

Abstract
Repetitive DNA sequences represent about half of the human genome. They have a central role in human biology, especially neurobiology, but are notoriously difficult to study. The purpose of this study was to quantify the transcription from repetitive sequences in a progerin-expressing cellular model of neuronal aging. Progerin is a nuclear protein causative of the Hutchinson–Gilford progeria syndrome that is also incrementally expressed during the normal aging process. A dedicated pipeline of analysis allowed to quantify transcripts containing repetitive sequences from RNAseq datasets oblivious of their genomic localization, tolerating a sufficient degree of mutational noise, all with low computational requirements. The pipeline has been applied to a published panel of RNAseq datasets derived from a well-established and well-described cellular model of aging of dopaminergic neurons. Progerin expression strongly downregulated the transcription from all the classes of repetitive sequences: satellites, long and short interspersed nuclear elements, human endogenous retroviruses, and DNA transposon. The Alu element represented by far the principal source of transcript originating either from repetitive sequences or from canonical coding genes; it was expressed on average at 192,493.5 reads per kilobase million (RPKM) (SE = 21,081.3) in the control neurons and dropped to 43,760.1 RPKM (SE = 5315.0) in the progerin-expressing neurons, being significant downregulated (p = 0.0005). The results highlighted a global perturbation of transcripts derived from repetitive sequences in a cellular model of aging and provided a direct link between progerin expression and alteration of transcription from human repetitive elements.
Electronic supplementary material
The online version of this article (10.1007/s11357-018-00050-2) contains supplementary material, which is available to authorized users.
Keywords: Alu, Repetitive sequences, Progerin, Retrotransposon, Satellites
Introduction
Biological roles of repetitive DNA sequences in human
Repetitive DNA sequences (RS) represent about half of the human genome. They have a role in almost every aspect of human biology (evolution, development, epigenetics, cancer transformation, infections, aging, neurodegenerative diseases), but their study have been hindered by their own repetitive nature. Being repetitive implies that they are difficult to be uniquely mapped in a reference genome. Moreover, during the evolution, they accumulated mutations and now they diverge into finely branched subfamilies. They are also often embedded in and transcribed together with coding genes or regulatory sequences. By all these reasons, quantifying or even identifying the transcripts derived from RS is often a very difficult task. A detailed description of their nature and the difficulties of their study is over the aim of this paper, but for detailed reviews, please refer to Treangen and Salzberg (2011) and Padeken et al. (2015).
Main classes of repetitive sequences
RS can be divided into (1) satellite repeats, which are short to medium tandem repeats that play a central role in dynamics and stability of telomeres, centromeres, and heterochromatin; (2) the long interspersed nuclear elements (LINE), that are non-long terminal repeat (non-LTR) retrotransposons among which are present the retrotransposition competent elements of the L1 family; (3) the small interspersed nuclear elements (SINE), which are non-LTR non-autonomous retrotransposons that may use L1 coded proteins to retrotranspose, the most abundant class of them in human consisting of the Alu elements; (4) the evolutive remnants of integrated LTR retroviruses, such as the human endogenous retrovirus (HERV) families; and (5) several families of DNA transposons.
The progerin-expressing aging cellular model
The analysis described in this paper follows the study published by J. D. Miller and colleagues (Miller et al. 2013). The authors induced the expression of progerin (a truncated form of lamin A causative of the Hutchinson Gilford Progeria Syndrome, HGPS) in a dopaminergic neuronal lineage derived from human induced pluripotent stem cells (iPSC) to obtain a model of aged neurons in order to study Parkinson’s disease (PD). The authors compared GFP (green fluorescent protein)-progerin-expressing iPSC-derived human midbrain dopamine neurons (iPSC-mDA-GFP-Progerin) with GFP-only expressing iPSC-derived human midbrain dopamine neurons as control (iPSC-mDA-GFP). The authors finely characterized the model and described how the progerin expression induced multiple age-related markers in the cells that recapitulate several features observed in aged neurons, such as degeneration of dendrite branching, breakdown of pre-existing neurites, and neuromelanin accumulation, a specific phenotype of aged dopaminergic neurons. The aged phenotype was further characterized and confirmed by canonical gene expression analysis by RNA-seq (Miller et al. 2013).
General features of progerin-expressing cells
Progerin protein is constitutively expressed in patients affected by HGPS, a premature aging syndrome, but its expression gradually increases also in normal aging individuals (Arancio et al. 2014; Prokocimer et al. 2013). By this point of view, progerin-expressing cells are usually considered a very promising aging model. Noteworthy, progerin-expressing cells show specific epigenetic, chromatin, nuclear architectural, and transcriptional alterations that resemble those of cells from older individuals (Arancio et al. 2014; Prokocimer et al. 2013). Coherently, the aging model used in this study recapitulated the vast majority of the characteristics associated to the aging phenotype caused by progerin expression (Miller et al. 2013).
The rationale of the study
A role for the alterations in the RS transcriptional landscape has been suggested in cellular and organismal aging, comprehending aging-related neurodegeneration. In particular, the activation of endogenous retroelements may lead to DNA damage, cellular senescence, and inflammatory response, all processes supposed to play a central role in human aging (Cardelli 2018).
Moreover, several studies suggested a diffuse L1 retrotransposition in human neurons both during the development and in the adult brain (Richardson et al. 2014), i.e., the human adult brain can be considered a genetic mosaic, strongly suggesting a role of RS both in normal neurobiology, as well as in neurological diseases. Nevertheless, data and observations on RS transcription in aging models are somewhat sparse and contradicting. This is due to the combined difficulties to use or sometimes even define a proper model of aging and to the intrinsic difficulties to map, classify, and quantify human RS. This study tries to partially overcome these issues. The model adopted here is a solid cellular model of neuronal aging, very well characterized both at the cellular and at the molecular level (Miller et al. 2013). Furthermore, the pipeline of analysis presented here is able to quantify the transcripts arising from RS independently from their genic (i.e., intergenic, exonic, or intronic) or genomic localization, copy number, and mutational landscape, thus giving an overview of the cellular content of RS transcripts before and after the induction of the aging phenotype via progerin expression.
A better knowledge of the effects of molecular aging (e.g., progerin-induced cellular aging) to the transcriptional profile of RS might not only help to gain a better understanding of the basic mechanisms of neuronal aging but might also pave the way for the experimentation of new diagnostic, prognostic, and therapeutic targets for age-related diseases.
Aim of the study
This study aimed to evaluate whether the progerin expression in this well characterized model might induce perturbation in RS transcriptional profile. A specific pipeline of analysis with low computational requirements has been designed to quantify RS expression. The study showed that RS transcription in this progerin-expressing model was globally strongly downregulated.
Results
Progerin expression downregulates the transcription from repetitive sequences
RS expression was analyzed by a dedicated pipeline of analysis as described in “Methods”. As proof of principle, the pipeline was validated comparing the results to those obtained by TEtools, an established pipeline of analysis for RS expression (Lerat et al. 2016). The concordance was very high: the comparison showed an angular coefficient (the slope) of the regression line of about 1 (1.06) with a very high determination coefficient (R2 = 0.998). Moreover, DESeq2 analysis (Love et al. 2014) did not highlight any significant difference between the results (Sup. Table 1). The results pointed out that the quantification obtained by the pipeline here described is comparable with those obtained by an established approach such as TEtools analysis.
RS expression in the model showed a striking downregulation of RS-derived transcripts after the expression of progerin (Sup. Table 2). The 35 most abundant RS transcripts (defined by the summation of the reads per kilobase million (RPKM) per element of the eight datasets) were statistically downregulated (Table 1).
Table 1.
The mean expression, and the relative standard error of the top 40 repetitive sequences. In the last column, the p value of the Student’s t test comparing progerin-expressing cells with the controls is reported
| RS name | Mean iPSC-mDA-GFP RPKM | SE iPSC-mDA-GFP RPKM | Mean iPSC-mDA-GFP-progerin RPKM | SE iPSC-mDA-GFP-progerin RPKM | p |
|---|---|---|---|---|---|
| ALU | 192,493.5 | 21,081.3 | 43,760.09 | 5315.012 | 0.00048 |
| GGAAT | 56,214.89 | 10,179.29 | 9725.601 | 6674.007 | 0.008768 |
| ALR_ | 9795.778 | 2684.152 | 1020.11 | 521.2053 | 0.018379 |
| HSATII | 6760.561 | 1631.868 | 756.4452 | 402.1595 | 0.01175 |
| ALR1 | 4741.626 | 1284.784 | 514.7198 | 284.585 | 0.018318 |
| SVA_A | 2614.239 | 292.1325 | 550.582 | 113.1218 | 0.000588 |
| BSR | 2748.718 | 816.4452 | 370.5756 | 235.6507 | 0.031223 |
| ALRb | 2490.633 | 647.849 | 268.4393 | 150.6021 | 0.015592 |
| ALR | 2166.722 | 575.391 | 208.7776 | 116.636 | 0.015709 |
| L1HS | 1322.683 | 156.4234 | 737.0422 | 46.15611 | 0.011492 |
| THE1B | 1331.944 | 286.5323 | 354.332 | 115.8635 | 0.019489 |
| L1PA4 | 1337.112 | 225.3016 | 261.4078 | 71.61118 | 0.003891 |
| L1 | 1188.609 | 287.7674 | 287.9438 | 79.55418 | 0.023497 |
| MER30 | 1310.19 | 146.5543 | 153.9609 | 11.77204 | 0.000224 |
| L1PA3 | 1155.169 | 167.4572 | 228.5632 | 55.37116 | 0.001913 |
| L1PA7 | 1106.177 | 208.5104 | 222.5417 | 62.93675 | 0.006673 |
| L1PA5 | 1096.145 | 182.0416 | 217.2718 | 60.54202 | 0.003766 |
| MSTA | 943.9478 | 186.9446 | 241.0317 | 74.92979 | 0.012982 |
| THE1C | 912.5423 | 203.1744 | 239.2613 | 74.72507 | 0.020845 |
| L1PA2 | 973.847 | 78.95267 | 169.308 | 33.49623 | 8.33E-05 |
| THE1_I | 856.1976 | 212.099 | 262.0294 | 65.75552 | 0.036745 |
| L1PA6 | 881.2831 | 144.4721 | 204.326 | 49.81518 | 0.004424 |
| L1PA8 | 917.5237 | 127.5013 | 166.3662 | 44.96404 | 0.001438 |
| THE1D | 841.4189 | 189.1453 | 221.2191 | 62.73324 | 0.020789 |
| THE1A | 720.706 | 151.264 | 307.1189 | 66.16459 | 0.046212 |
| SAR | 746.1229 | 123.8826 | 85.11794 | 42.71888 | 0.002347 |
| L1PREC1 | 662.7323 | 108.2127 | 162.8025 | 32.91405 | 0.004471 |
| L1PA16 | 520.017 | 83.52908 | 121.3695 | 27.21395 | 0.003942 |
| L1PA13 | 524.3263 | 84.54878 | 91.886 | 26.82222 | 0.00278 |
| MLT2A2 | 471.4167 | 109.0712 | 112.9857 | 35.11181 | 0.020373 |
| ALR2 | 521.5152 | 132.9471 | 54.01536 | 24.82073 | 0.013522 |
| L1PA10 | 463.2044 | 86.58916 | 102.7545 | 27.01867 | 0.007335 |
| ALRa_ | 499.6551 | 123.8754 | 51.12524 | 23.58908 | 0.011972 |
| L1PB1 | 426.6081 | 67.96866 | 88.70914 | 23.19274 | 0.003309 |
| L1PA7_5 | 361.1685 | 61.05436 | 87.46906 | 25.05514 | 0.00603 |
| LTR12C | 311.5778 | 74.67214 | 118.6743 | 40.09304 | 0.063144 |
| MLT2A1 | 347.5092 | 70.84894 | 78.20072 | 26.29989 | 0.011876 |
| TIGGER1 | 315.1914 | 46.63056 | 109.0498 | 18.28265 | 0.006246 |
| MLT1B | 313.7677 | 61.96006 | 84.95105 | 20.16838 | 0.012647 |
The angular coefficient (the slope) of the regression line for the comparison of RS expressions (x = expression values in iPSC-mDA-GFP; y = expression values in iPSC-mDA-GFP-Progerin) was 0.2205 (i.e., 0 < slope < 1 means that overall the x and y values increase both, but the x values grow more in comparison with the y values) with a very high determination coefficient (R2 = 0.9863) as shown in Fig. 1a, while the angular coefficient (the slope), of the regression line for the comparison of the expression values of the canonical set of coding genes devoid of LMNA transcripts (as defined in hg38_GENCODE_GENE_V19.bed), was 1.2752 (i.e., 1 < slope < ∞ means that overall the x and y values increase both, but the x values grow less in comparison with the y values) with a low coefficient of determination (R2 = 0.32) as shown in Fig. 1b.
Fig. 1.

Comparison of the expression levels of the repetitive sequences and controls. a The comparison of the expression levels of the repetitive sequences between iPSC-mDA-GFP and iPSC-mDA-GFP-Progerin cells expressed in logarithmic scale for RPKM > 1. b The comparison of gene expressions between iPSC-mDA-GFP and iPSC-mDA-GFP-Progerin cells expressed in logarithmic scale for RPKM > 1. c–h The comparison of expression of selected sequences showing the minimum, first quartile, median, third quartile, and maximum value of expression. Values are expressed in RPKM
The angular and the determination coefficients of the regression lines showed that the progerin expression did not globally downregulate the transcription but perturbed the transcription profile of the coding genes (Fig. 1b), as previously described (Miller et al. 2013). Progerin expression instead strongly downregulated the expression from RS (demonstrated by the angular coefficient of 0.2205), and this downregulation was consistent as demonstrated by the very high determination coefficient (Fig. 1a). These data strongly suggest that the global downregulation of RS transcription in this model of aging is highly specific.
An independent statistical analysis was performed via DESeq2 on the merged raw counts of the expression values of coding gene and RS (Fig. 2; Sup. Table 3), in order to identify the differentially expressed transcripts between the two conditions under investigation. The progerin-expressing neurons clearly differed from the control neurons, and overall, they cluster separately into two different groups (Fig. 2a). The analysis also confirmed that the vast majority of RS transcription was downregulated upon progerin expression in the model, while the coding genes differed on specific gene expression (Sup. Table 3).
Fig. 2.

The results of the DESeq2 analysis. a Heatmap of the sample to sample distance showing that the two different conditions cluster independently. b The estimation of the dispersion of the reads. c MA plot of the distribution of the mean of the normalized counts. d The comparison of expression of selected sequences
The analyses of archetypical and highly expressed repetitive sequences and controls
The Alu element
The most abundant SINE in the human genome, the Alu element (Treangen and Salzberg 2011; Padeken et al. 2015), was overall the most expressed sequence by far (Table 1; Sup. Table 3). It was expressed on average at 192,493.5 RPKM (SE = 21,081.3) in the controls and dropped to 43,760.1 RPKM (SE = 5315.0) in the iPSC-mDA-GFP-Progerin datasets (Fig. 1c), being significant downregulated (p = 0.0005). DESeq2 analysis reported that the normalized Alu raw counts were four-fold downregulated in the progerin-expressing neurons (Fig. 2d).
The L1 LINE
The retrotransposition competent L1 LINE (Treangen and Salzberg 2011; Padeken et al. 2015) was expressed on average at 1188.6 RPKM (SE = 287.8) in the controls and was downregulated in the progerin-expressing cells (287.9 RPKM, SE = 79.5), being significant downregulated (p = 0.0235, Fig. 1d). DESeq2 analysis reported that the normalized raw counts were 3.7 times downregulated in the progerin-expressing neurons (Fig. 2d).
HSATII
The human high-copy satellite II (HSATII) RS (Treangen and Salzberg 2011; Padeken et al. 2015), which has been reported to be upregulated in many cancers (Bersani et al. 2015), showed the same trend as before (average expression = 6760.6 RPKM, SE = 1631.9 in controls vs. average expression = 756.4 RPKM, SE = 402.1; p = 0.0117; Fig. 1e). DESeq2 analysis reported that the normalized raw counts were 6.5 times downregulated in the progerin-expressing neurons (Fig. 2d).
The HERV family
Analyzing the endogenous LTR retroviruses (Treangen and Salzberg 2011; Padeken et al. 2015), the most expressed family was HERVH, previously reported to regulate human pluripotency (Römer et al. 2017), which showed the same behavior as the previous ones (average expression = 126.0 RPKM, SE = 33.0 in controls vs. average expression = 27.2 RPKM, SE = 13.0; p = 0.0317; Fig. 1f). DESeq2 analysis reported that the normalized raw counts were 3.9 times downregulated in the progerin-expressing neurons (Fig. 2d).
The TIGGER1 transposon
The most expressed DNA transposon (Treangen and Salzberg 2011; Padeken et al. 2015) was TIGGER1; it belongs to the Tc1/Mariner family (Smit and Riggs 1996); again, it was downregulated in progerin-expressing cells in comparison with the controls (average expression = 315.2 RPKM, SE = 46.6 in controls vs. average expression = 109.0 RPKM, SE = 18.3; p = 0.0062; Fig. 1g). DESeq2 analysis reported that the normalized raw counts were 2.6 times downregulated in the progerin-expressing neurons (Fig. 2d).
The controls: the opposite behavior of beta actin
To avoid the possibility that the pipeline of analysis might introduce artifacts, the cDNA sequences of controls had been included in the reference files of the repetitive sequences, and their expressions have been analyzed concurrently in the same RS pipeline. Here the quantification of beta-actin cDNA (NM_001101.4) is reported. Beta actin showed an opposite behavior in comparison with the RS, probably due to the change in morphology caused by progerin overexpression. Beta actin was upregulated in iPSC-mDA-GFP-Progerin datasets (average expression = 264.5 RPKM, SE = 52.8 in controls vs. average expression = 1080.0 RPKM, SE = 203.4; p = 0.0082; Fig. 1h). DESeq2 analysis reported that the normalized raw counts were four-fold upregulated in the progerin-expressing neurons (Fig. 2d). Noteworthy, the expression level of beta-actin calculated by the RS pipeline and the one calculated by the coding-gene pipeline are comparable each other (Sup. Table 2), strongly suggesting that the pipeline of analysis applied was highly sensible. Several other housekeeping genes, as defined by Eisenberg and colleagues (Eisenberg and Levanon 2013) together with commonly used control genes (e.g., GAPDH), were also evaluated. The majority of them showed a behavior comparable to beta actin (Sup. Table 4). The negative control was always undetected (0 RPKM) in every dataset analyzed (data not shown), strongly suggesting that the pipeline of analysis applied was specific.
Comparative analysis of normal and HGPS fibroblasts
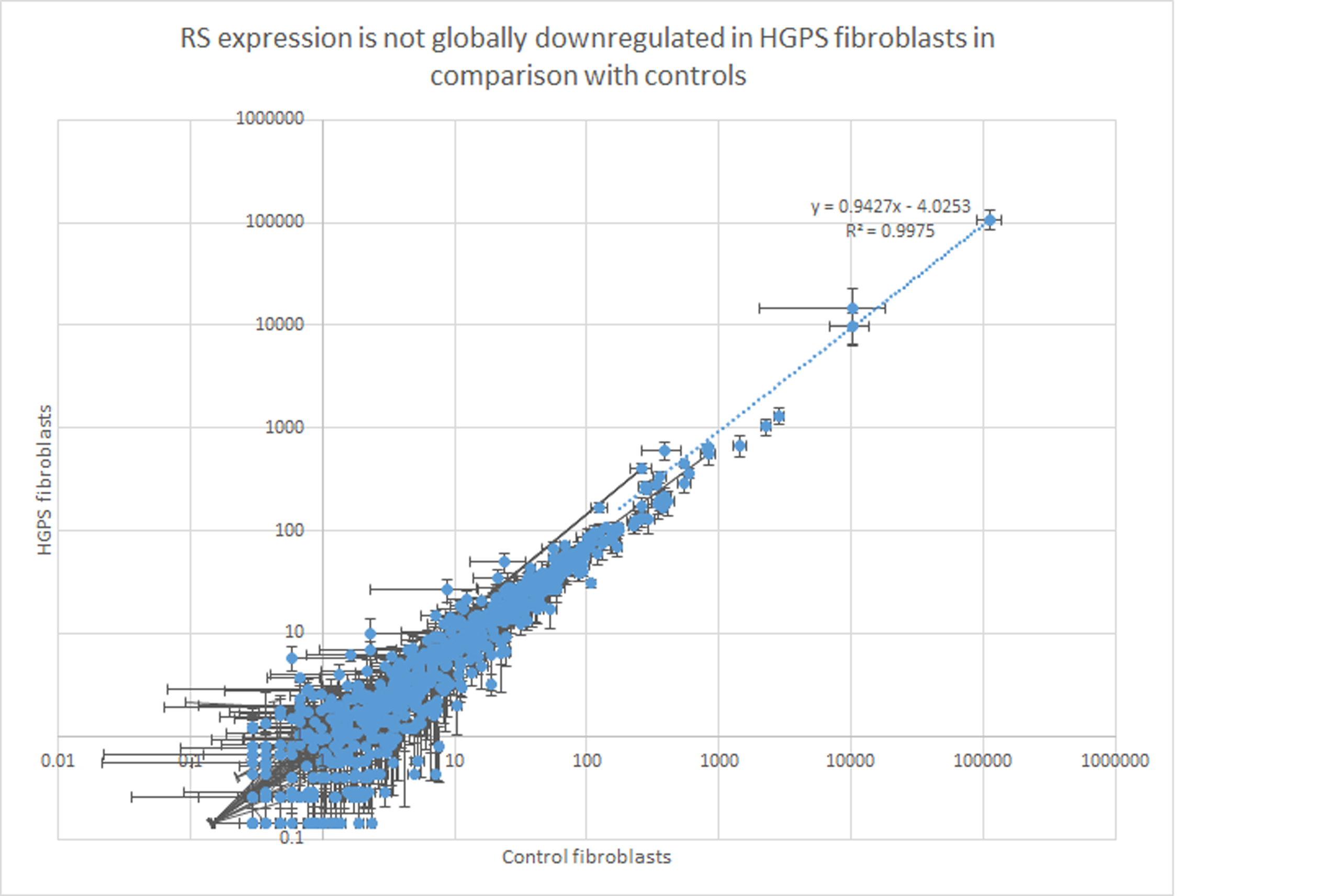
In order to investigate if the RS downregulation observed in the model after progerin expression could be a generalized phenomenon, RS expression was evaluated also in a panel of RNAseq datasets obtained from normal and HGPS fibroblasts (Kreienkamp et al. 2018; please refer to “Methods” for details). The results are controversial. RS expression is not globally downregulated in HGPS fibroblasts in comparison with controls. Indeed, the angular coefficient (the slope) of the regression line for the comparison of RS expressions (x = expression values in normal fibrobalst; y = expression values in HGPS fibrobalsts) was 0.9427 with a very high determination coefficient (R2 = 0.9975) as shown in Sup. Fig. 1, highlighting that the RS expressions were overall highly comparable between the two conditions. Nevertheless, analyzing in detail the RS expression, many archetypical and highly expressed RS are specifically downregulated in HGPS fibroblasts in comparison with controls (Sup. Table 5), that is, the case for L1 expression (1.03-fold decrease, p < 0.0001), HERVH (1.65-fold decrease, p < 0.0001), and TIGGER1 (0.55-fold decrease, p < 0.05). Interestingly, Alu expression was very high, as expected, but no significant difference was detected between HGPS and normal fibroblasts, while HSATII expression was very low both in normal and HGPS fibroblasts, in contrast with the reported expression in neurons.
Discussion
Transcripts derived from RS are usually unspliced and embedded in other transcripts. They are also present in multiple copies in the genome, presenting a constellation of point mutations and indels (Treangen and Salzberg 2011; Padeken et al. 2015). The pipeline of analysis presented here takes these features into account. In fact, the method allowed to identify and count transcripts containing RS from NGS datasets oblivious of their genomic localization and transcriptional landscape, tolerating a sufficient degree of mutational noise. In brief, a collection of the human RS was used to generate specific RS reference files. Then, a common aligner (Bowtie2) was used to find among the NGS data the matching sequences with those in the reference file, using very sensitive parameters, but allowing local alignment (respectively to tolerate several mismatches and identify RS transcripts embedded into others). Positive matches were counted and statistical analyses were performed thereafter. The pipeline used required very low computational time and capacity (the analysis required minutes to hours in the main Galaxy server), differently from other published methods, that can give more detailed results such as the clustering analysis of RS but might require days or even weeks of time (Novák et al. 2013). Moreover, the pipeline makes use of consolidated tools that are common in the field for the analysis of coding-gene expression. This permits to avoid the need to apply novel dedicated bioinformatics tools, for which the know-how might be lacking, but instead the analysis relies only on the customization of the reference files.
The pipeline of analysis was applied to a panel of published datasets (Miller et al. 2013) in order to investigate the expression of RS in a neuronal aging model where dopaminergic neurons derived from iPSCs have been engineered into progerin-expressing cells. Strikingly, the vast majority of RS analyzed was statistically downregulated after progerin expression (Figs. 1 and 2; Table 1; Sup. Tables 2 and 3). The downregulation was highly specific for the RS as shown by a canonical gene expression analysis (Fig. 1b) and internal controls (Fig. 1h). This analysis highlighted also that the transcription from RS is usually very high, especially the most transcribed families, as it can be easily inferred comparing for example Alu and L1 expression with that of beta-actin (Fig. 1c, d, h), that have been analyzed together in the same pipeline. The statistical analysis of the merged raw counts (Sup. Table 2) highlighted that the Alu element represents by far the principal source of transcript originating either from repetitive sequences or canonical coding genes (with the exception of transcripts that map on the LMNA locus in the progerin-expressing cells as experimental artifact: indeed, the LMNA locus codes for lamin-A and progerin).
Caution must be taken in interpreting these results, because even if the cellular model of aging has been well characterized, it is somehow extreme, because usually, lamin A production may be restricted in the central nervous system (Arancio et al. 2014; Jung et al. 2012; Miller et al. 2013; Prokocimer et al. 2013) and the global transcriptional downregulation from the RS here reported probably represents the end point of a complex modulation of the transcriptional output.
Moreover, when the same analysis was performed on HGPS and normal fibroblast, the generalized downregulation caused by progerin expression was not reported, while differential expression is reported on specific RS. Even if Alu sequences are always the most expressed RS, RS globally showed a different profile between neurons and fibroblast (data not shown), suggesting that progerin expression may alter RS expression in lineage-specific manner. This is not surprising, because several reports highlighted that HGPS, caused by progerin expression, is a “segmental disorder,” i.e., it affects multiple tissues and display many but not all symptoms of physiological aging, with tissue and organ specificities (Arancio et al. 2014; Jung et al. 2012; Vidak and Foisner 2016).
It is known that the induction of pluripotent state from differentiated cells might reactivate to some extent the endogenous retrotransposition (Klawitter et al. 2016). Even if both controls and experimental specimens are derived from iPSC, this effect must be taken into account during the interpretation of the model and, more generally, during every study regarding iPSCs; indeed, iPSCs are unique in bearing a second wave of endogenous retrotransposon activation.
RS activity in the brain has been widely demonstrated. In fact, the human brain is genomically heterogeneous. Genomic variations, including L1 and other mobile element retrotransposition, are diffused. Indeed, retrotransposition is particularly high in neuronal precursor cells (Macia et al. 2017). The reasons for brain mosaicism are not understood, although it seems functional to the differentiation process and cellular survival, and overall to brain structural and functional organization. However, uncontrolled mosaicism and RS activation could be a feature or even a possible causative agent for neurological and psychiatric diseases. (Bushman and Chun 2013; Bodea et al. 2018). Speculatively, a strongly downregulation of the activity of RS, as suggested by the results here presented, may be attributed to a safeguard mechanism that is functional to preserve the aged brain from unwanted genomic variation once the developmental program has been accomplished.
Alu elements have been recognized as important evolutionary drivers of primate brain evolution. Nevertheless, deleterious Alu activities have been associated with several neurological disorders and neurodegenerative diseases. In particular, the dysregulation of Alu elements can induce severe alterations of mitochondrial homeostasis and an increased expression of Alu transcripts contributes to inflammation within the nervous system. (Larsen et al. 2018). An interesting paper about Geographic atrophy (a form of age-related macular degeneration) showed evidence of accumulation of Alu RNA, caused by DICER1 deficiency, release of mitochondrial DNA, and inflammasome activation via the cytosolic DNA sensor cGAS. (Kerur et al. 2017). Coherently, the downregulation of Alu transcription in the model can be interpreted as a functional protection mechanism against inflammation.
Moreover, it is accepted that endogenous retrotransposition, especially driven by L1 coded proteins, can have a role in mutating genes and epigenetic dysregulation, thus contributing to tumorigenesis. (Goodier 2014; Rangasamy et al. 2015; Rodić and Burns 2013). By this perspective, RS downregulation in the model may be interpreted as a safeguard mechanism against transformation events.
Nevertheless, data are controversial. An interesting paper on the expression of the L1-coded ORF1 protein, supports the idea that ORF1 expression increases with aging, in vivo, especially in the hippocampal region, known for elevated rates of retrotransposition (Sur et al. 2017). Indeed, RS activation has been suggested as a feature of aging cells and the acquisition of the senescent phenotype (Pal and Tyler 2016).
Speculatively, the downregulation of RS transcription here reported could be interpreted as a safeguard mechanism for old neurons, which lose plasticity in favor of safety. The reported RS activity in neurodegenerative diseases and cancer transformation may be due to the overcame of this safeguard mechanism. Moreover, the differences reported in the RS expression between neurons and fibroblasts suggest that changes in RS expression during aging, and thus their influence in the aging process, may possess tissue specificity.
Methods
Identifying and quantifying the repetitive sequence expression
The FASTA file containing the collection of the human repetitive sequences was downloaded from REPBASE (Bao et al. 2015) updated to December 11, 2017. Then, the sequences of beta actin cDNA (NM_001101.4), 18S, and 5.8 ribosomal subunits have been manually appended as positive controls. The 1017 nucleotides long sequence of the locus EF191515 of Bacillus subtilis SMY strain has been manually appended as negative control. Then, a BED reference file has been generated from this revised FASTA file using the pyfaidx command in a Linux environment (faidx --transform bed name.fasta > name.bed). The analyses have been performed thereafter in a Galaxy environment (Afgan et al. 2016). The FASTQ raw sequences (obtained from the European Nucleotide Archive) have been uploaded, processed by FASTQ-Groomer (Galaxy Version 1.1.1) (Blankenberg et al. 2010) and then by Trimmomatic (Galaxy Version 0.36.5) (Bolger et al. 2014) with standard settings but for the minimum length = 40, and quality checked by FASTQC. Bowtie2 aligner (Galaxy Version 2.3.4.2) (Langmead et al. 2009; Langmead and Salzberg 2012) has been used on the paired-end sequences with very-sensitivity-local parameters (-D 20 -R 3 -N 0 -L 20 -i S,1,0.50) using as reference genome the previously generated FASTA file. At least, the generated BAM files have been compared using the BED file previously generated as reference. Raw data are reported in Sup. Table 2.
TEtools analysis
SRX378109 dataset was analyzed as above described, and the BAM file has been used as input for the TEcount tool of the TEtools bundle (Galaxy version 1.0.0) (Lerat et al. 2016) at the galaxy.prabi.fr server. The DESeq2 analysis (Galaxy Version 2.11.40.2) (Love et al. 2014) was performed in the Galaxy environment on the RS raw counts obtained by TEcounts and the custom pipeline.
Analysis of coding gene expression
The same raw FASTQ data processed by FASTQ-Groomer and by Trimmomatic have been aligned by the means of HISAT2 aligner (Galaxy Version 2.1.0) (Kim et al. 2015) using the Galaxy embedded hg38 as reference with default parameters. The generated BAM files have been compared using the hg38_GENCODE_GENE_V19.bed as reference (downloaded from https://sourceforge.net). The choice of this method was due to perform an analysis similar to the one used to identify and quantify the repetitive sequences.
Statistical analyses
The quantification of transcripts has been performed in RPKM, considering as “total reads number” for each dataset the count of reads that passed the quality checks, and not the mapped reads, due to the RS mapping strategy. Student’s t test, standard deviation and standard error calculations, regression analyses, and their graphical displays have been performed in the Microsoft Excel environment.
The DESeq2 analysis (Galaxy Version 2.11.40.2) (Love et al. 2014) was performed in the Galaxy environment on the merged raw counts of coding gene and RS.
Dataset used
The analyses were performed on datasets published by J. D. Miller et al. (2013). The authors induced pluripotent stem state in fibroblasts from young and old donors, and differentiated them into human midbrain dopamine neurons (iPSC-mDA), to mimicry a specific cell type that shows neurodegeneration in Parkinson’s disease. These cells were therefore engineered to overexpress GFP-progerin fusion protein to recapitulate the effect of aging. Deep sequencing was performed using RNA extracted from progerin-expressing cells and controls.
The analysis described here compare four RNAseq datasets of GFP-progerin-expressing iPSC-derived human midbrain dopamine neurons (iPSC-mDA-GFP-Progerin: SRX378111, SRX378112, SRX378115, SRX378116) with four datasets of GFP-only expressing iPSC-derived human midbrain dopamine neurons as control (iPSC-mDA-GFP: SRX378109, SRX378110, SRX378113, SRX378114).
HGPS (SRX2746782, SRX2746783, SRX2746784) and normal fibroblast (SRX2746788, SRX2746789, SRX2746790) datasets are described in Kreienkamp et al. (2018).
Datasets have been downloaded from the European Nucleotide Archive.
Electronic supplementary material
{kind=link}
(PNG 518 kb)
(XLSX 134 kb)
(XLSX 209 kb)
(XLSX 221 kb)
(XLSX 11 kb)
(XLSX 85 kb)
Acknowledgements
I want to thank the VICOR’s, Dr. S Genovese, and Prof. A.M. Puglia, Prof. G. Gallo, and all the members of their lab for their help in a difficult period of my personal and professional life.
Abbreviations
- HERV
human endogenous retrovirus
- HGPS
Hutchinson Gilford Progeria Syndrome
- HSAT
high copy satellite
- iPSC
induced pluripotent stem cell
- iPSC-mDA-GFP
GFP expressing iPSC-derived human midbrain dopamine neurons
- iPSC-mDA-GFP-Progerin
GFP-progerin-expressing iPSC-derived human midbrain dopamine neurons
- LINE
long interspersed nuclear element
- LTR
long terminal repeat
- PD
Parkinson’s disease
- RPKM
reads per kilobase million
- RS
repetitive sequence
- SE
standard error
- SINE
small interspersed nuclear elements
Compliance with ethical standards
Conflict of interest
The author declares that he has no conflict of interest to disclose, this research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors, that the data is not published or submitted elsewhere, and that approval of procedures and approval of the manuscript has been provided by all authors.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman-Jackson J, von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44(W1):W3–W10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancio W, Pizzolanti G, Genovese SI, Pitrone M, Giordano C. Epigenetic involvement in Hutchinson-Gilford progeria syndrome: a mini-review. Gerontology. 2014;60(3):197–203. doi: 10.1159/000357206. [DOI] [PubMed] [Google Scholar]
- Bao W, Kojima KK, Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6:11. doi: 10.1186/s13100-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersani F, Lee E, Kharchenko PV, Xu AW, Liu M, Xega K, MacKenzie OC, Brannigan BW, Wittner BS, Jung H, Ramaswamy S, Park PJ, Maheswaran S, Ting DT, Haber DA. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc Natl Acad Sci U S A. 2015;112(49):15148–15153. doi: 10.1073/pnas.1518008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Gordon A, Von Kuster G, Coraor N, Taylor J, Nekrutenko A, Galaxy Team Manipulation of FASTQ data with galaxy. Bioinformatics. 2010;26(14):1783–1785. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodea GO, McKelvey EGZ, Faulkner GJ. Retrotransposon-induced mosaicism in the neural genome. Open Biol. 2018;8(7):180074. doi: 10.1098/rsob.180074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman DM, Chun J. The genomically mosaic brain: aneuploidy and more in neural diversity and disease. Semin Cell Dev Biol. 2013;24(4):357–369. doi: 10.1016/j.semcdb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardelli M. The epigenetic alterations of endogenous retroelements in aging. Mech Ageing Dev. 2018;174:30–46. doi: 10.1016/j.mad.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. Human housekeeping genes, revisited. Trends Genet. 2013;29(10):569–574. doi: 10.1016/j.tig.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Goodier JL. Retrotransposition in tumors and brains. Mob DNA. 2014;5:11. doi: 10.1186/1759-8753-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HJ, Coffinier C, Choe Y, Beigneux AP, Davies BSJ, Yang SH, Barnes RH, Hong J, Sun T, Pleasure SJ, Young SG, Fong LG. Regulation of prelamin A but not lamin C by miR-9, a brain-specific microRNA. Proc Natl Acad Sci U S A. 2012;109(7):E423–E431. doi: 10.1073/pnas.1111780109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, Varshney A, Yasuma R, Fowler BJ, Baghdasaryan E, Marion KM, Huang X, Yasuma T, Hirano Y, Serbulea V, Ambati M, Ambati VL, Kajiwara Y, Ambati K, Hirahara S, Bastos-Carvalho A, Ogura Y, Terasaki H, Oshika T, Kim KB, Hinton DR, Leitinger N, Cambier JC, Buxbaum JD, Kenney MC, Jazwinski SM, Nagai H, Hara I, West AP, Fitzgerald KA, Sadda SVR, Gelfand BD, Ambati J. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2017;24(1):50–61. doi: 10.1038/nm.4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawitter S, Fuchs NV, Upton KR, Muñoz-Lopez M, Shukla R, Wang J, Garcia-Cañadas M, Lopez-Ruiz C, Gerhardt DJ, Sebe A, Grabundzija I, Merkert S, Gerdes P, Pulgarin JA, Bock A, Held U, Witthuhn A, Haase A, Sarkadi B, Löwer J, Wolvetang EJ, Martin U, Ivics Z, Izsvák Z, Garcia-Perez JL, Faulkner GJ, Schumann GG. Reprogramming triggers endogenous L1 and Alu retrotransposition in human induced pluripotent stem cells. Nat Commun. 2016;7:10286. doi: 10.1038/ncomms10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreienkamp R, Graziano S, Coll-Bonfill N, Bedia-Diaz G, Cybulla E, Vindigni A, Dorsett D, Kubben N, Batista LFZ, Gonzalo S. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. 2018;22(8):2006–2015. doi: 10.1016/j.celrep.2018.01.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods. Mar 4;9(4):357–9. 10.1038/nmeth.1923. PubMed PMID: 22388286; PubMed Central PMCID: PMC3322381. [DOI] [PMC free article] [PubMed]
- Larsen PA, Hunnicutt KE, Larsen RJ, Yoder AD, Saunders AM. Warning SINEs: Alu elements, evolution of the human brain, and the spectrum of neurological disease. Chromosom Res. 2018;26(1–2):93–111. doi: 10.1007/s10577-018-9573-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerat E, Fablet M, Modolo L, Lopez-Maestre H, Vieira C. TEtools facilitates big data expression analysis of transposable elements and reveals an antagonism between their activity and that of piRNA genes. Nucleic Acids Res. 2016;45(4):e17. doi: 10.1093/nar/gkw953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macia A, Widmann TJ, Heras SR, Ayllon V, Sanchez L, Benkaddour-Boumzaouad M, Muñoz-Lopez M, Rubio A, Amador-Cubero S, Blanco-Jimenez E, Garcia-Castro J, Menendez P, Ng P, Muotri AR, Goodier JL, Garcia-Perez JL. Engineered LINE-1 retrotransposition in nondividing human neurons. Genome Res. 2017;27(3):335–348. doi: 10.1101/gr.206805.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, Taldone T, Fusaki N, Tomishima MJ, Krainc D, Milner TA, Rossi DJ, Studer L. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13(6):691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novák P, Neumann P, Pech J, Steinhaisl J, Macas J. RepeatExplorer: a galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics. 2013;29(6):792–793. doi: 10.1093/bioinformatics/btt054. [DOI] [PubMed] [Google Scholar]
- Padeken J, Zeller P, Gasser SM. Repeat DNA in genome organization and stability. Curr Opin Genet Dev. 2015;31:12–19. doi: 10.1016/j.gde.2015.03.009. [DOI] [PubMed] [Google Scholar]
- Pal S, Tyler JK. Epigenetics and aging. Sci Adv. 2016;2(7):e1600584. doi: 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokocimer M, Barkan R, Gruenbaum Y. Hutchinson-Gilford progeria syndrome through the lens of transcription. Aging Cell. 2013;12(4):533–543. doi: 10.1111/acel.12070. [DOI] [PubMed] [Google Scholar]
- Rangasamy D, Lenka N, Ohms S, Dahlstrom JE, Blackburn AC, Board PG. Activation of LINE-1 retrotransposon increases the risk of epithelial-mesenchymal transition and metastasis in epithelial cancer. Curr Mol Med. 2015;15(7):588–597. doi: 10.2174/1566524015666150831130827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SR, Morell S, Faulkner GJ. L1 retrotransposons and somatic mosaicism in the brain. Annu Rev Genet. 2014;48:1–27. doi: 10.1146/annurev-genet-120213-092412. [DOI] [PubMed] [Google Scholar]
- Rodić N, Burns KH. Long interspersed element-1 (LINE-1): passenger or driver in human neoplasms? PLoS Genet. 2013;9(3):e1003402. doi: 10.1371/journal.pgen.1003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römer C, Singh M, Hurst LD, Izsvák Z. How to tame an endogenous retrovirus: HERVH and the evolution of human pluripotency. Curr Opin Virol. 2017;25:49–58. doi: 10.1016/j.coviro.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Smit AF, Riggs AD. Tiggers and DNA transposon fossils in the human genome. Proc Natl Acad Sci U S A. 1996;93(4):1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sur D, Kustwar RK, Budania S, Mahadevan A, Hancks DC, Yadav V, Shankar SK, Mandal PK. Detection of the LINE-1 retrotransposon RNA-binding protein ORF1p in different anatomical regions of the human brain. Mob DNA. 2017;8:17. doi: 10.1186/s13100-017-0101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011;13(1):36–46. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidak S, Foisner R. Molecular insights into the premature aging disease progeria. Histochem Cell Biol. 2016;145(4):401–417. doi: 10.1007/s00418-016-1411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 518 kb)
(XLSX 134 kb)
(XLSX 209 kb)
(XLSX 221 kb)
(XLSX 11 kb)
(XLSX 85 kb)