

Abstract
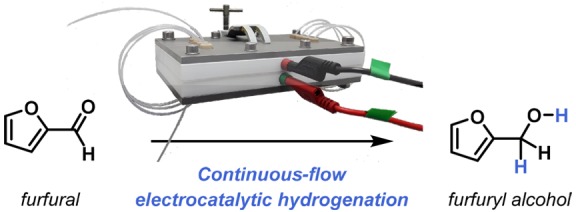
Furfural is considered to be an essential biobased platform molecule. Recently, its electrocatalytic hydrogenation is regarded as a more environmentally friendly process compared to traditional catalytic hydrogenation. In this study, a new, continuous-flow approach enabling furfural electrocatalytic reduction was developed. In an undivided multichannel electrochemical flow reactor at ambient temperature and pressure in basic reaction conditions, the yield of furfuryl alcohol reached up to 90% in only 10 min residence time. Interestingly, the faradaic efficiency was 90%, showing a good effectiveness of the consumed electrons in the generation of the targeted compound. Furthermore, the innovation lies in the direct electrolysis using the green solvent ethanol without the need for membrane separation or catalyst modification, which offers further proof for continuous and sustainable production in industry.
Keywords: electrochemistry, flow chemistry, furfural, furfuryl alcohol, microreactor, flow electrochemistry
Introduction
Furfural is one of the most significant industrial biobased chemicals, which is obtained via dehydration of sugars, as obtained from a variety of agricultural products such as corncobs and wheat bran.1 It possesses extensive application prospects as a renewable, nonpetroleum-based platform molecule.1−6 Consequently, furfuryl alcohol is widely used as a monomer for the synthesis of furan resins.5 These polymers are used in thermoset polymer matrix composites, cements, adhesives, and coatings. It is also recognized as a valuable intermediate in the fields of agriculture, medicine, and dyes.6 Compared to conventional high-pressure hydrogenation processes, the electrolysis of furfural offers not only a promotion for agricultural waste utilization7 but also constitutes an energy-saving green process.8 Li’s group recently reported an interesting electrocatalytic reduction of furfural to furfuryl alcohol in a divided cell using a cation exchange membrane.9 Recently, Koper et al. reviewed the state of the art for furfural electroreduction.10 The majority of the electrochemical reduction reactions were performed either with noble metal catalysts in batch electrochemical cells or required long residence times in flow-through electrochemical cells.11 Arguably, the remaining problems lie in the discontinuity of the batch process, the complexity of the utilized electrodes, and the limitation of the use of divided cells.
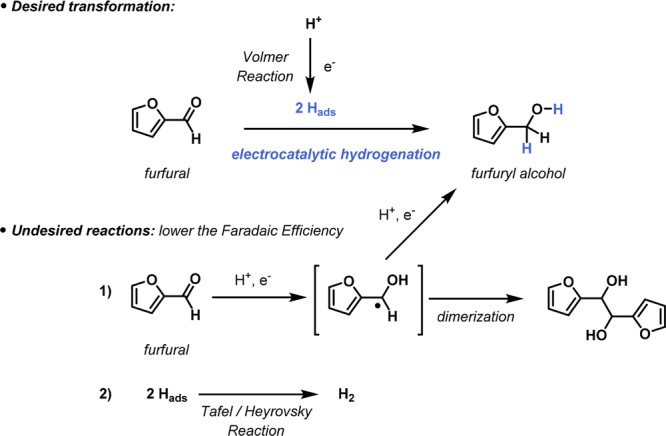
The electrocatalytic reduction is an intriguing example which enables the hydrogenation of molecules using adsorbed hydrogen at the electrode which originates from H+ or H2O via the Volmer reaction (Scheme 1).12 Hence, the reaction can be carried out at very mild reaction conditions (room temperature, atmospheric pressure, no H2 needed) and does not require expensive first or second row transition-metal-based catalysts. However, despite these apparent advantages in favor of electrocatalytic reductions, there are some challenges with regard to nonproductive competitive reactions (Scheme 1). One is the occurrence of hydrogen evolution where adsorbed hydrogen is consumed via the so-called Tafel and Heyrovsky reactions,13 which lowers the Faradaic efficiency of the transformation significantly.14 Another competitive transformation is the electroreduction of the carbonyl moiety to generate the respective radical. These radicals can consume another electron to generate the targeted furfuryl alcohol or can dimerize to generate undesired diols. Hence, efficient electrocatalytic reduction transformations require a careful balancing of all the different parameters (e.g., electrode material, reaction conditions) to favor the desired transformation over these unproductive pathways.
Scheme 1. Desired Electrocatalytic Hydrogenation of Furfural towards Furfuryl Alcohol versus Undesired Reactions.
Herein, we report a continuous-flow direct electrolysis approach for electroreduction of furfural to address the above-mentioned issues using an electrochemical flow reactor developed in our group (Figure 1).15 Key in the design of this electrochemical reactor is its flexible reactor volume, enabling two different operation modes, i.e. a serial (volume ranging from 88 μL/channel up to 704 μL) or a parallel mode (numbering-up or parallel screening of reaction conditions) (Figure 2).16 In this reactor, we screened both basic and acidic reaction conditions, and our results indicate that a basic environment is optimal not only to obtain a high yield of the targeted furfuryl alcohol but also to obtain high Faradaic efficiencies.
Figure 1.

Continuous multichannel electrochemical flow reactor which allows for a rapid screening of key reaction conditions in parallel.
Figure 2.

Serial mode (left) or parallel mode (right) operation of the electrochemical flow reactor.
Experimental Section
1. General Information
For the chemicals, furfural (99%), furfuryl alcohol (98%), 3-furancarboxaldehyde (97%), furan-3-methanol (99%), potassium hydroxide pellets (85%), potassium ethoxide (95%), sodium hydroxide pellets (98%), and tetrabutylammonium bromide (99%), were purchased from Sigma-Aldrich and used as received. Hydrochloric acid (37%), ethanol (99.5%), and acetonitrile (99%) were bought from VWR. Sulfuric acid (98%) was obtained from Merck. Ammoeng 110 Poly[oxy(methyl-1,2-ethanediyl)],a-[2-(diethylmethylammonio)ethyl]-w-hydroxy-,chloride (1:1), 95% was gained from Iolitec. One molar HCl and 0.5 M H2SO4 solution were prepared by dilution with deionized water (18.2 MΩ·cm).
2. Flow Setup
For the flow setup, microsyringe pumps (Fusion 720, Chemyx) were used to infuse the liquid solutions. Before each set of experiments, stock solutions were prepared according to the different conditions as described herein. The liquids were taken up into disposable plastic syringes (10 mL, BD Discardit II) and mounted on the syringe pumps. Microfluidic connections between the electrochemical flow reactor and the syringes consisted of perfluoroalkoxy capillary tubing (ID 0.75 mm or 1.58 mm, PFA) and were attained from APT. Microfluidic connectors (LT-115X) and ferrules (P-259X) were procured from IDEX. Electric power (0–30 V, 0–5 A) was delivered by a LABPS3005D power supply. Electrode material (i.e., stainless steel 316L, copper Cu-DHP, and graphite) were bought at Salomon’s Metalen B.V. The reactor body (reinforced iron plates and PTFE supporting containers) was manufactured by Equipment & Prototype Center at Eindhoven University of Technology. It should be noted that the electrodes did not display any corrosion under the given reaction conditions.15,17
3. Voltammogram
Prior to each experiment, a voltammogram was acquired which determines the suitable electrochemical reaction conditions.18 As shown in Figure 3, a plateau appeared at 2.80–3.00 V in acidic electrolyte, while it situated at 2.10–2.30 V in basic electrolyte. Therefore, 2.90 and 2.20 V were respectively chosen for subsequent optimization.
Figure 3.

Voltammogram of furfural at a residence time of 10 min in acidic (A) or basic (B) reaction conditions.
Results and Discussion
We commenced our investigations toward the electroreduction of furfural in 0.5 M sulfuric acid in combination with acetonitrile using a copper/stainless steel electrode pair (Table 1, entry 1). However, only a yield of 8% was obtained for the targeted furfuryl alcohol. When switching to a more protic solvent such as ethanol, an increase in yield toward 29% was witnessed (Table 1, entry 2). Interestingly, a further improvement was observed in the presence of hydrochloric acid (Table 1, entries 3 and 4). In contrast, changing the electrode material did not result into a further increase in yield (Table 1, entries 5–8), indicating that the combination of stainless steel/copper is optimal. Finally, we also checked the need for supporting electrolytes which are required to minimize the Ohmic drop in electrochemical processes. It is often claimed that microreactors do not require any supporting electrolytes due to small interelectrode gap.19,20 However, in our case, small amounts of electrolyte (0.05 M), such as the ionic liquid Ammoeng 110 or n-Bu4NBr, were beneficial to increase the overall yield.
Table 1. Optimization for Furfuryl Alcohol Production in Acidic Environmenta.

| entry | electrolyted | working electrodec | counter electrode | solvent | supporting electrolyte | yield (%)b |
|---|---|---|---|---|---|---|
| 1 | H2SO4 | copper | stainless steel | CH3CN | 8 | |
| 2 | H2SO4 | copper | stainless steel | EtOH | 29 | |
| 3 | HCl | copper | stainless steel | CH3CN | 33 | |
| 4 | HCl | copper | stainless steel | EtOH | 41 | |
| 5 | HCl | stainless steel | stainless steel | EtOH | 14 | |
| 6 | HCl | graphite | stainless steel | EtOH | 22 | |
| 7 | HCl | copper | graphite | EtOH | 1 | |
| 8 | HCl | copper | copper | EtOH | 10 | |
| 9 | HCl | copper | stainless steel | EtOH | Ammoeng 110 | 55 |
| 10 | HCl | copper | stainless steel | EtOH | n-Bu4NBr | 55 |
Reaction conditions: 0.1 M furfural, 0.1 M H+, 0.05 M supporting electrolyte, 10% H2O, flow rate 0.075 mL/min, residence time 10 min.
GC-yield using GC-FID with external standard calibration.
The SAE international stainless steel type is 316L; the copper type is Cu-DHP.
The molar concentration of HCl is 0.1 mol/L; the molar concentration of H2SO4 is 0.05 mol/L in stock solution.
Next, we turned our attention to basic reaction conditions using potassium ethoxide in ethanol.21,22 Interestingly, a substantial increase in the reaction yield was observed (Table 2). Varying the electrode material showed that the best results were obtained with a copper/graphite couple (Table 2, entry 3) yielding the targeted furfuryl alcohol in 90%. Other bases, such as NaOH and KOH, did not lead to any further improvement (Table 2, entries 6 and 7).
Table 2. Optimization for Furfuryl Alcohol Production in Basic Environmenta.

| entry | electrolyte | working electrodec | counter electrode | yield (%)b |
|---|---|---|---|---|
| 1 | C2H5OK | graphite | graphite | 25 |
| 2 | C2H5OK | stainless steel | graphite | 46 |
| 3 | C2H5OK | copper | graphite | 90 |
| 4 | C2H5OK | copper | stainless steel | 43 |
| 5 | C2H5OK | copper | copper | 10 |
| 6 | NaOH | copper | graphite | 41 |
| 7 | KOH | copper | graphite | 78 |
Reaction conditions: 0.1 M furfural, 0.05 M electrolyte, ethanol solvent, flow rate 0.075 mL/min, residence time 10 min.
GC-yield using GC-FID with external standard calibration.
The SAE international stainless steel type is 316L; the copper type is Cu-DHP. The counter electrode is graphite.
Given the importance of the use of acids or bases to effect the desired transformation, we next turned our attention to scan a wide variety of different concentrations (Table 3). For this specific experiment, our homemade electrochemical reactor design provided a clear and unique advantage as eight reaction conditions can be screened simultaneously in parallel in the eight different channels. Hence, we rapidly found that 0.10 M HCl and 0.05 M C2H5OK provided optimal results for electroreduction of furfural.
Table 3. Optimization of Electrolyte via 8-Channel Simultaneously Parallel Reactions.
| channela | electrolyte | yield (%)c | channelb | electrolyte | yield (%)c |
|---|---|---|---|---|---|
| 1 | 0.05 M HCl | 22 | 1 | 0.01 M C2H5OK | 38 |
| 2 | 0.07 M HCl | 35 | 2 | 0.02 M C2H5OK | 50 |
| 3 | 0.09 M HCl | 46 | 3 | 0.03 M C2H5OK | 62 |
| 4 | 0.10 M HCl | 55 | 4 | 0.04 M C2H5OK | 82 |
| 5 | 0.11 M HCl | 53 | 5 | 0.05 M C2H5OK | 90 |
| 6 | 0.13 M HCl | 49 | 6 | 0.06 M C2H5OK | 88 |
| 7 | 0.15 M HCl | 46 | 7 | 0.07 M C2H5OK | 86 |
| 8 | 0.17 M HCl | 45 | 8 | 0.08 M C2H5OK | 83 |
Reaction conditions: 0.1 M furfural, 2.90 V, 10% H2O, ethanol solvent, flow rate 0.075 mL/min, residence time 10 min.
Reaction conditions: 0.1 M furfural, 2.20 V, ethanol solvent, flow rate 0.075 mL/min, residence time 10 min.
GC-yield using GC-FID with external standard calibration.
Having determined the relative importance of the reaction partners, we investigated the influence of the reaction time (Figure 3). Interestingly, in both acidic and basic media, the optimal residence time to reach full conversion is only 10 min. We believe this very short residence time can be attributed to the large electrode surface-to-volume ratio and the short diffusion distances observed in the electrochemical microreactor. Indeed, when we carried out the reaction in a batch electrochemical cell, the yield dropped to 18% in acidic medium and 31% in basic conditions and requires 6 h of reaction time.
Next, we tested the influence of constant current or constant potential electrolysis conditions (Figure 4). Constant potential electrolysis offers a more stable and tunable selectivity,17 while constant current electrolysis provides a more controlled and constant reaction conversion.23 However, for the electroreduction of furfural, almost similar behavior was observed under these two operational regimes. This gives us a high level of flexibility: if higher selectivity is required, one can use the constant potential conditions, while galvanostatic conditions are preferred to establish an adequate chemical conversion in shorter time scales. However, it must be noted that even though we did use potentiostatic reaction conditions, the flow processing enables us the keep the conversion time very short as described above (tR = 10 min, see Figure 5).
Figure 4.

Constant-potential (A)(C) vs constant-current (B)(D) electrolysis.
Figure 5.
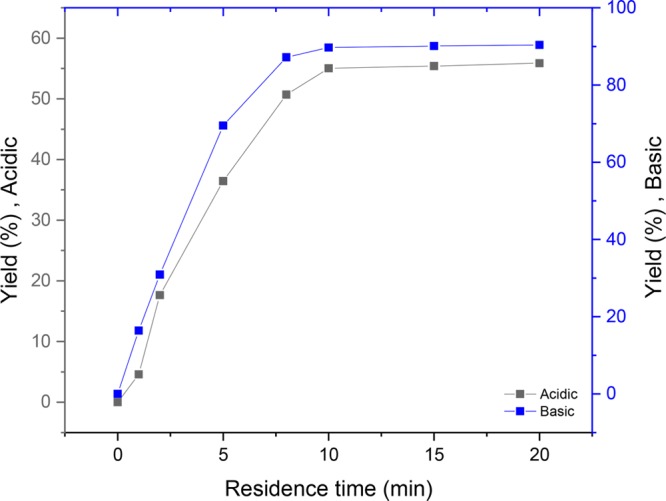
Influence of the residence time on the electroreduction of furfural to furfuryl alcohol in acidic and basic media.
Having the optimal reaction conditions for the electroreduction of 2-furfural in hand (Table 4, entries 1 and 2), we investigated if the conditions were also applicable to 3-furfural (Table 4, entries 5 and 6). Interestingly, the same trend was observed for 3-furfural with the most optimal conversion and yield observed in basic media (Table 4, entry 6). In addition, for all the different conditions, we calculated the Faradaic efficiency: high Faradaic efficiencies were obtained for the basic reaction conditions (90–92%), while in acidic media, very poor Faradaic efficiencies were observed (18–19%). This can be attributed to the generation of higher amounts of hydrogen, as the hydrogen evolution reaction competes more efficiently with the electrocatalytic hydrogenation reaction in acidic reaction conditions. Interestingly, the corresponding batch conditions were not only considerably slower but also provided lower yields and Faradaic efficiencies (Table 4, entries 3 and 4). We surmise that the slower reaction times are due to mass transfer limitations and lower electrode surface-to-volume ratios and give more time for the undesired hydrogen formation reaction to occur.
Table 4. Scope for Furfural Electroreduction in Flow and Comparison with Batch Reaction Conditions.
| entry | reactor type | substrate | electrolyte | yield (%)e | faradaic efficiency (%) |
|---|---|---|---|---|---|
| 1a | flow | furfural (2-furaldehyde) | HCl | 55 | 19 |
| 2b | flow | furfural (2-furaldehyde) | C2H5OK | 90 | 90 |
| 3c | batch | furfural (2-furaldehyde) | HCl | 18 | 12 |
| 4d | batch | furfural (2-furaldehyde) | C2H5OK | 31 | 60 |
| 3a | flow | 3-furfural (3-furaldehyde) | HCl | 40 | 18 |
| 4b | flow | 3-furfural (3-furaldehyde) | C2H5OK | 78 | 92 |
Reaction conditions: 0.1 M furfural, 2.90 V, 0.1 M electrolyte, 10% H2O, ethanol solvent, flow rate 0.075 mL/min, residence time 10 min.
Reaction conditions: 0.1 M furfural, 2.20 V, 0.05 M electrolyte, ethanol solvent, flow rate 0.075 mL/min, residence time 10 min.
Reaction conditions: 0.1 M furfural, 2.90 V, 0.1 M electrolyte, 10% H2O, ethanol solvent, residence time 300 min.
Reaction conditions: 0.1 M furfural, 2.20 V, 0.05 M electrolyte, ethanol solvent, residence time 300 min.
GC-yield using GC-FID with external standard calibration.
Finally, we investigated the steady state production of 2-furfuryl alcohol under these acidic and basic reaction conditions. We took reaction samples every 5 min and analyzed the obtained yield via GC-FID. The overall observed trend for both conditions showed a decline in yield over time. This decline can be attributed to erosion of the electrode efficiency. It was noteworthy, however, that the efficacy of the electrodes could be restored by incorporating an adequate cleaning and surface treatment step (Figure 6). This indicates that the reduced yield is caused by a deposition on the electrode surface which can be washed away effectively.24 To keep the reaction yield constant, we could place two reactors in parallel: one of them is in operation mode while the other one is in cleaning mode. When the yield drops, the modes can be switched and a “continuous” production of electrocatalytic reduction of furfural can be simulated.
Figure 6.
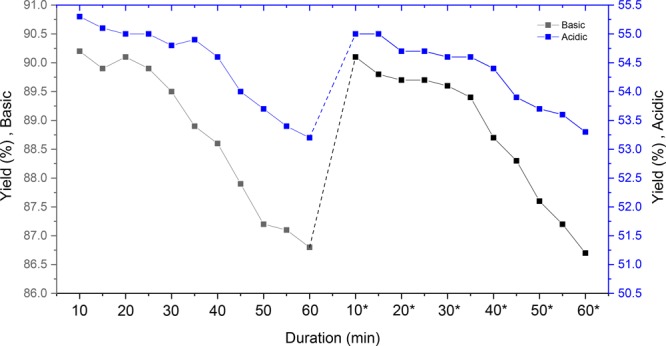
Two runs of stability test (60 min) for reactions (Table 4, entries 1 and 2) with 10 min residence time.
Conclusion
In conclusion, we developed a continuous-flow protocol for the electrocatalytic reduction of furfural. Both acidic and basic reaction conditions were evaluated. We found that the basic reaction conditions delivered the highest yield for the targeted furfuryl alcohol and provided a high Faradaic efficiency. The reaction could be completed in only 10 min and required only a minimum amount of supporting electrolyte, highlighting the benefits of the microreactor environment. While an efficient process was established in flow, a reduced efficiency was observed due to pollution of the electrodes. However, we found that a cleaning step could alleviate this issue effectively.
Acknowledgments
The authors acknowledge support from the China Scholarship Council (CSC).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.oprd.8b00428.
(i) Design details of the electrochemical batch and flow reactor; (ii) details on the calculation of the Faradaic efficiency; (iii) GC results for the optimal reaction conditions; (iv) detailed procedure for the cleaning of our electrodes (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Mariscal R.; Maireles-Torres P.; Ojeda M.; Sádaba I.; López Granados M. Furfural: A Renewable and Versatile Platform Molecule for the Synthesis of Chemicals and Fuels. Energy Environ. Sci. 2016, 9 (4), 1144–1189. 10.1039/C5EE02666K. [DOI] [Google Scholar]
- LaForge F. B. Furfural from Corncobs. Ind. Eng. Chem. 1923, 15 (5), 499–502. 10.1021/ie50161a032. [DOI] [Google Scholar]
- Riera F. A.; Alvarez R.; Coca J. Production of Furfural by Acid Hydrolysis of Corncobs. J. Chem. Technol. Biotechnol. 1991, 50 (2), 149–155. 10.1002/jctb.280500202. [DOI] [Google Scholar]
- Sánchez C.; Serrano L.; Andres M. A.; Labidi J. Furfural Production from Corn Cobs Autohydrolysis Liquors by Microwave Technology. Ind. Crops Prod. 2013, 42 (1), 513–519. 10.1016/j.indcrop.2012.06.042. [DOI] [Google Scholar]
- Gandini A.; Belgacem M. N. Furans in Polymer Chemistry. Prog. Polym. Sci. 1997, 22 (6), 1203–1379. 10.1016/S0079-6700(97)00004-X. [DOI] [Google Scholar]
- Eseyin E. A.; Steele H. P. An Overview of the Applications of Furfural and Its Derivatives. Int. J. Adv. Chem. 2015, 3 (2), 42–47. 10.14419/ijac.v3i2.5048. [DOI] [Google Scholar]
- Möhle S.; Zirbes M.; Rodrigo E.; Gieshoff T.; Wiebe A.; Waldvogel S. R. Modern Electrochemical Aspects for the Synthesis of Value-Added Organic Products. Angew. Chem., Int. Ed. 2018, 57 (21), 6018–6041. 10.1002/anie.201712732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamoulaud G.; Floner D.; Moinet C.; Lamy C.; Belgsir E. M. Biomass Conversion II: Simultaneous Electrosyntheses of Furoic Acid and Furfuryl Alcohol on Modified Graphite Felt Electrodes. Electrochim. Acta 2001, 46 (18), 2757–2760. 10.1016/S0013-4686(01)00507-2. [DOI] [Google Scholar]
- Chadderdon X. H.; Chadderdon D. J.; Matthiesen J. E.; Qiu Y.; Carraher J. M.; Tessonnier J.-P.; Li W. Mechanisms of Furfural Reduction on Metal Electrodes: Distinguishing Pathways for Selective Hydrogenation of Bioderived Oxygenates. J. Am. Chem. Soc. 2017, 139 (40), 14120–14128. 10.1021/jacs.7b06331. [DOI] [PubMed] [Google Scholar]
- Kwon Y.; Schouten K. J. P.; van der Waal J. C.; de Jong E.; Koper M. T. M. Electrocatalytic Conversion of Furanic Compounds. ACS Catal. 2016, 6 (10), 6704–6717. 10.1021/acscatal.6b01861. [DOI] [Google Scholar]
- Parpot P.; Bettencourt A. P.; Chamoulaud G.; Kokoh K. B.; Belgsir E. M. Electrochemical Investigations of the Oxidation-Reduction of Furfural in Aqueous Medium - Application to Electrosynthesis. Electrochim. Acta 2004, 49 (3), 397–403. 10.1016/j.electacta.2003.08.021. [DOI] [Google Scholar]
- Guena T.; Pletcher D. Electrosyntheses from Aromatic Aldehydes in a Flow Cell. Part I. The Reduction of Benzaldehyde. Acta Chem. Scand. 1998, 52, 23–31. 10.3891/acta.chem.scand.52-0023. [DOI] [PubMed] [Google Scholar]
- de Chialvo M. R. G.; Chialvo A. C. Hydrogen Evolution Reaction: Analysis of the Volmer-Heyrovsky-Tafel Mechanism with a Generalized Adsorption Model. J. Electroanal. Chem. 1994, 372 (1), 209–223. 10.1016/0022-0728(93)03043-O. [DOI] [Google Scholar]
- Yuan H.; He Z. Platinum Group Metal-Free Catalysts for Hydrogen Evolution Reaction in Microbial Electrolysis Cells. Chem. Rec. 2017, 17 (7), 641–652. 10.1002/tcr.201700007. [DOI] [PubMed] [Google Scholar]
- Laudadio G.; de Smet W.; Struik L.; Cao Y.; Noël T. Design and Application of a Modular and Scalable Electrochemical Flow Microreactor. J. Flow Chem. 2018, 8 (3), 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher D.; Green R. A.; Brown R. C. D. Flow Electrolysis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 2018, 118 (9), 4573–4591. 10.1021/acs.chemrev.7b00360. [DOI] [PubMed] [Google Scholar]
- Laudadio G.; Straathof N. J. W.; Lanting M. D.; Knoops B.; Hessel V.; Noël T. An Environmentally Benign and Selective Electrochemical Oxidation of Sulfides and Thiols in a Continuous-Flow Microreactor. Green Chem. 2017, 19, 4061–4066. 10.1039/C7GC01973D. [DOI] [Google Scholar]
- Nicholson R. S.; Shain I. Theory of Stationary Electrode Polarography. Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems. Anal. Chem. 1964, 36 (4), 706–723. 10.1021/ac60210a007. [DOI] [Google Scholar]
- Folgueiras-Amador A. A.; Philipps K.; Guilbaud S.; Poelakker J.; Wirth T. An Easy-to-Machine Electrochemical Flow Microreactor: Efficient Synthesis of Isoindolinone and Flow Functionalization. Angew. Chem., Int. Ed. 2017, 56 (48), 15446–15450. 10.1002/anie.201709717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folgueiras-Amador A. A.; Wirth T. Perspectives in Flow Electrochemistry. J. Flow Chem. 2017, 7 (3–4), 94–95. 10.1556/1846.2017.00020. [DOI] [Google Scholar]
- Bradley D. C. Metal Alkoxides. Adv. Chem. Ser. 1959, 23, 2–10. 10.1021/ba-1959-0023.ch002. [DOI] [Google Scholar]
- Msayib K. J.; Watt C. I. F. Ion Pairing and Reactivity of Alkali Metal Alkoxides. Chem. Soc. Rev. 1992, 21, 237–243. 10.1039/cs9922100237. [DOI] [Google Scholar]
- Wiebe A.; Riehl B.; Lips S.; Franke R.; Waldvogel S. R. Unexpected High Robustness of Electrochemical Cross-Coupling for a Broad Range of Current Density. Sci. Adv. 2017, 3 (10), eaao3920 10.1126/sciadv.aao3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville L.; Bareño J.; Jennings P.; McGordon A.; Lyness C.; Bloom I. The Effect of Pre-Analysis Washing on the Surface Film of Graphite Electrodes. Electrochim. Acta 2016, 206, 70–76. 10.1016/j.electacta.2016.04.133. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.