

Abstract
Histone lysine acetylation is critical in regulating transcription. Dysregulation of this process results in aberrant gene expression in various diseases, including cancer. The bromodomain, present in several proteins, recognizes promotor lysine acetylation and recruits other transcription factors. The bromodomain extra-terminal (BET) family of proteins consists of four conserved mammalian members that regulate transcription of oncogenes such as MYC and the NUT fusion oncoprotein. Targeting the acetyl-lysine-binding property of BET proteins is a potential therapeutic approach of cancer. Consequently, following the demonstration that thienotriazolodiazepine small molecules effectively inhibit BET, clinical trials were initiated. We thus discuss the mechanisms of action of various BET inhibitors and the prospects for their clinical use as cancer therapeutics.
Keywords: : BET inhibitors, BET protein, cancer, clinical trials, combination therapy, hematological malignancies, solid tumors, targeted therapy
Lay abstract
Addition of acetyl groups to histone proteins that are present in DNA is critical for proper copying of genetic information into producing proteins. This process is dysregulated in various diseases including cancer, resulting in continuous production of oncogenes – genes responsible for promoting cancer – such as NUT and MYC. Bromodomain extra-terminal (BET) proteins mediate acetyl addition, and as a result, transcription. BET inhibitors are small molecules that block the action of BET proteins and the transcription of oncogenes. In this review, we discuss the different available BET inhibitors, the way they act, and their potential use in clinical settings for cancer treatment.
Acetylation of histone lysine is arguably the most dynamic of the protein post-translational modifications, which participates in structural changes in chromatin and, in addition, regulates various cellular processes, including protein conformations and interactions [1]. Given that histone lysine acetylation has been associated with transcriptional activation [2], one would logically conclude that misregulation of histone acetylation could result in aberrant expression of oncogenes and, consequently, initiation and/or maintenance of cancer [3]. Three types of proteins regulate lysine acetylation and consequent effects, namely, bromodomain (BRD) proteins [4,5]; histone acetyltransferases; and histone deacetylases and sirtuins [6–9]. Acetyltransferases are the ‘writers’ of histone acetylation, and BRDs are the ‘readers’, whereas deacetylases and sirtuins are the ‘erasers’ [9].
The acetyl-lysine-binding BRDs belong to a family of evolutionarily conserved protein modules originally identified in chromatin-associated proteins and are a critical component in the functionality of histone acetyltransferases [10]. BRDs tether those to transcriptional sites and regulate the activities of multi-subunit chromatin remodeling complexes [11], allowing for involvement in diverse mechanisms underlying a range of cellular events [12].
Bromodomain and extra-terminal (BET) proteins belong to the BRD proteins family and share a common domain architecture comprising two N-terminal bromodomains (BD1 and BD2) that interact with acetylated lysine residues on histone tails and an extra-C terminal domain. The BET family consists of four conserved mammalian members, namely, BRD containing 2 (BRD2), BRD3, BRD4 and BRDT [13]. Filippakopoulos et al. [14] were the first to characterize a thieno-triazolo-1,4-diazepine histone-binding module inhibitor, JQ1, a cell-permeable small molecule that competitively and specifically binds to the BD1 and BD2 bromodomains with high affinity; this then paved the way for the further development of other BET inhibitors, now being assessed clinically across a range of cancers.
Biology of BET proteins
Although BRDT is primarily expressed in germ cells, BRD2, 3 and 4 are ubiquitously expressed and interact with acetylated histone tails to regulate transcription. Within the BRD family, sequence variations in the interhelical ZA and BC loops of the distinct structural ‘BRD fold’ result in different protein-binding sites and affinities [15]. BRD4 recruits PTEF-b to sites of active transcription of growth-promoting genes [16]. During elongation, Ser2 of RNA polymerase II is phosphorylated by PTEF-b, which depends on BRD4 for its nuclear localization and activation of its CDK9 kinase subunit [17]. Thus, BRD4 is involved in the conversion of basal transcription to high rates of active elongation by RNA polymerase II (Figure 1). In addition, the ET domain of BRD4 independently recruits transcriptional activators such as NSD3 (a histone methyl transferase), JMJD6 (a histone arginine demethylase) and CHD4 (the catalytic subunit of the NuRD chromatin remodeler) [18]. Unlike BRD4, BRD2 provides a scaffold on chromatin for histone acetyltransferases, histone deacetylases and E2F, thereby coupling histone acetylation to transcription in a PTEF-b-independent manner [16]. BRD3 regulates transcription via the E2F-Rb pathway and also targets GATA1 to chromatin [19].
Figure 1. . Biology of bromodomain and extra-terminal inhibitors and their role in cancer therapy.

BET protein binds to acetylated histones and recruits, via its BRD4 domain, PTEF-b to sites of active transcription of growth-promoting genes such as MYC and NUT. In addition, the ET domain of BRD4 independently recruits transcriptional activators such as NSD3, JMJD6 and CHD4 to further increase rates of transcription. BET inhibitors block the initial binding of BET proteins to acetylated histones, and thus halts the transcriptional cascade of oncogenes.
BET: Bromodomain and extra-terminal; PTEF-b: Positive transcriptional elongation factor complex.
BET proteins also act as mitotic bookmarks and regulators of the cell cycle. BRD4 remains associated with acetylated H4K5 on chromatin during mitosis, leading to rapid decompaction of adjoining chromatin and transcription [20]. BRD4 marks the transcription start sites of many M/G1 genes and promotes progression of the cell cycle from G1 to S and G2 to M phases [21,22]. In addition, E2F1 and E2F2, the key transcriptional regulator of S phase, are associated with BRD2 [23].
BET proteins in cancer
As mentioned previously, altered histone acetylation is associated with aberrant transcription of cancer-related genes. BET proteins promote aberrant expression of the MYC oncogene in various hematologic malignancies such as mixed-lineage leukemia, acute myeloid leukemia (AML), Burkitt's lymphoma and Burkitt-like lymphoma; in these cells, disruption of BET binding significantly reduces cellular proliferation and induces apoptosis [24–28]. MYC overexpression is also documented in a number of solid tumors including lung, ovary and breast cancer [29]. BRD4 recruits a histone methyltransferase to target genes in ER-positive cells, thus constitutively activating estrogen signaling, a critical pathway in breast cancer tumorigenesis [30]. BRD4 also forms super enhancer complexes with the Mediator complex, a multiprotein transcriptional regulator (containing the CDK8/CDK19/MED12/MED13 kinase module), via which it regulates the expression of oncogenic drivers such as MYC, a super-transcription factor deregulated in a majority of cancers [31]. BET proteins are overexpressed in glioblastoma and in primary and metastatic melanoma [32,33]. Consequently, inhibition of BET activity using small-molecule inhibitors might play a significant role in the treatment of these cancers. Furthermore, BET family proteins have been identified in oncogenic rearrangements comprising highly oncogenic fusion proteins that play critical roles in the development of several types of cancer. For example, the BRD-NUT in-frame fusion proteins (primarily t(15;19) involving BRD4) drive carcinogenesis in NUT midline carcinoma (NMC), an aggressive squamous cell carcinoma [34]. Notably, MYC is a downstream target of BRD/NUT [35]. Details regarding the involvement of BET proteins in different types of cancer and the efficacy of using BET inhibitors as cancer therapeutics will be discussed below.
Targeted therapy using BETs
Small-molecule BRD inhibitors were first identified based on structural characterization of the BRD acetyl-binding pocket and nuclear magnetic resonance spectroscopy-based screening of numerous candidate compounds [6,36]. These studies focused on the acetyltransferase CREB-binding protein, which acetylates and modulates p53 tumor-suppressor protein stability and function during DNA damage repair [36,37], and although they identified chemical compounds with low affinity for the BRD pocket and therefore unsuitable for clinical use, they did provide proof-of-principle that BRD inhibition was feasible [37]. Subsequently, multiple small-molecule higher-affinity inhibitors of BETs have been developed. The thienotriazolodiazepines, JQ1 and I-BET, both interact with NF-κB and induce apoptosis in drug-resistant leukemia [38]. I-BET762 mimics acetylated histones to disrupt chromatin complexes. PFI-1 is a highly selective dihydroquinazoline-2-one inhibitor, which blocks the interaction of BET bromodomains with acetylated histone tails. Picaud et al. [39] used cocrystal structures to show that the PFI-1 inhibitor acts as an acetyl-lysine mimetic that efficiently occupies the acetyl-lysine binding site in BRD2 and BRD4, which subsequently causes G1 arrest, downregulates MYC and Aurora B kinase, and induces apoptosis. The quinazolone RVX-208 is a BET inhibitor that shows highest selectivity for BD2 of BRD2 and BRD3. We will discuss the mechanism of action and clinical utilities of each of these inhibitors.
Mechanisms of action of BET inhibitors & preclinical studies
Chemical structures and inhibitory activities in preclinical studies of the various BET inhibitors are summarized in Table 1.
Table 1. . Preclinical (in vitro) inhibitory activity of bromodomain and extra-terminal inhibitors with their respective clinical structures.
| BET inhibitor | Ki† (nM) | IC50¶ (nM) | Chemical structure |
|---|---|---|---|
| JQ1 | 10.7 ± 1.1 | 77 | 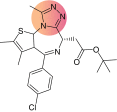 |
| OTX015 | 6.0 ± 0.3 | 75–650 | 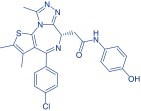 |
| BET-d246 | <1 | <10 | 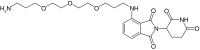 |
| ABBV-075 | 1-2.2 | <100 | 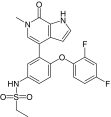 |
| I-BET 151 | 20–100 | 790 | 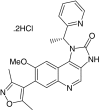 |
| I-BET 762 | 30.3 | 35 | 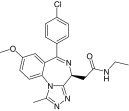 |
| CPI 203 | 50–90 | 37 | 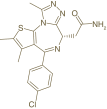 |
| PFI-1 | 49 × 103 | 220 | 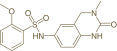 |
| RVX-208‡ | 195 | 510–600 | 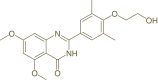 |
| Dinaciclib§ | 37 × 103 | 50–90 | 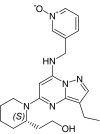 |
†to BD2 of BRD4 domain unless otherwise specified.
‡to BD2 of BRD3.
§to BRDT.
¶IC50 variation within ranges reflect variability of tested cell lines.
BET: Bromodomain and extra-terminal.
JQ1: a BRD2/4 inhibitor
Hematological malignancies
JQ1, a novel thieno-triazolo-1,4-diazepine, initially identified as a small molecule that strongly inhibits NMC, competitively binds to the acetyl-lysine binding motif effecting significant antiproliferative agent in BRD4-dependent cell lines. Screening of various inhibitors of BRD4-NUT showed higher activity of JQ1 compared with other clinical drugs in NMC, suggesting its translational potential [40].
As mentioned earlier, most common cancers (>50%) exhibit MYC deregulation [41]. Several studies have validated c-MYC as a therapeutic target [42–44], including transgenic mouse models where suppression of MYC expression resulted in tumor regression. Thus far, a direct targeting approach has been elusive. However, insofar as MYC transcription is associated with local and global changes in histone acetylation [45,46], a feasible, alternative, albeit indirect, way of targeting MYC transcription is through alteration of its histone acetylation status using a BET inhibitor (BETi). Significantly, using Raji cells, Mertz et al. showed BRD2 and BRD4 release from a site upstream of the P1 MYC promoter in conjunction with JQ1-mediated silencing of MYC [27]. In multiple myeloma (MM), characterized by dysregulation of multiple factors due in large part to gene rearrangements and translocations of MYC [47], BRD4 was found to be enriched at IgH (immune heavy chain) enhancers rearranged at the MYC locus. Furthermore, JQ1 exhibits considerable antiproliferative effect, cell cycle arrest and cellular senescence in three murine models of MM, emphasizing the importance of BET BRD inhibition in MM and in other malignancies with pathological c-MYC activation [48]. As to whether or not genomic alterations are sine qua non for BETi effectiveness, there are data demonstrating effective JQ1-mediated MYC silencing in both MYC amplified and unamplified cell lines [27]. Indeed, although MYC plays a central oncogenic role in T-ALL, genomic alterations are rarely seen. Rather, MYC overexpression is driven via NOTCH-driven transcriptional or PTEN/AKT/PI3K post-translational changes. Exposure of T-ALL cell lines to JQ1 resulted in downregulation of MYC RNA levels and protein expression [49]. The mechanism and effectiveness of BETi on MYC expression is highly cell type specific.
JQ1 can inhibit growth and induce apoptosis of human AML cells, including those expressing FLT3-ITD (FMS-like tyrosine kinase 3-internal tandem duplication), a mutant proto-oncogene. Cotreatment of JQ1 and a FLT3 inhibitor, FLT3-TKI, significantly reduces the expression of c-MYC, BCL2 and CDK4/6, while synergistically inducing apoptosis of cultured and primary CD34+ human AML blast progenitor cells. Furthermore, cotreatment with JQ1 and the pan-histone deacetylase inhibitor panobinostat synergistically induced apoptosis of FLT3-TKI-resistant cells [50]. Activation of intrinsic pathway caspase 3/7, but not extrinsic pathway caspase 8, after JQ1 treatment indicated the selective functional involvement of the former pathway [24].
The transcription factor STAT5 is constitutively active in most leukemia and drives the expression of genes involved in self-renewal, proliferation and survival. BRD2 is a critical mediator of STAT5 function and JQ1 possibly mediates its effect via STAT-5-dependent pathways. Indeed, JQ1 treatment reduced STAT-5-dependent transcription of heterologous reporters and endogenous genes and showed strong synergy with tyrosine kinase inhibitors (TKI) in inducing apoptosis in leukemia cells [51].
Ott et al. showed that JQ1 reduced the viability of B-ALL cell lines with high-risk cytogenetics, such as those with CRLF2 rearrangements, which are observed in approximately 10% of all B-ALL cases. In these cell lines, the IL7 receptor (IL7R) dimerizes with CRLF2 to drive proliferation and inhibit apoptosis via JAK1, JAK2 and STAT5 signaling. Administration of JQ1 downregulated MYC and inhibited BRD4 binding to the MYC promoter, while further depleting BRD4 from IL7R promoter, thereby downregulating IL7R expression and reducing JAK2 and STAT5 phosphorylation. Murine xenografts showed increased survival after JQ1 treatment that was concomitant with MYC suppression and inhibition of STAT5 phosphorylation [28].
As an example of MYC-independent targeting, genome-wide analysis of JQ1-induced transcriptional response in MYC-driven B-cell lymphoma revealed reduced BRD4 occupancy at the IFNγ PD-L1 ligand Cd274 without affecting MYC occupancy, resulting in a reduction of the CD274 mRNA level. Combined treatment with JQ1 and PD-L1 antibodies showed synergistic responses in mice with MYC-driven lymphoma [52].
Solid tumors
Nervous system tumors
Medulloblastoma is the most common malignant brain tumor in children and is characterized by amplification of MYC, MYCN and MYCL. JQ1 treatment significantly reduced cell proliferation and induced apoptosis and senescence in different human medulloblastoma cell lines (HD-BM3, ONS-76 and D-341) and effected inhibition of self-renewal and induction of apoptosis in stem cells [53,54]. JQ1 also reduced the expression of MYC-associated proteins such as Cyclin D1 and E2F1 [55]. Similar results were also observed in human neuroblastoma, where JQ1 targets BRD4 to regulate MYCN expression and induce cell death [52].
Glioblastoma multiforme (GBM), both highly aggressive and overwhelmingly lethal, is the most commonly seen brain tumor in adults. Cheng et al. reported that JQ1 induced marked G1 cell cycle arrest and apoptosis with significant changes in the expression of important GBM genes; c-MYC, p21CIP1/WAF1, hTERT, Bcl- 2 and BclxL. In vivo, orthotopic GBM tumors showed significant growth repression following treatment with JQ1 [56].
Diffuse Intrinsic Pontine Glioma (DIPG) is a fatal common brainstem tumor of children with limited effective treatment options. Owing to high MYCN levels, DIPG was treated sequentially with the bromodomain inhibitor JQ1, which targets MYCN, followed by MRK003, a potent and selective gamma secretase inhibitor that downregulates intracellular NOTCH1. This dual targeting inhibited DIPG growth and induced apoptosis, highlighting the efficacy of this therapeutic regimen [57].
Colorectal cancer
Colorectal carcinoma is currently the third leading cause of cancer-related deaths in the USA. The molecular prototype of oncogenic evolution, it arises in a step-wise manner from discrete genetic and epigenetic dysregulations including global hypermethylation (expressed as the CpG island methylator phenotype [CIMP]), which silences suppressor genes in 20% of patients (comprising 7% microsatellite stable tumors and 79% MSI tumors) [58,59]. CCAT1 is a 2628 nucleotide-IncRNA (long noncoding RNA) that appears to be transcribed off the MYC super-enhancer [60]. Although CCAT1 expression does not correlate with CIMP status (i.e., CIMP+ vs CIMP-), there is a correlation of CCAT1 RNA with BRD4 binding at the CCAT1 super-enhancer site [61] despite the lack of association with MYC expression. Importantly, JQ1 selectively reduces MYC expression in CCAT1 expressing cells. In vivo, a recent study by Zhang et al. treated the SW480 colon cancer mouse xenografts with JQ-1 [62] and demonstrated a significant reduction in tumor growth, improved mouse survival and induction of apoptosis. Interestingly, the study further showed that inhibition of proliferation was mediated via suppression of Wnt/β-catenin signaling and miR-21.
Breast cancer
JQ1 inhibits BRD3- and BRD4-mediated recruitment of WHSC1 to the ERα gene, thereby suppressing the classic estrogen receptor-α signaling pathway, resulting in the growth suppression of tamoxifen-resistant breast cancer cells in culture [30]. In addition, resistance to second-line everolimus (mTORC1 inhibitor) and exemestane (aromatase inhibitor) in women with ER+ breast cancer was shown to occur through upregulation of MYC driving mTORC1 resistance [63]. JQ1 restored everolimus sensitivity, and a combination of everolimus and JQ1 led to synergistic growth inhibition in 3D Matrigel cultures and xenograft models [63], suggesting at least partial MYC signaling pathway cooptation of the inhibited mTORC1 pathway.
Similarly, Borbely et al. [64] showed that JQ1 inhibits proliferation and induces apoptosis of both triple-negative and ER+ breast cancer cells. Treatment with JQ1 or the histone deacetylase inhibitor mocetinostat induced global changes in gene expression, resulting in suppression of genes involved in cell-cycle regulation. Combined treatment of JQ1 and mocetinostat synergistically reduced breast cancer cell viability. Furthermore, the combination treatment increased the expression of ubiquitin-specific protease 17 family of deubiquitinating enzymes, which subsequently attenuated the Ras/MAPK pathway and reduced cell viability [64]. JQ1 also synergistically reduced proliferation of triple-negative breast cancer (TNBC) cells with volasertib, an inhibitor of PLK1 (PLK1 is overexpressed in TNBC) and decreased the expression of stem cell markers [65]. Hypoxia is known to stimulate TNBC by triggering metabolic adaptation, angiogenesis and metastasis. da Motta et al. observed that JQ1 modulated the expression of hypoxia-inducible genes, including CA9 and VEGF-A, hindered HIF binding to CA9 promoter, inhibited TNBC growth and prevented xenograft vascularization [66].
JQ1 exerted antitumor activity in a transgenic mouse model of luminal B human breast cancer. The antitumor phenotype was accompanied by downregulation of oncogenes such as the breast carcinoma-amplified sequence 1 and the PDZ domain-containing 1 [67].
The HER2/ERBB2 oncogene is overexpressed in approximately 25% of breast cancer cases. HER2-positive breast cancer patients are treated with lapatinib, a small-molecule kinase inhibitor that prevents downstream MAPK signaling and AKT activation. However, patients develop resistance to therapy by adapting to lapatinib; this adaptation mechanism has been attributed to phosphorylation of ERBB3, and the subsequent induced expression of IGF1R, DDR1, MET and FGFRs. Stuhlmiller et al. [68] showed that growth of HER2-positive breast cancer cells was inhibited by both JQ1 and I-BET762. Treatment of SKBR-3 cells with JQ1 prevented lapatinib-induced phosphorylation and expression of ERBB3. JQ1 also suppressed lapatinib-induced expression of FGFR2, DDR1, IGF1R, pFAK, pSFK and pPKCd across multiple cell lines. Combination treatment with JQ1 and lapatinib enhanced the growth inhibitory effect.
The potent pan-BET bromodomain inhibitor JQ1 was key to delineating mechanism of action and establishing proof of principle of BET inhibition. However, JQ1 has a very short half-life and dose concentrations required to mediate single agent activity are above physiologic safety levels in vivo [69]. Furthermore, although its efficacy may be due in part to a considerable lack of discrimination between BD1 and BD2 across the BET family members, this broad target spectrum limits JQ1 specificity, and may narrow its therapeutic window. These considerations, specificity, affinity, potency and pharmacodynamics have led to development of alternative compounds.
OTX015
OTX015, an oral BET inhibitor targeting BRD2 and 4 [70], is now in Phase Ib clinical trial in patients with selected advanced solid tumors. In preclinical studies, OTX015 was observed to possess antiproliferative effects in lymphoma models and affects MYC, NFKB, JAK/STAT pathways, cell cycle regulation and chromatin structure; it increased HEXIM1 mRNA level in AML and ALL cell lines, which is responsible for sequestering the active form of PTEF-b [70]. Although cytostatic in function, OTX015 induced apoptosis in a genetically distinct subgroup of cells derived from activated B-cell-like diffuse large B-cell lymphoma. OTX015 showed in vitro synergism with several anticancer agents, especially with mTOR and BTK inhibitors [66,71] and with azacytidine and panobinostat, to retard the growth of leukemic cell lines [70].
BET-d246
TNBC remains clinically challenging owing to lack of effective targeted therapy. Recently, BET-d246, using a Proteolysis Targeting Chimera strategy conjugating JQ1 with a ligand for the Cullin-4A ligase, was found to exhibit superior potency and antitumor activity against human TNBC cells. MCL1 is a critical downstream effector for BET degraders, which synergized with small-molecule inhibitors of BCL-xL in triggering apoptosis. BETd-246, and BETd-260 (an analog), effectively depleted BET proteins in multiple murine xenograft models of human breast cancer tumors and exhibited strong antitumor activities at well-tolerated doses [72].
ABBV-075
ABBV-075 is a potent and selective BET family bromodomain inhibitor targeting both BDs of BRD2, BRD4 and BRDT that recently entered Phase I clinical trials to evaluate its safety and pharmacokinetics in subjects with advanced solid and hematological tumors. Preclinical evaluations indicated that this compound inhibits proliferation of a range of cancer types, including hematological malignancies and solid tumors. In addition to MYC deregulation, ABBV-075 triggers G1 phase arrest and apoptosis (via the intrinsic pathway) primarily in hematological malignancies, for example, AML, non-Hodgkin lymphoma and MM cells. It exhibits synergy with the BCL-2 inhibitor venetoclax in preclinical models of AML, including xenograft mouse models representing AML, MM and NHL, where ABBV-075 produced efficient antitumor efficacies. Its proapoptotic effectiveness in hematological versus solid tumors may depend on the quantitative levels of BCL2 and Bim as well as the BCL2/Bim ratio [73]. Unlike the lack of in vitro synergy between ABBV-075 and azacitidine, cytarabine or bortezomib, xenograft models of AML and MM exhibited enhanced activities of azacitidine and bortezomib with relatively low doses of ABBV-075. In fact, ABBV-075 at 0.25 mg/kg per day, when combined with bortezomib caused a deeper tumor response than bortezomib alone and a longer delay of tumor progression after treatment withdrawal. Similarly, combining azacitidine with ABBV-075 at 0.67 mg/kg per day resulted in a tumor response that was superior to monotherapies of either agent alone [73].
I-BET151
I-BET151, a pan-BET inhibitor, arrests cell cycle progression and decreases proliferation of GBM cells in vitro and in vivo. Single molecule sequencing revealed that BET proteins target the GBM-specific IncRNA, HOTAIR. I-BET151 decreases HOTAIR transcription and increases the expression of other GBM-downregulated noncoding RNAs [32]. I-BET151 also suppressed the Hedgehog activity-dependent growth of medulloblastoma cells, both in vitro and in vivo [74]. In addition, I-BET151 has been shown to exhibit significant antitumor activity in murine models of NUT midline carcinoma, MM, mixed-lineage leukemia and ALL, lung cancer and malignant brain tumor. In AML with mutations in NPM1 (encoding nucleophosmin), I-BET151 abrogates the HOX-independent transcription program [75], and represses the MYC-dependent program in myeloma, leading to strong antiproliferative effect in vitro and in vivo [76].
I-BET762
GSK525762A (I-BET762) was optimized from a chemical hit derived from a phenotypic screen attempting to identify small molecules that enhance ApoA1 expression. I-BET762 treatment of samples derived from patients with NMC resulted in terminal differentiation and proliferation arrest of malignant cells [77] and significantly reduced MYC expression in both prostate cancer cells and a patient-derived tumor model associated with growth retardation and reduction of tumor burden [78]. In neuroblastoma models, I-BET 762 triggered apoptosis via BET inhibition of both MYCN and Bcl-2 [79]. It was also shown to affect myeloma cell proliferation and resulted in a survival advantage in a systemic myeloma xenograft model [75].
CPI203
CPI203 is an analog of JQ1 with superior oral and intraperitoneal bioavailablity. Mantle cell lymphoma, a type of non-Hodgkin's lymphoma, is characterized by plasmacytic differentiation features such as IRF4 and Blimp-1 upregulation, and is usually treated with bortezomib. The development of bortezomib resistance in mantle cell lymphoma may limit its therapeutic efficacy and result in increased tumorigenicity. Combined treatment of bortezomib-resistant cells with CPI203 and lenalidomide downregulated MYC, an IRF4 target, and synergistically induced cell death [80]. Addition of CPI203 to lenalidomide therapy further decreased tumor burden in mice, which was accompanied by simultaneous MYC and IRF4 downregulation and induction of apoptosis [80]. Similar findings were described in in vitro and in vivo myeloma [81].
PFI-1
PFI-1, a novel dihydroquinazolinone BET chemical probe, binds to the BET bromodomain in a manner that is chemically distinct from those of previously reported BET inhibitors [39]. Aurora kinases are highly expressed in diverse cancer types and are also frequently upregulated in leukemia [82]. MYC and the critical mitotic entry Aurora A and B isoforms interact in a positive reciprocal feedback loop [39]. PFI-1 downregulates Aurora B kinase in leukemic cells and induces caspase-dependent apoptosis and differentiation. PFI-1 and JQ1 dissociate BRD4 from HOXA9, a marker of poor prognosis in patients with AML, and promotes differentiation [83].
BRD3 inhibitors (RVX-208)
BRD3 is primarily involved in recruiting GATA1 to chromatin in hematopoietic cells and regulating differentiation of erythroid, megakaryocyte and mast cell lineages. BRD3 inhibitors have not been extensively studied. However, pan-BET inhibitors such as JQ1 and I-BET151 have been observed to target BRD3 in NMC and leukemia. RVX-208, a derivative of the plant polyphenol resveratrol, binds to BD2 of BRD3 [84] and upregulates ApoA1 which plays an important role in hepatocellular carcinoma. Given that JQ1 strongly upregulates ApoA1 in HepG2 cells, it can be used in combination with RVX-208 as a multitarget inhibitor in hepatocellular carcinoma [85].
Dinaciclib (BRDT inhibitor)
Dinaciclib, an inhibitor of CDKs 1, 2, 5 and 9 is in Phase III clinical trial for leukemia [86]. Cocrystallization studies showed interaction with one of the BRDT BDs, providing support for further development of ATP site-directed kinase inhibitors and bromodomain inhibitors [87].
Clinical trials of BET inhibitors
The safety, tolerability, pharmacokinetics, pharmacodynamics and clinical activity of BET inhibitors are currently being investigated in clinical trials for several cancers, including NMC (OTX015 [NCT02259114, developed by OncoEthix] [71] and GSK525762/I-BET762 [NCT01587703, developed by GlaxoSmithKline]), refractory AML and elastodynamics syndrome ([NCT02308761], lymphoma [NCT01949883 for CPI-0610, developed by Constellation Pharmaceuticals], MM [NCT02157636 for CPI-0610, developed by Constellation Pharmaceuticals] and hematological malignancies [NCT01713582 for OTX015, developed by OncoEthix]). Additional single and combination agent trials in TNBC, ER+ breast cancers, small-cell and non-small-cell lung cancers, castration-resistant prostate cancer, pancreatic ductal adenocarcinoma, colorectal cancer, neuroblastoma and MYCN-driven solid tumors are summarized in Table 2.
Table 2. . Clinical trials of bromodomain and extra-terminal inhibitors.
| Target | BETi agent | Target population | Study Phase | Sponsor | Trial no. |
|---|---|---|---|---|---|
| BRD2/3/4, BRDT | ABBV-075 | Solid tumors, AML, MM, NSCLC, breast cancer | I | Abbvie | NCT02391480 |
| FT-1101 | AML, MDS | I | Forma Therapeutics/Celgene | NCT02543879 | |
| GSK525762/I-BET762 | Hematological malignancies (AML, MM, BCL-2/MYC-driven cohorts NMC, SCLC, NSCLC, CRC, NB, CRPC, TNBC, ER-positive breast cancer |
I I/II (open) |
GlaxoSmithKline GlaxoSmithKline |
NCT01943851 NCT01587703 |
|
| INCB057643 | Any advanced/recurrent malignancy | I/II | Incyte Corporation | NCT02711137 | |
| ZEN003694 | Metastatic CRPC Metastatic CRPC + enzalutamide |
I I |
Zenith Epigenetics |
NCT02705469 NCT02711956 |
|
| BRD2/3/4 | OTX015/MK-8628 | AML, DLBCL Solid tumors (NMC, TNBC, CRPC, NSCLC) GBM NMC harboring BRD4-NUT, TNBC, NSCLC harboring ALK-KRAS fusion, CRPC, PC AML, DLBCL, ALL, MM |
I I Phase II (Withdrawn) I I |
Merck/Mitsubishi Tanabe Merck/Mitsubishi Tanabe Merck/Mitsubishi Tanabe OncoEthix GmbH OncoEthix GmbH |
NCT02698189 NCT02698176 NCT02296476 NCT02259114 NCT01713582 |
| BRD2/4 | GSK2820151/I-BET151 | Solid tumors | I | GlaxoSmithKline | NCT02630251 |
| BRD2 | CC-90010 | Advanced solid tumors, relapsed/refractory NHL | I | Celgene | NCT03220347 |
| BRD4 | CPI-0610 | Lymphoma MM AML, ALL, CML Peripheral nerve sheath tumors II (not yet open) |
I I I I |
Constellation Pharmaceuticals/Roche |
NCT01949883 NCT02157636 NCT02158858 NCT02986919 |
| PLX51107 | Solid tumors, lymphoma, AML, MDS | I | Plexxicon | NCT02683395 | |
| ABBV-744 | AML and prostate cancer | I | Abbvie | NCT02391480 | |
| Undisclosed | BAY1238097 | Solid tumors and lymphoma | I (Terminated) | Bayer | NCT02369029 |
| BI 894999 | Solid tumors and NHL | I | Boehringer Ingelheim | NCT02516553 | |
| BMS-986158 | Solid tumors | I/II | Bristol-Myers Squibb | NCT02419417 | |
| GS-5829 | DLBCL, peripheral T-cell lymphoma, solid tumors ERþ breast cancer in combination with exemestane or fulvestrant Metastatic CRPC as single agent and in combination with enzalutamide |
I I/II I/II |
Gilead |
NCT02392611 NCT02983604 NCT02607228 |
|
| INCB054329 | Solid tumors, hematological malignancies | I/II | Incyte Corporation | NCT02431260 | |
| RO6870810/TEN-010 | MDS, AML | I | Hoffman-LaRoche | NCT02308761 | |
ALL: Acute lymphoblastic leukemia; AML: Acute myeloid leukemia; BETi: Bromodomain and extra-terminal inhibitor; CML: Chromic myeloid leukemia; CRPC: Castration-resistant prostate cancer; DLBCL: Diffuse large B-cell lymphoma; GBM: Glioblastoma multiforme; MDS: Elastodynamics syndrome; MM: Multiple myeloma; NB: Neuroblastoma; NHL: Non-Hodgkin lymphoma; NSCLC: Non-small-cell lung carcinoma; SCLC: Small-cell lung carcinoma; TNBC: Triple-negative breast cancer.
Trials to date have yielded mixed results. A Phase Ib trial of OTX015 in 47 patients with advanced solid tumors demonstrated four partial responses (PRs) and seven cases of stable disease (SD) for 4–8 months. Toxicities included Grade 3 and 4 thrombocytopenia, Grade 3 anemia and Grade 3 fatigue [88]. In another study, BAY1238097 treatment in eight patients with advanced solid tumors or NHL was stopped because of development of grade 3 headache, vomiting and low-back pain, respectively, at sub-therapeutic doses [89]. Of the four patients enrolled in an OTX015 trial for NMC, two achieved CT confirmed PRs, one of whom showed SUV normalization on PET scan and a third SD all with longer than expected survivals [90].
The seed product JQ1 is not being tested in clinical trials due to its short half-life. Open trials include an Incyte Corporation-sponsored Phase I/II of INCB054329 in subjects with advanced malignancies, and Tensha Therapeutics Phase I, multicenter, open-label study of TEN-010 (structurally similar to JQ1 but with longer half-life) for patients with advanced solid tumors (Table 2). GlaxoSmithKline has a dose escalation study of GSK2820151 in five patients with advanced or recurrent solid tumors, whereas Bayer has a Phase I study of BAY 1238097 (which targets the NFKB/TLR/JAK/STAT signaling pathways, MYC and E2F1-regulated genes, cell cycle regulation and chromatin structure [91] for patients with advanced malignancies). OTX015 has been tested in brain tumors in a nonrandomized, multicenter Phase IIa trial, which tested dose optimization in patients with recurrent GBM following failure of front-line therapy; the trial was terminated after 1 year (Table 2). The results of a first-in-human study of ABBV-075 (Mivebresib) in patients with relapsed/refractory AML have recently been published. Besides showing good tolerability in patients, the preliminary results also show antileukemic effects: bone marrow blast count was 50% of baseline in 4/10 evaluable patients, one patient reached complete remission and the median overall survival for all patients was 3.2 months [92].
Combination therapy
BET inhibitors have been successfully used in combination with epigenetic inhibitors [49,64] (SAHA, mocetinostat), kinase inhibitors [93], immune checkpoint inhibitors [52] (PD-L1), cell cycle inhibitors [94,95], DNA damage repair agents [96] and chemotherapeutic drugs such as vincristine [49], venetoclax [97], bortezomib [73], azacitidine [73] and lapatinib [68] (Table 3), some of which have been discussed earlier.
Table 3. . Combination therapy using bromodomain and extra-terminal inhibitors.
| Author (year) | Combination | Model used | Outcome |
|---|---|---|---|
|
BET inhibitor + epigenetic inhibitor Loosveld et al. (2014) [49] Borbely et al. (2015) [64] Stratikopoulos et al. (2015) [93] Fiskus et al. (2014) [50] Bauer et al. (2018) [99] |
HDAC inhibitor SAHA Mocetinostat Kinase inhibitor PI3K inhibitor + JQ1/MS417 JQ1 + FLT3-TK1 Ponatinib + JQ1/dBET1 |
Ex vivo screening assay Mice xenografted with human primary T-ALLs Breast cancer cells Broad range of cancer cell lines NOD/SCID mice injected with OCIAML3 or MOLM13 cells Colon (HCT116, HT29), breast (MCF-7, SKBR3) and ovarian (A2780, SKOV3) cancer cells |
Reduces MYC expression Reduced breast cancer cell viability via induction of ULP17 Combined treatment-induced cell death, tumor regression and clamped PI3K signaling in various cancer cell lines Significantly attenuates the expression of c-MYC, BCL2 and CDK4/6, while synergistically inducing apoptosis of cultured and primary CD34+ human AML blast progenitor cells Sensitizes colon, breast and ovarian cancer cells to JQ1 and dBET1; combination of these two types of inhibitors increased apoptosis and downregulated MYC |
|
BET inhibitor + cell cycle inhibitor Tontsch-Grunt et al. (2016) [94] Bolin et al. (2016) [95] |
BI 894999 and CDK9 inhibitors Alvocidib and LDC000067 Unidentified BET and CDK2 inhibitors |
Mice injected with MV-4-11, THP-1 and MOLM13 cells Mice transplanted with medulloblastoma |
Synergistic inhibition of tumor growth and MYC expression Synergistic effect on tumor growth inhibition and improvement of overall survival |
|
BET inhibitor + immune check point inhibitor Hogg et al. (2017) [52] |
JQ1 and PD-L1 antibodies |
Mice with Myc-driven lymphoma |
Synergistic response in mice with lymphoma |
|
BET inhibitor + DNA damage repair inhibitors Muralidharan et al. (2016) [96] |
RVX2135 and ATR inhibitor AZ20 |
λ820 and λ2749 murine Myc-induced lymphoma xenografts | Synergistic delay in tumor onset in λ820 xenografts; synergistic WBC reduction and improved survival in λ2749 xenografts |
|
BET inhibitor + chemotherapeutic drugs Loosveld et al. (2014) [49] Bui et al. (2017) [73] Bui et al. (2017) [73] Bui et al. (2017) [73] Stuhlmiller et al. (2015) [68] |
JQ1+Vincristine ABBV-075 +Venetoclax ABBV-075 +Bortezomib ABBV-075 + Azacitidine JQ1 + Lapatinib |
Ex vivo screening assay Mice xenografted with human primary T-ALLs AKM1 AML xenografts AKM1 AML xenografts AKM1 AML xenografts ERRB2-positive breast cancer cell lines resistant to lapatinib |
Reduces MYC expression Induced apoptosis via modulation of the intrinsic apoptotic pathway, tumor regression Synergistic effect on tumor regression with better tolerance by xenografts than BET inhibitor monotherapy Combination treatment with JQ1 and lapatinib showed stronger growth inhibitory effect of ERRB2-positive breast cancer, reduced levels of ERBB3, IGF1R, DDR1, MET and FGFR |
ALL: Acute lymphoblastic leukemia; AML: Acute myeloid leukemia; ATR: Ataxia Telangiectasia Mutated and RAD-3 related ; BET: Bromodomain and extra-terminal; FGFR: FGF receptor; HDAC: Histone deacetylase; MET: Tyrosine-protein kinase; NOD/SCID: Nodulation Factor/Severe Combined Immune Deficiency; SAHA: Suberanilohydroxamic acid; WBC: White Blood Cells.
Resistance to a Class I PI3K inhibitor in a PI3K and MYC-driven model of metastatic breast cancer was associated with feedback activation of tyrosine kinase receptors, AKT, mTOR and MYC. BET inhibitors alone could not control the growth of these tumors. However, combined PI3K and BET inhibition induced cell death, tumor regression and dampened inhibition of PI3K signaling in metastatic breast cancer cell lines as well as in ovarian, colorectal, GBM and prostate cancer cell lines, indicating a strategy for circumventing resistance to kinase inhibitor therapy [93] (Table 3). In this study, the combination of BET inhibition along with PI3K/AKT/PTEN pathway inhibition was shown to maximize the downregulation of c-myc expression and to overcome resistance to each of the single agents. In colon cancer cell lines with activating KRAS mutation, inhibition of the p110α subunit of PI3K using a PI3K inhibitor reduced viability and induced apoptosis or cell cycle arrest [98]. These findings altogether suggest that combining BETi and PI3K inhibitor could potentially act synergistically to prevent resistance to kinase inhibition. Furthermore, since the additive effect seems to be mediated via inhibition of signaling across the Ras pathway, the effect could potentially be more amplified by further inhibiting the Ras/MAPK pathway using an MEK inhibitor.
Ponatinib, a multikinase inhibitor that inhibits ABL and FGFR, sensitizes colon, breast and ovarian cancer cells to JQ1 and dBET1. Both single agent ponatinib and BET inhibitors show significant anticancer activity against several hematological malignancies unlike FGFR and/or MYC-driven solid tumors. Interestingly, a combination of the two inhibitors increased apoptosis and downregulated MYC [99] (Table 3).
Venetoclax and JQ1 acted synergistically in an in vitro drug screening in patient-derived primary leukemia specimens and cell lines (Table 3). This synergism was confirmed in vivo using a T-ALL cell line and patient-derived xenograft models. The therapeutic benefit of this drug combination might partially be mediated by acute induction of the proapoptotic factor BCL2L11 and concomitant loss of BCL-2 upon BET bromodomain inhibition (encoded by BCL2L11) to BCL-2 [97].
Resistance to BET inhibitors
Thus far, of the various mechanisms of resistance to BET inhibitors that have been detected, none of them are genetic target aberrations, in other words, BRD2/3/4 mutations. AML JQ1-resistant cells do not undergo apoptosis but show upregulation of Beclin1, increased LC3 lipidation and formation of autophagosomes. AMPK (p-Thr172)-ULK1 (p-Ser555) pathway activation was associated with JQ1-induced autophagy in the resistant cells independent of mTOR signaling. In support, pharmacological AMPK inhibition or knockdown of PRKAA/AMPKa suppressed autophagy and promoted JQ1-induced apoptosis in AML stem cells [100,101]. Similarly, bortezomib synergistically sensitized BETi-resistant cells to JQ1 treatment, and the JQ1-bortezomib combination induced G2/M arrest in colorectal cancer cells. Although inhibition of NF-κB by bortezomib or NF-κB inhibitor rendered BETi-resistant cells sensitive to JQ1 [102], bortezomib also effects phosphorylation of Bcl-2 and cleavage products that also led to G2/M arrest and apoptosis [103]. Other resistance mechanisms include compensatory upregulation of MYC through the WNT signaling pathway [104,105], hyperphosphorylation of BRD4 due to downregulation of the PP2A phosphatase [106] and increased expression of BCL2L1/BCL-XL [106]. Limits involved in resistance to small-molecule BET inhibitors may also be addressed or compensated via design and mechanism of the actual inhibition (i.e., proteolysis targeting chimera, methyl-acyl-pyrroles) [107]. Further study on a clinical basis, possibly in combination, will be needed to fully determine role of BET inhibitors in oncology.
Toxicities of clinically used BET inhibitors
A study that directly evaluated the side effects of sustained BET inhibition in the transgenic RNAi mouse model showed that BRD4-depleted mice displayed reversible hyperplasia, alopecia and stem cell depletion in the small intestine. Furthermore, intestines of BRD4-supressed mice showed impaired regeneration following irradiation, suggesting caution regarding the combination of BETi with radiation therapy [108]. In current clinical trials, more commonly seen toxicities are gastrointestinal (diarrhea, nausea, vomiting), fatigue, hyperbilirubinemia, thrombocytopenia and anemia.
In the first-in-human clinical trial (NCT02391480) for assessing the safety and pharmacokinetics of ABBV-075 in patients with advanced tumors, 71 patients (98.6%) reported ≥1 treatment-emergent adverse events (TEAEs); thrombocytopenia (56.9%), dysgeusia (48.6%), fatigue (43.1%) and nausea (34.7%) were the common side effects. Grade 3/4 TEAEs were reported in 52 patients (72.2%), among which thrombocytopenia and anemia were most common. Dose-limiting toxicities (DLTs) included thrombocytopenia, fatigue and elevation in aspartate aminotransferase levels, gastrointestinal bleeding and hypertension [92].
In the clinical trial NCT02516553 for BI894999, 21 patients with advanced solid tumors were treated with six dose levels of the drug daily (Arm A), whereas seven patients were administered intermittently with two dose levels of the drug (Arm B) for 3 weeks. The maximum tolerated dose was 1.5 mg in Arm A. Dose escalation was discontinued in Arm B because of cardiac changes in three, and elevated serum troponin level of eight patients. The most frequent (≥10%) TEAEs were fatigue, thrombocytopenia, decreased appetite, diarrhea, increased troponin T, dysgeusia, nausea, stomatitis, neutropenia and vomiting. DLTs included thrombocytopenia grade (G) 4, increased troponin (G3), hypophosphatemia G3 and multiple G2 events in one patient preventing adequate dose intensity in cycle 1. Thrombocytopenia prevented continuous dosing [109].
In the first-in-human Phase I study of CPI-0610 in patients with relapsed or refractory lymphomas (NCT01949883; n = 64), the most common TEAEs were thrombocytopenia, fatigue, nausea, decreased appetite and anemia. However, the thrombocytopenia was reversible and not cumulative. Nonetheless, five patients showed objective response, which included two complete responses (CRs) and three PRs; five patients had prolonged (>6 months) SD, indicating that CPI-0610 was a well-tolerated drug with clinical activity in patients with advanced lymphoma [110].
OTX015 (MK-8628) was clinically tested in a Phase IIa trial (NCT02296476) in patients with recurrent GBM. A traditional 3+3 dose escalation scheme was followed. Patients with first recurrence of GBM were orally administered MK-8628 at three dose levels (DL1: 80 mg QD, DL2: 120 mg QD, DL3: 160 mg QD) with 4-week cycles; the effects of the drug on the tumor was assessed using MRI after every two cycles. Three patients showed DLTs – one at DL1 (G3 thrombocytopenia >7 days) and two at DL3 (G3 thrombocytopenia >7 days without bleeding; G3 hyperbilirubinemia for 2 days). Other DLTs included G3 thrombocytopenia, G1-2 diarrhea and G2 myalgia. Overall, the drug was well-tolerated. However, owing to lack of detectable clinical activity the trial was close [111].
OTX015 was also tested clinically in a Phase I trial (NCT01713582) in patients with AML who had previously received two other lines of therapy. Six dose levels were tested and patients were administered one dose daily. No DLT was recorded until 160 mg/day, when one patient had G3 diarrhea and another had G3 fatigue. Common toxic effects for all OTX01 doses were fatigue (including G3 in three patients) and elevation in bilirubin concentration (including G3-4 in two patients). Nonetheless, three patients (receiving 40, 80 and 160 mg/day) achieved CR or CR with incomplete recovery of platelets lasting 2–5 months, and two additional patients had partial blast clearance [112]. Similarly, OTX015 was tested in another Phase I clinical trial involving patients with lymphoma and myeloma. In this trial, 80 mg once daily was selected as the tolerable dose as higher doses or multiple lower doses resulted in DLTs such as thrombocytopenia, anemia, neutropenia, diarrhea, fatigue and nausea [113].
A first-in-man 3+3 dose escalation study of PLX51107 (20–160 mg QD) in adult patients with relapsed or refractory solid tumors and AML was conducted to determine the recommended Phase II (NCT02683395). The most common toxicities (mainly G1-2) in ≥15% of patients included fatigue, vomiting, diarrhea, nausea, hyperbilirubinemia and increase in internal normalized ratio. Three TEAEs (one each of G3 nausea, G2 vomiting and G2 kidney injury) were observed, and patients with extensive hepatic metastases (>50% of the liver) demonstrated more TEAEs. DLT was observed at 20 mg (one G3 thrombocytopenia), 120 mg QD (one G3 nausea) and 160 mg QD (one G2 kidney injury) [114].
The Phase I trial of BAY 1238097 was characterized by the occurrence of two DLTs at the 80 mg/week dose level and all eight subjects included in the safety set experienced at least one TEAE. Six out of eight (75%) subjects had at least one serious adverse event, including at least one treatment-emergent event. Hence, this trial was discontinued (Bayer Clinical Study Synopsis, 2016).
Conclusion & future perspective
The increased understanding of the effect of genomic and epigenomic aberrations on the transcriptional process has led to a greater appreciation of the role of transcriptional deregulation in the initiation and maintenance of cancer [115]. Promoters regulate transcriptional gene activity. Sequences of lineage-associated transcription factors assemble as gene enhancers and function as an autoregulatory loop. Clusters of these enhancers processing cell identity and/or subserving cellular function are characterized as super-enhancers, which appear to have higher densities of Mediator (MED1) and BRD4 binding [13], particularly those that are oncogenic. Of further importance, although an oncogene may be common to multiple tumor types, the associated super-enhancers can be tumor-type specific. Thus, the super-enhancers play a major role in determining the cell-type-specific response to BETi.
Inhibitors of BET proteins are being extensively studied and many are currently in clinical trial such as OTX015 for hematological cancers and I-BET762 for NMC. With exception, single agent BETis have shown limited long-term effectiveness and multiple mechanisms of resistance have already emerged. Further investigations are necessary to expand our understanding of the function of the various isoforms of each BET protein (produced via alternative splicing), the differences in the expression levels of the BET proteins in different cancer types, and the lineage-related and contextual functionality of the BET-containing tumor molecular elements. The outcome of these investigations will contribute to individualization of therapy to maximize therapeutic effectiveness, minimize inefficient and costly (in terms of patient time and financial investment) application (i.e., by maximizing targeted population/candidate population) and contribute to a logical, fact-based and personalized approach to combinatory and combined modality therapy.
Executive summary.
The interaction between genomic and epigenetic aberration in cancer has led to investigation of transcriptional regulation as a potential therapeutic approach in treating malignancies.
Bromodomain and extra-terminal (BET) proteins are enriched at promoter sites, particularly upstream of oncogenes, where they recruit other transcriptional factors.
BET inhibitors disrupt this interaction and, through several preclinical studies, were shown to halt transcription of oncogenes, decrease cancer cell survival and induce apoptosis. Many trials are ongoing to further characterize the relevance of BET inhibitor use in a clinical setting.
Currently, and despite a relatively tolerable safety profile, clinical use of BET inhibitors monotherapy seems to be hindered by apparent resistance, mostly mediated by the emergence of alternative molecular pathways and pathway cross-talk.
Several studies have successfully established proof of concept for the potential use of BET inhibitors in combination therapy. Namely, combining epigenetic inhibition of BET with inhibition of relevant molecular pathways may not only contribute to clinical efficacy but also pave the way to a personalized approach to combined therapy involving BET inhibitors.
Footnotes
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Author contributions
All authors contributed to the design and implementation of the review and to the writing of the manuscript.
Open access
This work is licensed under the Creative Commons Attribution 4.0 License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]; • Comprehensive review of the role of lysine acetylation in various diseases.
- 2.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20(8):615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 3.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011;21(6):735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014;13(5):337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 5.Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol. Oncol. 2012;6(6):579–589. doi: 10.1016/j.molonc.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 7.Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014;6(4):a018713. doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Fu LL, Wen X, et al. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014;5:e1047. doi: 10.1038/cddis.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand P, Brown JD, Lin CY, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154(3):569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim. Biophys. Acta. 2014;1839(8):676–685. doi: 10.1016/j.bbagrm.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbieri I, Cannizzaro E, Dawson MA. Bromodomains as therapeutic targets in cancer. Brief. Funct. Genomics. 2013;12(3):219–230. doi: 10.1093/bfgp/elt007. [DOI] [PubMed] [Google Scholar]
- 12.Zhang G, Sanchez R, Zhou MM. Scaling the druggability landscape of human bromodomains, a new class of drug targets. J. Med. Chem. 2012;55(17):7342–7345. doi: 10.1021/jm3011977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu LL, Tian M, Li X, et al. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget. 2015;6(8):5501–5516. doi: 10.18632/oncotarget.3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 2002;22(11):3794–3802. doi: 10.1128/MCB.22.11.3794-3802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devaiah BN, Singer DS. Cross-talk among RNA polymerase II kinases modulates C-terminal domain phosphorylation. J. Biol. Chem. 2012;287(46):38755–38766. doi: 10.1074/jbc.M112.412015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahman S, Sowa ME, Ottinger M, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011;31(13):2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vollmuth F, Blankenfeldt W, Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009;284(52):36547–36556. doi: 10.1074/jbc.M109.033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Devaiah BN, Singer DS. Two faces of Brd4: mitotic bookmark and transcriptional lynchpin. Transcription. 2013;4(1):13–17. doi: 10.4161/trns.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao R, Nakamura T, Fu Y, Lazar Z, Spector DL. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011;13(11):1295–1304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer. 2012;12(7):465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herait PE, Berthon C, Thieblemont C, et al. Abstract CT231: BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at nontoxic doses: interim results of an ongoing Phase I trial in hematologic malignancies. Cancer Res. 2014;74(19 Suppl.):CT231–CT231. [Google Scholar]
- 24.Stewart HJ, Horne GA, Bastow S, Chevassut TJ. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med. 2013;2(6):826–835. doi: 10.1002/cam4.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Describes early identification of BRD4 as a potential target in cancer treatment.
- 27.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl Acad. Sci. USA. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Outlines the interaction between MYC and bromodomain and extra-terminal (BET), and describes the effects of BET inhibition on MYC levels of expression.
- 28.Ott CJ, Kopp N, Bird L, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852. doi: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Outlines the interaction between MYC and BET, and describes the effects of BET inhibition on MYC levels of expression.
- 29.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006;16(4):318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Feng Q, Zhang Z, Shea MJ, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24(7):809–819. doi: 10.1038/cr.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal. Transduct. Target Ther. 2018;3:5. doi: 10.1038/s41392-018-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pastori C, Daniel M, Penas C, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics. 2014;9(4):611–620. doi: 10.4161/epi.27906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein K, Kabala PA, Grabiec AM, et al. The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann. Rheum. Dis. 2016;75(2):422–429. doi: 10.1136/annrheumdis-2014-205809. [DOI] [PubMed] [Google Scholar]
- 34.Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011;13:e29. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grayson AR, Walsh EM, Cameron MJ, et al. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene. 2014;33(13):1736–1742. doi: 10.1038/onc.2013.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sachchidanand, Resnick-Silverman L, Yan S, et al. Target structure-based discovery of small molecules that block human p53 and CREB binding protein association. Chem. Biol. 2006;13(1):81–90. doi: 10.1016/j.chembiol.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 37.Borah JC, Mujtaba S, Karakikes I, et al. A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 2011;18(4):531–541. doi: 10.1016/j.chembiol.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ceribelli M, Kelly PN, Shaffer AL, et al. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl Acad. Sci. USA. 2014;111(31):11365–11370. doi: 10.1073/pnas.1411701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Picaud S, Da Costa D, Thanasopoulou A, et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET bromodomains. Cancer Res. 2013;73(11):3336–3346. doi: 10.1158/0008-5472.CAN-12-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beesley AH, Stirnweiss A, Ferrari E, et al. Comparative drug screening in NUT midline carcinoma. Br. J. Cancer. 2014;110(5):1189–1198. doi: 10.1038/bjc.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain M, Arvanitis C, Chu K, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297(5578):102–104. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 43.Soucek L, Helmer-Citterich M, Sacco A, Jucker R, Cesareni G, Nasi S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene. 1998;17(19):2463–2472. doi: 10.1038/sj.onc.1202199. [DOI] [PubMed] [Google Scholar]
- 44.Soucek L, Jucker R, Panacchia L, Ricordy R, Tato F, Nasi S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002;62(12):3507–3510. [PubMed] [Google Scholar]
- 45.Vervoorts J, Luscher-Firzlaff JM, Rottmann S, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. Embo. Rep. 2003;4(5):484–490. doi: 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frank SR, Parisi T, Taubert S, et al. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. Embo. Rep. 2003;4(6):575–580. doi: 10.1038/sj.embor.embor861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shou Y, Martelli ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc. Natl Acad. Sci. USA. 2000;97(1):228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loosveld M, Castellano R, Gon S, et al. Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget. 2014;5(10):3168–3172. doi: 10.18632/oncotarget.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiskus W, Sharma S, Qi J, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol. Cancer Ther. 2014;13(10):2315–2327. doi: 10.1158/1535-7163.MCT-14-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, Walker SR, Nelson EA, et al. Targeting STAT5 in hematologic malignancies through inhibition of the bromodomain and extra-terminal (BET) bromodomain protein BRD2. Mol. Cancer Ther. 2014;13(5):1194–1205. doi: 10.1158/1535-7163.MCT-13-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hogg SJ, Vervoort SJ, Deswal S, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18(9):2162–2174. doi: 10.1016/j.celrep.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henssen A, Thor T, Odersky A, et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget. 2013;4(11):2080–2095. doi: 10.18632/oncotarget.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venkataraman S, Alimova I, Balakrishnan I, et al. Inhibition of BRD4 attenuates tumor cell self-renewal and suppresses stem cell signaling in MYC driven medulloblastoma. Oncotarget. 2014;5(9):2355–2371. doi: 10.18632/oncotarget.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bandopadhayay P, Bergthold G, Nguyen B, et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014;20(4):912–925. doi: 10.1158/1078-0432.CCR-13-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng Z, Gong Y, Ma Y, et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 2013;19(7):1748–1759. doi: 10.1158/1078-0432.CCR-12-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Taylor IC, Hutt-Cabezas M, Brandt WD, et al. Disrupting NOTCH slows diffuse intrinsic pontine glioma growth, enhances radiation sensitivity, and shows combinatorial efficacy with bromodomain inhibition. J. Neuropathol. Exp. Neurol. 2015;74(8):778–790. doi: 10.1097/NEN.0000000000000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suzuki H, Gabrielson E, Chen W, et al. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat. Genet. 2002;31(2):141–149. doi: 10.1038/ng892. [DOI] [PubMed] [Google Scholar]
- 59.Vedeld HM, Merok M, Jeanmougin M, et al. CpG island methylator phenotype identifies high risk patients among microsatellite stable BRAF mutated colorectal cancers. Int. J. Cancer. 2017;141(5):967–976. doi: 10.1002/ijc.30796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiang JF, Yin QF, Chen T, et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24(5):513–531. doi: 10.1038/cr.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mccleland ML, Mesh K, Lorenzana E, et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Invest. 2016;126(2):639–652. doi: 10.1172/JCI83265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Tian S, Xiong J, Zhou Y, Song H, Liu C. JQ-1 inhibits colon cancer proliferation via suppressing Wnt/β-catenin signaling and miR-21. Chem. Res. Toxicol. 2018;31(5):302–307. doi: 10.1021/acs.chemrestox.7b00346. [DOI] [PubMed] [Google Scholar]
- 63.Bihani T, Ezell SA, Ladd B, et al. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget. 2015;6(4):2407–2420. doi: 10.18632/oncotarget.2964. [DOI] [PMC free article] [PubMed] [Google Scholar]; • One of the early studies to describe the impact of epigenetic and molecular pathways interaction in mediating resistance to cancer therapy.
- 64.Borbely G, Haldosen LA, Dahlman-Wright K, Zhao C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget. 2015;6(32):33623–33635. doi: 10.18632/oncotarget.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nieto-Jimenez C, Alcaraz-Sanabria A, Perez-Pena J, et al. Targeting basal-like breast tumors with bromodomain and extraterminal domain (BET) and polo-like kinase inhibitors. Oncotarget. 2017;8(12):19478–19490. doi: 10.18632/oncotarget.14465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vazquez R, Riveiro ME, Astorgues-Xerri L, et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget. 2017;8(5):7598–7613. doi: 10.18632/oncotarget.13814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perez-Salvia M, Simo-Riudalbas L, Llinas-Arias P, et al. Bromodomain inhibition shows antitumoral activity in mice and human luminal breast cancer. Oncotarget. 2017;8(31):51621–51629. doi: 10.18632/oncotarget.18255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stuhlmiller TJ, Miller SM, Zawistowski JS, et al. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 2015;11(3):390–404. doi: 10.1016/j.celrep.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016;19:45–50. doi: 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coude MM, Braun T, Berrou J, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6(19):17698–17712. doi: 10.18632/oncotarget.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boi M, Gaudio E, Bonetti P, et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015;21(7):1628–1638. doi: 10.1158/1078-0432.CCR-14-1561. [DOI] [PubMed] [Google Scholar]
- 72.Bai L, Zhou B, Yang CY, et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77(9):2476–2487. doi: 10.1158/0008-5472.CAN-16-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bui MH, Lin X, Albert DH, et al. Preclinical characterization of BET family bromodomain inhibitor ABBV-075 suggests combination therapeutic strategies. Cancer Res. 2017;77(11):2976–2989. doi: 10.1158/0008-5472.CAN-16-1793. [DOI] [PubMed] [Google Scholar]
- 74.Long J, Li B, Rodriguez-Blanco J, et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J. Biol. Chem. 2014;289(51):35494–35502. doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chaidos A, Caputo V, Gouvedenou K, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014;123(5):697–705. doi: 10.1182/blood-2013-01-478420. [DOI] [PubMed] [Google Scholar]
- 76.Dawson MA, Gudgin EJ, Horton SJ, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28(2):311–320. doi: 10.1038/leu.2013.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garnier JM, Sharp PP, Burns CJ. BET bromodomain inhibitors: a patent review. Expert Opin. Ther. Pat. 2014;24(2):185–199. doi: 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]
- 78.Wyce A, Degenhardt Y, Bai Y, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013;4(12):2419–2429. doi: 10.18632/oncotarget.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wyce A, Ganji G, Smitheman KN, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE. 2013;8(8):e72967. doi: 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moros A, Rodriguez V, Saborit-Villarroya I, et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia. 2014;28(10):2049–2059. doi: 10.1038/leu.2014.106. [DOI] [PubMed] [Google Scholar]
- 81.Diaz T, Rodriguez V, Lozano E, et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in in vitro and in vivo models of multiple myeloma by blockade of Ikaros and MYC signaling. Haematologica. 2017;102(10):1776–1784. doi: 10.3324/haematol.2017.164632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.De Paula Careta F, Gobessi S, Panepucci RA, et al. The Aurora A and B kinases are up-regulated in bone marrow-derived chronic lymphocytic leukemia cells and represent potential therapeutic targets. Haematologica. 2012;97(8):1246–1254. doi: 10.3324/haematol.2011.054668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Thorsteinsdottir U, Mamo A, Kroon E, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99(1):121–129. doi: 10.1182/blood.v99.1.121. [DOI] [PubMed] [Google Scholar]
- 84.Picaud S, Wells C, Felletar I, et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl Acad. Sci. USA. 2013;110(49):19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kempen HJ, Bellus D, Fedorov O, et al. Stimulation of hepatic apolipoprotein A-I production by novel thieno-triazolodiazepines: roles of the classical benzodiazepine receptor, PAF receptor, and bromodomain binding. Lipid Insights. 2013;6:47–54. doi: 10.4137/Lpi.s13258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fabre C, Gobbi M, Ezzili C, et al. Clinical study of the novel cyclin-dependent kinase inhibitor dinaciclib in combination with rituximab in relapsed/refractory chronic lymphocytic leukemia patients. Cancer Chemother. Pharmacol. 2014;74(5):1057–1064. doi: 10.1007/s00280-014-2583-9. [DOI] [PubMed] [Google Scholar]
- 87.Martin MP, Olesen SH, Georg GI, Schonbrunn E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013;8(11):2360–2365. doi: 10.1021/cb4003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Massard C, Soria JC, Stathis A, et al. A Phase Ib trial with MK-8628/OTX015, a small molecule inhibitor of bromodomain (BRD) and extra-terminal (BET) proteins, in patients with selected advanced solid tumors. Eur. J. Cancer. 2016;69:S2–S3. [Google Scholar]
- 89.Postel-Vinay S, Herbschleb K, Massard C, et al. First-in-human Phase I dose escalation study of the Bromodomain and Extra-Terminal motif (BET) inhibitor BAY 1238097 in subjects with advanced malignancies. Eur. J. Cancer. 2016;69:S7–S8. [Google Scholar]
- 90.Stathis A, Zucca E, Bekradda M, et al. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 2016;6(5):492–500. doi: 10.1158/2159-8290.CD-15-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bernasconi E, Gaudio E, Lejeune P, et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br. J. Haematol. 2017;178(6):936–948. doi: 10.1111/bjh.14803. [DOI] [PubMed] [Google Scholar]
- 92.Borthakur G, Wolff J, Aldoss I, et al. First-In-Human Study of ABBV-075 (Mivebresib), a pan-inhibitor of bromodomain and extra terminal (BET) proteins, in patients (Pts) with relapsed/refractory (RR) acute myeloid leukemia (AML): preliminary data. Clin. Lymphoma, Myeloma Leuk. 2018;18:S203. [Google Scholar]; • Results from Phase 1 study of the most recent BET inhibitor to enter clinical trials.
- 93.Stratikopoulos EE, Dendy M, Szabolcs M, et al. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell. 2015;27(6):837–851. doi: 10.1016/j.ccell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Solid in vivo testing providing proof of concept for the synergistic role of combining BET inhibitors and kinase inhibitors. Provides evidence for translating combinatory therapy to clinical trials.
- 94.Tontsch-Grunt U, Savarese F, Baum A, et al. Combination of the novel BET inhibitor BI 894999 with CDK9 inhibition suggests a promising regimen for the treatment of AML. Eur. J. Cancer. 2016;69:S89. [Google Scholar]
- 95.Bolin S, Borgenvik A, Persson C, et al. Abstract 2473: combined BET-bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Cancer Res. 2016;76(14 Suppl.):2473–2473. doi: 10.1038/s41388-018-0135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Muralidharan SV, Bhadury J, Nilsson LM, Green LC, Mclure KG, Nilsson JA. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene. 2016;35(36):4689–4697. doi: 10.1038/onc.2015.521. [DOI] [PubMed] [Google Scholar]
- 97.Peirs S, Frismantas V, Matthijssens F, et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31(10):2037–2047. doi: 10.1038/leu.2017.10. [DOI] [PubMed] [Google Scholar]
- 98.Sarker D, Ang JE, Baird R, et al. First-in-human Phase I study of pictilisib (GDC-0941), a potent pan-Class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. 2015;21(1):77–86. doi: 10.1158/1078-0432.CCR-14-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bauer K, Berger D, Zielinski CC, Valent P, Grunt TW. Hitting two oncogenic machineries in cancer cells: cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells. Oncotarget. 2018;9(41):26491–26506. doi: 10.18632/oncotarget.25474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jang JE, Eom JI, Jeung HK, et al. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy. 2017;13(4):761–762. doi: 10.1080/15548627.2016.1278328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jang JE, Eom JI, Jeung HK, et al. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin. Cancer Res. 2017;23(11):2781–2794. doi: 10.1158/1078-0432.CCR-16-1903. [DOI] [PubMed] [Google Scholar]
- 102.Wu T, Wang G, Chen W, et al. Co-inhibition of BET proteins and NF-kappaB as a potential therapy for colorectal cancer through synergistic inhibiting MYC and FOXM1 expressions. Cell Death Dis. 2018;9(3):315. doi: 10.1038/s41419-018-0354-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ling YH, Liebes L, Ng B, et al. PS-341, a novel proteasome inhibitor, induces Bcl-2 phosphorylation and cleavage in association with G2-M phase arrest and apoptosis. Mol. Cancer Ther. 2002;1(10):841–849. [PubMed] [Google Scholar]
- 104.Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525(7570):543–547. doi: 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Mechanistic description of acquired resistance to BET inhibition, including molecular pathways cross-signaling that provides transcriptional plasticity.
- 105.Kumar K, Raza SS, Knab LM, et al. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015;5:9489. doi: 10.1038/srep09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lucas X, Wohlwend D, Hugle M, et al. 4-Acyl pyrroles: mimicking acetylated lysines in histone code reading. Angew. Chem. Int. Ed. Engl. 2013;52(52):14055–14059. doi: 10.1002/anie.201307652. [DOI] [PubMed] [Google Scholar]
- 108.Bolden JE, Tasdemir N, Dow LE, et al. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. 2014;8(6):1919–1929. doi: 10.1016/j.celrep.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aftimos PG, Bechter O, Awada A, et al. Phase I first-in-man trial of a novel bromodomain and extra-terminal domain (BET) inhibitor (BI 894999) in patients (Pts) with advanced solid tumors. J. Clin. Oncol. 2017;35(15 Suppl.):2504–2504. [Google Scholar]
- 110.Blum KA, Abramson J, Maris M, et al. 41OA Phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann. Oncol. 2018;29(Suppl. 3):mdy048–mdy048. [Google Scholar]
- 111.Hottinger AF, Sanson M, Moyal E, et al. Dose optimization of MK-8628 (OTX015), a small molecule inhibitor of bromodomain and extra-terminal (BET) proteins, in patients (pts) with recurrent glioblastoma (GB) J. Clin. Oncol. 2016;34(15 Suppl.):e14123–e14123. [Google Scholar]
- 112.Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, Phase 1 study. Lancet Haematol. 2016;3(4):e186–e195. doi: 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- 113.Amorim S, Stathis A, Gleeson M, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, Phase 1 study. Lancet Haematol. 2016;3(4):e196–e204. doi: 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- 114.Patnaik A, Carvajal RD, Komatsubara KM, et al. Phase Ib/2a study of PLX51107, a small molecule BET inhibitor, in subjects with advanced hematological malignancies and solid tumors. J. Clin. Oncol. 2018;36(15 Suppl.):2550–2550. [Google Scholar]
- 115.Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168(4):629–643. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]