

Abstract
Human RPE65 mutations cause a spectrum of retinal dystrophies that result in blindness. While RPE65 mutations have been almost invariably recessively inherited, a c.1430A>G (p.(D477G)) mutation has been reported to cause autosomal dominant retinitis pigmentosa (adRP). To study the pathogenesis of this human mutation, we have replicated the mutation in a knock-in (KI) mouse model using CRISPR/Cas9-mediated genome editing. Significantly, in contrast to human patients, heterozygous KI mice do not exhibit any phenotypes in visual function tests. When raised in regular vivarium conditions, homozygous KI mice display relatively undisturbed visual functions with minimal retinal structural changes. However, KI/KI mouse retinae are more sensitive to light exposure and exhibit signs of degenerative features when subjected to light stress. We find that instead of merely producing a missense mutant protein, the A>G nucleotide substitution greatly affects appropriate splicing of Rpe65 mRNA by generating an ectopic splice site in comparable context to the canonical one, thereby disrupting RPE65 protein expression. Similar splicing defects were also confirmed for the human RPE65 c.1430G mutant in an in vitro Exontrap assay. Our data demonstrate that a splicing defect is associated with c.1430G pathogenesis, and therefore provide insights in the therapeutic strategy for human patients.
Keywords: Retina, retinal pigment epithelium, autosomal dominant, autosomal recessive Retinitis Pigmentosa, RPE65, RNA splicing
Graphical Abstract
Introduction
Retinitis pigmentosa (RP; MIM# 268000), a group of heterogeneously inherited conditions involving rod/cone dystrophies with subsequent loss of vision (Q. Wang et al., 2001), comprises the most common inherited retinal degeneration, with an estimated total of 1.5 million people affected worldwide (Hartong, Berson, & Dryja, 2006). 50–60% of RP is autosomal-recessive, while 30–40% is autosomal-dominant. 5% of RP is caused by genetic defects in retinal pigment epithelium (RPE), about half of which are associated with RPE65 gene (MIM# 180069; chr1: 68,428,821–68,450,321) mutations (Morimura et al., 1998). RPE65, abundantly expressed in the RPE, is the key retinol isomerase in the visual cycle that converts all-trans retinyl ester to 11-cis retinal chromophore (Jin, Li, Moghrabi, Sun, & Travis, 2005; Moiseyev, Chen, Takahashi, Wu, & Ma, 2005; Redmond, 2009; Redmond et al., 2005), providing a continuous supply of chromophore for the process of chromophore photon-capture and photoisomerization that initiate vision. In animal models, Rpe65 null mutations abolish chromophore production, leading to loss of visual function, and retinal degeneration (Redmond et al., 1998; Samardzija et al., 2008; Veske, Nilsson, Narfstrom, & Gal, 1999; Wright et al., 2014). In humans, RPE65 deficiency results in RP20 or early-onset severe retinal dystrophy (RP20/EOSRD; MIM# 613794) or Leber congenital amaurosis 2 (LCA2, MIM# 204100; Gu et al., 1997; Marlhens et al., 1998). Over 150 pathogenic RPE65 mutations have been identified, accounting for ~2% of patients with retinal dystrophy (Astuti et al., 2016; Thompson et al., 2000; see also http://www.retina-international.org/files/sci-news/rpe65mut.htm). Most of these introduce missense or nonsense mutations that affect isomerase function.
Almost all pathogenic RPE65 mutations are recessively inherited. However, a perplexing dominant-acting mutation in RPE65 in families of Irish heritage has been reported (Bowne et al., 2011; Hull, Mukherjee, Holder, Moore, & Webster, 2016). This is a single-nucleotide substitution (NM_000329.2:c.1430A>G) in exon 13 of RPE65. Most carriers of this mutation were shown to develop a slowly progressive visual disturbance along with an extensive choroid/retinal atrophy that mimicked aspects of choroideremia (Bowne et al., 2011; Hull et al., 2016). Unlike recessive RPE65 mutations, this mutation showed incomplete genetic penetrance such that disease severity and age of disease onset were quite variable, while some carriers are unaffected (Hull et al., 2016). Translation of the c.1430G mutation gives rise to a missense mutant protein, RPE65/D477G (NP_000320.1:p.(D477G)). Computational analysis indicates a moderate increase in aggregation propensity for RPE65/D477G, which may interfere with wild type (WT) RPE65 function and cause cellular toxicity (Choi et al., 2018). However, when forced-expressed in cultured cells, RPE65/D477G behaves as a functional RPE65 (Bowne et al., 2011), with regular subcellular localization, and adequate isomerization activity (Choi et al., 2018). Moreover, heterozygous RPE65/D477G knock-in (KI) mice produce sufficient chromophore and have relatively normal retinal structure and function (Choi et al., 2018; Shin, Moiseyev, Chakraborty, & Ma, 2017). Taken together, these data do not support the notion that RPE65/D477G acts as a gain-of-function missense mutant.
While knock-in (KI) mice have long been used to study human mutations, their generation heretofore relied on complicated and time-consuming classical homologous recombination, which introduces short foreign DNA sequences (i.e., loxP or FRT) into the mouse genome. If these impinge on recognition sites for splicing or other elements, they can be deleterious to target gene expression (Meier et al., 2010; Turlo, Gallaher, Vora, Laski, & Iruela-Arispe, 2010). Moreover, the mixed genetic background of mouse stem cells can further complicate downstream analysis (Choi et al., 2018; Shin et al., 2017). CRISPR/Cas9 genome editing enables production of transgenic animals in a considerably shorter time frame (Hsu, Lander, & Zhang, 2014; Zhang, Wen, & Guo, 2014), and importantly, without the necessity of foreign sequence insertion (Hsu et al., 2014), or complications of varying genetic background. Here, we describe a knock-in (KI) transgenic mouse model replicating the human c.1430A>G mutation created via CRISPR/Cas9 editing. In contrast to the prior studies (Choi et al., 2018; Shin et al., 2017), we found that, instead of merely being a missense mutation, c.1430A>G also disrupts regular splicing of the Rpe65 gene. In contrast to human c.1430A>G patients, heterozygous KI mice (WT/KI) do not exhibit a distinct phenotype. Visual functions of homozygous KI (KI/KI) mice are undisturbed with minimal changes in structure when raised in regular vivarium conditions. However, homozygous, but not heterozygous, KI mice are sensitive to light stress and display degenerative effects when exposed to intense light. Our data suggest that c.1430A>G does not result in a gain-of-function missense mutant protein, and that defective splicing associated with the c.1430G mutation likely contributes to the pathogenesis in human patients.
Materials and Methods
RPE65 Mutagenesis and transient transfection
To express in vitro the missense mutations (i.e., RPE65/D477G, RPE65/D477A, RPE65/D477E, RPE65/D477N), we followed our previously published methods (Redmond, Poliakov, Kuo, Chander, & Gentleman, 2010). First, site-directed mutagenesis of the dog RPE65 ORF cloned into the bicistronic expression vector pVitro2 (InvivoGen, San Diego, CA) along with CRALBP was done by using the QuikChange XL site-directed mutagenesis kit (Stratagene) and confirmed by sequencing. pVitro2 expression plasmids containing either wild-type RPE65 or RPE65 missense mutations were prepared by using QIAprep Maxi kits (Qiagen, Valencia, CA). Next, 30 μg each of pVitro2 plasmid (WT and/or mutant) and pVitro3 (expressing LRAT and RDH5; InvivoGen, San Diego, CA) were transfected into human HEK293-F FreeStyle suspension cells (Thermo Fisher Scientific, Rockford, IL, USA) using 40 μl of 293fectin transfection reagent (Thermo Fisher Scientific) as described in (Redmond et al., 2010). All-trans retinol (atROL) was added 24 h post-transfection, at a final concentration of 2.5 μM, close to the calculated physiological concentration in retina of 3.8 μM after a 2% rhodopsin bleach (Flannery, O’Day, Pfeffer, Horwitz, & Bok, 1990). Finally, cells were harvested at 6 h after substrate addition for downstream retinoid analysis. We also adopted similar methodology to test the in vitro isomerase activity of a truncated RPE65 protein (RPE65/E13AS) derived from an alternatively spliced RPE65 cDNA lacking the first 91 nucleotide of Exon 13. Specifically, the alternative spliced RPE65 cDNA construct was cloned using a Gibson Assembly Kit (New England Biolabs, Ipswich, MA) following the manufacturer’s instructions. For the Exontrap assay, site-directed mutagenesis of the human RPE65 genomic sequence spanning Intron 10 to Exon 14 was done using the QuikChange XL site-directed mutagenesis kit (Stratagene) and confirmed by sequencing. Exontrap vector (MoBiTec, Germany) with either wild-type RPE65, RPE65/c.1430A>G, RPE65/c.1430A>C, or RPE65/c.1430A>T was transfected into human 293T cells. Total RNAs were isolated 36 hours post-transfection for expression analysis.
Splice-site analysis
The strength of splicing signals was predicted using the Splice Site Prediction tool located at the Berkeley Drosophila Genome Project (BDGP: http://www.fruitfly.org/seq_tools/splice.html). This tool is based on a neural network model for eukaryotic gene structure (Reese, Eeckman, Kulp, & Haussler, 1997). We employed parameters specific for human genes. The output of the analysis is a score between 0 and 1 for potential splicing sites. A BDGP score of 0.4 is taken as the default minimum score. We also used a recent update of HUMAN SPLICE FINDER (v.3; http://www.umd.be/HSF3/index.html) that combines 12 different algorithms to identify and predict the effects of mutations on splicing motifs, including acceptor and donor splice sites, branch points and auxiliary sequences known to either enhance or repress splicing: Exonic Splicing Enhancers (ESE) and Exonic Splicing Silencers (ESS) (Desmet et al., 2009). The output of the analysis is an HSF score between 0 and 100 for potential splicing sites. An HSF score of 80 is taken as the default minimum score.
Generation of Rpe65 c.1430A>G Knockin Mice
All procedures concerning animals were in accordance with institutional regulations and with the US National Research Council’s ”Guide for the Care and Use of Laboratory Animals”, the US Public Health Service’s “Policy on Humane Care and Use of Laboratory Animals” and “Guide for the Care and Use of Laboratory Animals”, and with the statement of the Association for Research in Vision and Ophthalmology for the use of animals in research. Studies were carried out under an institutional Animal Study Protocol approved by the National Eye Institute, NIH, Animal Care and Use Committee. CRISPR-mediated genome editing was conducted in C57BL6/J mouse zygotes based on a protocol published by Wang et al (2013). Guide RNAs for Streptococcus pyogenes Cas9 (SpCas9) were designed using the online CRISPR tool (http://www.crisprscan.org) and synthesized by in vitro transcription (IVT) as described (Varshney et al., 2015). Briefly, A forward primer, containing the crRNA sequence (Supp. Table S1) following the T7 promoter, and a reverse primer, consisting of the last 20 nt of the tracrRNA sequence, were used to produce the templates for IVT by Polymerase chain reaction (PCR), which is then transcribed into crRNA-tracrRNA (single guide RNA or gRNA;Briner et al., 2014) using the MEGAshortscript T7 Transcription Kit (Thermo Fisher Scientific, Rockford, IL, USA). The donor DNA for homologous recombination repair of the CRISPR-induced double strand break is a single-strand oligodeoxynucleotide (ssODN; Integrated DNA Technologies, Coralville, IA) of 169 nt in length carrying the nucleotide substitutions (Supp. Table S1), which was designed to asymmetrically span the Cas9 cutting site (Richardson, Ray, DeWitt, Curie, & Corn, 2016). 100 ng/μl Cas9 mRNA, 50 ng/μl gRNA, and 100 ng/ml ssODNs were microinjected into zygotes from 3 weeks old superovulated C57/BL6J females (Jackson Laboratory). These were cultured in modified Krebs–Ringer bicarbonate or KSOM medium overnight, and the next day two-cell embryos were transferred into pseudopregnant recipient females. Pups carrying the knockin allele were identified by PCR amplification of the genomic sequence surrounding the mutation sites followed by Sanger sequencing of the amplicons. Primers used for the PCR are listed in Supp. Table S1. The identified F0 founders (genetic mosaics) were crossed with WT C57BL/6J for germline transmission of the KI allele. To mitigate the possibility of off-target effects caused by gRNA, F1 founders carrying the knockin allele were crossed to C57BL/6J WT mice for an additional three generations before heterozygous and homozygous knockin progeny were used in the studies. For a faster genotyping protocol, we designed a custom probe (Integrated DNA Technologies) for the knockin allele (FAM-labeled), in combination with a HEX-labeled reference probe outside the mutations. Genomic DNA isolated from mouse tails were analyzed using a QX200 Droplet Digital PCR (ddPCR) System (Bio‐Rad, Hercules, CA, USA). Sequences for primers and probes used in genotyping are listed in Supp. Table S1.
RNA analysis
Total RNA was isolated from the adult mouse eyecup (RPE-Choroid-Sclera) using miRNeasy Plus Mini Kit (Qiagen, Valencia, CA). 500 ng RNA was reverse-transcribed with cDNA synthesis kit (Thermo Fisher Scientific, Rockford, IL, USA). The quantification of transcripts was done using ddPCR. Approximately ten to fourteen thousand droplets, containing the template with the primers and probe, were generated. The measured fluorescence after PCR amplification was used to calculate the concentration of a transcript using Poisson statistics by the software QuantaSoft (Bio-Rad). Expression levels of a transcript were determined relative to Hprt expression. The sequences of the primers and probes are listed in Supp. Table S1.
For 3’RACE analysis, total RNAs from KI/KI mice eyecups (RPE-Choroid-Sclera) were reverse transcribed using a cDNA cloning primer (Integrated DNA Technology). The cDNA was amplified using a forward primer at Exon 6 and 3’ RACE PCR reverse primer. Amplicons were gel purified, cloned into TOPO TA cloning vector (ThermoFisher Scientific), and sequenced. Sequences of the primers used are listed in Supp. Table S1.
For RNA-seq analysis, RNA samples of eyecups (RPE-Choroid-Sclera) were independently processed from three WT, three WT/KI, and three KI/KI mice at 3-month of age. Total RNA (100 ng) was used with the TruSeq mRNA-seq Sample Preparation Kit (Illumina, San Diego, CA) to construct cDNA libraries. After quality confirmation of the libraries (DNA-1000 Kit, Agilent), 10 pM of each cDNA library was independently loaded into one flow cell lane, and single-read cluster generation proceeded using the TruSeq SR Cluster Generation Kit v5 (Illumina). Sequencing-by-synthesis (SBS) of 70-nucleotide length was performed on a Genome Analyzer IIx running SCS2.8 software using SBS v4 reagents (Illumina). Base calling and chastity filtering were performed using RTA (real-time analysis with SCS2.8). Raw reads that passed the chastity filter threshold were mapped using STAR (Dobin et al., 2013) to generate read alignments for each sample. Genomic annotations were obtained from Ensembl version 84. The transcript isoform level and gene level counts computed using Kallisto (Bray, Pimentel, Melsted, & Pachter, 2016) utility. A count per million (CPM) filtering cutoff ≥1.0 in all replicates of either the wild-type or heterozygous or homogenous knock-in samples was used to determine expressed transcripts. Differential transcript expression was then performed using the edgeR program (Robinson, McCarthy, & Smyth, 2010). Clusterprofiler R package (Yu, Wang, Han, & He, 2012) was used to perform the GO analysis.
Immunoblot analysis
Mouse eyecups were dissected and homogenized in PBS buffer with 0.15 mg/ml dodecyl maltoside and complete protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). 10–40 μg of protein extracts from mouse eye cup were analyzed by western blot using antibodies against RPE65 (“PETLET” antibody developed in this lab, 1:1000; Redmond & Hamel, 2000), GAPDH (Santa Cruz, 1:5000), SQSTM1 (Santa Cruz, 1:1000), and alpha-Actin (Santa Cruz, 1:5000). Relative expression levels were quantified using Li-Cor Odyssey Infrared Imaging system. RPE65 expression levels were analyzed using Image Studio (Li-Cor Biosciences, Lincoln, NE), normalized to the corresponding GAPDH expression levels. For a positive control of SQSTM1 expression, we used cell lysate from ARPE19 cells treated with vH+ATPase inhibitor bafilomycin A1 (BafA1) as previously described (Poliakov et al., 2014).
Histology
For morphological studies, enucleated eyeballs were fixed in 2% paraformaldehyde and 2.5% glutaraldehyde and embedded in methacrylate. Serial vertical sections were cut through the pupillary-optic nerve plane, stained with hematoxylin and eosin and analyzed by light microscopy. For RPE65 immunostaining, eyes were snap frozen in OCT media directly following enucleation. The fresh-frozen tissues were sectioned along the superior-inferior meridian at 10-μm thickness, incubated with anti-RPE65 antibody (1:300) and corresponding secondary antibody with fluorophore labels, and finally imaged on a confocal laser scanning microscope (Zeiss LSM 700, Carl Zeiss Inc., Thornwood, NJ). For immunostaining of opsins and actin bundles, mouse eyes were enucleated and dissected to remove the anterior segments. The resultant retina/eyecup was fixed in 4% formaldehyde at 4°C overnight and then embedded in 7% agarose. After tissue permeabilization and serum blocking, vibratome sections of 100 μm along the superior-inferior meridian were incubated with goat anti–OPN1SW (1:100, Santa Cruz, Santa Cruz, CA) for 24h at 4°C. Sections were subsequently incubated overnight with secondary antibody against goat, or fluorescent conjugated phalloidin and wheat germ agglutinin (Thermo Fisher Scientific). Stained retinal sections were imaged with confocal microscopy (FluoView 1000, Olympus, Center Valley, PA). For flat-mount TUNEL staining, mouse eyeballs were fixed in phosphate-buffered 4% paraformaldehyde (PFA) for 10 min at room temperature before dissection. Muscle and fat surrounding the eye globe were removed without puncturing the globe. The cornea was penetrated with a stab scalpel, followed by cutting with micro-scissors along the limbus to remove the cornea and lens. Subsequently, two incomplete circumferential cuts of the eyecup towards the optic nerve were made to facilitate neural retina removal and expose the RPE for subsequent staining. Finally, the optic nerve was snipped at its base. The dissected eye cup (sclera, choroid, and RPE) was refixed in 4% PFA for 20 minutes, washed three times in PBS-T (PBS with 0.2% Triton X-100), and permeabilized for 30 minutes at 65°C in PBS with 0.2% Triton X-100 and 0.1% Sodium Citrate. For positive controls, the eyecup was incubated with 100 μl 3U/μl Dnase I at room temperature for 20 minutes. After washing in PBS-T, the eye cup was subject to TUNEL staining using In Situ Cell Death Detection Kit, TMR red (Roche, Indianapolis, IN) according to manufacturer’s instruction. The eye cup was flattened on a precleaned slide, and a total of 8 straight cuts from the limbal edge were made to form a eight-petal flower-like structure. The eyecup was then mounted in Fluoro-Gel (Electron Microscopy Sciences, Hatfield, PA) with a 24×40 mm coverslip, and imaged by fluorescence microscopy.
Electroretinograms (ERGs)
Following dark adaptation for the indicated time period, the eyes of anesthetized mice were topically administered with 1% tropicamide and 2.5% phenylephrine for pupillary dilation, and 0.5% proparacaine hydrochloride for topical anesthesia. Body temperature was maintained at 37°C with a heating pad. Electroretinograms were recorded from both eyes using gold wire loop electrodes and an Espion e2 Visual Electrophysiology System (Diagnosys, Lowell, MA, USA). A gold wire loop placed in the mouth was used as the reference electrode. Dark-adapted ERGs were performed using flashes of intensities ranging from 0.0001 to 10 cd.s/m2. Light-adaptation was performed with background light at 20 cd/m2 for 2 min and the ERG response was recorded using flashes of intensities ranging from 0.3 to 100 cd.s/m2. Electroretinogram recordings were performed on at least six mice per age group. For photopic bleaching experiments, mice were illuminated under a Ganzfeld chamber (5,000 cd/m2) for 60 seconds after pupil dilation. A single-flash ERG at 10 cd.s/m2 was used to monitor recovery of photopic b-wave amplitude every 1 minute for 20 minutes. For c-wave recording, stimulus was presented for 10 seconds with intensities from 0.01 to 1000 cd/m2 with inter-stimulus interval of 2 – 5 minutes.
Optical Coherence Tomography (OCT) imaging
Mice were anesthetized, and their pupils dilated as described above. Artificial tears (Systane Ultra, Alcon, Fort Worth, TX) were used to maintain corneal hydration and clarity. OCT images were obtained using the Bioptigen Spectral Domain Ophthalmic Imaging System (Bioptigen Envisu R2200, Morrisville, NC). Image acquisition software was provided by the vendor. The thickness of the ONL was measured at a distance of 0.624 mm from the optic nerve head (ONH) using software provided by vendor.
Light Induced Retinal Damage
For acute light exposure, mice were dark-adapted for 7 days before subject to 15,000 lux of diffuse white fluorescent light for 60 minutes with dilated pupils (as described above). Following light exposure, the animals were kept in dark for 1 or 2 hours before euthanization and eyeball collection for retinoid analyses. Prior to the light exposure, the light intensity was measured and confirmed inside the cage with a digital light meter.
For chronic light exposure, the experimental mice were reared in a light cubicle for 6–7 months without pupil dilation. The light cubicle is equipped with white fluorescent light that operates in 12h light/dark cycles. During the light exposure time, a mock cage was placed in the light cubicle and the light meter measurements were taken on various positions at the bottom of the cage and average was taken as the light intensity to which the mice were exposed. The light measurements were repeated for three consecutive days. The average light intensity inside the amber cage is about 400–500 lux.
Retinoid Extraction, Saponification, and Retinoid Analysis.
All procedures were performed under red safelights. For analysis of WT and D477G RPE65 activity in HEK293-F cells, retinoids were extracted and saponified as previously described (Redmond et al., 2010) from cells harvested by centrifugation from 29 ml volumes of cultured transfected 293-F cells. Isomeric retinols were analyzed on 5 μm particle LiChrospher Si-60 (Alltech, Deerfield, IL) normal-phase columns (2 × 250 mm) on an Agilent 1100/1200 series HPLC system (Agilent Technologies, New Castle, DE), in hexane mobile phase containing ethyl acetate (11.2%):dioxane (2.0%):octanol (1.4%; HEDO) following Landers & Olson, 1988, as earlier modified by us (Redmond et al., 2010). Data were analyzed using ChemStation32 software (Agilent). For analysis of mouse ocular retinoids, mouse eye cups were homogenized in 0.5 ml of freshly made hydroxylamine buffer (50 mM MOPS, 10 mM NH2OH, pH 6.5) before addition of 1 ml hexane. The phases were separated by centrifugation at 3,000 × g for 3 min. The lower phase was extracted with a second 1 ml volume of hexane, and the upper phase from the two extractions were pooled and dried with argon. The dried samples were dissolved in 100 μl hexane. 20 μl sample aliquots, along with retinal oximes standards (Garwin & Saari, 2000), were separated using a 5 μm particle LiChrospher Si-60 normal-phase column on a Waters H-Class Acquity UPLC (Waters Corp., Milford, MA, USA) in HEDO (Landers & Olson, 1988) at a flow rate of 0.7 ml/min. Absorbance was monitored at 350 nm, and peak areas for 11-cis retinal oximes and retinyl esters were integrated and quantified using calibration curves based on external standards. Data were analyzed using Empower 3 software (Waters Corp.).
Statistical analysis
Unless otherwise specified, statistical significance of biochemical and gene expression experiments, as well as visual function tests between two groups, was assessed by student’s t-test. The level of significance was chosen at P = 0.05 using GraphPad Prism 7 (GraphPad Software, San Diego, CA).
Results
Expression of RPE65/D477G does not jeopardize isomerase activity in vitro
When transcribed and translated normally, the c.1430A>G substitution would give rise to the Asp477Gly (RPE65/D477G) missense mutation. To test whether RPE65/D477G is a dominant mutant interfering with wild type RPE65 function, we measured the isomerase activity of forced-expressed RPE65/D477G mutant protein in a minimal in vitro visual cycle (Lorenz et al., 2008; Redmond et al., 2005). Specifically, an expression vector carrying the canine RPE65 ORF containing the c.1430A>G mutation was transfected into HEK293-F cells. Following a 6-hour incubation with 2.5 μM all-trans retinol, transfected cells were harvested, and the levels of 11-cis retinol measured. Cells transfected with the RPE65/D477G mutant construct produced about 50% of the 11-cis retinol made by cells transfected with wild-type (WT) RPE65 (Figure 1), with comparable protein expression levels (data not shown). More importantly, when RPE65/D477G was co-transfected with the wild-type RPE65 construct at a 1:1 ratio, there was no interference of the mutant with WT RPE65 isomerase function. On the contrary, cells co-transfected with mutant and WT mixture produced higher level of 11-cis retinol compared to those transfected with the mutant alone- about 70% of that produced by WT RPE65 alone (Figure 1). Consistent with our findings, Choi et al (2018) found that a NIH3T3 cell line stably expressing the human RPE65/D477G mutant processed all-trans retinol and produced similar levels of 11-cis retinol to their control cell lines expressing WT RPE65. We conclude that the RPE65/D477G missense mutant has only moderately reduced isomerase activity and, furthermore, does not act as a dominant-negative mutant affecting WT protein function, but rather behaves similarly to, or even better than, other hypomorphic missense mutations of RPE65 that have been previously characterized (Li et al., 2015; Redmond et al., 2010; Samardzija et al., 2008).
Figure 1.

Isomerase activity of the missense mutation RPE65/D477G. (A) HPLC of retinoids from cell lysates incubated with all-trans retinol (peak 3), showing presence of 11-cis retinol (peak 1) and 13-cis retinol (peak 2). Inset shows representative chromatography for cells transfected with either canine WT RPE65, RPE65/D477G, or a mixture of WT and RPE65/D477G. (B) Comparison of total 11-cis retinol extracted from transfected cells (mean and SEM; n=4).
To further test the permissivity of aa477 of RPE65 in respect of amino acid variation, we also mutated D477 to other amino acids including alanine (D477A), glutamic acid (D477E), and asparagine (D477N) residues. Forced expression of these missense mutations in 293F cells had minimal impact on RPE65 isomerase activity (Supp. Figure S1), suggesting that a missense mutation of RPE65 at position 477 is not likely to cause deleterious effects on RPE65 function. It is of interest to note that in the assemblage of carotenoid oxygenase paralogs of RPE65 (animal and plant), the paralogous residue corresponding to D477 in RPE65 is most often G, with D, E, and N also occurring.
Generation of Rpe65 KI mice carrying the human c.1430A>G mutation
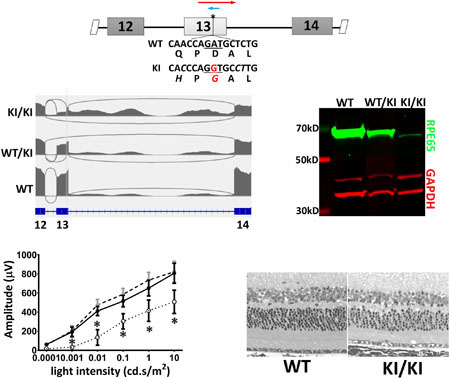
To study the pathogenic effect of the c.1430A>G mutation, we introduced this point mutation into the mouse genome via CRISPR/Cas9-mediated mutagenesis (Hsu et al., 2014; Zhang et al., 2014). In comparing the human genomic sequences with the mouse genome surrounding the c.1430A position, we found three differences in the WT sequences (Supp. Figure S2A). The c.1434T and c.1435C found in the mouse genome are silent substitutions for their human orthologs. The c.1425A gives rise to Q475 in mouse RPE65, as compared to H475 in the human sequence. Computational analysis predicts that H475Q substitution has minimal impact on protein function (http://genetics.bwh.harvard.edu/pph2/; Adzhubei, Jordan, & Sunyaev, 2013). In fact, alignment of mammalian RPE65 proteins shows that H is the predominantly conserved residue at aa475 across mammalian species, while only some rodents (including mouse and rat) bear Q475 (Supp. Figure S2B). Outside mammals, the identity of residue 475 is more variable. To make the DNA context of the c.1430A>G mutation a better representation of the human mutation, we included these three nucleotide substitutions into the mouse genome, along with the presumed pathogenic c.1430A>G mutation (Figure 2A). Founder mice with the KI allele were identified through sequencing (Figure 2B). To create an isogenic transgenic line and avoid off-target mutations of gRNA, the identified transgenic line was back-crossed three generations with wild-type C57BL/6J. As the overall background is on C57BL/6J, these mice express the aaM450 variant of RPE65 with lower isomerase activity (Wenzel, Reme, Williams, Hafezi, & Grimm, 2001). Heterozygous and homozygous progeny were identified through droplet digital PCR (ddPCR) analysis as shown in Figure 2C.
Figure 2.

Generation of c.1430A>G KI mouse. (A) Mouse genomic context for NM_029987.2:c.1430A>G mutation. DNA sequences around the mutation sites are specified along with the translated amino acid sequence in WT Rpe65. The nucleotides altered in the KI mouse model are in italics and the pathogenic A-to-G mutation is marked in red. Cas9 mRNA, gRNA targeting the point mutation site (blue arrow), and a 169-nt oligo (red arrow) carrying the nucleotide mutations were microinjected into mouse zygotes. (B) The knockin allele was identified by sequencing the PCR amplicon surrounding the mutation site (primer locations shown as green arrows in A). (C) Schemes for using ddPCR for genotyping the KI mice. A FAM-labeled probe (orange bar with T) centered on the mutation sites was designed to specifically recognize the mutant allele, while a HEX-labeled probe (green bar with R) located outside of the mutation sites served as a reference probe. The WT allele results in HEX-positive-only droplets (left panel) while the mutant allele produce double-positive droplets in both heterozygous (middle panel) and homozygous (right panel) KI mice.
Both heterozygous and homozygous KI mice show grossly normal retinal structures and visual functions under regular husbandry.
Heterozygous (WT/KI) and homozygous (KI/KI) KI mice were raised under regular vivarium light conditions (less than 100 lux), along with their wild-type control siblings (WT). The retinal integrity of the WT/KI and KI/KI mice were constantly monitored via both optical coherence tomography (OCT) and hematoxylin and eosin (H&E) staining of mouse eyeballs at various ages (3–12 months old). OCT shows no detectable changes in retinal integrity for either WT/KI or KI/KI mice at the age of 11 months old (Figure 3A). Consistently, H&E staining of the methacrylate sections also shows comparable thickness of various retinal layers in both the WT/KI and KI/KI to the WT (Figure 3B). However, we consistently observed structural anomalies (apparent vacuoles; black arrows in Figure 3B) in the outer segment (OS) layer of the KI/KI retina.
Figure 3.

Phenotypic characterization of heterozygous and homozygous KI mice. (A-B) Retinal structure of KI mice. Retinal integrity of 11-month WT/KI and KI/KI mice was monitored by both in vivo OCT imaging (A) and H&E staining of retina methacrylate sections (B). While there are no differences in retinal structure between WT/KI (middle panel in B) and their WT siblings (left panel in B), KI/KI mice show the presence of vacuoles in the outer segment layer (arrow in right panel of 2B). Scale bar=50 μm. (C-D) Visual function analysis show that both the rod (C) and cone (D) functions of the WT/KI and KI/KI mice are comparable to the WT sibling controls; n=10–12 for each genotype group. (E-F) HPLC analysis of the retinoid content in the KI mice following 24-hour dark adaptation. The retinae of the WT/KI and KI/KI mice contain similar levels of 11-cis retinal as those of the WT (E). However, the retinyl ester levels in the KI/KI mice eyes are significantly increased compared to those in the WT eyes (F); n=3 for each genotype group in the HPLC analysis. INL: inner nuclear layer; ONL: outer nuclear layer; OS: outer segment; and RPE: retinal pigment epithelium.
Rod and cone functions in the WT/KI and KI/KI retinae were tested by both scotopic and photopic electroretinogram (ERG) after a full (overnight) dark adaptation. The ERG responses were monitored at different ages (3–12 month-old). Consistent with grossly normal retinal structure, ERG responses of both the WT/KI and the KI/KI mice exhibited similar amplitudes as their WT siblings under both dark-adapted (Figure 3C) and light-adapted (Figure 3D) conditions, indicating mutation does not affect rod or cone photoreceptor sensitivities. Furthermore, when chromophore contents were analyzed via HPLC, both the WT/KI and the KI/KI retinae revealed comparable amounts of 11-cis retinal (Figure 3E) to the WT. However, there was a significant increase in retinyl ester accumulation in the RPE of KI/KI mice (Figure 3F). Taken together, this suggests an adequate yet less efficient chromophore production in the KI/KI mouse retinae.
We further investigated RPE cell function and integrity in our KI mouse models. Flat-mount eyecups prepared from the WT/KI and KI/KI mice were subject to TUNEL staining and surveyed for cell death in the RPE layer. As shown in Supp. Figure S3A, the density of nuclei of RPE appears normal in 4-month-old WT/KI and KI/KI mice, when compared to their WT siblings. More importantly, we did not detect any obvious apoptosis of RPE cells in either the heterozygous or the homozygous KI mice, suggesting that the RPE is largely healthy. We also examined the ionic channel activity of RPE plasma membrane in our KI mouse models by measuring the slow ocular potentials of the ERG c-wave after a full dark adaptation (Nilsson, 1985; Steinberg, 1985). The c-wave amplitudes of the 4-month-old KI/KI and WT/KI mice are comparable to those of their WT siblings (Supp. Figure S3B), indicative of a grossly physiologically functional RPE in heterozygous and homozygous KI mice.
It has been suggested that missense mutation of RPE65/D477G may exerts toxicity via protein aggregation and subsequent clearance via ubiquitination (Choi et al., 2018; Kiser et al., 2018). We tested the possibility that c.1430A>G, p.(D477G) may form extensive aggresomes and thus induce expression of stress-related proteins in the RPE. The scaffolding adaptor protein sequestosome-1/p62 (Sqstm1) is a stress-inducible protein that mediates autophagic clearance of aggregated proteins via ubiquitination (L. Wang, Cano, & Handa, 2014). It provides an important compensatory mechanism in age-related dysfunction of the RPE (Cano et al., 2014). We examined Sqstm1 expression in the RPE/choroid of the KI/KI and WT/KI mice at age of 10 month. As shown in Supp. Figure S3C, we did not detect expression of this stress-induced protein in the RPE of the aged KI mice.
KI/KI mice are sensitive to light stress and bright light exposure.
To determine how the KI/KI mice respond to light stress, we subjected mice raised in normal vivarium light conditions (less than 100 lux) to a shorter than usual dark adaptation period of 20 minutes before ERG testing. Following shortened dark adaptation, KI/KI mice exhibited a 40–60% decrease in both the a-wave (Figure 4A) and b-wave (Figure 4B) amplitudes of the dark-adapted ERG compared to their WT siblings, suggesting a slower rod function recovery in KI/KI animals. In comparison, the photopic response of the KI/KI mice is undisturbed by the short dark adaptation (Supp. Figure S4). Heterozygous WT/KI mice, on the other hand, behave similarly to WT controls in both scotopic and photopic ERG tests and hence do not exhibit dominant traits under the testing conditions (Figure 4 and S4).
Figure 4.

Recovery of rod function and chromophore generation are impaired in the KI/KI mice following light exposure. (A-B) Scotopic ERG recordings were performed following dark adaptation of 20 minutes. Both the a-wave (A) and b-wave (B) response of the KI/KI animals are significantly lower compared to those of the WT animals. The WT/KI animals, on the other hand, behave similarly to the WT mice. Asterisks denote that the differences are statistically significant with p<0.05 analyzed by student’s t-test; n=8–12 for each genotype group. (C-D) 11-cis retinal recoveries and retinyl ester reduction are slower in KI/KI mice following strong light exposure. Dark-adapted mice were exposed to light of 15,000 lux for 60 minutes and returned to the dark until eyeballs were collected. Retinoid analysis by HPLC at different time points were performed to quantify amounts of 11-cis retinal (C) and retinyl esters (D) at 1-hour and 2-hour post light exposure. Error bars indicate the S.D. of the mean (n=3); *: p<0.05, student’s t-test.
Impaired rod recovery after a short dark adaptation suggests that KI/KI mice have an inefficient visual cycle that cannot provide sufficient chromophore in times of light stress. To test this hypothesis, we depleted the 11-cis retinal pool by subjecting the KI/KI mice to a strong light bleach (15,000 lux for 1 hour) before measuring chromophore regeneration. One hour after light treatment, the exhausted 11-cis retinal pool has already been replenished in WT retinae (Fig. 4C). In contrast, the rate of synthesis of chromophore in KI/KI mice fell far behind with more than 10-fold less 11-cis retinal content one hour after light bleaching and with no significant accumulation beyond that two hours later (Figure 4C). On the other hand, the level of retinyl esters in the KI/KI RPE was 4-fold more than that in the WT RPE one hour post light treatment and about 8-fold more two hours later as the WT mice far more efficiently regenerated their chromophore than KI/KI mice (Fig. 4D).
We next investigated whether KI/KI mice were more susceptible to chronic exposure to increased light intensity. KI/KI and WT littermates were subject to a cyclic light cycle (12-hr light/dark) at about 500 lux for over 6 months (Figure 5). Scotopic responses of the KI/KI mice at the end of the 6-month light exposure were tested after full overnight dark adaptation. The KI/KI mice displayed similar scotopic amplitudes compared to their WT siblings (Supp. Figure S5), suggesting that the chronic light exposure did not affect the rod sensitivities of the KI/KI mice. However, when the cone functions were tested after a moderate visual pigment bleach (5000 cd/m2 for 60s), the recovery rate of photopic ERG response to a 10 cd.s/m2 stimuli was slowed down significantly in the KI/KI mice (Figure 5D). The recovery rate of heterozygous WT/KI mice on the other hand, is similar to that of the WT mice. Interestingly, when mice housed in regular vivarium light conditions (less than 100 lux) are subjected to the moderate light bleach of 5000 cd/m2 for 60 seconds, the recovery rate of photopic responses of KI/KI mice is as fast as their WT siblings (Supp. Figure S6), suggesting a role of long-term chronic light exposure in the slowed cone recovery in KI/KI mice. Furthermore, two weeks after the moderate bleaching, the KI/KI mouse retinae showed a mild yet consistent decrease in the retinal thickness (Figure 5A) as compared to the age-matched WT controls without bleaching (-LE). In contrast, WT siblings that went through the same moderate bleaching (+LE) showed normal retinal thickness in OCT analysis, similar to the WT controls (Figure 5A). Measurement from 4–8 biological replicates of OCT images showed that the ONL thickness at 0.624 mm from the optic nerve head (ONH) is significantly reduced in the KI/KI mouse retinae compared to WT(Figure 5C). The temporal retina is more severely damaged than nasal retina on the T–N meridian, a typical feature for light-induced retinal degeneration (Tanito, Kaidzu, Ohira, & Anderson, 2008; X. Wang et al., 2017). Further histopathologic examination revealed that the KI/KI retinae display subtle yet persistent degenerative features, including more frequent vacuole formation in the RPE layer and moderate decrease in the width of ONL and photoreceptor OS layer (Figure 5B).
Figure 5.

Integrity of the retinal structure, as well as cone ERG recovery, is impaired in the KI/KI mice after moderate light exposure. (A) KI/KI mice, along with their WT siblings, were housed in light cubicle (light intensity roughly 500 lux) for 6 months (+LE). Two weeks after brief light bleaching, OCT imaging shows a slight decrease in retinal thickness in the KI/KI mice, particularly in the ONL. The WT retinal structure remained normal with (+LE) or without (-LE) light bleaching. (B) H&E staining of the eyes shows that KI/KI display degenerative features including thinner ONL and photoreceptor layer, as well as presence of vacuoles in the RPE layer. The arrowhead in the inset marks the position of the retinal structures presented in B. Scale bar=50 μm. (C) Measurement of the ONL thickness, from OCT imaging at 0.624 mm distance from the optic nerve head, shows a decrease of ONL thickness in the KI/KI mice, particularly at the temporal side of the retinae; n=4–8 for each group. (D) The cone ERG recovery after light bleaching is slowed in KI/KI mice subjected to chronic light exposure. Asterisks denotes the differences are statistically significant with p<0.05 analyzed by student’s t-test; ONL: outer nuclear layer; OS: outer segment; and RPE: retinal pigment epithelium.
KI/KI mice express minimal RPE65 in RPE and exhibit mild opsin mislocalization in retinae.
When the ORF encoding the RPE65/D477G missense mutant is force-expressed in cultured cells, the protein expression level is comparable to that of the WT cDNA clones. However, the expression of RPE65 protein was barely detectable in KI/KI mouse eye cups (Figure 6A). When quantified and normalized to GAPDH level, RPE65 protein levels in KI/KI eye cups were decreased to less than 3% of wild type levels (Figure 6A). Similarly, immunofluorescence staining of KI/KI mouse eye cross-sections showed the mutant protein was correctly localized in RPE at a greatly reduced level, but not absent as in the homozygous Rpe65 knockout (KO) mice (Figure 6B). The expression level of the mutant protein did not alter as the transgenic animals aged but remained at about 3% of the wild type control from age of 12 weeks (data not shown) to 7 months (Figure 6A).
Figure 6.

Expression level of the mutant RPE65 is greatly reduced in the KI/KI mice. (A) western blot analysis shows the RPE65 expression level in 7-month-old KI/KI mice is less than 3% of the age-matched WT siblings. Asterisks denote the differences are statistically significant with p<0.05 analyzed by student’s t-test; n=3. (B) immunostaining of the mouse eye cup frozen sections shows minimal expression of the mutant RPE65 protein in the KI/KI mouse. Arrowheads denote the position for RPE layer. (C) Despite the low RPE65 expression, integrity of the retinal outer segment layer remained intact in the KI/KI mice, as demonstrated by actin (green) and WGA (red) staining. On the other hand, while the majority of the short-wave opsin (OPN1SW) is localized at the outer segment in the KI/KI mice, as seen in the WT, we consistently observe a small yet persistent mislocalization of the OPN1SW near the basal side of the outer nuclear layer (indicated by white arrowhead). Scale bar=50 μm. ONL: outer nuclear layer; OS: outer segment; and IS: inner segment.
RPE65 expression is critical to cone survival and proper localization of cone opsins, particularly the short-wave cone opsin (OPN1SW; Rohrer et al., 2005). Therefore, we investigated whether the minimal expression of RPE65 in KI/KI mice affects the expression of cone opsins. Figure 6C shows typical OPN1SW staining of the KI/KI mouse retinae raised in dim light vivarium conditions. In age-matched WT retinae, OPN1SW was detected almost exclusively in OS, with minimal staining at the apical side of the outer nuclear layer. In the age-matched Rpe65 KO mice, OPN1SW expression was greatly reduced and a significant portion of OPN1SW was mislocalized to the inner segment (IS), throughout the outer nuclear layer (ONL), and at the level of the synaptic pedicles. In comparison, the majority of OPN1SW labeling was localized to the OS in the 4-month-old KI/KI retina as were the opsins in the WT control. However, we observed an increased incidence of OPN1SW mislocalization at the basal side of the ONL in KI/KI retinae (arrowheads in Figure 6C). On the other hand, the overall integrity of the retinal outer segments, examined through WGA and phalloidin staining, is normal in the KI/KI mice as compared to the greatly reduced signals in the Rpe65 KO/KO mice (Figure 6C).
The c.1430A>G mutation impairs RPE65 mRNA splicing
To investigate why KI/KI mice express such low levels of RPE65 protein, we analyzed the Rpe65 transcript levels of the KI/KI mice using Taqman probes located either at the 5’ end of the transcript (spanning Exon 3 and Exon 4), or at the 3’ end of the transcript (Exon 14; Figure 7A). Expression of the KI/KI Rpe65 mRNA varied with the locations of the probes. Transcript levels measured by the 5’-probe showed a decrease of 50% compared to WT for the KI/KI mice, whereas KI/KI mRNA levels measured using the 3’-probe were less than 10% of the WT RPE65 level (Figure 7B). The difference in mutant Rpe65 transcript levels detected by the different probes suggests that the mutation may result in alternative splicing products that are no longer bearing the regular Exon 13 and 14. To test this hypothesis, we performed 3’-RACE analysis using a poly(dT)-based 3’ cDNA cloning primer, together with a 5’-primer located on Exon 6 (Figure 7A; for details, see Methods). 3’-RACE of a WT cDNA yielded a major amplicon of 1.2 kb (Figure 7C), which we subsequently cloned, sequenced and confirmed as authentic Rpe65 cDNA. The 3’-RACE products from the KI/KI cDNA, however, appear more heterogeneous and smeared compared to the WT controls (Figure 7C). 3’-RACE products from the KI/KI mouse total RNA were gel purified, cloned, and sequenced. Out of 53 sequenced clones, we were able to identify various abnormal RPE65 transcripts, of which three examples are listed in Figure 7D. We cloned a normally spliced Rpe65 transcript (E13E14) that would give rise to a mutant RPE65 protein with the missense Gly mutation at aa477. We identified alternative splice variants where the c.1430G-mutated Exon 13 sequence acts as a splice site acceptor and works with the 3’ donor sequence of Exon 12 to create a truncated Exon 13 (E13-AS). We also obtained cDNA clones that skipped the entire Exon 13 such that the Exon 12 is spliced directly to Exon 14 (E13-DEL). Apart from the alternative spliced transcripts, we also detected a truncated mRNA species that is polyadenylated within either Intron 12 or Intron 13 and stops prematurely (data not shown). Unlike the sequences containing the missense mutation, all the alternative splicing events identified in the RACE analysis lead to a premature truncation of the protein, via frameshift and introduction of downstream stop codons.
Figure 7.

The c.1430A>G mutation generates an alternative splicing site in the mouse KI allele. (A) Scheme of Rpe65 mRNA and 3’-RACE. The asterisk marks the c.1430A>G mutation. Pink bars denote positions of the Taqman assays for mRNA level quantification. Following reverse transcription, 3’-RACE was performed using forward primer (green arrow) located in Exon 6. (B) ddPCR analysis showed that Rpe65 mRNA levels are significantly lower in the KI/KI mice than in the WT. However, the expression level difference between the WT and KI/KI varies with the locations of the Taqman probes. Asterisks denote the differences are statistically significant with p<0.05 analyzed by student’s t-test; n=3. (C) Electrophoresis of the 3’-RACE products. In the WT animals, 3’-RACE yields a major band of 1.2 kb. In contrast, the 3’-RACE for the KI/KI mice result in multiple bands of lower intensity. Arrows indicate the PCR amplicons from 3’-RACE that were subsequently gel-purified and sequenced. (D) Examples of alternatively spliced transcripts cloned from the 3’-RACE of the KI/KI mice. (E) The novel transcripts identified by RACE were confirmed by ddPCR using Taqman probes specifically targeting different variants (shown as pink bars in D).
To confirm the novel transcripts identified by 3’-RACE, we designed Taqman assays to detect alternatively spliced variants of the E13E14, E13-AS, and E13-DEL (Figure 7D). The cDNA pools from KI/KI, WT/KI, and WT mouse eyecups were analyzed by ddPCR quantification using a number of probe assays (Figure 7E). As shown in Figure 7E, novel aberrant transcripts like E13-AS and E13-DEL are readily detected in the KI/KI but not in the WT mice. The level of novel aberrant transcripts detected in WT/KI are about 50% of the KI/KI mice, exhibiting a dosage-dependent expression of the mutant allele. As ddPCR quantification is based on binomial statistics (Quan, Sauzade, & Brouzes, 2018), we were able to directly compare the expression levels between different transcripts in the KI/KI mouse eye cup. The number of copies of the missense mRNA transcript (E13E14), is about twice that of the E13-AS transcript and 7 times more than the E13-DEL transcript (Table S1). However, the sum of various splicing variants tested only comprises about 1/3 of the total copies of the overall RPE65 cDNA, as detected by the E3E4 assay, suggesting that many different splicing variants may be generated by the mutant allele, in addition to the tested variants.
Transcriptome profiling of KI/KI mice bearing c.1430A>G mutation.
We next compared gene expression profiles for the eyecups of the WT, WT/KI, and KI/KI mice. Specifically, three mice from each genotype raised under regular husbandry were killed at age three months, their eyecups (RPE-Choroid-Sclera) were dissected separately, and total RNAs for individuals were isolated and analyzed via RNA-seq. The correlation plot (Figure 8A) shows similar expression values for transcripts between biological replicates and the difference between different biological groups are minimal. A volcano plot of the data shows that 13 genes are differentially expressed (at fold change >1.5; FDR< 0.10) in KI/KI eye cups compared with WT (Figure 8B; Table S2). Most of these differentially expressed genes are involved in either regulation of the cell proliferation/apoptosis (e.g., Zbtb8a, Naip, and Arg1), or cytoskeletal organization (e.g., Mid1ip1, Gbf1, and Dnah9). The subtle changes in gene expression profile are consistent with the minimal phenotypic changes observed in the KI/KI mice raised under regular mouse husbandry (Figure 3). Except for two genes (i.e., Cr2 and Xist), the gene expression profile of WT/KI mouse eyecup is similar to that of WT (Figure 8C, Table S2). Thus, WT/KI mice raised under usual vivarium conditions do not manifest any dominant effect on gene expression, consistent with their non-dominant phenotypic characterization (Figure 3).
Figure 8.

RNA-seq analysis of mouse eyecup RNA (RPE-Choroid-Sclera). (A) Sample correlation matrix displaying the overall similarity in expression profile for each sample from WT, WT/KI, and KI/KI groups (n=3). Increasing sample correlation is represented by darker grey whereas lighter grey indicates decreasing sample correlation. Dendrogram clustering on the X and Y-axis indicates the overall similarity of these samples. (B-C) Volcano plot of unadjusted p values in relation to expression fold changes between the KI/KI and WT (B) or WT/KI and WT (C) transcriptomes. Open dots denote differentially expressed genes with FDR-adjusted p<0.1. (D) Sashimi plots showing RNA-seq reads mapping to Rpe65 Exons 12–14 locus in WT, WT/KI, and KI/KI mice. Heights of the bars represent overall read coverage. Arcs represent major splicing events detected in the RNA-seq reads. The number of sequence reads aligned on Intron 13 is greatly increased in the KI/KI mice. Splice junctions supported by >10 split reads are displayed as loops. Arrowheads denote the novel splicing reads corresponding to the E13-AS and E13-DEL in Figure 7D. (E) Zoom-in of the dotted rectangle on Sashimi plot areas (D) shows that reads increase right at the pathogenic c.1430A>G site in both KI/KI and WT/KI mice, suggesting that the c.1430G can work as a novel acceptor site for novel splicing. Italic letters denote mutated nucleotide sequences in the KI/KI mice while the pathogenic c.1430G in KI/KI mice is italicized and underlined.
Among the differentially expressed genes, the total number of normalized reads for Rpe65 is decreased by 2-fold in the KI/KI mice compared to that in the WT. To characterize the Rpe65 transcripts synthesized in KI/KI mice, Rpe65 reads were mapped to the corresponding genomic location and visualized by Integrative Genomics Viewer (IGV). The resulting Sashimi plot revealed that, compared to the reads for Exon 13 in the WT RPE65 transcript, there is a significant reduction in reads at the 5’-half of the Rpe65 Exon 13 in the KI/KI transcript (Figure 8D). The read accumulation in the KI/KI Exon 13 increases at the mutation site of c.1430G (Figure 8E), consistent with our 3’-RACE analyses that mutation of c.1430A>G can act as a novel splicing acceptor and splice with the E12 (Figure 7D). In fact, alignment of the RNA-seq reads detected two major alternative splicing events (arrowheads in Figure 8D) between Exon 12–14 in the KI/KI transcriptome that correspond to the E13-AS and E13-DEL transcripts (Figure 7D). Despite the lower total reads at Exon 13, there is a significant increase in intron retention in the KI/KI mice between Exon 13 and 14 compared to the WT Rpe65 transcript (Figure 8D). An increase in intron retention is also observed in the WT/KI Rpe65 reads, albeit to a much lesser extent.
Human RPE65 c.1430A>G mutation leads to splicing defects similar to the KI mouse.
We next investigated whether the RPE65 c.1430G mutation impacts canonical splicing of the human RPE65 gene as observed in the transgenic mice. We first compared the RPE65 genomic sequence between mouse and human. Using the BDGP program (Reese et al., 1997), we found, in both the mouse and human genomes, that Exon 13 of RPE65, where the pathogenic mutation resides, has weak donor and acceptor sites with the scores below the default 0.4 threshold for effective splicing (Figure 9A). Similarly, using the HUMAN SPLICE FINDER algorithm, the corresponding acceptor and donor scores are also close to the threshold value 80 (82.67 and 80.87). The relatively weak native splicing signals of Exon 13 may, therefore, be more susceptible to splicing anomalies caused by the c.1430G mutation. To test the hypothesis that only the pathogenic c.1430G mutation, but not other nucleic acid substitutions, generate a cryptic splicing site, we performed an in vitro splicing analysis of the human RPE65 gene using the Exontrap system (Duyk, Kim, Myers, & Cox, 1990). A minigene consisting of human RPE65 genomic sequence spanning Intron 11-Exon 14 was cloned into the exontrap vector; site-directed mutagenesis was used to introduce into Exon 13 various mutations including c.1430G, c.1430C, and c.1430T. The WT (c.1430A) and mutant RPE65 minigenes were separately transfected into HEK293T cells. Total RNA was isolated 48 hours post-transfection and reverse transcribed. The resulting cDNA pools were subjected to ddPCR analysis using Taqman probes targeting the potential human splicing variants E13-AS, as well as the read-through RPE65 transcript E13E14 with regular splicing (Figure 9B). The read-through transcript can be readily detected in both the WT- and various mutant- transfected cells. In contrast, the E13-AS variant was greatly enriched in the cells that were transfected with c.1430G mutant (Figure 9C) but not in cells transfected with either the WT-, c.1430C- or c.1430T- minigenes. Therefore, the c.1430G mutation also results in alternative splicing in the human RPE65 gene as found for the KI allele in transgenic mice.
Figure 9.

Defects in splicing are readily detected in human RPE65 minigene carrying the c.1430G mutation. (A) Scores of splicing signals strength for Exons 9–14 of WT RPE65 in mouse and human. High score indicates a strong splicing signal with the default cut-off score for an effective splicing site being 0.4. Note that in both mouse and human, Exon 13 has a weak splicing strength as the scores are below 0.4 and are shown in italic bold fonts. (B-E) minigene constructs that include the human genomic sequence of either WT (c.1430A) or mutants (c.1430G, c.1430C, c.1430T) were transfected into HEK293T cells and the transcribed variants were analyzed via ddPCR. One dimensional ddPCR plots for quantification of the regular spliced read-through transcript E13-E14 (B) , and the alternatively spliced variant E13-AS (C) that lacks the first 91 nucleotide of Exon 13. Data were derived from duplicates of construct transfections. Each point represents a single droplet, which is scored as positive (blue colored) or negative (grey) depending on the fluorescent amplitude. The expression levels were normalized to the expression of reference gene HPRT1 (D and E). While the read-through transcript E13-E14 was readily detected in both the cells expressing WT and the mutant minigenes (B and D), the alternative spliced E13-AS was only enriched in the cells expressing mutant minigene (C and E). The asterisk marks the c.1430A>G mutation. Pink bars denote positions of the Taqman probes for mRNA level quantification.
We further tested the possibility that alternatively spliced transcripts give rise to truncated RPE65 that may serve as a neomorphic mutant. An expression vector carrying the canine RPE65 mutant with an alternatively spliced exon 13 (E13-AS) was transfected into HEK293F cells and force-expressed. Protein expression was examined by western blot; and the isomerase activity of forced-expressed protein was measured in the minimal in vitro visual cycle system. No protein was detectable (Supp. Figure S7A) using an antibody (PETLET) that recognizes an epitope expected to be preserved in the truncated protein. Furthermore, expression of the truncated protein resulted in no 11-cis retinol formation after transfection and atROL addition, indicating a lack of isomerase activity (Supp. Figure S7B). Interestingly, when WT and the truncated mutant were co-expressed at a ratio of 1:1, we did not observe interference of the mutant with WT RPE65 isomerase function. Therefore, truncated RPE65 mutants from alternative splicing are likely subject to protein degradation due to improper folding and not serve as a neomorphic mutant.
Discussion
RPE65 c.1430A> G mutation causes aberrant alternative splicing in both transgenic mice and human cells
In this paper we present the characterization of a KI mouse line bearing the c.1430A>G mutation reported to underlie an autosomal dominant retinitis pigmentosa (adRP) in several families of Irish heritage (Bowne et al., 2011; Hull et al., 2016). We show in our KI mouse model that, rather than solely acting as the expected p.D477G missense mutation, the c.1430A>G exerts its major effect by disrupting the canonical splicing of the Rpe65 gene and giving rise to aberrant mRNA transcripts. Various novel Rpe65 transcripts were cloned from the cDNA pool of the KI/KI eye cup via 3’-RACE, and later confirmed by both ddPCR (Figure 7E) and RNA-seq analysis (Figure 8D). Quantitation of several novel transcripts shows that there is no major transcript raised from the alternative splicing events in KI/KI mice. Paradoxically, instead of working as a strong cryptic splicing site, the mutation appears to impair the recognition of the correct acceptor site at the 3’-end of Intron 12, thus, apparently, forcing the spliceosome to search for an alternate acceptor site in a rather undefined fashion (Figure 9). The disruption by the c.1430A>G mutation of the customary splicing signals results in a significant loss of the normally spliced Rpe65/D477G transcript, as shown by ddPCR quantitation (Figure 7E). While the mutant allele generates novel mRNA transcripts, it does not appear to greatly affect overall mRNA stability, as demonstrated by quantitation of the mRNA level from the 5’-end of the Rpe65 gene (Figure 7B), as well as from the RNA-seq analysis (Table S2). The great reduction in Rpe65 mRNA level is only revealed when PCR analysis is done over the region containing the mutation. The importance of the location of the quantitation assays may explain why previous studies have reported comparable levels of mRNA generated in the c.1430G KI mice as that in WT controls (Choi et al., 2018; Shin et al., 2017). Similar novel splicing events are also found when the human RPE65 genomic sequence carrying the c.1430G mutation is transcribed in cultured cells, suggesting that the pathogenesis associated with the c.1430G mutation may also contribute to defects in splicing in human.
It is increasingly appreciated that nucleotide substitutions (that are not otherwise nonsense mutations) in exons of a protein-coding gene do not necessarily only lead to missense mutations altering protein function; rather, they can influence pre-mRNA splicing and/or mRNA stability (Cartegni, Chew, & Krainer, 2002), with even synonymous substitutions causing this outcome (Livingstone et al., 2017). Some of these exonic mutations disrupt canonical splice sites and lead to exon skipping (Messiaen, Callens, De Paepe, Craen, & Mortier, 1997). Others disrupt or create splicing regulatory elements (SREs) which facilitate exon-intron boundary recognition (Shiga et al., 1997), resulting in aberrant splicing. There are as yet others that result in intronic retentions forming a hybrid mRNA containing both exon and intron (Marquez, Hopfler, Ayatollahi, Barta, & Kalyna, 2015). In fact, one study reported that up to 10% (513/4,964) of known disease-associated missense variants alter mRNA splicing (Soemedi et al., 2017). We show that the c.1430A>G mutation disrupts mRNA splicing in both mouse and human RPE65 genes. Similarly, an RPE65 mutation in human Intron 3 has recently been identified that creates a cryptic splice site, resulting in a premature stop codon via frameshift (Tucker et al., 2015).
Minimal expression of RPE65/D477G protein leads to subtle changes in retinal structures and visual functions in KI/KI mice.
Defective splicing of Rpe65 in the c.1430G mutant gives rise to aberrant mRNA species that in turn results in frameshift, leading to premature truncation of the translated products. These variant transcripts of the Rpe65 mutant allele are prone to be eliminated through nonsense-mediated mRNA decay (Lykke-Andersen & Jensen, 2015). In western blot analysis of KI/KI RPE, antibody to RPE65 recognizes a very faint band of similar molecular weight to WT RPE65 (Figure 6A), representing the RPE65/D477G missense mutant translated from properly spliced mRNA. The expressed RPE65/D477G accounts for the residual yet functional RPE65 protein that is sufficient to sustain a normal amount of chromophore under the dim light conditions of regular mouse husbandry (Figure 3E). The sustained level of chromophore leads to relatively functional retinae in the KI/KI mice, comparable to those in the age-matched WT mice, except for subtle structural changes (Figure 3B). However, the visual cycle is significantly less efficient in the KI/KI mice, as demonstrated in the decreased scotopic ERG in KI/KI mice when the period of dark adaptation is shortened from overnight (~16 hr) to 20 min (Figure 4 A and B). The less efficient chromophore regeneration makes the KI/KI mice more susceptible to bright light challenge. When exposed to the acute light challenge (15,000 lux for 60 min), chromophore and retinyl ester levels are drastically reduced and recover extremely slowly (Figure 4 C and D). This is comparable with our prior results with the P25L Rpe65 missense mutation (Li et al, 2015) but required significantly higher light levels (15,000 lux for 60 min vs. 20,000 lux for 30 min) due to the use of the C57BL/6J background (M450) for the D477G mutant as opposed to the 129/Sv background (L450) in the P25L mutant (Wenzel et al., 2001). When subject to chronic moderate light exposure (~500 lux for 6–7 months), a brief moderate light bleach (5000 lux for 1 min) leads to degenerative changes in the KI/KI mouse retinae including decreased ONL thickness, somewhat less tightly packed retinal OS, and mild disorganization of the RPE (Figure 5B).
The expression level of RPE65/D477G in the KI/KI mice is comparable to a previously reported RPE65/R91W KI mouse model (Samardzija et al., 2008) at about 3–5% of WT RPE65 detected in the WT mice. However, the isomerase activity of RPE65/D477G is far higher than that of the R91W missense mutation (~50% WT vs. less than 1%, when force-expressed). The greater isomerase activity of the RPE65/D477G protein is likely to account for the more subtle and milder retinal phenotype of c.1430A>G KI/KI mice, as compared to the early onset progressive retinal degeneration associated with the R91W mutation (Samardzija et al., 2008).
Defective splicing may contribute to adRP pathogenesis associated with c.1430G mutation
Retinitis pigmentosa pathogenesis is complex and heterogenous, and a significant portion of characterized RP disease-causing genes (27 of >80) are dominantly inherited (Ezquerra-Inchausti et al., 2017). For many adRP genes, mutations lead to a gain-of-function end-product that interferes with either protein folding or transport of the WT gene product. The most prevalent mutations in this category are the dominant mutations in the RHO gene (Gargini, Novelli, Piano, Biagioni, & Strettoi, 2017). Such dominant mutations are usually challenging for gene therapy, as success is contingent on increasing the expression of the functional gene product while simultaneously suppressing the abnormal one. Another common cause of adRP involves a group of genes coding for core spliceosome proteins, including pre-mRNA splicing factors and RNA helicases (Ezquerra-Inchausti et al., 2017). While expressed ubiquitously, most if not all of the mutations are associated exclusively with autosomal dominant pathogenesis in retina/RPE. Phenotypic characterization of KI/KO mouse models generated for such spliceosome genes unanimously found that pathological changes in RPE precede retinal abnormalities (Farkas et al., 2014; Graziotto et al., 2011; Graziotto, Inglehearn, Pack, & Pierce, 2008) , pointing to RPE as a primary target for adRP caused by splicing defects. In the case of our studies on the c.1430A>G mutation, we do not find evidence in support of a gain-of-function protein being produced, as shown by both in vitro functional assays as well as the phenotypic and molecular characterization of the WT/KI mice. Instead, a typical splicing defect is readily detectable both in our KI mouse model and the human minigene construct with the point mutation. Therefore, the pathogenicity of c.1430A>G mutation is likely to arise, at least in part, from defective splicing, rather than from a missense mutant protein. The fact that the c.1430A>G mutation does not operate via a gain-of-function mutant protein opens the possibility that somatic RPE65 gene therapy is feasible for patients carrying this mutation. Alternatively, but not mutually exclusively, the c.1430G mutation may give rise to a truncated RPE65 protein in human but not in mice, which leads to a gain-of-function and dominant phenotype in patients. In the later scenario, therapeutic avenues for the patients would still require the correction of the mutant allele. Test of these hypotheses and possibilities requires further study of the mutation in human iPS-derived RPE cells.
Phenotypic differences between mouse knock-in models and human patients
While c.1430A>G is identified as a dominant mutation associated with RP in human patients (Bowne et al., 2011; Hull et al., 2016), our KI mice do not manifest a dominant phenotype for visual functions. On the contrary, a functional retina with minimal structural changes was observed even in the homozygous mice. The failure to completely recapitulate human pathology in the transgenic mouse models is not an uncommon result in the study of retinal degenerations (Chrispell et al., 2009; Kim et al., 2005; Li et al., 2015). In particular, KI mouse models carrying splicing factor mutations associated with adRP, including PRPF3-T494M (NM_004698.3:c.1481C>T (p.Thr494Met)) and PRPF8-H2309P (NM_006445.3(PRPF8):c.6926A>C (p.His2309Pro)), manifest no major photoreceptor degeneration and minimal defects in visual functions when raised in ambient mouse room lighting (Graziotto et al., 2011). As RPE65 mutations exert their effects on retinal function by affecting the rate of chromophore generation, the photic environment to which carriers of the mutations are exposed to may be crucial for the manifestation of retinal phenotypes. In fact, the light-induced retinal degeneration in the KI/KI mice indicates the role of light intensity in triggering pathogenesis (Figure 5). While strong light exposures are required for pathogenesis in the gain of function Rhodopsin adRP mutations Tvrm1 and Tvrm4 (Budzynski et al., 2010) and the canine Rhodopsin T4R mutation (Cideciyan et al., 2005), increased light in the c.1430A>G mutant mice may simply exacerbate the effect on cone photoreceptor viability of drastically reduced chromophore (i.e., due to loss of function of RPE65) by increased photoisomerization and, thus, enhanced loss of chromophore. Further studies on KI mice under various light intensities may help develop this aspect of the phenotypic characterization of the KI mouse model.
Alternatively, the failure to recapitulate a dominant effect of c.1430A>G in the mouse model may suggest an overlooked genetic modifier closely linked to the RPE65 locus in human patients. The original reports of variable genetic penetrance associated with the human c.1430A>G mutation (Bowne et al., 2011; Hull et al., 2016), particularly the complete lack of symptoms in some carriers of the mutation, indicate a possibility that additional mutations in/modifier effects by other genes may help modulate ocular phenotypes induced by the c.1430A>G mutation. Further genomic analysis may help elucidate possible linked mutation(s) in human patients.
Supplementary Material
Acknowledgments
We thank Jingqi Lei, Carl Haugen, and Steve Lee (Genetic Engineering Core, NEI) for assistance with knock-in mouse generation. We also wish to thank Dr. Alexander N. Gubin (CIT, NIH) for valuable advice on construction of the exontrap vector, the advice of Dr. Robert N. Fariss (Biological Imaging Core, NEI) on microscopic imaging, and of Dr. Maria M. Campos (Histopathology Core, NEI) for pathological characterization. R.F. and A.R. were supported by the Post-baccalaureate IRTA program of the NIH. This research was supported by the Intramural Research Programs of the National Eye Institute and the National Library of Medicine, National Institutes of Health.
Funding Statement
This research was supported by the Intramural Research Programs of the National Eye Institute and the National Library of Medicine, National Institutes of Health.
References
- Adzhubei I, Jordan DM, & Sunyaev SR (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet, Chapter 7, Unit7 20. doi: 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti GD, Bertelsen M, Preising MN, Ajmal M, Lorenz B, Faradz SM, … Cremers FP (2016). Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur J Hum Genet, 24(7), 1071–1079. doi: 10.1038/ejhg.2015.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS, Kenna PF, Tam LC, Kiang AS, … Humphries P (2011). A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet, 19(10), 1074–1081. doi: 10.1038/ejhg.2011.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, & Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol, 34(5), 525–527. doi: 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Briner AE, Donohoue PD, Gomaa AA, Selle K, Slorach EM, Nye CH, … Barrangou R (2014). Guide RNA functional modules direct Cas9 activity and orthogonality. Mol Cell, 56(2), 333–339. doi: 10.1016/j.molcel.2014.09.019 [DOI] [PubMed] [Google Scholar]
- Budzynski E, Gross AK, McAlear SD, Peachey NS, Shukla M, He F, … Nishina PM (2010). Mutations of the opsin gene (Y102H and I307N) lead to light-induced degeneration of photoreceptors and constitutive activation of phototransduction in mice. J Biol Chem, 285(19), 14521–14533. doi: 10.1074/jbc.M110.112409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano M, Wang L, Wan J, Barnett BP, Ebrahimi K, Qian J, & Handa JT (2014). Oxidative stress induces mitochondrial dysfunction and a protective unfolded protein response in RPE cells. Free Radical Biology and Medicine, 69, 1–14. doi: 10.1016/j.freeradbiomed.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, & Krainer AR (2002). Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet, 3(4), 285–298. doi: 10.1038/nrg775 [DOI] [PubMed] [Google Scholar]
- Choi EH, Suh S, Sander CL, Hernandez CJO, Bulman ER, Khadka N, … Kiser PD (2018). Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum Mol Genet. doi: 10.1093/hmg/ddy128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrispell JD, Feathers KL, Kane MA, Kim CY, Brooks M, Khanna R, … Thompson DA (2009). Rdh12 activity and effects on retinoid processing in the murine retina. J Biol Chem, 284(32), 21468–21477. doi: 10.1074/jbc.M109.020966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, … Aguirre GD (2005). In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci U S A, 102(14), 5233–5238. doi: 10.1073/pnas.0408892102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, & Beroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res, 37(9), e67. doi: 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, … Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15–21. doi: 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyk GM, Kim SW, Myers RM, & Cox DR (1990). Exon trapping: a genetic screen to identify candidate transcribed sequences in cloned mammalian genomic DNA. Proc Natl Acad Sci U S A, 87(22), 8995–8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezquerra-Inchausti M, Barandika O, Anasagasti A, Irigoyen C, Lopez de Munain A, & Ruiz-Ederra J (2017). High prevalence of mutations affecting the splicing process in a Spanish cohort with autosomal dominant retinitis pigmentosa. Sci Rep, 7, 39652. doi: 10.1038/srep39652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas MH, Lew DS, Sousa ME, Bujakowska K, Chatagnon J, Bhattacharya SS, … Nandrot EF (2014). Mutations in pre-mRNA processing factors 3, 8, and 31 cause dysfunction of the retinal pigment epithelium. Am J Pathol, 184(10), 2641–2652. doi: 10.1016/j.ajpath.2014.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery JG, O’Day W, Pfeffer BA, Horwitz J, & Bok D (1990). Uptake, processing and release of retinoids by cultured human retinal pigment epithelium. Exp Eye Res, 51(6), 717–728. [DOI] [PubMed] [Google Scholar]
- Gargini C, Novelli E, Piano I, Biagioni M, & Strettoi E (2017). Pattern of retinal morphological and functional decay in a light-inducible, rhodopsin mutant mouse. Sci Rep, 7(1), 5730. doi: 10.1038/s41598-017-06045-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwin GG, & Saari JC (2000). High-performance liquid chromatography analysis of visual cycle retinoids. Methods Enzymol, 316, 313–324. [DOI] [PubMed] [Google Scholar]
- Graziotto JJ, Farkas MH, Bujakowska K, Deramaudt BM, Zhang Q, Nandrot EF, … Pierce EA (2011). Three gene-targeted mouse models of RNA splicing factor RP show late-onset RPE and retinal degeneration. Invest Ophthalmol Vis Sci, 52(1), 190–198. doi: 10.1167/iovs.10-5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziotto JJ, Inglehearn CF, Pack MA, & Pierce EA (2008). Decreased levels of the RNA splicing factor Prpf3 in mice and zebrafish do not cause photoreceptor degeneration. Invest Ophthalmol Vis Sci, 49(9), 3830–3838. doi: 10.1167/iovs.07-1483 [DOI] [PubMed] [Google Scholar]
- Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, … Gal A (1997). Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet, 17(2), 194–197. doi: 10.1038/ng1097-194 [DOI] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, & Dryja TP (2006). Retinitis pigmentosa. Lancet, 368(9549), 1795–1809. doi: 10.1016/S0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, & Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157(6), 1262–1278. doi: 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull S, Mukherjee R, Holder GE, Moore AT, & Webster AR (2016). The clinical features of retinal disease due to a dominant mutation in RPE65. Mol Vis, 22, 626–635. [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li S, Moghrabi WN, Sun H, & Travis GH (2005). Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell, 122(3), 449–459. doi: 10.1016/j.cell.2005.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TS, Maeda A, Maeda T, Heinlein C, Kedishvili N, Palczewski K, & Nelson PS (2005). Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J Biol Chem, 280(10), 8694–8704. doi: 10.1074/jbc.M413172200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiser PD, Zhang J, Sharma A, Angueyra JM, Kolesnikov AV, Badiee M, … Palczewski K (2018). Retinoid isomerase inhibitors impair but do not block mammalian cone photoreceptor function. The Journal of general physiology, 150(4), 571–590. doi: 10.1085/jgp.201711815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landers GM, & Olson JA (1988). Rapid, simultaneous determination of isomers of retinal, retinal oxime and retinol by high-performance liquid chromatography. J. Chromatogr, 438(2), 383–392. [DOI] [PubMed] [Google Scholar]
- Li Y, Yu S, Duncan T, Li Y, Liu P, Gene E, … Redmond TM (2015). Mouse model of human RPE65 P25L hypomorph resembles wild type under normal light rearing but is fully resistant to acute light damage. Hum Mol Genet, 24(15), 4417–4428. doi: 10.1093/hmg/ddv178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone M, Folkman L, Yang Y, Zhang P, Mort M, Cooper DN, … Zhou Y (2017). Investigating DNA-, RNA-, and protein-based features as a means to discriminate pathogenic synonymous variants. Hum Mutat, 38(10), 1336–1347. doi: 10.1002/humu.23283 [DOI] [PubMed] [Google Scholar]
- Lorenz B, Poliakov E, Schambeck M, Friedburg C, Preising MN, & Redmond TM (2008). A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Invest Ophthalmol Vis Sci, 49(12), 5235–5242. doi: 10.1167/iovs.07-1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, & Jensen TH (2015). Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol, 16(11), 665–677. doi: 10.1038/nrm4063 [DOI] [PubMed] [Google Scholar]
- Marlhens F, Griffoin JM, Bareil C, Arnaud B, Claustres M, & Hamel CP (1998). Autosomal recessive retinal dystrophy associated with two novel mutations in the RPE65 gene. Eur J Hum Genet, 6(5), 527–531. doi: 10.1038/sj.ejhg.5200205 [DOI] [PubMed] [Google Scholar]
- Marquez Y, Hopfler M, Ayatollahi Z, Barta A, & Kalyna M (2015). Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res, 25(7), 995–1007. doi: 10.1101/gr.186585.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier ID, Bernreuther C, Tilling T, Neidhardt J, Wong YW, Schulze C, … Schachner M (2010). Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J, 24(6), 1714–1724. doi: 10.1096/fj.09-140749 [DOI] [PubMed] [Google Scholar]
- Messiaen L, Callens T, De Paepe A, Craen M, & Mortier G (1997). Characterisation of two different nonsense mutations, C6792A and C6792G, causing skipping of exon 37 in the NF1 gene. Hum Genet, 101(1), 75–80. [DOI] [PubMed] [Google Scholar]
- Moiseyev G, Chen Y, Takahashi Y, Wu BX, & Ma JX (2005). RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A, 102(35), 12413–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura H, Fishman GA, Grover SA, Fulton AB, Berson EL, & Dryja TP (1998). Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A, 95(6), 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson SE (1985). Electrophysiology in pigment epithelial changes. Acta Ophthalmol Suppl, 173, 22–27. [DOI] [PubMed] [Google Scholar]
- Poliakov E, Strunnikova NV, Jiang JK, Martinez B, Parikh T, Lakkaraju A, … Redmond TM (2014). Multiple A2E treatments lead to melanization of rod outer segment-challenged ARPE-19 cells. Mol Vis, 20, 285–300. [PMC free article] [PubMed] [Google Scholar]
- Quan PL, Sauzade M, & Brouzes E (2018). dPCR: A Technology Review. Sensors (Basel), 18(4). doi: 10.3390/s18041271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM (2009). Focus on Molecules: RPE65, the visual cycle retinol isomerase. Exp Eye Res, 88(5), 846–847. doi: 10.1016/j.exer.2008.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, & Hamel CP (2000). Genetic analysis of RPE65: from human disease to mouse model. Methods Enzymol, 316, 705–724. [DOI] [PubMed] [Google Scholar]
- Redmond TM, Poliakov E, Kuo S, Chander P, & Gentleman S (2010). RPE65, visual cycle retinol isomerase, is not inherently 11-cis-specific: support for a carbocation mechanism of retinol isomerization. J Biol Chem, 285(3), 1919–1927. doi: 10.1074/jbc.M109.027458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, & Gentleman S (2005). Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci U S A, 102(38), 13658–13663. doi: 10.1073/pnas.0504167102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, … Pfeifer K (1998). Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat Genet, 20(4), 344–351. doi: 10.1038/3813 [DOI] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, & Haussler D (1997). Improved splice site detection in Genie. J Comput Biol, 4(3), 311–323. doi: 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, & Corn JE (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol, 34(3), 339–344. doi: 10.1038/nbt.3481 [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, & Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Lohr HR, Humphries P, Redmond TM, Seeliger MW, & Crouch RK (2005). Cone opsin mislocalization in Rpe65−/− mice: a defect that can be corrected by 11-cis retinal. Invest Ophthalmol Vis Sci, 46(10), 3876–3882. doi: 10.1167/iovs.05-0533 [DOI] [PubMed] [Google Scholar]
- Samardzija M, von Lintig J, Tanimoto N, Oberhauser V, Thiersch M, Reme CE, … Wenzel A (2008). R91W mutation in Rpe65 leads to milder early-onset retinal dystrophy due to the generation of low levels of 11-cis-retinal. Hum Mol Genet, 17(2), 281–292. doi: 10.1093/hmg/ddm304 [DOI] [PubMed] [Google Scholar]
- Shiga N, Takeshima Y, Sakamoto H, Inoue K, Yokota Y, Yokoyama M, & Matsuo M (1997). Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. The Journal of clinical investigation, 100(9), 2204–2210. doi: 10.1172/JCI119757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Moiseyev G, Chakraborty D, & Ma JX (2017). A Dominant Mutation in Rpe65, D477G, Delays Dark Adaptation and Disturbs the Visual Cycle in the Mutant Knock-In Mice. Am J Pathol, 187(3), 517–527. doi: 10.1016/j.ajpath.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, … Fairbrother WG (2017). Pathogenic variants that alter protein code often disrupt splicing. Nat Genet, 49(6), 848–855. doi: 10.1038/ng.3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg RH (1985). Interactions between the retinal pigment epithelium and the neural retina. Doc Ophthalmol, 60(4), 327–346. [DOI] [PubMed] [Google Scholar]
- Tanito M, Kaidzu S, Ohira A, & Anderson RE (2008). Topography of retinal damage in light-exposed albino rats. Exp Eye Res, 87(3), 292–295. doi: 10.1016/j.exer.2008.06.002 [DOI] [PubMed] [Google Scholar]
- Thompson DA, Gyurus P, Fleischer LL, Bingham EL, McHenry CL, Apfelstedt-Sylla E, … Gal A (2000). Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest Ophthalmol Vis Sci, 41(13), 4293–4299. [PubMed] [Google Scholar]
- Tucker BA, Cranston CM, Anfinson KA, Shrestha S, Streb LM, Leon A, … Stone EM (2015). Using patient-specific induced pluripotent stem cells to interrogate the pathogenicity of a novel retinal pigment epithelium-specific 65 kDa cryptic splice site mutation and confirm eligibility for enrollment into a clinical gene augmentation trial. Transl Res, 166(6), 740–749 e741. doi: 10.1016/j.trsl.2015.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlo KA, Gallaher SD, Vora R, Laski FA, & Iruela-Arispe ML (2010). When Cre-mediated recombination in mice does not result in protein loss. Genetics, 186(3), 959–967. doi: 10.1534/genetics.110.121608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney GK, Pei W, LaFave MC, Idol J, Xu L, Gallardo V, … Burgess SM (2015). High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res, 25(7), 1030–1042. doi: 10.1101/gr.186379.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veske A, Nilsson SE, Narfstrom K, & Gal A (1999). Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics, 57(1), 57–61. doi: 10.1006/geno.1999.5754 [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, & Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell, 153(4), 910–918. doi: 10.1016/j.cell.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cano M, & Handa JT (2014). p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochimica Et Biophysica Acta-Molecular Cell Research, 1843(7), 1248–1258. doi: 10.1016/j.bbamcr.2014.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chen Q, Zhao K, Wang L, Wang L, & Traboulsi EI (2001). Update on the molecular genetics of retinitis pigmentosa. Ophthalmic Genet, 22(3), 133–154. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhao L, Zhang Y, Ma W, Gonzalez SR, Fan J, … Wong WT (2017). Tamoxifen Provides Structural and Functional Rescue in Murine Models of Photoreceptor Degeneration. J Neurosci, 37(12), 3294–3310. doi: 10.1523/JNEUROSCI.2717-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Reme CE, Williams TP, Hafezi F, & Grimm C (2001). The Rpe65 Leu450Met variation increases retinal resistance against light-induced degeneration by slowing rhodopsin regeneration. J Neurosci, 21(1), 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CB, Chrenek MA, Feng W, Getz SE, Duncan T, Pardue MT, … Nickerson JM (2014). The Rpe65 rd12 allele exerts a semidominant negative effect on vision in mice. Invest Ophthalmol Vis Sci, 55(4), 2500–2515. doi: 10.1167/iovs.13-13574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, & He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS, 16(5), 284–287. doi: 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wen Y, & Guo X (2014). CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet, 23(R1), R40–46. doi: 10.1093/hmg/ddu125 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.