

Abstract
Lead identification and optimization are essential steps in the development of a new drug. It requires cost-effective, selective and sensitive chemical tools. Here, we report a novel method using nanobodies that allows the efficient screening for potent ligands. The method is illustrated with the cystic fibrosis transmembrane conductance regulator inhibitory factor (Cif), a virulence factor secreted by the opportunistic pathogen Pseudomonas aeruginosa. 18 nanobodies selective to Cif were isolated by bio-panning from nanobody-phage library constructed from immunized llama. 8 out of 18 nanobodies were identified as potent inhibitors of Cif enzymatic activity with IC50s in the range of 0.3–6.4 μM. A nanobody VHH219 showed high affinity (KD=0.08nM) to Cif and the highest inhibitory potency, IC50=0.3 μM. A displacement sandwich ELISA (dsELISA) with VHH219 was then developed for classification of synthetic small molecule inhibitors according their inhibitory potency. The developed assay allowed identification of new inhibitor with highest potency reported so far (0.16±0.02 μM). The results from dsELISA assay correlates strongly with a conventional fluorogenic assay (R= 0.9998) in predicting the inhibitory potency of the tested compounds. However, the novel dsELISA is an order of magnitude more sensitive and allows the identification and ranking of potent inhibitors missed by the classic fluorogenic assay method. These data were supported with Octet biolayer interferometry measurements. The novel method described herein relies solely on the binding properties of the specific neutralizing nanobody, and thus is applicable to any pharmacological target for which such a nanobody can be found, independent of any requirement for catalytic activity.
Keywords: competitive sandwich ELISA, Pseudomonas aeruginosa, screening, inhibitory nanobody, drug
Graphical Abstract
1. Introduction
The bacterial species Pseudomonas aeruginosa is a common opportunistic pathogen found widely in the environment. It is responsible for morbidity and high mortality of critically ill or immunocompromised patients with infections following surgery, with implanted medical devices, catheters and wounds.1,2 A key bacterial survival strategy involves the formation of biofilms that confer on P. aeruginosa high levels of resistance to antibiotics. Once established, biofilms of P. aeruginosa are very difficult to eliminate and require costly treatments, making them a target of therapeutic development.3 P. aeruginosa is particularly dangerous to patients with underlying airway diseases, since it can establish chronic lung infections.4 The bacterium causes ~50% of acute exacerbations in chronic obstructive pulmonary disease (COPD), which affects about 24 million US patients.5 COPD is the 4th leading cause of death in US, and treatment costs exceed $40 billion per year.6 P. aeruginosa is also the leading cause of fatal episodes of ventilator-associated pneumonia, with mortality rates > 70%, and it is thought to cause 23–65% of cases of community-acquired pneumonia.7 P. aeruginosa eventually colonizes >60% of individuals with cystic fibrosis (CF) and is a major contributor to respiratory failure in most of these patients. The CF patient population is estimated at 30,000 in the US and 70,000 worldwide, with average treatment costs per patient of over $ 94,000 per year.8 To establish and maintain infections, the bacterium secretes a variety of virulence factors. One of them triggers degradation of the cystic fibrosis transmembrane conductance regulator (CFTR).9 The CFTR inhibitory factor (Cif), which has putative orthologs in several opportunistic airway pathogens,10 is an epoxide hydrolase. Its catalytic activity affects host mucociliary and antiviral defenses and appears to facilitate infection of the lungs.11,12 It also degrades a host factor that promotes pro-resolution signaling.13 Interestingly, Cif has also been implicated in eye infections mediated by P. aeruginosa.14 As a result, targeting Cif could weaken the ability of the bacterium to establish infections and exacerbate inflammatory responses, thus reducing patient burden.
Small-molecule inhibitors for Cif have been developed using crystal structures and medicinal-chemistry approaches.15 The obtained compounds are potent (IC50 < 300nM) and display at least 25-fold selectivity for Cif over off-target proteins such as the human epoxide hydrolases and thyroid hormone receptor. However, further improvements in inhibitory potency towards the target are potentially needed, as well as optimization of in vivo half-life of the inhibitors, bioavailability and accessibility to target enzyme. A fluorescence-generating assay was employed to evaluate the potency of the inhibitors.11,16 Unfortunately, because of the low rate of turnover by Cif, the assay requires a protein concentration of at least 0.6 μM, and thus has correspondingly low sensitivity: it cannot distinguish among inhibitors with IC50 < 0.3 μM.15 Methods such as surface plasmon resonance (SPR), bio-layer interferometry (ForteBio Octet) or LC-MS/MS detection have lower detection limits; however, they are laborious, time-consuming and involve expensive instrumentation. Thus, throughput is low and constrains quick evaluation of new compounds. Scintillation proximity assay17,18, fluorescence resonance energy transfer (FRET)19 and fluorescence polarization20 approaches are successfully used for screening and determining the potency of drug candidates for proteins with low activity or no activity, such as transporters or receptors. However, development of an appropriate reporter substrate can be a challenging and labor-intensive task, very similar to the stages of scaffold identification and affinity optimization in the development of a drug candidate. Therefore, there is a need of more efficient methods for inhibitors identification and ranking according to their inhibitory potencies suitable for slow enzymes and proteins without catalytically properties, like receptors, transporters … Development of such assay is the objective of this work.
Compared to small-molecule ligands, antibodies generally bind more tightly and with higher selectivity towards their targets.21,22 While antibodies can be readily obtained through the affinity maturation process in a host animal, they are often limited in applications by their size, stability or purity. Nanobodies or VHHs (variable heavy domain on heavy chain only antibodies) are very small recombinant antibody fragments that offer the advantages of both small-molecule ligands (ease of production, purity, stability, and solubility) and antibodies (high potency, ease of labeling with reporter molecules such as a fluorescent probe).22–24 We hypothesize that inhibitory nanobodies may be a suitable tool to study protein-ligand interaction. Here, we report the use of nanobodies in a novel assay format as a tool for the screening of small-molecule inhibitors. As a model of a slow turnover enzyme system, we used Cif. The idea of using an inhibitory nanobody to displace a small-molecule inhibitor from the active site is simple but innovative.
2. Materials and Methods.
All chemicals were of analytical grade and were purchased either from Fisher Scientific Co. (Chicago. Il, USA) or from Sigma-Aldrich Co (St. Louis. MO, USA) unless otherwise stated. All the reagents and instruments were purchased from USA companies.
2.1. Buffers and other reagents.
All buffers and water solutions were prepared with ultrapure deionized water; phosphate-buffered saline (PBS, 10mM, pH 7.5); wash buffer PBST (PBS containing 0.05% Tween 20); coating buffer (14 mM Na2CO3, 35 mM NaHCO3, pH 9.8); blocking buffer (1% BSA w/v in PBST); substrate buffer (0.1 M sodium citrate/acetate buffer, pH 5.5). Substrate solution contained 0.2 mL of 0.6% 3,3’,5,5’-tetramethylbenzidine (TMB in pure dimethyl sulfoxide, DMSO w/v), 0.05 mL of 1% H2O2 in 12.5mL of substrate buffer. Stop solution was 2M H2SO4.
The detailed procedures for inhibitors synthesis, their characterization and other facts are given in supporting information.
2.2. Immunization and library construction.
An alpaca was immunized with the Cif protein (200 μg per injection) in complete Freund’s adjuvant and boosted 4 times with the same dose in incomplete Freund’s adjuvant. Blood was drawn every two weeks to assess the antibody titer. Alpaca immunizations and blood handling were performed by Triple J Farms (Bellingham, WA). A VHH phage-display library was constructed as previously described.25 The mRNA was extracted using LeukoLOCK™ Total RNA Isolation System (Thermo Fisher) and transcribed to cDNA with SuperScript III RT System (Invitrogen). The cDNA was then used as template for PCR amplification of the VHH genes in the first step using primers Call001 and Call002, and in the second step using the forward primers, VH1, VH3, VH4, VHH1 Back, VHH 6 Back, F and F2D and the reverse primer JH (Table S1).25–27 The amplified DNA was digested with SfiI and ligated into pComb3X (gift from Dr. Carlos Barbas, The Scripps Research Institute, La Jolla, USA). The resulting vectors were electroporated into electrocompetent cells E. coli ER2738. The cells were cultured and the phage library displaying the VHH repertoire was created by superinfection with helper phage M13KO7. Library diversity was evaluated through sequencing of 20 random clones.
2.3. Phage library panning for Cif selective clones.
The panning procedure was performed based on protocols described by Barbas 28 Two isolation approaches were used. Approach 1: A library was panned against Cif bound to a purified polyclonal anti-Cif antibody (pAb).13 Two wells of a microtiter plate were coated with pAb (0.3 μg/well) in coating buffer at room temperature (RT) for 1 h. These two wells, plus an additional four wells, were blocked/coated with 200 μL of 1% w/v BSA in PBS for at least 2 h at RT. One of the wells coated with pAb was then incubated with a decreasing amount of Cif at each round of panning. An aliquot of the phage library (100 μL) was added to a well coated with pAb only and incubated for 1 h at RT to remove non-specific pAb binders. All unbound phage was transferred to the well coated only with BSA, 25 μL per well, to remove nonspecific BSA binders. After another 1 h incubation, unbound phages were transferred to a well coated with both pAb and Cif and incubated for 1 h at RT. After washing with PBST, this well was eluted with 100 μL of trypsin at 10 mg/mL in PBS at 37°C for 30 min. Approach 2: The library was panned against Cif directly bound to the plate. The procedure is similar to Approach 1 but does not involve the pAb steps. Details on panning conditions are provided in Table S2. The eluent was collected and amplified with addition of the M13KO7 helper phage (1×1012 cfu/mL). The titer was assessed for the output library after panning and for the input library after amplification. ER2738 E. coli were infected with the eluted or amplified phages and titered on LB-carbenicillin agar plates. 100 μL of the amplified phages was employed again in the next round of panning. For the second, third, and fourth rounds, the same procedure was used, except the concentration of Cif was gradually decreased. After the final round of panning, individual clones were screened to identify positive clones by performing a phage ELISA.
2.4. Expression and purification of Cif VHHs.
From the agar plate containing the fourth elution output, 54 individual clones from Approach 1 and Approach 2 were randomly selected and grown individually in cultures overnight preliminary induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cultures for each clone were spun down at 3,000 × g for 10 min at 4 °C. For the protein extraction, the bacterial protein extraction reagent kit (B-PER) was employed, and the obtained protein was further characterized by phage ELISA (for details see the SI). For positive clones the plasmid was extracted from cultures with the Qiagen Mini Prep kit and the sequences were submitted to the UC Davis DNA Sequencing Facility. The plasmids pComb3X, containing positive VHH clones, were transformed by heat shock into Top 10F’ cells.
The expression and purification of VHHs was performed as previously described.28–30 A 1-mL aliquot of overnight culture was diluted in 100 mL of Super Broth with 50 03BCg/mL carbenicillin. After the OD600 reached 0.5–1, the culture was induced with 1 mM IPTG and incubated in a shaker at 37 °C overnight. The culture was centrifuged, and the cell pellet was lysed with B-PER lysis buffer at 4 mL/g pellet containing protease inhibitors (cocktail, Thermo Fisher) at ambient temperature for 10 min. The cell lysate supernatants were clarified by centrifugation at 13000 × g for 10 min, followed by purification on a 1-mL Ni-NTA resin column. The column was equilibrated and washed with 40 mM imidazole (dissolved in 10 mM PBS, pH 7.4). The VHH was eluted with 150 mM imidazole, and the purified VHH was stored at −20 °C after desalting on Zeba desalting column (Thermo Fisher). The size and purity of the proteins were verified on NuPAGE 12% Bis-Tris Protein gel (Thermo Fisher, USA) according to the manufacturer protocol.
2.5. High yield expression of inhibitory VHHs.
The VHH genes were cloned in the pET 28a (+) vector, flanked by the coding sequences of the ompA signal peptide at the 5′ end, and 6 x His and the HA epitope coding sequences (vector generously provided by Dr. Gonzalez-Sapienza, UDELAR, Uruguay). The vector was transformed into BL21(DE3) E. coli, and individual clones were grown on LB-kanamycin (40 μg/mL) plates. The culture of single clones was incubated until the OD600 reached 0.5 and nanobody expression was induced with 10 μM IPTG during 4 h at 37 °C. Cells were pelleted and the periplasmic proteins were extracted by osmotic shock as described previously.31 Nanobody purification was performed on Ni-NTA columns in the FPLC purification system (Bio-Rad) according to the manufacturer’s instructions.
2.6. VHH-based assay.
The optimal concentration of the coating anti-Cif pAb was determined as a minimal amount of the pAb that produces a saturating signal with secondary antibody, and it corresponded to 0.3 μg/well (Fig. S1A). The optimal concentrations of Cif and VHHs for both types of assay were determined from a checkerboard titration (Fig.S1 B and C). While for simplicity the following assays are performed at room temperature (RT ≈ 23°C), we did not observe any change in performance between RT and 37°C incubations. All washing steps were performed with the automated washing system Aqua Max 2000 (Molecular Devices) following the vendor protocol.
1). Immunoassay.
A 96-well plate was coated with 100 μL per well of pAb at 3 μg/mL at RT for 1 h. The plate was blocked with 1 % w/v skimmed milk in PBST at RT for at least 2 h. Cif diluted in PBS was loaded on the plate at increasing concentrations and incubated for 1 h at RT. Following 5 consecutive washes with PBST, VHH was loaded at 0.5 μg/mL in PBS, 100 μL per well (or 50 ng/well). The plate was incubated for 1 h at RT and then washed 5 times with wash buffer. Rat monoclonal anti-HA-HRP conjugate was added at 100 μL/well in a 1:3000 or 0.8 ng per well (Roche, clone 3F10, Sigma-Aldrich). The plate was incubated for 1 h at RT and washed 5 times. The plate was developed for 10 min with substrate solution added at 100 μL/well. The reaction was stopped by addition of 2M H2SO4 (50 μL/well) and absorbance was read at 450 nm. SigmaPlot 11.0 software was used for curve fitting and data analysis.
2). . Displacement sandwich ELISA.
A 96-well plate was coated with pAb at 3 μg/mL, 100 μL per well at RT for 1 h. The plate was blocked with 1 % w/v skimmed milk in PBST at RT for at least 2 h. Cif diluted in PBS was loaded at 20 ng/mL, 100 μL/well and incubated for 1 h at RT. Following 5 consecutive washes with PBST, a serial dilution of small-molecule synthetic inhibitors in PBS containing 20% v/v methanol at 50 μL/well was loaded, followed by 50 μL/well of VHH at 0.1 μg/mL (or 5 ng/well) in PBS and incubated for 1 h at RT. Rat anti-HA-HRP conjugate was added at 100 μl/well in a 1:3000 fold dilution. The plate was incubated for 1 h at RT and washed 5 times. The plate was developed for 10 min with substrate solution added at 100 μL/well. The reaction was stopped by addition of 2M H2SO4 (50 μL/well) and absorbance was read at 450 nm. SigmaPlot 11.0 software was used for curve fitting and data analysis.
3. Results
3.1. Selection of anti-Cif VHHs.
Two approaches to panning for Cif-selective nanobodies were used. Panning in both approaches was performed with decreasing Cif concentrations in each round thus promoting the selection of clones with higher affinity (Table S2). For the first approach, in an effort to preserve Cif’s three-dimensional structure, rabbit polyclonal anti-Cif antibody (pAb) was used to present Cif to the phage library of nanobodies. To ensure that the selected nanobodies recognized only Cif, the library was counter-screened with pAb and BSA to remove non-Cif binders. The phages were collected and used in the following panning steps. After four rounds of panning, an ELISA performed with an amplified enriched phage library showed a strong signal selective to Cif as compared to a non-panned library. This first approach of course reduces the chance of finding a nanobody which will bind to epitopes recognized by the pAb but increases the chance of finding a nanobody that will be valuable in a sandwich assay with the pAb. For the second approach, the screening plate was directly coated with the Cif protein, which can result in deformed and partially denatured protein. While the selective signal to Cif increased after two rounds of panning, it then decreased dramatically approaching non-specific signal as in a non-panned library (data not shown).
Overall, 24 individual clones were picked after four rounds of panning from the first approach, and 30 clones were picked after two rounds of panning from the second approach. All selected phage clones showed recognition of Cif in the ELISA. The DNA of corresponding clones was isolated. Among the 24 clones obtained from the first approach, there were nine unique sequences. However, three of the nine unique clones did not show satisfactory protein expression in later experiments, leaving six for further analysis. Among the 30 clones obtained in the second approach, three had already been identified in the first approach. Among the remaining 27 clones, there were 12 new unique sequences. Thus, in total, we identified 18 unique clones selective for Cif. The corresponding sequences are shown in Figures 2 and S2. The phagemid vectors containing DNA of unique clones were transformed in TOP10 F’ cells and expressed VHHs were purified using Ni-NTA affinity chromatography. The size and purity of the proteins were verified on 12% SDS-PAGE gels with a major band at the expected MW of approximately 17000 (Image S1).
Figure 2.

Sequences of the selected nanobodies. FR indicates framework, or conserved domain of the nanobody; CDR stands for complementarity-determining region, a variable domain of the nanobody responsible for selective recognition of the target analyte; TAG includes 6xHis tag and hemagglutinin (HA) tag.
To evaluate the resulting nanobodies, a classical sandwich ELISA was performed using purified rabbit polyclonal anti-Cif antibodies as a capturing antibody (Fig. 1). pAb at 0.3 μg/well was selected by checkerboard titration as a minimal amount of antibody necessary to maximize the signal from the secondary anti-rabbit antibody conjugated to HRP. Cif protein was loaded in a serial dilution range of 0.05–100 ng/mL, and 50 ng of VHHs were added to each well. The concentration of VHHs used in the assay was chosen based on previously published literature and was not further optimized.29,30 The sensitivity of the assays resulting from these clones varied by one order of magnitude with IC50 values ranging from 6 to 51 ng/mL (Fig. 1). Further optimization of the assay with selected clones for the development of analytical tool for Cif quantification in biological samples will be the subject of separate study.
Figure 1.

ELISA curves for 18 positive clones. The sensitivity of the assay varied in the range of 6 to 50 ng/mL. Conditions: pAb anti-Cif 0.3 μg/well; serial dilution of Cif in PBS; VHH 50 ng/well in PBS; anti-HA mAb dilution 1:3000 in PBST.
3.2. Identification and characterization of inhibitory nanobodies.
The fluorescent enzyme activity assay was used to test whether the nanobodies had any inhibitory activity toward Cif. The small molecule inhibitor KB2115 (commercial name) was used as a positive control, while a non-Cif nanobody was used as a negative control. Eight nanobodies were identified as having significant inhibitory properties (Table S3). Nanobodies 113 (from the first panning approach) and 219 (from the second panning approach) were subsequently identified as the most potent inhibitors of Cif having IC50 values of 0.4 and 0.3 μM respectively (Table 1). It should be noted that potencies of both nanobodies reached the lower limit of assay sensitivity equal to half the Cif concentration (0.3 μM), explaining multi-orders of magnitude difference between IC50 values and dissociation constants. In addition, these two biologicals had IC90 values around 1 μM, confirming their high potency against Cif. These two VHHs were selected for further investigation of their potency.
Table 1.
Inhibitory potency of VHHs selective to Cif measured with fluorescence-based assay.
| Nanobody # | IC50 (μM) | IC90 (μM) |
|---|---|---|
| KB2115* | 14.8±3.8 | > 100 |
| 113 | 0.4±0.0 | 1±0.1 |
| 219 | 0.3±0.0 | 0.9±0.1 |
| 313 | 1.5±0.0 | 4.1±0.1 |
| 114 | 0.9±0.0 | 2.4±0.1 |
| 212 | 0.6±0.0 | 1.6±0.1 |
| 101 | 6.4±0.3 | >14.7 |
| 222 | 0.6±0.1 | 2.0±0.6 |
| 214 | 0.5±0.0 | 1.6±0.1 |
Small molecule inhibitor, commercial name, positive control, see Table 3 for structure; Conditions: [Cif] = 0.6 μM, substrate [MNCG] = 25 μM, 37°C, mean ± SD, n = 3. IC50 corresponds to the concentration that leads to 50% decrease (inhibition) of signal, and IC90 corresponds to the concentration leading to 90% decrease of signal.
Single-cycle binding kinetics were measured by SPR with increasing concentrations of the VHHs. The VHHs showed very tight binding to the Cif-protein evidenced by long dissociation times (Fig. S4). Therefore, the rate of association (ka) was measured from 5 forward reactions where contact time was set at 5 min each, and the dissociation rate (kd) was measured from one reverse reaction with data collection for 20 min for a smooth fit. The curve was fitted to a 1 : 1 binding model, the simplest model for the description of the interaction between antigen and a monovalent antibody, allowing the calculation of the equilibrium dissociation rate constant (KD) as a ratio of kd/ka (Table 2, Fig. S4). Both nanobodies have very high ka and very slow kd values, resulting in very tight KD values, in the mid-picomolar range. The equilibrium dissociation constant for VHH113 (0.13 nM) was roughly twice as large (i.e. half as potent) as the value for VHH219 (0.08 nM), which was due to both a larger ka and a smaller kd (Table 2). These are relatively high affinities compared to the published literature on affinities of nanobodies for their protein substrates, with values usually in the range of 10–500 nM.32–35 Interestingly, Rossotti et al. 34 reported nanobodies to human sEH with slightly weaker KD values around 0.5 nM using a similar panning protocol.
Table 2.
Kinetic and equilibrium parameters of two nanobodies binding to Cif obtained from surface plasmon resonance experiments
| Clone | ka (1/Ms) | kd (1/s) | KD (nM) |
|---|---|---|---|
| VHH113 | 5.3×105 | 6.9×10−5 | 0.13 |
| VHH219 | 7.4×105 | 5.9×10−5 | 0.08 |
3.3. Development of a nanobody-based screening assay.
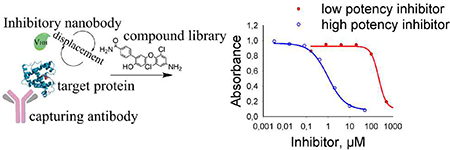
To screen for potent small molecule chemical inhibitors of Cif, a sandwich displacement assay was designed (Fig. 3A). In this assay, the plate was first coated with anti-Cif pAb to capture the Cif protein. Inhibitory anti-Cif VHH in the role of a reporter was added at constant concentration together with a serial dilution of a tested inhibitor. The mixture was incubated at room temperature for 1 h to allow the competition between the inhibitor of interest and the reporting VHH ligand to proceed. The non-bound VHH was washed away, and the remaining VHH attached to Cif was detected and quantified with a secondary antibody against HA tag labeled with HRP as reporting system (Fig. 3B). The concentration of the inhibitor that resulted in a 50% decrease of the HRP-generated signal was named the EC50 concentration, which is characteristic of each small molecule inhibitor in this system and is inversely proportional to the potency of the compounds tested.
Figure 3.

Displacement sandwich ELISA (dsELISA). A. Workflow scheme of the assay: the plate is coated with anti-Cif pAb and Cif, followed by incubation with the small molecule inhibitor and the nanobody. Unbound material is washed out and the bound nanobody is reveled with secondary anti-HA antibody labeled with HRP. Conditions: anti-Cif pAb 0.3 μg/well; Cif 20 ng/mL in PBS; VHH 5 ng/well in PBS; anti-HA mAb dilution 1:3000 in PBST. B. Inhibition curves for the tested compounds reveal high competitive capacity of the compound 8, followed by 7 and 6. Blue arrows indicate the order of increasing potency, where compound 8 is the most potent and compound 1 is the less potent. Compounds with higher potency have inhibitory curves shifted to the left of the graph.
In this assay, the amount of anti-Cif pAb loaded in the plate was first optimized. For this purpose, a serial dilution of pAb was loaded onto the plate, and after the incubation period, the amount of pAb bound to the plate was detected with an excess of secondary antibody. The minimum level of coating pAb per well that produced a saturating signal was 0.3 μg. Any further increase in antibody load did not produce further increases in signal (Fig. S1A). Second, the amount of Cif protein captured on pAb was optimized in a similar way as the first experiment; 20 ng/mL of Cif was sufficient to obtain a signal reaching a plateau. The quantity of nanobody in the assay (5 ng/well) was also optimized to provide the maximum signal possible while avoiding excess of the reagent (Fig.S1B and C). Both nanobodies VHH113 and VHH219 were evaluated in the inhibitory assay. Even though the concept of an inhibitory sandwich assay worked with both nanobodies, VHH113 consistently showed less good performance with lower data fit correlation coefficient (Figure S3. Therefore, further experiments were performed with VHH219 only.
The sandwich competitive assay with VHH219 was used to analyze a range of small molecule inhibitors previously reported by Kitamura et al.15 plus two additional inhibitors synthesized and reported in this work (Table 3, Table S4). Figure 3B shows the curves obtained with the sandwich ELISA while Table 3 provides structures of the tested compounds as well as both EC50 values obtained with the VHH-based assay and IC50 values obtained with the fluorescent assay. The sandwich competitive assay and enzyme activity assay have different principles of interaction, thus absolute values for IC50 and EC50 are different. Nevertheless, IC50 values from the enzyme activity assay correlated well with the EC50 values from the sandwich competitive assay (Pearson Correlation Coefficient= 0.9998, r2=0.9996). Most interestingly, while the fluorescent assay cannot distinguish the potencies of compounds 2–6 and 7–8 (IC50 values covered only a 2-fold range), the dsELISA EC50 values for the same compounds were distributed over a 15-fold range, yielding clear information for structure-activity relationships and future inhibitor design. For example, the VHH-based assay clearly identified compound 8 as far more potent toward Cif than the rest of the compounds tested.
Table 3.
Potency comparison of small molecule inhibitors in VHH-based (dsELISA) and fluorescent assays, and equilibrium parameter (KD) obtained from bio-layer interferometry experiments.
| Cmpd. # | Structure | dsELISA, EC50,μM | Fluo. assay, IC50,μM | KDa (μM) |
|---|---|---|---|---|
| 1T | 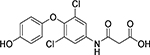 |
>200 | >50 | |
| KB2115 | 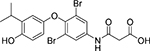 |
13±1 | 2.6 | |
| 2 T | 3.1±1.0 | 0.46 | ||
| 3 T | 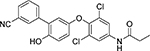 |
3.0±1.1 | 0.58 | |
| 4 T | 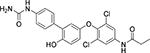 |
2.5±0.9 | 0.29 | 2.1 ± 0.7 |
| 5 T | 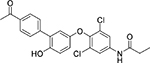 |
1.5±0.7 | 0.35 | |
| 6 T | 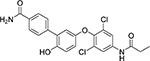 |
1.2±0.3 | 0.35 | 0.5 ± 0.1 |
| 7* | 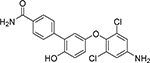 |
0.7±0.3 | 0.55 | 0.9 ± 0.1 |
| 8* | 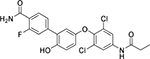 |
0.2±0.1 | 0.47 | 0.16 ± 0.02 |
Results are shown as mean ± SD (n = 2 or 3).
Data calculated based on steady state analysis
synthesized and reported in 15
synthesized and reported in this paper.
To verify these findings, we measured the affinity toward Cif protein of the new 8 and 7 compounds, as well as 6 and 4, as controls, with the Octet (Octet Red96, ForteBio). Results are reported in Table 3. The affinities follow the same pattern as the one observed for the EC50 values, with 8 as the most potent compound with a KD of 0.16±0.02 μM. Therefore, even though there was no way to verify directly the accuracy of the developed assay, the Octet experiment confirmed the ability of the VHH-based assay to differentiate among inhibitors according to their inhibitory potency. This is indirect validation of the accuracy of the dsELISA.
To further evaluate the assay performance, inter- and intra-day variation was assessed as well as the reproducibility of the results. To determine the intra-day variation, we analyzed results obtained on the same plate and day in duplicates and tested on two different days. The intra-day variation was generally below 20% (Figure S3). Inter-day variation was calculated by using results obtained within one week, in addition to results obtained over a five-month period. From these data variations in absolute EC50 value were found to be up to 45% (Fig. S5).
4. Discussion
A number of assay formats are successfully used in the drug development process, screening and determining the potency of drug candidates. Such assays, including scintillation proximity assay,17,18 FRET,19 and fluorescence polarization,20 are quantitative and satisfy criteria for high-throughput screening. They are performed in homogeneous conditions, where no incubation and washing are needed. Therefore, they are amenable to automation and are thus time and cost effective. However, these methods rely on use of an appropriate reporter substrate. In turn, the development of such a reporter is a separate challenging and labor-intensive task, similar to the initial stages of lead development. The use of inhibitory nanobodies as ligands in a competitive sandwich format can serve as an effective alternative to existing screening methods. The dsELISA determines an EC50 value for the molecule of interest corresponding to its inhibitory potency. The ranking of compounds potency is based on comparison of these values. The ranking procedure is similar to established fluorescence based assay. However, in dsELISA EC50 values vary in a larger range compared to IC50 values obtained with fluorescence assay for the same compounds. In addition, the dsELISA allowed the identification and ranking of potent inhibitors where fluorescence-based assay reached its limit of detection (0.5 μM). Therefore, dsELISA shows better sensitivity for inhibitors identification and ranking. The limit of detection (LOD) of the assay should indicate what would be the highest inhibitory potency of the compound that the assay could detect. It is hard to estimate this value without actual compounds (so far we reached potency of 0.2 μM with compound 8). However, based on the fact that dsELISA in principle is competitive ELISA for small molecule, we may assume that LOD will be similar to LOD in ELISA for small molecule that is low nM range.36,37
Similar to a classic antibody, the nanobody benefits from naturally occurring immune-maturation in the animal, after which desired potent clones are isolated by established techniques.27 However, the key to developing a powerful assay is to select a potent nanobody. Classically, for proteins, panning procedures involve constant high concentrations of proteins (100 μg/mL). Herein, an approach used for small molecules was deployed: starting with a low Cif concentration (10 μg/mL) and then further decreasing this concentration and increasing the number of washing cycles and stringency to enrich the pool with phages selective to the target analyte.25 In addition, coating the plate first with polyclonal anti-Cif antibody followed by the Cif protein was more efficient at selecting high-affinity nanobodies than directly coating the protein to the plate, in agreement with a previous observation.38 However, as noted earlier each approach offers advantages. When applied to Cif, these improvements in the panning procedure contributed significantly to the successful isolation of potent clones. Further, assisted presentation may be particularly useful for panning directed to a specific part of the molecule (e.g. binding pockets, catalytic sites etc.), when the site of interest is better exposed to the phage while the other part of the molecule is hidden.
By combining a classical sandwich ELISA with a competitive ELISA (see Fig. S6 for details), we created a novel inhibition assay that has the advantages and limitations from both. As a sandwich ELISA, the assay uses a primary antibody to present Cif. In this study, we used a polyclonal antibody, so the orientation of Cif was not controlled. It certainly selects for nanobodies useful in a sandwich assay with the pAb used. However, one can envision using a nanobody binding the protein from the side opposite to the drug candidate binding site to better present it. This selective orientation may result in higher binding capacity of the layer and thus overall increased sensitivity of the assay. As in a competitive ELISA, the core of the assay is a competition between two ligands: the reporting nanobody and the small molecule being tested. Therefore, the potency of a compound is read as a decrease from a high signal. This always results in lower sensitivity compared to the appearance of the signal from low background. Another limitation of the assay reported here is its heterogeneous format, requiring washing steps and defined incubation time. Therefore, it might be challenging to automate for a high throughout processing. The assay presented in this article is a proof of concept, and further optimization of the assay conditions may result in more stable assay performance. However, these limitations did not affect the rank of the compounds’ potencies. Once compounds are ranked, an absolute quantitative value can be measured by more precise analytical methods like SPR or Octet on a limited number of identified leads, as well as confirmation assays such as LC-MS of the substrate or the product.
There are three possible mechanisms of Cif inhibition by the nanobodies selected herein. Nanobodies are known to have the CDR3 regions unusually long, and they have been shown to possess the extraordinary capacity to form convex extensions that reach the cavities like active sites of enzymes, binding pockets of the transporters or receptors. A number of recent studies successfully demonstrated that nanobodies block the enzymatic35,39,40 and cytotoxic activity41 of proteins and modulate receptor functioning.42 Nanobodies also have the ability to inhibit enzymes through an allosteric modulation of enzymatic activity.43–45 The binding to an allosteric site, a site different from the active catalytic site of the enzyme, may induce conformational changes within the protein structure and thus conformation change of the catalytic site resulting in decreased substrate recognition. Finally, the nanobodies may create steric hindrance at the entrance to the active site thus preventing the substrate diffusion to the active site, like non-competitive inhibitors. For example, Zhu et al.35 demonstrated that a nanobody to furin inhibited the cleavage of furin natural substrates; however, smaller surrogate peptide substrates could still be hydrolyzed suggesting changes to the catalytic site access. In our study, the possible mechanisms of inhibition are still unknown.
To conclude, the displacement sandwich ELISA format described here represents a novel kind of assay to screen for small-molecule inhibitors. Interestingly, there is a good correlation between the efficacy measured with the VHH-based assay and the binding potency measured with the Octet biolayer interferometry measurements. The assay reported here is an improvement over the currently existing fluorescence based assay since it provides higher sorting efficiency with at least 10 times higher sensitivity. This assay can also be used as relatively high-throughput and cost-efficient technique for enzymes with slow turn over, receptors, cell membrane proteins, and other relevant protein targets. This study sheds light on the unexplored potential of nanobodies as multi-purpose ligands for drug discovery and development.
Supplementary Material
Highlights.
novel method using nanobodies to screen for potent ligands of a protein
displacement sandwich ELISA (dsELISA) for direct KD measurement
results with dsELISA correlate with Octet biolayer
dsELISA is an order of magnitude more sensitive in identifying potent small molecule inhibitors than kinetic assay
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants: R01-AI091699 and P20-GM113132 [DRM], R01-ES002710 and P42-ES004699 [BDH]. The research was also supported by National Institute of Environmental Health Sciences, Superfund Research Program, P42 ES04699. We thank Dr. Ian Wilson at The Scripps Research Institute for providing access to Octet instrumentation.
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
SUPPORTING INFORMATION
Additional information including extended methods, five tables and three figures are available in the supporting information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- (1).Mittal R; Aggarwal S; Sharma S; Chhibber S; Harjai K J Infect Public Health 2009, 2, 101–111. [DOI] [PubMed] [Google Scholar]
- (2).Church D; Elsayed S; Reid O; Winston B; Lindsay R Clin Microbiol Rev 2006, 19, 403–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Al-Wrafy F; Brzozowska E; Gorska S; Gamian A Postepy higieny i medycyny doswiadczalnej 2017, 71, 78–91. [DOI] [PubMed] [Google Scholar]
- (4).Gellatly SL; Hancock RE W. Pathog Dis 2013, 67, 159–173. [DOI] [PubMed] [Google Scholar]
- (5).Montero M; Dominguez M; Orozco-Levi M; Salvado M; Knobel H Infection 2009, 37, 16–19. [DOI] [PubMed] [Google Scholar]
- (6).Guarascio AJ; Ray SM; Finch CK; Self TH ClinicoEconomics and outcomes research : CEOR 2013, 5, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Alp E; Voss A Annals of clinical microbiology and antimicrobials 2006, 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Agrawal A; Agarwal A; Mehta D; Sikachi RR; Du D; Wang J Intractable Rare Dis Res 2017, 6, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bahl CD; Morisseau C; Bomberger JM; Stanton BA; Hammock BD; O’Toole GA; Madden DR Journal of bacteriology 2010, 192, 1785–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bahl CD; Madden DR Protein and peptide letters 2012, 19, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bahl CD; Hvorecny KL; Bomberger JM; Stanton BA; Hammock BD; Morisseau C; Madden DR Angew Chem Int Edit 2015, 54, 9881–9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hvorecny KL; Dolben E; Moreau-Marquis S; Hampton TH; Shabaneh TB; Flitter BA; Bahl CD; Bomberger JM; Levy BD; Stanton BA; Hogan DA; Madden DR Am J Physiol-Lung C 2018, 314, L150–L156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Flitter BA; Hvorecny KL; Ono E; Eddens T; Yang J; Kwak DH; Bahl CD; Hampton TH; Morisseau C; Hammock BD; Liu XY; Lee JS; Kolls JK; Levy BD; Madden DR; Bomberger JM P Natl Acad Sci USA 2017, 114, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bahl CD; Laurent S; D. J; Karthikeyan RSG; Priya JL; Prajna L; Zegans ME; Madden DR Cornea 2017, 36, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kitamura S; Hvorecny KL; Niu J; Hammock BD; Madden DR; Morisseau C J Med Chem 2016, 59, 4790–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Morisseau C; Bernay M; Escaich A; Sanborn JR; Lango J; Hammock BD Anal Biochem 2011, 414, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wu S; Liu B BioDrugs 2005, 19, 383–392. [DOI] [PubMed] [Google Scholar]
- (18).Glickman JF; Schmid A; Ferrand S Assay Drug Dev Technol 2008, 6, 433–455. [DOI] [PubMed] [Google Scholar]
- (19).Tian H; Ip L; Luo H; Chang DC; Luo KQ British journal of pharmacology 2007, 150, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lea WA; Simeonov A Expert Opin Drug Discov 2011, 6, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hajduk PJ J Med Chem 2006, 49, 6972–6976. [DOI] [PubMed] [Google Scholar]
- (22).Beghein E; Gettemans J Front Immunol 2017, 8, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bever CS; Dong JX; Vasylieva N; Barnych B; Cui Y; Xu ZL; Hammock BD; Gee SJ Anal Bioanal Chem 2016, 408, 5985–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Muyldermans S Annu Rev Biochem 2013, 82, 775–797. [DOI] [PubMed] [Google Scholar]
- (25).Tabares-da Rosa S; Rossotti M; Carleiza C; Carrion F; Pritsch O; Ahn KC; Last JA; Hammock BD; Gonzalez-Sapienza G Anal Chem 2011, 83, 7213–7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kim HJ; McCoy MR; Majkova Z; Dechant JE; Gee SJ; Tabares-da Rosa S; Gonzalez-Sapienza GG; Hammock BD Anal Chem 2012, 84, 1165–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pardon E; Laeremans T; Triest S; Rasmussen SGF; Wohlkonig A; Ruf A; Muyldermans S; Hol WGJ; Kobilka BK; Steyaert J Nat Protoc 2014, 9, 674–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Barbas CF Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press, 2001. [Google Scholar]
- (29).Bever CRS; Majkova Z; Radhakrishnan R; Suni I; McCoy M; Wang YR; Dechant J; Gee S; Hammock BD Anal Chem 2014, 86, 7875–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang J; Bever CRS; Majkova Z; Dechant JE; Yang J; Gee SJ; Xu T; Hammock BD Anal Chem 2014, 86, 8296–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lorimer IAJ; Keppler-Hafkemeyer A; Beers RA; Pegram CN; Bigner DD; Pastan I P Natl Acad Sci USA 1996, 93, 14815–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ghannam A; Kumari S; Muyldermans S; Abbady AQ Plant Mol Biol 2015, 87, 355–369. [DOI] [PubMed] [Google Scholar]
- (33).Schmitz KR; Bagchi A; Roovers RC; Henegouwen PMPVE; Ferguson KM Structure 2013, 21, 1214–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rossotti MA; Pirez M; Gonzalez-Techera A; Cui YL; Bever CS; Lee KSS; Morisseau C; Leizagoyen C; Gee S; Hammock BD; Gonzalez-Sapienza G Anal Chem 2015, 87, 11907–11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zhu JJ; Declercq J; Roucourt B; Ghassabeh GH; Meulemans S; Kinne J; David G; Vermorken AJM; Van de Ven WJM; Lindberg I; Muyldermans S; Creemers JWM Biochem J 2012, 448, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pirez-Schirmer M; Rossotti M; Badagian N; Leizagoyen C; Brena BM; Gonzalez-Sapienza G Anal Chem 2017, 89, 6800–6806. [DOI] [PubMed] [Google Scholar]
- (37).He T; Wang Y; Li P; Zhang Q; Lei J; Zhang Z; Ding X; Zhou H; Zhang W Anal Chem 2014, 86, 8873–8880. [DOI] [PubMed] [Google Scholar]
- (38).Cui YL; Li DY; Morisseau C; Dong JX; Yang J; Wan DB; Rossotti MA; Gee SJ; Gonzalez-Sapienza GG; Hammock BD Anal Bioanal Chem 2015, 407, 7275–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Buelens K; Hassanzadeh-Ghassabeh G; Muyldermans S; Gils A; Declerck PJ J Thromb Haemost 2010, 8, 1302–1312. [DOI] [PubMed] [Google Scholar]
- (40).Menzel S; Rissiek B; Haag F; Goldbaum FA; Koch-Nolte F The FEBS journal 2013, 280, 3543–3550. [DOI] [PubMed] [Google Scholar]
- (41).Unger M; Eichhoff AM; Schumacher L; Strysio M; Menzel S; Schwan C; Alzogaray V; Zylberman V; Seman M; Brandner J; Rohde H; Zhu K; Haag F; Mittrucker HW; Goldbaum F; Aktories K; Koch-Nolte F Sci Rep-Uk 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jahnichen S; Blanchetot C; Maussang D; Gonzalez-Pajuelo M; Chow KY; Bosch L; De Vrieze S; Serruys B; Ulrichts H; Vandevelde W; Saunders M; De Haard HJ; Schols D; Leurs R; Vanlandschoot P; Verrips T; Smit MJ P Natl Acad Sci USA 2010, 107, 20565–20570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Barlow JN; Conrath K; Steyaert J BBA-Proteins and Proteomics 2009, 1794, 1259–1268. [DOI] [PubMed] [Google Scholar]
- (44).Sohier JS; Laurent C; Chevigne A; Pardon E; Srinivasan V; Wernery U; Lassaux P; Steyaert J; Galleni M Biochem J 2013, 450, 477–486. [DOI] [PubMed] [Google Scholar]
- (45).Oyen D; Srinivasan V; Steyaert J; Barlow JN J Mol Biol 2011, 407, 138–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.