

Abstract
Recent evidence suggests hypertension may be secondary to chronic inflammation that results from hypoactive neuro-immune regulatory mechanisms. To further understand this association, we used systemic lupus erythematosus (SLE) as a model of inflammation-induced hypertension. In addition to prevalent inflammatory kidney disease and hypertension, SLE patients suffer from dysautonomia in the form of decreased efferent vagal tone. Based on this, the cholinergic anti-inflammatory pathway, an endogenous vagus-to-spleen mechanism that, when activated results in decreases in systemic inflammation, may be compromised in SLE. We hypothesized that stimulation of the cholinergic anti-inflammatory pathway via pharmacological potentiation of the efferent vagus nerve would reduce inflammation and halt the development of hypertension and renal injury in SLE. Female NZBWF1 mice, an established model of murine SLE, and female control mice were treated with galantamine (4 mg/kg daily ip), an acetylcholinesterase inhibitor, or saline for 14 days. At the end of therapy, carotid catheters were surgically implanted and were used to measure mean arterial pressure before the animals were euthanized. Chronic galantamine administration attenuated both splenic and renal cortical inflammation, which likely explains why the hypertension and renal injury (i.e., glomerulosclerosis and fibrosis) typically observed in murine SLE was attenuated following therapy. Based on this, the anti-inflammatory, antihypertensive, and renoprotective effects of galantamine may be mediated through activation of the cholinergic anti-inflammatory pathway. It is possible that dysfunction of the cholinergic anti-inflammatory pathway exists in SLE at the level of the efferent vagus nerve and promoting restoration of its activity through central cholinergic receptor activation may be beneficial.
Keywords: galantamine, hypertension, lupus nephritis, neuroimmune, renal inflammation
INTRODUCTION
Hypertension affects nearly one-half of the adult population in the United States, and this figure will only increase in the coming decades (67). Despite growing awareness of the numerous etiologies of hypertension, at least 15–20% of hypertensive patients are resistant to current treatment modalities (50). It is, therefore, necessary to investigate novel mechanisms involved, to develop new therapeutic strategies to target the disease.
Recent studies support the role of dysregulated immune cell activity and chronic inflammation in the development and maintenance of hypertension. In general, elevated circulating proinflammatory cytokines [i.e., TNF-α (5), IL-6 (5), and B cell activating factor (BAFF) (11), among others] are correlated with hypertension, indicating that peripheral inflammation may mechanistically contribute to chronic increases in blood pressure (7, 10, 37). Renal inflammation, in particular, also directly affects kidney processes that may promote increases in blood pressure. TNF-α in the kidney, for example, has been shown to increase renal vascular resistance (56), as well as, prompt immune cell-mediated renal damage (52, 68) Blockade of TNF-α, in turn, attenuates both renal injury and hypertension in a variety of experimental animal models (16, 17, 63, 64). In response, the finding that immunomodulatory drugs are therapeutic in both hypertensive animal models (60) and in human patients with underlying chronic inflammation (24) warrants further investigation into the mechanisms that regulate renal inflammation, in particular, due to the role of the kidneys in the long-term control of blood pressure.
Endogenous nervous system/immune system interactions are known to control inflammation both acutely (46) and chronically (15, 32). Tracey and colleagues have demonstrated that the vagally mediated cholinergic anti-inflammatory pathway is capable of attenuating splenic inflammation on an immediate basis, in the context of an acute inflammatory stressor (3, 6, 8, 31). However, the role of this nerve-to-spleen pathway in chronic, or even inherent inflammatory states, in the case of autoimmunity, is not as well delineated. We have identified the autoimmune inflammatory disease systemic lupus erythematosus (SLE) as an appropriate model to investigate neuroimmune mechanisms that regulate chronic inflammation, which, if left unchecked, can lead to hypertension (38, 54). Indeed, the prevalence of hypertension in SLE is exceedingly high, especially in its target demographic of reproductive-age women who are normally protected from hypertension and cardiovascular disease (54). Dysautonomia is also associated with SLE, and these patients have decreased heart rate variability in the high-frequency power domain, which indicates impaired efferent vagal tone (61). A functioning efferent vagus nerve is thought to be necessary for proper neuroimmune communication in the cholinergic anti-inflammatory pathway, and since efferent vagal tone is decreased in SLE, perhaps the efficacy of this pathway in reducing inflammation is compromised.
Galantamine is a reversible acetylcholinesterase inhibitor capable of crossing the blood-brain barrier and can act upon M1 muscarinic receptors centrally to potentiate efferent vagus nerve activity (48). In the present study, we hypothesized that pharmacological stimulation of the efferent vagus nerve, using galantamine, would enhance the cholinergic anti-inflammatory pathway and attenuate systemic inflammation, thus protecting from renal inflammation, renal injury, and hypertension in a mouse model of SLE.
MATERIALS AND METHODS
Animals
Female SLE (NZBWF1) and control (NZW/LacJ) mice were obtained from Jackson Laboratories (Bar Harbor, ME) at 5–6 wk of age. All mice were maintained on a 12-h light/dark cycle in temperature-controlled rooms with access to food and water ad libitum. Mice were monitored starting at 29 wk of age, an age at which SLE mice have already developed autoantibodies and renal disease. All animal studies were approved by the University of North Texas Health Science Center Institutional Animal Care and Use Committee and were in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Acute vagus nerve recording.
Vagus nerve recordings were performed on anesthetized animals. SLE and control mice were weighed and intraperitoneally injected with α-chloralose (80 mg/kg) and urethane (1,000 mg/kg). Fur was shaved off the cervical region and mice were instrumented with a model SPR 671 Millar catheter in the left carotid artery to continuously record blood pressure and heart rate. The right cervical vagus nerve was localized and placed on a hooked platinum-iridium electrode (36 gauge). The electrode was encased in QuikSil silicone gel to minimize noise and interference from the environment. Electric signals were amplified (×200,000) and filtered (band pass: 10–10,000 Hz) to represent vagus nerve activity. A 10-min baseline was recorded in Spike2 (Cambridge Electronic Design), and then a single dose of galantamine (4 mg/kg dissolved in saline) was intraperitoneally injected, with the following vagus nerve activity recorded. Vagal activity was allowed to recover to baseline, and this process was repeated.
Chronic galantamine administration.
At 32–33 wk of age, animals were randomly divided into four groups: control/vehicle, control/galantamine, SLE/vehicle, and SLE/galantamine. Animals were injected intraperitoneally with galantamine (4 mg·kg−1·day−1; Sigma, St. Louis, MO) (48) or the vehicle, normal saline, for 14 consecutive days.
Blood pressure measurements.
At 34–35 wk of age (following 2 wk of galantamine injections), catheters were implanted into the left carotid artery as previously described (38–43). Mean arterial pressure was then measured in conscious mice via pressure transducers for 1.5 h for 2 consecutive days postsurgery. At the end of the study, anesthetized mice were perfused with 2% heparinized saline and then euthanized. Plasma and tissues (i.e., kidneys and spleen) were harvested and stored for biochemical analysis.
Autoantibody measurements.
Plasma double-stranded DNA (dsDNA) autoantibodies, an index of SLE disease severity, were quantified via commercial ELISA kits (Alpha Diagnostics), as previously described (18, 22, 40–42, 65).
Inflammatory mediators.
Upon euthanasia of anesthetized mice, the spleen was dissected from each mouse and was homogenized at 4°C in RIPA buffer. The renal cortex was dissected from the right kidney and was also homogenized in RIPA buffer. Aortas were stripped from the spine of each mice and then flash frozen in liquid nitrogen to quickly crush with a plastic plunger to homogenize with an appropriate amount of RIPA buffer (i.e., 8 times the weight of the aorta). Protein concentrations of the splenic, renal cortical, and aortic supernatants were quantified via bicinchoninic acid assay (ThermoFisher).
Standard Western blotting techniques were performed as previously described using splenic, cortex-rich renal, and aortic homogenates (18) to measure TNF-α (catalog no. sc-52746; Santa Cruz Biotechnology, Santa Cruz, CA); high mobility group box protein 1 (HMGB-1) (catalog no. sc-26351; Santa Cruz Biotechnology); and BAFF (catalog no. MAB1357–100; R&D Systems, Minneapolis, MN), all well-accepted markers of proinflammation. Proteins of interest were visualized using horseradish peroxidase-conjugated donkey anti-mouse, donkey anti-rabbit, or goat anti-mouse secondary antibodies, as appropriate. Western blots were imaged using the ChemiDoc MP Imaging System (Bio-Rad Laboratories; Hercules, CA) following chemiluminescent detection via Clarity Western ECL (Bio-Rad) and analyzed using Image Lab software version 5.1. Data are reported as volume intensity, which refers to the cytokine densitometry of the protein band normalized to total lane protein, which was determined by the stain-free total protein method as previously described (23). For TNF-α, both the 26- and 51-kDa bands were analyzed, the former comprising the transmembrane form and the latter, constituting the active, secreted trimeric form (57). To emphasize the mechanistic role of renal inflammation upon blood pressure, only data from animals that also had blood pressure measurements were included in the analysis of renal proinflammatory cytokine measurements.
Indices of renal injury.
Albumin excretion rate was determined following 24-h urine collection via metabolic cages and quantification of urinary albumin by ELISA (Alpha Diagnostics) as previously described (18, 22, 38–43, 65). Formalin-fixed sections of left kidney were embedded in paraffin and then sliced at 6 μm and stained with Periodic-Acid Schiff and Masson’s Trichrome, and 30 glomeruli from each subject were blindly scored, as previously reported (43).
Statistical analysis.
All data are calculated as means ± SE and statistical analysis were performed using SigmaPlot 11.0 (Systat, Richmond, CA). Statistical differences (all P < 0.05) between multiple groups were determined by two-way ANOVA, with or without repeated measures, followed by the Holm-Sidak post hoc test, as specified in the figures and accompanying figure legends.
RESULTS
Galantamine increases efferent vagus nerve activity.
To confirm the ability of peripherally administered galantamine to potentiate efferent vagus nerve activity, anesthetized SLE and control mice were injected with galantamine (4 mg/kg ip), and the immediate response of the vagus nerve was recorded. Galantamine produced a robust increase in vagus nerve activity that was mirrored by a decrease in heart rate in control and SLE mice (n = 4/group; Fig. 1, B and D). In comparison, intraperitoneal injection of normal saline did not affect vagus nerve activity or heart rate in control or SLE mice (n = 3/group, Fig. 1, A and C).
Fig. 1.

Galantamine increases efferent vagus nerve activity. Representative vagus nerve activity (V, voltage) recorded from the cervical vagus, and corresponding heart rate (BPM, beats per minute) tracings at baseline and immediately after administration of normal saline or galantamine (4 mg/kg ip) in anesthetized control (A and B) and systemic lupus erythematosus (SLE, C and D) mice (n = 3–4/group).
Galantamine has no effect on body weight.
SLE mice treated with vehicle and galantamine had higher body weight than control mice treated with vehicle and galantamine throughout the study (P < 0.001) (Fig. 2). In addition, body weight was significantly reduced throughout the study (P < 0.001). However, there was no significant interaction between treatment group and time (P = 0.274).
Fig. 2.
Galantamine does not alter body weight. There was a natural significant reduction in body weight (g, grams) in all mice used in the study between weeks 32 and 34. Both vehicle- and galantamine-treated systemic lupus erythematosus (SLE) mice had greater body weight than control mice throughout the study. Body weight did not differ between vehicle-treated SLE mice and galantamine-treated SLE mice or between vehicle-treated control mice and galantamine-treated control mice. Values are presented as means ± SE. A two-way ANOVA with repeated measures was used to make statistical comparisons (n = 11–13/group).
Galantamine decreased plasma concentrations of dsDNA autoantibodies in female SLE mice.
Plasma dsDNA autoantibodies are a commonly accepted diagnostic and prognostic indicator of the severity of SLE in both human patients and animal models. Female SLE mice had elevated plasma dsDNA autoantibodies (3.0e5 ± 5.8e4 vs. 5.8e4 ± 1.8e4 activity units; P < 0.001) compared with controls (Fig. 3). Galantamine attenuated plasma concentrations of dsDNA autoantibodies in SLE mice (1.4e5 ± 3.0e4; P < 0.001) but had no significant effect in control mice (1.5e4 ± 4.1e3; P = 0.733).
Fig. 3.
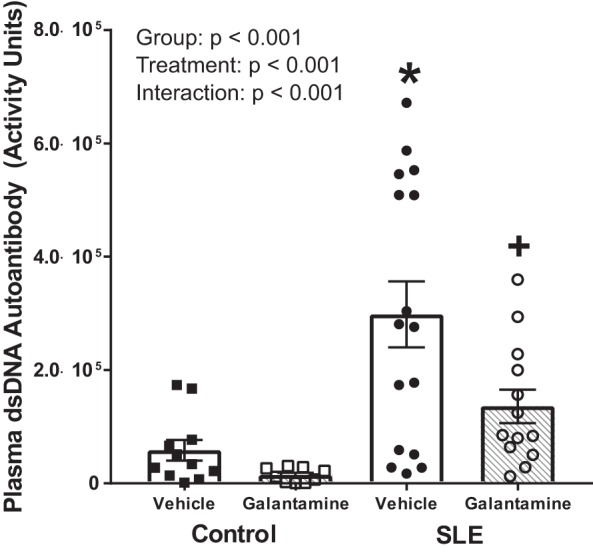
Galantamine decreases SLE disease severity. Anti-double-stranded DNA (dsDNA) autoantibodies are specific to systemic lupus erythematosus (SLE) and used to diagnose the condition, as well as gauge the severity of disease. Female SLE mice have elevated anti-dsDNA autoantibodies in their plasma compared with control mice. Galantamine-treated SLE mice had attenuated anti-dsDNA autoantibody concentrations in their plasma compared with vehicle-treated SLE mice. Values are presented as means ± SE. A two-way ANOVA was conducted to detect statistical differences. P values and accompanying symbols were determined using the results of Holm-Sidak post hoc analysis. (n = 11–13/group; *P vs. control/vehicle; +P vs. SLE/vehicle).
Galantamine reduces splenic cytokines in female SLE mice.
As proposed, the cholinergic anti-inflammatory pathway results in a reduction in cytokine release from the spleen on stimulation of the vagus nerve. To confirm that galantamine alters splenic cytokines through modulation of the cholinergic anti-inflammatory pathway, we measured inflammatory mediators in the spleen following after galantamine therapy. Many of the splenic cytokines measured [e.g., both the 26-kDa (transmembrane) and 51-kDa (trimeric) forms of TNF-α and BAFF] appeared to be reduced after galantamine therapy in SLE mice; however, because of the variability of disease in the animals, significance was not reached following a two-way ANOVA (Fig. 4, A, B, and D). However, splenic HMGB-1 was elevated in SLE mice compared with controls (6.8e6 ± 1.3e6 vs. 1.0e6 ± 2.1e5 intensity units; P < 0.001) and decreased in galantamine-treated SLE mice compared with vehicle-treated SLE mice (2.0e6 ± 2.6e5 vs. 6.8e6 ± 1.3e6 intensity units; P = 0.008) (Fig. 4C). Galantamine had no effect on splenic HMGB-1 in controls (8.7e5 ± 1.7e5 intensity units; P = 0.898). Finally, splenic IL-1β was not different among control and SLE groups treated with galantamine or vehicle (Fig. 4E).
Fig. 4.

Galantamine reduces splenic proinflammatory mediator in female SLE mice. A and B: Western blots of tumor necrosis factor (TNF)-α. C: high-mobility group box protein 1 (HMGB-1). D: B cell activating factor (BAFF). E: interleukin (IL)-1β in the spleen. Splenic TNF-α (at 26 kDa, the transmembrane form), TNF-α (at 51 kDa, the trimeric form), BAFF and IL-1β were not significantly altered. Splenic HMGB-1 was elevated in vehicle-treated systemic lupus erythematosus (SLE) mice compared with controls and galantamine markedly attenuated its expression. All values are presented as means ± SE. Statistical comparisons were determined using a two-way ANOVA. P values and accompanying symbols were determined using the results of Holm-Sidak post hoc analysis. (n = 5–6/group; *P vs. control/vehicle; +P vs. SLE/vehicle).
Galantamine prevents the rise in blood pressure and decreases renal injury typically observed in female SLE mice.
To determine whether chronically enhancing the cholinergic anti-inflammatory pathway with galantamine effects SLE hypertension, we measured blood pressure following galantamine therapy. While control mice were normotensive, SLE mice were hypertensive at 34–35 wk of age (115 ± 2 vs. 141 ± 6 mmHg; P < 0.001) (Fig. 5A). Galantamine-treated SLE mice had ~12 mmHg lower mean arterial pressure than vehicle-treated SLE mice at 34–35 wk (128 ± 3 vs. 141 ± 6 mmHg; P = 0.024). Galantamine did not significantly affect blood pressure in control mice (124 ± 4 mmHg; P = 0.085).
Fig. 5.

Galantamine decreases blood pressure and dampens the severity of renal injury in female SLE mice. A: mean arterial pressure (mmHg) measured in conscious mice via indwelling carotid catheters. Systemic lupus erythematosus (SLE) mice had higher blood pressure compared with control mice. Galantamine-treated SLE mice were protected from the rise in blood pressure that the vehicle-treated SLE mice experienced. Galantamine had no statistical effect on mean arterial pressure in controls. (n = 6–8/group) B: albumin excretion rate (AER) was based on results from albumin ELISA on urine and the amount of time spent in metabolic cages. Although there are obviously higher AERs in SLE mice, there was no significant difference among groups possibly due to variability of the disease. (n = 10–13/group) C: representative histological images and quantification of glomerulosclerosis in renal cortical glomeruli stained with Periodic Acid Schiff. Thirty glomeruli from each subject were blindly scored and assigned one of the following scores: 0: no glomerulosclerosis, hypercellularity, mesangial cell expansion, intact glomerular basement membrane; 1: 0 to 25% of the cross-sectional area is characterized by glomerulosclerosis, nodules, and hypercellularity; 2: 25 to 50% of the cross-sectional area is characterized by glomerulosclerosis, nodules, and hypercellularity; 3: 50–75% of the cross-sectional area is characterized by glomerulosclerosis, nodules, and hypercellularity; or 4: >75% of the cross-sectional area is characterized by glomerulosclerosis, nodules, and hypercellularity. SLE mice had increased glomerulosclerotic and hypercellular cross-sectional area compared with controls. Galantamine-treated SLE mice had decreased glomerulosclerosis compared with vehicle-treated SLE mice. (n = 3–7/group). All values are presented as means ± SE. Statistical comparisons were determined using a two-way ANOVA. P values and accompanying symbols were determined using the results of Holm-Sidak post hoc analysis. (*P vs. control/vehicle; +P vs. SLE/vehicle).
Although there were no significant differences in albumin excretion rate among treatment groups (Fig. 5B), SLE mice predictably had increased glomerulosclerotic injury compared with controls (P = 0.010). Galantamine-treated SLE mice had decreased renal glomerulosclerotic injury compared with vehicle-treated SLE mice (P = 0.019) (Fig. 5C). Galantamine did not have a statistical effect on glomerulosclerosis in control mice (P = 0.212).
Galantamine decreases renal proinflammatory cytokine expression in female SLE mice.
To determine whether a reduction in renal inflammation ultimately protected SLE animals from hypertension and renal injury, we measured inflammatory mediators in the renal cortex. When compared with control mice, SLE mice had higher renal cortical TNF-α, both in the 26-kDa (transmembrane) and 51-kDa (trimeric) forms (4.4e6 ± 7.5e5 vs. 6.7e5 ± 1.3e5 intensity units, P < 0.001 for 26 kDa; 4.4e6 ± 6.3e5 vs. 8.8e5 ± 2.4e5 intensity units for 51 kDa, P < 0.001), whereas galantamine-treated SLE mice had decreased expression of this cytokine compared with vehicle-treated SLE mice (2.4e6 ± 7.7e5 vs. 4.4e6 ± 7.5e5 intensity units, P = 0.028 for 26 kDa; 2.5e6 ± 6.5e5 vs. 4.4e6 ± 6.3e5 intensity units, P = 0.025 for 51 kDa) (Fig. 6, A and B). Galantamine had no effect on transmembrane or trimeric TNF-α in controls (1.2e6 ± 4.6e5, P = 0.476 for 26 kDa; 1.4e6 ± 5.8e5, P = 0.534 for 51 kDa).
Fig. 6.

Galantamine reduces renal cortical proinflammatory mediators in female SLE mice. A and B: Western blotting for tumor necrosis factor (TNF)-α. C: high-mobility group box protein 1 (HMGB-1). D: B cell activating factor (BAFF). E: IL-1β in the renal cortex. Renal cortical TNF-α (at 26 kDa, the transmembrane form) and TNF-α (at 51 kDa, the trimeric form) were elevated in vehicle-treated systemic lupus erythematosus (SLE) mice compared with controls, and galantamine markedly attenuated its expression. Renal cortical HMGB-1 and IL-1β were also increased in vehicle-treated SLE mice compared with controls; however, galantamine did not significantly alter expression. Statistical comparisons were determined using a two-way ANOVA. P values and accompanying symbols were determined using the results of Holm-Sidak post hoc analysis. (n = 5–7/group; *P vs. control/vehicle; +P vs. SLE/vehicle).
Renal cortical HMGB-1 and IL-1β were elevated in SLE mice compared with controls (3.8e6 ± 1.1e6 vs. 9.6e5 ± 2.2e5 intensity units, P = 0.010 for HMGB-1; 1.7e7 ± 1.8e6 vs. 4.9e6 ± 5.7e5 intensity units, P < 0.001 for IL-1β). Galantamine did not have a significant effect on HMGB-1 or IL-1β in SLE (1.7e6 ± 4.6e5 intensity units, P = 0.068 for HMGB-1; 1.3e7 ± 2.8e6 intensity units, P = 0.083 for IL-1β) or control mice (2.6e6 ± 7.9e5 intensity units, P = 0.118 for HMGB-1; Fig. 6, C and E). Finally, renal cortical BAFF was not different (based on the results of the two-way ANOVA) among SLE and control groups treated with galantamine or vehicle (Fig. 6D).
Galantamine does not affect aortic expression of TNF-α.
Vascular inflammation is also implicated in the development of hypertension, so we investigated whether galantamine had any effect on vascular inflammation. Neither the transmembrane nor the trimeric forms of TNF-α were different between groups based on the results of a two-way ANOVA (Fig. 7, A and B).
Fig. 7.

Galantamine does not alter aortic proinflammatory mediators in female SLE mice. Western blot analysis of aortic TNF-α at 26 kDa (A) and 51 kDa (B) showed no differences between groups. SLE, systemic lupus erythematosus. Statistical comparisons were determined using a two-way ANOVA. P values and accompanying symbols were determined using the results of Holm-Sidak post hoc analysis (n = 5–7/group).
DISCUSSION
We demonstrated that 2 wk of daily galantamine therapy 1) reduced SLE disease severity, 2) attenuated splenic inflammation and renal inflammation, and 3) reduced blood pressure and renal injury in SLE mice. Our findings suggest that the prevention of SLE hypertension may be the result of the effects of galantamine on systemic and renal inflammation, as well as SLE disease severity: 2 wk of galantamine treatment decreased both splenic (Fig. 4) and renal cortical (Fig. 6) proinflammatory cytokine expression and significantly reduced dsDNA titer in SLE mice (Fig. 3). The results of this study indicate that potentiation of the efferent vagus nerve and enhancement of the cholinergic anti-inflammatory pathway may protect from renal inflammation, hypertension, and renal injury in SLE. Our findings increase curiosity of the protective effects of central cholinergic stimulation in diseases of chronic inflammation like hypertension and SLE.
Galantamine is a reversible acetylcholinesterase inhibitor that crosses the blood-brain barrier and prolongs acetylcholine expression to promote activity of cholinergic neurons (20, 36), especially clinically in Alzheimer’s disease (55). Other clinical applications of galantamine include protection from glaucoma (2) and general neuroprotection (4, 9). Galantamine is known to potentiate efferent vagus nerve traffic because of its ability to prolong the life of acetylcholine at M1 muscarinic channels centrally (55). In rodents, galantamine was also demonstrated to act on central M2 muscarinic receptors to potentiate efferent vagus nerve activity (31). Others have suggested that galantamine may act peripherally by preventing cholinesterase activity in the vicinity of nicotinic receptors (51). We confirmed in this study that galantamine increases efferent vagus nerve activation (Fig. 1), and we propose that galantamine mediates its anti-inflammatory, therapeutic effects (also demonstrated in this study) by stimulating the cholinergic anti-inflammatory pathway and inhibiting downstream inflammation (Figs. 4 and 6). The cholinergic anti-inflammatory pathway is an intricate communicative mechanism that begins with efferent vagal transmission to the celiac ganglion, which prompts splenic nerve release of norepinephrine in the spleen, which activates specialized T cells to synthesize and release acetylcholine. This T cell-generated acetylcholine then binds to α7-subunit of the nicotinic acetylcholine receptor (α7-nAChR) on splenic immune cells, ultimately inhibiting proinflammatory cytokine release (3, 6, 47, 49, 62). The present study will help elucidate to the importance of this pathway in regulating inflammation in chronic inflammatory diseases.
The anti-inflammatory effects of galantamine being protective in murine SLE are not unusual, considering that immunomodulatory and immunosuppressive agents are the most common treatment for SLE patients as they delay the disease course and consequently lessen the severity of the many of the systemic complications of SLE (24, 45). The most common cause of death in SLE is cardiovascular disease (21), for which hypertension is a known risk factor. However, clinical trials for immunosuppressive drugs do not usually investigate the efficacy of these treatments against hypertension specifically, and other anti-inflammatory agents, especially exogenous glucocorticoids (33), worsen hypertension (35). Donepezil, another acetylcholinesterase inhibitor that crosses the blood-brain barrier like galantamine, also improved the inflammatory profile in a cecal ligation and puncture mouse model of sepsis (30). Although the anti-inflammatory actions of acetylcholinesterase inhibitors are well documented (25, 27, 31), not much is known about the effects of chronic galantamine therapy on blood pressure regulation. An early study demonstrated that intravenous galantamine infusion results in an unusual acute hypertensive response in rats (12); however, by contrast and as demonstrated in our present study, chronic galantamine administration has the opposite effect (Fig. 5A).
In our study, we discovered that galantamine therapy mediated an ~12 mmHg drop in blood pressure in the NZBWF1 disease model of SLE hypertension (Fig. 5A). Others have found that acetylcholinesterase inhibition prevented the development of hypertension and decreased plasma inflammatory markers in spontaneously hypertensive rats, although their treatments were prophylactically administered for a total duration of 16 wk (35). In the present study, the implications of such a generous blood pressure difference after only 2 wk of treatment are exciting, since galantamine therapy began when inflammatory end-organ damage in the kidneys and blood vessels had already manifested in SLE mice. The reduction in renal injury that we discovered may help explain this drop in blood pressure, since renal inflammation promotes hypertension. In addition to decreasing both renal (and splenic) inflammation (Figs. 4 and 6), galantamine attenuated other autoimmune inflammatory processes in SLE mice (Fig. 3). Future studies may examine the effects of other clinically used acetylcholinesterase inhibitors in experimental hypertension to determine whether this therapeutic decrease is specific to galantamine or a property of other agents from its class.
In spontaneously hypertensive rats, donepezil decreased blood pressure and systemic inflammation while pyridostigmine (an acetylcholinesterase inhibitor that does not cross the blood-brain barrier) did not, indicating that central acetylcholinesterase inhibition specifically contributes to anti-inflammatory processes (35). Since galantamine crosses the blood-brain barrier, we likewise found that central acetylcholinesterase inhibition mediates a powerful attenuation of autoimmune processes in SLE, as treatment with galantamine significantly reduced plasma dsDNA autoantibody concentration (Fig. 3), the most commonly accepted diagnostic and prognostic indicator of the severity of human SLE (19). The phenomenon that only two weeks of galantamine treatment significantly attenuated dsDNA autoantibody titer in SLE mice is exciting in terms of potential translational application to human SLE patients. Galantamine has additionally shown to be efficacious in nonobese diabetic (NOD) type 1 diabetic mice, another form of autoimmune disease resulting from antibodies produced against pancreatic islet cells (27). After 2 wk of daily injections, female NOD mice had a lower anti-insulin autoantibody titer, and both the incidence of hyperglycemia and diabetes was prevented in these mice (27). Galantamine also decreased plasma, liver, and muscle inflammation through its central mechanism of action, as indicated by dose-dependent decreases in brain acetylcholinesterase activity, in the n5-streptozocin rat model of diabetes type II (1).
Clinical studies have shown that SLE patients suffer from dysautonomia in the form of decreased efferent vagal tone, which manifests as decreased heart rate variability (59). Decreased heart rate variability is also noted in other chronic inflammatory states and may be indicative of a hypoactive cholinergic anti-inflammatory pathway (14). For instance, in rheumatoid arthritis, patients with heightened heart rate variability indicating higher vagus nerve tone respond better to anti-inflammatory treatment (34). Our rationale for using galantamine in SLE is to correct a hypoactive or dysfunctional cholinergic anti-inflammatory pathway in the pathogenesis of lupus hypertension due to lessened vagus nerve firing, as galantamine potentiates vagus nerve firing centrally (31). The effects of galantamine on potentiating, and therefore functionally increasing efferent vagal outflow, (31) call into mind treatment modalities such as electrical vagus nerve stimulation, a novel therapeutic modality for conditions as varied as epilepsy, to rheumatoid arthritis (70), although pharmalogical agents like galantamine and other acetylcholine-bolstering agents portend fewer cardiac side effects and are less invasive (29). Treatment could also be ceased more readily, if necessarily. However, reversible acetylcholinesterase inhibition is also known to cause systemic side effects, especially in the gastrointestinal system (13). No gastrointestinal side effects were noted in any of the animal groups treated with galantamine, and ultimately this manifested as no difference in body weight throughout the study (Fig. 2). In sum, our discovery that central vagus nerve potentiation using the acetylcholinesterase inhibitor galantamine protected from renal inflammation, hypertension, and renal injury in murine SLE shows that increasing cholinergic anti-inflammatory pathway activity may be protective in this autoimmune disease. This observation can also be taken to mean that the cholinergic anti-inflammatory pathway is hypoactive in SLE. Future studies will investigate dysfunction at other areas of the pathway in SLE, as well as possible deficiencies in other neuroimmune pathways mediated by the vagus nerve.
The results from this study are also promising in the context of therapeutics for essential hypertension. Chronic inflammation may be implicated in other etiologies in hypertension, as it is becoming increasingly known that systemic, low-grade inflammation is correlated with high blood pressure (66). Additionally, the loss of renal function, stemming from etiologies as diverse as SLE to diabetes, also accompanies and exacerbates longstanding hypertension (58). Renal proinflammatory cytokine expression has been mechanistically linked to renal injury in the pathogenesis of hypertension. In fact, TNF-α in the kidney contributes to angiotensin-II-induced experimental hypertension (68) and inhibition of TNF-α with etanercept decreases renal inflammation and prevents hypertension in autoimmune- (64) and angiotensin II-induced hypertension (26), as well as in fructose- (63), high-salt- (17), and deoxycorticosterone acetate-fed (16) rats. Other anti-inflammatory agents yield therapeutic effects in hypertension, especially the immunosuppressive agent mycophenolate mofetil, an agent that inhibits T and B cell growth, which prevents salt-sensitive hypertension in angiotensin II-treated Sprague-Dawley rats while decreasing infiltrating renal immune cells (53). A similar phenomenon was demonstrated in a mouse model of SLE, where mycophenolate mofetil reduces renal immune cells and renal injury while also preventing hypertension (60). Mycophenolate mofetil also has been shown to lower blood pressure in hypertensive rheumatoid and psoriatic arthritis patients after three months of treatment (28). Clearly, anti-inflammatory agents mediate protective effects in both experimental animal models of hypertension and in hypertensive patients with autoimmune disease. Likewise, we discovered that galantamine treatment reduced glomerular injury in SLE mice (Fig. 5C), although it did not significantly impact urinary albumin excretion following drug administration (Fig. 5B). This finding may have been due to the time course of renal injury in SLE mice; perhaps the galantamine administration was able to decrease the severity of structural and glomerular injury in tandem with decreasing blood pressure, but glomerular basement injury continued to facilitate the loss of albumin in the urine. Additionally, the female NZBWF1 model of SLE presents variably in its phenotype, and it is not unusual for several mice from a cohort to undergo fulminant renal failure or other flare-ups of the disease, similarly to human lupus patients (44); this phenomenon may help explain the lack of significance we obtained in certain areas. Our study addresses both renal inflammation and injury in the pathogenesis and maintenance of hypertension, and supports the concept that targeting a higher, neural target that regulates inflammation (e.g., the vagus nerve) may be therapeutic, even in the context of established disease. Galantamine’s central mechanism of action may help to elucidate novel targets in the treatment of both hypertension and SLE.
Perspectives and Significance
Our study demonstrates that galantamine, a centrally acting acetylcholinesterase inhibitor, may be protective in the chronic autoimmune disease systemic lupus erythematosus. We and others have demonstrated that galantamine directly enhances efferent vagus nerve activity, which can subsequently activate the cholinergic anti-inflammatory pathway and inhibit inflammation downstream. When administered chronically, we have shown that galantamine attenuates splenic and renal inflammation, decreases pathogenic autoantibodies, and ultimately decreases blood pressure in a murine model of SLE. The findings from this study demonstrate that chronic interventions that enhance vagally-mediated neuroimmune pathways may lead to protective functional results. This, in turn, may identify additional novel targets for the treatment of chronic inflammatory diseases like hypertension, SLE, and other autoimmune diseases.
GRANTS
This study was partially funded by the American Heart Association (Grant 14SDG18320033 to K. W. Mathis and Grant16PRE29910012 to G. S. Pham), the National Institutes of Health (Grant 1K01HL139859 to K. W. Mathis) and the Lupus Research Alliance (Grant 550778 to K. W. Mathis).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.W.M. conceived and designed study; G.S.P., L.A.W., and K.W.M. performed experiments; G.S.P., L.A.W., and K.W.M. analyzed data; G.S.P., L.A.W., and K.W.M. interpreted results of experiments; G.S.P., L.A.W., and K.W.M. prepared figures; G.S.P. drafted manuscript; G.S.P., L.A.W., and K.W.M. edited and revised manuscript; G.S.P., L.A.W., and K.W.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Amber Fairley and Daniel Fancher for technical assistance and Sarika Chaudhari, for editorial insights in the preparation of this manuscript.
REFERENCES
- 1.Ali MA, El-Abhar HS, Kamel MA, Attia AS. Antidiabetic effect of galantamine: novel effect for a known centrally acting drug. PLoS One 10: e0134648, 2015. doi: 10.1371/journal.pone.0134648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almasieh M, Zhou Y, Kelly ME, Casanova C, Di Polo A. Structural and functional neuroprotection in glaucoma: role of galantamine-mediated activation of muscarinic acetylcholine receptors. Cell Death Dis 1: e27, 2010. doi: 10.1038/cddis.2009.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andersson U, Tracey KJ. Neural reflexes in inflammation and immunity. J Exp Med 209: 1057–1068, 2012. doi: 10.1084/jem.20120571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes CA, Meltzer J, Houston F, Orr G, McGann K, Wenk GL. Chronic treatment of old rats with donepezil or galantamine: effects on memory, hippocampal plasticity and nicotinic receptors. Neuroscience 99: 17–23, 2000. doi: 10.1016/S0306-4522(00)00180-9. [DOI] [PubMed] [Google Scholar]
- 5.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 19: 149–154, 2005. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 6.Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey KJ. Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 195: 781–788, 2002. doi: 10.1084/jem.20011714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int 63: 1791–1800, 2003. doi: 10.1046/j.1523-1755.2003.00929.x. [DOI] [PubMed] [Google Scholar]
- 8.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405: 458–462, 2000. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 9.Capsoni S, Giannotta S, Cattaneo A. Nerve growth factor and galantamine ameliorate early signs of neurodegeneration in anti-nerve growth factor mice. Proc Natl Acad Sci USA 99: 12432–12437, 2002. doi: 10.1073/pnas.192442999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension 38: 399–403, 2001. doi: 10.1161/01.HYP.38.3.399. [DOI] [PubMed] [Google Scholar]
- 11.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, Tipping P, Vinh A, Samuel CS, Peter K, Guzik TJ, Kyaw TS, Toh B-H, Bobik A, Drummond GR. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension 66: 1023–1033, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05779. [DOI] [PubMed] [Google Scholar]
- 12.Chrusciel M, Varagić V. The effect of galantamine on the blood pressure of the rat. Br J Pharmacol Chemother 26: 295–301, 1966. doi: 10.1111/j.1476-5381.1966.tb01908.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colović MB, Krstić DZ, Lazarević-Pašti TD, Bondžić AM, Vasić VM. Acetylcholinesterase inhibitors: pharmacology and toxicology. Curr Neuropharmacol 11: 315–335, 2013. doi: 10.2174/1570159X11311030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper TM, McKinley PS, Seeman TE, Choo T-H, Lee S, Sloan RP. Heart rate variability predicts levels of inflammatory markers: Evidence for the vagal anti-inflammatory pathway. Brain Behav Immun 49: 94–100, 2015. doi: 10.1016/j.bbi.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist 10: 40–52, 2004. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- 16.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-alpha inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol 294: R76–R83, 2008. doi: 10.1152/ajpregu.00466.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension 47: 557–562, 2006. doi: 10.1161/01.HYP.0000198545.01860.90. [DOI] [PubMed] [Google Scholar]
- 18.Fairley AS, Mathis KW. Cholinergic agonists reduce blood pressure in a mouse model of systemic lupus erythematosus. Physiol Rep 5: e13213, 2017. doi: 10.14814/phy2.13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Förger F, Matthias T, Oppermann M, Becker H, Helmke K. Clinical significance of anti-dsDNA antibody isotypes: IgG/IgM ratio of anti-dsDNA antibodies as a prognostic marker for lupus nephritis. Lupus 13: 36–44, 2004. doi: 10.1191/0961203304lu485oa. [DOI] [PubMed] [Google Scholar]
- 20.Geerts H, Guillaumat P-O, Grantham C, Bode W, Anciaux K, Sachak S. Brain levels and acetylcholinesterase inhibition with galantamine and donepezil in rats, mice, and rabbits. Brain Res 1033: 186–193, 2005. doi: 10.1016/j.brainres.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 21.Giannelou M, Mavragani CP. Cardiovascular disease in systemic lupus erythematosus: a comprehensive update. J Autoimmun 82: 1–12, 2017. doi: 10.1016/j.jaut.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Gilbert EL, Mathis KW, Ryan MJ. 17β-Estradiol protects against the progression of hypertension during adulthood in a mouse model of systemic lupus erythematosus. Hypertension 63: 616–623, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilda JE, Gomes AV. Stain-free total protein staining is a superior loading control to β-actin for Western blots. Anal Biochem 440: 186–188, 2013. doi: 10.1016/j.ab.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ginzler EM, Dooley MA, Aranow C, Kim MY, Buyon J, Merrill JT, Petri M, Gilkeson GS, Wallace DJ, Weisman MH, Appel GB. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med 353: 2219–2228, 2005. doi: 10.1056/NEJMoa043731. [DOI] [PubMed] [Google Scholar]
- 25.Gowayed MA, Refaat R, Ahmed WM, El-Abhar HS. Effect of galantamine on adjuvant-induced arthritis in rats. Eur J Pharmacol 764: 547–553, 2015. doi: 10.1016/j.ejphar.2015.07.038. [DOI] [PubMed] [Google Scholar]
- 26.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanes WM, Olofsson PS, Kwan K, Hudson LK, Chavan SS, Pavlov VA, Tracey KJ. Galantamine attenuates type 1 diabetes and inhibits anti-insulin antibodies in nonobese diabetic mice. Mol Med 21: 702–708, 2015. doi: 10.2119/molmed.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17, Suppl 3: S218–S225, 2006. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 29.Howes LG. Cardiovascular effects of drugs used to treat Alzheimer’s disease. Drug Saf 37: 391–395, 2014. doi: 10.1007/s40264-014-0161-z. [DOI] [PubMed] [Google Scholar]
- 30.Jeremias IC, Victorino VJ, Barbeiro HV, Kubo SA, Prado CM, Lima TM, Soriano FG. The role of acetylcholine in the inflammatory response in animals surviving sepsis induced by cecal ligation and puncture. Mol Neurobiol 53: 6635–6643, 2016. doi: 10.1007/s12035-015-9538-y. [DOI] [PubMed] [Google Scholar]
- 31.Ji H, Rabbi MF, Labis B, Pavlov VA, Tracey KJ, Ghia JE. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol 7: 335–347, 2014. doi: 10.1038/mi.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kane D, Lockhart JC, Balint PV, Mann C, Ferrell WR, McInnes IB. Protective effect of sensory denervation in inflammatory arthritis (evidence of regulatory neuroimmune pathways in the arthritic joint). Ann Rheum Dis 64: 325–327, 2005. doi: 10.1136/ard.2004.022277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasturi S, Sammaritano LR. Corticosteroids in lupus. Rheum Dis Clin North Am 42: 47–62, 2016. doi: 10.1016/j.rdc.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Koopman FA, van Maanen MA, Vervoordeldonk MJ, Tak PP. Balancing the autonomic nervous system to reduce inflammation in rheumatoid arthritis. J Intern Med 282: 64–75, 2017. doi: 10.1111/joim.12626. [DOI] [PubMed] [Google Scholar]
- 35.Lataro RM, Silva CA, Tefé-Silva C, Prado CM, Salgado HC. Acetylcholinesterase inhibition attenuates the development of hypertension and inflammation in spontaneously hypertensive rats. Am J Hypertens 28: 1201–1208, 2015. doi: 10.1093/ajh/hpv017. [DOI] [PubMed] [Google Scholar]
- 36.Lilienfeld S. Galantamine–a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev 8: 159–176, 2002. doi: 10.1111/j.1527-3458.2002.tb00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margretardottir OB, Thorleifsson SJ, Gudmundsson G, Olafsson I, Benediktsdottir B, Janson C, Buist AS, Gislason T. Hypertension, systemic inflammation and body weight in relation to lung function impairment-an epidemiological study. COPD 6: 250–255, 2009. doi: 10.1080/15412550903049157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathis KW. An impaired neuroimmune pathway promotes the development of hypertension in systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol 309: R1074–R1077, 2015. doi: 10.1152/ajpregu.00143.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathis KW, Broome HJ, Ryan MJ. Autoimmunity: an underlying factor in the pathogenesis of hypertension. Curr Hypertens Rep 16: 424, 2014. doi: 10.1007/s11906-014-0424-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathis KW, Venegas-Pont M, Flynn ER, Williams JM, Maric-Bilkan C, Dwyer TM, Ryan MJ. Hypertension in an experimental model of systemic lupus erythematosus occurs independently of the renal nerves. Am J Physiol Regul Integr Comp Physiol 305: R711–R719, 2013. doi: 10.1152/ajpregu.00602.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathis KW, Venegas-Pont M, Masterson CW, Wasson KL, Ryan MJ. Blood pressure in a hypertensive mouse model of SLE is not salt-sensitive. Am J Physiol Regul Integr Comp Physiol 301: R1281–R1285, 2011. doi: 10.1152/ajpregu.00386.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathis KW, Venegas-Pont M, Masterson CW, Stewart NJ, Wasson KL, Ryan MJ. Oxidative stress promotes hypertension and albuminuria during the autoimmune disease systemic lupus erythematosus. Hypertension 59: 673–679, 2012. doi: 10.1161/HYPERTENSIONAHA.111.190009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mathis KW, Wallace K, Flynn ER, Maric-Bilkan C, LaMarca B, Ryan MJ. Preventing autoimmunity protects against the development of hypertension and renal injury. Hypertension 64: 792–800, 2014. doi: 10.1161/HYPERTENSIONAHA.114.04006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakagawa P, Masjoan-Juncos JX, Basha H, Janic B, Worou ME, Liao TD, Romero CA, Peterson EL, Carretero OA. Effects of N-acetyl-seryl-asparyl-lysyl-proline on blood pressure, renal damage, and mortality in systemic lupus erythematosus. Physiol Rep 5: e13084, 2017. doi: 10.14814/phy2.13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK-M, Thomas M, Kim H-Y, León MG, Tanasescu C, Nasonov E, Lan J-L, Pineda L, Zhong ZJ, Freimuth W, Petri MA; BLISS-52 Study Group . Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 377: 721–731, 2011. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 46.Ohki T, Kamimura D, Arima Y, Murakami M. Gateway reflexes: A new paradigm of neuroimmune interactions. Clin Exp Neuroimmunol 8: 23–32, 2017. doi: 10.1111/cen3.12378. [DOI] [Google Scholar]
- 47.Olofsson PS, Rosas-Ballina M, Levine YA, Tracey KJ. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol Rev 248: 188–204, 2012. doi: 10.1111/j.1600-065X.2012.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pavlov VA, Parrish WR, Rosas-Ballina M, Ochani M, Puerta M, Ochani K, Chavan S, Al-Abed Y, Tracey KJ. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav Immun 23: 41–45, 2009. doi: 10.1016/j.bbi.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pavlov VA, Tracey KJ. Neural circuitry and immunity. Immunol Res 63: 38–57, 2015. doi: 10.1007/s12026-015-8718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pimenta E, Calhoun DA. Resistant hypertension: incidence, prevalence, and prognosis. Circulation 125: 1594–1596, 2012. doi: 10.1161/CIRCULATIONAHA.112.097345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pohanka M. Inhibitors of acetylcholinesterase and butyrylcholinesterase meet immunity. Int J Mol Sci 15: 9809–9825, 2014. doi: 10.3390/ijms15069809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramseyer VD, Garvin JL. Tumor necrosis factor-α: regulation of renal function and blood pressure. Am J Physiol Renal Physiol 304: F1231–F1242, 2013. doi: 10.1152/ajprenal.00557.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, Parra G, Herrera-Acosta J, Gómez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001. doi: 10.1046/j.1523-1755.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- 54.Sabio JM, Vargas-Hitos JA, Navarrete-Navarrete N, Mediavilla JD, Jiménez-Jáimez J, Díaz-Chamorro A, Jiménez-Alonso J; Grupo Lupus Virgen de las Nieves . Prevalence of and factors associated with hypertension in young and old women with systemic lupus erythematosus. J Rheumatol 38: 1026–1032, 2011. doi: 10.3899/jrheum.101132. [DOI] [PubMed] [Google Scholar]
- 55.Samochocki M, Höffle A, Fehrenbacher A, Jostock R, Ludwig J, Christner C, Radina M, Zerlin M, Ullmer C, Pereira EFR, Lübbert H, Albuquerque EX, Maelicke A. Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J Pharmacol Exp Ther 305: 1024–1036, 2003. doi: 10.1124/jpet.102.045773. [DOI] [PubMed] [Google Scholar]
- 56.Shahid M, Francis J, Majid DS. Tumor necrosis factor-alpha induces renal vasoconstriction as well as natriuresis in mice. Am J Physiol Renal Physiol 295: F1836–F1844, 2008. doi: 10.1152/ajprenal.90297.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith RA, Baglioni C. The active form of tumor necrosis factor is a trimer. J Biol Chem 262: 6951–6954, 1987. [PubMed] [Google Scholar]
- 58.Stehouwer CD, Gall M-A, Twisk JW, Knudsen E, Emeis JJ, Parving H-H. Increased urinary albumin excretion, endothelial dysfunction, and chronic low-grade inflammation in type 2 diabetes: progressive, interrelated, and independently associated with risk of death. Diabetes 51: 1157–1165, 2002. doi: 10.2337/diabetes.51.4.1157. [DOI] [PubMed] [Google Scholar]
- 59.Stein KS, McFarlane IC, Goldberg N, Ginzler EM. Heart rate variability in patients with systemic lupus erythematosus. Lupus 5: 44–48, 1996. doi: 10.1177/096120339600500109. [DOI] [PubMed] [Google Scholar]
- 60.Taylor EB, Ryan MJ. Immunosuppression with mycophenolate mofetil attenuates hypertension in an experimental model of autoimmune disease. J Am Heart Assoc 6: 1–12, 2017. doi: 10.1161/JAHA.116.005394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thanou A, Stavrakis S, Dyer JW, Munroe ME, James JA, Merrill JT. Impact of heart rate variability, a marker for cardiac health, on lupus disease activity. Arthritis Res Ther 18: 197, 2016. doi: 10.1186/s13075-016-1087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tracey KJ. The inflammatory reflex. Nature 420: 853–859, 2002. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 63.Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem 330: 219–228, 2009. doi: 10.1007/s11010-009-0136-z. [DOI] [PubMed] [Google Scholar]
- 64.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, Ryan MJ. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 56: 643–649, 2010. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venegas-Pont M, Mathis KW, Iliescu R, Ray WH, Glover PH, Ryan MJ. Blood pressure and renal hemodynamic responses to acute angiotensin II infusion are enhanced in a female mouse model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol 301: R1286–R1292, 2011. doi: 10.1152/ajpregu.00079.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Virdis A, Dell’Agnello U, Taddei S. Impact of inflammation on vascular disease in hypertension. Maturitas 78: 179–183, 2014. doi: 10.1016/j.maturitas.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 67.Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD, Wright JT Jr. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71: e13–e115, 2018. doi: 10.1161/HYP.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, Sparks MA, Jin H, Wu M, Lin EE, Crowley SD. Tumor necrosis factor-α produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension 64: 1275–1281, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zitnik RJ. Treatment of chronic inflammatory diseases with implantable medical devices. Ann Rheum Dis 70, Suppl 1: i67–i70, 2011. doi: 10.1136/ard.2010.138677. [DOI] [PubMed] [Google Scholar]