

Abstract
A number of outstanding problems in genomics, such as identifying structural variations and sequencing through centromeres and telomeres, stand poised to benefit tremendously from emerging long-read genomics technologies such as nanopore sequencing and genome mapping in nanochannels. However, optimal application of these new genomics technologies requires facile methods for extracting long DNA from cells. These sample preparation tools should be amenable to automation and minimize fragmentation of the long DNA molecules by shear. We present one such approach in a poly(dimethylsiloxane) device, where gel-based high molecular weight DNA extraction and continuous flow purification in a 3D cell culture-inspired geometry is followed by electrophoretic extraction of the long DNA from the miniaturized gel. Molecular combing reveals that the device produces molecules that are typically in excess of 100 kilobase pairs in size, with the longest molecule extending up to 4 megabase pairs. The microfluidic format reduces the standard day-long and labor-intensive DNA extraction process to 4 hours, making it a promising prototype platform for routine long DNA sample preparation.
Introduction
As healthcare advances into the precision medicine era,1 genomic analysis promises to become an important diagnostic technology for identifying the DNA modifications that lie at the heart of the majority of genetic diseases.2 These new clinical approaches will be enabled in part by next-generation sequencing (NGS) technologies that have transformed human DNA sequencing into an affordable and accessible tool.3 The short reads (~100 base pairs) produced by NGS are ideal for identifying single nucleotide polymorphisms. However, NGS encounters significant challenges when analyzing the kilobase (kbp) to megabase (Mbp) long genomic aberrations, known as structural variations, that are associated with diseases such as cancer.4,5 NGS also struggles with some regions of the genome, for example sequencing through centromeres.6 Even when sufficient NGS data can be obtained, aligning tens of millions of short reads during de novo genome assembly poses a substantial computational challenge.7
Over the last five years, various commercial long-read technologies such as nanopore sequencing,8 single molecule real time sequencing,9 and nanochannel-based genome mapping,10 as well as droplet barcoding techniques such as linked-read sequencing,11 have emerged to assist (and, perhaps, supplant) NGS and improve genome sequencing.12 Their read lengths, reaching up to several hundreds of kilobases,13,14 have enabled the resolution of important structural variations,13,15 closing of gaps in the human genome,16 identification of long repetitive regions,17 and sequencing of various complex species.18 The success of these long-read technologies in turn relies on robust sample preparation methods that prevent long DNA from fragmentation during processing in order to achieve the high read lengths.
The standard method to extract long DNA from cells is by agarose immobilization,19 which uses the mechanical stability of the gel to maintain the integrity of the long, genomic DNA during extraction and sample purification steps. The purified DNA is then recovered by enzymatically digesting the gel and performing drop dialysis. Although robust and simple to implement, the agarose immobilization protocol requires long times for diffusion of reagents and cellular waste into and out of the gel plug, making the process tedious and hands-on. Furthermore, the gel digestion and drop dialysis induce DNA fragmentation due to shear caused by flow in the liquid phase. The sample preparation process has been sped up and automated in recent commercially available platforms that perform electrophoretic purification of the DNA.20 However, the gain in speed in these systems is obtained by performing cell lysis in the liquid-phase, which has the potential to fragment the DNA by shearing.21
Microfluidics can offer a significant reduction in process time by exploiting the mismatch in the size of human cells and the macroscopic gel used in the plug lysis method. In addition to reducing diffusion time, microfluidic processing is ideal for cases where a small number of cells are available, for example from a needle biopsy.22 One microfluidic option is to physically trap cells and DNA by either employing optical tweezers23 or a microfabricated array of posts.24 While genomic DNA has been extracted from cells in such devices, these approaches are either fairly involved with respect to external equipment use or require careful operation at low flow rates to avoid shearing long molecules. The resultant DNA also elongates and gets highly entangled around the posts during flow-based sample purification, making it difficult to recover long molecules out of the device. A gel-based microfluidic approach that can maintain the integrity of the long DNA while reducing the overall processing time via small diffusion lengths, and then allow for non-deleterious recovery of DNA out of the gel, can address this outstanding problem in long DNA sample preparation. One such approach has been recently reported where alginate microparticle encapsulation achieves high-throughput extraction of long, genomic DNA from single cells.25 Despite the many advantages of this technique, this droplet-based multi-platform approach has microfluidic complexities that cannot yet match the simplicity and robustness of the state-of-the-art plug lysis method. We posit that microfluidic agarose immobilization of cells and employment of large-pore gel electrophoresis can provide a more straightforward approach for facile extraction of ultra-long DNA.
We show here that long DNA sample preparation from cells can be performed in the simple microfluidic device of Fig. 1 by leveraging a hydrogel-pinning technique previously employed for three-dimensional cell culture,26 thereby retaining both the advantages and the familiarity of the agarose plug method19 while reducing processing time. Our device utilizes simultaneous diffusion and continuous flow to purify the DNA extracted from the lysed cells. The long genomic DNA are recovered electrophoretically from a very low concentration (0.2 wt%) agarose, whose mechanical stability makes it difficult to handle outside a microfluidic setting,27 thereby avoiding the need to digest the gel and thus minimizing shear in the liquid phase. Typical DNA molecules eluted from the device are in excess of 100 kbp, in line with the needs of long-read genomics methods,17 with molecules frequently in the Mbp range to redefine the limits of ultra-long read sequencing.14 The simple format of the device offers opportunities for integration both upstream (via cell culture with environmental control) and downstream (via direct integration with genomics methods) to make a total analysis system,28 as well as the potential to reduce the labor requirement via automation of the fluid flow. The prototype device presented here produces 10 ng DNA from 2000 human cells. Genomics technologies can now readily work with such small quantities of starting DNA material, but they require error-free long-range DNA amplification. Instead, with straightforward multiplexing,29 our approach could be used for facile extraction of high-concentration long genomic DNA from cells, reducing the present day-long sample preparation process to a few hours.
Fig. 1.
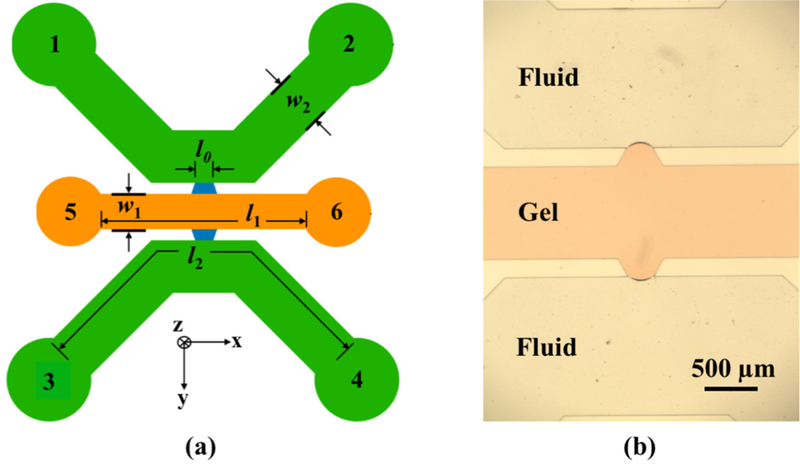
Microfluidic device for long DNA extraction from cells. (a) Schematic illustration of the device indicating the various reservoirs, gel channel (orange), fluidic channels (green) and communicating trapezoidal channels (blue). The gel channel length is l1 = 6 mm and its width is w1 = 1 mm. The fluid channel contour length is l2 = 10 mm and its width is w2 = 1.5 mm. The overlap length l0 = 250 µm is defined by the short edge of the trapezoid. The device depth is 100 µm. (b) Microscope image of the communicating trapezoidal channels in the PDMS device filled with orange-colored agarose in the central gel channel.
Results
Device Design
The PDMS device features a central gel microchannel for embedding cells and immobilizing DNA during lysis and sample purification steps, communicating with two parallel fluidic channels on either side of the gel via the trapezoidal microchannels. The communicating channels facilitate diffusive molecular exchange between the gel and the fluid flowing through the side channels, and later aid in electrophoretic extraction. The design of the DNA extraction device required considering two aspects: (i) the hydrodynamic resistance during gel loading and fluid exchange; and (ii) the electric field strength in different parts of the device.
Balancing the hydrodynamic resistance during gel loading is crucial to ensure that the gel remains in the central channel. This is achieved by contact line pinning, a physical principle previously exploited in microfluidic devices for 3D cell culture.30 Since PDMS is hydrophobic, a trapezoid-shaped communicating channel with a 60° angle supplements the PDMS-gel contact angle to establish a radius of curvature that provides a high Laplace pressure. To prevent gel from bursting into the fluid channel, the hydraulic pressure drop in the gel channel while loading agarose should not exceed the surface tension-sustained pressure differential at the agarose-air interface, where the latter depends on the radii of curvature in the z (along the device depth) and x (along the gel channel length) directions.31 This effect has been optimized for 3D cell culture by choosing appropriate channel depth and trapezoid dimensions to obtain the required Laplace pressure.26 In the cell culture application, it is important to have deep channels to maintain three-dimensionality. For DNA extraction, in theory, the ultimate lower bound on channel depth is governed by the cell size. However, in practice, the resultant Laplace pressure also needs to be considered. Since this pressure is independent of the direction of gravity, we interchanged the channel depth and trapezoid dimensions in our design so that we obtain similar gel caging as 3D cell culture using relatively shallow channels, which obviates the need to handle highly viscous photoresists during fabrication.
The extraction of DNA out of the gel is maximized by employing electrophoresis both along (lateral) and across (transverse) the gel channel. Choosing a small gel length (l1) reduces hydrodynamic resistance during gel loading, provides a high electric field along the gel for a given applied potential and ensures a small electrophoresis migration distance. To induce a high electric field in the overlapping region of the gel, the remaining channel dimensions need to be chosen such that a significant potential drop occurs across the width of the gel. To achieve this, we used Kirchoff’s laws as a preliminary design step. Here, we estimate the resistance R of a channel of length l and cross-sectional area A as R = σl/A. The resistivity σ for the fluid and gel channels can be approximated as equal because the gel is 99.8% buffer. Since all channels in the device have the same depth, the ratio of length to width of each section determines their relative resistance. We calculated the potential drop in the different channels along the transverse electrophoretic path by first considering an elementary resistor network shown in Fig. 2a, and then using a more accurate COMSOL model by including the lateral electrophoresis effects too (Fig. 2b). The equivalent circuit has two parallel fluid channels in series with a gel and another fluid channel. The goal here was to maximize the gel resistance R1 relative to the other resistances in the equivalent circuit, and hence the potential drop across the gel. Apart from this, other noteworthy points during dimension selection were that (i) using wide (w2) and short (l2) fluid channels provides low resistance during continuous flow reagent exchange, and ensures a small potential drop in the fluid channels; (ii) a small gel width (w1) ensures a small diffusion length and a small electrophoresis migration distance; and (iii) a small overlap length (l0) equivalent to a single trapezoid communicating channel ensures uniform and almost straight electric field lines in the transverse direction, and provides a high gel resistance because it is the effective gel width during the electrical resistance calculation. Based on these considerations, we identified an approximate working regime for all dimensions by applying Kirchoff and Ohm’s laws to the equivalent circuit model, and then arrived at a final set of dimensions by modeling an equivalent geometry in COMSOL to study the potential drop and electric field lines. The gel channel length l1 was ultimately adjusted to obtain similar electric field magnitude in both lateral and transverse electrophoresis directions in the gel. In our 100 µm deep device, the gel channel has a length l1 = 6 mm, width w1 = 1 mm and 2 mm diameter reservoirs. The fluid channels have a length l2 = 10 mm, width w2 = 1.5 mm and 3 mm diameter reservoirs. The trapezoidal short edge defining the overlap length is l0 = 250 µm, with the longer edge being 500 µm.
Fig. 2.
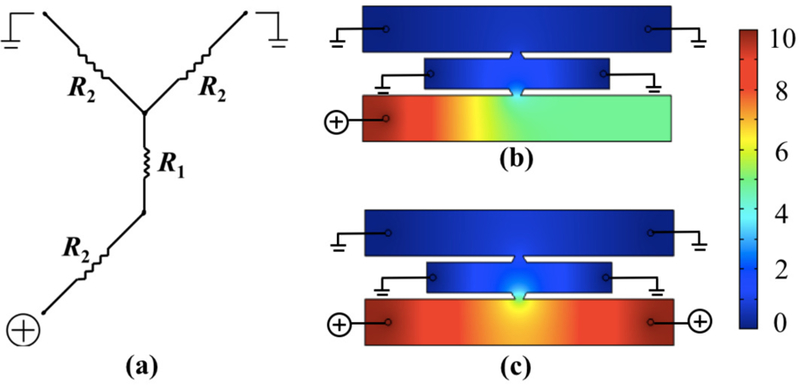
(a) Equivalent Kirchoff circuit for transverse electrophoresis with fluid channel resistance R2 ~ l2/2w2 and gel channel resistance R1 ~ w1/l0. (b,c) COMSOL computation of electric potential drop in an equivalent device geometry with (b) single-anode configuration and (c) double-anode configuration, for an applied external potential of 10 V. Color bar represents electric potential in V.
The finite element calculation in COMSOL exhibited a strong electric field in the gel in both electrophoresis directions, for an applied voltage as little as 10 V, with almost straight field lines minimizing the electrophoretic path of DNA (Fig. 3). The modeling also revealed a benefit of having one anode instead of two. While the potential drop across the gel is the same in the one anode and two anode cases, the electric field in the anodic fluid channel is stronger in the single anode configuration, leading to faster motion of DNA to the reservoir (Fig. 2b-c). The single anode also drives all the DNA to one reservoir, rather than splitting the DNA yield into two streams, simplifying DNA recovery.
Fig. 3.
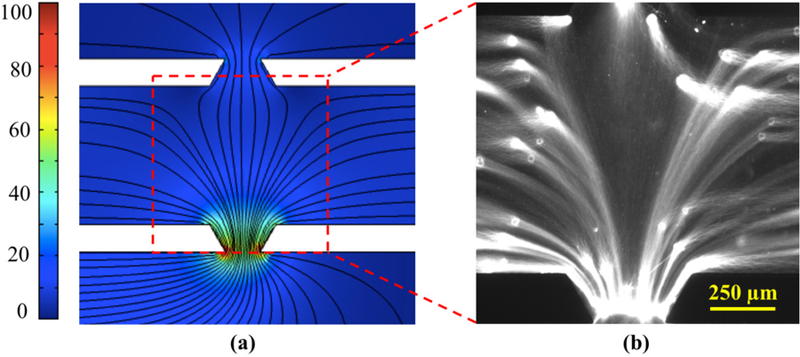
(a) Computed electric field lines in the gel corresponding to the single-anode configuration of Fig. 2b at 10 V potential. Color bar represents electric field magnitude in V/cm. (b) Fluorescent image of YOYO-stained whole genomic DNA, extracted from MCF-7 cells in the device, following the field lines during electrophoresis.
DNA Extraction
To extract DNA from MCF-7 cells in the device, we loaded cells embedded in a 0.2 wt% agarose gel at a density of 1500 cells/µL in the gel channel. The cells were lysed diffusively at 37 °C for 1.5 hours by filling the two fluid channels with a detergent lysis solution containing SDS and Proteinase K. An illuminating component of our lysis solution is the cell membrane-impermeant DNA intercalating dye YOYO-1, which fluoresces on the completion of cell lysis. We also demonstrate a blind (YOYO-free) extraction at the conclusion of our paper, as applications such as nanopore sequencing do not permit the use of these fluorescent labels. Detergent in chemical lysis solutions causes cellular membrane degradation, which is instantaneous, and the small detergent molecules are not diffusion-limiting. While typical plug lysis protocols use stronger detergents (1% SDS),32 the choice of a milder detergent (0.17% SDS) in this work was guided by the requirement of simultaneous YOYO-labeling, which is not effective in the presence of stronger detergents.33 The lysis temperature and the combination of buffers in the lysis solution were chosen for effective degradation of the proteins by Proteinase K.34 After lysis, the cellular debris, digested proteins, and other salts and contaminants were allowed to diffuse out of the gel into the fluid channels, which were continuously replenished with fresh wash buffer at ~ 4 µL/min for 1.5 hours. Since all contaminant molecules are smaller than the pore size for 0.2 wt% agarose,35 effective washing of the gel was achieved by molecular diffusion. Moreover, continuous flow in the fluidic channels facilitated immediate elimination of the cellular waste once it diffused to the gel-fluid interface.
The time required for cell lysis and sample purification is based on the diffusion of different reagent molecules through the gel. To estimate this time, we performed a control experiment to characterize the diffusion of fluorescein across the gel channel when loaded with MCF-7 cells embedded in agarose (Fig. S1†). The results of this experiment allowed us to readily scale up the lysis time using the diffusivity of Proteinase K, which is the biggest molecule in the lysis solution, based on the inverse dependence of process time on the diffusivity of the molecule (details in SI†). The sample purification time was estimated similarly to remove all the Proteinase K from the gel after its role in protein digestion was complete. While completion of cell lysis was marked by illumination of the DNA by YOYO, complete digestion of histones by Proteinase K also was verified in a separate experiment by measuring the fluorescence of histones in Histone 2B-GFP transfected cells before and after cell lysis using our lysis protocol (Fig. S2†).
In the miniaturized gel, the electric field is strong even for low applied potentials (Fig. 3).36 This helps us to accomplish fast electrophoretic extraction of DNA without encountering high voltage ramifications like fluid evaporation from the reservoirs or Joule heating.37 After purification, the DNA was extracted electrophoretically at 10 V into the anodic reservoir 3 following the electrode configuration of Fig. 2b. Owing to the large pore size of the 0.2 wt% agarose gel and the indentations created in the gel at cellular locations, rapid electrophoretic extraction of long DNA was achieved without encountering irreversible trapping of DNA within the gel at the 10 V operating electric potential.
During extraction, we observed that some DNA molecules were, however, trapped at the gel-fluid interface and ultimately fragmented during constant voltage electrophoresis. We attribute this holdup to the entanglements between the DNA and the dangling fibers at the end of the gel. Since the exiting DNA molecule has a higher extension than when in the gel,38 the transmission of tension along the extended chain to the trapping point causes DNA fragmentation under a constant voltage. To ameliorate this problem, we implemented a voltage loop: 10 V for 18 s followed by 0 V for 2 s in LabVIEW to assist in periodic chain relaxation for releasing the transient tension along the molecule length.39 The 2 s period for relaxation was determined based on the experimentally observed recoiling time associated with the overhanging chain during electrophoresis. We propose that the pulsing allows the released DNA to relax any entanglements with the dangling fibers when compared to the constant voltage case. To expedite the migration of DNA towards reservoir 3 after extraction from the gel, a potential of 20 V was applied along the lower channel of Fig. 1a for 3 minutes after each 7-minute long extraction protocol. The total electrophoresis time of 60 minutes, comprising 6 electrophoresis cycles, was selected based on the fluorescence intensity reduction in the gel, which was recorded by time-lapse imaging of the DNA every 10 minutes (Fig. 4). The small diffusion and electrophoresis time in the miniaturized gel helped to complete the entire DNA extraction from cells in 4 hours, which is significantly faster than both conventional plug lysis (24 hours), as well as commercial platforms performing sample preparation for optical mapping such as the Aurora system (30 hours)40.
Fig. 4.

Effect of electrophoresis time on DNA extraction out of the gel. (a) Fluorescent image of DNA released from cells after the lysis and washing step. (b) Fluorescence in the gel after 3 electrophoresis cycles, and (c) after 6 electrohoresis cycles. One 10-minute cycle comprises of 7 minutes of gel electrophoresis at a pulsed external potential: 10 V for 18 s and then 0 V for 2 s, followed by 3 minutes of enhanced electrophoretic driving of extracted DNA to the anodic reservoir 3 at 20 V.
Device Performance
The DNA recovered from the device and any protein contaminants were quantified using fluorometry in a Qubit fluorometer (Table S1†-S2†). Since the fluorometer reads the fluorescent intensity of molecules bound with specific dyes, we accounted for the YOYO-labeling of the DNA by calibrating with a control solution comprising a mixture of YOYO-labeled λ DNA (16.67 ng/µL) and human histone H4 (16.67 ng/µL) in 1x TBE buffer. The output dsDNA concentration of this control solution in the fluorometer was 46.01 ng/µL, and consequently all DNA readings obtained from the Qubit fluorometer were scaled down by a factor of 2.76. The DNA concentrations of the samples recovered from two different devices were 0.671 ng/µL and 0.674 ng/µL. The volume of DNA sample collected from each device was 15 µL, giving a yield of 10.05 ng DNA per device. For the starting 12.34 ng DNA seeded in the gel channel in the form of <2000 cells, we obtained 81.4% extraction of DNA out of the gel (details in SI†).
To verify the removal of proteins from the gel, we first checked for the complete digestion of histones during cell lysis (Fig. S2†), and then measured the amount of protein in the DNA sample collected from the device by fluorometry. Since the presence of YOYO-labeled DNA can increase the protein signal in the fluorometer due to the broad emission spectra of the dyes, we used the control solution for calibration. The protein concentration of the control solution containing 16.67 ng/µL of histones was measured as 52.75 ng/µL by the Qubit, and consequently all protein readings were scaled down by a factor of 3.16. The protein concentration of the samples recovered from two different devices was 11.14 ng/µL and 7.25 ng/µL. To assess how our sample purity compares with DNA prepared from traditional methods, we evaluated the DNA:protein ratio for samples prepared in our device and for the DNA prepared using conventional plug lysis. The protein reading for the conventional DNA sample diluted to 2.2 ng/µL was 13.83 ng/µL, giving a DNA:protein ratio of 0.159. The corresponding ratio for the DNA sample prepared in our device was 0.073.
The DNA recovered from the device were analyzed by molecular combing in microchannels on activated glass coverslips as shown in Fig. 5. (Stitched images from other molecular combing channels are available as Supplementary files MC1 - MC11†.) The use of microchannels for loading DNA during size analysis helps to orient the long molecules in the direction of capillarity, avoiding random overlap of molecules.41 To estimate the size of the molecules based on their pixel length, we stretched λ DNA (48.5 kbp) in the combing device to yield a calibration factor of 3.13 kbp/µm (Fig. S3†). The weight distribution of the representative DNA molecules in Fig. 5 is shown in Fig. 6. The characterized DNA lengths are much in excess of the typical sizes in SMRT sequencing (10–50 kbp)42 and nanopore sequencing (10–100 kbp).14 Several molecules in the DNA sample exhibit extensions corresponding to molecular weights of more than 500 kbp, with the longest observed molecule in a separate combing channel being 4 Mbp (Fig. S4†).
Fig. 5.

Fluorescent image of YOYO-stained, device-extracted DNA, stretched on silanized glass in 100 µm wide and 5 µm deep PDMS channel. The image is stitched to cover 8 ROIs of an Andor Zyla camera at 100x magnification. The 32 µm scale bar corresponds to 100 kbp.
Fig. 6.
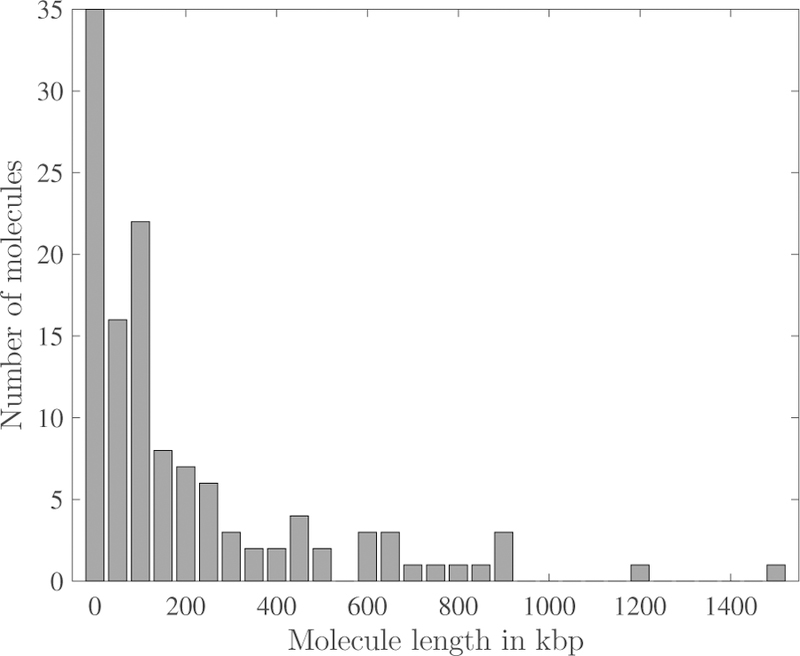
Molecular weight distribution of the 122 combed DNA molecules captured in the 8 fields of view of Fig. 5.
Blind Extraction
The sample preparation chemistry for certain genomics applications requires working with DNA molecules where the backbone is not fluorescently labeled.43 To demonstrate the utility of our device for such applications, we performed “blind” sample preparation by not including YOYO in the lysis solution. The DNA were electrophoretically extracted out of the gel and driven to the reservoir 3 using our electrophoresis protocol without visual verification. The recovery of DNA was verified subsequently by adding 2 µL of 0.01 mM YOYO to the anodic reservoir 3 to stain the DNA, and then stretching these molecules in the combing device (Fig. S5†). Apart from successful blind DNA extraction out of the microgel, this experiment also demonstrates the ability to perform downstream biochemistry on long DNA in the anodic reservoir.
Discussion
Our device utilizes a miniaturized gel to perform long DNA extraction from human cells, reducing the standard day-long protocol to 4 hours. We recognize that all-liquid phase DNA extraction from cells is much faster than diffusion-limited DNA extraction in a gel.44,45 Unfortunately, liquid phase DNA extraction typically leads to DNA fragmentation due to shearing during processing. Despite all other advantages of the commercially available semi-automated platforms, this particular shortcoming still persists.20 Our agarose-based device eliminates liquid phase shear on the DNA except for the final chip-to-world step, which is necessary at present to interface with genomics technologies.
The DNA purification in our device is accomplished by diffusive molecular exchange, which is enhanced by continuous flow in the fluid channels. The underlying diffusion approach to sample purification is inspired by conventional plug lysis. The cellular debris and digested proteins, being much smaller than genomic DNA, follow the continually replenished concentration gradient to escape out of the gel, and are immediately eliminated out of the device by bulk flow. Due to the use of electrophoresis for DNA purification from cell lysate in commercial sample purification equipments, 20 impurities in the gel in the form of small acidic peptides having low isoelectric points cannot be ruled out completely.46 After complete gel washing by diffusion, we extract the DNA in electrophoresis buffer, which theoretically gives pure DNA in buffer while eliminating all possible contaminants like chemical remnants from gel digestion or small negatively charged molecules. On assessment, the DNA:protein ratio of our sample is not dramatically different from that obtained by conventional plug lysis.
Post-processing of the extracted long DNA such as nick-labeling for genome mapping and adapter ligation for nanopore sequencing involve complex and sequential chemistry steps.43,47 In conventional protocols, typically there is no elimination of past reagents from the genomic sample, which can interfere with the genomic analysis. Microfluidic platforms demonstrating enzymatic labeling, concentration and purification of DNA have been reported;48 however they take DNA as input, and to date have been shown to work with relatively small λ DNA (48.5 kilobase pairs). The temperature-sensitive chemical lysis of cells, YOYO labeling of the released genomic DNA, and cellular waste elimination from our device constitute a proof-of-concept demonstration of not only the ability to execute chemistry on cells as well as DNA in the gel, but also the efficient elimination of unwanted reagents after their role in sample processing is complete. The extracted DNA product can then be directly loaded into genomics chips.
Our microfluidic device has a simple design and employs reusable external electrodes for DNA extraction by electrophoresis. The incorporation of on-chip electrophoresis in our method demonstrates the tunability of our device to drive the extracted DNA around without manually probing it. World-to-chip interfacing to transfer long DNA samples to genomics chips without molecule fragmentation and sample loss is a serious and yet unaddressed problem in long-read sequencing. In current tube-based protocols, use of a pipette for transfer is inevitable. Being a microfluidic approach, our simple design has the potential to be integrated with genomics chips, serving as a powerful tool to deliver ultra-long DNA directly from cells to genomic analysis technologies without any human intervention. Although single-cell DNA isolation for either linearization in nanochannels49 or optical mapping in nanoslits50 have been demonstrated on a single device, there is yet no generic sample preparation platform that can be used upstream of any genomic analysis technology.
The DNA extracted from our device are hundreds of kilobases long, in line with the requirement of current long-read genomics technologies. Many long DNA preparation techniques extract native DNA out of cells, but the fragment size is successively reduced throughout the process. In this work, we have recovered DNA molecules as long as 4 Mbp out of the device and demonstrated their integrity in a secondary environment. At present, it is challenging to establish the overall molecular weight distribution produced by the device. Due to the typical molecular weights observed, sample sizing techniques are limited to pulsed-field gel electrophoresis or molecule extension either by nanochannel confinement or molecular combing. While pulsedfield requires very high concentration input DNA, nanochannel confinement requires sophisticated nanofluidics chips. Molecular combing in microchannels is relatively straightforward for visualization and elementary DNA sizing if performed at a low DNA concentration to avoid molecular overlap, but it requires high-throughput machine-vision51 to analyze hundreds of fields of view across many combing channels and obtain a statistically significant molecular weight distribution. The manually obtained distribution in Fig. 6 is for the 122 molecules in the 8 fields of view stitched in Fig. 5, which, although illustrative of the presence of long DNA, represents a very small fraction of the total DNA recovered and combed from the device. Long-read genomics technologies such as nanopore sequencing and genome mapping in nanochannels capture the DNA fragment size during each run to report their observed read lengths, and we anticipate that these end-applications will ultimately prove to be a more accurate validation of our sample preparation approach.
The presented approach produces tens of nanograms of DNA from a few thousand cells as input, in line with the emerging inclination towards using genomics technologies for personalizing treatment via analysis of patient samples.52 While it is possible to sequence small amounts of DNA in long-read technologies, typical protocols currently employ amplification of the low abundance input sample, which will be challenging to implement accurately for very long molecules. The most straightforward approach to overcome this challenge is to pool samples from multiple devices. Additionally, extensive device multiplexing can be implemented and all the DNA can be eluted in a common outlet to produce high concentration samples.29 To increase the DNA yield from a single device, the gel-fluid overlap region, demarcated by the trapezoidal channels, can be elongated by patterning multiple parallel trapezoidal posts similar to cell culture devices. For such geometries, the potential drop across the gel can be increased by fabricating deeper fluid channels while leaving the gel channel depth, and hence the Laplace pressure, unaltered.53 Alternatively, in the case of a longer gel overlap, electrodes can either be injected into another side channel,54 or be patterned on the glass slide such that after PDMS bonding, they align parallel to the gel channel on either side to ensure a strong electric field in the overlapping gel region.55 The DNA can then be directed to either reservoir by using external reservoir electrodes.
Conclusions
In this paper, we have successfully demonstrated quick and facile microfluidic long DNA extraction from human breast cancer cells in a simple PDMS device. The key phenomena exploited in the sample preparation process are contact line pinning, molecular diffusion and DNA electrophoresis. The microfluidic size ensures rapid diffusion and small migration distance during electrophoresis, making the process considerably faster than conventional methods. The device design produces a strong electric field in the high porosity gel, enabling electrophoretic DNA extraction at a low applied potential. The device could be optimized for extracting different-sized genomes by adjusting the electrophoresis parameters, as well as sample preparation from various cell types by modifying the lysis protocol. The DNA extraction process is fairly automated with a continuous flow purification setup, which maintains a high concentration gradient at all times with immediate waste elimination from the device. Further automation is possible by using a programmable syringe pump for cartridge-based reagent delivery to eliminate any interim labor requirement.
The device makes small-scale sample preparation possible, with a few thousand cells and microliter reagents required as starting material to produce nanograms of DNA. With a multiplexed design, the extraction concept can be used to produce high concentration DNA. The hundreds of kilobases-long DNA recovered from the device are commensurate with the size requirement for long-read single molecule technologies which are instrumental in identifying large-scale structural variations. Future work will focus on performing more involved sample preparation biochemistry on the genomic DNA prior to its electrophoretic extraction from the device, followed by demonstration of long-read genomics applications. We envision this unit operation to be easily integrated upstream of nanochannel-based genome mapping chips, and possibly long-read sequencing technologies, to make a total analysis system. This device, being easy to fabricate and operate, and requiring minimal process equipment, should be useful for users outside the microfluidics community to prepare DNA samples for subsequent analysis using the long-read genomics technologies that are gaining significant interest in the genomics community.
Materials and Methods
Device Fabrication
The silicon master mold was fabricated using standard photolithography.56 SU8 2050 (MicroChem) was used to pattern the 100 µm deep channels. Devices were made by replica molding using degassed 10:1 poly(dimethylsiloxane) (PDMS) (Sylgard 184, Dow Corning) and cured at 75 °C for 2 hours. Reservoirs were punched in the gel and fluid channels, and the devices were bonded to glass microscope slides after oxygen plasma treatment for 2 minutes. The devices were heated at 75 °C for 36 hours to completely restore the hydrophobicity of the PDMS.
Cell Culture and Seeding
MCF-7 cells derived from human breast adenocarcinoma (ATCC HTB-22) were cultured at 37 °C and 5% CO2 in a 24-well plate in high glucose Dulbecco’s Modified Eagle’s Medium (Sigma), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific) and 1% penicillin-streptomycin (Thermo Fisher Scientific). After confluence, cells were trypsinized and centrifugally washed two times with 1x phosphate buffered saline (PBS) (Thermo Fisher Scientific). These 6 × 105 cells were re-suspended in 200 µL PBS to produce a cell density of ~3000 cells/µL. 20 µL of this cell solution was pipette-mixed with 20 µL of molten 0.4 wt% pulsed-field certified agarose (BioRad) prepared in 1x TBE buffer. The final 0.2 wt% agarose mixture having a cell density of 1500 cells/µL was loaded into the gel channel using a pipette, avoiding spillage into the fluidic channel. The device was equilibrated at room temperature for 5 minutes, and then cooled at 4 °C for 30 seconds by placing it in a petri dish to solidify the agar plug.
Cell Lysis and DNA Purification
Cells were lysed with a lysis solution containing 70 µL RIPA buffer (Thermo Fisher Scientific; 0.1% SDS), 10 µL pH 8 TE buffer (Thermo Fisher Scientific), 10 µL SDS lysis buffer (Sigma; 1% SDS), Proteinase K (Qiagen) at a concentration of 2 mg/mL, and YOYO-1 (Thermo Fisher Scientific) at a concentration of 4 µM. This lysis solution was filled in both fluidic channels, and the device was incubated at 37 °C for 1.5 hours. The completion of cell lysis was verified by examining YOYO-labeled DNA in the gel channel on an inverted epifluorescence microscope (Leica DMI 4000B).
After cell lysis, DNA was purified by flowing 1x wash buffer (10 mM Tris-HCl pH 8, 50 mM EDTA) in the fluidic channels for 45 minutes, followed by TE buffer for 45 minutes. Gravity-driven continuous flow at ~ 4 µL/min was established in the fluid channels by maintaining a height difference of ~ 5 mm between the inlet and outlet fluid reservoirs.
Histone Labeling
For cell transfection and selective labeling of histone 2B with GFP, 15 µL of CellLight Histone 2B-GFP, BacMam 2.0 construct (Thermo Fisher Scientific) was added to 50,000 cells in a 24-well plate and incubated for 16 hours. Cells were then washed, mixed with agarose and seeded in the device. These cells were lysed for hours at 37 °C using our lysis solution without YOYO.
DNA Extraction and Visualization
For DNA extraction by electrophoresis, both fluid channels and the two gel channel reservoirs were filled with 1x TBE buffer (Tris-HCl pH 8, boric acid, EDTA). The hydrophobicity of PDMS was exploited to make fluidic contact between reservoirs 1 and 5, and 2 and 6 via pendant droplets of TBE buffer (see Fig. 1a). Platinum electrodes connected to a DC power supply (Keithley 2230G-30–1) were immersed in reservoirs 1, 2 and 3. Reservoirs 1, 2, 5 and 6 were grounded, reservoir 3 served as the anode and reservoir 4 was floating. The DC power supply was programmed using LabVIEW to apply 10 V for 18 s followed by 0 V for 2 s. This electrophoresis loop was carried out for 7 minutes. The electrode configuration was then changed to have reservoirs 1, 2, 5 and 6 floating, 3 as the anode and 4 as the ground. A constant potential of 20 V was applied for 3 minutes. This total 10-minute electrophoresis protocol was repeated 6 times. The electrophoretic DNA extraction out of the gel was visualized and verified by fluorescence microscopy using an sCMOS camera (video in SI). Following the extraction step, DNA was collected from the anodic reservoir 3 for analysis.
DNA and Protein Quantitation
The concentration of DNA eluted from the gel was measured by fluorometry using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific). 15 µL of DNA sample was collected from reservoir 3 using a pipette, and mixed with 185 µL of working solution from the ds-DNA broad range assay kit. DNA samples from 2 different devices were collected, and 3 measurements were made per sample. To calculate the sample purity based on the DNA:protein ratio, the protein content of the sample was measured in the Qubit fluorometer. 15 µL of sample each was collected from two more devices from reservoir 3, and mixed with 185 µL of working solution from the protein assay kit. Each of the two samples was read 3 times in the fluorometer.
The control DNA solution was prepared by adding 2.5 µL of stock λ DNA (500 µg/mL, New England Biolabs), YOYO at a concentration of 0.5 µM, and 1.25 µL of stock human histone H4 (1000 µg/mL, BioRad) to 71 µL 1x TBE buffer. The plug prepared DNA at 110 ng/µL was diluted to 2.2 ng/µL in 1x TBE buffer, and labeled with YOYO at a concentration of 0.5 µM.
Molecular Combing
To demonstrate the ability to process the recovered DNA in a separate genomic method, the YOYO-stained DNA extracted from the device were sized using molecular combing. For linearly stretching the DNA, 5 µm deep, 100 µm wide and 10 mm long channels were fabricated in PDMS.41 After plasma treatment of the PDMS surface, the combing device was bonded to a glass coverslip activated by silanization.43 Briefly, 22 × 22 mm2 glass coverslips were stacked in a coverslip drying rack and incubated for 7 hours in a 2:1 (v/v) mixture of 70% nitric acid and 37% hydrochloric acid to clean and hydrolyze the glass surface. After washing the coverslips with ultrapure water and drying, they were immersed in a premixed solution of 200 µL N-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride and 53 µL of vinyltrimethoxysilane in 80 mL ultrapure water and incubated for 17 hours at 65 °C. The coverslips were washed with water and ethanol, and used immediately for PDMS bonding or stored for up to one week in ethanol at 4 °C. 2.5 µL of the DNA solution extracted from the anodic reservoir 3 of the device was used to fill 15 channels on a single combing device using capillarity. The stretched single DNA molecules were imaged with an sCMOS camera (ANDOR Zyla 4.2) using a 100x oil objective mounted on an epifluorescence microscope (Leica DMI 4000B). For analyzing the molecules that extended beyond a single image ROI, consecutive images were overlapped using the ImageJ stitching plugin.57
Plug Lysis
To extract DNA from cells in conventional agarose plugs, 2 × 106 cells were washed two times and resuspended in cell suspension buffer (CHEF mammalian DNA extraction kit, BioRad). Molten 2% low melting point CleanCut agarose (BioRad) was added to cells at a final concentration of 0.7%, and the mixture was cast into plugs using plug molds. Plugs were solidified at 4 °C for 5 minutes, and then incubated with lysis buffer (100 mM Tris-HCl pH 8, 50 mM EDTA, 100 mM NaCl and 1% SDS) and Proteinase K (2 mg/mL) at 50 °C for 4 hours and then overnight. Plugs were rinsed three times with 1x wash buffer and then washed four times with 10 mL wash buffer by shaking at 180 rpm for 15 minutes. Plugs were then washed four times with 10 mL TE buffer by shaking at 180 rpm for 10 minutes. The dried plugs were melted at 70 °C for 2 minutes, equilibrated at 43 °C for 5 minutes, and then digested at 43 °C for 45 minutes by adding agarase (Sigma). The digested plugs were then subjected to drop dialysis against TE buffer using 0.22 µm dialysis membranes for 2 hours. 120 µL of viscous DNA solution was recovered for analysis.
Supplementary Material
Acknowledgements
We thank Yuval Ebenstein (Tel Aviv University), Jeffrey G. Reifenberger (BioNano Genomics, Inc.), Michael T. Bowser and Pranav Agrawal for useful discussions that informed the content of this manuscript. We also thank Samira Azarin for providing the MCF7 cell line and cell culture facilities. This work was supported by NIH (R21-HG009208). Portions of this work were conducted in the Minnesota Nano Center, which is supported by the National Science Foundation through the National Nano Coordinated Infrastructure Network (NNCI) under Award Number ECCS-1542202. COMSOL modeling was performed using the resources at Minnesota Supercomputing Institute.
Footnotes
Electronic Supplementary Information (ESI) available: Details of diffusion characterization, concentration measurements, extraction efficiency, Fig. S1-S5, DNA extraction Video1, and stitched molecular combing files MC1-MC11. See DOI: 10.1039/b000000x/
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Collins FS and Varmus H, New Engl. J. Med, 2015, 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aronson SJ and Rehm HL, Nature, 2015, 526, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mardis ER, Trends Genet, 2008, 24, 133–141. [DOI] [PubMed] [Google Scholar]
- 4.Feuk L, Carson AR and Scherer SW, Nat. Rev. Genet, 2006, 7, 85–97. [DOI] [PubMed] [Google Scholar]
- 5.Alkan C, Sajjadian S and Eichler EE, Nat. Methods, 2011, 8, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eichler EE, Clark RA and She X, Nat. Rev. Genet, 2004, 5, 345–354. [DOI] [PubMed] [Google Scholar]
- 7.Treangen TJ and Salzberg SL, Nat. Rev. Genet, 2012, 13, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howorka S, Cheley S and Bayley H, Nat. Biotechnol, 2001, 19, 636–639. [DOI] [PubMed] [Google Scholar]
- 9.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B et al. , Science, 2009, 323, 133–138. [DOI] [PubMed] [Google Scholar]
- 10.Lam ET, Hastie A, Lin C, Ehrlich D, Das SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M et al. , Nat. Biotechnol, 2012, 30, 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng GX, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM et al. , Nat. Biotechnol, 2016, 34, 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, Stütz AM, Stedman W, Anantharaman T, Hastie A et al. , Nat. Methods, 2015, 12, 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao H, Hastie AR, Cao D, Lam ET, Sun Y, Huang H, Liu X, Lin L, Andrews W, Chan S et al. , GigaScience, 2014, 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT et al. , Nat. Biotechnol, 2018, 36, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stancu MC, Roosmalen MJ, Renkens I, Nieboer MM, Middelkamp S, Ligt J, Pregno G, Giachino D, Mandrile G, Valle-Inclan JE et al. , Nat. Commun, 2017, 8, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D et al. , Genome Res, 2017, 27, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, Paten B, Haussler D, Willard HF, Akeson M and Miga KH, bioRxiv, 2018, 170373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staňková H, Hastie AR, Chan S, Vrána J, Tulpová Z, Kubaláková M, Visendi P, Hayashi S, Luo M, Batley J et al. , Plant Biotechnol. J, 2016, 14, 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwartz DC and Cantor CR, Cell, 1984, 37, 67–75. [DOI] [PubMed] [Google Scholar]
- 20.Zaremba-Niedzwiedzka K, Caceres EF, Saw JH, Bäckström D, Juzokaite L, Vancaester E, Seitz KW, Anantharaman K, Starnawski P, Kjeldsen KU et al. , Nature, 2017, 541, 353–358. [DOI] [PubMed] [Google Scholar]
- 21.Kovacic RT, Comai L and Bendich AJ, Nucleic Acids Res, 1995, 23, 3999–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, Abdulwahab S, Choi K, Lafrenière NM, Mudrik JM, Gomaa H, Ahmado H, Behan L-A, Casper RF and Wheeler AR, Anal. Chem, 2015, 87, 4688–4695. [DOI] [PubMed] [Google Scholar]
- 23.Oana H, Nishikawa K, Matsuhara H, Yamamoto A, Yamamoto TG, Haraguchi T, Hiraoka Y and Washizu M, Lab Chip, 2014, 14, 696–704. [DOI] [PubMed] [Google Scholar]
- 24.Benítez JJ, Topolancik J, Tian HC, Wallin CB, Latulippe DR, Szeto K, Murphy PJ, Cipriany BR, Levy SL, Soloway PD et al. , Lab Chip, 2012, 12, 4848–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimny P, Juncker D and Reisner W, Biomicrofluidics, 2018, 12, 024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farahat WA, Wood LB, Zervantonakis IK, Schor A, Ong S, Neal D, Kamm RD and Asada HH, PloS One, 2012, 7, e37333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fangman WL, Nucleic Acids Res, 1978, 5, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Johnson M, Hill P and Gale BK, Integr. Biol, 2009, 1, 574–586. [DOI] [PubMed] [Google Scholar]
- 29.Paegel BM, Blazej RG and Mathies RA, Curr. Opin. Biotechnol, 2003, 14, 42–50. [DOI] [PubMed] [Google Scholar]
- 30.Huang CP, Lu J, Seon H, Lee AP, Flanagan LA, Kim H-Y, Putnam AJ and Jeon NL, Lab Chip, 2009, 9, 1740–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batchelor GK, An Introduction to Fluid Dynamics, Cambridge University Press, 2000. [Google Scholar]
- 32.Gabrieli T, Sharim H, Fridman D, Arbib N, Michaeli Y and Ebenstein Y, Nucleic Acids Res, 2018, 46, e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Åkerman B and Tuite E, Nucleic Acids Res, 1996, 24, 1080–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hilz H, Wiegers U and Adamietz P, FEBS J, 1975, 56, 103–108. [DOI] [PubMed] [Google Scholar]
- 35.Narayanan J, Xiong J-Y and Liu X-Y, J. Phys. Conf. Ser, 2006, 28, 83–86. [Google Scholar]
- 36.Brahmasandra SN, Ugaz VM, Burke DT, Mastrangelo CH and Burns MA, Electrophoresis, 2001, 22, 300–311. [DOI] [PubMed] [Google Scholar]
- 37.Stone HA, Stroock AD and Ajdari A, Annu. Rev. Fluid Mech, 2004, 36, 381–411. [Google Scholar]
- 38.Underhill PT and Doyle PS, Phys. Rev. E, 2007, 76, 011805. [DOI] [PubMed] [Google Scholar]
- 39.Gurrieri S, Smith SB and Bustamante C, Proc. Natl. Acad. Sci. USA, 1999, 96, 453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maydan J, Thomas M, Tabanfar L, Mai L, Poon H-L, Pe J, Hahn K, Goji N, Amoako K, Marziali A et al. , J. Biomol. Tech, 2013, 24, S57. [Google Scholar]
- 41.Dimalanta ET, Lim A, Runnheim R, Lamers C, Churas C, Forrest DK, de Pablo JJ, Graham MD, Coppersmith SN, Goldstein S et al. , Anal. Chem, 2004, 76, 5293–5301. [DOI] [PubMed] [Google Scholar]
- 42.Berlin K, Koren S, Chin C-S, Drake JP, Landolin JM and Phillippy AM, Nat. Biotechnol, 2015, 33, 623–630. [DOI] [PubMed] [Google Scholar]
- 43.Gabrieli T, Sharim H, Nifker G, Jeffet J, Shahal T, Arielly R, Levy-Sakin M, Hoch L, Arbib N, Michaeli Y et al. , bioRxiv, 2018, 260166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prinz C, Tegenfeldt JO, Austin RH, Cox EC and Sturm JC, Lab Chip, 2002, 2, 207–212. [DOI] [PubMed] [Google Scholar]
- 45.Brown RB and Audet J, J. R. Soc. Interface, 2008, 5, S131–S138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kozlowski LP, Nucleic Acids Res, 2016, 45, D1112–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo J-S, Rhie A, Kim J, Lee S, Sohn M-H, Kim C-U, Hastie A, Cao H, Yun J-Y, Kim J et al. , Nature, 2016, 538, 243–247. [DOI] [PubMed] [Google Scholar]
- 48.Marie R, Pedersen JN, Mir KU, Bilenberg B and Kristensen A, Nanoscale, 2018, 10, 1376–1382. [DOI] [PubMed] [Google Scholar]
- 49.Yu M, Hou Y, Song R, Xu X and Yao S, Small, 2018, 14, 1800229. [DOI] [PubMed] [Google Scholar]
- 50.Marie R, Pedersen JN, Brlocher L, Koprowska K, Pødenphant M, Sabatel C, Zalkovskij M, Mironov A, Bilenberg B, Ashley N et al. , Proc. Natl. Acad. Sci. USA, 2018, 201804194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Teague B, Waterman MS, Goldstein S, Potamousis K, Zhou S, Reslewic S, Sarkar D, Valouev A, Churas C, Kidd JM et al. , Proc. Natl. Acad. Sci. USA, 2010, 107, 10848–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murtaza M, Dawson S-J, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin S-F, Kingsbury Z, Wong AS et al. , Nature, 2013, 497, 108–112. [DOI] [PubMed] [Google Scholar]
- 53.Fonslow BR, Barocas VH and Bowser MT, Anal. Chem, 2006, 78, 5369–5374. [DOI] [PubMed] [Google Scholar]
- 54.Pavesi A, Adriani G, Tay A, Warkiani ME, Yeap WH, Wong SC and Kamm RD, Sci. Rep, 2016, 6, 26584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ugaz VM, Lin R, Srivastava N, Burke DT and Burns MA, Electrophoresis, 2003, 24, 151–157. [DOI] [PubMed] [Google Scholar]
- 56.Xia Y and Whitesides GM, Angew. Chem. Int. Ed, 1998, 37, 550–575. [DOI] [PubMed] [Google Scholar]
- 57.Preibisch S, Saalfeld S and Tomancak P, Bioinformatics, 2009, 25, 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.