

Abstract
Adaptive responses that counter starvation have evolved over millennia to permit organismal survival, including changes at the level of individual organelles, cells, tissues, and organ systems. In the past century, a shift has occurred away from disease caused by insufficient nutrient supply towards over-nutrition, leading to obesity and diabetes, atherosclerosis, and cardiometabolic disease. The burden of these diseases has spurred interest in fasting strategies that harness physiological responses to starvation, thus limiting tissue injury during metabolic stress. Insights gained from animal and human studies suggest that intermittent fasting and chronic caloric restriction extend lifespan, decrease risk factors for cardio-metabolic and inflammatory disease, limit tissue injury during myocardial stress, and activate a cardioprotective metabolic program. Acute fasting activates autophagy, an intricately orchestrated lysosomal degradative process that sequesters cellular constituents for degradation, and is critical for cardiac homeostasis during fasting. Lysosomes are dynamic cellular organelles that function as incinerators to permit autophagy, as well as degradation of extracellular material internalized by endocytosis, macropinocytosis and phagocytosis. The last decade has witnessed an explosion of knowledge that has shaped our understanding of lysosomes as central regulators of cellular metabolism and the fasting response. Intriguingly, lysosomes also store nutrients for release during starvation; and function as a nutrient sensing organelle to couple activation of mTOR to nutrient availability. This article reviews the evidence for how the lysosome, in the guise of a janitor, may be the ‘undercover boss’ directing cellular processes for beneficial effects of intermittent fasting and restoring homeostasis during feast and famine.
Introduction
Diseases affecting cardiac function are the most common cause of mortality and morbidity in the United States today (237). The transition from a more frugal, intermittent food supply to nutrient over-supply has resulted in an epidemic of cardiometabolic disease, including increased obesity rates, an epidemic of diabetes, non-alcoholic fatty liver disease, and atherosclerotic vascular disease; culminating in myocardial infarction, ischemic heart disease, and heart failure. Whether dietary strategies can prevent or even reverse cardiometabolic disease has profound implications for public health. In particular, strategies that emphasize fasting, or various regimens of abstinence from food, have emerged as a way to prevent or even reverse cardiometabolic disease (39, 72, 119, 205).
The heart is a unique organ, consuming 10% of the body’s total oxygen uptake, in an effort to pump 7,200 liters of blood each day in a pulsatile fashion to maintain survival (215). Even small interruptions or decline in functional capacity have lethal consequences. The ability of the myocardium to adapt metabolically allows preservation of cardiac function during physiological stress. In fact, intracellular ATP levels are maintained at a constant level, with production and utilization of 35 kg of ATP over a 24 hour period (17). Under homeostatic conditions, β-oxidation of fatty acids is the main source of myocardial ATP, whereas the heart can shift substrate utilization towards more abundant energy sources as they become available. For example, during starvation or in conditions of end-stage heart failure, the myocardium turns to metabolism of ketone bodies (16, 22). Furthermore, the activation of compensatory, evolutionarily conserved pathways such as autophagy and mitophagy may prevent deleterious changes in the cardiac myocytes, as well as the onset of cardiac dysfunction (50).
Nutrient deprivation has been a dominant evolutionary pressure from primordial life forms to humans. While cells with high replicative potential can dynamically alter cellular number to match nutrient supply and functional demands, terminally differentiated post-replicative cells (i.e. cardiac myocytes and neurons) cannot. In both the brain and the heart, each cell serves a critical function and is treated as an ultra-long-lived non-replaceable unit. Therefore, each cell requires mechanisms to ensure survival and integrated adaptive mechanisms that combat metabolic stress. For instance, in cardiac myocytes, under continually varying nutrient supply and contractile loading, maintenance of a continuous unchanging ATP supply as well as optimal sarcomeric function is critical to driving each successful heartbeat to sustain life. In this model, each cell needs to be able to independently respond to cues in the external milieu to maintain optimal function without disruption of the functional cardiac syncytium. The lysosomes play a central role in integrating these signals with organelle and sarcomere quality control in cardiac myocytes, while maintaining the balance between nutrient excess and nutrient deficiency in physiology.
The lysosomal nutrient sensing complex couples nutrient supply to mTOR (mammalian target of rapamycin) activation, and is critical in both controlling metabolic decision making to ensure energy supply while simultaneously remodeling organelles and cellular architecture to provide optimal function (116). mTOR, a serine-threonine kinase, is the master regulator of protein synthesis to drive anabolic growth and hypertrophy, and is critical for embryonic cardiac growth and post-natal development (180). Nonetheless, sustained alterations in cardiac metabolism can lead to prolonged activation of mTOR with adverse consequences (reviewed in Sciarretta et al (180)). While unopposed stimulation of the LYNUS (Lysosomal Nutrient Sensing complex)-mTOR axis can provoke contractile dysfunction in cardiometabolic disease states (182, 198, 250), accumulating experimental evidence from our lab and others indicates that cyclic activation and inactivation of this pathway during periods of feeding and fasting may restore cellular homeostasis, in a manner that depends on intact autophagy and lysosomal function (72) (13, 119), Indeed, studies from C. elegans to humans support the notion that fasting strategies prolong lifespan, prevent life-threatening illnesses such as cancer, stimulate cellular regeneration and enhance the ability of organisms to endure subsequent stress (25, 39, 76, 88, 229, 240). In addition, markers of aging and senescence also appear to decrease in models of intermittent fasting (25). In contrast to most pharmacological and/or genetic approaches to disease management, intermittent fasting offers a novel, and well tolerated and sustainable strategy to improve outcomes in cardiac disease as well as increase cardiac health. In addition, the salutary benefits with regards to systemic metabolic changes may play a significant role in the pathogenesis of many cardiac diseases.
From the original discovery of the lysosome as a membrane-limited bag of hydrolytic enzymes by Christian de Duve in the early 1950’s (174) to beginning of the 21st century, our understanding of the role of lysosomes was largely informed by monogenic disorders that led to deficiency of individual lysosomal enzymes or proteins (62). Collectively termed lysosomal storage diseases (LSDs), these disorders taken together are among the most common genetic diseases in children with a prevalence of ~1 in 5000–8000 births, second only to cystic fibrosis (104, 132). Since lysosomes are ubiquitous organelles, LSDs affect multiple organ systems. Notably, the central nervous system (CNS) and the heart, two highly metabolically active organs, are affected in the overwhelming majority of patients, resulting in severe impairments and early death (163). Following the discovery of TFEB (177), and related proteins, TFE3 and MiTF (all members of the MiT/TFE3 family of transcriptional activators) as master regulators of the lysosome biogenesis program, our understanding of the role of lysosomes in homeostasis has evolved dramatically. Coupled with the emerging evidence that lysosome function is impaired in a wide range of diseases, such as myocardial infarction, heart failure, neurodegeneration and metabolic disorders (114, 123, 188), these discoveries have catapulted the role of ‘acquired lysosome dysfunction’ in the spotlight as a underlying mechanism for common diseases. Ongoing work in our program and other laboratories is actively investigating strategies to therapeutically target lysosome function in highly prevalent cardio-metabolic (72, 119, 185, 204) and neurodegenerative diseases (164, 238, 239).
In this review, we discuss cardiac physiology in the context of fasting and discuss our rapidly advancing understanding of the role of the lysosome as the critical cellular organelle in maintaining cardiac myocyte metabolism. We will also explore the short and long term intracellular homeostatic changes that link nutrient deprivation to salutary cellular processes coordinated by lysosomes. While these mechanistic studies have largely been conducted in non-cardiac myocytes, the pathways uncovered are of broad relevance to cellular metabolism in the heart as well as in other tissues, and therefore the focus of intense investigation in cardiovascular biology and pathophysiology. Finally, we review the current animal and clinical models of intermittent fasting and caloric restriction, with a critical analysis of potential clinical benefits. Please see Table 1 for definitions of various terms used frequently throughout this manuscript.
Table 1:
Definitions for terminology employed in the article
| Number | Term | Definition |
|---|---|---|
| 1. | Calorie restriction (CR) | Dietary modification to limit caloric intake, typically without affecting basal homeostatic requirements, in vivo. |
| 2. | Starvation | Complete or partial deprivation of nutrients (individually or in combination) to below levels required to sustain homeostasis, in vitro. While, unfortunately, observed in vivo in nature and human society, it cannot be studied in an ethical manner in the laboratory. |
| 3. | Fasting | Dietary modification to limit overall dietary intake, often to zero for a defined period of time. Differentiated from starvation by an absence of an overall impact on homeostasis. |
| 4. | Intermittent fasting | Periods of fasting interspersed with ad-lib access to food. |
| 5. | Time-restricted feeding | Restriction of food intake to specific time periods during a 24 hours cycle. |
| 6. | Efficiency | Efficiency is defined as the ratio of useful work over the energy input. In terms of cardiac metabolism, this is defined as the rate of substrate utilization (i.e. number of molecules of ATP generated per carbon atom metabolized). |
| 7. | Autophagy | Evolutionarily conserved and vital intracellular process to reuse, repurpose and renew cellular resources via creation of double-membraned vesicles to engulf and subsequently, digest intracellular organelles, large proteins and structures to increase availability of nutrients for energy generation and protein synthesis; and recycle intracellular constituents. |
| 8. | Autophagosome | Double membrane bound vesicles, created by activation of specific proteins binding to donor membranes from intracellular organelles. These engulf large intracellular proteins and structures for autophagy as defined above. |
| 9. | Lysosome | Single membrane bound intracellular vesicles formed by enrichment of endosomes with hydrolytic enzymes thus creating a contained intracellular digestion site for lipids, proteins and carbohydrates. These ancient organelles are capable of acting as nutrient sensors as well as regulating intracellular metabolism, both by regulating nutrient supply as well as control of metabolic gene expression. Lysosomes also integrate multiple cellular processes such as autophagy, endocytosis, macropinocytosis, phagocytosis and exocytosis. |
| 10. | LYNUS | Lysosomal Nutrient Sensing machinery consists of a complex lysosomal membrane bound multiprotein structure on the consisting of mTORC1 complex, Ragulator and the V-ATPase channel as well as several regulatory proteins that can be altered by changes in lysosomal flux of specific metabolites eventually resulting in regulation of lysosomal metabolic responses. |
| 11. | Mitophagy | Targeted degradation of damaged and dysfunctional mitochondria following ubiquitination and subsequent trafficking via binding to adaptor proteins into autophagosomes and, subsequently, lysosomes. |
Cardiac energetics, substrate metabolism and pathophysiology under fasting states.
Cardiac myocytes are highly durable cells that mechanically shorten and relax ~3 billion times over the lifetime in a human with the average life expectancy of 80 years, and dynamically adapt to a variety of stresses over this period. A variety of processes confer this stress resistance. Innately, cardiac myocytes are rich in mitochondria, the powerhouses of the cell that generate energy, as well as the sarcoplasmic reticulum and peroxisomes that control protein quality and lipid metabolism, respectively. They also have storage reserves of energy rich compounds, such as lipid droplets and glycogen that are able to replenish ATP in the face of complete nutrient and oxygen deprivation. Contemporary work has focused upon the role of lysosomes in ensuring nutrient supply as well as maintaining organelle quality to counter stress. This section summarizes the state of our knowledge of cardiac metabolism and highlights the role of lysosomal pathways, therein.
Cardiac energetics
Generation of ATP is critical to all cellular processes. Cardiac myocytes have extremely high energy requirements as a result of the unique properties of excitation-contraction coupling, metabolism and transmembrane ionic fluxes that are key to generation of membrane potential and automaticity. As the ability of cardiac myocytes to store ATP is limited, metabolic processes result in near complete turnover of the entire myocardial ATP pool every 10 seconds (reviewed in (215)). During aerobic conditions, >90% of myocardial ATP production occurs from oxidative phosphorylation resulting from production of reducing equivalents by cytosolic glycolysis, as well as mitochondrial β-oxidation of fatty acids, the tricarboxylic acid cycle and pyruvate oxidation (206, 213). Cardiac myocytes utilize glucose, fatty acids and ketone bodies for normal aerobic respiration. Of these, while the greatest efficiency appears to be oxidation of glucose, fatty acids form the bulk of the substrates utilized for physiologic energy generation (213). However, the ratio of fatty acid to glucose oxidation as a source of ATP production is variable and depends on a number of regulators and disease states. Furthermore, fatty acid and glucose oxidation undergo inverse regulation, with increased functioning of one resulting in decrease in the other pathway (213). In physiology, under fasting conditions, fatty acids and ketone bodies are the primary source for energy generation while circulating glucose and insulin levels are low.
Fat metabolism
Roughly ~70% of cardiac myocyte ATP production is sourced from fatty acid β-oxidation (215). Substrate utilization appears to occur in a competitive fashion with a reciprocal relationship between fatty acids and glucose. Free fatty acids are transported in from extracellular sources via specific cell membrane transporter proteins, CD36 and the fatty acid binding protein (FABP) (102). Free fatty acid (FFA) uptake via CD36 can be regulated via PPAR α/β/γ agonists, insulin stimulation and β-adrenoreceptor antagonists. Once in the cytosol, a coenzyme A (CoA) group is added by fatty acyl coA synthetase (ACSL1) (102). Thus activated, the carnitine palmitoyl tranferase1 (CPT1) and the carnitine translocase transport the fatty acyl CoA to the mitochondrial matrix (63). The conjugated fatty acids are then oxidized via fatty acid oxidation as well as the TCA cycle to produce reactive species to drive the electron transport chain and finally the F0/F1 ATPase resulting in energy production. 5’-AMP activated protein kinase (AMPK), which also plays a role in other nutrient sensing metabolic pathway, indirectly controls fatty acid oxidation via regulating production of malonyl CoA (12). In addition to FFA transported into the cell, intracellular lipid stores in fat droplets formed from stored fatty acids (in the form of triacylglycerols (TAG)) can undergo lipolysis and represent a flux to maintain constant FFA supply in the setting of varying external sources. An important consideration here is the concept of ‘efficiency’ of energy generation (214). In contrast to glucose which requires less oxygen (6 moles of oxygen for a net gain of 38 high energy phosphate bonds resulting in 6.3 high energy phosphate bonds per mole of oxygen), oxidative phosphorylation of fatty acids is less efficient and requires more oxygen (for example, 129 high energy phosphate bonds per 23 moles of oxygen resulting in 5.6 high energy phosphate bonds per mole of oxygen) (92). Also, unlike glucose and pyruvic acid, free fatty acids are known to promote actions of mitochondrial uncoupling proteins that can reduce the efficiency of ATP production (112). Interestingly, although glucose metabolism is more efficient for ATP production, a single fatty acid molecule subjected to β-oxidation results in multiple acetyl CoA moieties, thus overcoming the energetic cost of fatty acid oxidation in comparison to glucose (106). The effect of reduced efficiency of fatty acid metabolism in cardiac myocytes is best highlighted by the decreased cardiac function noted in animal models of a high fat diet (153). In these, increased CD36 translocation to the sarcolemma drives increased fatty acid oxidation, and appears to result in a cardiomyopathy (reviewed in (71)). Inhibition of the CD36 translocation rescues this cardiomyopathy (244). Conversely, FFAs may play a role in augmenting their own absorption as well as β-oxidation. Work in myocytes indicates that the CD36 molecule is located in lipid rafts in complex with AMPK kinase LKB1 (liver kinase b1) and a src kinase (FYN) (176). When exposed to fatty acids, Fyn is dissociated from this complex resulting in AMPK phosphorylation by LKB1 and resultant activation. It is interesting to speculate that AMPK activation triggers autophagic degradation of lipids in cardiac myocytes during fasting (99, 199) as has been shown for other tissues; which is coordinated by transcriptional induction of the lysosome biogenesis program (187) (vide infra for details).
Lysosomal processing of triglycerides and cholesteryl esters by lysosomal acid lipase plays a critical role in homeostatic lipid metabolism in the body, as its deficiency results in Wolman’s disease as well as cholesteryl ester storage disease (210). The role of lysosomal acid lipase in cardiac metabolism in the resting state and during fasting stress remains to be explored. It is also intriguing to note that while fatty acids are the dominant source for energy generation in cardiac myocytes during fasting, the rapid rate of influx of fatty acids exceeds their utilization resulting in increased lipid storage and myocardial lipid accumulation (234); suggesting that availability of lipid stores is not the rate-limiting step for fatty acid oxidation in cardiac myocytes during fasting. In contrast, during the fed state, with energetic sufficiency, absorbed fatty acids as well as by products of fatty acid synthesis are largely conjugated to form triacylglycerols for membrane synthesis as well as storage. Interestingly, de novo synthesis of fatty acids via fatty acid synthase (FASN) is also important for homeostatic maintenance of cardiac function with aging, and protection against pressure overload stress (167).
Carbohydrate metabolism
In contrast to fatty acids, glucose forms a more efficient source of energy production in cardiac myocytes. Glucose transporters on the cell surface translocate from cytosol to the sarcolemma to facilitate glucose absorption. GLUT1 and GLUT4 are the dominant glucose transporters in the heart, with GLUT1 localized to the sarcolemma - thus carrying out basal absorption of glucose , while the intracellular vesicle bound GLUT4 being the dominant transporter following trafficking to the cell membrane and facilitating insulin driven glucose absorption in the fed state(reviewed in PMID 26756635 ). Once intracellular, glucose is trapped in the cytosol by hexokinase (HK) mediated phosphorylation (HK1 in prenatal and HK2 in adult life). Upregulation of the glucose transporter GLUT1 during myocardial hypertrophy results in increased glycolytic activity and resultant changes in myocardial efficiency. GLUT4 is normally located on endocytic vesicles that are trafficked to the sarcolemma by AMPK activation via the Akt substrate protein (AS160) (190). Along with the increased trafficking to the sarcolemma, AMPK activation appears to decrease cycling, thus increasing the GLUT4 density on the cell surface in order to maximize glucose uptake. AMPK also inhibits glycogen synthase activation, both reducing ATP consumption as well as increasing substrate availability for glucose oxidation (190) Cytosolic glycolysis results in generation of pyruvic acid or lactic acid (92). It also results in both utilization as well as generation of high energy phosphates in the cytosol as well as reducing equivalents for oxidative phosphorylation. The rate limiting step of glycolysis is phosphofructokinase-1 (PFK1) activation. This critical step is indirectly regulated by AMPK directed phosphofructokinase-2 which produces fructose 2, 6–bis-phosphate that is an allosteric stimulant of PFK1. Endogenous substrates in cardiac myocytes are stored in the form of glycogen and triacylglycerol. Significant cardiac reserves of glycogen (branched polymer of glucose with a-1,4 and a-1,6-glycosidic linkages with up to 55,000 residues) are noted (reviewed in (3)). During states of ATP deficiency, glycogenolysis takes place in the cytosol as well as the lysosome. The latter appears to play a role in postnatal cardiac function. Lysosomal alpha-glucosidase deficiency (Pompe disease (GSD III)) results in reduced autophagic flux with presence of persistent glycogen containing vacuoles with appearance of a fatal cardiomyopathy (particularly in the infantile form of the disease) (reviewed in (3)). Analogously, mutations in LAMP2, a lysosomal membrane protein result in hypertrophic cardiomyopathy, heart failure necessitating cardiac transplantation and premature death; and manifest with accumulation of glycogen in the myocardium (147, 218). Abnormal glycogen accumulation is also observed in other lysosome disorders such as Fabry’s disease, wherein the characteristic hypertrophic cardiomyopathy phenotype mimics the cardiac pathology observed in humans with mutations in non-lysosomal metabolic pathways resulting in glycogen accumulation in the heart (11, 37). Recent studies indicate that cardiac glycogen breakdown by autophagy (termed ‘glycophagy’) is an important contributor to sex-specific differences in myocardial glycogen content (168), the pathophysiologic ramifications of which remain to be experimentally determined.
Pyruvate metabolism
Pyruvate is a critical metabolite at the intersection of multiple substrate utilization pathways (92). Glycolysis is an important source of cytosolic pyruvate via pyruvate kinase-catalyzed dephosphorylation of phosphoenolpyruvate into pyruvate during the final, irreversible step of glycolysis. One molecule of glucose is broken down into two molecules of pyruvate and two ATP molecules via glycolysis. Pyruvate in transported into the mitochondria under aerobic conditions via a mitochondrial pyruvate carrier, where the pyruvate dehydrogenase (PDH) complex catalyzes its oxidative de-carboxylation yielding acetyl-CoA. Acetyl-CoA is metabolized through the TCA cycle to yield ATP or is shuttled into anabolic production of fatty acids, cholesterol and acetylcholine. Another fate of pyruvate is carboxylation to oxaloacetate by pyruvate carboxylase or to malate by malic enzyme; and pyruvate acts as an anaplerotic substrate for the Krebs cycle via this mechanism. Based upon the reduced efficiency observed with fatty acid oxidation in cardiomyopathy and heart failure, pharmacologic stimulation of the pyruvate dehydrogenase complex has been explored as a therapeutic option to increase to stimulate glucose oxidation with demonstrable benefits. By contrast the physiologic metabolism in the normal heart is very plastic and rapidly adapts to chronic pyruvate dehydrogenase inhibition via upregulation of fatty acid metabolism (36). Pyruvate is predominantly converted reversibly to lactate by lactate dehydrogenase, which is abundantly generated by the skeletal muscle under anerobic conditions (such as with exercise) and utilized by the heart (92). Lactate metabolism in cancer cells has been implicated in controlling lysosomal acidification via generation of protons (28). The interplay between pyruvate metabolism and lysosome function in the heart remains to be determined.
Ketone body metabolism
The heart is the single largest consumer of ketone bodies (215). The ability of the heart to use multiple energy substrates is well illustrated by ketone body oxidation. By switching to ketone body utilization at the expense of fatty acid oxidation and glucose oxidation, during nutrient and hemodynamic stress, cardiac myocytes are able to take advantage of greater metabolic efficiency. During starvation as well as when supplied high ketone diets, use of ketone bodies also has salutary roles with regards to reducing the oxidative stress engendered by metabolites of fatty acid oxidation as well as maximizing energy extraction. Ketone body utilization is an adaptive phenomenon and both in acute and long term use have no negative implications with regards to cardiac contractile function. Ketone bodies are produced in the liver from surplus acetylCoA from fatty acid β-oxidation. Circulating as acetates, acetoacetates and β-hydroxybutyrate, ketone bodies are produced by the liver during states of glucose deprivation, impaired insulin signaling and fatty acid abundance. Interestingly, autophagy and lysosome function are essential for formation of ketone bides in the liver and kidney during fasting (217). They are absorbed by cardiac myocytes via monocarboxylate transporters (MCT1/Slc16A1) and undergo ketone oxidation with a fatty acid and glucose sparing effect. Transport of ketone bodies via MCT1/Slc16a1 across the plasma membrane appears to be affected by changes in the NADH/NAD+ ratio (reviewed in (80)). Once intracellular, oxidation by β-hydoxy butyrate dehydrogenase −1 (BDH1) results in formation of acetoacetate. Subsequently, the enzyme succinyl-CoA:3-oxoacid-CoA transferase (SCOT) is a key step in generation of acetyl CoA to drive the TCA. By themselves, ketone bodies also appear to target the metabolism and cardiac function via changes in nuclear transcription by acting as inhibitors of HDAC1, resulting in protection from oxidative stress (192). Mice fed ketogenic diets also show decreases in mTOR activity with increases in Sirt1 and HIF-1 as well as AMPK activity (145, 171). Besides states of nutritional deprivation, ketone bodies appear to play a significant role in cardiac metabolism in the post natal remodeling (43) as well as in models of heart failure (16, 22). Ketone bodies also drive acetylation reactions, which may explain its benefits on extending lifespan and improving healthspan seen when mice are maintained on a ketogenic diet chronically (171) or intermittently (145). In addition, among TCA cycle intermediates, oxaloacetate, and a-ketoglutarate appear to play roles outside their role in mitochondrial acetyl CoA/TCA metabolism. By inhibition of the F1F0 ATP synthase, lifespan in C. elegans is increased with decreased O2 consumption, which resulted in mTOR inhibition and increased autophagy (41). Intriguingly, long term persistent administration of ketogenic diet resulted in impaired glucose tolerance (171) and reduced beta cell mass in mice (59), an area of concern that will need to be examined with intermittent ketogenic diet feeding protocols (145) to evaluate its safety for clinical application in humans, long term.
Amino acid metabolism
Amino acids appear to play an important role in regulation of cardiac myocyte metabolism (209) and, in some cases, serve as direct substrates to yield energy. As a unique source of non-oxidative metabolism, with a low potential for intracellular acidification, amino acids play a role in energy production during states of nutrient and oxygen deprivation (215). Amino acids also play a significant role in generation of glucose (gluconeogenesis), as well as in synthesis of acetyl CoA which can be utilized in energy generation, fatty acid synthesis and ketone body generation (92). Hepatic gluconeogenesis is critical for generation of glucose which is a required metabolite for neuronal metabolism (31). Reduced availability of amino acids results in inhibition of mTOR, as well as activation of AMPK (251). Amino acids are usually released via proteasomal digestion. However, autophagy of intracellular proteins appears to result in amino acid production during nutrient deprivation that acts to both prevent further autophagy in a feedback loop via mTORC1 as well as serve as a direct and indirect energy source for cell survival (Reviewed in Wolfson and Sabatini, Cell Metab 26 (2) 2017 301–309 (233). Similarly, increased levels of acetyl CoA (e.g. during abundance of fatty acids and glucose), especially in the nucleocytosolic compartment results in increased protein acetylation and inhibit further autophagy (57, 126, 179). Recent studies have uncovered inhibition of mTOR signaling in skeletal muscle of mice modeled for Pompe’s disease (117) which could be re-activated by feeding arginine-supplemented diet resulting in reversal of muscle atrophy and autophagy impairment observed secondary to the lysosome dysfunction in this model. Curiously, while mTOR activation has been shown to TFEB activity by phosphorylation and cytosolic retention (vide infra) (189), adenoviral transduction of TFEB was also sufficient to ameliorate the autophagic impairment and glycogen accumulation by enhancing lysosomal exocytosis of un-degraded lysosomal substrate (204). The abundance of amino acids likely regulates signaling through the LYNUS complex during cardiac physiology and can be harnessed to therapeutic advantage; a paradigm that needs to be explored in experimental model systems. Germane to this discussion is the recent observation that cholesterol delivery via lipoproteins to the lysosomes also regulates mTOR via the LYNUS (34), engendering the speculation that multiple classes of nutrients may regulate lysosome function, a premise that needs to examined in vivo in future studies. Indeed, prior observations have implicated flux through glucose metabolic pathways, specifically the accumulation of glucose-6-phosphate (G6P) in activating myocardial mTOR signaling in heart failure (183); which was observe to reverse upon mechanical unloading of failing human hearts.
Mitochondrial oxidative phosphorylation
As the name suggests, the essential adjunct to oxidative phosphorylation is a steady supply of oxygen. Unlike many organs, the heart extracts 70–80% of delivered oxygen to drive the large energy demand discussed above (215). Maintenance of cardiac energetics is driven by nutrient and oxygen supply, as well as neurohumoral signaling, calcium metabolism, and local factors driving oxygen demand including chronotropic and inotropic states, preload and resistance to cardiac output. A bulk of energy production is directed to contractile function of the heart. As NADH and FADH2 are generated from both fatty acid oxidation as well as the TCA cycle (and lesser extent from glycolysis), protons are transported through the electrochemical gradient between the mitochondrial matrix and the intermembrane space via the F0F1 ATPase and storage of the resultant chemical energy as ATP bound to membrane-associated cytoskeletal elements (79).
In the absence of oxygen, there is a resultant accumulation of reducing equivalents in the cell and mitochondria (92). This results in influx of calcium (Ca2+) as a combined effect of the Na+/H+ and the Na+/Ca2+ channels at the cell surface. Ca2+ overload has several deleterious roles in the cell, reinforcing the importance of maintaining a flow of oxidative phosphorylation. Ca2+-mediated calpain activation can result in maladaptive inhibition of autophagy by calpain-mediated cleavage of ATG5 (248), resulting in apoptotic cell death. Reactive oxygen species are a potentially toxic byproduct of oxidative phosphorylation and have been implicated in a variety of deleterious consequences. Inhibition of ROS generation by sirtuin-mediated deacetylation of mitochondrial proteins appears to be a mechanism of cardioprotection during calorie restriction (242). In cardiac myocytes, the organization of mitochondria, and the interactions with the cytoskeleton appear to play a role in regulating energetics. Mitochondria are located in a intermyofibrillar pattern between sarcomeres, adjacent to the sarcoplasmic reticulum T-tubules that supply the mitochondria with oxygen. This compartmentalization of myocardial energetic units helps optimize the efficiency of nutrient and oxygen delivery as well as rapidly regenerating ATP from ADP produced by the contractile elements rather than being dependent on diffusion gradients and a diffuse distribution throughout the cell. ATP and ADP, themselves, appear to be attached to cytoskeletal structures rather than free in the cytosol (79). This is important with regards to being able to control the availability of high-energy phosphates to the cardiac myofilaments. Experimental data with isolated mitochondria has demonstrated that association of mitochondrial outer membrane proteins (VDAC, mtCK and ANT) with tubulin in the cytoskeleton appears to be required for both ADP affinity as well as deltaΨm (mitochondrial membrane potential) (10). The autophagy-lysosome pathway plays a critical role in mitochondrial quality control in cardiac myocytes (52, 75) (also see section on mitophagy below); and is therefore a major determinant of the contribution of mitochondrial oxidative phosphorylation to cardiac energetics.
By and large, mammals are bulk feeders as compared with continuous feeding behavior in various lower organisms and diurnal variations in food intake regulate myocardial nutrient availability. In a carefully designed set of experiments, Brewer et al. mapped out the temporal responses to food intake and fasting in mice over the course of the dark (active) phase and the light (sleep phase), and correlated myocardial energetics with rates of substrate utilization and autophagy (as a measure of lysosome function) in these nocturnal animals (27). Their observations define a natural fasting period in mice during the light (sleep phase), which is sandwiched between two dominant peaks of food intake and correlates directly with physical activity, energy expenditure and respiratory exchange ratio (RER, a measure of substrate utilization that decreases with fat utilization and increases with carbohydrates as the dominant energy source). Fasting, initiated early in the light (sleep) phase, provoked increased physical activity during the dark (active phase) while lowering energy expenditure and the RER, which was accompanied by reduced glucose utilization and increased fat utilization in the myocardium. This was accompanied by concordant changes in gene expression. These studies provide a framework to understand the benefits of sleep phase fasting on myocardial nutrient utilization; and protein and organelle quality control and will spur further investigation in these areas.
A summary of the nutrient source-specific metabolic processes in cardiac myocytes in physiology and under pathological states is provided in Figure 1. These wide variety of nutrient choices underscore the description of cardiac myocytes as ‘omnivores’ (215) and highlight the dynamic nature of resource utilization to maintain energy generation in these unique ‘constantly working’ cell-types. The next section discusses the central role of lysosomes in this process.
Figure 1: Cardiac metabolism in health and disease:

During normal physiology, the vast majority of myocardial ATP production is from oxidative phosphorylation. Of this, a majority of this is from β-oxidation of fatty acids, with a smaller contribution from glucose oxidation. By contrast, glycolysis, ketone body oxidation and amino acid metabolism contribute very small amounts to overall ATP generation. During pathological states, cardiac myocytes use alternate preferences for energy generation (reviewed in (215) (213); please see text under the section ‘Cardiac energetics, substrate metabolism and pathophysiology under fasting states’ for details).
A. Metabolic changes during nutrient deprivation vary depending the duration (ST= short term and LT= long term). During short term fasting, fatty acid oxidation is unchanged while glucose metabolism is suppressed. In addition, marked increases in ketone body oxidation and amino acid metabolism contribute to myocardial energetics. During prolonged fasting, however, lipid oxidation is increased and serves as the main energetic source.
B. During ischemic syndromes (I= ischemia and IR= ischemia-reperfusion), energetic choices are driven by oxygen availability. While ischemia is associated with reduction in fatty acid and glucose oxidation, glycolysis serves as a far more significant source of ATP. When reperfused, glycolysis rates decline and glucose oxidation is still suppressed, while fatty acid oxidation increases. In most ischemic syndromes, TCA (tricarboxylic acid cycle)-independent amino acid metabolism is augmented.
C. Cardiac myocyte dysfunction in cardiomyopathy is often accompanied by abnormalities in myocardial metabolism and energetics. Prominent among these is a switch from a preference for glucose oxidation as opposed to lipid oxidation.
D. During aging, similar to cardiomyopathy, fatty acid oxidation is decreased with increases in glucose oxidation and glycolysis driving myocardial energetics. E. Fatty acid utilization is increased in the diabetic myocardium (with type 2 diabetes due to insulin resistance) with reduction in glucose utilization. The diabetic heart increasingly utilizes ketone bodies as a nutrient source. ‘’ indicates upregulation, whereas ‘’ indicates downregulation.
Lysosomes as regulators of cardiac homeostasis and the fasting response.
The earliest studies on cardiac myocyte lysosome function observed the distribution of lysosomal contents in ischemic hearts before and after reperfusion, and discovered evidence for leakage of lysosomal enzymes into the cytosol with prolonged hypoxia (47, 49). These observations spurred the conclusion that lysosomes participate in cardiac myocyte death with prolonged ischemia. Simultaneously, these studies also observed autophagic vacuoles with shorter ischemic duration as well as with fasting in rabbit hearts and concluded that lysosomes were playing a role in repair via the process of ‘autophagy’ following injury or starvation (48), predating the discovery of molecular mechanisms of autophagy by over a decade (138). Subsequent work elucidated the pathologic involvement of the myocardium in nearly 2/3rds of all known lysosome storage disorders (reviewed by Platt et al. (163)), and uncovered defects in autophagy and cardiac metabolism in mouse models of Danon disease (218), Pompe’s disease (95) and mucopolysaccharidoses (235) as potential underlying mechanisms for the cardiovascular pathology observed in humans afflicted with these LSDs. Recent studies from our lab and others have begun to characterize the role of lysosomes in the myocardial response to fasting (72, 83, 96) and its ramifications towards understanding its role in cardiomyopathy and heart failure.
Nutrient sensing pathways
Intracellular metabolite sensors play a key role in metabolic switches to adapt to both changes in nutrition as well as physiologic situations like exercise and remodeling and pathological situations i.e. ischemia-reperfusion and heart failure. These metabolite-responsive proteins have direct short term effects in the form of changes in cytosolic metabolite preferences, uncoupling of mitochondrial oxidative phosphorylation, and intracellular substrate generation via autophagy and alterations of lysosomal function. Indirect intermediate and long term effects of these proteins are reflected by nuclear changes in gene expression. Adenosine-monophosphate-kinase (AMPK) senses the energetic state of the cell (151). In cardiac myocytes, ATP deficiency and increased AMP abundance results in AMPK activation. By contrast, increased amino acid and nutrient abundance activates mTOR (mammalian target of rapamycin), a kinase that exists in two distinct complexes, namely mTORC1 and mTORC2 to control cell growth and metabolism (178). Activated mTORC1 has multiple inhibitory influences on protein catabolism as well as autophagy (54, 189). Fatty acid levels play a role in activation of the PPAR family of nuclear hormone receptors (228). Alteration in the NADH/NAD+ ratio is detected by the metabolic sensor SIRT1 (Sirtuin-1 (Silent mating type Information Regulation 2 homolog)). In addition to the effect of ketone bodies on class I HDACs, this NAD-dependent class III HDAC, results in intermediate and long term changes in regulation of proteins involved in lipid and carbohydrate metabolism (reviewed in (247)). Besides its role as a chromatic deacetylase, SIRT1 has wider effects on metabolism as it serves as a protein deacetylase to inhibit mTORC2 (227) affecting gluconeogenesis, activates PGC1-α (172) and FOXO-1 (gluconeogenesis) (29, 140); and FXR (autophagy) (100). SIRT1 also impairs PPAR-α signaling, impairing fatty acid oxidation (165). Recently, SIRT1 was shown to de-acetylate and activate TFEB resulting in increased autophagy (19). In C. elegans, dietary restriction appears to result in delay of aging in a Sirtuin-dependent mechanism, indicating a key role for this nutrient-sensing pathway in longevity (236). Interestingly, suppression of the estrogen-receptor related (ERR) transcriptional pathway by PPARα/SIRT1 was observed as a physiological response to fasting in the heart, but also implicated in development of pressure overload-induced cardiac hypertrophy and failure (152). An emerging body of literature has also begun to uncover the roles of extranuclear sirtuins in cytosol (SIRT2) and mitochondria (SIRT3, SIRT4, and SIRT5) in shaping the cardiac response to stress, by regulating AMPK and PGC1α activity as well as antioxidant signaling (reviewed in (58)). The hexosamine biosynthetic pathway links multiple substrates to formation of UDP-GlcNAc (uridine diphosphate-N-acetylglucosamine) through acetyl CoA, which is also postulated to be a ‘nutrient sensing’ mechanism by coupling nutrient availability to signaling via covalent O-GlcNAc linkage (O-GlcNAcylation), a highly prevalent posttranslational modification in the heart (reviewed in (46)). In addition to these molecular nutrient sensors, the lysosome has emerged as the key organelle for integration of nutrient signaling in the cell.
Much like the key role of mitochondria in ATP production, lysosomes play a key role in integration of nutrient sensing as well as marshaling of intracellular resources to maintain a steady substrate supply and thus a constant level of ATP. Although previously viewed as “intracellular garbage disposal”, an emerging body of work indicates that lysosomes are a highly active site for integration of the metabolome (1). A key lysosomal function, namely autophagy, is not just a process of garbage collection and removal in the cell, but a key cellular mechanism for energetic homeostasis as well as maintenance of cell survival in the cardiovascular system (reviewed in (134) and (68)). Our studies have identified impaired lysosome function or insufficient transcriptional upregulation of lysosome function as a driver of impaired cardiac function under stress (72, 124). Furthermore, abnormal lysosomal function results in inadequate constitutive turnover of cellular components and accumulation of dysfunctional organelles, misfolded proteins, lipid droplets with severe impairment of the cell’s ability to withstand metabolic stress (122–124).
Autophagy, a key lysosomal function in nutrient regulation
Autophagy is a conserved cellular function in all eukaryotes from yeast through humans (reviewed in (137)). In this process, intracellular proteins, organelles, and other cellular components are degraded within lysosomes and their components either used a source of nutrients or recycled as simple building blocks for reuse. In mammalian systems, autophagy can be broadly classified under three distinct types: macroautophagy, micro-autophagy and chaperone mediated autophagy (86). While a role for the latter two forms of autophagy is yet to be characterized in the cardiovascular system, macroautophagy (from here on referred to as autophagy) is by far the most clearly delineated pathway in cardiac biology mammalian systems (reviewed in (68)). This improved understanding of autophagy has been accompanied by advances in lysosomal biology. Previously considered passive membrane bound repositories of enzymes, lysosomes function as the central node as nutrient sensors, metabolic regulators and in combination with cytosolic proteins, participate in gene regulation in autophagy and metabolism (162). As a whole, autophagy and lysosomal function have significant implications with regards to cell viability, and aging as well as serving critical roles in mammalian cardiac homeostasis (66).
The most common representation of autophagy is characterized by appearance of double membrane autophagosomes that serve as bags for isolating various cellular components targeted for intracellular degradation. This unique phenomenon of “self-eating” is highly conserved across species from yeasts to humans and is essential for homeostasis. This primordial cellular process is operative at basal rates in homeostasis but also responds dynamically occurs in response to changes in most stress states, but most exquisitely during decreases in nutrient supply (139).
The stages of autophagy can be divided into initiation mechanisms, formation of the autophagophore, autophagosome assembly and capture of intracellular components and, finally, fusion with lysosomes and degradation of the captured components (reviewed extensively in (137)). Lysosomes contain a highly acidic environment as a result of energy dependent lysosome specific ion channels (162). Autophagosome formation is initiated and propagated by a highly orchestrated interplay of an evolutionarily conserved family of ubiquitin-like conjugating enzymatic proteins (ATG proteins, see Figure 2) (144). Assembly of the ATG protein complex on the donor membrane from either ER or plasma membrane (166) helps to create a vacuole that surrounds the targeted cellular components. This helps to not only generate nutrients from breakdown of superfluous intracellular contents, but also as a mechanism to degrade misfolded proteins and dysfunctional organelles (137), which are targeted to the autophagosomes by both selective and non-selective mechanisms (197). Specific adaptor proteins bind ubiquitinated targets and sequester them within the developing autophagosome by interacting with LC3 family of proteins that are lipidated and embedded in the inner autophagosome membrane. While short-term regulation of autophagy in cell culture experiments is primarily via post-translational mechanisms, in vivo regulation of the autophagy-lysosome machinery involves careful orchestration of transcriptional, post-transcription and post-translational regulatory pathways to titrate autophagic flux to the physiologic demand (61).
Figure 2: Mechanisms for execution of autophagy as a lysosomal degradative pathway:

Nutrient deprivation and organelle damage trigger macro-autophagy in cardiac myocytes (reviewed in (68)). Assembly of the phagophore (the initiation of the double membrane) is activated by class III phosphatidylinositol 3-kinase (PtdIns3K) catalyzed generation of phosphatydil inositol 3-phosphate (PtdIns3P) to recruit PtdIns3P-binding proteins. A protein complex is formed comprised of Vps15 homolog phosphoinositide-3-kinase, regulatory subunit 4 (PIK3R4), the Vps34 homolog phosphatidylinositol 3-kinase, catalytic subunit type 3 (PIK3C3), and the Vps30/Atg6 homolog Beclin 1. This interacts with AMBRA1 (not shown) and ATG14, and also contain the BECN1-interacting protein UV radiation resistance associated (UVRAG, not shown) and SH3-domain GRB2-like endophilin B1 (SH3GLB1/Bif-1, not shown) to promote autophagosome formation. Autophagosome formation has been shown to be initiated at multiple sites including the mitochondria-associated membrane, endoplasmic reticulum, the ER-golgi interface and the plasma membrane. PtdIns3P at the ER triggers the recruitment of the PtdIns3P-binding protein ZFYVE1/DFCP1 (zinc finger, FYVE domain containing 1) protein and WD repeat domain, phosphoinositide interacting 2 protein (WIPI2) to from a PtdIns3P-enriched ER-associated structure termed the omegasome for its Ω-like shape. Elongation and expansion of the phagophore membrane takes place by the Atg12–Atg5-Atg16 and Atg8 conjugation systems, which are ubiquitin like conjugation systems. The Atg12–Atg5-Atg16 complex catalyzes the lipidation of Atg8 by Atg4-mediated cleavage followed by covalent linkage to phosphatidylethanolamine. Mammalian Atg8 homologs, namely, the microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) and GABA (A) receptor-associated protein (GABARAP) subfamily proteins are involved in membrane expansion and also interact with adaptor proteins that bind ubiquitinated proteins (as cargo) to sequester them within the autophagosomes. Adaptor proteins such as p62, NBR1, NDP52, VCP and optineurin interact with ubiquitinated proteins through a ubiquitin-binding domain (UBA) and with LC3 family of proteins via a LC3-interacting region (LIR). After closure of the double membrane, autophagosome-lysosome fusion occurs in a manner dependent upon lysosomal pH (requires acidified lysosomes) and regulated by Rab7, SNARE and HOPS complex proteins. This is followed by intralysosomal degradation of autophagosome contents with recycling of basic building blocks back to the cytosol, completing the process of autophagy. Lysosomes harbor the LYNUS complex on their cytosolic face, which is comprised of mTOR complexed with the lysosomal proton pump, Ragulator, Rag GTPases, GAP (GTPase activating) and GEF (GTP exchange factor) proteins and Rheb GTPase. The LYNUS complex activates mTOR during the nutrient replete state which phosphorylates the TFEB family of transcription factors (reviewed in (162, 188)). Phosphorylated TFEB on lysosome membrane exists in equilibrium with a pool that is bound to cytosolic 14–3-3 proteins. Activated mTOR inactivates autophagy via phosphorylation of ULK1 and ATG13. Nutrient depletion induces the LYNUS complex to unravel, whereby mTOR becomes inactive relieving the constitutive phosphorylation of TFEB. Simultaneously, release of lysosome calcium via activation of the mucolipin channel activates calcineurin, which dephosphorylates TFEB to unmask its nuclear nuclear localization signal. Nuclear TFEB activates transcription of autophagy and lysosome genes, and induces autophagosome formation and lysosome biogenesis to upregulate flux through the macro-autophagy pathway. These lysosomal degradative processes generate nutrients that provide energy and support various metabolic functions (reviewed in (98)), and destroy invading organisms in immune cells via the process of xenophagy and enable antigen presentation to mount an immune response (reviewed in (136).
In the initiation phase, nutrient deprivation results in mTORC1 inactivation and activation of the AMPK cascade. A direct target of this change is the activation of the uncoordinated protein 51 like kinase (ULK) (Figure 2). In addition to phosphorylating mTORC1 and further inhibiting it, ULK binds ATG13 and FIP200 in an initiation complex. This translocates to the endoplasmic reticulum to initiate activation of the phosphatidyl- inositol 3 kinase (PI3K) complex. During nutrient deprivation, JNK1 phosphorylates Bcl-2. This results in dissociation from Beclin-1, thus preventing the inhibition of Beclin-1. In combination with vps34, ATG14, vps15 and Ambra1, Beclin (ATG6) forms the Class III PI3 kinase on the surface of the ER that is activated by ULK1 in the initiation complex. This results in production of PI3P on the recruited membrane. Simultaneously, ATG9L recruits donor membranes (from sarcolemma, ER, golgi and nuclear as well as mitochondrial membranes) to form a vesicle. When the vesicle comes in contact with the initiation complex and the PI3P, it results in formation of the pre-autophagosomal complex. Activation of the ATG5 complex by ATG 12 and ATG16L1 results in covalent modification of the LC3 (ATG8 homolog) with phosphatidylethanolamine on the membrane surface to form LC3 II. Activated LC3 II directs binding of the PAS to the membranes of dysfunctional organelles via direct interation as well as via the adaptor protein p62 to ubiquitinated proteins. This sequesters the proteins (and organelles marked with ubiquitinated proteins such as damaged mitochondria, ER and lysosomes) into the autophagosome. ATG14 interacts with Q-SNAREs (Soluble N-ethlymaleimide-sensitive factor Attachment REceptor proteins) syntaxin and SNAP29 on the autophagosomal membrane as well as VAMP8 on the lysosomal membrane leading to fusion. Nutrient-dependent covalent modification of SNAP29 with an O-linked b-N-acetyl glucosamine group appears to link this process to the cellular nutrient status (78). Once fused, autophagosome contents are denatured in the high acidified milieu of the lysosomes and digested by a combination of lysosomal enzymes including lipases, protease (cathepsin), glycosidases, acid phosphatases, nucleases and sulfatases. Following digestion into simple nutrients, the resultant substrates are actively and passively transported in the cytosol for utilization. Failure of acidification of the lysosomes during biogenesis or lysosomal enzyme deficiencies result in impairment of autophagic flux and lysosomal storage disorders with deleterious cellular consequences (reviewed in Platt et al (163)).
Baseline levels of autophagy are seen in all cells and appear to be important for optimal function (82, 139, 143). Genetic disruption of autophagy with germline ablation of ATG5 resulted in neonatal lethality with evidence for myocardial ischemia, and depleted amino acid and energy stores in the myocardium (109) whereas inducible cardiac myocyte-specific ablation of ATG5 resulted in rapid induction of cardiomyopathy (143) highlighting the critical need for cardiac autophagy signaling in maintenance of myocardial homeostasis. That the neonatal lethality in ATG5 null mice could be partially rescued by forced feeding (109) or completely corrected by restoration of sucking behavior by re-expression of ATG5 in the ATG5 null background (246) strongly indicates that autophagy plays a critical role in maintaining cellular nutrient homeostasis in both fed and starved conditions. Indeed, the critical requirement for intact autophagy-lysosome machinery is borne out by the fact that >70% of all lysosome storage disorders, provoked by mutations in lysosomal proteins in humans, result in cardiac pathology.(163) Prominent examples of these diseases are Danon disease secondary to mutations in lysosomal membrane protein, LAMP2; Fabry’s disease due to alpha-galactosidase deficiency; and mucolipidosis due to N-acetyl glucosamine phosphoryl transferase α/β deficiency; which result in cardiac hypertrophy, cardiomyopathy and heart failure, often necessitating heart transplant at an early age (163). Accumulation of undigested substrates within autophagic vacuoles is a hallmark of global lysosome dysfunction these disorders, implicating an essential role for lysosomal degradation in maintenance of myocardial homeostasis. Furthermore, our studies show that repetitive fasting in the setting of lysosomal dysfunction worsened autophagic flux impairment resulting in accumulation of autophagsoeomes; and accelerated development of cardiomyopathy in Lamp2 null mice (72).
Lysosomes and nutrient sensing
Regulation of autophagy in mammalian cells is largely driven by metabolic triggers, both directly via protein kinase cascades as well as indirectly via genetic regulatory networks (Figure 3; also reviewed in (98)). The key protein cascade linking nutrient sensing and acute regulation of autophagy and lysosomal function is the master growth regulator, mTORC1, an ancient serine-threonine kinase. At the junction of nutrient signaling, energy levels and cellular growth pathways, mTORC1 controls cell size, regulates cell function and cell division, and the response to stress (reviewed in (178)). Physical association of lysosomes and mTORC1 is required for these functions, and mTOR kinase is active on the lysosomal membrane in a LYNUS complex (Lysosomal Nutrient Sensing Complex) in the fed state, signaling cellular growth (251). This activity is maintained via amino acid fluxes across the lysosome membrane directed into the lysosomes and activity of the proton pump (38). An important target of mTORC1 kinase activity at the LYNUS complex is the basic helix-loop-helix transcription factor TFEB, the master regulator of lysosomal biogenesis and autophagy (177, 189). In the fed state, TFEB is phosphorylated which masks its nuclear localization signal and results in its binding to 14–3-3 proteins and actin and retention within the cytosol (186). With starvation, the amino acid fluxes reverse, insulin-mediated tonic activation of mTOR kinase via loss of inhibitory TSC signal on Rheb GTPase is removed and the mTORC-Ragulator complex disassembles, thereby relieving the tonic phosphorylation of TFEB (251). This is coupled with lysosomal calcium release via the mucolipin channel resulting in activation of calcineurin phosphatase which drives de-phosphorylation and nuclear translocation of TFEB (131), stimulating TFEB-activated transcription. TFEB binds to preferred E-box sequences, that have been termed as CLEAR sites (for coordinated lysosomal expression and regulation network), to activate transcription of its cognate genes (154). TFEB activation not only drives the autophagy-lysosome genes, but upregulates transcription of metabolic activators, PPARα and PGC1α, thereby coordinately driving mitochondrial and peroxisomal biogenesis and stimulation of lipolysis to access intracellular lipid stores (186). Recent work has demonstrated that TFEB activation also drives transcriptional upregulation of RAG-D GTPase, which is critical for re-activation of mTOR with re-feeding (51). Conceivably, this ensures that the starved cell is prepared to utilize the nutrient supply when it becomes available after a period of starvation, to ensure viability. Our studies demonstrated that fasting was sufficient to activate TFEB in the myocardium, mirroring TFEB activation by starvation with complete nutrient deprivation in cardiac myocytes in vitro (72). The delayed time scale of TFEB activation with fasting in the myocardium (up to 24 hours) contrasts with the rapid nuclear translocation with starvation in vitro (within 30 minutes) (72) suggesting that fasting-induced metabolite shifts are sensed differently, in vivo, than a complete deficiency of exogenous source of nutrients, in vitro, in cardiac myocytes. In these studies, fasting-induced TFEB activation resulted in transcriptional stimulation of autophagy-lysosome machinery as well as stimulation of autophagic flux in myocardium.
Figure 3: Regulation of Autophagy-Lysosome Machinery:

Various cell-intrinsic and extrinsic inputs regulate the autophagy-lysosome machinery. Organelle damage and impaired protein quality control with dysfunction of the ubiquitin-proteasome system activates selective autophagy to remove the damaged organelles (mitochondria, ER, golgi and lysososomes) and protein aggregates (reviewed in (69)). Aging induces ROS upregulation and accumulation of DNA damage, which induce senescence in cardiac progenitors through telomere attrition and DNA damage (reviewed in (195). While SIRT1 expression is increased, experimental evidence indicates that further stimulation of SIRT1 function enhances autophagy via activation of FOXO1. Serum levels of GDF11, a TGFβ family member, decreases with age in the serum and increasing GDF11 levels beneficially affects age-related cardiac phenotypes. While GDF11 stimulates autophagy in skeletal muscle; its effects on the heart have not been studied. miR-216a increases in the aging heart and is known to downregulate Beclin-1, a protein essential for autophagy induction. Nicotinamide phosphoribosyl transferase (Nampt), a key enzyme in the salvage pathway of NAD+ synthesis in cardiomyocytes is down regulated with aging, and restoring Nampt and NAD+ levels restores autophagic flux in the heart. Multiple transcriptional pathways intricately regulate the autophagy-lysosome machinery (reviewed in (61)). The FOXO family of transcription factors are autophagy activators and are activated by phosphorylation effected by AMPK, JNK and MST1 kinases. However, Akt-mediated phosphorylation of FOXO1 at a different site inactivates FOXO1 and holds it in the cytosol. Activation of the stress sensor ERN1/IRE1α signaling also holds FOXO activity in check. TP53 (tumor protein p53), a transcription factor that regulates cell cycle activates autophagy when localized to the nucleus by directly activating transcriprion of various ATG genes, but inhibits autophagy when in the cytosol. Lysosomal lipases are held in check by mxl3 in C. elegans and by its orthologue MAX in mice. Both are transcriptional repressors that bind to the CLEAR response element where TFEB binds to inhibit lipophagy and lipolysis for generation of energy (reviewed in (98). CREB, or cAMP response element-binding protein is held in check by its interaction with FXR, farnesoid X receptor in the fed state. Fasting induced FXR degradation and binding of CREB to CRTC2, its co-activator to induce TFEB transcription. Fasting also causes LYNUS machinery to deactivate mTOR which results in dephosphorylation and activation of TFEB. TFEB stimulates transcription of autophagy and lysosome pathway genes. Circadian rhythm plays a central role in regulation of the fasting feeding responses and controls autophagy-lysosome machinery. Emerging evidence points to counter-regulation of TFEB family of transcription factors by circadian rhythm regulators either indirectly via mTOR activity or directly via TFEB binding to Per genes (reviewed in (155). Numerous metabolic influences also regulate autophagy in the heart, as shown (and reviewed in (98)). Please see section titled ‘Cardiac energetics, substrate metabolism and pathophysiology under fasting states’ for a detailed discussion of cardiac metabolism under stress. ‘’ indicates , whereas ‘’ indicates inhibition.
Nuclear translocation of TFEB results in expression of proteins involved in autophagy, metabolic enzymes (via induction of other transcription factors), and lysosomal biogenesis. Long term transcriptional changes affecting the autophagy-lysosome pathway are tightly coupled into transcriptional networks that govern fatty acid mediated signaling. In these pathways, PPARα and CREB appear to activate multiple autophagy genes, both directly and indirectly, supporting optimal levels of autophagy and lipophagy that promote survival. In the fed state, the FXR (Farnesoid X receptor) complexes with CREB, thereby preventing CREB-induced TFEB transcription (184). Simultaneously, basal transcription of autophagy-lysosome genes is speculated to be suppressed by MAX (98) (experimental proof for which is currently lacking), a Myc super-family transciption factor that bears homology to mxl-3 in C. elegans, a transcriptional repressor that has been demonstrated to bind to CLEAR sites and prevent binding of HLH-30 (the worm orthologue for TFEB) at these sites (150). With nutrient deprivation, FXR is inactivated which permits CREB binding to its co-activator CRTC2 and drives transcription of autophagy-lysosome genes. Additionally, fasting also removes the inhibitory effect of FXR on PPARα, and results in PPARα-mediated transcriptional induction of autophagy pathway genes (113).
Early studies have begun to characterize the role of the lysosome nutrient sensing complex in cardiac physiology. Mice expressing a constitutively active form of RAG-A (in a GTP-locked state) instead of wild-type RAG-A protein, demonstrate a defect in inactivation of mTOR resulting in inability to induce autophagy and markedly increased mortality in the neonatal period of physiologic starvation prior to establishment of maternal milk supply (53). This was accompanied by profound decrease in plasma glucose and amino acid concentrations, which could be rescued by administration of glucose or gluconeogenic amino acids resulting in prolongation of survival. This study also demonstrated that mTORC1 is resistant to glucose deprivation in constitutively active RAG-A mutant expressing mice and that glucose, like amino acids, controls mTOR recruitment to the lysosomal surface which is the site of mTORC1 activation. Importantly, these findings recapitulated prior observations of neonatal lethality in ATG5 null mice that cannot activate autophagy during the period of physiologic starvation, and demonstrate marked reductions in circulating and cardiac amino acid levels resulting in cardiac ischemia and death (109). Conversely, studies have demonstrated that insulin receptor signaling is critical to suppress cardiac autophagy signaling upon re-feeding, and persistence of markedly elevated autophagic flux resulting in cardiac myocyte death, cardiomyopathy and heart failure which could be rescued by suppression of autophagic flux with concomitant haplo-insufficiency of Beclin-1 (169). Persistent activation of mTOR signaling pathways has also been demonstrated to be detrimental, when experimentally induced in cardiac myocytes. Combined loss of RAG-A and RAG-B with experimental genetic ablation in cardiac myocytes resulted in a hypertrophic cardiomyopathy phenotype, with impaired autophagic flux and accumulation of autophagosomes and autolysosomes; and reduced expression of lysosomal proton pump despite persistent activation of TFEB (103). Also, cardiac myocyte-specific ablation of TSC2 (tuberous sclerosis complex 2 protein that complexes with TSC1 to inhibit RHEB, a GTPase that activates mTOR) resulted in suppressed autophagic flux, cardiomyopathy and premature mortality. Additionally, overexpression of DDITL4, a stress-responsive endosomal protein was sufficient to drive persistent activation of mTORC1 complex resulting in enhanced autophagic flux and cardiomyopathic dysfunction in a, which could be reversed by suppressing its expression or haplo-insufficiency of concomitant Beclin-1 (198).
Taken together, these data point to a critical role for the LYNUS complex in regulating cardiac myocyte metabolism in both the fed and the fasted state, a premise that needs to be experimentally explored and targeted for therapeutic benefit in the heart. Indeed, administration of trehalose, a safe non-reducing sugar has been demonstrated to be effective in activating TFEB in macrophages (185) and cardiac myocytes (181); and trehalose treatment attenuated atherosclerosis as well as post-myocardial infarction remodeling in an autophagy-dependent fashion raising hopes for developing lysosome-targeted therapies for cardiovascular disease.
Lysosomal control of organelle quality
To maintain cellular homeostasis, damaged and/or dysfunctional cellular organelles such as mitochondria, endoplasmic reticulum, peroxisomes, lysososomes and even bits of the nucleus are actively recycled in autophagic processes termed mitophagy, ER-phagy, pexophagy, lysophagy and nucleophagy, respectively (8). Mitochondrial autophagy or mitophagy has received much attention given the critical role of mitochondria in cardiac myocyte energy generation, and the central role for damaged mitochondria in triggering cardiac myocyte cell death under pathologic conditions (26). Dysfunctional and/or damaged mitochondria have been described to activate PINK1, a serine threonine kinase which recruits PARKIN, a E3- ubiquitin ligase which ubiquitinates mitochondrial outer membrane proteins for targeting mitochondria to the autophagosomes and subsequent lysosomal degradation. Various studies have evaluated the role of PINK1 and PARKIN in cardiac myocytes, and while germline ablation of PINK1 provoked cardiomyopathy with abnormal mitochondria (24); PARKIN has been ascribed a specific role in mitophagy during the perinatal period (75) without evidence of mitophagy dysfunction with PARKIN deficiency in the unstressed adult heart (108, 202). Interestingly, PARKIN deficiency predisposes to impaired mitophagy under stress after myocardial infarction triggering increased cardiac myocyte death (108). Conversely, PINK1 deficient hearts and isolated cardiac myocytes demonstrated PARKIN recruitment to the mitochondria after myocardial infarction or mitochondrial uncoupler treatment, raising further doubts after the obligatory roles, if any, for the PINK1-PARKIN axis in mitophagy in cardiac myocytes (107). These observations have prompted further investigations in the mechanisms for basal mitophagy in the heart; and work from our lab has identified TRAF2 (245), an adaptor protein in innate immunity signaling with E3-ubiquitin ligase activity, that participates in ubiquitination of mitochondrial proteins in cardiac myocytes and targets mitochondria for autophagic degradation. Emerging evidence also points to a RAB5-dependent endosomal pathway for lysosomal degradation of sequestered mitochondria that is independent of autophagosome engulfment of mitochondria but may depend upon key autophagy proteins for its execution (81). Overall, the emerging consensus from these studies is that basal and stress-induced mitophagy play a critical role in maintaining cardiac structure and function, and could be therapeutically targeted to achieve cardioprotection under stress (reviewed in (142)). Indeed, studies indicate suppression of mitophagy with prolonged cardiac stress, such as pressure overload, which can be rescued by stimulating autophagosome formation with Tat-Beclin-1 transduction (196) or enhancing AMPK2α-mediated PINK1 phorphorylation (225). Molecular players that mediate mitochondrial dynamics, namely mitofusins (MFN1 and MFN2) that mediate mitochondrial fusion; and Drp1, that mediates mitochondrial fission, play a critical role in maintaining mitochondrial quality control and regulating mitophagy in the heart (201, 203). Mitochondrial fission via Drp-1 was shown to be essential for basal mitophagy and heterozygous disruption of Drp1 in cardiac myocytes was sufficient to induce cardiomyopathic dysfunction with fasting (91), indicating a critical need for mitophagy under nutrient deprivation conditions (55). Oral supplementation with a nautral polyamine, spermidine, was shown to activate mitophagy and macro-autophagy in the heart among its primary mechanisms for conferring cardioprotection and delaying progression to heart failure in a model of salt-sensitive hypertension (56). Whether fasting entrains mitophagy to transduce salutary benefits in cardiovascular pathology, remains to be determined.
Recent studies have documented activation of TFEB (as well as its family members, TFE3 and MiTF) and the lysosomal biogenesis program upon induction of mitophagy, revealing a coordinated regulation of lysosomal biogenesis program together an organelle selective autophagy process (146). Relevant to the role of autophagy under fasting stress, studies have observed that starvation induces mitochondrial damage to activate mitochondrial autophagy (55). In myocardial ischemia, during states of nutrient and/or oxygen deprivation, the oxidative phosphorylation machinery is halted. This, results in accumulation of both reducing equivalents and metabolites, resulted in both electrochemical changes affecting membrane potential as well as osmotic effects on the mitochondrial matrix. This results in an increase in potentially dysfunctional, and even injured, mitochondria. Activation of autophagy, concurrently during such states, provides for the cell’s ability to isolate and remove dysfunctional mitochondria, in an effort to ensure adequate nutrient supply to the maximally functioning organelles. Accordingly, mitophagy has been demonstrated to play a critical role in protecting against cardiac myocyte cell death in myocardial infarction (108).
Our studies demonstrate that intermittent fasting improves mitochondrial quality control in the myocardium as the mechanism for the reconditioning effects on the myocardium (72). Indeed, mitochondrial structure and polarization were compromised with deficiency of Beclin-1 and LAMP2, with intermittent fasting in vivo and repetitive starvation in vitro, respectively, in cardiac myocytes; which was associated with lack of preconditioning to injury. In subsequent work, we made similar observations in pancreatic β-cells in an obesity-induced diabetes milleu (119) attesting to a critical role for the autophagy-lysosome machinery in organelle quality control with intermittent fasting. Figure 4 depicts the proposed role for LYNUS machinery in cardiac myocyte homeostasis and response to stress.
Figure 4: Proposed mechanism for the effects of intermittent fasting on LYNUS-TFEB-mTORC1 axis in cardiac myocyte pathophysiology:

Cardiac myocytes in pathological states are likely to be affected by the functioning of the Lysosomal NUtrient Sensing complex (see reference (162) for a detailed review of signaling via the LYNUS complex, and text under the sub-section titled ‘Lysosomes and Nutrient Sensing’). Unopposed activation of mTORC1 as a result of nutrient oversupply, aging, lysosomal dysfunction and diabetes mellitus, is expected to result in a sustained synthetic state. This would result in overload of the ER-translational machinery precipitating ER stress and the unfolded protein response due to accumulation of misfolded entities. This pathological synthetic state is expected to provoke decreased recycling of organelles as well sarcomere renewal and repair. Furthermore, the overall metabolic reserve of the cell would be reduced due to a decrease in the ability of cells to activate catabolic pathways. Similarly, during starvation or with continuous mTOR antagonism, the translation machinery is expected to be suppressed while proteolysis is increased. This would drive breakdown of organelles and sarcomeres with suppression of organelle and sarcomere biogenesis. In addition, progressive depletion of cellular components by catabolic pathways is expected to result in a similar decrease in metabolic reserve. In contrast, intermittent fasting drives cyclic activation of both TFEB and mTORC1, which is expected to optimize protein turnover. This is accompanied by decreased ER stress, increased organelle turnover, where accelerated organelle/sarcomere breakdown is accompanied by increase biogenesis and optimal availability of cellular components and metabolic pathways to increase metabolic reserve. As compared to cardiac myocyte dysfunction seen with sustained activation of either mTORC1 or TFEB, acceleration of the cyclic activation of mTORC1 and TFEB is expected to results in cardiac myocyte rejuvenation. Similar mechanisms are proposed to drive the beneficial effects of exercise, intermittent rapamycin, circadian feeding and sirtuin action.
Interplay of lysosome function with the metabolic circadian axis
Regulation of the circadian rhythm
From primitive life forms to humans, the terrestrial day-night cycle results in unique environmental challenges. Specifically, this results in diurnal variation in access to nutrition as well as external threats. Individuals and species may utilize these variations for evolutionary advantage. In order to adapt to this, all cells appear to be controlled by external light sensing clocks as well internal nutrient/ATP sensing cellular clocks that regulate homeostasis (155). While, in nocturnal animals (like mice), night results in heightened activity, diurnal species have an opposite effect. In this paradigm, waking is associated with feeding, increased resource utilization and autophagy, sleep is associated with the post-fed state associated with physical inactivity and increased synthetic function as well as limited autophagy (35). Nutrient sensing appears to play a reciprocal role in the circadian pathways, just as the circadian advantage allows for optimizing nutrient availability. Nearly half of the mammalian genome appears to be subject to circadian variability (156), but only a handful of genes form a core oscillator that appears to interact with tissue specific elements to give rise to appropriate tissue specific changes in gene expression. In this circadian paradigm, the core oscillator results in cyclic changes in gene expression corresponding to a terrestrial day. While this oscillator is normally entrained by environmental cues, even in the absence of these, cyclic changes in core oscillator genes persist. In eukaryotic systems, activity of two transcription factors, CLOCK and BMAL1 appear to be key to this process (reviewed in (35)). In heterodimeric form, these proteins bind to a variety of transcription factors as well as chromatin modifying factors to result in changes of gene expression. The CLOCK/BMAL1 heterodimer also results in a negative feedback loop by binding to the E-boxes of their own repressors. These include the cryptochromes (CRY1 and CRY2) and their partners, the Period genes (Per-1,−2, and, −3). The CLOCK/BMAL1 heterodimer also appears to drive the expression of retinoic acid-related orphan receptors (ROR) i.e. RORα, RORβ, and RORγ (transcriptional activators) as well as the REV-ERB (transcriptional repressors) group of proteins that regulate the expression of CRY and PER proteins as well as of BMAL1 in a rhythmic feedback loop with the circadian periodicity. A variety of factors appear to affect this core oscillator. These include other transcription factors (HSF1, PGC1α), protein kinases (AMPK, mTORC1), PARP-1, and simple nutrients (NAD, glucose, calcium, AMP, Sterols and glucocorticoids) (15). Circadian function appears to play roles in control of organelle biogenesis, protein folding and secretion as well as the unfolded protein response, and oxidative phosphorylation as well as stress responses to oxidative and hypoxic stresses (reviewed in (35)). While data are beginning to emerge attesting to circadian variation of autophagy with fasting/feeding cycles in the heart (27), much work remains to be done to understand the regulation of lysosome biogenesis and function by the circadian clock.
Role of lysosomes in regulation of circadian rhythms
While central light sensitive clocks are guided via the suprachiasmatic nuclei in the hypothalamus, the commonality of circadian mechanisms in all life forms requires the presence of individual cellular clocks which use a combination of metabolic and neurohumoral cues to synchronize locally and regionally as well as with the central light sensing clock. While nuclear control of gene expression via transcription translational feedback loops (TTFL) seems an obvious control point for circadian physiology,(155) it is interesting to note that in mammalian non nucleated RBCs (148), as well as in the transcription-limited eukaryotic algae, Ostreococcus tauri, circadian oscillations persist (149). This observation begs the question whether the TTFL represents the orchestra to an unknown cellular conductor. In fact, in animals with lesions of the suprachiasmatic nuclei, circadian rhythmicity of behavior can be induced by time-restricted feeding via a ‘food-entrainable oscillator (FEO)’ (45). Interestingly, the FEO appears to be independent of a functional molecular clock, and is functional in Bmal1 deficient (159) and mice deficient in Per (Period) genes animals when treated with time restricted feeding (160).
There is an abundance of data that indicate that a variety of cues appear to regulate the core oscillator function. As the organelle controlling nutrient sensing responses as well as cellular remodeling, lysosomes are an attractive target as master regulators of circadian physiology as well. While ablation of circadian genes have ramifications in all cell types resulting in severe systemic disease, conversely, interference in TOR signaling also appears to affect circadian function. Germline ablation of bHLH-PAS transcription factor, CLOCK results in hyperphagia and obesity (222) and beta-cell specific targeting of circadian genes results in onset of diabetes mellitus (type 2, secondary to increased insulin resistance) (125, 161). Conversely, inhibition of mTOR via TSC2 results in ablation of the peripheral circadian clock. In fact, mTOR appears to regulate BMAL1 via S6K1 mediated phosphorylation, (118) and control the central circadian clock via regulating the activity of the translational repressor, eukaryotic translational initiation factor 4E binding protein 1 (4E-BP1) (32) Intriguingly, restricting food intake to the active period (for 8 hours, at night time) in mice simultaneously restored circadian variations in TOR activity and prevented the dampening of circadian gene oscillations in mice fed high fat diet, with complete prevention of adverse metabolic consequences (85), and these findings recapitulate independent observations by other groups in murine models with varying dietary composition and timing of feeding (158, 221). Indeed, time-restricted feeding activated circadian cycling of autophagic flux in various tissues (128) and the associated metabolic effects were critically dependent on autophagy. Given prior observation that fasting induced AMPK-activation targets circadian repressor CRY for degradation (111), while feeding-induced TOR activation modulates Per gene expression (70) these observations point to intricate counter-regulation of lysosome function with circadian rhythm in driving metabolic consequences. Intriguingly, studies with cardiac myocyte-specific ablation of BMAL1 and CLOCK revealed persistent activation of Akt and TOR, with suppression of cardiac myocyte autophagy (130) and mTOR inhibition with rapamycin restored cardiac autophagy to attenuate pathologic cardiac hypertrophy. Accordingly, the LYNUS may be key to control of circadian changes in metabolism as well as other aspects of cellular function. Intriguingly, the MiTF/TFE3 family of transcription factors may regulate expression of Per genes to directly regulate circadian biology (120, 154). Furthermore, control of multiple cells using the same cues of control for each individual LYNUS complex via simple nutrients and ATP, would allow for synchronized control across an entire organ and possibly, the organism; a premise that needs to be experimentally tested in the laboratory. Figure 5 depicts the proposed role for the complex interaction between regulation of the LYNUS complex with circadian rhythms in cardiac myocytes, which needs to be experimentally dissected in future studies.
Figure 5: Proposed LYNUS-TFEB-mTORC1 cues to entrain the circadian rhythm.
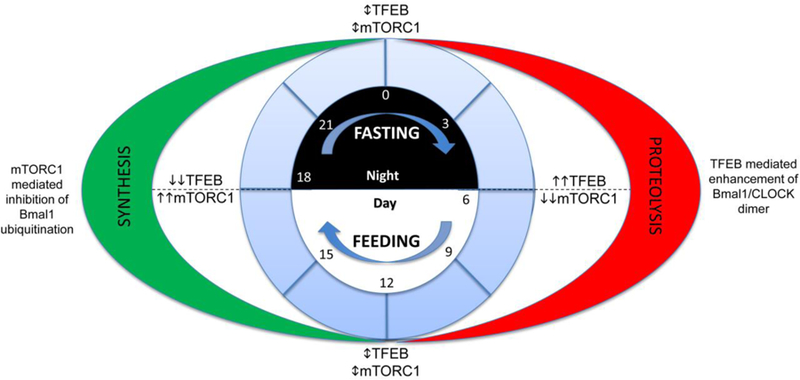
Dietary factors have been noted to affect circadian physiology. In this proposed mechanism, diurnal variation in feeding behavior is accompanied by the effect of mTORC1 and TFEB on the Bmal1 and Clock heterodimer resulting in synchronization of the circadian clock with nutrient supply. High TFEB levels at waking (“Day” in diurnal species) enhance the Bmal1/CLOCK heterodimer, thus driving circadian genes. In contrast, peak mTORC1 activation at the onset of “night” phase, when feeding is complete affects Bmal1 ubiquitination and alters the circadian gene expression, coincident with the peak synthetic phase. As opposed to these two phases, the transition from synthesis to proteolysis (and vice versa) at ‘0’ and ‘12’, is also accompanied by combined effects of TFEB and mTORC1 on the Bmal1/CLOCK proteins. By adhering to a fixed pattern, it may be thus possible to synchronize the metabolic and biologic clocks, and thus maximize cellular repair and renewal while optimizing metabolism. Please also see text under the section titled ‘Interplay of lysosome function with the metabolic circadian axis’.
Effects of caloric restriction on cardiac physiology
Systemic caloric restriction and fasting clearly play a significant role in development of protective adaptations in cardiac myocytes. While calorie restriction and fasting are markedly different in intent with calorie restriction implying a sustained reduction in the amount of calories ingested, whereas fasting indicating a complete cessation of caloric intake over a period of time (see Table 1) without restriction in food intake during the feeding periods, the effects observed on cardiovascular physiology are overlapping with important differences as noted below. In behavioral terms, the differences in outcomes over the long term are also likely to reflect the ability to sustain either strategy over prolonged period of time for maintenance of the beneficial effects; a premise that needs to be carefully studied in epidemiologic studies in humans. Indeed, while most studies of calorie restriction in humans demonstrate it to be an effective strategy for weight loss, the Achilles’ heel of all weight control strategies is that most subjects regain body weight, even with continued participation in the weight management programs with progressive attenuation of the initial weight loss and its beneficial effects (175, 231). In contrast, intermittent fasting is routinely practiced by different cultures around the world without any described untoward consequences, and has been employed in clinical trials for prolonged periods of time with excellent adherence to the regimen (84). Therefore, understanding the effects of both strategies on cardiovascular physiology is critical to harnessing them for their preventive and therapeutic potential.
Most scientific studies addressing these questions have been performed on small mammals such as rodents. It is important to recognize that given the profound differences in lifespan and cardiovascular parameters (such as heart rate) between rodents and humans, the parameters of calorie restriction and duration of fasting may not be interchangeably applied between the species. Nevertheless, these animal studies have generated critical insights which have been mirrored by observations in humans (see next section). Calorie restriction was first demonstrated to have survival benefits in mice in 1946 (33). Subsequent studies have also demonstrated that intermittent fasting prologs life-span in mice (76). The survival benefit of caloric restriction appears to be related to an effect on aging mechanisms (42, 121). Similarly, caloric restriction was found to improve Doppler parameters of diastolic function in B6D2-F1 hybrid mice, a mouse model of senescent diastolic dysfunction, without an impact on systolic parameters (216). In another study that implemented graded regimen for persistent calorie restriction (progressing to 40% reduction as compared with ad-lib) in male C57BL/6J mice beginning at 3, 12 and 19 months of age, the older two age groups demonstrated attenuated age-related cardiac myocyte hypertrophy and fibrosis, accompanied by reduction in markers of fetal gene expression program typically associated with pathologic cardiac hypertrophy; whereas the youngest group of mice paradoxically manifest reduced systolic performance (191). Interestingly, calorie restriction in older mice (and not younger ones) was associated with induction of AMPK signaling, upregulation of FOXO genes and markers indicating increased autophagic flux. In contrast, intermittent calorie restriction or fasting started early in the adult life has not been demonstrated to have any adverse effects on cardiac structure and function in mice and rats (5, 9, 72).
In a rat model of myocardial infarction when animals were subjected to coronary artery ligation after 3 months of intermittent fasting, there was a decrease in infarct size, improved remodeling (both immediate and delayed), accompanied by reductions in apoptosis as compared with ad-lib fed animals (4). Furthermore, chronic intermittent fasting starting 2 weeks after complete LAD ligation and myocardial infarction, markedly reduced mortality and attenuated parameters of adverse LV remodeling in rats (97). These studies did not examine the role of autophagy in the cardioprotection. We demonstrated that autophagic flux was upregulated in a pulsed fashion (during the fasted days, with myocardial TFEB activation) in 8 week old adult C57Bl6J male mice subjected to intermittent fasting (every other day feeding); and intermittently fasted mice demonstrated reduction in infarct size following ischemia reperfusion as compared to ad-lib controls (72); and this cardioprotection was critically dependent upon an intact autophagy-lysosome pathway. Indeed, prior studies have demonstrated that administration of rapamycin, a mTOR inhibitor and inducer of TFEB transcriptional program and the autophagy-lysosome pathway, attenuated post-MI remodeling in rats (30). Rapamycin treatment initiated late in life (at 24 months of age in female mice) resulted in attenuation of age-related cardiac hypertrophy, improving ejection performance and reduced markers of inflammatory signaling in the heart (64). Also, rapamycin treatment restored mitochondrial quality control via enhancing both mitochondrial autophagy and biogenesis to reverse age-related reduction in fatty acid oxidation to restore metabolism to a more youthful state (40), accompanied by improved protein quality control mimicking the effects of calorie restriction (44).
Studies have suggested that other pathways apart from the autophagy-lysosome system may be instrumental in the protective benefits of calorie restriction, too. In a rat model of ischemia reperfusion (isolated heart), use of calorie restriction was associated with cardio-protection with concurrent increase in AMPK phosphorylation, matching data in lower species (193). By contrast, in a model of prolonged calorie restriction, similar salutary effects with regards to infarct size and functional recovery were noted – without concurrent differences in AMPK or AMPK phosphorylation levels (194). In this model, increases in SIRT1 activation were noted. Subsequent work indicates that the protective role of prolonged caloric restriction is lost in SIRT1 null mice (241). These data indicate that caloric restriction has benefits mediated by different pathways depending on the duration of the restriction. Interestingly, the presence of a favorable phenotype, similar to one engendered by calorie restriction in a knockout model of adenylyl cyclase 5 (AC5), is associated by a loss of the protective effects of calorie restriction (243). In the AC5 knockout mouse, as seen in mice undergoing calorie restriction, lower body mass, glycogen stores, fat content and blood glucose are seen. However, these mice have a normal food intake and metabolic rate. When these mice are subjected to calorie restriction, they are noted to starve and die. A salutary change related to the knockout of adenylyl cyclase 5 appears to be the upregulation of sirtuins (SIRT1) and the beneficial effects thereof (reviewed above) (242).
In humans, calorie restriction is associated with improvement in blood pressure, serum markers of inflammation and improvement in age related cardiac diastolic function (133, 170). Furthermore, autonomic indices like heart rate and heart rate variability also appear to improve with caloric restriction (207). Long term calorie restriction (over a period of 6 years) is associated with reduced atherosclerotic risk in humans (65). In this study, as compared to matched individuals, calorie restricted human subjects were noted to have leaner body types with markedly lower levels of total cholesterol, LDL-C, high-sensitivity CRP, fasting insulin, platelet derived growth factor-AB, and systolic/diastolic blood pressures with higher levels of HDL-cholesterol. Notably, carotid artery intimal –media thickness was 40% less than in the control group. Decline in lysosomal function and autophagy with accumulation of misfolded proteins is a hallmark of senescence in many organ systems. As diastolic dysfunction is often the earliest marker of deposition diseases in the heart, e.g. cardiac amyloidosis, senescent changes in diastolic dysfunction may represent an early lysosomal deposition disease. Caloric restriction is associated with increased expression of a variety of autophagic markers (including ATG7, ATG9 and lipidated LC3) in the myocardium of senile rats (232). The improvement in aging related diastolic dysfunction seen in both animal and human models of disease with caloric restriction may indicate that upregulation of lysosomal function and, possibly, autophagy may be a novel mechanism to preserve cardiac function in rapidly aging human populations, worldwide. While reversing the effects of aging is a desirable goal, the decline in markers of autophagy with age may be a reflection of metabolic changes that also predict a reduced ability to tolerate ischemia and/or reperfusion. While calorie restriction reduces potential risks of atherosclerosis and ischemic events by the potential effects on insulin, glucose, lipids and blood pressure, it also has the potential to reduce the impact of myocardial ischemic syndromes by increasing the degree of cardiac myocyte resilience to ischemic stress via effects on both integrated lysosomal metabolic pathways and substrate/organelle recycling with autophagy.
Differential effects of intermittent fasting have been observed in male and female mice on regulation of the hypothalamo-hypophysial-gonadal axis, with reduced fertility in rats (110). Similar findings were also in a observed with sustained and severe calorie restriction in female mice, where 40% calorie restriction caused the females to become emaciated and cease menstrual cycling accompanied with hormonal changes of masculinization and improved cognitive performance; effects that were markedly in contrast to the male mice (127). Directionally similar changes, albeit of reduced intensity were also observed in intermittently fasted females in this study in contrast to the male mice. Importantly, in our studies we observed a comparable degree of intermittent fasting-induced cardioprotection and preconditioning effect (to myocardial infarction); as well as intermittent fasting-induced preservation of β-cell mass in obesity-induced diabetes in both male and female mice subjected to intermittent fasting (72, 119). Future studies will need to carefully catalog the sex-specific effects of various fasting regimens and characterize the molecular mechanisms for the observed differences to guide development of clinically-relevant therapeutic strategies.
Intermittency as a protective mechanism.
Despite the benefits of caloric restriction, starvation, if unremittent, results in atrophy and, ultimately death. Similarly, benefits from exercise accrue with periods of recovery (21). From a nutrient perspective, exercise is, in fact, a state of relative caloric restriction (18). Re-feeding, in this paradigm, following caloric restriction can thus be considered the equivalent of the recovery phase of an exercise regimen, and vice versa. Similar parallels can be drawn between the sleep-wake cycle of the circadian rhythm. It is reasonable to posit that lysosomal function has two facets of function: TFEB-driven pathways during supply demand mismatch and mTORC1 driven pathways during states with matched supply and demand (Figure 4). In the first paradigm, lysosomal function results in harvesting of cellular resources with an emphasis of removing dysfunctional structures, and redundant organelles to remodel the cell to best deal with demands. Conversely, during recovery and states of nutritional repletion, mTORC1 results in activation of synthetic pathways resulting in synthesis of new proteins and organelles. Thus, cycling between the two states results in cellular renewal while activation of either state results in cellular dysfunction. We hypothesize that failure of this ability of a cell to renew itself, is manifested as senescence, and aging, rather than the other way around. Resultantly, as indicated by data in aging studies (reviewed in Shirakabe et al (195)), processes inducing cycling between remodeling and synthesis maintain cellular youth while those that prevent lysosomal cycling between these two functional states result in the appearance of aging. This suggests that the lysosome is not merely an executor in cellular metabolic and remodeling pathways but is the main controller of cellular homeostasis via the balance between the activities of mTOR and MiT/TFE3 family of transcription factors. Furthermore, while it is often difficult to cause synchronous control of the metabolic functioning of multiple cell types in metazoan tissue, use of metabolic cues allow the lysosomes to be arbiters of cellular control in a coordinated manner for optimal resource utilization, while maximizing function of tissues, organs and organisms.
Indeed, intermittent fasting has been shown to require TOR and its upstream activator, RHEB-1 GTP-ase, in its lifespan prolonging effects in worms (88). Interestingly, median lifespan extension (as well as slowing of the decline in locomotion of aging worms, a surrogate marker of health-span) was greater with intermittent fasting than persistent calorie restriction in worms, and mechanistically transduced via suppression of insulin signaling and upregulation of DAF16 (the worm orthologue of FOXO) (88). Subsequent studies demonstrate that remodeling of mitochondrial networks with fasting and re-feeding plays a critical role in the metabolic effects of intermittent fasting, as worms deficient in mitochondrial fusion (fzo-1) and fission (drp-1) genes did not derive the lifespan extension benefits of intermittent fasting, despite being long-lived under nutrient replete conditions (230). Our studies with intermittent fasting in a mouse model of obesity-induced diabetes (a model of type 2 diabetes with insulin resistance) demonstrated preservation of β-cell mass and insulin-secretory capacity despite continued high fat diet feeding, due to attenuated β-cell death and a dramatic upregulation of endocrine progenitor markers in the islet and per-islet tissue suggesting β-cell regeneration (119). Intriguingly, these salutary effects were entirely lost in mice with deficiency of BECLIN-1, a protein critical to autophagosome formation and LAMP2, a lysosomal membrane protein essential to lysosome function. Furthermore, experiments with human islets from healthy subjects and those with type 1 diabetes demonstrated evidence for β-cell regeneration with pharmacologic inactivation of mTOR and PKA (protein kinase A) signaling and treatment with serum from humans enrolled in a calorie restricted diet trial (39). Intriguingly, re-feeding following periods of fasting was essential in these studies to evoke β-cell regeneration, attesting to the critical need for cycling of TOR signaling in the β-cell regenerative response. Interestingly, intermittent rapamycin administration was shown to specifically inhibit mTORC1 signaling while avoiding mTORC2 inhibition as a strategy to overcome the negative metabolic effects of continuous rapamycin on worsening insulin resistance as well as worsened β-cell function (13); and intermittent rapamycin started at 20 months of age was sufficient to extend the lifespan of female mice (14).
Interestingly, in the study by Brewer et al., diurnal variation in food intake did not alter mTOR activity or protein synthesis in the mouse heart in the unstressed (physiologic) state (27). However, fasting attenuated myocardial mTOR activity and as well reduced the rate of protein synthesis early with a rebound back to non-fasted levels despite continued fasting. This is consistent with prior observations in vitro (249), documenting mTOR inhibition with acute starvation followed by re-activation induced by nutrients released by autophagy. Fasting-induced reduction in mTOR activity was associated with reduced protein synthesis and induction of autophagy in the mouse heart, as evidenced by increased numbers of autophagosomes and reduced p62, an autophagic substrate. Moreover, these studies also demonstrated that myocardial autophagy was more responsive to fasting during the light (sleep phase) than the dark (active phase). In an elegant series of experiments, mice were exposed to various paradigms of calorie restriction, either time-restricted feeding or sustained calorie restriction (see Table 1 for definitions) and monitored for behavioral changes in feeding.(2) While ad-lib fed mice were nocturnal in feeding behavior and demonstrated highest levels of activity during that period, mice restricted to 70% of calorie intake (imposed from onset of the nocturnal period) behaviorally limited their food intake to the first 2 hours after onset of nocturnal period and increased their levels of activity during the day-time, pointing to calorie restriction-mediated regulation of circadian rhythms. Importantly, these data suggest that a self-imposed fasting period may play a mechanistic role in the benefits of sustained calorie restriction.(2) Taken together, these findings suggest that intermittent fasting may exert favorable effects via enhancement of the cyclic variation of normal changes in autophagic flux.
Indeed, in our studies, we have found that intermittent fasting results in cyclical induction of autophagic flux in the myocardium, with upregulation of autophagy-lysosome genes driven by TFEB activation (nuclear translocation) with each fasting episode, all of which return back to baseline (fed levels) with re-feeding (72). Indeed, this cyclical induction of autophagic flux translated into restoration of autophagic flux and levels of active (nuclear) TFEB in the islets of mice modeled for diet-induced obesity, wherein autophagic flux and TFEB activation was inhibited as compared with chow-fed controls (119). As with the observations in the islets, these salutary effects of intermittent fasting required an intact autophagy-lysosome machinery, as intermittent fasting did not precondition the myocardium to ischemia-reperfusion injury (72) in mice with haplo-insufficiency of autophagy protein BECLIN-1 or LAMP2, which did not demonstrate a phenotype in the unstressed state. Intriguingly, intermittent fasting provoked cardiac myocyte death and hastened the onset of cardiomyopathy observed in the LAMP2-deficient mice (208); as well as provoked β-cell loss in mice with complete absence of LAMP2 (72, 119). These data point to a critical role for lysosomes in integrating circadian variations in nutrient supply to autophagy and mTOR activation, in maintaining homeostasis and responding to stress in the heart. Whether intermittent fasting stimulates and/or restores autophagic flux in models of cardiomyopathy and heart failure is the subject of active investigation.
Benefits of intermittent fasting in animal models on cardiometabolic disease
As discussed above, various fasting regimens have been tested by investigators in mice to determine their benefits on metabolism and life-span. In a variation of time-restricted feeding paradigm, Martinez-Lopez et al tested the effect of feeding two times a day during two fixed 2 hour-long windows to institute two mandatory periods of fasting every day. Mice fed chow diet twice a day ingested similar calories per day as ad-lib fed mice, whereas mice fed a high fat diet twice a day were resistant to weight gain (128); mimicking prior observations with every other day fasting regimens in young adult mice on similar diets in our hands (72, 119). Importantly, twice a day time-restricted feeding led to reduced fat mass with commensurate increase in lean body mass, and a twice a day peak in autophagic flux in the liver versus the typical circadian pattern of highest autophagic flux observed at mid-day in ad-lib fed mice during their ‘sleep’ cycle (128). This was accompanied by increased anti-inflammatory signaling in adipose tissue macrophages, and salutary global metabolic effects as improved glucose tolerance, reduced hepatic gluconeogenesis, reduced hepatic lipogenesis and increased lipophagy under high fat diet conditions. Furthermore, the authors studies tissue-specific loss-of-function models of ATG7, a protein critical for autophagosome formation, and demonstrate the critical need for autophagy signaling in individual tissues for the specific benefits. Intermittent fasting has also been demonstrated to prevent doxorubicin-induced cardiomyopathy in a mouse model of repetitive doxycycline exposure resulting in cardiomyopathy (7). Intermittent fasting rescued the impaired autophagic flux in this setting, which was worsened by concomitant deficiency of UVRAG, a protein essential for autophagosome formation.
Intermittent fasting in diabetes
Interestingly, intermittent fasting regimens have been found to alter the gut microbiome as a mechanism for driving beneficial effects in models of retinopathy in db/db mice, a genetic model of obesity and diabetes (23); as well as for induction of beige fat biogenesis and reversing obesity-induced diabetes (115). In the latter study, the molecular mechanism for the observed beige fat induction was ascribed to generation of bacterial fermentation products, acetate and lactate and the elective upregulation of monocarboxylate transporter 1 expression, which was prevented by microbiota depletion and restored by transfer of microbiota from intermittently fasted mice. Indeed, as described in the previous section, we observed weight loss in mice with diet-induced obesity subjected to intermittent fasting with preservation of β-cell mass despite continued high fat feeding and restoration of β-cell function (119). In another study of chronic intermittent fasting in both young and aged mice, an every other day fasting regimen prolonged lifespan, reduced left ventricular mass and attenuated ageing-induced cardiac hypertrophy, and improved glucose tolerance (240), but did not affect other metabolic parameters in young mice, pointing to an important interaction between age at which the intermittent fasting regimen was initiated and metabolic outcomes. An important note of caution is the observation for increased insulin resistance with every other day fasting regimens in young obese mice in our studies (119), which was mirrored by a trend towards increased circulating insulin levels in young mice (240) as well as evidence for increased hepatic insulin resistance in rats (157). These findings suggest that the duration of fasting may be a stress, and will need to be carefully titrated depending upon the species (as 24 hours of fasting in a mouse is perhaps equivalent to a much longer period of fast in humans given differences in lifespan). Obesity and diabetes induce a plethora of myocardial changes that culminate in a clinically observed entity of diabetic cardiomyopathy (93) which is characterized by myocardial lipid overload (200). Whether these beneficial effects of intermittent fasting are also observed in the diabetic myocardium and are the subject of active investigation.
As discussed above, the mechanisms whereby calorie restriction and fasting confer beneficial effects on cellular and organ physiology to ameliorate disease have just begun to be elucidated. Our studies, as well as those from other labs have ascribed a prominent role for autophagy-lysosome machinery in transducing these benefits in intermittent fasting and sustained calorie restriction paradigms in animal models. In the next section, we will review the extant literature on the beneficial effects of these paradigms on cardiovascular disease prevention. Whether these paradigms extend to a therapeutic benefit in patients with established cardiovascular disease, and the potential for a mechanistic role for the autophagy-lysosome pathway in conferring these benefits, is the subject of ongoing investigation.
Intermittent fasting as a tool to regulate cardiometabolic pathophysiology in humans
Intermittent fasting in myocardial disease
Dietary intervention studies in animal models support direct myocardial protective effects of (IF) on ischemia/reperfusion injury and heart failure. Prospective, controlled clinical trials in humans have only examined the role of IF in cardiometabolic risk factor modification, rather than hard clinical endpoints. However, clinical interest in fasting regimens has increased in light of observational studies suggesting that fasting reduces the risk of clinical events including coronary artery disease and diabetes. Based on initial observations made during studies of smoking cessation, it was found that amongst a group of 448 Mormon subjects in Utah, those who practiced routine fasting had significantly reduced risk of coronary disease compared to those who did not fast (adjusted OR: 0.46; 95% CI: 0.27, 0.81; P = 0.007 (89)). These findings were also applicable to those study participants of other religious affiliations. During further post-hoc analysis, the same investigators secondarily found that fasting was associated with lower risk of diabetes. Interestingly, sustained voluntary calorie restriction in middle-aged adults was associated with marked reductions in cardiometabolic risk factors namely total cholesterol, LDL cholesterol, triglycerides, fasting glucose, fasting insulin, CRP, PDFG-AB, and systolic and diastolic BP whereas HDL cholesterol was significantly higher than age-matched healthy individuals on typical American diets (65). This translated into a 40% reduction in carotid intima-media thickness demonstrating favorable effects on attenuation of atherosclerosis. Whether intermittent fasting affects development and/or progression of atherosclerotic lesions, or changes their composition remains to be characterized.
In a second follow-up study by the same group, patients undergoing routine coronary angiography (n=200) were surveyed for fasting behaviors by dietary questionnaire. This study corroborated the earlier findings that fasting was associated with lower risk of diabetes, coronary disease, and lower BMI (90). These findings are certainly provocative, but limited by virtue of the fact that patients who fast likely benefit from other healthy habits that are difficult to adjust for in multivariate analyses. Nonetheless, the studies of Horne et al. are suggestive of benefits to IF on risk of coronary heart disease. There are currently no randomized trials that have evaluated whether IF may improve these hard clinical endpoints.
Intermittent fasting and cardiometabolic risk factors
Fasting can take many forms, including acute or chronic fasting, intermittent fasting vs caloric restriction, religious fasting during specified times of year, and the degree of abstinence or restriction on fasting days. As opposed to caloric restriction, which involves reducing caloric intake by 20–50% every day, IF regimens only require individuals to restrict feeding anywhere from 1–3 days per week. One strategy of IF is alternate day fasting (ADF), where fasting days (25% of caloric needs) alternate with ad-libitum feeding. ADF may be as effective as traditional IF in terms of risk factor modification. A detailed compilation of the effects of various fasting strategies on cardiovascular prevention is reviewed by St-Onge et al. (205).
Clinical trials of both caloric restriction and IF regimens have suggested improvements in markers of pre-diabetes, which frequently leads to diabetes (type 2, secondary to insulin resistance) (77, 84). Among adults greater than 20 years of age, approximately 35% have pre-diabetes, 20% of who will develop diabetes in the next 5 years (173). The long-term sequelae of diabetes include increased risk of myocardial infarction, stroke, and peripheral vascular disease. The role of weight loss in prevention of diabetes progression is clear, but many individuals find weight loss difficult to achieve and sustain. In terms of dietary strategies to achieve weight loss, chronic caloric restriction is particularly effective and difficult to maintain, while IF is a strategy that has shown early clinical promise and may improve compliance.
A strategy of time-restricted feeding has also been evaluated in individuals with pre-diabetes, who were assigned into a group that ate between 8AM-2PM and fasted for the remaining 18 hours and compared them with individuals who followed their routine pattern of food intake between 8AM-8PM with a period of 12 hours fasted in between (211). Time-restricted feeding resulted in dramatically lowered insulin levels and improved insulin sensitivity and β cell responsiveness, and lower blood pressure without altering circulating lipoprotein levels or markers of inflammation, without adverse effects. Taken together, while these clinical studies have demonstrated a beneficial effect of intermittent fasting in the setting of diabetes and pre-diabetes, it remains unclear if this strategy is sustainable (as the longest clinical trial lasted one year (220)) and if the beneficial effects are durable. It is also important to note that fasting strategies may be detrimental in individuals with type 1 diabetes which is characterized by primary beta cell failure and insulin deficiency, and provoke potentially fatal hypoglycemia; and therefore not a subject of clinical investigation.
Effects of fasting regimens on weight loss
Multiple studies have examined the effects of IF and ADF on weight loss, with most studies supporting that both regimens result in weight loss proportional to the number of fasting days (reviewed in (129)). On average, IF regimens were associated with 0.25 kg/week of weight loss whereas ADF regimens were associated with 0.75 kg/week of weight loss. Additionally, studies in which a small meal replacement was given on the fasting days (<400 kCal) resulted in the most significant weight loss(20). Based on these studies, authors have suggested recommending IF for overweight, at risk patients who wish to achieve slower weight loss, while recommending ADF for patients who wish to achieve faster weight loss. These studies have been informative with regards to the degree of caloric restriction with fasting, insofar as consuming a small meal on fasting days was surprisingly associated with greater weight loss. In studies of both IF and ADF, weight loss correlated with visceral fat loss(20). Findings from trials of IF and ADF consistently show weight loss of 3–8%, with similar visceral fat loss in the range of 4–7%. One key limitation of these studies is that visceral fat was assessed by measurement of waist circumference rather more robust metrics for visceral fat determination, such as magnetic resonance imaging, which can assess the number of kilograms of visceral fat lost more precisely.
Effects of fasting regimens on glucose and insulin levels
The ability to prevent progression of pre-diabetes (defined as hemoglobin A1C 5.7–6.4% or fasting glucose level of 100–125 mg/dL) to overt diabetes via lifestyle modifications is well established. In studies of IF in pre-diabetic individuals, consistent yet small reductions in fasting glucose on the order of 5% have been observed. This was accompanied by 20–30% reduction in insulin levels, indicative of improved insulin sensitivity(105). In multiple studies, the overall percentage reduction in insulin levels were correlated with the total reduction in caloric consumption, rather than the number of fasting days (223, 224). This suggests that the overall reductions in insulin levels may be attributable to the overall caloric restriction that occurs with fasting. That reductions in fasting glucose were accompanied with decreased insulin levels suggests improvements in insulin sensitivity in patients after intermittent fasting. IF has been shown to decrease insulin resistance as measured by homeostatic model assessment (HOMA-IR)(212). Thus, the overall metabolic effects of IF are more significant with respect to weight loss and increased insulin sensitivity, with less pronounced effects on glucose levels.
Comparative analyses of studies with persistent calorie restriction versus intermittent fasting (define as reduced calorie intake 1–3 times per week) of alternate day fasting (with >75% reduction in calorie intake alternating with ad-lib feeding) in humans have revealed greater weight loss with persistent calorie restriction but comparable reductions in circulating glucose, insulin, and insulin resistance (assessed with HOMA-IR; reviewed in (20)). While some sex-specific effects of intermittent fasting were noted on specific metabolic parameters in a study comparing the effects of alternate day fasting in non-obese men versus women (87), the salutary metabolic effects have been and large been uniformly observed in both sexes (252).
IF in hypertension and inflammatory disease
Hypertension is a major risk factor for stroke, myocardial infarction, and subsequent heart failure. In addition to metabolic regulation of glucose homeostasis, periodic fasting may decrease blood pressure. In a study of medically supervised water-only fasting in 68 patients, the average blood pressure reduction was 20/7 mmHg (74), with pressures remaining lower after subjects were subsequently fed a vegan diet. An earlier study from the same group in frankly hypertensive subjects also observed substantial reductions in blood pressure in 168 subjects (73). Both studies were limited by the lack of a proper control group.
IF regimens have been tested in inflammatory and autoimmune diseases including asthma and rheumatoid arthritis (RA) (94, 135). In one small study, alternate day fasting resulted in significant reductions in inflammatory markers in asthmatic patients compared with controls (94). Furthermore, in four studies, symptoms of RA were reduced by at least one week of IF, although reductions in symptoms only persisted if a vegan diet was instituted after the fasting period (141). Interestingly, in a small study of healthy volunteers, Traba et al. recently showed that acute fasting decreases inflammasome activation (219), suggesting a possible mechanism for improvements in inflammasome-mediated disease observed in human clinical trials. Interestingly, in this study, oxygen consumption rates were higher and associated with lower reactive oxygen species levels in peripheral blood mononuclear cells isolated from fasted as compared with fed state, suggesting improved mitochondrial metabolism and/or mitochondrial quality conferred by fasting. Overall, relatively small studies show promise for utilization of fasting regimens in the treatment of hypertension and inflammatory diseases, although larger prospective randomized clinical trials are needed to address this important issue.
Intermittent fasting: most recent trials
Unsurprisingly, IF has gained popularity as a dietary strategy amongst otherwise healthy individuals. A recent non-randomized trial of IF (24 hours/week) in healthy volunteers found significant reductions in fat mass compared to a control group, but no other changes in metabolic biomarkers or parameters, including insulin, glucose, insulin resistance, insulin-like growth factor-1, lipids, liver enzymes, or hemoglobin A1c (101). In general, this study supports the concept that once weekly intermittent fasting is safe and well tolerated in healthy individuals. Recently, ADF (25% of energy needs on fasting days, 125% on feast days) and chronic caloric restriction (75% of energy needs daily) were compared in a randomized controlled clinical trial of 100 patients (220). Overall, although both groups were associated with weight loss compared with control patients, there were no significant differences between ADF and chronic caloric restriction, although caloric restriction was better tolerated, with fewer drop outs. The authors concluded that ADF did not produce superior weight loss, weight maintenance, adherence, or cardioproteciton compared to chronic caloric restriction. In this randomized study, those randomized to ADF tended to consume more than their prescribed calorie goals on fasting days and less on feast days, highlighting the difficulties of randomization in dietary intervention studies.
Development of fasting-mimicking diets to harness the benefits of intermittent fasting
Recognizing the potential challenges on adherence to various regimens of intermittent fasting, researchers have developed a fasting mimicking diet (FMD), which involves calorie restriction to <800 calories on average for 5 days in a row, every month while consuming a plant-based diet rich in fats and complex carbohydrates (25, 229). Three cycles of the FMD could be reliably implemented by otherwise healthy humans with desired compliance in >70% of individuals (229). Importantly, 3 cycles of FMD and were effective in reducing body weight, trunk and total body fat, waist circumference, both systolic and diastolic blood pressure, and IGF-1 in comparison to a normal ad-lib diet. In addition, 3 cycles of FMD were effective in reducing total cholesterol, LDL and HDL cholesterol, and relative lean body mass was increased, and levels of circulating C-reactive protein, a potent marker for cardio-metabolic risk was reduced to normal levels in subjects with elevated CRP at baseline (and sustained for 3 months) indicating a favorable effect on global cardio-metabolic risk factors (229).
One outstanding question that remains unanswered is what is the significance of dietary content and exercise in the setting of fasting or caloric restriction. Recently, one study tested the efficacy of a high-protein, calorie-restricted diet with and without resistance training in older adults. These authors found that resistance training was required to maintain lean body mass, even in the setting of a high-protein diet (6), suggesting that resistance training exercise can prevent loss of muscle mass that occurs despite high protein intake in the face of caloric restriction.
As discussed above, intermittent fasting regimens and exercise-based interventions (that are reviewed elsewhere) have tremendous preventive benefits and potential for development as therapeutic modalities in humans, to facilitate healthspan and fight disease. Given a central role for the lysosome machinery as the sub-cellular orchestrators of these beneficial effects, it remains to be seen whether administration of small moleculae activators of the lysosome biogenesis program (60, 226) may also confer similar benefits in humans with an accceptable safety profile.
Conclusion
With discovery of transcriptional activators and repressors of lysosomal biogenesis, as well as development of tools to target with genetic and pharmacologic approaches, the last decade has witnessed an explosion of knowledge in lysosome biology. The emerging paradigm defines the lysosome as a highly dynamic and multi-functional organelle that plays critical roles cellular nutrient regulation, trafficking pathways and organelle quality control. These processes ensure cellular survival. Conversely lysosomes can also be effectors of cell death via lysosomal permeabilization with leakage of the degradative enzymes in the cytosol, or inappropriate activation of lysosomal processes such as autophagy or autosis (see Figure 6). Studies from our lab and others point to intermittent fasting as an attractive strategy to harness lysosome function to prevent and treat cardiovascular pathology. Indeed, intermittent fasting is a feasible and sustainable approach that is being actively evaluated in human clinical trials, and may be ready for translation in the near future. Developing small molecules to target the lysosomal machinery and/or the lysosome biogenesis program is an area of actively investigation. Whether the efficacy and safety of these small molecule based approaches will compare favorably to intermittent fasting and other calorie restriction based strategies, remains to be seen.
Figure 6: Lysosomal regulation of life and death.
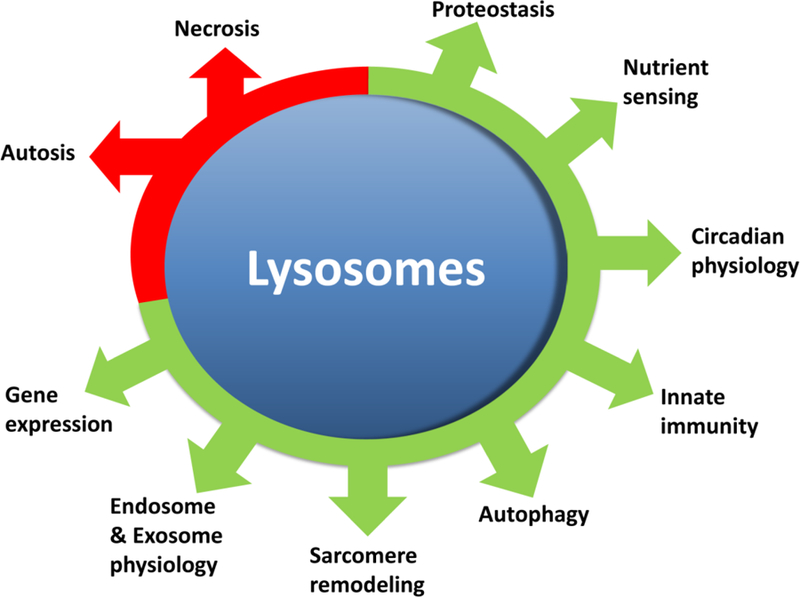
Lysosomes play multiple salutary roles as well as participate in orchestrating cell death programs. Necrosis and autosis (or autophagic cell death) are both characterized by lysosomal digestion of cellular contents. Please see reference (67) for a comprehensive review of the role of lysosomes and autophagy in cell death. In contrast to this destructive role, lysosomes play critical roles in both synthesis and non-proteasomal breakdown of proteins. By virtue of the Lysosomal NUtrient Sensing mechanisms, they control cellular metabolic choices as well as circadian physiology. Lysosomes are also vital for cellular innate immunity against pathogens as well as dysfunctional organelles, sarcomeres and even, pathologically aggregated proteins. Through effects on TFEB and mTORC1, lysosomes can control gene expression as well as the translational machinery. Finally, processing of both intracellular and extracellular messages, via exosomes and endosomes, is regulated via lysosomal function.
Didactic Synopsis.
Major teaching points:
Lysosomes play a critical role in cardiac myocyte homeostasis and preserving normal cardiac structure and function.
Lysosomes regulate substrate supply to maintain cardiac energetics and maintain organelle quality by degrading damaged organelles via autophagy, to promote cardiac myocyte homeostasis in both fed and fasted states.
Lysosome nutrient sensing complex (LYNUS) plays a critical role in transcriptional control of autophagy in cardiac myocytes.
Intermittent fasting stimulates the lysosome machinery to precondition the myocardium, and attenuate effects of injurious stimuli as well as attenuate development of adverse ventricular remodeling in response.
Intermittent fasting benefits cardiometabolic parameters in healthy humans, as well as those suffering from diabetes or other risk factors for development of cardiac diseases.
Cross References.
Intracellular digestion: lysosomes and cellular injury (legacy)
Mitophagy as a protective mechanism against myocardial stress
Intermittent fasting regulation of cardiac function
Cardiac metabolism in perspective
Circadian rhythms (legacy)
Acknowledgements
This work was supported by grants from National Institutes of Health (HL107594), the Department of Veterans Affairs (I01BX000448, 1I01BX001969, I01BX004235) and Children’s Discovery Institute of Washington University School of Medicine and the St. Louis Children’s Hospital to A.D.; from the NIH (K08HL138262–01) to A.J.; and a seed grant from the John Cochran VA Medical Center to K.M.
Bibliography
- 1.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358: 807–813, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acosta-Rodriguez VA, de Groot MHM, Rijo-Ferreira F, Green CB, and Takahashi JS. Mice under Caloric Restriction Self-Impose a Temporal Restriction of Food Intake as Revealed by an Automated Feeder System. Cell Metab 26: 267–277 e262, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adeva-Andany MM, Gonzalez-Lucan M, Donapetry-Garcia C, Fernandez-Fernandez C, and Ameneiros-Rodriguez E. Glycogen metabolism in humans. BBA Clin 5: 85–100, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmet I, Wan R, Mattson MP, Lakatta EG, and Talan M. Cardioprotection by intermittent fasting in rats. Circulation 112: 3115–3121, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Ahmet I, Wan R, Mattson MP, Lakatta EG, and Talan M. Cardioprotection by intermittent fasting in rats. Circulation 112: 3115–3121, 2005. [DOI] [PubMed] [Google Scholar]
- 6.Amamou T, Normandin E, Pouliot J, Dionne IJ, Brochu M, and Riesco E. Effect of a High-Protein Energy-Restricted Diet Combined with Resistance Training on Metabolic Profile in Older Individuals with Metabolic Impairments. J Nutr Health Aging 21: 67–74, 2017. [DOI] [PubMed] [Google Scholar]
- 7.An L, Hu XW, Zhang S, Hu X, Song Z, Naz A, Zi Z, Wu J, Li C, Zou Y, He L, and Zhu H. UVRAG Deficiency Exacerbates Doxorubicin-Induced Cardiotoxicity. Sci Rep 7: 43251, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anding AL, and Baehrecke EH. Cleaning House: Selective Autophagy of Organelles. Developmental cell 41: 10–22, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anson RM, Guo Z, de CR, Iyun T, Rios M, Hagepanos A, Ingram DK, Lane MA, and Mattson MP. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. ProcNatlAcadSciUSA 100: 6216–6220, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Appaix F, Kuznetsov AV, Usson Y, Kay L, Andrienko T, Olivares J, Kaambre T, Sikk P, Margreiter R, and Saks V. Possible role of cytoskeleton in intracellular arrangement and regulation of mitochondria. Exp Physiol 88: 175–190, 2003. [DOI] [PubMed] [Google Scholar]
- 11.Arad M, Maron BJ, Gorham JM, Johnson WH Jr., Saul JP, Perez-Atayde AR, Spirito P, Wright GB, Kanter RJ, Seidman CE, and Seidman JG Glycogen storage diseases presenting as hypertrophic cardiomyopathy. NEnglJMed 352: 362–372, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Arad M, Seidman CE, and Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res 100: 474–488, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, and Lamming DW. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 15: 28–38, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arriola Apelo SI, Pumper CP, Baar EL, Cummings NE, and Lamming DW. Intermittent Administration of Rapamycin Extends the Life Span of Female C57BL/6J Mice. J Gerontol A Biol Sci Med Sci 71: 876–881, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asher G, and Sassone-Corsi P. Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell 161: 84–92, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, and Kelly DP. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 133: 698–705, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balaban RS, Kantor HL, Katz LA, and Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science 232: 1121–1123, 1986. [DOI] [PubMed] [Google Scholar]
- 18.Bales CW, and Kraus WE. Caloric restriction: implications for human cardiometabolic health. J Cardiopulm Rehabil Prev 33: 201–208, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bao J, Zheng L, Zhang Q, Li X, Zhang X, Li Z, Bai X, Zhang Z, Huo W, Zhao X, Shang S, Wang Q, Zhang C, and Ji J. Deacetylation of TFEB promotes fibrillar Abeta degradation by upregulating lysosomal biogenesis in microglia. Protein Cell 7: 417–433, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnosky AR, Hoddy KK, Unterman TG, and Varady KA. Intermittent fasting vs daily calorie restriction for type 2 diabetes prevention: a review of human findings. Transl Res 164: 302–311, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Batacan RB Jr., Duncan MJ, Dalbo VJ, Connolly KJ, and Fenning AS Light-intensity and high-intensity interval training improve cardiometabolic health in rats. Appl Physiol Nutr Metab 41: 945–952, 2016. [DOI] [PubMed] [Google Scholar]
- 22.Bedi KC Jr., Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, and Rame JE Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133: 706–716, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beli E, Yan Y, Moldovan L, Vieira CP, Gao R, Duan Y, Prasad R, Bhatwadekar A, White FA, Townsend S, Chan L, Ryan CN, Morton D, Moldovan EG, Chu FI, Oudit GY, Derendorf H, Adorini L, Wang XX, Evans-Molina C, Mirmira RG, Boulton ME, Yoder MC, Li Q, Levi M, Busik JV, and Grant MB. Restructuring of the Gut Microbiome by Intermittent Fasting Prevents Retinopathy and Prolongs Survival in db/db Mice. Diabetes 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billia F, Hauck L, Konecny F, Rao V, Shen J, and Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. ProcNatlAcadSciUSA 108: 9572–9577, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandhorst S, Choi IY, Wei M, Cheng CW, Sedrakyan S, Navarrete G, Dubeau L, Yap LP, Park R, Vinciguerra M, Di Biase S, Mirzaei H, Mirisola MG, Childress P, Ji L, Groshen S, Penna F, Odetti P, Perin L, Conti PS, Ikeno Y, Kennedy BK, Cohen P, Morgan TE, Dorff TB, and Longo VD. A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab 22: 86–99, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bravo-San Pedro JM, Kroemer G, and Galluzzi L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res 120: 1812–1824, 2017. [DOI] [PubMed] [Google Scholar]
- 27.Brewer RA, Collins HE, Berry RD, Brahma MK, Tirado BA, Peliciari-Garcia RA, Stanley HL, Wende AR, Taegtmeyer H, Rajasekaran NS, Darley-Usmar V, Zhang J, Frank SJ, Chatham JC, and Young ME. Temporal partitioning of adaptive responses of the murine heart to fasting. Life Sci 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brisson L, Banski P, Sboarina M, Dethier C, Danhier P, Fontenille MJ, Van Hee VF, Vazeille T, Tardy M, Falces J, Bouzin C, Porporato PE, Frederick R, Michiels C, Copetti T, and Sonveaux P. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 30: 418–431, 2016. [DOI] [PubMed] [Google Scholar]
- 29.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, and Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303: 2011–2015, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M, Izumo S, Katus HA, and Hardt SE. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. JAmCollCardiol 54: 2435–2446, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Camandola S, and Mattson MP. Brain metabolism in health, aging, and neurodegeneration. The EMBO journal 36: 1474–1492, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao R, Robinson B, Xu H, Gkogkas C, Khoutorsky A, Alain T, Yanagiya A, Nevarko T, Liu AC, Amir S, and Sonenberg N. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron 79: 712–724, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson AJ, and Hoelzel F. Apparent prolongation of the life span of rats by intermittent fasting. The Journal of nutrition 31: 363–375, 1946. [DOI] [PubMed] [Google Scholar]
- 34.Castellano BM, Thelen AM, Moldavski O, Feltes M, van der Welle RE, Mydock-McGrane L, Jiang X, van Eijkeren RJ, Davis OB, Louie SM, Perera RM, Covey DF, Nomura DK, Ory DS, and Zoncu R. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 355: 1306–1311, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaix A, Zarrinpar A, and Panda S. The circadian coordination of cell biology. The Journal of cell biology 215: 15–25, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers KT, Leone TC, Sambandam N, Kovacs A, Wagg CS, Lopaschuk GD, Finck BN, and Kelly DP. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. JBiolChem 286: 11155–11162, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chandramouli C, Varma U, Stevens EM, Xiao RP, Stapleton DI, Mellor KM, and Delbridge LM. Myocardial glycogen dynamics: new perspectives on disease mechanisms. Clin Exp Pharmacol Physiol 42: 415–425, 2015. [DOI] [PubMed] [Google Scholar]
- 38.Chantranupong L, Wolfson RL, and Sabatini DM. Nutrient-sensing mechanisms across evolution. Cell 161: 67–83, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng CW, Villani V, Buono R, Wei M, Kumar S, Yilmaz OH, Cohen P, Sneddon JB, Perin L, and Longo VD. Fasting-Mimicking Diet Promotes Ngn3-Driven beta-Cell Regeneration to Reverse Diabetes. Cell 168: 775–788 e712, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiao YA, Kolwicz SC, Basisty N, Gagnidze A, Zhang J, Gu H, Djukovic D, Beyer RP, Raftery D, MacCoss M, Tian R, and Rabinovitch PS. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY) 8: 314–327, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, and Huang J. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 510: 397–401, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, and Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325: 201–204, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cotter DG, Schugar RC, Wentz AE, d’Avignon DA, and Crawford PA. Successful adaptation to ketosis by mice with tissue-specific deficiency of ketone body oxidation. Am J Physiol Endocrinol Metab 304: E363–374, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ, and Rabinovitch PS. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13: 529–539, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, and Schibler U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes & development 14: 2950–2961, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dassanayaka S, and Jones SP. O-GlcNAc and the cardiovascular system. Pharmacol Ther 142: 62–71, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Decker RS, Poole AR, Crie JS, Dingle JT, and Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. II. Immunohistochemical and biochemical changes in cathepsin D. AmJPathol 98: 445–456, 1980. [PMC free article] [PubMed] [Google Scholar]
- 48.Decker RS, Poole AR, and Wildenthal K. Distribution of lysosomal cathepsin D in normal, ischemic, and starved rabbit cardiac myocytes. Circ Res 46: 485–494, 1980. [DOI] [PubMed] [Google Scholar]
- 49.Decker RS, and Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. The American journal of pathology 98: 425–444, 1980. [PMC free article] [PubMed] [Google Scholar]
- 50.Delbridge LMD, Mellor KM, Taylor DJ, and Gottlieb RA. Myocardial stress and autophagy: mechanisms and potential therapies. Nat Rev Cardiol 14: 412–425, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, Di Giovannantonio LG, Nusco E, Platt N, Guida A, Ogmundsdottir MH, Lanfrancone L, Perera RM, Zoncu R, Pelicci PG, Settembre C, and Ballabio A. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356: 1188–1192, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Diwan A Regulation of the transcription factor EB-PGC1alpha axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress. Autophagy 35: 956–976, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, and Sabatini DM. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493: 679–683, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Egan D, Kim J, Shaw RJ, and Guan KL. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7: 643–644, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, and Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Carmona-Gutierrez D, Buttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, Horsch M, Moreth K, Beckers J, Fuchs H, Gailus-Durner V, Neff F, Janik D, Rathkolb B, Rozman J, de Angelis MH, Moustafa T, Haemmerle G, Mayr M, Willeit P, von Frieling-Salewsky M, Pieske B, Scorrano L, Pieber T, Pechlaner R, Willeit J, Sigrist SJ, Linke WA, Muhlfeld C, Sadoshima J, Dengjel J, Kiechl S, Kroemer G, Sedej S, and Madeo F. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nature medicine 22: 1428–1438, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola F, Harger A, Zimmermann A, Moustafa T, Sprenger A, Jany E, Buttner S, Carmona-Gutierrez D, Ruckenstuhl C, Ring J, Reichelt W, Schimmel K, Leeb T, Moser C, Schatz S, Kamolz LP, Magnes C, Sinner F, Sedej S, Frohlich KU, Juhasz G, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, and Madeo F. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab 19: 431–444, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Elkhwanky MS, and Hakkola J. Extranuclear Sirtuins and Metabolic Stress. Antioxid Redox Signal 2017. [DOI] [PubMed] [Google Scholar]
- 59.Ellenbroek JH, van Dijck L, Tons HA, Rabelink TJ, Carlotti F, Ballieux BE, and de Koning EJ. Long-term ketogenic diet causes glucose intolerance and reduced beta- and alpha-cell mass but no weight loss in mice. Am J Physiol Endocrinol Metab 306: E552–558, 2014. [DOI] [PubMed] [Google Scholar]
- 60.Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, and Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arteriosclerosis, thrombosis, and vascular biology 34: 1942–1952, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng Y, Yao Z, and Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends in cell biology 25: 354–363, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferreira CR, and Gahl WA. Lysosomal storage diseases. Transl Sci Rare Dis 2: 1–71, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fillmore N, and Lopaschuk GD. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochimica et biophysica acta 1833: 857–865, 2013. [DOI] [PubMed] [Google Scholar]
- 64.Flynn JM, O’Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, Zykovich A, Mooney SD, Strong R, Rosen CJ, Kapahi P, Nelson MD, Kennedy BK, and Melov S. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 12: 851–862, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fontana L, Meyer TE, Klein S, and Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proceedings of the National Academy of Sciences of the United States of America 101: 6659–6663, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fontana L, Vinciguerra M, and Longo VD. Growth factors, nutrient signaling, and cardiovascular aging. Circ Res 110: 1139–1150, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, and Kroemer G. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death and differentiation 25: 486–541, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gatica D, Chiong M, Lavandero S, and Klionsky DJ. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res 116: 456–467, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gatica D, Lahiri V, and Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nature cell biology 20: 233–242, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giebultowicz J, and Kapahi P. Circadian clocks and metabolism: the nutrient-sensing AKT and TOR pathways make the link. Current biology : CB 20: R608–609, 2010. [DOI] [PubMed] [Google Scholar]
- 71.Glatz JF, Nabben M, Heather LC, Bonen A, and Luiken JJ. Regulation of the subcellular trafficking of CD36, a major determinant of cardiac fatty acid utilization. Biochimica et biophysica acta 1861: 1461–1471, 2016. [DOI] [PubMed] [Google Scholar]
- 72.Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, Crosby SD, Saftig P, and Diwan A. Repetitive stimulation of autophagy-lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy 11: 1537–1560, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goldhamer A, Lisle D, Parpia B, Anderson SV, and Campbell TC. Medically supervised water-only fasting in the treatment of hypertension. J Manipulative Physiol Ther 24: 335–339, 2001. [DOI] [PubMed] [Google Scholar]
- 74.Goldhamer AC, Lisle DJ, Sultana P, Anderson SV, Parpia B, Hughes B, and Campbell TC. Medically supervised water-only fasting in the treatment of borderline hypertension. J Altern Complement Med 8: 643–650, 2002. [DOI] [PubMed] [Google Scholar]
- 75.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, and Dorn GW, 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350: aad2459, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goodrick CL, Ingram DK, Reynolds MA, Freeman JR, and Cider N. Effects of intermittent feeding upon body weight and lifespan in inbred mice: interaction of genotype and age. MechAgeing Dev 55: 69–87, 1990. [DOI] [PubMed] [Google Scholar]
- 77.Gow ML, Garnett SP, Baur LA, and Lister NB. The Effectiveness of Different Diet Strategies to Reduce Type 2 Diabetes Risk in Youth. Nutrients 8: 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo B, Liang Q, Li L, Hu Z, Wu F, Zhang P, Ma Y, Zhao B, Kovacs AL, Zhang Z, Feng D, Chen S, and Zhang H. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nature cell biology 16: 1215–1226, 2014. [DOI] [PubMed] [Google Scholar]
- 79.Guzun R, Kaambre T, Bagur R, Grichine A, Usson Y, Varikmaa M, Anmann T, Tepp K, Timohhina N, Shevchuk I, Chekulayev V, Boucher F, Dos Santos P, Schlattner U, Wallimann T, Kuznetsov AV, Dzeja P, Aliev M, and Saks V. Modular organization of cardiac energy metabolism: energy conversion, transfer and feedback regulation. Acta Physiol (Oxf) 213: 84–106, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Halestrap AP. Monocarboxylic acid transport. Compr Physiol 3: 1611–1643, 2013. [DOI] [PubMed] [Google Scholar]
- 81.Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, Li H, Kitsis RN, Dorn Ii GW, Sadoshima J, Ellisman MH, and Gustafsson AB. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 8: 14050, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, and Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889, 2006. [DOI] [PubMed] [Google Scholar]
- 83.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, and Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. CircRes 107: 1470–1482, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harvie MN, Pegington M, Mattson MP, Frystyk J, Dillon B, Evans G, Cuzick J, Jebb SA, Martin B, Cutler RG, Son TG, Maudsley S, Carlson OD, Egan JM, Flyvbjerg A, and Howell A. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. IntJObes(Lond) 35: 714–727, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JA, Ellisman MH, and Panda S. Time-Restricted Feeding without Reducing Caloric Intake Prevents Metabolic Diseases in Mice Fed a High-Fat Diet. Cell Metab 15: 848–860, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He C, and Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43: 67–93, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Heilbronn LK, Smith SR, Martin CK, Anton SD, and Ravussin E. Alternate-day fasting in nonobese subjects: effects on body weight, body composition, and energy metabolism. The American journal of clinical nutrition 81: 69–73, 2005. [DOI] [PubMed] [Google Scholar]
- 88.Honjoh S, Yamamoto T, Uno M, and Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 457: 726–730, 2009. [DOI] [PubMed] [Google Scholar]
- 89.Horne BD, May HT, Anderson JL, Kfoury AG, Bailey BM, McClure BS, Renlund DG, Lappe DL, Carlquist JF, Fisher PW, Pearson RR, Bair TL, Adams TD, Muhlestein JB, and Intermountain Heart Collaborative S. Usefulness of routine periodic fasting to lower risk of coronary artery disease in patients undergoing coronary angiography. Am J Cardiol 102: 814–819, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Horne BD, Muhlestein JB, May HT, Carlquist JF, Lappe DL, Bair TL, Anderson JL, and Intermountain Heart Collaborative Study G. Relation of routine, periodic fasting to risk of diabetes mellitus, and coronary artery disease in patients undergoing coronary angiography. Am J Cardiol 109: 1558–1562, 2012. [DOI] [PubMed] [Google Scholar]
- 91.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, and Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116: 264–278, 2015. [DOI] [PubMed] [Google Scholar]
- 92.Jeremy M Berg JLT, and Lubert Stryer. Biochemistry 2002. [Google Scholar]
- 93.Jia G, Hill MA, and Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res 122: 624–638, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Johnson JB, Summer W, Cutler RG, Martin B, Hyun DH, Dixit VD, Pearson M, Nassar M, Telljohann R, Maudsley S, Carlson O, John S, Laub DR, and Mattson MP. Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic Biol Med 42: 665–674, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kamphoven JH, Stubenitsky R, Reuser AJ, Van Der Ploeg AT, Verdouw PD, and Duncker DJ. Cardiac remodeling and contractile function in acid alpha-glucosidase knockout mice. Physiol Genomics 5: 171–179, 2001. [DOI] [PubMed] [Google Scholar]
- 96.Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, Li L, Kawamura I, Takeyama T, Kawaguchi T, Nagashima K, Fujiwara T, Fujiwara H, Seishima M, and Minatoguchi S. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. AmJPathol 174: 1705–1714, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Katare RG, Kakinuma Y, Arikawa M, Yamasaki F, and Sato T. Chronic intermittent fasting improves the survival following large myocardial ischemia by activation of BDNF/VEGF/PI3K signaling pathway. JMolCell Cardiol 46: 405–412, 2009. [DOI] [PubMed] [Google Scholar]
- 98.Kaur J, and Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nature reviews Molecular cell biology 16: 461–472, 2015. [DOI] [PubMed] [Google Scholar]
- 99.Kaushik S, and Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 12: 432–438, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kemper JK, Xiao Z, Ponugoti B, Miao J, Fang S, Kanamaluru D, Tsang S, Wu SY, Chiang CM, and Veenstra TD. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab 10: 392–404, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kessler CS, Stange R, Schlenkermann M, Jeitler M, Michalsen A, Selle A, Raucci F, and Steckhan N. A nonrandomized controlled clinical pilot trial on 8 wk of intermittent fasting (24 h/wk). Nutrition 46: 143–152 e142, 2018. [DOI] [PubMed] [Google Scholar]
- 102.Kim TT, and Dyck JR. The role of CD36 in the regulation of myocardial lipid metabolism. Biochimica et biophysica acta 1861: 1450–1460, 2016. [DOI] [PubMed] [Google Scholar]
- 103.Kim YC, Park HW, Sciarretta S, Mo JS, Jewell JL, Russell RC, Wu X, Sadoshima J, and Guan KL. Rag GTPases are cardioprotective by regulating lysosomal function. Nat Commun 5: 4241, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kingma SD, Bodamer OA, and Wijburg FA. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract Res Clin Endocrinol Metab 29: 145–157, 2015. [DOI] [PubMed] [Google Scholar]
- 105.Klempel MC, Kroeger CM, Bhutani S, Trepanowski JF, and Varady KA. Intermittent fasting combined with calorie restriction is effective for weight loss and cardio-protection in obese women. Nutr J 11: 98, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kolwicz SC Jr., Purohit S, and Tian R Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113: 603–616, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kubli DA, Cortez MQ, Moyzis AG, Najor RH, Lee Y, and Gustafsson AB. PINK1 Is Dispensable for Mitochondrial Recruitment of Parkin and Activation of Mitophagy in Cardiac Myocytes. PloS one 10: e0130707, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, and Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. JBiolChem 288: 915–926, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, and Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036, 2004. [DOI] [PubMed] [Google Scholar]
- 110.Kumar S, and Kaur G. Intermittent fasting dietary restriction regimen negatively influences reproduction in young rats: a study of hypothalamo-hypophysial-gonadal axis. PloS one 8: e52416, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF, Vasquez DS, Juguilon H, Panda S, Shaw RJ, Thompson CB, and Evans RM. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 326: 437–440, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lange LG, and Sobel BE. Mitochondrial dysfunction induced by fatty acid ethyl esters, myocardial metabolites of ethanol. The Journal of clinical investigation 72: 724–731, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, and Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 516: 112–115, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG, and Hill JA. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 133: 1668–1687, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li G, Xie C, Lu S, Nichols RG, Tian Y, Li L, Patel D, Ma Y, Brocker CN, Yan T, Krausz KW, Xiang R, Gavrilova O, Patterson AD, and Gonzalez FJ. Intermittent Fasting Promotes White Adipose Browning and Decreases Obesity by Shaping the Gut Microbiota. Cell Metab 26: 801, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lim CY, and Zoncu R. The lysosome as a command-and-control center for cellular metabolism. The Journal of cell biology 214: 653–664, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lim JA, Li L, Shirihai OS, Trudeau KM, Puertollano R, and Raben N. Modulation of mTOR signaling as a strategy for the treatment of Pompe disease. EMBO molecular medicine 9: 353–370, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lipton JO, Yuan ED, Boyle LM, Ebrahimi-Fakhari D, Kwiatkowski E, Nathan A, Guttler T, Davis F, Asara JM, and Sahin M. The Circadian Protein BMAL1 Regulates Translation in Response to S6K1-Mediated Phosphorylation. Cell 161: 1138–1151, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu H, Javaheri A, Godar RJ, Murphy J, Ma X, Rohatgi N, Mahadevan J, Hyrc K, Saftig P, Marshall C, McDaniel ML, Remedi MS, Razani B, Urano F, and Diwan A. Intermittent Fasting Preserves Beta-Cell Mass in Obesity-induced Diabetes via the Autophagy-Lysosome Pathway. Autophagy [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Luo W, Ma S, Yang Y, Wang C, Zhang D, Zhang Q, Liu Y, and Liu Z. TFEB regulates PER3 expression via glucose-dependent effects on CLOCK/BMAL1. The international journal of biochemistry & cell biology 78: 31–42, 2016. [DOI] [PubMed] [Google Scholar]
- 121.Ma S, and Gladyshev VN. Molecular signatures of longevity: Insights from cross-species comparative studies. Semin Cell Dev Biol 70: 190–203, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, and Diwan A. Autophagy is impaired in cardiac ischemia-reperfusion injury. Autophagy 8: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, and Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 125: 3170–3181, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, Weinheimer CJ, Barger PM, and Diwan A. Regulation of TFEB-PGC1alpha Axis by BECLIN-1 Controls Mitochondrial Quality and Cardiomyocyte Death under Stress. Molecular and cellular biology 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, Crosby SD, JeBailey L, Wang X, Takahashi JS, and Bass J. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466: 627–631, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, Enot DP, Fernandez AF, Martins I, Kepp O, Senovilla L, Bauvy C, Morselli E, Vacchelli E, Bennetzen M, Magnes C, Sinner F, Pieber T, Lopez-Otin C, Maiuri MC, Codogno P, Andersen JS, Hill JA, Madeo F, and Kroemer G. Regulation of autophagy by cytosolic acetyl-coenzyme A. Molecular cell 53: 710–725, 2014. [DOI] [PubMed] [Google Scholar]
- 127.Martin B, Pearson M, Kebejian L, Golden E, Keselman A, Bender M, Carlson O, Egan J, Ladenheim B, Cadet JL, Becker KG, Wood W, Duffy K, Vinayakumar P, Maudsley S, and Mattson MP. Sex-dependent metabolic, neuroendocrine, and cognitive responses to dietary energy restriction and excess. Endocrinology 148: 4318–4333, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinez-Lopez N, Tarabra E, Toledo M, Garcia-Macia M, Sahu S, Coletto L, Batista-Gonzalez A, Barzilai N, Pessin JE, Schwartz GJ, Kersten S, and Singh R. System-wide Benefits of Intermeal Fasting by Autophagy. Cell Metab 26: 856–871 e855, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mattson MP, Longo VD, and Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res Rev 39: 46–58, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.McGinnis GR, Tang Y, Brewer RA, Brahma MK, Stanley HL, Shanmugam G, Rajasekaran NS, Rowe GC, Frank SJ, Wende AR, Abel ED, Taegtmeyer H, Litovsky S, Darley-Usmar V, Zhang J, Chatham JC, and Young ME. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin-mediated processes in the heart. Journal of molecular and cellular cardiology 110: 80–95, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, and Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nature cell biology 17: 288–299, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Meikle PJ, Hopwood JJ, Clague AE, and Carey WF. Prevalence of lysosomal storage disorders. JAMA 281: 249–254, 1999. [DOI] [PubMed] [Google Scholar]
- 133.Meyer TE, Kovacs SJ, Ehsani AA, Klein S, Holloszy JO, and Fontana L. Long-term caloric restriction ameliorates the decline in diastolic function in humans. Journal of the American College of Cardiology 47: 398–402, 2006. [DOI] [PubMed] [Google Scholar]
- 134.Mialet-Perez J, and Vindis C. Autophagy in health and disease: focus on the cardiovascular system. Essays Biochem 61: 721–732, 2017. [DOI] [PubMed] [Google Scholar]
- 135.Michalsen A, Riegert M, Ludtke R, Backer M, Langhorst J, Schwickert M, and Dobos GJ. Mediterranean diet or extended fasting’s influence on changing the intestinal microflora, immunoglobulin A secretion and clinical outcome in patients with rheumatoid arthritis and fibromyalgia: an observational study. BMC Complement Altern Med 5: 22, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mitchell G, and Isberg RR. Innate Immunity to Intracellular Pathogens: Balancing Microbial Elimination and Inflammation. Cell Host Microbe 22: 166–175, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mizushima N, and Komatsu M. Autophagy: renovation of cells and tissues. Cell 147: 728–741, 2011. [DOI] [PubMed] [Google Scholar]
- 138.Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, and Ohsumi Y. A protein conjugation system essential for autophagy. Nature 395: 395–398, 1998. [DOI] [PubMed] [Google Scholar]
- 139.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. MolBiolCell 15: 1101–1111, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, and Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563, 2004. [DOI] [PubMed] [Google Scholar]
- 141.Muller H, de Toledo FW, and Resch KL. Fasting followed by vegetarian diet in patients with rheumatoid arthritis: a systematic review. Scand J Rheumatol 30: 1–10, 2001. [DOI] [PubMed] [Google Scholar]
- 142.Nah J, Miyamoto S, and Sadoshima J. Mitophagy as a Protective Mechanism against Myocardial Stress. Compr Physiol 7: 1407–1424, 2017. [DOI] [PubMed] [Google Scholar]
- 143.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, and Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nature medicine 13: 619–624, 2007. [DOI] [PubMed] [Google Scholar]
- 144.Nakatogawa H, Suzuki K, Kamada Y, and Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature reviews Molecular cell biology 10: 458–467, 2009. [DOI] [PubMed] [Google Scholar]
- 145.Newman JC, Covarrubias AJ, Zhao M, Yu X, Gut P, Ng CP, Huang Y, Haldar S, and Verdin E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab 26: 547–557 e548, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nezich CL, Wang C, Fogel AI, and Youle RJ. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. The Journal of cell biology 210: 435–450, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, Sue CM, Yamamoto A, Murakami N, Shanske S, Byrne E, Bonilla E, Nonaka I, DiMauro S, and Hirano M. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406: 906–910, 2000. [DOI] [PubMed] [Google Scholar]
- 148.O’Neill JS, and Reddy AB. Circadian clocks in human red blood cells. Nature 469: 498–503, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.O’Neill JS, van Ooijen G, Dixon LE, Troein C, Corellou F, Bouget FY, Reddy AB, and Millar AJ. Circadian rhythms persist without transcription in a eukaryote. Nature 469: 554–558, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.O’Rourke EJ, and Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nature cell biology 15: 668–676, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, and Kemp BE. AMPK is a direct adenylate charge-regulated protein kinase. Science 332: 1433–1435, 2011. [DOI] [PubMed] [Google Scholar]
- 152.Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, and Sadoshima J. PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab 14: 598–611, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ouwens DM, Boer C, Fodor M, de Galan P, Heine RJ, Maassen JA, and Diamant M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 48: 1229–1237, 2005. [DOI] [PubMed] [Google Scholar]
- 154.Palmieri M, Impey S, Kang H, di RA, Pelz C, Sardiello M, and Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. HumMolGenet 20: 3852–3866, 2011. [DOI] [PubMed] [Google Scholar]
- 155.Panda S Circadian physiology of metabolism. Science 354: 1008–1015, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, and Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109: 307–320, 2002. [DOI] [PubMed] [Google Scholar]
- 157.Park S, Yoo KM, Hyun JS, and Kang S. Intermittent fasting reduces body fat but exacerbates hepatic insulin resistance in young rats regardless of high protein and fat diets. J Nutr Biochem 40: 14–22, 2017. [DOI] [PubMed] [Google Scholar]
- 158.Peliciari-Garcia RA, Goel M, Aristorenas JA, Shah K, He L, Yang Q, Shalev A, Bailey SM, Prabhu SD, Chatham JC, Gamble KL, and Young ME. Altered myocardial metabolic adaptation to increased fatty acid availability in cardiomyocyte-specific CLOCK mutant mice. Biochimica et biophysica acta 1861: 1579–1595, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pendergast JS, Nakamura W, Friday RC, Hatanaka F, Takumi T, and Yamazaki S. Robust food anticipatory activity in BMAL1-deficient mice. PloS one 4: e4860, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pendergast JS, Wendroth RH, Stenner RC, Keil CD, and Yamazaki S. mPeriod2 (Brdm1) and other single Period mutant mice have normal food anticipatory activity. Sci Rep 7: 15510, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, Peek CB, Hong H, Huang W, Omura C, Allred AL, Bradfield CA, Dinner AR, Barish GD, and Bass J. Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 350: aac4250, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Perera RM, and Zoncu R. The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol 32: 223–253, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Platt FM, Boland B, and van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. JCell Biol 199: 723–734, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM, Sardiello M, Ballabio A, and Zheng H. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO molecular medicine 6: 1142–1160, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, and Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9: 327–338, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ravikumar B, Moreau K, Jahreiss L, Puri C, and Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature cell biology 12: 747–757, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Razani B, Zhang H, Schulze PC, Schilling JD, Verbsky J, Lodhi IJ, Topkara VK, Feng C, Coleman T, Kovacs A, Kelly DP, Saffitz JE, Dorn GW 2nd, Nichols CG, and Semenkovich CF Fatty acid synthase modulates homeostatic responses to myocardial stress. The Journal of biological chemistry 286: 30949–30961, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Reichelt ME, Mellor KM, Curl CL, Stapleton D, and Delbridge LM. Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. Journal of molecular and cellular cardiology 65: 67–75, 2013. [DOI] [PubMed] [Google Scholar]
- 169.Riehle C, Wende AR, Sena S, Pires KM, Pereira RO, Zhu Y, Bugger H, Frank D, Bevins J, Chen D, Perry CN, Dong XC, Valdez S, Rech M, Sheng X, Weimer BC, Gottlieb RA, White MF, and Abel ED. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. The Journal of clinical investigation 123: 5319–5333, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Riordan MM, Weiss EP, Meyer TE, Ehsani AA, Racette SB, Villareal DT, Fontana L, Holloszy JO, and Kovacs SJ. The effects of caloric restriction- and exercise-induced weight loss on left ventricular diastolic function. American journal of physiology Heart and circulatory physiology 294: H1174–1182, 2008. [DOI] [PubMed] [Google Scholar]
- 171.Roberts MN, Wallace MA, Tomilov AA, Zhou Z, Marcotte GR, Tran D, Perez G, Gutierrez-Casado E, Koike S, Knotts TA, Imai DM, Griffey SM, Kim K, Hagopian K, Haj FG, Baar K, Cortopassi GA, Ramsey JJ, and Lopez-Dominguez JA. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab 26: 539–546 e535, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, and Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434: 113–118, 2005. [DOI] [PubMed] [Google Scholar]
- 173.Rowley WR, Bezold C, Arikan Y, Byrne E, and Krohe S. Diabetes 2030: Insights from Yesterday, Today, and Future Trends. Popul Health Manag 20: 6–12, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sabatini DD, and Adesnik M. Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge. Proceedings of the National Academy of Sciences of the United States of America 110: 13234–13235, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sacks FM, Bray GA, Carey VJ, Smith SR, Ryan DH, Anton SD, McManus K, Champagne CM, Bishop LM, Laranjo N, Leboff MS, Rood JC, de Jonge L, Greenway FL, Loria CM, Obarzanek E, and Williamson DA. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. The New England journal of medicine 360: 859–873, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, Stahl PD, and Abumrad NA. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes 64: 353–359, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sardiello M, Palmieri M, di RA, Medina DL, Valenza M, Gennarino VA, Di MC, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, and Ballabio A. A gene network regulating lysosomal biogenesis and function. Science 325: 473–477, 2009. [DOI] [PubMed] [Google Scholar]
- 178.Saxton RA, and Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 168: 960–976, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schroeder S, Pendl T, Zimmermann A, Eisenberg T, Carmona-Gutierrez D, Ruckenstuhl C, Marino G, Pietrocola F, Harger A, Magnes C, Sinner F, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, and Madeo F. Acetyl-coenzyme A: a metabolic master regulator of autophagy and longevity. Autophagy 10: 1335–1337, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sciarretta S, Forte M, Frati G, and Sadoshima J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ Res 122: 489–505, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, Tong M, Del Re DP, Vecchione C, Schirone L, Forte M, Rubattu S, Shirakabe A, Boppana VS, Volpe M, Frati G, Zhai P, and Sadoshima J. Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction. Journal of the American College of Cardiology 71: 1999–2010, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, and Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 125: 1134–1146, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, and Taegtmeyer H. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. Journal of the American Heart Association 2: e004796, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, Ma J, Kemper B, and Kemper JK. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 516: 108–111, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, Javaheri A, Crowley JR, Ballabio A, Schilling JD, Epelman S, Weihl CC, Diwan A, Fan D, Zayed MA, and Razani B. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun 8: 15750, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Settembre C, and Ballabio A. Lysosome: regulator of lipid degradation pathways. Trends in cell biology 24: 743–750, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Settembre C, De CR, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Jurgen KT, Wollenberg AC, Di BD, Chan L, Irazoqui JE, and Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. NatCell Biol 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Settembre C, Fraldi A, Medina DL, and Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nature reviews Molecular cell biology 14: 283–296, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, and Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. The EMBO journal 31: 1095–1108, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shao D, and Tian R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr Physiol 6: 331–351, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sheng Y, Lv S, Huang M, Lv Y, Yu J, Liu J, Tang T, Qi H, Di W, and Ding G. Opposing effects on cardiac function by calorie restriction in different-aged mice. Aging Cell 16: 1155–1167, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr., de Cabo R, Ulrich S, Akassoglou K, and Verdin E Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339: 211–214, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Shinmura K, Tamaki K, and Bolli R. Short-term caloric restriction improves ischemic tolerance independent of opening of ATP-sensitive K+ channels in both young and aged hearts. Journal of molecular and cellular cardiology 39: 285–296, 2005. [DOI] [PubMed] [Google Scholar]
- 194.Shinmura K, Tamaki K, Sano M, Murata M, Yamakawa H, Ishida H, and Fukuda K. Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. Journal of molecular and cellular cardiology 50: 117–127, 2011. [DOI] [PubMed] [Google Scholar]
- 195.Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, and Sadoshima J. Aging and Autophagy in the Heart. Circ Res 118: 1563–1576, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, and Sadoshima J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 133: 1249–1263, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, and Kroemer G. Organelle-Specific Initiation of Autophagy. Molecular cell 59: 522–539, 2015. [DOI] [PubMed] [Google Scholar]
- 198.Simonson B, Subramanya V, Chan MC, Zhang A, Franchino H, Ottaviano F, Mishra MK, Knight AC, Hunt D, Ghiran I, Khurana TS, Kontaridis MI, Rosenzweig A, and Das S. DDiT4L promotes autophagy and inhibits pathological cardiac hypertrophy in response to stress. Science signaling 10: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ. Autophagy regulates lipid metabolism. Nature 458: 1131–1135, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sletten AC, Peterson LR, and Schaffer JE. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J Intern Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Song M, Franco A, Fleischer JA, Zhang L, and Dorn GW 2nd., Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab 26: 872–883 e875, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, and Dorn GW 2nd., Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ Res 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Song M, Mihara K, Chen Y, Scorrano L, and Dorn GW 2nd., Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–286, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G, Ballabio A, and Raben N. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO molecular medicine 5: 691–706, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.St-Onge MP, Ard J, Baskin ML, Chiuve SE, Johnson HM, Kris-Etherton P, Varady K, American Heart Association Obesity Committee of the Council on L, Cardiometabolic H, Council on Cardiovascular Disease in the Y, Council on Clinical C, and Stroke C. Meal Timing and Frequency: Implications for Cardiovascular Disease Prevention: A Scientific Statement From the American Heart Association. Circulation 135: e96–e121, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Stanley S, Wynne K, McGowan B, and Bloom S. Hormonal regulation of food intake. Physiological reviews 85: 1131–1158, 2005. [DOI] [PubMed] [Google Scholar]
- 207.Stein PK, Soare A, Meyer TE, Cangemi R, Holloszy JO, and Fontana L. Caloric restriction may reverse age-related autonomic decline in humans. Aging Cell 11: 644–650, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Stypmann J, Janssen PM, Prestle J, Engelen MA, Kogler H, Lullmann-Rauch R, Eckardt L, von FK, Landgrebe J, Mleczko A, and Saftig P. LAMP-2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic ResCardiol 101: 281–291, 2006. [DOI] [PubMed] [Google Scholar]
- 209.Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, and Wang Y. Catabolic Defect of Branched-Chain Amino Acids Promotes Heart Failure. Circulation 133: 2038–2049, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sun Y, Xu YH, Du H, Quinn B, Liou B, Stanton L, Inskeep V, Ran H, Jakubowitz P, Grilliot N, and Grabowski GA. Reversal of advanced disease in lysosomal acid lipase deficient mice: a model for lysosomal acid lipase deficiency disease. Mol Genet Metab 112: 229–241, 2014. [DOI] [PubMed] [Google Scholar]
- 211.Sutton EF, Beyl R, Early KS, Cefalu WT, Ravussin E, and Peterson CM. Early Time-Restricted Feeding Improves Insulin Sensitivity, Blood Pressure, and Oxidative Stress Even without Weight Loss in Men with Prediabetes. Cell Metab 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Svendsen PF, Jensen FK, Holst JJ, Haugaard SB, Nilas L, and Madsbad S. The effect of a very low calorie diet on insulin sensitivity, beta cell function, insulin clearance, incretin hormone secretion, androgen levels and body composition in obese young women. Scand J Clin Lab Invest 72: 410–419, 2012. [DOI] [PubMed] [Google Scholar]
- 213.Taegtmeyer H, Lam T, and Davogustto G. Cardiac Metabolism in Perspective. Compr Physiol 6: 1675–1699, 2016. [DOI] [PubMed] [Google Scholar]
- 214.Taegtmeyer H, Wilson CR, Razeghi P, and Sharma S. Metabolic energetics and genetics in the heart. Annals of the New York Academy of Sciences 1047: 208–218, 2005. [DOI] [PubMed] [Google Scholar]
- 215.Taegtmeyer H, Young ME, Lopaschuk GD, Abel ED, Brunengraber H, Darley-Usmar V, Des Rosiers C, Gerszten R, Glatz JF, Griffin JL, Gropler RJ, Holzhuetter HG, Kizer JR, Lewandowski ED, Malloy CR, Neubauer S, Peterson LR, Portman MA, Recchia FA, Van Eyk JE, Wang TJ, and American Heart Association Council on Basic Cardiovascular S. Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circ Res 118: 1659–1701, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Taffet GE, Pham TT, and Hartley CJ. The age-associated alterations in late diastolic function in mice are improved by caloric restriction. J Gerontol A Biol Sci Med Sci 52: B285–290, 1997. [DOI] [PubMed] [Google Scholar]
- 217.Takagi A, Kume S, Kondo M, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Haneda M, Chano T, Matsusaka T, Nagao K, Adachi Y, Chan L, Maegawa H, and Uzu T. Mammalian autophagy is essential for hepatic and renal ketogenesis during starvation. Sci Rep 6: 18944, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, and Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406: 902–906, 2000. [DOI] [PubMed] [Google Scholar]
- 219.Traba J, Kwarteng-Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, Waclawiw MA, Han K, Pelletier M, Sauve AA, Siegel RM, and Sack MN. Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. The Journal of clinical investigation 125: 4592–4600, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Trepanowski JF, Kroeger CM, Barnosky A, Klempel MC, Bhutani S, Hoddy KK, Gabel K, Freels S, Rigdon J, Rood J, Ravussin E, and Varady KA. Effect of Alternate-Day Fasting on Weight Loss, Weight Maintenance, and Cardioprotection Among Metabolically Healthy Obese Adults: A Randomized Clinical Trial. JAMA Intern Med 177: 930–938, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Tsai JY, Villegas-Montoya C, Boland BB, Blasier Z, Egbejimi O, Gonzalez R, Kueht M, McElfresh TA, Brewer RA, Chandler MP, Bray MS, and Young ME. Influence of dark phase restricted high fat feeding on myocardial adaptation in mice. Journal of molecular and cellular cardiology 55: 147–155, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, and Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308: 1043–1045, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Varady KA, Bhutani S, Church EC, and Klempel MC. Short-term modified alternate-day fasting: a novel dietary strategy for weight loss and cardioprotection in obese adults. Am J Clin Nutr 90: 1138–1143, 2009. [DOI] [PubMed] [Google Scholar]
- 224.Varady KA, Bhutani S, Klempel MC, Kroeger CM, Trepanowski JF, Haus JM, Hoddy KK, and Calvo Y. Alternate day fasting for weight loss in normal weight and overweight subjects: a randomized controlled trial. Nutr J 12: 146, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, Zou MH, Chen C, and Wang DW. AMPKalpha2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ Res 122: 712–729, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wang C, Niederstrasser H, Douglas PM, Lin R, Jaramillo J, Li Y, Olswald NW, Zhou A, McMillan EA, Mendiratta S, Wang Z, Zhao T, Lin Z, Luo M, Huang G, Brekken RA, Posner BA, MacMillan JB, Gao J, and White MA. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat Commun 8: 2270, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, and Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. The Journal of clinical investigation 121: 4477–4490, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Warren JS, Oka SI, Zablocki D, and Sadoshima J. Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. American journal of physiology Heart and circulatory physiology 313: H584–H596, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wei M, Brandhorst S, Shelehchi M, Mirzaei H, Cheng CW, Budniak J, Groshen S, Mack WJ, Guen E, Di Biase S, Cohen P, Morgan TE, Dorff T, Hong K, Michalsen A, Laviano A, and Longo VD. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Science translational medicine 9: 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, and Mair WB. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab 26: 884–896 e885, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wing RR. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four-year results of the Look AHEAD trial. ArchInternMed 170: 1566–1575, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, and Dunn WA Jr., Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res 10: 281–292, 2007. [DOI] [PubMed] [Google Scholar]
- 233.Wolfson RL, and Sabatini DM. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab 26: 301–309, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wolins NE, Quaynor BK, Skinner JR, Tzekov A, Croce MA, Gropler MC, Varma V, Yao-Borengasser A, Rasouli N, Kern PA, Finck BN, and Bickel PE. OXPAT/PAT-1 is a PPAR-induced lipid droplet protein that promotes fatty acid utilization. Diabetes 55: 3418–3428, 2006. [DOI] [PubMed] [Google Scholar]
- 235.Woloszynek JC, Kovacs A, Ohlemiller KK, Roberts M, and Sands MS. Metabolic adaptations to interrupted glycosaminoglycan recycling. The Journal of biological chemistry 284: 29684–29691, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, and Sinclair D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430: 686–689, 2004. [DOI] [PubMed] [Google Scholar]
- 237.Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, and Stroke Statistics S. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 133: e38–360, 2016. [DOI] [PubMed] [Google Scholar]
- 238.Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, Diwan A, and Lee JM. Enhancing astrocytic lysosome biogenesis facilitates abeta clearance and attenuates amyloid plaque pathogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience 34: 9607–9620, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Xiao QY P; Ma X; Liu H; Perez R; Zhu A; Gonzales E; Tripoli DD; Czerniewski L; Ballabio A; Cirrito JR; Diwan A; Lee JM. Neuronal-targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Aβ Generation and Amyloid Plaque Pathogenesis. Journal of Neuroscience 35: 12137–12151, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Xie K, Neff F, Markert A, Rozman J, Aguilar-Pimentel JA, Amarie OV, Becker L, Brommage R, Garrett L, Henzel KS, Holter SM, Janik D, Lehmann I, Moreth K, Pearson BL, Racz I, Rathkolb B, Ryan DP, Schroder S, Treise I, Bekeredjian R, Busch DH, Graw J, Ehninger G, Klingenspor M, Klopstock T, Ollert M, Sandholzer M, Schmidt-Weber C, Weiergraber M, Wolf E, Wurst W, Zimmer A, Gailus-Durner V, Fuchs H, Hrabe de Angelis M, and Ehninger D. Every-other-day feeding extends lifespan but fails to delay many symptoms of aging in mice. Nat Commun 8: 155, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Yamamoto T, Tamaki K, Shirakawa K, Ito K, Yan X, Katsumata Y, Anzai A, Matsuhashi T, Endo J, Inaba T, Tsubota K, Sano M, Fukuda K, and Shinmura K. Cardiac Sirt1 mediates the cardioprotective effect of caloric restriction by suppressing local complement system activation after ischemia-reperfusion. American journal of physiology Heart and circulatory physiology 310: H1003–1014, 2016. [DOI] [PubMed] [Google Scholar]
- 242.Yan L, Park JY, Dillinger JG, De Lorenzo MS, Yuan C, Lai L, Wang C, Ho D, Tian B, Stanley WC, Auwerx J, Vatner DE, and Vatner SF. Common mechanisms for calorie restriction and adenylyl cyclase type 5 knockout models of longevity. Aging Cell 11: 1110–1120, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, and Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 130: 247–258, 2007. [DOI] [PubMed] [Google Scholar]
- 244.Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, and Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res 100: 1208–1217, 2007. [DOI] [PubMed] [Google Scholar]
- 245.Yang KC, Ma X, Liu H, Murphy J, Barger PM, Mann DL, and Diwan A. Tumor necrosis factor receptor-associated factor 2 mediates mitochondrial autophagy. Circulation Heart failure 8: 175–187, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yoshii SR, Kuma A, Akashi T, Hara T, Yamamoto A, Kurikawa Y, Itakura E, Tsukamoto S, Shitara H, Eishi Y, and Mizushima N. Systemic Analysis of Atg5-Null Mice Rescued from Neonatal Lethality by Transgenic ATG5 Expression in Neurons. Developmental cell 39: 116–130, 2016. [DOI] [PubMed] [Google Scholar]
- 247.Yoshino J, Baur JA, and Imai SI. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, and Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nature cell biology 8: 1124–1132, 2006. [DOI] [PubMed] [Google Scholar]
- 249.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, and Lenardo MJ. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J, Vagnozzi RJ, Lal H, and Force T. GSK-3alpha is a central regulator of age-related pathologies in mice. The Journal of clinical investigation 123: 1821–1832, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334: 678–683, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Zuo L, He F, Tinsley GM, Pannell BK, Ward E, and Arciero PJ. Comparison of High-Protein, Intermittent Fasting Low-Calorie Diet and Heart Healthy Diet for Vascular Health of the Obese. Front Physiol 7: 350, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]