLA A*0201 restricted class I epitope. Depicted here are the epitopes used in this study.
Results
SLP design
Our aim was to design aSLPs of 25–35aa long (a previously reported length for SLP1) that could be efficiently processed by DC to generate both defined class I and class II restricted T cell epitopes. The rationale for designing our aSLPs was as follows: we decided to place the class II-restricted epitope first, then the protease-sensitive linker and then the class I-restricted epitope (Figure 1a). We figured that with this design, even if protease cleavage generated a class I epitope elongated at the N-terminus, the trimming necessary for loading into HLA class I molecules would be performed by the physiological ERAD system in DC.21 On the other hand, the production of a slightly elongated class II-restricted epitope should not prevent its loading into class II molecules since class II HLA molecules are much more permissive in terms of epitope length than class I HLA molecules.22 The next critical choice which constitutes the core of this work was the amino-sequence of the linker peptide. Considering that SLP are internalized and processed by DC through the endosomal route,4 the main enzymes involved in SLP initial processing are cathepsins. They represent a large family with multiple cathepsins involved in antigen processing by DC among which the main endopeptidases are cathepsins S, L and D (for review23). Using the MEROPS on-line database (merops@ebi.ac.uk24), we designed our linker sequences so that they could presumably be cut by at least one of these three cathepsins considering that they show some redundancy in their recognition sequence.
Separating overlapping epitopes increases DC presentation
The first situation that we wanted to explore was the case of a natural overlap between a class II epitope and a class I epitope i.e. in which the processing of the class II epitope should result in the destruction of the class I epitope. In our MELOE-1 antigen, this is precisely the case between the DRB01*11-restricted epitope CPPWHPSERISSTL and the HLA-A*0201-restricted TLNDECWPA (Figure 1b). We figured that the competition for processing between the two epitopes could possibly be alleviated by separating these two epitopes in a synthetic long peptide. We designed two such aSLPs (31aa) (table S1) where the two epitopes where linked either with a control GGGG linker or with a potential cathepsin-sensitive linker LVGS. We then assessed the efficiency of DC processing and presentation of these aSLPs at various concentrations by measuring the dose-dependent activation of a DRB01*11-restricted specific T cell clone through classical presentation (Figure 2a) and the dose-dependent activation of a CD8 HLA-A*0201-restricted specific T cell clone (Figure 2b) through cross-presentation. This was compared to DC loaded with either full-length MELOE-1 (46 aa) or a 31 aa-long peptide that contains the native overlapping epitopes (table S1).
Figure 2.
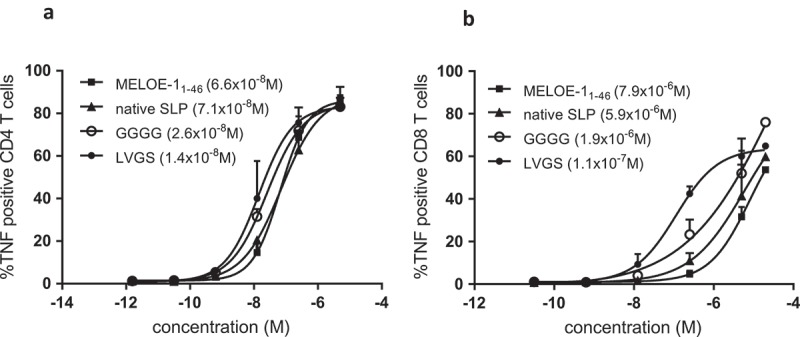
Panel A. TNFα production by a MELOE-124-37 specific HLA DRB1*11 restricted CD4 T cell clone activated by mature DC loaded with increasing concentrations of SLP (MELOE-124-37 xxxx MELOE-136-44) containing linker GGGG or LVGS, or the native SLP MELOE-111-46 or the full-length MELOE-11-46. Panel B. TNFα production by a MELOE-136-44 specific HLA A*0201 restricted CD8 T cell clone activated by mature MΦ-DC loaded as in panel A. Calculated EC50 are indicated in brackets.
For the presentation of the DRB01*11-restricted epitope, there was no difference of efficacy between the full-length MELOE-1 antigen and the 31 aa-long native long peptide (EC50 around 7 × 10–8 M for both). The presentation was improved when the two epitopes were separated by the GGGG linker (EC50 = 2.6 × 10–8 M) and further improved with the LVGS linker (1.4 × 10–8 M) (n = 2) (Figure 2a).
For the cross-presentation of the HLA-A*0201-epitope, differences between aSLP were more striking. In fact, full-length MELOE-1 and the natural long peptide did not differ significantly in terms of cross-presentation (7.9 × 10–6 M vs 5.9 × 10–6 M, respectively) while separating the two epitopes with the GGGG linker improved cross-presentation (EC50 = 1.9 × 10–6 M). The linker LVGS, designed as a target for cathepsins, markedly improved cross-presentation (1.1 × 10–7 M) (n = 2) (Figure 2b). This observation prompted us to test a variety of linkers for their ability to favor cross-presentation.
Strong influence of the linker on cross-presentation of the HLA class I epitope
To assess the influence of the linker sequence on cross-presentation, we designed another series of aSLPs containing the previously described DRB1*0101-restricted epitope MELOE11-2317 linked to the HLA-A*0201-restricted MELOE-1 epitope. The choice of a DRB1*0101-restricted epitope was motivated by the fact that this HLA haplotype is frequent in the population and DRB1*0101 transgenic mice are available for in vivo studies. As presented in Figure 3, we first focused on cross-presentation and observed major differences in the presentation of the HLA-A*0201-restricted MELOE-1 epitope depending on the linker used. In three independent experiments, using full length MELOE-1 as reference (EC50 = 2.5 × 10–7 M), we observed that some linkers induced poorer or similar cross-presentation (GGGG, 1.2 × 10–6 M and GSGS, 4.3 × 10–7 M), ten-fold better presentation (ASLG, 1.3 × 10–8 M and the previously tested LVGS, 3.4 × 10–8 M) or 100-fold better presentation (PIVG, 3.2 × 10–9 M; LLSV, 2.8 × 10–9 M; VLSVG, 2.1 × 10–9 M).
Figure 3.
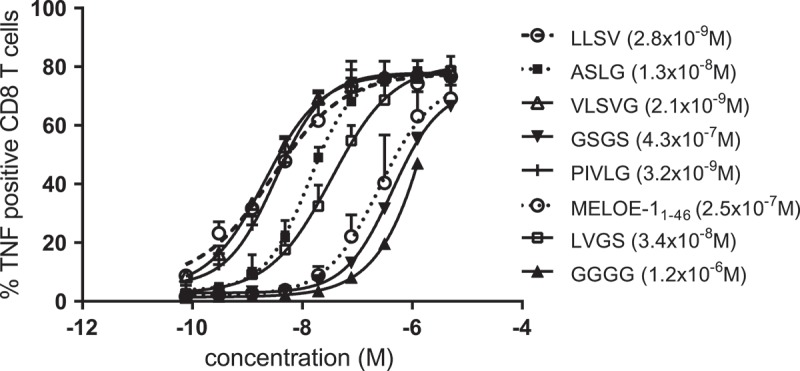
TNFα production by a MELOE-136-44 specific HLA A*0201 restricted CD8 T cell clone activated by mature DC loaded with increasing concentrations of SLP. (MELOE-111-23 xxxx MELOE-136-44) containing different linkers. Calculated EC50 are indicated in brackets.
In the next series of experiments, we designed aSLPs with a newly identified DRB1*0101-restricted epitope, MELOE-113-27 (Figure 1a), obtained after in vitro T cell stimulation and cloning from a DRB1*0101 blood donor (data not shown). We wanted to assess the effect of changing the C-terminus of the class II epitope on SLP cross-presentation i.e. how it may affect processing at the level of the linker. According to the MEROPS database, the C-terminus of MELOE-113-27 (PPW) should be less favorable for cutting at the linker level than the C-terminus of MELOE-111-23 (AAC). Indeed, when we tested the two linkers previously identified as the most favorable ones for cross-presentation (LLSV and VLSVG) with this new upstream class II epitope MELOE-113-27, they were still better than full-length MELOE-1 but only tenfold so, instead of hundredfold (Table 1). This suggested than the aa sequence contributed by the C terminus of the class II epitope can affect processing efficiency at the linker level. Considering this observation, we decided to lengthen the linker to 6 aa and explored variations in the aa sequence around the LSV core that was favorable for processing (Table 1). SLP containing the linker LLSVGG was the most efficiently processed and cross-presented one in this experiment.
Table 1.
Evaluation of different linkers to promote cross-presentation of the aSLP MELOE-113-27 xxxx MELOE-136-44.
| |
MELOE-113-27 xxxx MELOE-136-44 |
|
|
|---|---|---|---|
| code | Sequence | EC50 ratioa | Nb |
| P13 | ----- GGGG ----- | <0,1 | 5 |
| P15 | ----- LLSV ----- | 6–14 | 5 |
| P28 | ----- VLSVG ----- | 3–13 | 2 |
| P43 | ----- LLSVG ----- | 10–20 | 3 |
| P37 | ----- PLSVII ----- | 2.3–10 | 2 |
| P40 | ----- LLSVGG ----- | 12–56 | 3 |
| P25 | ----- VLSVGG ----- | 1.5–2 | 2 |
| P32 | ----- GLSVGG ----- | 1 | 2 |
| P33 | ----- GLSVVV ----- | 1 | 2 |
| P34 | ----- SLSVAA ----- | 7–8 | 2 |
| P35 | ----- SLSVGG ----- | 1–8 | 3 |
| P36 | ----- ALSVGG ----- | 1–3 | 2 |
aRange of EC50 ratio defined as EC50 of MELOE-111-46/EC50 of aSLP.
EC50 were determined by the TNFα response curves of a MELOE-111-46 specific CD8 T cell clone to SLP-loaded DC.
bNumber of independent experiments.
In the same line of thought, we wanted to check whether the linkers defined as favorable for cross-presentation of the HLA-A*0201-restricted MELOE-1 would remain so if we changed the class I epitope. We replaced the MELOE-136-44 epitope by the HLA-A*0201-restricted Melan-A A27L epitope (ELAGIGLTV), thus changing the aa downstream of the linker from TLND to ELAG and assessed DC cross-presentation to a Melan-A/A*0201 specific T cell clone. The long peptide Melan-A16-40 A27L that can cross-present the HLA-A*0201-restricted Melan-A A27L epitope25 was used as reference in these experiments. As shown in Figure 4, this epitope replacement resulted in dramatic changes in terms of linker efficiency to promote cross-presentation. The aSLP containing the linker LLSV became one of the worst aSLPs in terms of cross-presentation (EC50 = 2.0 × 10–5 M) suggesting that cleavage downstream of the linker was favored and thus destroyed the Melan-A epitope. With an additional G, the linker LLSVG did slightly better (8.1 × 10–6 M) but was less efficient than the native Melan-A sequence (2.5 × 10–6 M). Finally, with the longer linker LLSVGG, very efficient cross-presentation was restored (1.5 × 10–7 M) suggesting that with this longer sequence, cleavage was favored within the linker.
Figure 4.
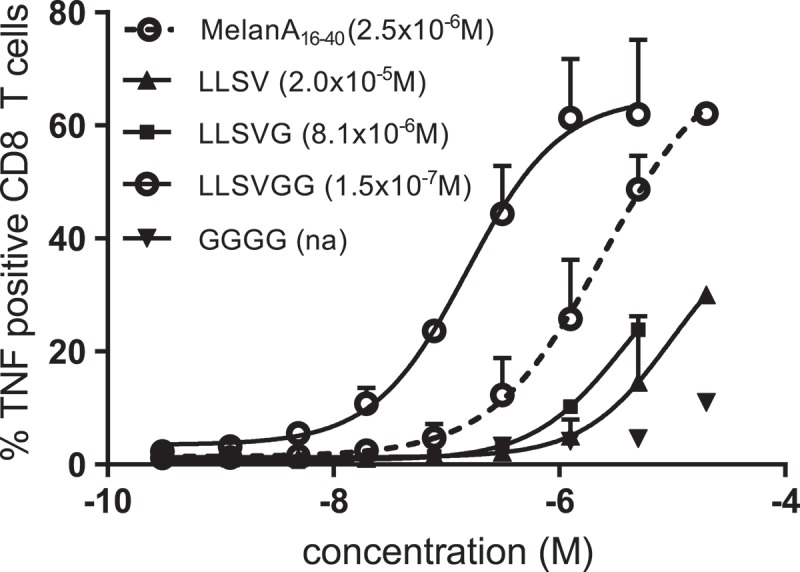
TNFα production by a MELOE-136-44 specific HLA A*0201 restricted CD8 T cell clone activated by mature MΦ-DC loaded with increasing concentrations of SLP (MELOE-113-27 xxxx MelanA26-35A27L) containing different linkers. Calculated EC50 are indicated in brackets.
No major influence of the linker on CD4 antigen presentation
In parallel, aSLPs containing MELOE-113-37 and MELOE-136-44 with linkers GGGG, LLSV, LLSVG or LLSVGG were tested for presentation to a CD4 MELOE-113-37-specific T cell clone to assess the influence of the linker sequence on HLA class II presentation by DC.
MELOE-111-46 native long peptide and aSLP with the linker GGGG were used as controls. As shown on Figure 5, the SLP with linker GGGG was poorly presented in comparison to the native long peptide (EC50: 3.5 × 10–8 M vs 8.4 × 10–9 M) whereas all the other linkers tested were as efficient (LLSVG, 6.8 × 10–9 M) or slighty better (LLSV, 2.7 × 10–9 M; LLSVGG, 3.8 × 10–9 M) than the native sequence in terms of processing for class II presentation.
Figure 5.
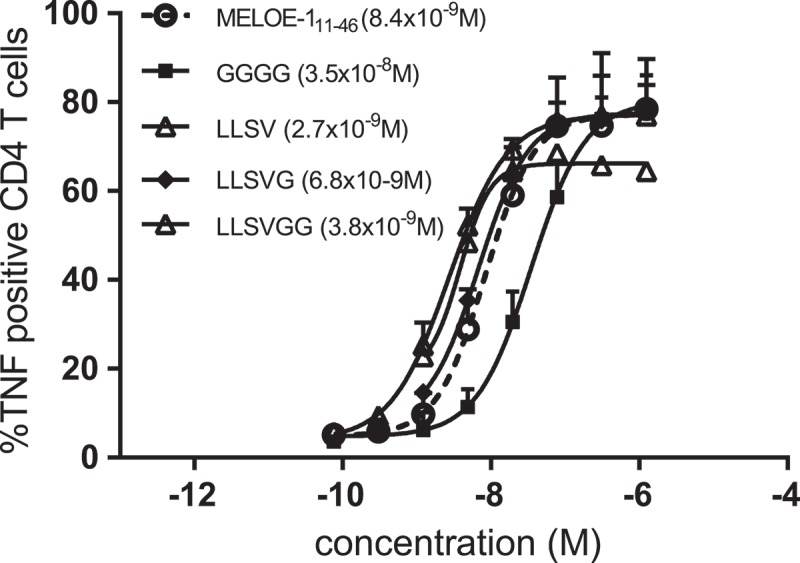
TNFα production by a MELOE-113-27 specific HLA DRB1*01 restricted CD4 T cell clone activated by mature MΦ-DC loaded with increasing concentrations of relevant SLP (MELOE-113-27 xxxx MELOE-136-44) containing different linkers. Calculated EC50 are indicated in brackets.
Increased specific CD8 T cell expansion in vitro
We then tested the ability of aSLP containing MELOE-113-37 and MELOE-136-44 with linkers GGGG, LLSV or LLSVGG to reactivate and expand CD8-specific T cells within PBMC from HLA-A*0201 healthy donors in vitro. For this purpose, PBMC were treated for 24 h with GM-CSF and IL-4 to accelerate the differentiation of monocytes into DC together with SLP at 5 µM and then TNFα, IL-1β and PGE2 were added as maturation agents as previously described26 (see M&M). After 21 days, the percentage of CD8-specific T cells in each microculture was assessed by tetramer staining (Figure S1). A typical experiment is presented in Figure 6a showing the number of positive wells and the percentages of tetramer-positive T cells per well (threshold of positivity, 0.5%) following stimulation of PBMC from healthy donor 1 (HD1) with the aSLP containing either the linker LLSV, the linker GGGG or the native MELOE-1 sequence. Stimulation and in vitro expansion of MELOE-136-44 -specific T cells with the aSLP containing LLSV was more efficient than with the aSLP containing the GGGG linker (27/96 vs 8/96, p < 0.01) and also than the native sequence MELOE-111-46 (27/96 vs 18/96) although not significantly so. Similar results were obtained with donor 2 (HD2) while two donors (HD3 and HD4) displayed too few positive microcultures (reflecting low frequencies of circulating CD8+ MELOE-1 specific T cells) to assess differences in aSLP cross-presentation (summarized in Table 2).
Figure 6.
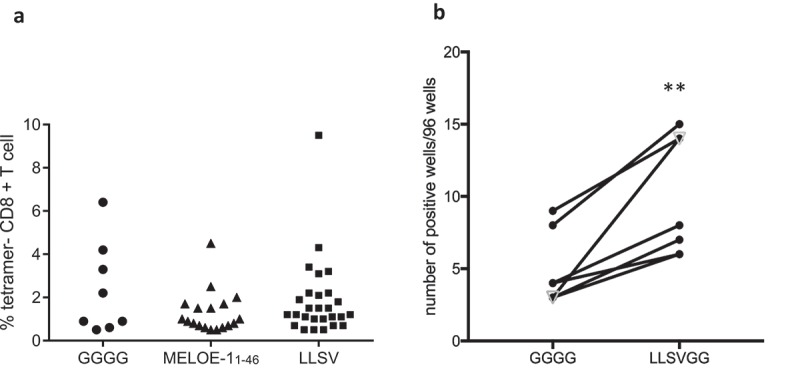
Panel A. Typical example of an in vitro stimulation of PBMC from a healthy donor with MELOE-1 SLP (MELOE-113-27 xxxx MELOE-136-44) containing various linkers or the native MELOE-111-46 followed by the assessment of CD8 responses (number of positive microcultures and frequency of positive tetramer+/CD8+ lymphocytes per well). PBMC were stimulated in 96 well plates with SLP (5 µM) and cytokines (see M&M) for 21 days. Microcultures were screened by tetramer and CD8 double staining (threshold 0.5% of positive cells). Panel B. Comparison of in vitro stimulation with the same aSLP as in A containing either the linker GGGG or the linker LLSVGG with PBMC from six healthy donors and one melanoma patient (open triangle). **p = 0.004, paired t-test.
Table 2.
Assessment of CD8 responses in 4 healthy donors after PBMC stimulation in vitro with the aSLP MELOE-113-27 -xxxx- MELOE-136-44.
| HD1 | HD2 | HD3 | HD4 | |
|---|---|---|---|---|
| GGGG | 8/96 | 21/96 | 0/96 | 3/96 |
| LLSV | 27/96** | 38/96* | 4/96 | 5/96 |
| MELOE-111-46 | 18/96 |
Number of positive CD8+ microcultures (threshold: 0.5% tetramer positive cells).
*p < 0.05, **p < 0.01 for GGGG vs. LLSV, Fischer exact test.
We next assessed our optimal linker LLSVGG using PBMC from seven healthy donors and two melanoma patients. Within this group, one healthy donor and one patient were excluded from the analysis for lack of any T cell response against MELOE-1. With the remaining six donors and one patient, we showed that stimulation with aSLP containing the linker LLSVGG reactivated significantly more specific responses then aSLP containing the linker GGGG (p = 0.004, paired t-test) (Figure 6b).
Increased in vivo immunogenicity
We explored the immunogenicity of our aSLPs in vivo in HLA-DRB1*0101/HLA-A*0201 transgenic mice. Mice were immunized subcutaneously with 100 µg of aSLP in IFA and poly I:C and boosted at D14 and D28 with 50 µg of aSLP with the same adjuvants (see M&M). We focused on cross-presentation and thus assessed CD8 T cell responses by ELIspot-IFN-γ after re-stimulation of sorted CD8+ splenocytes with the short MELOE-136-44 epitope or with medium alone. A mouse was considered responder when ELIspots were above ten spots and at least twice the background. A typical experiment is presented in Figure 7 comparing immunization with aSLP containing MELOE-113-37 and MELOE-136-44 with linker GGGG (panel A) or LLVSGG (panel B). Immunization with aSLP containing GGGG generated moderate CD8 responses in three out of five mice while immunization with aSLP with LLSVGG was efficient in all five mice with two mice showing robust CD8 responses. Previous experiments performed with the shorter linkers LLSV and LLSVG also supported the hypothesis that our synthetic linkers favored cross-presentation when compared to either the linker GGGG or the native sequence (Table 3). In contrast, CD4 responses evaluated ex vivo towards the HLA-DRB1*0101 epitope were very low (Figure 7c). This suggested that the mouse T cell repertoire against this epitope is scarce as compared to the human T cell repertoire and thus T cell help may not have contributed much to the CD8 responses in those mice.
Figure 7.
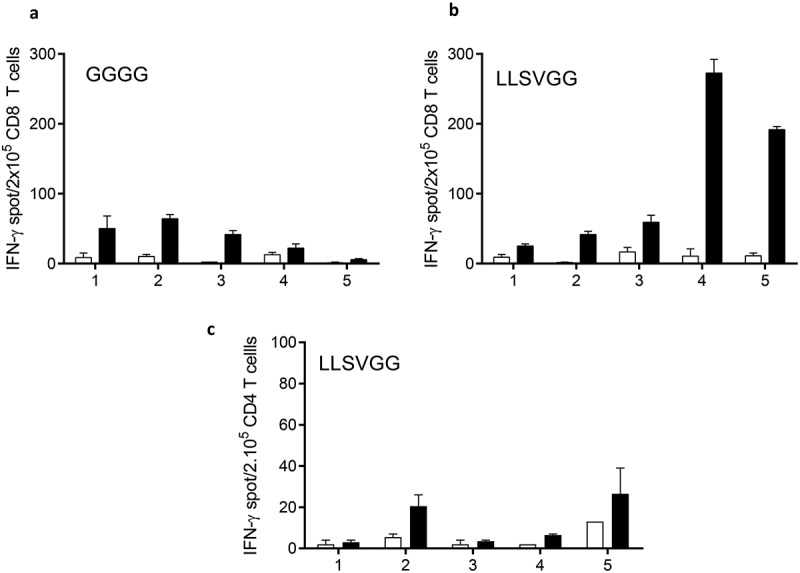
Immunization of HLA DRB1*01/HLA A*0201 transgenic mice with SLP (MELOE-113-27 xxxx MELOE-136-44) containing either the linker GGGG or LLSVGG. Mice received a priming injection of 100 µg of SLP followed by 2 boosts of 50 µg emulsified in IFA with addition of 50 µg of PolyI:C. At day 28, splenocytes were harvested, CD8+ T cells (panel A and B) or CD4+ T cells (panel C) were sorted and re-stimulated with (black bars) or without (white bars) the class I epitope MELOE-136-44 (A and B) or the class II epitope MELOE-113-27 (panel C) . IFN-γ production was assessed by ELIspot.
Table 3.
CD8 responses in HLA-DRB1*0101/HLA-A*0201 transgenic mice following immunization with aSLP MELOE-113-27 xxxx MELOE-136-44.
| Exp#1 | Exp#2 | Exp#3 | |
|---|---|---|---|
| MELOE-111-46 | 0/5 | ||
| GGGG | 1/3 | 3/5 | |
| LLSV | 2/3 | ||
| LLSVG | 2/5 | ||
| LLSVGG | 5/5 |
CD8+ splenocytes from immunized mice were tested by INFγ-ELIspot following restimulation with the short epitope MELOE-136-44. Mice were considered positive if the number of spots after restimulation was over ten spots and above twice the background level.
Antitumor effect in vivo triggered by aSLP vaccination
To evaluate the anti-tumor potential of vaccination with aSLP, we used the previously described SARC-A2 cell line27 transduced with a cDNA encoding the whole MELOE-1 antigen. The ability of this transduced cell line to present the HLA-A*0201-restricted MELOE-136-44 epitope was confirmed in vitro by its ability to stimulate a MELOE-1 specific CD8 T cell clone (Figure 8a). Following tumor engraftment, mice were vaccinated at day 6 by a prime injection of 100 µg of aSLP with the LLSVGG linker plus adjuvant, or adjuvant alone or PBS, followed by a boost 14 days later. Mice were monitored until the end point at day 36. Comparison of median tumor sizes at endpoint in the three groups of mice is shown in Figure 8b. At that time, setting the threshold at 50 mm2, all tumors were growing in the seven untreated mice despite variability in tumor size while tumor growth was inhibited in four out of eight mice (0/7 vs 4/8, p = 0.0513, Fisher’s exact test) (Figure 8c and d).
Figure 8.
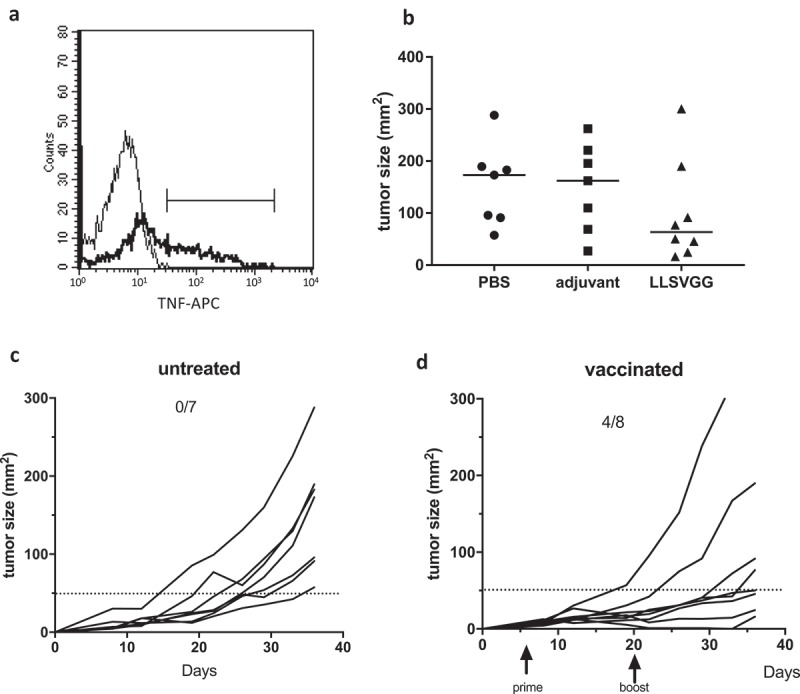
Panel A. Recognition of the SARC-A2 cell line transfected or not with MELOE-1 cDNA by a MELOE-136-44-specific HLA-A*0201-restricted CD8 T cell clone. TNFα production was assessed by intracellular staining following incubation of the clone with tumor cells for 5 h at a 1:1 ratio. Panel B. Comparison of tumor sizes at day 36 in the three groups of mice, vaccinated with PBS, or adjuvant alone or aSLP containing the LLSVGG linker. Bars indicate median of each group. Panel C and D. Monitoring of the growth of subcutaneous SARC-A2-MELOE-1 cells (2x105 cells) in PBS-treated mice (panel C) or vaccinated mice (panel D). Prime vaccination and boost are indicated by arrows.
Discussion
In the field of cancer vaccination with SLP, two strategies can be applied: the first strategy consists in vaccinating patients with pools of peptides from native antigen sequences containing potential class I and class II T cell epitopes, considering that, whatever the HLA context, some epitopes will be processed and presented to CD4 and/or CD8 T lymphocytes. This was the strategy followed by the team of Melief and coll. who vaccinated patients with cervical cancer or vulvar neoplasia with pools of overlapping peptides covering the whole sequences of HPV16 oncogenic proteins E6 and E7.7,28 This was also the case in the recent publication of Ott and coll8 in which they describe vaccination of eight melanoma patients with pools of 20 personal tumor neoantigens containing predicted class I and class II T cell epitopes. The convincing clinical responses that they reported strongly validate the targeting of neoantigens by peptide vaccination. However, it is noteworthy that CD8 responses were scarce as compared to CD4 responses not only in terms of percentage of targeted neoantigens (16% vs 60%, respectively) but also in terms of frequency of reactive T cells since neoantigen-reactive CD8 T cells could not be evidenced directly ex vivo but required in vitro restimulation. Among the different possible factors responsible for this predominance of CD4 responses, one hypothesis is that the SLP used were not very efficiently cross-presented by dendritic cells. In addition, we reckon that vaccination with pools of peptides may result in unforeseen competition for DC processing as previously reported,28 thereby further decreasing the presentation of a given CD8 epitope.
For all these reasons, the alternative strategy in peptide vaccination that we describe in this work consists in selecting immunodominant class I and class II epitopes and connect them with a cathepsin-sensitive linker to design specific aSLP that can be efficiently presented by DC to stimulate both CD4 and CD8 T cells. Nishimura and coll. already reported the use of such artificial SLP made of peptides containing class I and class II epitopes connected via an artificial linker and documented enhanced CD4 and CD8 T cell responses in vitro9 and in vivo in a mouse model.10 In both publications, they chose a five glycine linker to design their SLP which was better than a five alanine or five proline linker.
In this line of thought, we explored the vaccination potential of aSLP composed of defined class II and class I epitopes from two melanoma antigens, MELOE-1 and Melan-A, linked together by various short linker sequences selected for their potential cathepsin sensitivity. We first used macrophage-derived DC to present increasing concentrations of aSLP to specific CD4 or CD8 T cell clones. In our experimental setting, DC was loaded in medium without serum and washed before presentation to T cells. Therefore, the probability that SLP may be degraded in the culture medium and loaded directly onto HLA molecules was minimal and thus presentation, and especially cross-presentation critically depended on intracellular processes. Our results demonstrated that in those conditions, the linker sequence had a major influence on aSLP cross-presentation to CD8 T cell clones but much less on classical presentation to CD4 clones. We first provided evidence that separating overlapping class I and class II epitopes, especially with a cathepsin-sensitive linker, enhanced cross-presentation of the class I epitope. This finding is particularly relevant in the field of cancer vaccination since this overlap between class II and class I epitopes is frequently observed in tumor antigens such as, for example, gp100, Melan-A, LAGE and MAGE antigens (https://cancerresearch.org/scientists/meetings-and-resources/peptide-database). This overlap will result in competition between the two processing pathways. Separating the two epitopes and favoring cleavage in the linker likely increased the amount of class I epitope that could be translocated to the cytoplasm, processed by the proteasome and presented in HLA class I molecules. This route of SLP processing for cross-presentation has been reported previously4 and using the proteasome inhibitors epoxomycin or MG132, we confirmed that proteasome was critical for cross-presentation of our aSLPs (data not shown).
To determine the optimal linker sequence for cross-presentation, we tested a number of linker sequences predicted to be recognized by the endocathepsins present in DC endosomes i.e. mainly cathepsins L, S and D.23 Our results showed an unexpected wide range in cross-presentation efficiency among them with some aSLP being 100 fold more efficient then the native antigen sequence. In addition, we demonstrated that the aa sequences provided by the class II and class I epitopes upstream and downstream of the linker could critically affect the processing. The most striking example was the abrogation of cross-presentation of the Melan-A A27L epitope when short linkers where used. We thus decided to extend the linker to a 6 aa length to favor cleavage within the linker sequence.
A few selected aSLP were then further evaluated for their ability to expand CD8-specific T cells from healthy donors in vitro. It should be pointed out that in those experiments, aSLP were used at high concentrations (5 μM) and persisted in the culture for many days. Therefore, extracellular partial digestion of the aSLP by surface or secreted proteases may have occurred and somewhat blurred differences between the different linkers. Nonetheless, even in those conditions, we confirmed that aSLP containing linkers LLSV and LLSVGG were more efficient than the native MELOE-111-46 or the aSLP with a GGGG linker to expand MELOE-136-44 specific CD8 T lymphocytes. In addition, we confirmed in vivo in transgenic mice that vaccination with aSLP designed with cathepsin-sensitive linkers were the most efficient to trigger CD8+ specific T cell responses. However, our data showed that mouse CD4 T cell responses against the HLA-DRB1*0101-restricted MELOE-1 epitope were weak and thus probably provided little help to the CD8 T cell responses. Such differences in T cell repertoire between those HLA-DRB1*0101/HLA-A*0201 transgenic mice and humans have been previously reported before by some of us: in fact among the four HLA-DRB1*0101-restricted epitopes derived from telomerase reverse transcriptase (hTERT) that are immunogenic in humans (coined UCP 1, 2, 3 and 4), only UCP2 and UCP3 triggered significant CD4 T cell responses and provided help for the CD8 responses in those mice.13 Thus, we reckon that with our aSLPs in this mouse model, we mainly assessed the linker-dependent efficiency of in vivo cross-presentation of the class I epitope with little influence of the class II epitope.
Finally, vaccination with our optimal aSLP inhibited the growth of transplanted SARC-A2 tumors expressing MELOE-1 antigen. Although vaccination inhibited tumor growth in only four out of eight mice, we believe that the vaccination protocol could be further improved in the future. Indeed, in the recent publication of Ott et al. showing impressive clinical results in melanoma patients following vaccination with long peptides, priming consisted in five injections followed by two boosts8 while we only injected our mice twice.
In conclusion, our data provide evidence that (i) designing aSLPs with defined class II and class I epitopes connected by a cathepsin-sensitive linker is a valid approach to allow presentation of both CD4 and CD8 epitopes by DCs and (ii) choosing the optimal linker can significantly enhance cross-presentation to CD8+ T cells and (iii) vaccination with such aSLP can trigger an anti-tumor response in vivo. This approach should now be evaluated in vaccination trials in cancer patients.
Materials and methods
Peptides
Designed peptides were purchased at >90% purity (ProteoGenix, Schiltigheim, France and GeneCust, Ellange, Luxemburg). Lyophilized powder was resuspended at 10mM stock in DMSO and stored at −80°C. Purity was assessed by HPLC, and precise quantification was calculated based on protein content analysis.
Mice
HLA-DRB1*0101/HLA-A*0201 transgenic mice (A2/DR1 mice) have been previously described.20 Mice were bred and housed at Animalerie centrale UFC/UFR ”SMP” Besançon. Female mice 6–10 weeks old were used in the experiments. All experiments were performed according to the good laboratory practices after agreement #58 by the local ethical committee.
Tumor cell line
The SARC-L1 cell line which spontaneously occurred in A2/DR1 mice was genetically modified to over-express HLA-A*0201 as previously described.27 SARC-A2 cells were then transduced with a gammaretroviral dicistronic vector encoding the whole MELOE-1 antigen and, downstream of an IRES element, a puromycin N-acetyltransferase allowing selection of the transduced cells with puromycin (5μg/mL). HLA-A*0201 expression was checked by flow cytometry. The capacity of the MELOE-1-transduced SARC-A2 (SARC-A2-MELOE-1) cells to present the A2-restricted MELOE-1 epitope was confirmed by their recognition by an HLA-A*0201-restricted T cell clone (Figure 8a).
Monocyte–derived dendritic cells (MΦ-DC) generation
Monocytes were purified from PBMC of HLA-A*0201 and/or HLA-DRB1*0101 healthy donors (DTC core facility, CIC BT1413, Nantes, France) by negative selection using magnetic sorting (Stem Cell, France). Immature dendritic cells were generated by culturing monocytes in RPMI supplemented with 2% albumin, 1000 IU/mL of GM-CSF and 200 IU/mL of IL-4 (Cellgenix, Freiburg, Germany) for 5 days. Then, DC were pulsed with a concentration range of the various aSLP tested and matured with 20 ng/mL of TNFα and 50 µg/mL of Poly I:C (Sigma–Aldrich, France) for 16 h at 37°C.
T cell clone assay
Once loaded and matured (MΦ-DC) were washed twice in RPMI-2% albumin and co-cultured with T cell clones at a 1:1 ratio for 5 h in the presence of brefeldinA (Sigma–Aldrich, 10 µg/mL). TNFα production was assessed by flow cytometry after fixation with 4% paraformaldehyde, permeabilization with 0,1% saponin and intracellular staining (clone Mab11, 5 µg/mL, Biolegend, San Diego, USA). The percentage of TNFα positive cells was assessed amongst CD8+ cells (clone RPA-T8, BioLegend) or CD4+ cells (clone RPA-T4, BioLegend) to formally exclude DC.
In vitro stimulation
At day 0, PBMCs were plated in 96 wells plate at 2 × 105 cells/wells in RMPI 1640 medium containing 8% human serum, 50 IU/mL IL-2 (Proleukin, Novartis) and stimulated with 5 µM of various aSLP tested. Medium was supplemented according to the accelerated DC (acDC) protocol26 by addition of 1000 U/mL of GM-CSF and 500 UI/mL of IL-4. After 24 h TNFα (1000IU/mL), IL-1β (10 ng/mL) and prostaglandin E2 (1 µM) (BioTechne, France) were added as DC maturation agents. After 21 days, the number of microcultures containing specific CD8+ T cells was evaluated by surface staining with anti-CD8 mAb and tetramer staining (HLA A*0201-MELOE-136-44 or MelanAA27L26-35) (recombinant protein facility, P2R, SFR Santé, Nantes). The threshold of positivity was set at 0.5% tetramer-positive cells.
In vivo immunization
HLA-DRB1*0101/HLA-A*0201 transgenic mice (A2/DR1 mice20) were used to evaluate aSLP immunogenicity. All experiments were carried out in accordance with the French regulations on animal experimentation. Group of 5 female A2/DR1 mice (6–10 weeks) were immunized subcutaneously with 100 µg of each aSLP emulsified in incomplete Freund adjuvant (v/v) and 50 µg of polyI:C. At day 14 and day 28 boost vaccinations were performed with 50 µg of each aSLP along with the same adjuvants. Specific immune responses were evaluated at day 10 after the last boost by ELISpot-IFN-γ (Diaclone, France). Briefly, freshly isolated and sorted CD8+ splenocytes (CD8+ T Cell Isolation Kit, Miltenyi Biotec) were incubated (2.105 cells per well) during 15–20 hours in ELIspot plate in the presence of the class I MELOE-1 epitope (5 µg/mL final) or medium (X-vivo, as negative control). Ex vivo CD4 T cell responses were evaluated on the CD8 negative fraction as above with the HLA-DRB1*0101-restricted epitope MELOE-113-27.
Spots were revealed according to the supplier’s recommendations. Spot-forming cells were counted using the « C.T.L. Immunospot » system (Cellular Technology Ltd). Results were considered positive when the ELIspot counts were >10 spots and above 2× the background (medium).13
Anti-tumor vaccination
A2/DR1 mice were subcutaneously injected with 2 × 105 SARC-A2-MELOE-1 cells in 100 µl in the right flank. The first vaccination with aSLP (100 µg in IFA, as above) or adjuvant alone or PBS started when tumor became detectable (around 10 mm2). A boost vaccination with 50 µg of aSLP was performed at day 20. Tumor growth was evaluated twice a week using a caliper and mice were euthanized when their tumor exceeded 300 mm2.
Statistical analysis
Curve fitting (log (agonist) vs. response, four parameters) and EC50 determination were performed with GraphPad Prism v6.01. Comparison of a number of positive microcultures after stimulation with aSLP in each subject was evaluated by Fisher’s exact test and as a group using paired t- test (α = 0.5%). The efficiency of anti-tumor vaccination was evaluated by Fisher’s exact test (α = 0.5%).
Funding Statement
This work was carried out with the support of the LabEx IGO project (n° ANR-11-LABX-0016–01) funded by the «Investissements d’Avenir» French Government program, managed by the French National Research Agency (ANR).
Acknowledgments
We thank the Cytometry Facility “CytoCell” (SFR Santé) and the DTC core facility, CIC BT 1413, for expert technical assistance.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH.. Therapeutic cancer vaccines. J Clin Invest. 2015;125:3401–3412. doi: 10.1172/JCI80323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pol J, Sevko A, Heide J, Dams M, Rupp A-K, Macas J, Starmann J, Tjwa M, Plate KH, Sültmann H, et al. Trial watch: peptide-based anticancer vaccines. Oncoimmunology. 2015;4:e974411–13. doi: 10.1080/2162402X.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bijker MS, van Den Eeden SJF, Franken KL, Melief CJM, Offringa R, van der Burg SH.. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol. 2007;179:5033–5040. doi: 10.4049/jimmunol.179.8.5033. [DOI] [PubMed] [Google Scholar]
- 4.Rosalia RA, Rebmann V, Lemaoult J, Rodriguez C, Horn PA, Díaz-Lagares A, Echeveste JI, González A. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur J Immunol. 2013;43:2554–2565. doi: 10.1002/eji.201343318. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Hong H, Li D, Ma S, Di Y, Stoten A, Haig N, Di Gleria K, Yu Z, Xu X-N, et al. Comparing pooled peptides with intact protein for accessing cross-presentation pathways for protective CD8+ and CD4 + T Cells. J Biol Chem. 2009;284:9184–9191. doi: 10.1074/jbc.M809456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wada H, Isobe M, Kakimi K, Mizote Y, Eikawa S, Sato E, Takigawa N, Kiura K, Tsuji K, Iwatsuki K, et al. Vaccination with NY-ESO-1 overlapping peptides mixed with Picibanil OK-432 and montanide ISA-51 in patients with cancers expressing the NY-ESO-1 antigen. J Immunother. 2014;37:84–92. doi: 10.1097/CJI.0000000000000017. [DOI] [PubMed] [Google Scholar]
- 7.Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG, Berends-van der Meer DMA, Vloon APG, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009;361:1838–1847. doi: 10.1056/NEJMoa0810097. [DOI] [PubMed] [Google Scholar]
- 8.Ott PA, Jacobs Z, Marwick B, Fullagar R, Wallis L, Smith M, Roberts RG, Hayes E, Lowe K, Carah X, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohtake J, Ohkuri T, Togashi Y, Kitamura H, Okuno K, Nishimura T. Identification of novel helper epitope peptides of Survivin cancer-associated antigen applicable to developing helper/killer-hybrid epitope long peptide cancer vaccine. Immunol Lett. 2014;161:20–30. doi: 10.1016/j.imlet.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Masuko K, Wakita D, Togashi Y, Kita T, Kitamura H, Nishimura T. Artificially synthesized helper/killer-hybrid epitope long peptide (H/K-HELP): preparation and immunological analysis of vaccine efficacy. Immunol Lett. 2015;163:102–112. doi: 10.1016/j.imlet.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 11.Daftarian P, Mansour M, Benoit AC, Pohajdak B, Hoskin DW, Brown RG, Kast WM. Eradication of established HPV 16-expressing tumors by a single administration of a vaccine composed of a liposome-encapsulated CTL-T helper fusion peptide in a water-in-oil emulsion. Vaccine. 2006;24:5235–5244. doi: 10.1016/j.vaccine.2006.03.079. [DOI] [PubMed] [Google Scholar]
- 12.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/S1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 13.Dosset M, Godet Y, Vauchy C, Beziaud L, Lone YC, Sedlik C, Liard C, Levionnois E, Clerc B, Sandoval F, et al. Universal cancer peptide-based therapeutic vaccine breaks tolerance against telomerase and eradicates established tumor. Clin Cancer Res. 2012;18:6284–6295. doi: 10.1158/1078-0432.CCR-12-0896. [DOI] [PubMed] [Google Scholar]
- 14.Godet Y, Moreau-Aubry A, Guilloux Y, Vignard V, Khammari A, Dreno B, Jotereau F, Labarriere N. MELOE-1 is a new antigen overexpressed in melanomas and involved in adoptive T cell transfer efficiency. J Exp Med. 2008;205:2673–2682. doi: 10.1084/jem.20081356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobinet M, Vignard V, Florenceau L, Lang F, Labarriere N, Moreau-Aubry A, Slominski AT. Overexpression of meloe gene in melanomas is controlled both by specific transcription factors and hypomethylation. PLoS One. 2013;8:e75421. doi: 10.1371/journal.pone.0075421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carbonnelle D, Vignard V, Sehedic D, Moreau-Aubry A, Florenceau L, Charpentier M, Mikulits W, Labarriere N, Lang F, Slominski AT. The melanoma antigens MELOE-1 and MELOE-2 are translated from a bona fide polycistronic mRNA containing functional IRES sequences. PLoS One. 2013;8. doi: 10.1371/journal.pone.0075233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogel A, Vignard V, Bobinet M, Labarriere N, Lang F. A long peptide from MELOE-1 contains multiple HLA class II T cell epitopes in addition to the HLA-A*0201 epitope: an attractive candidate for melanoma vaccination. Cancer Immunol Immunother. 2011;60:327–337. doi: 10.1007/s00262-011-1011-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bobinet M, Vignard V, Rogel A, Khammari A, Dreno B, Lang F, Labarriere N, Le Gall S. MELOE-1 antigen contains multiple HLA class II T cell epitopes recognized by Th1 CD4+ T cells from melanoma patients. PLoS One. 2012;7:e51716. doi: 10.1371/journal.pone.0051716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godet Y, Desfrançois J, Vignard V, Schadendorf D, Khammari A, Dreno B, Jotereau F, Labarrière N. Frequent occurrence of high affinity T cells against MELOE-1 makes this antigen an attractive target for melanoma immunotherapy. Eur J Immunol. 2010;40:1786–1794. doi: 10.1002/eji.200940132. [DOI] [PubMed] [Google Scholar]
- 20.Pajot A, van Veelen PA, de Ru AH, Hensbergen PJ, Mizuno K, Koerten HK, Koning F, Tensen CP, Mommaas AM. A mouse model of human adaptive immune functions: hLA-A2.1-/HLA-DR1-transgenic H-2 class I-/class II-knockout mice. Eur J Immunol. 2004;34:3060–3069. doi: 10.1002/eji.200324241. [DOI] [PubMed] [Google Scholar]
- 21.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien C, Flower DR, Feighery C. Peptide length significantly influences in vitro affinity for MHC class II molecules. Immunome Res. 2008;4:6–7. doi: 10.1186/1745-7580-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman HA. Endosomal proteases in antigen presentation. Curr Opin Immunol. 2006;18:78–84. doi: 10.1016/j.coi.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Rawlings ND, Barrett AJ, Finn R. Twenty years of the MEROPSdatabase of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016;44:D343–D350. doi: 10.1093/nar/gkv1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chauvin JM, Larrieu P, Sarrabayrouse G, Prevost-Blondel A, Lengagne R, Desfrancois J, Labarriere N, Jotereau F. HLA anchor optimization of the melan-A-HLA-A2 epitope within a long peptide is required for efficient cross-priming of human tumor-reactive T cells. J Immunol. 2012;188:2102–2110. doi: 10.4049/jimmunol.1101807. [DOI] [PubMed] [Google Scholar]
- 26.Martinuzzi E, Hackbarth J, Schneider PA, Peterson KL, Meng XW, Dai H, Witzig TE, Kaufmann SH. acDCs enhance human antigen-specific T-cell responses. Blood. 2011;118:2128–2137. doi: 10.1182/blood-2011-02-334870. [DOI] [PubMed] [Google Scholar]
- 27.Rangan L, Galaine J, Boidot R, Hamieh M, Dosset M, Francoual J, Beziaud L, Pallandre J-R, Joseph ELM, Asgarova A, et al. Identification of a novel PD-L1 positive solid tumor transplantable in HLA-A*0201/DRB1*0101 transgenic mice. Oncotarget. 2017;8:48959–48971. doi: 10.18632/oncotarget.v8i30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG, Berends-van der Meer DMA, Vloon APG, Drijfhout JW, Wafelman AR, Oostendorp J, Fleuren GJ, et al. Phase I immunotherapeutic trial with long peptides spanning the E6 and E7 sequences of high-risk human papillomavirus 16 in end-stage cervical cancer patients shows low toxicity and robust immunogenicity. Clin Cancer Res. 2008;14:169–177. doi: 10.1158/1078-0432.CCR-07-1881. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.