

Abstract
Coiled-coil-helix-coiled-coil-helix domain containing protein 2 (CHCHD2) mutations were linked with autosomal dominant Parkinson’s disease (PD) and recently, Alzheimer’s disease/frontotemporal dementia. In the current study, we generated isogenic human embryonic stem cell (hESC) lines harboring PD-associated CHCHD2 mutation R145Q or Q126X via clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) method, aiming to unravel pathophysiologic mechanism and seek potential intervention strategy against CHCHD2 mutant-caused defects. By engaging super-resolution microscopy, we identified a physical proximity and similar distribution pattern of CHCHD2 along mitochondria with mitochondrial contact site and cristae organizing system (MICOS), a large protein complex maintaining mitochondria cristae. Isogenic hESCs and differentiated neural progenitor cells (NPCs) harboring CHCHD2 R145Q or Q126X mutation showed impaired mitochondria function, reduced CHCHD2 and MICOS components and exhibited nearly hollow mitochondria with reduced cristae. Furthermore, PD-linked CHCHD2 mutations lost their interaction with coiled-coil-helix-coiled-coil-helix domain containing protein 10 (CHCHD10), while transient knockdown of either CHCHD2 or CHCHD10 reduced MICOS and mitochondria cristae. Importantly, a specific mitochondria-targeted peptide, Elamipretide/MTP-131, now tested in phase 3 clinical trials for mitochondrial diseases, was found to enhance CHCHD2 with MICOS and mitochondria oxidative phosphorylation enzymes in isogenic NPCs harboring heterozygous R145Q, suggesting that Elamipretide is able to attenuate CHCHD2 R145Q-induced mitochondria dysfunction. Taken together, our results suggested CHCHD2–CHCHD10 complex may be a novel therapeutic target for PD and related neurodegenerative disorders, and Elamipretide may benefit CHCHD2 mutation-linked PD.
Introduction
Numerous pathogenic genes and susceptibility loci have been associated with the common neurodegenerative disease, Parkinson’s disease (PD). Among them, missense mutations (Thr61Ile and Arg145Gln) of coiled-coil-helix-coiled-coil-helix domain containing protein 2 (CHCHD2) (located on chromosome 7q11.2) were identified in inherited late-onset autosomal dominant PD cases (1). Four missense CHCHD2 variants including three amino acid substitutions (p.Ala32Thr, p.Pro34Leu and p.Ile80Val) were reported in additional studies based on PD patients with European ancestry (2). A nonsense heterozygous variant of CHCHD2 (c.376C>T, p.Gln126X), leading to a truncated protein, was then identified in a German PD patient (age at onset >40 years) (3). Recently, a heterozygous mutation of CHCHD2 (c.196G>A, p.Val66Met) was identified in a patient with multiple system atrophy (4); missense variants were identified in patients with Alzheimer’s disease (5) and frontotemporal dementia (FTD) (6). Besides, a 27-month-old boy was reported with psychomotor delay that is linked to a de novo 393 kb microdeletion of 7p11.2 covering CHCHD2 (7). A patient with a 47 kb deletion of this region including CHCHD2 was reported to have developmental delay and intellectual disability (8). Collectively, heterozygous CHCHD2 mutations or deletions harboring CHCHD2 have been linked with human neuronal dysfunction.
CHCHD2 is a member of a family of proteins containing coiled-coil-helix-coiled-coil-helix (CHCH) domain, locating in the mitochondria and the nuclear (9). CHCHD2 promotes mitochondrial oxygen consumption and is consistently co-expressed with other nuclear-encoded structural oxidative phosphorylation (OXPHOS) subunits (10). It promotes mitochondrial oxygen consumption, and knockdown of CHCHD2 reduces the activity of complex IV and I (10). Downregulation of CHCHD2 increases cellular reactive oxygen species (9). CHCHD2 is found to be an inhibitor of apoptosis by binding to Bcl-xl (11) and/or by binding to cytochrome c along with MICS1, a member of Bax inhibitor-1 superfamily (12). However, the biological role of CHCHD2 in the mitochondria and how disease-related CHCHD2 mutations are linked to mitochondria dysfunction remains largely elusive.
Mitochondria are cellular energy-generating machines, exhibiting a complicated topology with inner (IM) and outer membrane (OM). The IM runs parallel to the OM and engulfs into mitochondria matrix forming cristae structures, providing extended membrane for OXPHOS enzymes to produce Adenosine triphosphate (ATP). Such delicate mitochondria cristae are maintained by protein modulators and unique phospholipid in inner mitochondria membrane such as cardiolipin (13). Among identified protein modulators of cristae structure, mitochondrial contact site and cristae organizing system (MICOS) plays a central role in the biogenesis and maintenance of the cristae junction. MICOS is a large protein complex, evolutionary conserved from yeast to mammals. Currently, seven mammalian MICOS subunits have been identified: Mitofilin/Mic60, CHCHD3/Mic19, CHCHD6/Mic25, APOO/Mic26, APOOL/Mic27, QIL1/Mic13 and MINOS1/Mic10 with two distinct MICOS subcomplexes marked by the core components Mitofilin and MINOS1, respectively (14). Complete absence or impairment of any MICOS components causes drastic alternation of mitochondria cristae and subsequently, mitochondria dysfunction (15–18). In many cases, loss of one subunit of MICOS also prevents the stable accumulation of other MICOS components in the mitochondria (19–21). Current understanding of molecular architecture of MICOS and components of its subcomplexes is still limited, and multiple studies suggest additional unknown subunits of MICOS in mammals (22–24).
Coiled-coil-helix-coiled-coil-helix domain containing protein 10 (CHCHD10), a homologue of CHCHD2, was reported to associate with MICOS (25). CHCHD10 mutations were identified in patients with FTD, motor neuron disease, cerebellar ataxia and mitochondria myopathy (26), FTD-ALS amyotrophic lateral sclerosis clinical spectrum (27–32), SMAJ (late-onset spinal motor neuropathy) (33) and Charcot–Marie–Tooth disease type 2 (34). Disease-associated CHCHD10 mutations promote loss of mitochondria cristae junctions (25). CHCHD10 was reported to bind the key molecular of MICOS, Mitofilin (25), but following studies failed to replicate so (35,36), leading to the debate on the biological role of CHCHD10 (37,38).
Human pluripotent stem cells, such as human embryonic stem cells (hESCs), have been used to study diseases, such as late-onset neurodegenerative diseases including PD (39). We used clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) method to generate isogenic hESC lines harboring different PD-linked CHCHD2 mutations. By using these isogenic hESCs and derived neural progenitor cells (NPCs), along with human dopaminergic cell lines, human post-mortem brain lysates and mouse brain sections, we identified the pathogenic mechanism of PD-associated CHCHD2 mutations, highlighting an overlapping pathophysiologic link across a wide spectrum of neurological disorders, showing potential therapy targeting this link.
Results
Generation of isogenic hESC lines harboring PD-linked CHCHD2 mutation
CHCHD2 T61I, Q126X and R145Q were associated with PD. Q126X is a C-terminal truncation mutation leading to loss of CHCH domain, while R145Q is a point mutation in CHCH domain (Fig. 1A). We employed CRISPR-Cas9 system (40) to create isogenic lines carrying PD-related CHCHD2 mutations on H9 hESC. Briefly, mRNA of gRNA for each mutation (Fig. 1B) and Cas9 was electroporated into H9 hESC together with single-strand oligo donor. hESC colonies formed by single cell were screened and validated by Sanger sequencing. For Q126X mutation, 3 positive clones, Q10, Q13 and Q35, out of 75 colonies were identified to carry heterozygous Q126X+/−, with no homozygous Q126X−/− clones found (Fig. 1C). For R145Q mutation, 1 heterozygous R4 (R145Q+/−) and 2 homozygous R10 (R145Q−/−) and R17 (R145Q−/−) lines were identified from 280 colonies (Fig. 1D). For T61I mutations, we failed to obtain any clones harboring homozygous/heterozygous mutations.
Figure 1.
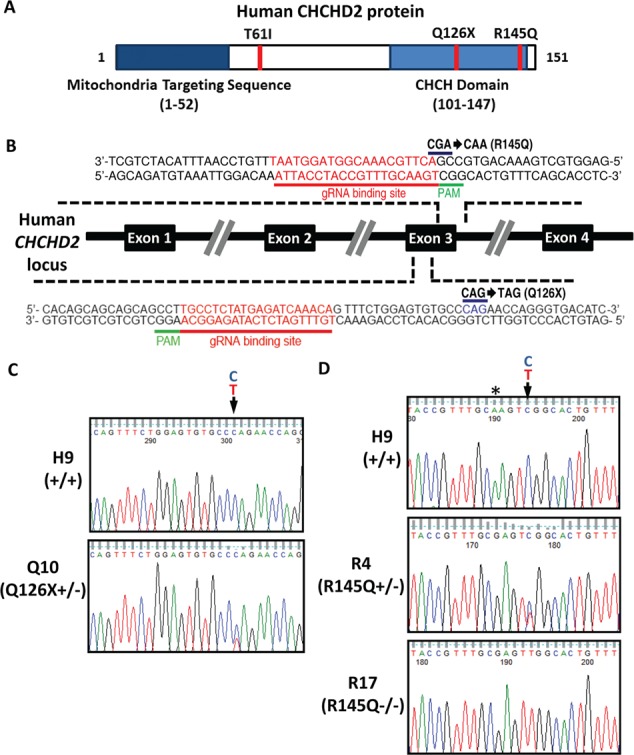
Generation of human isogenic lines harboring PD-related CHCHD2 mutants. (A) PD-associated CHCHD2 mutants T61I, Q126X and R145Q on functional domains of human CHCHD2 protein. CHCH domain. (B) Schematic overview depicting human CHCHD2 locus and Cas9 targeting strategy with designed guide RNA recognized exon 3 sequences in human CHCHD2 locus. (C) Sanger sequencing chromatogram showing the heterozygous point mutation leading to Q126X+/−. Representative results from Q10 were shown. (D) Sanger sequencing chromatogram on the reverse DNA strand showing the heterozygous point mutation leading to R145Q+/− (R4), the homozygous point mutant leading to R145Q−/− (R17) at the targeting locus of H9 (+/+) hESCs. *the blocking nonsense mutation to enhance the specificity of Cas9 system.
Isogenic cell lines carrying Q126X or R145Q displayed typical hESC morphology, with a high nucleus/cytoplasm ratio and prominent nucleoli. These lines can be maintained for more than 30 passages in vitro. The pluripotency of isogenic cells was validated by immunofluorescence assay with positive staining of Oct-4, Nanog or SSEA-4 by in vivo teratoma formation assay and karyotyping (Supplementary Material, Fig. S1A–C). Potential off-target sites of CRISPR-Cas9 were recruited (http://crispr.mit.edu) and validated by Sanger sequencing. No off-target mutagenesis was found in isogenic lines by using top 20 sets of primers for potential off-target sites spanning through whole genome (Supplementary Material, Table S1). We differentiated hESC to NPCs (Supplementary Material, Fig. S1D). All isogenic hESC lines, like H9, can be differentiated into NPC without major difference, as revealed by immunostaining of NPC markers Pax6, Sox2 and Nestin (Supplementary Material, Fig. S1E). Collectively, we generated three hESC lines carrying Q126X+/−, one line carrying R145Q+/− and two lines carrying R145Q−/− on H9 hESC. Those lines maintained pluripotency and differentiation capacity of parental H9 hESC.
Mitochondria dysfunction caused by PD-associated CHCHD2 mutations
To investigate whether PD-associated CHCHD2 mutations caused mitochondria dysfunction, we determined oxygen consumption rate (OCR) of isogenic hESCs harboring Q126X and R145Q sequentially treated with the inhibitor of ATP synthase Oligomycin, then with the proton ionophore, carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP) and lastly a mixture of a mitochondrial complex I inhibitor Rotenone with a mitochondrial complex III inhibitor Antimycin. Isogenic hESCs carrying R145Q as R4 (R145Q+/−), R10 (R145Q−/−) and R17 (R145Q−/−) showed ~30–50% reduction in basal OCR, ATP turnover and maximum respiratory capacity when compared with H9 cells (Fig. 2A). These measured parameters were also reduced in isogenic hESCs carrying Q126X as Q13 and Q35 (Fig. 2B). Similarly, transient CHCHD2 knockdown reduced ~30–40% OCR in human dopaminergic SK-N-SH cells (Fig. 2C). In addition, a reduction in the protein abundance of OXPHOS complexes, especially Complex I, III and IV/V, was observed in Q10 (Q126X+/−), R4 (R145Q+/−) and R17 (R145Q−/−) hESCs or NPCs as compared with control cells (Fig. 2D and E).
Figure 2.
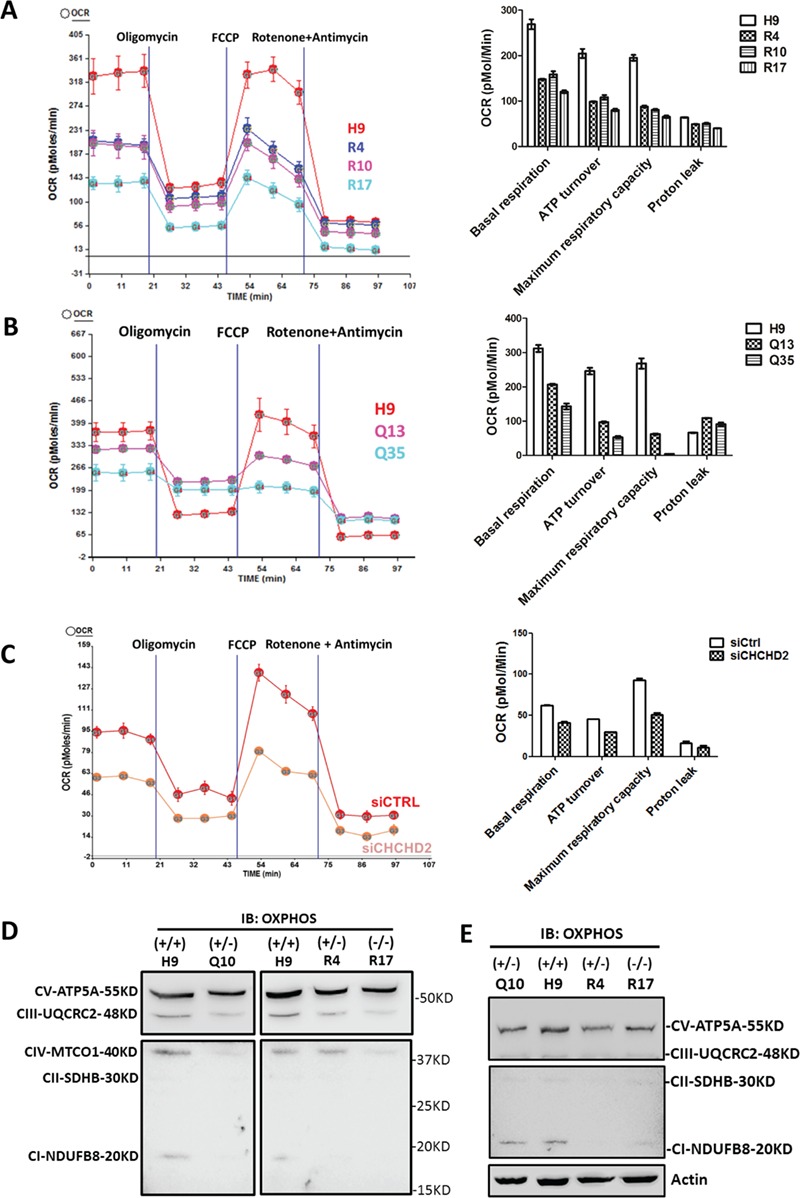
CHCHD2 Q126X/R145Q mutants caused mitochondria dysfunction as transient CHCHD2 knockdown. (A–C) The function of mitochondria electron respiration complex was reflected by OCR on isogenic R4 (R145Q+/−) or R10, R17 (R145Q−/−) hESCs (A), on isogenic Q13, Q35 (Q126X+/−) hESCs (B) and on dopaminergic SK-N-SH cells transiently transfected with CHCHD2 siRNA (C). Quantification of basal respiration, ATP turnover, maximum respiration capacity and proton leak were shown. Representative results from independent biological replicates were shown. (D) The abundance of OXPHOS enzymes of H9 (+/+), Q10 (Q126X+/−), R4 (R145Q+/−) and R17 (R145Q−/−) hESCs shown by total OXPHOS antibody cocktail. Complex I–V were labeled accordingly. Equal amount of protein was loaded in each lane. (E) The abundance of OXPHOS enzymes of H9 (+/+), Q10 (Q126X+/−), R4 (R145Q+/−) and R17 (R145Q−/−) NPCs shown by total OXPHOS antibody cocktail. Actin served as loading control.
The distribution of CHCHD2 in MICOS complex of mitochondria
To investigate how CHCHD2 mutations cause mitochondria dysfunction, we examine CHCHD2 subcellular localization in human dopaminergic SK-N-SH cells by an antibody that recognizes C-terminus of CHCHD2. The specificity of this antibody was confirmed by western blotting and immunofluorescence staining (Supplementary Material, Fig. S2A and B). It showed endogenous CHCHD2 localized in both mitochondria and nucleus (Fig. 3A). Overexpressed non-tagged CHCHD2 showed a similar pattern as endogenous CHCHD2 with less nucleus distribution (Fig. 3A). CHCHD2 showed little overlap with endoplasmic reticulum (ER, shown by KDEL antibody) or with autophagy–lysosomal system (shown by LAMP1 antibody) (Fig. 3A).
Figure 3.
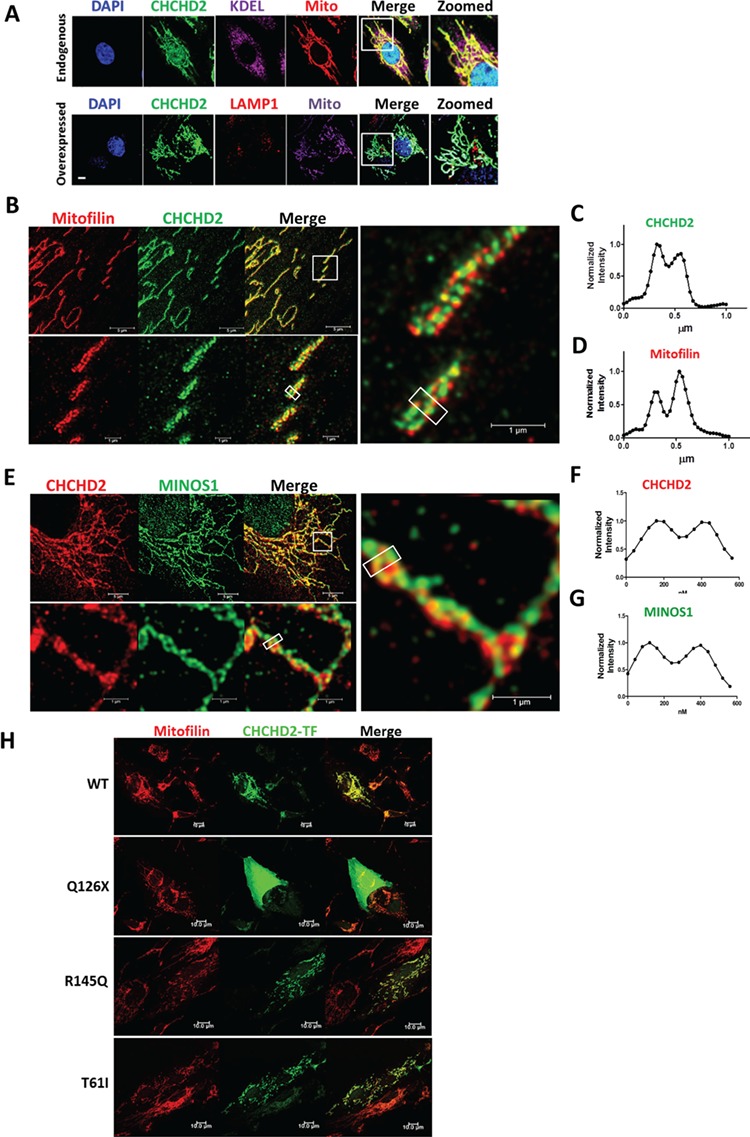
Super-resolution microscope revealed CHCHD2 localized in the MICOS complex of mitochondria. (A) Upper panel: immunostaining of endogenous CHCHD2 (green) in SK-N-SH cells co-stained with ER marker KDEL (purple), MitoTracker (Mito, red) and DAPI (blue). Lower panel: immunostaining of overexpressed non-tagged CHCHD2 (green) in SK-N-SH cells co-stained with autophagy–lysosomal marker LAMP1 (red), MitoTracker (Mito, purple) and DAPI (blue). The boxed regions were zoomed to show the details. Scale bar, 10 μm. (B–D) Immunostaining of the mitochondria network of SK-N-SH cells shown by two-color STED microscopy. Endogenous CHCHD2 (green) and Mitofilin (red) was shown. The boxed region of the upper panel was enlarged and shown in the lower panel and right panel. Averaged intensity profiles for CHCHD2 (green) and Mitofilin (red) across the indicated mitochondrial tubule sections within the respective boxed area in the magnification lower panel were shown in (C) and (D). Scale bar, 5 μm (upper panel) and 1 μm (lower panel). (E–G) Immunostaining of the mitochondria network of SK-N-SH cells by dual-color STED microscopy. Endogenous CHCHD2 (red) and MINOS1 (green) were shown. The boxed region of the upper panel was enlarged and shown in the lower panel and right panel. Averaged intensity profiles for CHCHD2 (red) and MINOS1 (green) across the indicated mitochondrial tubule sections within the respective boxed area were shown in (F) and (G). Scale bar, 5 μm (upper panel) and 1 μm (lower panel). (H) Immunostaining of SK-N-SH cells transiently transfected with non-tagged CHCHD2 WT/T61I/R145Q/Q126X plasmids, co-stained with Mitofilin (red) and CHCHD2 (green) by a CHCHD2 antibody against a middle region of CHCHD2 (labeled as CHCHD2-TF). Scale bar, 10 μm.
Super-resolution microscopy stimulated emission depletion (STED), which provides an optical resolution well below the diffraction limit, showed that CHCHD2 was not homogenously distributed along the mitochondria but concentrated into individual clusters (Fig. 3B). Those CHCHD2 clusters were often arranged at the rim of mitochondrial tubules (Fig. 3C). Such characteristic localization is very similar to ‘discontinuous rail-like distribution’ of MICOS components such as Mitofilin, MINOS1 and CHCHD3, which was previously revealed by STED (41). A dual-color STED microscope revealed similar dot-like discontinuous distribution for both CHCHD2 and Mitofilin, a core protein in MICOS (Fig. 3B). Average intensity profile across the indicated mitochondria tubule sections within the box showed both CHCHD2 and Mitofilin were enriched in the mitochondria rim and with similar distance to each other (Fig. 3C and D). Similar distribution of CHCHD2 such as MINOS1, another core component of MICOS, was also observed (Fig. 3E–G). Collectively, the specific distribution pattern of CHCHD2 along mitochondria and its proximity to both Mitofilin and MINOS1, cores of two subcomplexes of MICOS, strongly suggested a biological role of CHCHD2 in MICOS. Meanwhile, only partial CHCHD2 co-localized with Mitofilin or MINOS1, as shown yellow in the merge panel of green and red fluorescence in the enlarged super-resolution microscopy imaging (Fig. 3B and E).
We transiently overexpressed non-tagged CHCHD2 wild-type (WT)/ PD-linked mutations in SK-N-SH cells. Immunostaining showed CHCHD2 T61I and R145Q had similar mitochondria distribution as CHCHD2 WT with Mitofilin, whereas Q126X showed cytoplasmic distribution (Fig. 3H). As Q126X (loss of the C-terminus of CHCHD2) cannot be recognized by the CHCHD2 antibody against its C-terminus epitope, this immunostaining was stained by an antibody recognizing a middle region of CHCHD2, labeled as CHCHD2-TF (tested in Supplementary Material, Fig. S2D).
PD-related CHCHD2 mutations disrupted MICOS, similar as CHCHD2 knockdown
NPCs derived from isogenic hESC display more mature mitochondria morphology than hESC (42); therefore, we investigated mitochondria morphology in derived NPCs from isogenic hESC. Transmission electronic microscope (TEM) showed ultrastructure of mitochondria in isogenic NPCs. Q10 (Q126X+/−), R4 (R145Q+/−) or R17 (R145Q−/−) exhibited frequently nearly hollow appearance with few cristae compared with control (Fig. 4A). The quantification based on images revealed abnormal mitochondria that has few cristae or detached cristae was ~40% in R4 (R145Q+/−) and R17 (R145Q−/−) compared with 10% in H9 (+/+) NPCs (Supplementary Material, Fig. S3A). We observed a similar disturbed mitochondria ultrastructure in CHCHD2 transient knockdown cells (Fig. 4B). Next we analyzed MICOS components and found CHCHD2 knockdown reduced ~80% Mitofilin and ~50% MINOS1 protein in SK-N-SH cells (Fig. 4C–G). Reduced MICOS components, such as Mitofilin, MINOS1, CHCHD3, CHCHD6, were also found in isogenic R4 (R145Q+/−), R17 (R145Q−/−) and Q10 (Q126X+/−) hESCs (Fig. 4H). Moreover, lower CHCHD10 expression was found in transient CHCHD2 knockdown cells (Fig. 4C and G) and in all mutant isogenic cells R4, R17 and Q10 as compared with their controls (Fig. 4H and I). Besides, CHCHD2 protein itself was reduced ~40–90% in isogenic hESCs or NPCs harboring R145Q or Q126X (Fig. 4H–J; Supplementary Material, Fig. S3B). Reduced MICOS components were also found in isogenic hESCs or NPCs (Fig. 4H–J; Supplementary Material, Fig. S3C). Hsp60, a mitochondria chaperone, remains almost constant in different isogenic NPCs (Fig. 4J), suggesting CHCHD2 mutations specifically reduced MICOS components in the mitochondria. Transient CHCHD2 knockdown did not reduce the mass of mitochondria by staining mitochondria chaperones Grp75 (data not shown). Collectively, cells with CHCHD2 knockdown or cells harboring disease-linked CHCHD2 mutation showed reduced MICOS and loss of mitochondria cristae, suggesting CHCHD2 plays a crucial role to maintain the integrity of MICOS.
Figure 4.
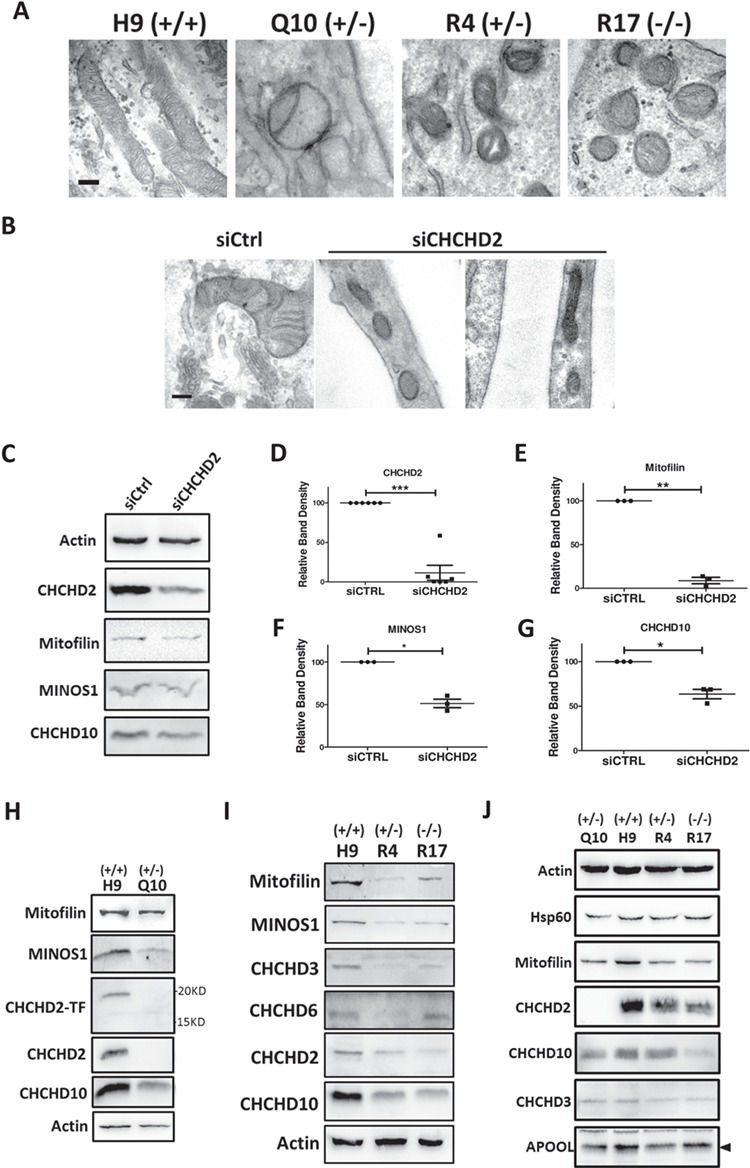
PD-related CHCHD2 mutants disrupted MICOS as CHCHD2 knockdown. (A) Ultrastructure analysis by transmission electronic microscopy of isogenic NPCs harboring Q126X or R145Q. Representative results from H9 (+/+), Q10 (Q126X+/−), R4 (R145Q+/−) and R17 (R145Q−/−) were shown. Scale bar, 200 nm. (B) Ultrastructure analysis by transmission electronic microscopy of SK-N-SH cells transiently transfected with CHCHD2 siRNA and control siRNA. Scale bar, 200 nm. (C) Expression of CHCHD2, CHCHD10 and MICOS components Mitofilin, MINOS1 in SK-N-SH cells transiently transfected with CHCHD2 siRNA and control siRNA. Actin served as loading control. (D–G) Quantification of protein abundance of CHCHD2 (D), Mitofilin (E), MINOS1 (F) and CHCHD10 (G) in SK-N-SH cells transiently transfected with CHCHD2 siRNA and control siRNA. Actin served as internal control. Data are presented as mean ± SEM. n = 3. Paired t-test was used. (D), ***P = 0.0002; (E), **P = 0.0015; (F), *P = 0.0478; (G), *P = 0.0141. (H and I) Expression of CHCHD2 and MICOS components in hESCs of H9, isogenic lines harboring CHCHD2 Q126X or R145Q. Actin served as loading control. CHCHD2-TF, antibody against the middle-epitope of CHCHD2; CHCHD2, antibody against C-terminus epitope of CHCHD2. (J) Expression of CHCHD2 and MICOS components in derived NPCs from H9, isogenic lines harboring CHCHD2 Q126X or R145Q. Actin served as loading control. Representative blots were shown.
PD-linked CHCHD2 mutations showed reduced binding to CHCHD10
To understand how CHCHD2 mutations reduce MICOS components, we searched for binding partner(s) of CHCHD2 within MICOS. In dopaminergic SK-N-SH cells, we found endogenous CHCHD2 pulled down endogenous CHCHD10 (Fig. 5A) and vice versa (Fig. 5B), but we did not detect any interaction between endogenous CHCHD2 and other known MICOS components, such as Mitofilin, MINOS1 or CHCHD3 (Supplementary Material, Fig. S4A–C). CHCHD2–CHCHD10 interaction was also detected in healthy human brain lysates (Fig. 5C) and hESC H9 (Fig. 5E). CHCHD2 shares 58% sequence identity with CHCHD10. We used antibodies that specifically recognize endogenous CHCHD2 or CHCHD10 with no cross-reaction with the other homologue in SK-N-SH and H9 hESCs (Supplementary Material, Fig. S2C and D).
Figure 5.
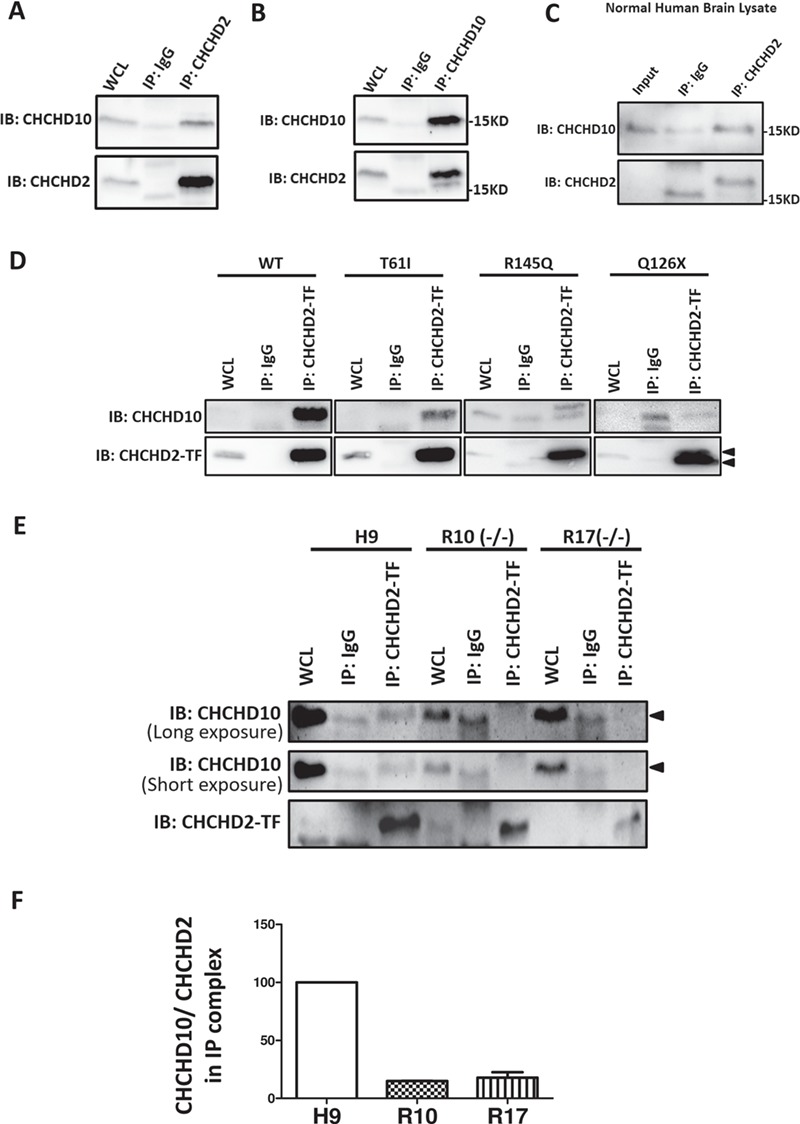
PD-linked CHCHD2 mutants showed reduced binding to CHCHD10. (A) Co-immunoprecipitation of endogenous CHCHD2 by antibody against CHCHD2 in SK-N-SH cells. WCL: whole cell lysate, 5% total protein used in co-IP experiment. (B) Co-immunoprecipitation of endogenous CHCHD10 by antibody against CHCHD10 in SK-N-SH cells. WCL: whole cell lysate, 5% total protein used in co-IP experiment. (C) Co-immunoprecipitation of endogenous CHCHD2 by antibody against CHCHD2 in human brain tissue lysates. (D) Co-immunoprecipitation of CHCHD2 by a CHCHD2 antibody against middle region of CHCHD2 (labeled as CHCHD2-TF) in SK-N-SH cells transfected with non-tagged CHCHD2 WT/T61I/R145Q/Q126X. WCL: whole cell lysate, 5% total protein used in co-IP experiment. Lower arrow pointed to CHCHD2 Q126X and higher arrow pointed to full length CHCHD2. (E) Co-immunoprecipitation of CHCHD2 by a CHCHD2 antibody against middle region of CHCHD2 (labeled as CHCHD2-TF) in hESCs of H9 and isogenic lines harboring homozygous R145Q (−/−).WCL: whole cell lysate, 5% total protein used in co-IP experiment. Arrow pointed to CHCHD10. Representative results from R10 and R17 (R145Q−/−) were shown. (F) Quantification of protein abundance of CHCHD10 normalized with CHCHD2 on the co-immunoprecipitation complex from isogenic hESC lysates.
We next examined the role of PD-linked CHCHD2 mutations in CHCHD2–CHCHD10 interaction. Endogenous CHCHD10 can be pulled down by non-tagged CHCHD2 WT but not by C-terminal myc-tagged CHCHD2 WT (Supplementary Material, Fig. S4D), indicating that small C-terminal myc tag on CHCHD2 may obstruct CHCHD2–CHCHD10 interaction. These data suggested C-terminus of CHCHD2 is important for CHCHD2’s association with CHCHD10. Non-tagged CHCHD2 WT/T61I/R145Q/Q126X plasmids were then used in subsequent immunoprecipitation assay. By using the antibody against the middle region epitope of CHCHD2 (CHCHD2-TF), WT CHCHD2 was shown to bind to endogenous CHCHD10, while PD-associated mutations T61I, Q126X and R145Q had impaired binding with CHCHD10 (Fig. 5D). Q126X, a truncation mutation, is 25 amino acids shorter than WT, so Q126X can be distinguished from the endogenous CHCHD2 by a band shift in western blot (Fig. 5D). Moreover, we detected the interaction of CHCHD2 R145Q with CHCHD10 by using isogenic R10 and R17 cell lines harboring homozygous R145Q−/− that produced CHCHD2 R145Q with no CHCHD2 WT protein. About 90% reduction of CHCHD2–CHCHD10 interaction in these lines as compared with H9 control was observed (Fig. 5E and F).
CHCHD2–CHCHD10 complex maintained MICOS integrity
We examined the localization of CHCHD2 and CHCHD10. CHCHD10 showed strong staining on mitochondria with CHCHD2 (Fig. 6A). STED imaging showed both CHCHD2 and CHCHD10 displayed ‘rail-like’ distribution of mitochondria (Fig. 6A). Average intensity profile across the indicated mitochondria tubule sections within the box showed both CHCHD2 and CHCHD10 were enriched in the mitochondria rim (Fig. 6B and C). As mutations of CHCHD2 or CHCHD10 are linked with neurological disorders, we investigated the localization of CHCHD2 and CHCHD10 in mouse brain, specifically in the substantial nigra region. Immunostaining on mouse brain sections showed most CHCHD2 and CHCHD10-immunoreactive cells in the pars compacta of substantial nigra (Fig. 6D). A few positively stained cells were found in the pars reticulata. Co-localization of CHCHD2 with CHCHD10 in the cytoplasm of cells was clearly visible (Fig. 6D, zoomed in images in insets). Such expression pattern suggested CHCHD2 and CHCHD10 co-localize in dopaminergic neurons in the substantial nigra. To prove this, we stained mouse brain sections with a dopaminergic neuron marker tyrosine hydroxylase (TH), along with CHCHD2 or CHCHD10. CHCHD2 (Fig. 6E) was shown to highly overlap with dopaminergic neuron marker TH, whereas CHCHD10 (Fig. 6F), with a broader staining pattern in the mouse brain section, partially co-localized with TH.
Figure 6.
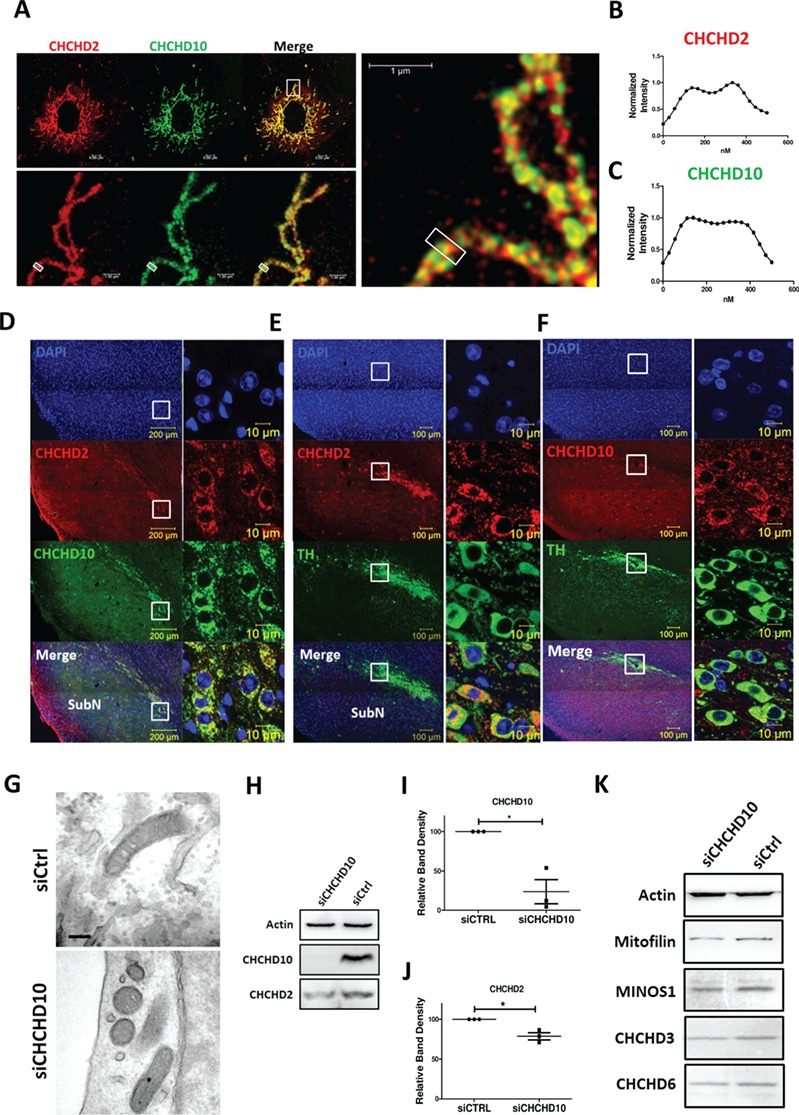
CHCHD2 and CHCHD10 maintained MICOS intergrity. (A–C) Immunostaining of the mitochondria by two-color super-resolution STED microscopy on of SK-N-SH cells transfected with non-tagged CHCHD2 WT (red), co-stained with CHCHD10 (green). The boxed region of the upper panel was enlarged and shown in the lower panel. Averaged intensity profiles for CHCHD10 (green) (B) and CHCHD2 (red) (C) across the indicated mitochondrial tubule sections within the respective boxed area in the magnification lower panel were shown. Scale bar, 5 μm (upper panel) and 1 μm (lower panel). (D) Immunostaining of CHCHD2 (red) and CHCHD10 (green) in substantial nigra (SubN) region of mouse brain slides. Boxed region was zoomed to show in right panel. Scale bar, 100 μm (left panel) and 10 μm (right panel). (E) Immunostaining of CHCHD2 (red) and dopaminergic neuron marker TH (green) in substantial nigra (SubN) region of mouse brain slides. Boxed region was zoomed to show in right panel. Scale bar, 100 μm (left panel) and 10 μm (right panel). (F) Immunostaining of CHCHD10 (red) and dopaminergic neuron marker TH (green) in substantial nigra region of mouse brain slides. Boxed region was zoomed to show as right panel. Scale bar, 100 μm (left panel) and 10 μm (right panel). (G) Ultrastructure analysis by transmission electronic microscopy of SK-N-SH cells transiently transfected with CHCHD10 siRNA and control siRNA. Scale bar, 200 nm. (H–J) Expression of CHCHD10 and CHCHD2 in SK-N-SH cells transiently transfected with CHCHD10 siRNA and control siRNA. The quantification of protein abundance of CHCHD10 (I) and CHCHD2 (J) were shown. Data are presented as mean ± SEM. Actin served as internal control. Results were from three independent biological replicates. Paried t-test was used. (I), *P = 0.038; (J), *P = 0.0410. (K) Expression of MICOS components Mitofilin, MINOS1 and CHCHD10 in SK-N-SH cells transiently transfected with CHCHD10 siRNA and control siRNA. Actin served as loading control. The blots were representative from three independent biological replicates.
CHCHD10 was reported to bind Mitofilin in MICOS, but we did not detect a direct CHCHD10–Mitofilin interaction. To ensure CHCHD2 mutations-caused MICOS defects is due to impaired CHCHD2–CHCHD10 interaction, we examined the biological role of CHCHD10 in MICOS by knocking down CHCHD10. TEM showed ultrastructure of mitochondria with less or no cristae in CHCHD10 transient knocked-down cells (Fig. 6G), similar as CHCHD2 (Fig. 4A). Western blot analysis revealed 70 and 75% reduction in the expression of CHCHD10 and CHCHD2, respectively, in CHCHD10 knockdown cells as compared with control (Fig. 6H–J). Meanwhile, MICOS components Mitofilin, MINOS1, CHCHD3 and CHCHD6 were reduced (Fig. 6K) with CHCHD10 knockdown. Taken together, those results proved CHCHD10’s role in MICOS and highlighted the importance of CHCHD2–CHCHD10 interaction in maintaining MICOS integrity.
Elamipretide attenuated CHCHD2 mutations-caused mitochondria dysfunction
MICOS not only controls mitochondria cristae but also maintains mitochondria via mitochondria transcription factor A (TFAM), a key transcriptional factor in mitochondria transcription and mitochondria DNA (mtDNA) maintenance (15,43). Reduced TFAM protein was observed in all isogenic hESCs carrying R145Q or Q126X (Fig. 7A and B). Similarly, CHCHD2 knockdown caused ~40% reduction in the expression of TFAM (Fig. 7C) but not TFAM mRNA (Supplementary Material, Fig. S5A). There was no significant change in the mtDNA copy number in isogenic cells or cells with CHCHD2 knockdown (Supplementary Material, Fig. S5B and C). Average intensity profile based on super-resolution microscopy showed TFAM or mtDNA localized at the interior of mitochondria while CHCHD2 at the rim of mitochondria (Fig. 7D and E) with little overlap with TFAM or mtDNA.
Figure 7.
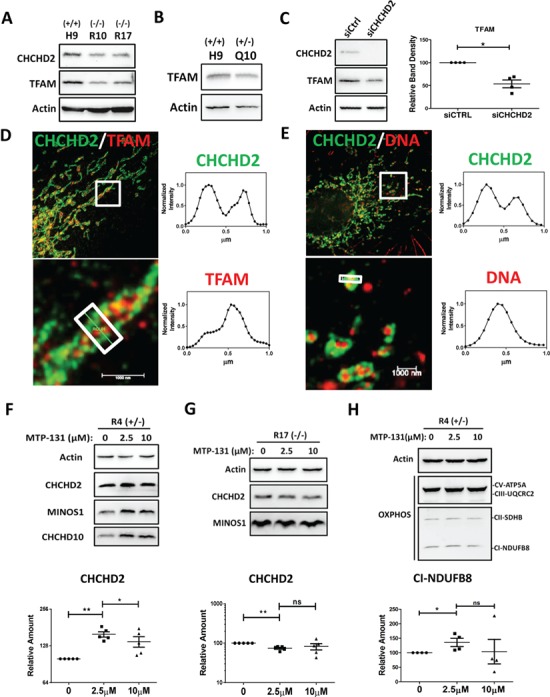
CHCHD2 mutants reduced mitochondria transcription factor TFAM and mitochondrial targeted peptide Elamipretide/MTP-131 boosted CHCHD2 and MICOS components in isogenic R4 (R145Q+/−) NPCs. (A and B) Expression of TFAM and CHCHD2 in isogenic R10 (R145Q−/−), R17 (R145Q−/−) hESCs (A) and isogenic Q10 (Q126X+/−) hESCs (B). (C) Expression of CHCHD2 and TFAM in SK-N-SH cells transiently transfected with CHCHD2 siRNA and control siRNA. Actin served as loading control. Quantification of band density of TFAM normalized with Actin in SK-N-SH cells transiently transfected with CHCHD2 siRNA and control siRNA. Actin served as loading control. Data are presented as mean ± SEM. *P = 0.0121, n = 4. (D and E) Immunostaining of the mitochondria of SK-N-SH cells by dual-color super-resolution STED microscopy. Endogenous CHCHD2 (green) and TFAM (red) were shown in D, and endogenous CHCHD2 (green) and DNA (red) was shown in E. The boxed regions were enlarged and shown. Averaged intensity profiles for CHCHD2 (green) and TFAM/DNA (red) across the indicated mitochondrial tubule sections within the respective boxed area in the magnification lower panel were shown in (D) and (E). Scale bar, 1 μm (lower panel). (F) Expression of CHCHD2, CHCHD10 and MINOS1 in isogenic NPCs R4 (R145Q+/−) treated with Elamipretide/MTP-131 for 24 h. Quantification of CHCHD2 protein abundance was shown. Actin served as internal control. Data are presented as mean ± SEM. Five independent biological replicates were shown. Paired t-test was used. **P = 0.0017; *P = 0.0368. (G) Expression of CHCHD2 and MINOS1 in isogenic NPCs R17 (R145Q−/−) treated with Elamipretide/MTP-131 for 24 h. Quantification of CHCHD2 protein abundance was shown. Actin served as internal control. Data are presented as mean ± SEM. Five independent biological replicates were shown. Paired t-test was used. **P = 0. 0029; ns = not significant. (H) The abundance of OXPHOS enzymes on R4 (R145Q+/−) NPC cells treated with Elamipretide/MTP-131 for 24 h. Actin served as loading control. Data are presented as mean ± SEM. Representative blots from four independent biological replicates were shown. The quantification of Complex I-NDUFB8, normalized with actin was shown. *P = 0.043; ns = not significant.
Based on our identified CHCHD2’s role in maintaining MICOS complex and mitochondria cristae, we then sought for potential strategy to correct defects caused by CHCHD2 mutations. Elamipretide/MTP-131 is a cell-permeable and mitochondria-targeting peptide (44) that protects mitochondrial cristae by interacting with phospholipid in inner mitochondrial membrane named cardiolipin (45). Elamipretide/MTP-131 is currently tested in clinical trials for ischemia reperfusion injury, mitochondrial myopathies (46) and heart failure (47). We hypothesized Elamipretide/MTP-131 could restore CHCHD2 mutations-induced defects by protecting the mitochondria cristae. To test this, we used isogenic human NPCs as a cellular model for human diseases (42). As expected, Elamipretide/MTP-131 enhanced CHCHD2 in control H9 NPC (Supplementary Material, Fig. S6A). In heterozygous isogenic cells R4 (R145Q+/−), 2.5 μm Elamipretide /MTP-131 significantly enhanced CHCHD2 level to ~150% of control (Fig. 7F). A total of 10 μm Elamipretide/MTP-131, compared with 2.5 μm, did not further boost CHCHD2 protein expression. Quantitative reverse transcription PCR (RT-qPCR) analysis revealed Elamipretide/MTP-131 did not enhance CHCHD2 or TFAM mRNA expression in H9 or R4 (R145Q+/−) NPCs (Supplementary Material, Fig. S6B and C). In homozygous isogenic R17 (R145Q−/−) NPCs, Elamipretide/MTP-131 did not enhance CHCHD2 level at 2.5 or 10 μm (Fig. 7G). Like its effect on CHCHD2, Elamipretide/MTP-131 enhanced the abundance of CHCHD10, MINOS1 and OXPHOS Complex V and I in R4 (R145Q+/−) NPCs (Fig. 7F and H). Quantification showed OXPHOS Complex I was enhanced ~25% by 2.5 μm Elamipretide/MTP-131. Collectively, these results suggested Elamipretide/MTP-131 attenuated mitochondria dysfunction in cells harboring heterozygous CHCHD2 R145Q.
Taken together, we showed CHCHD2 localized in close proximity to MICOS, and CHCHD2 preserved the integrity of MICOS and mitochondria cristae. PD-related CHCHD2 mutations caused mitochondria dysfunction that includes the reduction of OXPHOS complexes, TFAM protein and impairment of oxygen consumption of mitochondria. PD-related CHCHD2 mutations lost their association with CHCHD10, leading to loss of mitochondria cristae, reduced MICOS complex and CHCHD2 itself. Elamipretide, a mitochondrial targeting peptide to restore mitochondria cristae, was able to enhance CHCHD2 level, abundance of MICOS and OXPHOS enzymes thus protect cells harboring heterozygous PD-associated R145Q (Fig. 8).
Figure 8.
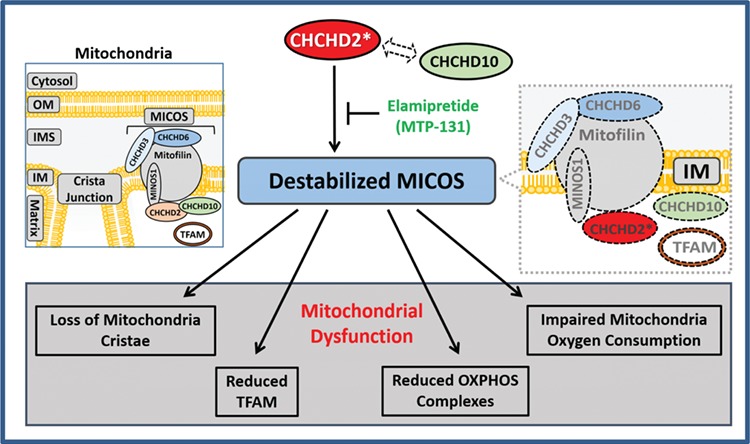
Illustration of the pathophysiological role of CHCHD2 in MICOS. Mitochondria are separated from cytosol by mitochondria OM. Mitochondria IM separates the matrix from inter-membrane space. MICOS complex composing of Mitofilin, CHCHD3, CHCHD6 and MINOS1, was responsible for cristae morphology and maintenance in the mitochondria inner member. CHCHD2 binds to CHCHD10. However, PD-linked CHCHD2 mutants (showed in red with ‘*’) have impaired binding with CHCHD10, causing a reduced MICOS, with loss of mitochondria cristae, loss of mitochondria OXPHOS complexes, reduced oxygen consumption of mitochondria and decreased mitochondria transcription factor TFAM. A mitochondrial targeted peptide Elamipretide/MTP-131 was able to enhance CHCHD2, CHCHD10 and MICOS components with the abundance of mitochondria OXPHOS enzymes in cells harboring heterozygous R145Q.
Discussion
Mitochondria dysfunction is a key pathological hallmark of PD and other neurological disorders. Herein, we delineated a novel mechanistic link between PD-related CHCHD2 mutations and mitochondria dysfunction. First, by using STED super-resolution microscopy providing optical resolution below the diffraction limit of light, we demonstrated CHCHD2 had distinct feature of ordered ‘rail-like’ pattern and clustered along mitochondria as MICOS (41). Second, dual-color STED images identified CHCHD2 to be in proximity to both Mitofilin and MINOS1, two key components of MICOS, suggesting a physiological role of CHCHD2 related to MICOS and cristae maintenance. Third, TEM analysis revealed less or no cristae in the mitochondria from either cells with CHCHD2 knockdown or isogenic NPCs harboring PD-associated Q126X or R145Q. Fourth, transient CHCHD2 knockdown cells and isogenic hESCs and NPCs showed reduced expression of protein components of MICOS including Mitofilin, MINOS1, CHCHD3, CHCHD10 and CHCHD6. Similar cristae distortion and MICOS reduction have been reported in multiple experimental models such as human cells/flies/yeast when MICOS were disrupted (15,18,43,48,49). Therefore, our results suggest CHCHD2 plays a significant role in maintaining MICOS and mitochondria cristae, whereas PD-associated Q126X and R145Q disrupt MICOS and impair mitochondria function. This is also the first evidence that CHCHD2 is essential to maintain both Mitofilin subcomplex and MINOS1 subcomplex of MICOS (14,22), highlighting CHCHD2’s role for proper mitochondria function.
Reduction of CHCHD2 protein itself, along with reduction of MICOS components, was observed in isogenic cells harboring Q126X or R145Q. This suggested that CHCHD2 could be reduced with MICOS as a protein associated with MICOS. However, we did not detect direct interaction between endogenous CHCHD2 and Mitofilin, MINOS1, CHCHD3 or CHCHD6. Though CHCHD10 was shown to associate with MICOS via binding to Mitofilin directly (25), we and others failed to replicate a direct CHCHD10–Mitofilin interaction (35,36). By using specific antibody against each protein (Supplementary Material, Fig. S2), we showed an interaction between endogenous CHCHD2 and CHCHD10 in multiple human neuronal models including dopaminergic SK-N-SH cells and healthy human brain tissue lysates. Recent studies in HEK293 cells or human cancer cells corroborated our finding (35,36,50,51). Furthermore, we showed co-localization of CHCHD2 and CHCHD10 in mouse brain sections, thus highlighting a functional role of CHCHD2–CHCHD10 in the neuronal system. Super-resolution microscopy showed both CHCHD2 and CHCHD10 appeared as ‘rail-like’ clusters at the rim of mitochondria, suggesting both proteins were in proximity to MICOS. As current understanding of MICOS components is still limited and additional unknown subunits of mammalian MICOS have been suggested (22–24), we could not rule out the possibility that some unidentified MICOS components bridges CHCHD2–CHCHD10 complex to known MICOS components. Further study is needed to extend our understanding of MICOS.
We found transient CHCHD10 knockdown cells exhibited impaired mitochondria cristae and reduced MICOS, similar as transient CHCHD2 knockdown in dopaminergic cells. We ruled out the possibility of such phenotypes was caused by off-target effects of siRNA by testing on four different siRNAs targeting different exons (data not shown). Moreover, we reported knockdown of CHCHD2 led to reduction of CHCHD10 protein and vice versa, which could explain why impairment of either CHCHD2 or CHCHD10 disrupts MICOS. This also suggested CHCHD2 and CHCHD10 play overlapping role in maintaining MICOS, and they may compensate with the other when one is ablated. Of note, Huang et al. (37) recently reported CHCHD2 and CHCHD10 are partially functionally redundant and are not essential for MICOS stability based on CHCHD2/CHCHD10 single and double knockout HEK293 stable cell lines. Besides the difference in cell lines (HEK293 versus dopaminergic cells), the major difference between this and the current study was in the strategy of gene manipulation. Huang et al. constructed stable completed knockout cell lines, while we used transiently knockdown of target protein via siRNA transfection. During gene silencing, loss of one gene could be compensated by another gene with overlapping function to maintain fitness of the living organisms (52,53). When generating stable lines, chronic cultured cells could gradually activate cellular mechanisms to compensate for the lack of the target gene while transient knockdown of targeted gene shows instant cellular responses upon losing the target gene; therefore, the results of stable gene silencing may be compromised compared with those of transient genetic manipulation. As ours and Huang et al.’s study both pointed to the possibility of functionally redundancy to each other, it is not surprising for us to find CHCHD2 was enhanced in stable CHCHD10 knockout lines and vice versa in Huang et al.’s study. This may also explain why CHCHD2-knockout MEFs showed no obvious defects in the mitochondria cristae (12). With transient knockdown of CHCHD2 or CHCHD10 in dopaminergic cells, our results highlighted the importance of maintaining both CHCHD2 and CHCHD10 for proper mitochondria function, which also correlates with that mutations of either CHCHD2 or CHCHD10 are associated with neurological diseases in human.
MICOS maintains not only mitochondria cristae morphology but also mtDNA organization via TFAM (15,54). Loss of TFAM caused early-onset Parkinsonism and aging phenotype in mice (55). We found reduced TFAM in cells with transient knockdown of CHCHD2 and in isogenic cells carrying PD-associated CHCHD2 mutations. This suggests loss of CHCHD2 function causes impaired MICOS, leading to reduced TFAM, highlighting a positive association between CHCHD2 and TFAM. With significantly lower TFAM protein in isogenic hESCs harboring PD-associated CHCHD2 mutations, however, we did not detect any significant change in the mtDNA copy number in isogenic cells or cells with CHCHD2 knockdown. It is possible that mtDNA defects in isogenic CHCHD2 mutation cells could be obvious upon chronic stress, as shown in the previous CHCHD10 study (25). TFAM and mtDNA also drive mitochondria biogenesis. We did not detect significant change of the expression of key genes in mitobiogenesis, as NRF1 or PLOG (data not shown), suggesting CHCHD2 does not interfere mitobiogenesis.
CHCHD2 mutations were identified in autosomal dominant PD (1). Mitochondria dysfunction was observed in isogenic hESCs/NPCs harboring only one copy of mutated gene. This indicated that one copy of mutant could drive mitochondria defects, highlighting its association with autosomal dominant PD. Besides, genetic deletion harboring CHCHD2 was found in human patients with neuronal dysfunction (7,8). Isogenic cells carrying PD-associated CHCHD2 mutations had few mitochondria cristae and impaired mitochondria function, like the cells with transient CHCHD2 knockdown. CHCHD2 mutations, T61I, R145Q and Q126X, have reduced or loss of binding to CHCHD10. Collectively, we showed PD-related CHCHD2 mutations mediate a loss-of-function mechanism in various human models, consistent with the previous CHCHD2 study in the drosophila model (12).
Based on the above mechanistic studies, we further searched for potential therapy against CHCHD2-induced mitochondria dysfunction. We found a small mitochondria-targeting peptide Elamipretide that was able to attenuate CHCHD2 R145Q-caused mitochondrial defects. Elamipretide crosses the mitochondrial OM and localizes to the IM where it associates with cardiolipin, a phospholipid exclusively expressed on the inner mitochondrial membrane. Cardiolipin plays a crucial role in cristae formation, OXPHOS and mtDNA integrity (13,56). Here we demonstrated Elamipretide was able to enhance CHCHD2, MICOS components and OXPHOS enzymes in isogenic R4 (R145Q+/−) NPCs. Elamipretide attenuated mitochondria defects and enhance CHCHD2 and CHCHD10 in isogenic cells harboring heterozygous R145Q but not cells harboring homozygous R145Q, suggesting one copy of WT CHCHD2 is required for Elamipretide to protect the mitochondria. This implicated one copy of WT CHCHD2 may partially preserve cardiolipin function, allowing Elamipretide to mitigate mitochondria dysfunction induced by heterozygous CHCHD2 mutation. The founding that Elamipretide ameliorated CHCHD2 mutant-caused defects also prove CHCHD2’s role in maintaining mitochondria cristae. Of note, The US Food and Drug Administration has granted Stealth BioTherapeutics (Newton, MA) fast track designation for Elamipretide as a treatment for people with rare mitochondrial diseases (2017–2018). Considering the good safety profile and known pharmacokinetics and pharmacodynamics of Elamipretide, our study suggests that Elamipretide could be used as a potential treatment for CHCHD2-mutation related neuronal disorders. Since our study revealed a similar role of CHCHD2 and CHCHD10 in maintaining MICOS, we suggest that Elamipretide may also be beneficial for patients with the broader spectrum of neurological disorders associated with CHCHD10 mutations.
Besides PD, CHCHD2 has been associated with multiple neurological disorders such as Alzheimer’s disease, FTD and multiple system atrophy. We showed CHCHD2 loss-of-function caused distortion of mitochondria cristae, impaired MICOS complex and CHCHD10 binding, leading to mitochondria dysfunction. Our study suggested that CHCHD2 is important in maintaining proper mitochondria function, highlighting the significance of mitochondria in neurodegenerative disorders. Importantly, Elamipretide, a mitochondria-targeting peptide now in phase 3 clinical trial for mitochondria diseases, attenuated CHCHD2 R145Q-caused mitochondria dysfunction, suggesting that Elamipretide could benefit patients carrying either CHCHD2 or CHCHD10 mutation.
Materials and Methods
Genomic editing by CRISPR-Cas9 to generate isogenic hESCs
To construct gRNA for Q126X, oligos 5′-CACCGTGTTTGATCTCATAGAGGCA-3′ and 5′-AAACTGCCTCTATGAGATCAAACAC-3′ were used. To construct gRNA for R145Q, oligos 5′-GATCGATTACCTACCGTTTGCAAGTG-3′ and 5′-AAAACACTTGCAAACGGTAGGTAATC-3′ were used. Oligos were annealed and ligated with pre-digested pT7-Guide-IVT vector (Origene, Rockville, MD), followed with in vitro transcription by MeGAshortscript T7 kit (Thermo Fisher Scientific, Waltham, MA) to produce gRNA mRNA. Capped and polyadenylated Cas9 mRNA was obtained from pT7-Cas9 vector (Origene) by mMESSAGE Mmachine T7 ULTRA kit (Thermo Fisher Scientific). H9 cells were electroporated with Cas9 mRNA, gRNA mRNA and single-strand oligo donor (Q126X:5′ATGATTTTCTTTCTCAGCAATGTTGAATGTTTTGCCTTTGCAGGAGCCTCAGGGAACCCAGCCAGCACAGCAGCAGCAGCCTTGTCTTTATGAGATCAAACAGTTTCTGGAGTGTGCCTAGAACCAGGGTGACATCAAGCTCTGAGGGTTTCAATGAGGTGCTGAAACAGTTGCAAACGGTAGGTAATTTG-3′; R145Q: 5′-ATGAGATCAAACAGTTTCTGGAGTGTGCCCAGAACCAGGGTGA.
CATCAAGCTCTGTGAGGGTTTCAATGAGGTGCTGAAACAGTGCCAACTCGCAAACGGTAGGTAATTTGTCCAATTTACATCTGC-3′) by Neon Transfection System (Thermo Fisher Scientific). Cells were then re-seeded at 1 cell/well. Clones were screened after 7–10 days by sequencing (R145Q forward: 5′-TTTTTGGAGGTGGAGTCTCG-3′, reverse: 5′-TGTGCCTGGCCTATACCTTC-3′; Q126X forward: 5′-TTCTTTCTCAGCAATGTTGAATG-3′, reverse: 5′-CCTAAGTTCTACAGGATCCAGGAAT-3′).
hESC and NPC culture
hESC H9 (WiCell, Madison, WI) was cultured in mTeSR™1 medium (StemCell Technologies, Vancouver, BC, Canada) on plates coated with Geltrex™ (Thermo Fisher Scientific). H9 cells and isogenic cell lines were harvested using a scraper and cultured in suspension as embryoid bodies (EBs) for 8 days in StemPro® defined medium (Thermo Fisher Scientific) minus FGF2. EBs were cultured for additional 2–3 days in neural induction medium containing DMEM/F12 with Glutamax, non-essential amino acids (NEAAs), N2 and FGF2. Neural rosettes were formed 2–3 days later and were isolated manually using stretched glass Pasteur pipette. Rosettes were dissociated into single cells using Accutase™ (StemCell Technologies) and replated onto culture dishes to obtain a homogeneous population of NPCs. The NPCs were expanded in Neurobasal media containing NEAA, glutamine, B27 and FGF2 (57).
Cell culture, animals and reagents
SK-N-SH cells (ATCC, Manassas, VA) were maintained in Minimal Essential Medium Eagle (Sigma, St. Louis, MO) supplemented with 10% Fetal Bovine Serum (FBS) (Hyclone, Waltham, MA), NEAA and sodium pyruvate (Gibco, Waltham, MA). siRNA or plasmid transfection was mediated by TurboFect™ (Thermo Fisher Scientific). Non-tagged and C-terminal myc-tagged CHCHD2 plasmids were from Origene. T61I, R145Q and Q126X mutations of CHCHD2 were constructed using Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Human CHCHD2 siRNAs and non-targeting siRNA control were from Ambion® (Thermo Fisher Scientific). Antibodies against CHCHD2 (C-terminus epitope) was from Sigma HPA027407; most experiments were used by this antibody unless indicated otherwise. Antibody against CHCHD2 (middle region epitope, labeled as CHCHD2-TF) was from Invitrogen (Waltham, MA) PA5–23564. Antibody against CHCHD10 was from Proteintech (Rosemont, IL) 25671-1-AP. Antibody against Mitofilin (ab110329), CHCHD3 (ab99491) were from Abcam (Cambridge, MA). Antibody against CHCHD6 was from ProSci (Poway, CA) (7133). Antibody against DNA was from Progen (Heidelberg, Germany) AC-30-10. TFAM antibody was from Cell Signaling Technology (Danvers, MA) 7495S. Antibody against TH (mouse) was from Immunostar Hudson, WI. Antibody against TH (rabbit) was from Millipore, St.Louis, MO. Human brain tissue lysates were from Novus Centennial, CO. Elamipretide was from MedChemExpress Monmouth Junction, NJ. Adult (5–6 weeks old) male C57BL/6 mice (n = 3) were purchased from SingHealth Experimental Medicine Centre, Singapore and kept at 22 ± 2°C with 12 h light–dark cycle. All animal procedures and applicable regulations of animal welfare were in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines and approved by SingHealth IACUC, Singapore.
Immunoblotting and immunohistochemistry
Cell or tissue lysates were subjected to electrophoresis on 6–15% SDS-PAGE gels, and proteins were electrophoretically transferred to nitrocellulose membranes, which were blocked with 5% milk and then incubated with primary antibodies followed by Horseradish peroxidase(HRP)-conjugated secondary antibodies. Signals were detected by UVITEC Imaging system (Cleaver Scientific, Warwickshire, United Kingdom). Immunocytochemistry was performed as previously (58). In brief, mice were anaesthetized and perfused transcardially with saline, followed by 4% paraformaldehyde. Sagittal sections at 40 μm thickness were cut using a cryostat. Fixed sections were incubated in primary antibody with blocking buffer, followed by Alexa Fluor®-conjugated secondary antibodies. Sections were then mounted to glass slides. Images were acquired on a Zeiss (Oberkochen, Germany) LSM 710 confocal microscope.
Confocal and STED super-resolution microscopy
Cells were fixed by 4% paraformaldehyde and incubated with anti-CHCHD2 antibody followed by Alexa Fluro®488 conjugated anti-rabbit IgG secondary antibody (Invitrogen) and/or with anti-Mitofilin/DNA/TFAM antibody followed by Alexa Fluro®568 conjugated anti-mouse IgG secondary antibodies (Invitrogen). Conventional microscopy was performed with a Leica (Wetzlar, Germany) TCS SP8 confocal microscope. Dual-color STED images were recorded with a custom-built STED microscope, which combines two pairs of excitation and STED laser beams (597 nm, 660 nm), all derived from a single supercontinuum laser source. Using these microscopes, a resolution of 40–50 nm in the STED images was achieved. STED images were deconvoluted with Huygens Professional software (Hilversum, The Netherlands) by default setting.
Transmission electron microscope
Cells in dishes were fixed with 3% paraformaldehyde, 1% glutaraldehyde and then scraped off. Cells were collected as pellets and sealed in 2% low melting agarose, followed with osmication, dehydration in alcohols and acetone then embedded in Exopy812 resin. Sections were then cut with Leica ultracut microtome, picked up on carbon-coated copper slot grids and imaged under Jeol (Tokyo, Japan) transmission electron microscope.
Immunoprecipitation
Cells were harvested with lysis buffer containing 1% Triton. Lysates were then pre-cleared with Protein A/G Sepharose beads (Thermo Fisher Scientific). The flow through was incubated with either control IgG or specific antibodies at 4°C, followed with fresh Sepharose beads. Unspecific binding proteins were removed by washing. The immunoprecipitated complex was then subject to immunoblotting.
Measurement of mitochondria respiration capacity
Mitochondria oxygen consumption was measured using an XF24 extracellular analyzer (Seahorse Bioscience, Santa Clara, CA). Isogenic hESCs or SK-N-SH cells (40 000) transected with either CHCHD2 or negative control siRNA for 48 h were seeded in 24 well plates. Cells were changed to unbuffered DMEM and incubated at 37°C in a non-CO2 incubator for 1 h. OCR was accessed via serial injection of 1 μm Oligomycin, 1 μm FCCP and 0.5 μm Antimycin with 0.5 μm Rotenone. Every point represents an average of five different wells. Final reading for each spot was normalized with total protein content of the well.
Statistical analysis
Data were shown as mean ± standard error of the mean (SEM) of three independent experiments. All data were analyzed using a student’s t-test by GraphPad Prism 5.0 with P-values indicated in figure legends.
Supplementary Material
Acknowledgements
We thank Keshmarathy D/O Sacadevan and Wong Wei Juan at the Advanced Bioimaging Center of SingHealth for their outstanding assistance with electron microscopy and super-resolution microscopy.
Conflict of Interest statement. None declared.
Funding
Parkinson’s disease translational clinical program (NMRC/TCR/013-NNI/2014); STaR award (NMRC/STaR/014/2013 and NMRC/STaR/0030/2018 to E.-K.T.).
References
- 1. Funayama M., Ohe K., Amo T., Furuya N., Yamaguchi J., Saiki S., Li Y., Ogaki K., Ando M., Yoshino H. et al. (2015) CHCHD2 mutations in autosomal dominant late-onset Parkinson’s disease: a genome-wide linkage and sequencing study. Lancet Neurol., 14, 274–282. [DOI] [PubMed] [Google Scholar]
- 2. Jansen I.E., Bras J.M., Lesage S., Schulte C., Gibbs J.R., Nalls M.A., Brice A., Wood N.W., Morris H., Hardy J.A. et al. (2015) CHCHD2 and Parkinson’s disease. Lancet Neurol., 14, 678–679. [DOI] [PubMed] [Google Scholar]
- 3. Koschmidder E., Weissbach A., Brüggemann N., Kasten M., Klein C. and Lohmann K. (2016) A nonsense mutation in CHCHD2 in a patient with Parkinson disease. Neurology, 86, 577–579. [DOI] [PubMed] [Google Scholar]
- 4. Nicoletti G., Gagliardi M., Procopio R., Iannello G., Morelli M., Annesi G. and Quattrone A. (2018) A new CHCHD2 mutation identified in a southern Italy patient with multiple system atrophy. Parkinsonism Relat. Disord., 47, 91–93. [DOI] [PubMed] [Google Scholar]
- 5. Liu X., Jiao B., Zhang W., Xiao T., Hou L., Pan C., Tang B. and Shen L. (2018) Identification of CHCHD2 mutations in patients with Alzheimer’s disease, amyotrophic lateral sclerosis and frontotemporal dementia in China. Mol. Med. Rep., 18, 461–466. [DOI] [PubMed] [Google Scholar]
- 6. Che X.Q., Zhao Q.H., Huang Y., Li X., Ren R.J., Chen S.D., Guo Q.H. and Wang G. (2018) Mutation screening of the CHCHD2 gene for Alzheimer’s disease and frontotemporal dementia in Chinese mainland population. J. Alzheimers Dis., 61, 1283–1288. [DOI] [PubMed] [Google Scholar]
- 7. Varvagiannis K., Papoulidis I., Koromila T., Kefalas K., Ziegler M., Liehr T., Petersen M.B., Gyftodimou Y. and Manolakos E. (2014) De novo 393 kb microdeletion of 7p11.2 characterized by aCGH in a boy with psychomotor retardation and dysmorphic features. Meta Gene, 2, 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coulter M.E., Miller D.T., Harris D.J., Hawley P., Picker J., Roberts A.E., Sobeih M.M. and Irons M. (2011) Chromosomal microarray testing influences medical management. Genet. Med., 13, 770–776. [DOI] [PubMed] [Google Scholar]
- 9. Aras S., Bai M., Lee I., Springett R., Hüttemann M. and Grossman L.I. (2015) MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion, 20, 43–51. [DOI] [PubMed] [Google Scholar]
- 10. Baughman J.M., Nilsson R., Gohil V.M., Arlow D.H., Gauhar Z. and Mootha V.K. (2009) A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet., 5, e1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y., Clegg H.V., Leslie P.L., Di J., Tollini L.A., He Y., Kim T.H., Jin A., Graves L.M., Zheng J. and Zhang Y. (2015) CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ., 22, 1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meng H., Yamashita C., Shiba-Fukushima K., Inoshita T., Funayama M., Sato S., Hatta T., Natsume T., Umitsu M., Takagi J. et al. (2017) Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat. Commun., 8, 15500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikon N. and Ryan R.O. (2017) Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta., 1859, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wollweber F., Malsburg K. and Laan M. (2017) Mitochondrial contact site and cristae organizing system: a central player in membrane shaping and crosstalk. Biochim. Biophys. Acta., 1864, 1481–1489. [DOI] [PubMed] [Google Scholar]
- 15. Li H., Ruan Y., Zhang K., Jian F., Hu C., Miao L., Gong L., Sun L., Zhang X., Chen S. et al. (2016) Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ., 23, 380–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guarani V., Jardel C., Chrétien D., Lombès A., Bénit P., Labasse C., Lacène E., Bourillon A., Imbard A., Benoist J.F. et al. (2016) QIL1 mutation causes MICOS disassembly and early onset fatal mitochondrial encephalopathy with liver disease. Elife, 5, e17163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harner M.E., Unger A.K., Izawa T., Walther D.M., Ozbalci C., Geimer S., Reggiori F., Brügger B., Mann M., Westermann B. and Neupert W. (2014) Aim24 and MICOS modulate respiratory function, tafazzin-related cardiolipin modification and mitochondrial architecture. Elife, 3, e01684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guarani V., McNeill E.M., Paulo J.A., Huttlin E.L., Fröhlich F., Gygi S.P., Van Vactor D. and Harper J.W. (2015) QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. Elife, 4, e06265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ott C., Ross K., Straub S., Thiede B., Götz M., Goosmann C., Krischke M., Mueller M.J., Krohne G., Rudel T. and Kozjak-Pavlovic V. (2012) Sam50 functions in mitochondrial intermembrane space bridging and biogenesis of respiratory complexes. Mol. Cell. Biol., 32, 1173–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bohnert M., Zerbes R.M., Davies K.M., Mühleip A.W., Rampelt H., Horvath S.E., Boenke T., Kram A., Perschil I., Veenhuis M. et al. (2015) Central role of Mic10 in the mitochondrial contact site and cristae organizing system. Cell Metab., 21, 747–755. [DOI] [PubMed] [Google Scholar]
- 21. Hoppins S., Collins S.R., Cassidy-Stone A., Hummel E., Devay R.M., Lackner L.L., Westermann B., Schuldiner M., Weissman J.S. and Nunnari J. (2011) A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria. J. Cell Biol., 195, 323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laan M., Horvath S.E. and Pfanner N. (2016) Mitochondrial contact site and cristae organizing system. Curr. Opin. Cell Biol., 41, 33–42. [DOI] [PubMed] [Google Scholar]
- 23. Kozjak-Pavlovic V. (2017) The MICOS complex of human mitochondria. Cell Tissue Res., 367, 83–93. [DOI] [PubMed] [Google Scholar]
- 24. Rampelt H., Zerbes R.M., Laan M. and Pfanner N. (2017) Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta., 1864, 737–746. [DOI] [PubMed] [Google Scholar]
- 25. Genin E.C., Plutino M., Bannwarth S., Villa E., Cisneros-Barroso E., Roy M., Ortega-Vila B., Fragaki K., Lespinasse F., Pinero-Martos E. et al. (2016) CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med., 8, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bannwarth S., Plutino M., Bannwarth S., Villa E., Cisneros-Barroso E., Roy M., Ortega-Vila B., Fragaki K., Lespinasse F., Pinero-Martos E. et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain, 137, 2329–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chaussenot A., Le Ber I., Ait-El-Mkadem S., Camuzat A., Septenville A., Bannwarth S., Genin E.C., Serre V., Augé G., French research network on FTD and FTD-ALS et al. (2014) Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging, 35, 2884.e1–4. [DOI] [PubMed] [Google Scholar]
- 28. Johnson J.O., Glynn S.M., Gibbs J.R., Nalls M.A., Sabatelli M., Restagno G., Drory V.E., Chiò A., Rogaeva E. and Traynor B.J. (2014) Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain, 137, e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chio A., Mora G., Sabatelli M., Caponnetto C., Traynor B.J., Johnson J.O., Nalls M.A., Calvo A., Moglia C., Borghero G. et al. (2015) CHCHD10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging, 36, 1767.e3–1767.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kurzwelly D., Krüger S., Biskup S. and Heneka M.T. (2015) A distinct clinical phenotype in a German kindred with motor neuron disease carrying a CHCHD10 mutation. Brain, 138, e376. [DOI] [PubMed] [Google Scholar]
- 31. Ronchi D., Riboldi G., Del Bo R., Ticozzi N., Scarlato M., Galimberti D., Corti S., Silani V., Bresolin N. and Comi G.P. (2015) CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain, 138, e372. [DOI] [PubMed] [Google Scholar]
- 32. Zhang M., Xi Z., Zinman L., Bruni A.C., Maletta R.G., Curcio S.A., Rainero I., Rubino E., Pinessi L., Nacmias B. et al. (2015) Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain, 138, e380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Penttila S., Jokela M., Bouquin H., Saukkonen A.M., Toivanen J. and Udd B. (2015) Late onset spinal motor neuronopathy is caused by mutation in CHCHD10. Ann. Neurol., 77, 163–172. [DOI] [PubMed] [Google Scholar]
- 34. Auranen M., Ylikallio E., Shcherbii M., Paetau A., Kiuru-Enari S., Toppila J.P. and Tyynismaa H. (2015) CHCHD10 variant p.(Gly66Val) causes axonal Charcot–Marie–Tooth disease. Neurol. Genet., 1, e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Straub I.R., Janer A., Weraarpachai W., Zinman L., Robertson J., Rogaeva E. and Shoubridge E.A. (2018) Loss of CHCHD10–CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet., 27, 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Burstein S.R., Valsecchi F., Kawamata H., Bourens M., Zeng R., Zuberi A., Milner T.A., Cloonan S.M., Lutz C., Barrientos A. and Manfredi G. (2018) In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet., 27, 160–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang X., Wu B.P., Nguyen D., Liu Y.T., Marani M., Hench J., Bénit P., Kozjak-Pavlovic V., Rustin P., Frank S. and Narendra D.P. (2018) CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet., 27, 3881–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Genin E.C., Bannwarth S., Lespinasse F., Ortega-Vila B., Fragaki K., Itoh K., Villa E., Lacas-Gervais S., Jokela M., Auranen M. et al. (2018) Loss of MICOS complex integrity and mitochondrial damage, but not TDP-43 mitochondrial localisation, are likely associated with severity of CHCHD10-related diseases. Neurobiol. Dis., 119, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Avior Y., Sagi I. and Benvenisty N. (2016) Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol., 17, 170–182. [DOI] [PubMed] [Google Scholar]
- 40. Liang X., Potter J., Kumar S., Zou Y., Quintanilla R., Sridharan M., Carte J., Chen W., Roark N., Ranganathan S. et al. (2015) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol., 208, 44–53. [DOI] [PubMed] [Google Scholar]
- 41. Jans D.C., Wurm C.A., Riedel D., Wenzel D., Stagge F., Deckers M., Rehling P. and Jakobs S. (2013) STED super-resolution microscopy reveals an array of MINOS clusters along human mitochondria. Proc. Natl. Acad. Sci. U. S. A., 110, 8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lorenz C., Lesimple P., Bukowiecki R., Zink A., Inak G., Mlody B., Singh M., Semtner M., Mah N., Auré K. et al. (2017) Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell, 20, 659–674. [DOI] [PubMed] [Google Scholar]
- 43. Yang R.F., Zhao G.W., Liang S.T., Zhang Y., Sun L.H., Chen H.Z. and Liu D.P. (2012) Mitofilin regulates cytochrome c release during apoptosis by controlling mitochondrial cristae remodeling. Biochem. Biophys. Res. Commun., 428, 93–98. [DOI] [PubMed] [Google Scholar]
- 44. Zhao K., Zhao G.M., Wu D., Soong Y., Birk A.V., Schiller P.W. and Szeto H.H. (2004) Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem., 279, 34682–34690. [DOI] [PubMed] [Google Scholar]
- 45. Birk A.V., Liu S., Soong Y., Mills W., Singh P., Warren J.D., Seshan S.V., Pardee J.D. and Szeto H.H. (2013) The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol., 24, 1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karaa A., Haas R., Goldstein A., Vockley J., Weaver W.D. and Cohen B.H. (2018) Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology, 90, e1212–e1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Daubert M.A., Yow E., Dunn G., Marchev S., Barnhart H., Douglas P.S., O’Connor C., Goldstein S., Udelson J.E. and Sabbah H.N. (2017) Novel mitochondria-targeting peptide in heart failure treatment: a randomized, placebo-controlled trial of elamipretide. Circ. Heart Fail., 10, e004389. [DOI] [PubMed] [Google Scholar]
- 48. John G.B., Shang Y., Li L., Renken C., Mannella C.A., Selker J.M., Rangell L., Bennett M.J. and Zha J. (2005) The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol. Biol. Cell., 16, 1543–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ding C., Wu Z., Huang L., Wang Y., Xue J., Chen S., Deng Z., Wang L., Song Z. and Chen S. (2015) Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci. Rep., 5, 16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Floyd B.J., Wilkerson E.M., Veling M.T., Minogue C.E., Xia C., Beebe E.T., Wrobel R.L., Cho H., Kremer L.S., Alston C.L. et al. (2016) Mitochondrial protein interaction mapping identifies regulators of respiratory chain function. Mol. Cell, 63, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Purandare N., Somayajulu M., Hüttemann M., Grossman L.I. and Aras S. (2018) The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem., 293, 6517–6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barabasi A.L. and Oltvai Z.N. (2004) Network biology: understanding the cell’s functional organization. Nat. Rev. Genet., 5, 101–113. [DOI] [PubMed] [Google Scholar]
- 53. El-Brolosy M.A. and Stainier D.Y.R. (2017) Genetic compensation: a phenomenon in search of mechanisms. PLoS Genet., 13, e1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang R.F., Sun L.H., Zhang R., Zhang Y., Luo Y.X., Zheng W., Zhang Z.Q., Chen H.Z. and Liu D.P. (2015) Suppression of Mic60 compromises mitochondrial transcription and oxidative phosphorylation. Sci. Rep., 5, 7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ekstrand M.I., Terzioglu M., Galter D., Zhu S., Hofstetter C., Lindqvist E., Thams S., Bergstrand A., Hansson F.S., Trifunovic A. et al. (2007) Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. U. S. A., 104, 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kameoka S., Adachi Y., Okamoto K., Iijima M. and Sesaki H. (2018) Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol., 28, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M. and Studer L. (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol., 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ma D., Yoon S.I., Yang C.H., Marcy G., Zhao N., Leong W.Y., Ganapathy V., Han J., Van Dongen A.M., Hsu K.S. et al. (2015) Rescue of methyl-CpG binding protein 2 dysfunction-induced defects in newborn neurons by pentobarbital. Neurotherapeutics, 12, 477–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.