

Abstract
Extensive research in the past decade has brought mRNA closer to the clinical realization of its therapeutic potential. One common structural feature for all cellular messenger RNAs is a poly(A) tail, which can either be brought in cotranscriptionally via the DNA template (plasmid- or PCR-based) or added to the mRNA in a post-transcriptional enzymatic process. Plasmids containing poly(A) regions recombine in E. coli, resulting in extensive shortening of the poly(A) tail. Using a segmented poly(A) approach, we could significantly reduce recombination of plasmids in E. coli without any negative effect on mRNA half-life and protein expression. This effect was independent of the coding sequence. A segmented poly(A) tail is characterized in that it consists of at least two A-containing elements, each defined as a nucleotide sequence consisting of 40–60 adenosines, separated by a spacer element of different length. Furthermore, reducing the spacer length between the poly(A) segments resulted in higher translation efficiencies compared to homogeneous poly(A) tail and reduced recombination (depending upon the choice of spacer nucleotide). Our results demonstrate the superior potential of segmented poly(A) tails compared to the conventionally used homogeneous poly(A) tails with respect to recombination of the plasmids and the resulting mRNA performance (half-life and translational efficiency).
Keywords: poly(A) tail, plasmid recombination, mRNA therapeutics, transcript therapy
INTRODUCTION
The past decade has witnessed the emergence and rapid application of in vitro transcribed messenger RNA (mRNA) as a therapeutic molecule. Compared to classical gene therapy with DNA-based vectors, use of mRNA offers several advantages, such as transient expression (Kapeli and Yeo 2012; Lodish 2012), lack of necessity to enter the nucleus, and no risk of chromosomal integration (Cannon and Weissman 2002; Hacein-Bey-Abina et al. 2003; Bernal 2013). After overcoming the intrinsic limitations of the mRNA, namely immunogenicity and instability, using chemically modified nucleotides (Karikó et al. 2005, 2008; Kormann et al. 2011), researchers have significantly improved the mRNA molecule by optimizing the structural elements, namely cap (Grudzien-Nogalska et al. 2013; Ziemniak et al. 2013; Kowalska et al. 2014), 5′- and 3′-UTRs (Ferizi et al. 2016; Schrom et al. 2017; Trepotec et al. 2018), and coding sequence(s) (Thess et al. 2015; Schrom et al. 2017). Increasing its stability and translational yields has led to a progress from the first use of encoding a potentially therapeutic protein (Wolff et al. 1990) to preclinical (Bahl et al. 2017; Richner et al. 2017) and clinical applications (Mullard 2016).
Regardless of the target protein or tissue, all cellular protein encoding RNAs with some exceptions (e.g., histones) share a common structural feature, that is, poly(A) tail. Inside the cell nucleus, the poly(A) tail is added to the mRNA in a post-transcriptional manner downstream from the gene-encoded polyadenylation signal (AATAAA). The poly(A) tail is essential for the stability (Sachs 1990; Oliveira and McCarthy 1995) and translation (Sachs 1990; Wells et al. 1998) of the mRNA. The poly(A) tail in the mRNA is recognized by poly(A)-binding protein (PABP) which in turn interacts with eIF4G of the translation initiation complex, thereby forming a closed loop (Mangus et al. 2003; Goldstrohm and Wickens 2008) and the resulting messenger ribonucleoprotein particle.
In the case of in vitro-transcribed mRNA, the poly(A) tail can be either encoded into the DNA template (PCR product- or plasmid-based) or added enzymatically to the mRNA in a separate step after in vitro transcription. Each of the abovementioned approaches has its own set of limitations. While PCR offers the ease of high throughput and is widely used for small-scale mRNA production (up to a few hundred milligrams), high production costs, and risk of mutagenesis during PCR amplification (compared to plasmid production in bacteria) limit its usefulness for large-scale production (several grams). Plasmid production on the other hand is well established, can be performed under GMP conditions, has lower production costs and risks of mutations (in the coding sequence) when compared to a PCR-based approach. However, plasmid DNA encoded homopolymeric stretches [e.g., poly(A)] recombine during bacterial amplification of the plasmid DNA. Previous studies reported generation of spontaneous deletion mutants during amplification of plasmids starting with ∼100 bp of poly(dA:dT) sequences (Preiss et al. 1998). For longer poly(A)s, for example, poly(A)150, the instability is too high to allow isolation of any single positive clone (Grier et al. 2016). Despite this limitation, template-encoded poly(A) offers certain advantages over enzymatic post-polyadenylation of mRNA, such as defined and reproducible poly(A) length resulting in a homogenous product (Holtkamp et al. 2006). Although enzymatic post-polyadenylation of mRNA warrants sufficiently long poly(A) tails (Cao and Sarkar 1992; Martin and Keller 1998), the composition of the final product due to different poly(A) lengths is difficult to control and therefore might not meet regulatory requirements (Weissman 2015).
In addition to the above listed advantages of cotranscriptional polyadenylation, the reduced number of steps in RNA production are likely to translate to lower production costs. Moreover, enzymatic polyadenylation of mRNA needs to be carried out under alkaline conditions, as the enzyme poly(A) polymerase has highest activity at pH >7.5. mRNA is highly susceptible to alkaline hydrolysis which in turn results in poorer mRNA quality, especially with longer transcripts (>3 kb). (Voet and Voet 2011)
As an alternative to circular plasmids, Grier et al. (2016) have proposed the use of a linear plasmid system, pEVL, which allows stable cloning of poly(A)s of up to 500 bp. A similar linear vector-based system (pJAZZ) is commercially available from Lucigen but suffers from limitations of large vector size (>12 kb), limited choice for cloning enzymes (available only as either SmaI or NotI predigested vector) and is a very low copy vector. Having a plasmid template-encoded poly(A) tail which is not prone to recombination but still supports mRNA stability and translational efficiency in comparable manner to a conventional natural poly(A) would be an ideal solution to the abovementioned limitations.
The main aim of the present study was to investigate if segmentation of the poly(A) tail could reduce recombination of a high copy plasmid vector in E. coli. For this, the most widely used but relatively unstable poly(A) tail of ∼120 A's [poly(A)120] was split into either two or three segments of 40A's [poly(A)3 × 40] or 60A's [poly(A)2 × 60], respectively. The segmentation scheme was designed keeping in mind the functional “PABP footprint” on mRNA. While PABP requires a minimum of 12 adenosines to bind, protein oligomers can bind to the same poly(A) stretch, thereby forming a repeating unit of ∼27–30 nt (Baer and Kornberg 1980, 1983; Wang et al. 1999). Moreover, the work by Preiss et al. showed that a single PABP molecule bound to mRNA, while interacting with the 5′ cap structure was not sufficient for promoting translation (Preiss et al. 1998). Based on these data, our constructs were designed to enable at least one oligomeric stretch of PABP per segment (30 nt). Besides recombination, translation efficiency and mRNA half-life measurements were made to compare the effect on segmentation on these critical attributes of the molecule. Here too, poly(A)120 was used as a benchmark as it has been shown in previous studies to result in high protein expression (Holtkamp et al. 2006; Bangel-Ruland et al. 2013). We show that segmentation of the poly(A) tail, does not negatively affect translational yield and mRNA half-life, but eases the technical difficulties connected with recombination of homopolymeric poly(A) stretches in plasmid vectors.
RESULTS AND DISCUSSION
The current study on segmentation of poly(A) into smaller fragments separated by spacer elements was prompted by technical challenges often met while designing and producing plasmid DNA templates for use in in vitro transcription to produce mRNA for use as transcript therapies.
Design of modified/segmented poly(A) tails
In order to produce recombinant RNA transcripts with segmented poly(A) tails, the corresponding DNA sequences were cloned into a plasmid vector downstream from the gene of interest (GOI). Figure 1 schematically shows the composition of different poly(A) tails and their spacer separators. The most conventionally used standard poly(A) tail in plasmid vectors (Holtkamp et al. 2006; Kormann et al. 2011; Vallazza et al. 2015; Balmayor et al. 2016, 2017; Ferizi et al. 2016) containing ∼120 A's [poly(A)120] was split either into three segments, each comprising 40 A's separated by a NsiI restriction site of 6 nt [poly(A)3 × 40_6] or into two equal segments of 60 A's, also separated by 6 nt [poly(A)2 × 60_6]. This way, we could control the size of each segment and still have a physical separator among adenosines. According to previous literature (Baer and Kornberg 1980, 1983; Wang et al. 1999), a minimum of 12 adenosines are needed for the binding of a single PABP molecule. However, a single PABP bound to poly(A) is not enough to support translation, even though it can interact with the eIF4 complex (Preiss et al. 1998). The segments of poly(A)3 × 40_6 and poly(A)2 × 60_6 are long enough to ensure binding of more than three copies of PABP per segment. In order to investigate the role of the spacer length between the two A60 segments, besides the one separated by 6 nt, five additional constructs were synthesized with a spacer length of either 12 [poly(A)2 × 60_12], 24 [poly(A)2 × 60_24] nt, or 1 nt [poly(A)2 × 60_C, poly(A)2 × 60_G, poly(A)2 × 60_T].
FIGURE 1.

Schematic representation of all combinations of poly(A) modifications tested in the current study. Different homo- or heteropolymeric poly(A) stretches were inserted downstream from the gene of interest (GOI).
Segmented poly(A) tails reduce recombination of poly(A)-containing plasmids in E. coli
Instability of poly(A)-containing plasmids in E. coli has been previously reported (Kühn and Wahle 2004; Godiska et al. 2009) and is a major risk of failure when using such poly(A)-containing plasmids for large-scale mRNA production. We examined whether the use of segmented poly(A) affected the recombination efficiency of plasmids post-transformation into E. coli. To test this, coding regions for different proteins (d2EGFP, luciferase, and hEPO) were cloned upstream of these poly(A) formats [poly(A)120, poly(A)2 × 60_6, poly(A)3 × 40_6] into a pUC57-Kanamycin (GenScript) vector. Post-transformation into E. coli, clones were screened for insert and positive clones (containing the desired insert) were additionally screened for the length of the poly(A) region. For each of the poly(A) formats, the poly(A) region was digested with restriction enzymes and the digestions were resolved on Fragment Analyzer (capillary gel electrophoresis) to measure the size of the poly(A) fragment. As expected, recombination in the poly(A) region was observed for more than 50% of the clones containing a homologous poly(A), poly(A)120. The proportion of recombination observed with the poly(A)120 format was sequence-independent and comparable to the values reported by Grier et al. (2016). By splitting the poly(A) into either poly(A)3 × 40 or poly(A)2 × 60, recombination in E. coli could be reduced with most stable clones (<20% recombination) obtained with plasmids containing poly(A)2 × 60_6 (Fig. 2). This trend was observed for all the tested sequences indicating this reduction in recombination to be sequence-independent.
FIGURE 2.
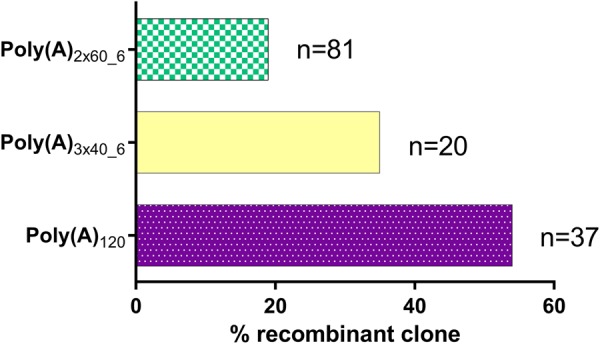
Quantification of poly(A) tail recombination for A120 and segmented poly(A) tails of poly(A)3 × 40_6 and poly(A)2 × 60_6. (n) Total number of clones of d2EGFP, luciferase, and hEPO sequences tested with a particular poly(A) format.
Effects of poly(A) segmentation on mRNA productivity
Encouraging results of reduced recombination prompted us to investigate the performance of our segmented poly(A) tails with respect to mRNA stability and expression. For this, the coding region of d2EGFP (destabilized EGFP with a relatively short protein half-life) was cloned into our different poly(A)-containing vectors and chemically modified mRNA (modification 1) was produced using previously described protocols (Kormann et al. 2011; Trepotec et al. 2018). The resulting mRNAs were transfected into A549 cells and at different time points post-transfection (4, 24, 48, and 72 h), both d2EGFP protein and mRNA were quantified using FACS and real-time reverse transcriptase PCR, respectively (Fig. 3). Comparable levels of d2EGFP protein were observed for segmented poly(A) constructs compared to the control A120 at all four time points. Comparable levels of d2EGFP mRNA were observed for all poly(A) formats, except at 24-h post-transfection, where lower mRNA amounts were quantified for the segmented poly(A) formats. Similar to our previously published work (Ferizi et al. 2016), we calculated the mRNA productivity, defined as the amount of protein (d2EGFP median fluorescence intensity) normalized to the amount of mRNA (quantified via qPCR) for the three poly(A) formats. Surprisingly, higher mRNA productivities were observed for segmented poly(A) constructs at earlier time points (4 and 24 h) compared to A120 format.
FIGURE 3.

Determination of d2EGFP protein expression and mRNA quantification of different poly(A)-containing d2EGFP mRNAs post-transfection in A549 cells. (A) Mean MFI (median fluorescence intensity) at 4-, 24-, 48-, and 72-h post-transfection, measured by FACS in A549 cells. (B) d2EGFP mRNA quantification in A549 cells. (C) mRNA productivity was calculated by dividing the mean MFI (FACS data; A) by the mRNA amounts (real-time PCR data; B) and normalizing these ratios to those observed with poly(A)120 construct. Values represent mean ± SD of three replicates. Statistical significance was assessed by two-way ANOVA test with P-values: (*) P < 0.5, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
Similar to the experiments with d2EGFP, luciferase protein and mRNA quantification was investigated in A549 cells at 24-h post-transfection with luciferase-encoding mRNA, containing either of the poly(A) formats [poly(A)2 × 60_6, poly(A)3 × 40_6 vs. poly(A)120]. Furthermore, to address the effect of nucleotide modifications, luciferase mRNAs were produced using either unmodified nucleotides, modification 1 or modification 2 nucleotides. These modifications have been described previously (Trepotec et al. 2018). Briefly, modification 1 included 25% 5-methylcytidine and 25% 2-thiouridine. As for the modification set 2, 35% 5-iodouridine and 7.5% 5-iodocytidine were used in the IVT reaction for RNA production. As an additional benchmark, a previously published construct (Thess et al. 2015) comprising homopolymeric A stretch (A63), homopolymeric C stretch (C31) and histone stem–loop (ACH; Fig. 1) was used. As histone mRNAs lack poly(A) tails, the functions of poly(A), that is, stability and translation efficiency are performed by the conserved stem–loop (Williams and Marzluff 1995; Zanier et al. 2002). Use of segmented poly(A)2 × 60_6 construct significantly increased protein levels post-transfection in a modification-independent manner when compared to poly(A)120 and ACH benchmarks (Fig. 4). No drastic differences were observed between the mRNA amounts for the different poly(A) format containing luciferase mRNAs across modifications. Poly(A)2 × 60_6 construct was more productive than any other poly(A) format when using modification sets 1 or 2. The construct with hairpin structures (ACH) expressed significantly less amounts of luciferase compared to all other constructs containing 120 adenosines despite these three constructs being present in the cell in substantial amounts (qPCR data). The reduced translation efficiency of hairpin-containing constructs is further confirmed by calculating the mRNA productivities.
FIGURE 4.

Determination of luciferase expression and mRNA quantification of different poly(A)-containing luciferase mRNA 24-h post-transfection in A549 cells. (A) Luciferase activity, measured as relative light units (RLU: arbitrary units) in protein lysates from A549 cells transfected with different poly(A)-containing luciferase RNA measured 24-h post-transfection. (B) Luciferase mRNA quantification in A549 cells. (C) mRNA productivity was calculated by dividing the luciferase expression values (RLU; A) by the mRNA amounts (real-time PCR data; B) and normalizing these ratios to those observed with poly(A)120 construct. Values represent mean ± SD of six replicates. Statistical significance was assessed by two-way ANOVA test with P-values: (*) P < 0.5, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
Effects of poly(A) segmentation on translation of physiological targets
The initial results of reduced recombination and comparable/higher mRNA productivity compared to poly(A)120, with intracellular reporter proteins (d2EGFP and luciferase), prompted us to further test the poly(A)2 × 60_6 format with additional physiological targets. The selected targets varied in the length of their mRNAs and cellular localization of the protein: human erythropoietin (0.9 kb) as a prototype of a secretory protein and human cystic fibrosis transmembrane conductance regulator (CFTR; 4.5 kb) as a prototype of a membrane protein.
The codon optimized sequence encoding hEPO was cloned into pUC57-Kanamycin vector upstream of either poly(A)120 or poly(A)2 × 60_6. mRNA was produced for each of the constructs using either unmodified, modification 1 or modification 2 sets of nucleotides. Since EPO is primarily secreted by kidney cells, transfection experiments, at two doses, were performed in human HEK293 cells. Protein concentrations were determined via ELISA at 24-, 48-, and 72-h post-transfection (Fig. 5). With some exceptions (e.g., unmodified RNA at 24 h and 72 h and modification 1 at 72 h), no significant differences were observed between the compared poly(A) formats at any of the compared doses, time points or modifications.
FIGURE 5.

Quantification of secreted human erythropoietin protein levels as measured via ELISA in supernatants from HEK293 cells transfected either with poly(A)120- or poly(A)2×60-containing EPO mRNA at 24 h (A), 48 h (B), and 72 h (C) post-transfection. Values represent mean ± SD of three replicates. Statistical significance was assessed by two-way ANOVA test with P-values: (**) P < 0.01, (***) P < 0.001, n = 3.
To further investigate the relationship between physiological gene expression and poly(A) tail segmentation, we focused on mRNA constructs encoding human CFTR furnished with either poly(A)2×60_6 or poly(A)120. Both constructs were produced with unmodified set of nucleotides, and transfection experiments with CFTR mRNA were performed in 16HBE14o- cells. Only unmodified CFTR mRNA was used, as a previous study (Bangel-Ruland et al. 2013) has demonstrated functional restoration of CFTR in human CF airway epithelia after transfection with unmodified CFTR mRNA containing a poly(A) tail of 120A's.
At 24 h and 48 h post-transfection, cells were lysed and western blot was performed for the CFTR protein. Hsp90 was used as a housekeeper. Similar to our previous results with d2EGFP, luciferase and hEPO, use of segmented poly(A)2×60_6 did not negatively affect the resulting protein amounts post-transfection when compared to the conventionally used poly(A)120 (Fig. 6).
FIGURE 6.

Relative quantification of CFTR protein in 16HBE14o- lysates as measured via western blot. (A) 16HBE14o- cells were transfected either with poly(A)120- or poly(A)2×60_6-containing hCFTR mRNA and protein lysates analyzed at 24 and 48 h post-transfection. (B) Densitometry analysis of western blot images. Values represent mean ± SD of two replicates. Statistical significance was assessed by paired t-test with P-values: ns = P > 0.5.
Spacer region expansion in poly(A)2×60
Reduced recombination with segmented poly(A)2×60_6 with comparable (d2EGFP, EPO, CFTR) or higher (luciferase) translation without significant effects on mRNA stability prompted us to further investigate the spacer length of this specific poly(A) format. For the ease of experimental feasibility, two new luciferase constructs were made with longer spacers [12 and 24 nt: construct poly(A)2 × 60_12 and poly(A)2 × 60_24 in Figure 1]. The different luciferase mRNAs (unmodified, modification 1 and modification 2) for the three poly(A) formats [poly(A)2 × 60_6, poly(A)2 × 60_12, and poly(A)2 × 60_24] were transfected into A549 cells. At 24 h post-transfection, significantly lower luciferase expression was observed with longer spacers using unmodified and modification 1 containing mRNA (Fig. 7). These modification-specific effects could be due to the spacer region, upon incorporation of chemically modified nucleotides affecting the binding of PABP to the two segments of poly(A). With two exceptions [poly(A)2 × 60_24 unmodified and poly(A)2 × 60_6 modification 1], comparable levels of luciferase mRNA could be quantified in the cells. Therefore, increasing the spacer length to more than 6 nt in segmented poly(A)2 × 60_6 tail did not result in any significant advantage, neither in translation nor in mRNA stability.
FIGURE 7.

Determination of luciferase expression and mRNA quantification of different poly(A)-containing luciferase mRNA at 24 h post-transfection in A549 cells. (A) Luciferase activity, measured as relative light units (RLU: arbitrary units), in protein lysates from A549 cells transfected with different poly(A)-containing luciferase mRNA. (B) Luciferase mRNA quantification in A549 cells transfected with different poly(A)-containing luciferase mRNA. (C) mRNA productivity was calculated by dividing the luciferase expression values (RLU; A) by the mRNA amounts (real-time PCR data; B) and normalizing these ratios to those observed with poly(A)120 construct. Values represent mean ± SD of six replicates. Statistical significance was assessed by two-way ANOVA test with P-values: (*) P < 0.5, (**) P < 0.01, (****) P < 0.0001.
Spacer region reduction in poly(A)2 × 60
The next set of experiments was addressed to examining the effect of reducing the spacer length to a single nucleotide in poly(A)2 × 60 segmented poly(A) tail on protein expression and mRNA productivity. All three possible constructs (with C, T, or G as a spacer) were made. For each construct, unmodified, modification 1 and modification 2 containing mRNAs were produced. The different luciferase mRNAs were compared in A549 cells. As a benchmark, standard poly(A)120 was used. Independent of the used spacer nucleotide/modifications, all three segmented poly(A) constructs resulted in significantly higher luciferase expression when compared to poly(A)120 (Fig. 8). With a few exceptions, mRNA levels were comparable for the different luciferase mRNAs. Irrespective of the spacer nucleotide, segmented poly(A) constructs were more productive than the standard poly(A)120. Among the three spacer nucleotides, no significant differences could be observed. Since the mRNA stability was not affected, it is very likely that segmented poly(A) with a single nucleotide spacer augmented translation. Besides increasing translation, incorporation of a single G as a spacer further reduced recombination from 20% [as observed with poly(A)2 × 60_6] to zero (Fig. 9). A spacer with a single nucleotide of T recombined in 10% of cases, and the one with a C as a spacer nucleotide recombined in 50% of cases, which in turn is comparable to recombination observed with A120 (Fig. 2). Identification of the mechanisms underlying the observed reduced recombination and enhanced translation with segmented poly(A)2 × 60_1 compared to classical poly(A)120 will be the subject of future studies.
FIGURE 8.

Determination of luciferase expression and mRNA quantification of different poly(A)-containing luciferase mRNA at 24 h post-transfection in A549 cells. (A) Luciferase activity, measured as relative light units (RLU: arbitrary units), in protein lysates from A549 cells transfected with different poly(A)-containing luciferase mRNA. (B) Luciferase mRNA quantification in A549 cells transfected with different poly(A) containing luciferase mRNA. (C) mRNA productivity was calculated by dividing the luciferase expression values (RLU; A) by the mRNA amounts (real-time PCR data; B) and normalizing these ratios to those observed with poly(A)120 construct. Values represent mean ± SD of six replicates. Statistical significance was assessed by two-way ANOVA test with P-values: (*) P < 0.5, (****) P < 0.0001.
FIGURE 9.
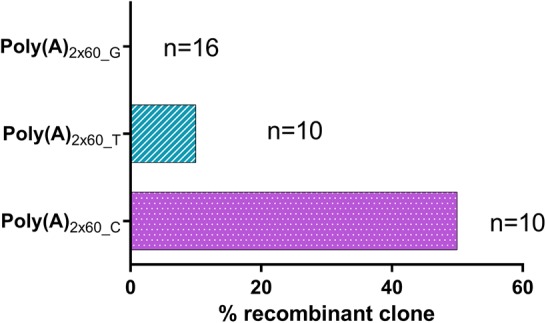
Quantification of poly(A) tail recombination rate for segmented poly(A) tails with a single nucleotide spacer. (n) Total number of clones of luciferase tested with a particular poly(A) format.
These results allow us to recommend a segmented poly(A) region [poly(A)2 × 60] with either a 6 or a single nucleotide (G/T) spacer for use in plasmid-based vectors for RNA production. Using such a segmented poly(A) did not have any negative effect on protein expression and mRNA half-life but reduced recombination of plasmids in E. coli.
MATERIALS AND METHODS
Plasmid preparation
The synthetic poly(A) sequences were introduced to the vector backbone either as annealed complementary oligonucleotides or fragments created by PCR (Table 1). For sequences comprising of 2 × 60, 3 × 40, and ACH, specific sets of complementary oligonucleotides were synthesized and annealed. The synthetic poly(A) fragments of A120, 2 × 60_1, 2 × 60_12, and 2 × 60_24 were created by PCR.
TABLE 1.
Segmented poly(A) sequences and their corresponding cloning strategy using either PCR primer sets or oligonucleotides

Annealing of complementary oligonucleotides was performed as follows: 100 µM of each oligonucleotide were mixed with 40 µL annealing buffer (10 mM Tris-HCl, 50 mM NaCl, 1 mM EDTA, pH 7.5) and incubated for 5 min at 95°C. Subsequently, the mixture was let to cool down to room temperature before proceeding with restriction digestion (BglII-BstBI).
For the high performance of PCR reaction, Phusion High-fidelity PCR master mix (Thermo Fisher Scientific) was used. To the mastermix, which contains 2× Phusion DNA Polymerase, nucleotides and optimized reaction buffer including MgCl2, 0.5 µM of forward and reverse primer, 3% DMSO, and 1 ng of template DNA were added to the reaction. The total volume of 25 µL per reaction was initially denatured at 98°C for 30 sec, following by 30 cycles at 98°C for 10 sec, annealing at 72°C for 30 sec, and extension at 72°C for 30 sec/kb. The final extension was performed at 72°C for 10 min. The size of the PCR product was confirmed on 1% agarose gel and the desired band was purified using NucleoSpin Gel and PCR clean-up kit (Macherey Nagel). Purified PCR product was digested with NheI-BstBI and stored at −20°C till further use.
Digested products of annealed oligonucleotides and PCR products were cloned into accordingly digested pUC57-Kana vector (GenScript) containing the desired coding sequences (firefly luciferase, d2EGFP, human EPO, and human CFTR).
Generation of mRNA
To generate in vitro transcribed mRNA, plasmids were linearized by BstBI (Thermo Fisher Scientific) digestion and purified by chloroform extraction and ethanol precipitation. Purified linear plasmids were used as a template for in vitro transcription. Plasmid templates (0.5 µg/µL) were subjected to in vitro transcription using 3 U/µL T7 RNA polymerase (Thermo Fisher Scientific), transcription buffer II (Ethris GmbH), 1 U/µL RiboLock Rnase inhibitor (Thermo Fisher Scientific), 0.015 U/µL inorganic pyrophosphatase 1 (Thermo Fisher Scientific) with a defined choice of natural and chemically modified ribonucleotides (Jena Biosciences).
The modification set 1 was synthetized using 5-methylcytidine (25%) and 2-thiouridine (25%), in addition to unmodified nucleotides. For modification set 2, instead of 5-methylcytidine (25%) and 2-thiouridine (25%), 5-iodouridine (35%), and 5-iodocytidine (7.5%) were used. The complete IVT-mix was incubated at 37°C for 2 h. Afterwards, 0.01 U/µL DNase I (Thermo Fisher Scientific) was added for an additional 45 min at 37°C to remove the plasmid template. RNA was precipitated with ammonium acetate at a final concentration of 2.5 mM, followed by two washing steps with 70% ethanol. The pellet was re-suspended in aqua ad injectabilia. A C1-m7G cap structure was added enzymatically by 0.5 mM Vaccinia Virus Capping Enzyme (New England Biolabs) to the 5′ end of the previously denatured transcript (1 mg/mL) at 80°C for 5 min. The capping reaction mix also contained 1× capping buffer (New England Biolabs), 0.5 mM GTP (New England Biolabs), 0.2 mM S-methyladenosine (New England Biolabs), 2.5 U/µL mRNA Cap 2′-O-Methyltransferase (New England Biolabs), and 1 U/µL RiboLock RNase Inhibitor (Thermo Fisher Scientific). The capping mixture was incubated for 60 min at 37°C, followed by RNA precipitation with ammonium acetate at a final concentration of 2.5 mM and two washing steps with 70% ethanol. The pellet was re-suspended in aqua ad injectabilia.
RNA quality and concentration were measured spectrophotometrically on a NanoDrop2000C (Thermo Fisher Scientific). Its correct size and purity were determined via automated capillary electrophoresis (Fragment Analyzer, Advanced Analytical).
Cell culture
A549 (ACC-107) and HEK293 (ACC-305) cells were purchased from DSMZ. 16HBE14o- cells were kindly provided by Professor Weber (University of Muenster, Germany).
All cells were cultivated in minimum essential media (MEM) with Glutamax (Gibco/Life Technologies). Media were supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco/Life Technologies) and 1% penicillin/streptomycin (Gibco/Life Technologies). Cells were cultured in a humidified 5% CO2 incubator at 37°C.
In vitro transfection
A549 and HEK293 cells were seeded at the density of 2 × 104 cells/well and 4 × 104 cells/well, respectively, in a 96-well plate, for the purpose of firefly luciferase, FACS measurements and EPO ELISA assay. 16HBE14o- cells were seeded in a 6-well plate at the density of 7.5 × 105 cells/well, for the purpose of western blot analysis. At 24-h post-seeding, cells were transfected using the commercial transfection reagent Lipofectamine 2000 (Thermo Fischer Scientific). Complexes were prepared at a ratio of 2 µL Lipofectamine 2000 per 1 µg mRNA. A549 and HEK293 cells were transfected with 250 ng/well and 250 and 125 ng/well mRNA, respectively. For experiments in A549 and HEK293 cells, required amounts of mRNA were diluted in water and the needed amounts of Lipofectamine 2000 in serum-free MEM. mRNA was added to the Lipofectamine 2000 solution followed by 20 min incubation at RT. The concentration of the final mRNA/Lipofectamine 2000 solution was 25 ng/µL. Ten microliters of the complex solution was added to the cells and cells were incubated for 24 h. For every mRNA construct, replicates of three or six were prepared. For 16HBE14o- cells, Lipofectamine MessengerMax was used due to its superior transfection efficiency (data not shown). For transfection, 7.5 µg mRNA was diluted in 125 µL water, and 11.25 µL Lipofectamine MessengerMax separately in 125 µL serum-free MEM. The mRNA solution was added to the Lipofectamine MessengerMax solution followed by 5 min incubation time at RT. A total volume of 250 µL of the lipoplex solution was added to the cells containing 2 mL normal growth media. The media was changed 4 h after transfection.
Flow cytometry analysis for d2EGFP
Cells were washed with PBS, detached with TrypLE (Gibco/Life Technologies), and re-suspended in flow cytometry buffer (PBS supplemented with 10% FBS). Shortly before measurement, cells were stained with propidium iodide for discrimination between live and dead cells (1 µg/mL; Sigma Aldrich). Analysis was performed on an Attune Acoustic Focusing Cytometer (Life Techologies) with Attune Cytometric Software (version 2.1; Life Technologies) and FlowJo (version 10).
Firefly luciferase assay
For detection of firefly luciferase activity, the assay was performed 24 h post-transfection. Cells were washed with PBS, followed by addition of 100 µL lysis buffer (25 mM Tris-HCl, 0.1% TritonX-100, pH 7.4). Cells were shaken for 20 min at room temperature. After lysis, 50 µL of the cell lysate was used to measure luciferase activity via photon luminescence emission for 5 sec using InfiniteR 200 PRO (Tecan). The protein amount in each sample was quantified in 5 µL of the cell lysate with Bio-Rad protein assay (Bio-Rad), using bovine serum albumin as a standard. Luciferase values were normalized to the protein concentrations.
Western blot analysis for human CFTR
Cells were lysed with RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% sodium dodecyl sulfate). Total protein amount was determined by the bicinchoninic acid (BCA) assay, following manufacturer's instructions (Thermo Fisher Scientific). Cell lysates were separated on 3%–8% TRIS-Acetate gels (Thermo Fisher Scientific) and transferred to polyvinylidene fluoride (PVDF) blotting membranes (Bio-Rad). Membranes were blocked in Western Breeze blocking buffer (Thermo Fisher Scientific) and probed with antibodies against CFTR (R&D Systems MAB25031; 1:2.000) and Hsp90 (Origene TA500494; 1:15.000). Horseradish peroxidase (HRP)-conjugated anti-mouse antibody (1:10.000; ab6820; Abcam) was used as secondary antibody. Blots of CFTR were developed using Super Signal West Femto (Thermo Fischer Scientific), and of Hsp90 using Luminata Forte Western HRP Substrate (Millipore).
Enzyme-linked immunosorbent assay for hEPO
Quantification of hEPO protein in cell supernatants was performed using human Erythropoietin Quantikine IVD ELISA kit (R&D Systems) following manufacturer's instructions.
RNA isolation and reverse transcription
RNA was isolated at different time points post-transfection using Single Shot Cell Lysis kit (Bio-Rad) following manufacturer's protocol. Prior to RNA extraction, the cell culture media was removed and cells were washed twice with PBS before being lysed in respective RNA isolation buffer. From the lysates (1 µg of RNA), cDNA was synthesized using iScript Select cDNA Synthesis kit (Bio-Rad) with oligo(dT) primers following manufacturer's instructions. The synthesized cDNA was stored at −20°C.
Quantitative real-time polymerase chain reaction (qPCR)
Real-time qPCR was performed with short hydrolysis probes for d2EGFP and Luciferase targets (Universal Probe Library #37 and #29; Roche) on a Roche Light Cycler 96 (Roche Diagnostics). For d2EGFP, the following primers were used: 5′-cctgaagttcatctgcacca-3′ and 5′-ctcgtgaccaccctgacc-3′. Luciferase mRNA was quantified using the following primers, 5′-acgccgagtacttcgagatg-3′ and 5′-attcagcccatagcgcttc-3′. Absolute mRNA values were calculated by interpolation from the standard curve.
Statistical analysis
Each experiment was performed with at least three technical replicates per sample. Results are shown as means ± SD unless otherwise stated. Statistical analysis was performed using GraphPad Prism software (version 6). Data were tested for normal distribution using D'Agostino-Pearson omnibus normality test. Multiple comparisons were conducted by two-way ANOVA, followed by Sidak's test (pairwise comparison) or Dunnett's test (many-to-one comparison). A P-value ≤ 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
The project was supported by the NIM Excellence Cluster.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.069286.118.
REFERENCES
- Baer BW, Kornberg RD. 1980. Repeating structure of cytoplasmic poly(A)-ribonucleoprotein. Proc Natl Acad Sci 77: 1890–1892. 10.1073/pnas.77.4.1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer BW, Kornberg RD. 1983. The protein responsible for the repeating structure of cytoplasmic poly(A)-ribonucleoprotein. J Cell Biol 96: 717–721. 10.1083/jcb.96.3.717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, Laska ME, Smith M, Almarsson Ö, Thompson J, et al. 2017. Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol Ther 25: 1316–1327. 10.1016/j.ymthe.2017.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmayor ER, Geiger JP, Aneja MK, Berezhanskyy T, Utzinger M, Mykhaylyk O, Rudolph C, Plank C. 2016. Chemically modified RNA induces osteogenesis of stem cells and human tissue explants as well as accelerates bone healing in rats. Biomaterials 87: 131–146. 10.1016/j.biomaterials.2016.02.018 [DOI] [PubMed] [Google Scholar]
- Balmayor ER, Geiger JP, Koch C, Aneja MK, van Griensven M, Rudolph C, Plank C. 2017. Modified mRNA for BMP-2 in combination with biomaterials serves as a transcript-activated matrix for effectively inducing osteogenic pathways in stem cells. Stem Cells Dev 26: 25–34. 10.1089/scd.2016.0171 [DOI] [PubMed] [Google Scholar]
- Bangel-Ruland N, Tomczak K, Fernández Fernández E, Leier G, Leciejewski B, Rudolph C, Rosenecker J, Weber WM. 2013. Cystic fibrosis transmembrane conductance regulator-mRNA delivery: a novel alternative for cystic fibrosis gene therapy. J Gene Med 15: 414–426. 10.1002/jgm.2748 [DOI] [PubMed] [Google Scholar]
- Bernal JA. 2013. RNA-based tools for nuclear reprogramming and lineage-conversion: towards clinical applications. J Cardiovasc Transl Res 6: 956–968. 10.1007/s12265-013-9494-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon G, Weissman D. 2002. RNA based vaccines. DNA Cell Biol 21: 953–961. 10.1089/104454902762053882 [DOI] [PubMed] [Google Scholar]
- Cao GJ, Sarkar N. 1992. Identification of the gene for an Escherichia coli poly(A) polymerase. Proc Natl Acad Sci 89: 10380–10384. 10.1073/pnas.89.21.10380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferizi M, Aneja MK, Balmayor ER, Badieyan ZS, Mykhaylyk O, Rudolph C, Plank C. 2016. Human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. Sci Rep 6: 39149 10.1038/srep39149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godiska R, Mead D, Dhodda V, Wu C, Hochstein R, Karsi A, Usdin K, Entezam A, Ravin N. 2009. Linear plasmid vector for cloning of repetitive or unstable sequences in Escherichia coli. Nucleic Acids Res 38: e88 10.1093/nar/gkp1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstrohm AC, Wickens M. 2008. Multifunctional deadenylase complexes diversify mRNA control. Nat Rev Mol Cell Biol 9: 337–344. 10.1038/nrm2370 [DOI] [PubMed] [Google Scholar]
- Grier AE, Burleigh S, Sahni J, Clough CA, Cardot V, Choe DC, Krutein MC, Rawlings DJ, Jensen MC, Scharenberg AM, et al. 2016. pEVL: a linear plasmid for generating mRNA IVT templates with extended encoded poly(A) sequences. Mol Ther Nucleic Acids 5: e306 10.1038/mtna.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudzien-Nogalska E, Kowalska J, Su W, Kuhn AN, Slepenkov S V, Darzynkiewicz E, Sahin U, Jemielity J, Rhoads RE. 2013. Synthetic mRNAs with superior translation and stability properties. In Synthetic messenger RNA and cell metabolism modulation: methods and protocols, methods in molecular biology (ed. Rabinovich PM), Vol. 969, pp. 55–72. Springer Science+Business Media, New York. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, et al. 2003. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348: 255–256. 10.1056/NEJM200301163480314 [DOI] [PubMed] [Google Scholar]
- Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, Tureci O, Sahin U. 2006. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108: 4009–4017. 10.1182/blood-2006-04-015024 [DOI] [PubMed] [Google Scholar]
- Kapeli K, Yeo GW. 2012. Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front Neurosci 6: 1–15. 10.3389/fnins.2012.00144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karikó K, Buckstein M, Ni H, Weissman D. 2005. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23: 165–175. 10.1016/j.immuni.2005.06.008 [DOI] [PubMed] [Google Scholar]
- Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, Weissman D. 2008. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16: 1833–1840. 10.1038/mt.2008.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kormann MSD, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, Huppmann M, Mays LE, Illenyi M, Schams A, et al. 2011. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol 29: 154–157. 10.1038/nbt.1733 [DOI] [PubMed] [Google Scholar]
- Kowalska J, Del Nogal AW, Darzynkiewicz ZM, Buck J, Nicola C, Kuhn AN, Lukaszewicz M, Zuberek J, Strenkowska M, Ziemniak M, et al. 2014. Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res 42: 10245–10264. 10.1093/nar/gku757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn U, Wahle E. 2004. Structure and function of poly(A) binding proteins. Biochim Biophys Acta 1678: 67–84. 10.1016/j.bbaexp.2004.03.008 [DOI] [PubMed] [Google Scholar]
- Lodish HF. 2012. Translational control of protein synthesis: the early years. J Biol Chem 287: 36528–36535. 10.1074/jbc.X112.420356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangus DA, Evans MC, Jacobson A. 2003. Poly(A)-binding proteins: multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol 4: 223 10.1186/gb-2003-4-7-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W. 1998. Tailing and 3′-end labeling of RNA with yeast poly(A) polymerase and various nucleotides. RNA 4: 226–230. [PMC free article] [PubMed] [Google Scholar]
- Mullard A. 2016. mRNA-based drug approaches Phase I milestone. Nat Rev Drug Discov 15: 595 10.1038/nrd.2016.182 [DOI] [PubMed] [Google Scholar]
- Oliveira CC, McCarthy JEG. 1995. The relationship between eukaryotic translation and mRNA stability. J Biol Chem 270: 8936–8943. 10.1074/jbc.270.15.8936 [DOI] [PubMed] [Google Scholar]
- Preiss T, Muckenthaler M, Hentze MW. 1998. Poly(A)-tail-promoted translation in yeast: implications for translational control. RNA 4: 1321–1331. 10.1017/S1355838298980669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, et al. 2017. Modified mRNA vaccines protect against Zika virus infection. Cell 169 10.1016/j.cell.2017.03.016 [DOI] [PubMed] [Google Scholar]
- Sachs A. 1990. The role of poly(A) in the translation and stability of mRNA. Curr Opin Cell Biol 2: 1092–1098. 10.1016/0955-0674(90)90161-7 [DOI] [PubMed] [Google Scholar]
- Schrom E, Huber M, Aneja M, Dohmen C, Emrich D, Geiger J, Hasenpusch G, Herrmann-Janson A, Kretzschmann V, Mykhailyk O, et al. 2017. Translation of angiotensin-converting enzyme 2 upon liver- and lung-targeted delivery of optimized chemically modified mRNA. Mol Ther Nucleic Acids 7: 350–365. 10.1016/j.omtn.2017.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thess A, Grund S, Mui BL, Hope MJ, Baumhof P, Fotin-Mleczek M, Schlake T. 2015. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol Ther 23: 1456–1464. 10.1038/mt.2015.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepotec Z, Aneja M, Geiger J, Hasenpusch G, Plank C, Rudolph C. 2018. Maximizing the translational yield of mRNA therapeutics by minimizing 5′-UTRs. Tissue Eng Part A 25: 69–79. 10.1089/ten.TEA.2017.0485 [DOI] [PubMed] [Google Scholar]
- Vallazza B, Petri S, Poleganov MA, Eberle F, Kuhn AN, Sahin U. 2015. Recombinant messenger RNA technology and its application in cancer immunotherapy, transcript replacement therapies, pluripotent stem cell induction, and beyond. Wiley Interdiscip Rev RNA 6: 471–499. 10.1002/wrna.1288 [DOI] [PubMed] [Google Scholar]
- Voet D, Voet JG. 2011. Biochemistry. 4th ed. John Wiley & Sons, New York. [DOI] [PubMed] [Google Scholar]
- Wang Z, Day N, Trifillis P, Kiledjian M. 1999. An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol Cell Biol 19: 4552–4560. 10.1128/MCB.19.7.4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman D. 2015. mRNA transcript therapy. Expert Rev Vaccines 14: 265–281. 10.1586/14760584.2015.973859 [DOI] [PubMed] [Google Scholar]
- Wells SE, Hillner PE, Vale RD, Sachs AB. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2: 135–140. 10.1016/S1097-2765(00)80122-7 [DOI] [PubMed] [Google Scholar]
- Williams AS, Marzluff WF. 1995. The sequence of the stem and flanking sequences at the 3′ end of histone mRNA are critical determinants for the binding of the stem-loop binding protein. Nucleic Acids Res 23: 654–662. 10.1093/nar/23.4.654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J, Malone R, Williams P, Chong W, Acsadi G, Jani A, Felgner P. 1990. Direct gene transfer into mouse muscle in vivo. Science 247: 1465–1468. 10.1126/science.1690918 [DOI] [PubMed] [Google Scholar]
- Zanier K, Luyten I, Crombie C, Muller B, Schümperli D, Linge JP, Nilges M, Sattler M. 2002. Structure of the histone mRNA hairpin required for cell cycle regulation of histone gene expression. RNA 8: 29–46. 10.1017/S1355838202014061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemniak M, Strenkowska M, Kowalska J, Jemielity J. 2013. Potential therapeutic applications of RNA cap analogs. Future Med Chem 5: 1141–1172. 10.4155/fmc.13.96 [DOI] [PubMed] [Google Scholar]