

The causative progeria lamin A mutation inhibits CK2 enzyme activity to accelerate aging in laboratory mice.
Abstract
Defective nuclear lamina protein lamin A is associated with premature aging. Casein kinase 2 (CK2) binds the nuclear lamina, and inhibiting CK2 activity induces cellular senescence in cancer cells. Thus, it is feasible that lamin A and CK2 may cooperate in the aging process. Nuclear CK2 localization relies on lamin A and the lamin A carboxyl terminus physically interacts with the CK2α catalytic core and inhibits its kinase activity. Loss of lamin A in Lmna-knockout mouse embryonic fibroblasts (MEFs) confers increased CK2 activity. Conversely, prelamin A that accumulates in Zmpste24-deficent MEFs exhibits a high CK2α binding affinity and concomitantly reduces CK2 kinase activity. Permidine treatment activates CK2 by releasing the interaction between lamin A and CK2, promoting DNA damage repair and ameliorating progeroid features. These data reveal a previously unidentified function for nuclear lamin A and highlight an essential role for CK2 in regulating senescence and aging.
INTRODUCTION
Lamin A is a major component of the nuclear lamina. It is predominantly located beneath the inner nuclear membrane and partially extends to the nuclear interior. The mechanical properties of lamin A enable it to help maintain nuclear structure, organize chromatin, regulate gene expression, and ensure genome stability (1, 2). Lamin A is encoded by the LMNA gene; it is first synthesized as prelamin A, which is processed by ZMPSTE24, a zinc metallopeptidase, to the mature form. Mutations in either LMNA or ZMPSTE24 are associated with progeria syndromes, such as Hutchinson-Gilford progeria syndrome (HGPS) and restrictive dermopathy. These syndromes are characterized by truncated prelamin A accumulation in cell nuclei (3, 4). Consistently, Zmpste24-deficient mice exhibit premature aging features resembling progeria, including hair loss, growth retardation, osteoporosis, and premature mortality (3). Although progeria caused by lamin A defect is rare, studies over the past decade have shown that progeria models offer new possibilities to understand the mechanisms of aging and age-related diseases.
Truncated prelamin A accumulation is found in tissues and fibroblasts of healthy, elderly people and is a risk marker for cardiovascular disease in an age-dependent manner (5). Mechanistically, truncated prelamin A leads to a thickening of the nuclear lamina, dramatic changes in nuclear morphology (6), and widespread alterations to chromatin structure, which is a main epigenetic feature of progeria (7). Progeroid fibroblasts and Zmpste24−/− cells exhibit genomic instability due to slow DNA repair kinetics (8), especially heterochromatin DNA damage repair. These mechanisms are increasingly recognized as key drivers of cellular senescence and organismal aging (9). Despite such advances in knowledge, the precise mechanisms as to how lamin A functionally links to DNA damage repair and aging are unclear.
Casein kinase 2 (CK2) is a ubiquitous serine/threonine protein kinase involved in various cell processes, including proliferation and survival, differentiation and development, apoptosis, and DNA repair (10). CK2 typically functions as a tetrameric complex consisting of two catalytic subunits (CK2α/α′) and two regulatory subunits (CK2β); it acts as a constitutive enzyme to phosphorylate hundreds of proteins both in vitro and in vivo (11). High CK2 activation underscores the pathogenic potential of various human cancers, and CK2 overexpression is oncogenic in transgenic mouse models (12, 13). Therefore, great efforts have been made to search for efficient and specific CK2 inhibitors to incorporate into cancer treatments.
Most of the currently known CK2 inhibitors are adenosine 5′-triphosphate (ATP) competitors and thus suffer from nonspecific targeting to multiple ATP kinases (14). A few endogenous protein factors, however, have been found to inhibit CK2 kinase activity, such as p53, p21WAF1, eIF2β, and Nopp140. p53, p21WAF1, and eIF2β bind to the regulatory CK2β subunit, while nucleolar Nopp140 interacts with the catalytic CK2α subunit (15–17). Notably, most of these endogenous proteins that regulate CK2 are involved in cellular senescence pathways (18). Furthermore, pharmacological CK2 inhibitors used in cancer treatments cause cellular senescence, and down-regulation of CK2β activity induces cellular senescence in Caenorhabditis elegans (19). Together, these data imply that CK2 activity may be affected by aging-associated molecules.
CK2 binds to the nuclear lamina and nuclear matrix (NM) (20), chromatin- and NM-bound CK2 can be rapidly modulated by mitogenic signals (21). These findings, although mechanistically unclear, raise the question of whether a decline in CK2 indeed underlies organismal aging and progeria. In this study, we demonstrate that proper CK2α nuclear localization and kinase activity relies on lamin A. Accumulation of prelamin A increases CK2 NM tethering, inhibits CK2 enzymatic activity, and accelerates cellular senescence. Moreover, pharmaceutical activation of CK2α with spermidine rescues cellular senescence, ameliorates progeroid features, and extends life span in progeria mice. Our findings reveal an essential role for lamin A–mediated CK2 activity in regulating senescence and organismal aging. Therapeutics designed around CK2 may be a beneficial strategy for progeria.
RESULTS
Lamin A mediates proper CK2 NM localization
CK2 resides in the cytoplasm and, most predominantly, in the NM, where it regulates phosphorylation-mediated signaling transduction of nuclear proteins (22). To examine whether lamin A is involved in the association between CK2 and the NM, we generated enhanced green fluorescent protein (EGFP)–tagged CK2 (CK2α-GFP) and DsRed-tagged lamin A (lamin A–Red) constructs and studied their localization in transfected human skin fibroblasts (HSFs). By immunofluorescent confocal microscopy, we observed that the majority of singly transfected CK2α-GFP localized to the nucleus (Fig. 1A). When cotransfected with lamin A–Red, CK2α-GFP accumulated at the nuclear lamina, where it colocalized with lamin A (observed in ~87% of the cells). When cotransfected with DsRed-tagged prelamin A (prelamin A–Red) constructs, CK2α accumulated at the nuclear lamina and colocalized with prelamin A–Red in a clear ring structure (observed in ~89% of the cells) (Fig. 1A). The same phenomenon was observed in cells cotransfected with the CK2α′ isoform and lamin A or prelamin A (fig. S1A).
Fig. 1. Prelamin A increases CK2 accumulation in the NM.
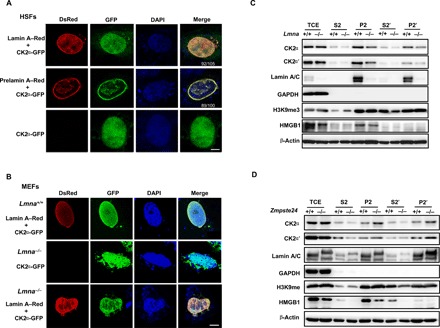
(A) Representative immunofluorescence confocal microscopy images of CK2α-EGFP and lamin A/prelamin A–DsRed in human skin fibroblasts (HSFs). The majority of CK2α-GFP distributes in the nucleus and colocalizes with lamin A/prelamin A. Of 105 cells successfully cotransfected with lamin A and CK2α, 92 showed a similar localization pattern. Representative images are shown. In the case of prelamin A and CK2α, 89 of 100 transfected cells showed a similar localization pattern. Scale bar, 5 μm. (B) Representative immunofluorescence confocal microscopy images of CK2α-EGFP and lamin A–Red transfected into Lmna−/− mouse embryonic fibroblasts (MEFs). The localization of CK2α was restored to the NM in Lmna−/− MEFs. Scale bar, 5 μm. (C) Representative Western blots showing the subcellular distribution of CK2α/CK2α′ and lamin A/prelamin A in Lmna−/− MEFs. CK2α mainly localized in the P2′ fraction in Lmna−/− and WT littermate control MEFs but to a lesser degree in Lmna−/− MEFs. HMGB1, H3K9me3, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as protein markers for euchromatin, heterochromatin, nuclear, and cytoplasm, respectively. TCE (total cell extracts), S2 (nucleoplasm), P2 (chromatin + NM), S2′ (nucleoplasm + nuclease-susceptible chromatin), and P2′ (NM + nuclease-resistant chromatin) are as described in Materials and Methods. (D) Representative Western blots showing the subcellular distribution of CK2α/CK2α′ in Zmpste24−/− MEFs. A higher abundance of CK2α was found in the P2′ fraction in Zmpte24−/− compared to WT littermate control MEFs. All MEFs were analyzed at passage 4. Data represent one of three independently derived MEF cultures in separate experiments.
To verify that CK2 NM localization requires lamin A, we transfected CK2α-GFP and/or lamin A–Red into Lmna−/− MEFs. Loss of A types of lamins compromised CK2α localization in both the nucleoplasm and the nuclear membrane, resulting in diffuse cytoplasmic staining. This aberrant localization pattern was restored upon overexpressing wild-type (WT) lamin A (Fig. 1B, bottom). By contrast, overexpressing a mutant form of lamin A–Red, in which the nuclear localization sequence (NLS) (amino acid, 417 to 422) was deleted, failed to restore CK2α nuclear localization (fig. S1B).
To show that lamin A mediates CK2 NM tethering, we fractionated the cellular lysate of Lmna−/− MEFs into S2, P2, S2′, and P2′ portions (see Materials and Methods) and monitored CK2 protein expression. Here, we found that the amount of CK2α and CK2α′ subunit was notably lower in the NM-associated fraction (P2′) of Lmna−/− MEFs than in Lmna+/+ MEFs (Fig. 1C). These data support the idea that lamin A mediates CK2 distribution on the NM. Last, to determine whether CK2 distribution is also affected by prelamin A, we repeated the subcellular fractionation assay, this time using Zmpste24−/− MEFs. We found more CK2α present in the NM (P2′) fraction of prelamin A–expressing Zmpste24−/− MEFs than WT MEFs. CK2α′ exhibited a distribution pattern similar to that of CK2α in Zmpste24−/− MEFs (Fig. 1D). Meanwhile, immunofluorescence staining showed that endogenous prelamin A led to increased CK2α accumulation at the nuclear periphery and maldistribution in the nuclear interior in Zmpste24−/− MEFs (fig. S1C). Together, these data suggest that lamin A mediates proper CK2 nuclear localization and tethering to the NM.
The lamin A C terminus physically interacts with the CK2 catalytic domain
We next examined whether CK2 interacts with lamin A by performing coimmunoprecipitation (Co-IP) experiments in human embryonic kidney (HEK) 293 cells overexpressing FLAG–lamin A and hemagglutinin (HA)–CK2α. We detected HA-CK2α in anti-FLAG immunoprecipitates, with increased binding capacity to prelamin A than to lamin A (Fig. 2A). Lamin A and prelamin A were found in anti-HA immunoprecipitates in the same pattern (Fig. 2B). CK2α′ and CK2β also interacted with lamin A and showed elevated binding capacity to prelamin A (fig. S2, A to C). Endogenous CK2α and the lamin A complex mutually precipitated each other in MEFs (Fig. 2C).
Fig. 2. The lamin A C terminus binds the CK2α active region.
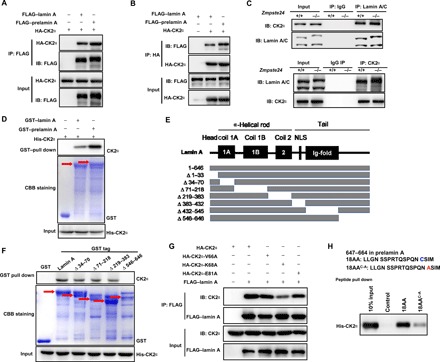
(A and B) Representative Co-IP and Western blots showing transfected HA-CK2α and FLAG–lamin A/prelamin A protein levels in HEK293 cells using an anti-FLAG (A) or anti-HA (B) antibody, respectively. The interactions between ectopically expressed lamin A/prelamin A and CK2α were detected in the immune complexes. IB, immunoblot. (C) Representative Co-IP and Western blots showing the interaction between CK2α and lamin A/prelamin A in Zmpste24+/+ and Zmpste24−/− cells in vivo. CK2α was pulled down by anti–lamin A/C immunoprecipitates (top) and lamin A/prelamin A was pulled down by anti-CK2α immunoprecipitates (bottom). (D) GST pull down detecting CK2α protein levels in vitro with purified GST–lamin A/prelamin A (top) and Ni-NTA pull down with 6× His-CK2α (bottom). Coomassie brilliant blue (CBB) staining was performed to detect GST or GST–lamin A/prelamin A levels. Red arrows indicate the corresponding protein bands. (E) Schematic of the lamin A mutants. NLS, nuclear localization sequence. (F) Truncated lamin A and lamin A mutant peptides were subjected to His-CK2α pull down. Western blotting was performed to detect CK2α protein levels, and CBB staining was performed to detect GST or GST lamin A and lamin A mutant-tagged proteins. The red arrows indicate the corresponding protein bands. (G) Representative Co-IP and Western blots between CK2α kinase–dead mutations and lamin A. HEK293 cells were cotransfected with either FLAG–lamin A and HA-CK2 (WT) or FLAG–lamin A and mutant HA-CK2 (V66A, K68A, or E81A) for 48 hours. FLAG–lamin A immunoprecipitates were subjected to Western blotting with the indicated antibodies. (H) Western blot analysis of the unique C-terminal 18–amino acid (647 to 664) prelamin A peptide pull down with His-CK2α and the WT and 18AAC-A (C661A) mutant peptides. The above experiments were repeated three times with consistent results.
We then performed a glutathione S-transferase (GST) pull-down assay to verify whether the interaction between CK2 and lamin A is direct. Both CK2α and CK2β directly interacted with lamin A and prelamin A in vitro (Fig. 2D and fig. S2D). Notably, endogenous and transfected CK2 levels were higher in the prelamin A immunoprecipitates compared to the lamin A immunoprecipitates, supporting that CK2 binds to prelamin A with a higher affinity than to lamin A.
Lamin C has identical sequence homology with lamin A but lacks a 98–amino acid sequence in its C terminus. Thus, to map the domains that mediate CK2α and lamin A interaction, we first assessed binding between CK2α and lamin C. Lamin C was hardly detectable in the anti-CK2α immunoprecipitates from WT and Zmpste24−/− MEFs (Fig. 2C, bottom), implying that CK2α binds to the lamin A C terminus that is lacking in lamin C. To confirm this observation, we generated various lamin A truncation mutants (Fig. 2E). CK2α was not detectable in the GST pull down using a 546– to 646–amino acid–deleted lamin A construct, thus supporting the idea that the lamin A C terminus provides the CK2α binding site (Fig. 2F). To map the reciprocal lamin A binding site in CK2α, we generated the CK2α kinase–dead site trunctation mutants (K68A, E81A, and V66A) (23, 24). We found that the K68A point mutation in the CK2α enzymatic core reduced the binding with lamin A (Fig. 2G).
To explore the structural characteristics of the strong binding between prelamin A and CK2, we synthesized a peptide corresponding to the 18 amino acids of the prelamin A C terminus (amino acids 647 to 664) and narrowed down the essential amino acids required for CK2 binding. By performing peptide pull-down assays, we found that the 18–amino acid peptide exhibited strong binding to CK2α (Fig. 2H). Because an early study found that CK2 anchors to the NM via a sulfhydryl group (25), we mutated C661 to A661. This mutation dramatically down-regulated the binding capacity of CK2 to the prelamin A (Fig. 2H). These findings indicate that the C-terminal C661 site is important for providing an additional binding site for prelamin A and CK2α. Together, our data demonstrate that lamin A and CK2 physically interact and that prelamin A enhances this interaction via the additional 18 amino acids found in its C terminus.
Lamin A is an endogenous inhibitor of CK2 kinase
The physical binding of lamin A to the enzymatic core of the CK2α subunit prompted us to examine whether CK2 kinase activity is modulated by lamin A. To this end, we used a CycLex CK2 kinase assay to determine CK2α/β (holoenzyme) activity by incubating CK2 with recombinant human lamin A/prelamin A protein [rh(pre)laminA] in vitro (26). Here, we found that “rhlamin A” significantly inhibited CK2 activity in a dose-dependent manner (Fig. 3A); this inhibition was even stronger with “rhprelamin A.” To validate these findings, we measured CK2 activity in crude NM extracts from Lmna−/−, Zmpste24−/−, and WT MEFs. Consistently, CK2α/β activity was much higher in Lmna−/− MEFs (Fig. 3B), suggesting that lamin A modulates CK2 activity in the NM. By contrast, CK2α/β activity in Zmpste24−/− cells was significantly lower than that in WT cells. Notably, the CK2α protein and mRNA levels did not differ between Zmpste24−/− and WT cells (fig. S3, A and B), implying that lamin A directly affects CK2 activity.
Fig. 3. Prelamin A preferentially down-regulates CK2 activity over lamin A.
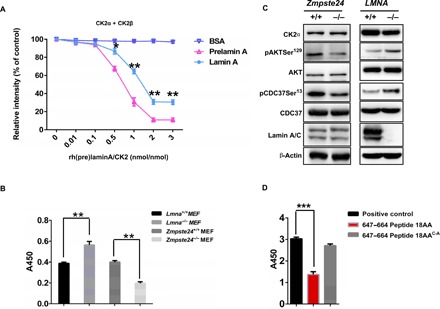
(A) CycLex CK2 assay to detect CK2 enzyme activity. rh(pre)laminA protein was incubated with CK2 in vitro. Bovine serum albumin (BSA) was used as a control. Statistical analysis showing lamin A inhibits CK2α/β activity in a dose-dependent manner. The effect of prelamin A on inhibiting CK2α/β activity is stronger than that of lamin A. The data (means ± SEM) represent three independent experiments. *P < 0.05, **P < 0.01, two-tailed Student’s t test. (B) CK2 enzyme activity in Zmpste24−/− MEFs and Lmna A−/− MEFs. CK2 enzyme activity was decreased in Zmpste24−/− MEFs and increased in Lmna−/− MEFs compared to WT littermate controls. The data (means ± SEM) represent three independently derived lines of MEFs in separate experiments. **P < 0.01. (C) Representative Western blots showing protein levels of CK2α and its downstream target proteins, pAKTSer129 and pCDC37Ser13, which reflect CK2 activity in vivo, in Zmpste24−/− MEFs and WT littermate controls. (D) CycLex CK2 assay to detect CK2 enzyme activity by incubation with synthesized WT prelamin A (amino acids 647 to 664) peptide and C661A mutant peptide. All MEFs were used at passage 4. Data represent three independently derived lines of MEFs in separate experiments. ***P < 0.001.
To validate this observed dysregulated CK2 activity in Lmna−/− and Zmpste24−/− MEFs, we assessed the phosphorylation status of pAKTSer129 and pCDC37Ser13, which are CK2 substrates and widely considered as reliable reporters of CK2 in vivo activity (27, 28). The results from this assay confirmed our hypothesis, showing that pAKTSer129 and pCDC37Ser13 phosphorylation levels increased in Lmna−/− MEFs but decreased in Zmpste24−/− MEFs (Fig. 3C). As prelamin A has the additional 18 amino acids in the C terminus, we reasoned that this extra “tail” might enhance the inhibitory function of lamin A on CK2 activity. To test this hypothesis, we incubated the WT and mutated 18–amino acid (amino acids 647 to 664) prelamin A peptides with CK2α/β in kinase assay buffers. Here, we found that, while the WT peptide was sufficient to inhibit CK2 activity, the C661A mutant peptide compromised this inhibition (Fig. 3D). These data confirm that the C661 site is essential not only for CK2 binding but also for modulating CK2 activity. Furthermore, these data suggest that lamin A is an endogenous inhibitor of CK2 holoenzyme activity, and prelamin A gains additional inhibitory function in Zmpste24−/− MEFs.
Reduced CK2 activity underlies premature senescence in progeria
The above data showed that CK2 activity is compromised in Zmpste24−/− MEFs; whether compromised CK2 activity is a causal factor of premature aging is thus an important question. To address this point, we explored whether silencing CK2α accelerates cellular senescence in MEFs. When we knocked down CK2α using small interfering RNA (siRNA), senescence-associated β-galactosidase (SA-β-gal) activity significantly increased, supporting the notion that CK2 decline is a causal factor of cellular senescence (Fig. 4, A and B). Next, we asked whether forced CK2α expression could rescue premature senescence in progeria cells. We generated a CK2αTG transgenic mouse line and crossed these mice to Zmpste24+/− mice to isolate compound-mutant Zmpste24−/−CK2αTG MEFs for analysis. When CK2α was overexpressed, the percentage of β-gal–positive cells dropped from an average of 51.42% in Zmpste24−/− MEFs to <15% in Zmpste24−/−CK2αTG MEFs (Fig. 4, C and D). Moreover, Western blotting analysis showed that the level of pAKTSer129 and pCDC37Ser13 increased, while the level of p16Ink4a, a widely applied senescence marker, decreased in Zmpste24−/−CK2αTG MEFs compared to Zmpste24−/− MEFs (Fig. 4E). These data support the idea that CK2 activity regulates cell senescence.
Fig. 4. Declined CK2 activity underlies premature senescence in progeria.
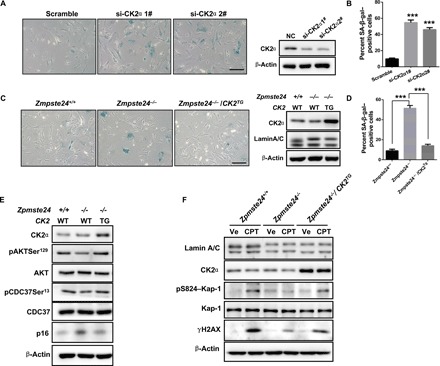
(A) SA-β-gal staining (left) and Western blots (right) of MEFs treated with si-CK2α or scrambled siRNA. Scale bar, 50 μm. (B) Quantification of SA-β-gal–positive staining from 10 randomly chosen fields of view for each group. The data represent the means ± SEM. ***P < 0.001. (C) SA-β-gal staining (left) and Western blots (right) in Zmpste24−/−, Zmpste24−/− CK2αTG, and WT littermate control MEFs. (D) Quantification of SA-β-gal–positive staining from 10 randomly chosen fields of view for each group. The data represent the means ± SEM. ***P < 0.001. (E) Western blots showing the phosphorylation levels of CK2 target proteins pAKTSer129 and pCDC37Ser13 and the senescence marker p16 in MEFs isolated from intercrossed Zmpste24+/− CK2TG (TG) mice embryos. (F) Representative Western blots showing γH2AX, pS824–Kap-1 and CK2α protein levels in Zmpste24−/− CK2αTG MEFs and their WT littermate controls. Note that the response to DNA repair is more sensitive in Zmpste24−/− CK2αTG MEFs because of higher γH2AX and pS824–Kap-1 expression. MEFs were treated with 4 μM CPT for 1 hour or vehicle (Ve; DMSO) and then analyzed by Western blotting. Data represent three independently derived lines of MEFs in separate experiments.
As genomic instability and a defective ATM–Kap-1–mediated DNA damage response (DDR) are characteristics of Zmpste24−/− MEFs (8), and because CK2 is an essential regulator of the DDR and DNA repair, we examined the DDR in Zmpste24−/− CK2αTG MEFs. We incubated these cells with 4 μM camptothecin (CPT) for 1 hour to induce DNA damage and then monitored γH2AX and pS824–Kap-1 expression. γH2AX and pS824–Kap-1 levels were low in Zmpste24−/− MEFs but were restored in Zmpste24−/− CK2αTG MEFs (Fig. 4F), suggesting that CK2 decline contributes to a defective DDR in progeroid cells. Collectively, these findings suggest that excessive inhibition of CK2 activity by prelamin A contributes to premature senescence.
Spermidine activates CK2 to alleviate senescence in Zmpste24−/− MEFs
Considering the compromised CK2 activity that we observed in Zmpste24−/− cells, we moved to search for activators of CK2 with potential anti-aging applications. Previous reports have shown that spermidine activates CK2 (29). We confirmed this finding by showing a dose-dependent relationship between increasing spermidine concentrations and absorbance, indicative of an increase in CK2 kinase activity in vitro (Fig. 5A). Treating Zmpste24−/− and WT MEFs with 4 μM spermidine caused a significant increase in CK2 activity (Fig. 5B). Western blot analysis showed that spermidine increased pAKTSer129 and pCDC37Ser13 levels in both WT and Zmpste24 MEFs, reflecting effective CK2 activation in vivo. Simultaneously, spermidine treatment considerably reduced p16Ink4a expression in Zmpste24−/− MEFs, almost to the same level as WT cells, indicative of reduced senescence (Fig. 5C).
Fig. 5. Spermidine activates CK2 and thus alleviates senescence in Zmpste24−/−cells.
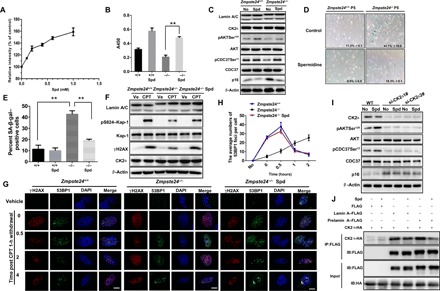
(A) Relative CK2 enzyme activity under 0 to 1 mM spermidine (Spd) treatment in vitro. The data (means ± SEM) represent three independent experiments. (B) Relative CK2 enzyme activity in 4 μM Spd-treated Zmpste24−/– MEFs in vivo. (C) Representative Western blots showing pAKTSer129, pCDC37Ser13, and p16 protein levels in Zmpste24−/− MEFs and WT littermate controls treated with 4 μM Spd or mock (ddH2O; Ctr). (D) Representative images of SA-β-gal activity showing blue-stained senescent cells in Zmpste24−/− MEFs and WT littermate controls treated with 4 μM Spd or mock ddH2O (control) at passage 5. Scale bars, 100 μM. (E) Percentage of senescent cells. The data represent the means ± SEM. (F) Representative Western blots showing γH2AX and pS824-Kap-1 protein levels in Zmpste24−/− MEFs treated with 4 μM Spd for 24 hours and exposed to 4 μM CPT treatment for 1 hour to induce DNA damage or vehicle DMSO. At least three independent experiments were performed. (G) Representative immunofluorescence confocal microscopy images of γH2AX and 53BP1 immunofoci recruitment in response to 4 μM CPT-induced DNA damage in Spd-treated Zmpste24−/− MEFs. Spd rescued γH2AX and 53BP1 recruitment at the indicated time after CPT treatment (0, 0.5, 2, and 4 hours). Scale bars, 10 μm. A representative image for each condition is shown. (H) Quantification of immunofoci recruitment from nine randomly selected fields of view for each group. At least 100 cells were counted. The data presented in the line chart (means ± SEM) represent three independent experiments. *P < 0.05; **P < 0.01. (I) Representative Western blots showing pAKTSer129, pCDC37Ser13, and p16 protein levels in siRNA-CK2–transfected WT MEFs treated with 4 μM Spd or mock treated with ddH2O (No). MEFs were assayed at passage 4. (J) Representative Co-IP and Western blots showing transfected HA-CK2α and FLAG–lamin A/prelamin A protein levels using an anti-FLAG antibody in HEK293 cells treated with 4 μM Spd or mock for 6 hours. The interactions between ectopically expressed lamin A/prelamin A and CK2α were detected in the immune complexes. The above experiments were repeated three times with similar results.
We then examined SA-β-gal activity in spermidine-treated Zmpste24−/− MEFs and found that the percentage of positively stained cells dropped from 42.1 to 18.3% after spermidine treatment (Fig. 5, D and E). These results suggest that spermidine indeed ameliorates the senescence phenotype of Zmpste24−/− MEFs. We also assessed the effects of spermidine treatment on the DDR and repair in Zmpste24−/− MEFs. Here, pS824–Kap-1 and γH2AX levels peaked ~1 hour after CPT treatment; this peak was dampened in Zmpste24−/− MEFs but rescued upon spermidine incubation (Fig. 5F and fig. S4A). Consistently, spermidine treatment promoted 53BP1 recruitment to sites of DNA damage in Zmpste24−/− MEFs (Fig. 5, G and H).
To exclude the possibility of off-target effects elicited by spermidine, we knocked down CK2α in WT MEFs using siRNA. Here, we found that spermidine treatment could not restore pAKTSer129, pCDC37Ser13, or p16Ink4a levels (Fig. 5I and fig. S4B). In addition, spermidine treatment could not rescue the increased SA-β-gal activity in CK2α siRNA-treated WT cells (fig. S4, C and D), indicating that spermidine treatment alleviates premature senescence in a CK2-dependent manner. Of note, spermidine treatment promoted CK2 activity in Lmna−/− MEFs to a pattern similar to that observed in WT MEFs. Specifically, we detected elevated pAKTSer129 levels (fig. S4E), suggesting that the action of spermidine on CK2 kinase activity is not directly affected by lamin A. Our results also showed that spermidine does not alter CK2α nuclear localization (fig. S5). Last, we assessed whether spermidine affects the interaction between CK2 and lamin A/prelamin A in transfected HEK293 cells. We found that the interaction between CK2α and lamin A, and more significantly between prelamin A and CK2α, indeed decreased following spermidine treatment (Fig. 5J), implying that spermidine activates CK2 by reducing its capacity to bind lamin A and prelamin A. Collectively, these results demonstrate that spermidine activates CK2 and thus can alleviate senescence in Zmpste24−/− cells.
Spermidine treatment ameliorates progeroid features and extends life span in progeria mice
Last, we monitored whether spermidine can rescue premature aging in Zmpste24−/− mice. To test this, we supplemented the drinking water with spermidine (3 mM/liter) (30). The treatment was initiated 4 weeks after birth, and control mice received normal drinking water (vehicle). Spermidine treatment increased body weight in Zmpste24−/− mice (Fig. 6, A and B). Life span was extended in spermidine-treated Zmpste24−/− mice (Fig. 6C); Kaplan-Meier analysis revealed that the median life span increased from 16 (untreated mice) to 24 weeks (spermidine-treated mice). Spermidine-treated mice also showed fewer rib fractures adjacent to the costovertebral joints than untreated mice (Fig. 6, D and E), which might be explained by a significant increase in bone density [determined by micro–computed tomography (micro-CT)] in spermidine-fed Zmpste24−/− mice (Fig. 6F). Collectively, these results demonstrate that spermidine treatment ameliorates progeroid features and extends life span in the progeria Zmpste24−/− mouse model.
Fig. 6. Spermidine ameliorates progeroid features and extends life span in Zmpste24−/−mice.
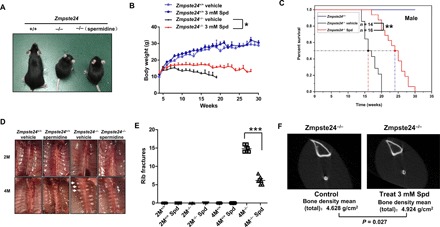
(A) Images of Zmpste24+/+, Zmpste24−/−, and spermidine-fed Zmpste24−/− mice at 4 months of age. Spd (3 mM/liter) or vehicle (ddH2O) was dissolved in drinking water daily. (B) Body-weight curves of male, Spd-fed Zmpste24+/+ (n = 14) and Zmpste24−/− (n = 16) mice were compared with vehicle controls (n = 16 and n = 14, respectively). The data represent the means ± SEM. *P < 0.05. (C) The survival and life span of Spd-fed Zmpste24−/− mice (n = 16) were compared with vehicle controls (n = 14) by Kaplan-Meier analysis and found to be statistically significant using the log-rank test (**P < 0.01). Median life span was observed, extending from 16 weeks in vehicle-treated mice to 24 weeks in Spd-fed Zmpste24−/− mice. (D) Ventral view of spinal columns from 2-month-old (2M) and 4-month-old (4M) Zmpste24+/+ and Zmpste24−/− mice treated with Spd or vehicle. Arrowheads indicate rib fractures at the costovertebral joints. (E) Comparison of the number of rib fractures in Zmpste24+/+ (n = 9), Zmpste24−/− (n = 9), Spd-fed Zmpste24+/+, and Zmpste24−/− mice (n = 16). Spd-fed mice showed fewer rib fractures compared to vehicle-treated mice. The data represent the means ± SEM, ***P < 0.001. (F) Bone density analysis in Zmpste24−/− mice and Spd-fed Zmpste24−/− mice by micro–computed tomography (micro-CT). The micro-CT shows a slight but statistically significant increase in bone density in the Spd-fed Zmpste24−/− mice. The data are representative of at least three independent experiments.
DISCUSSION
Although constitutive CK2 activation leads to malignant transformation (12, 13), the endogenous activating mechanism is poorly understood. Accumulating data suggest that CK2 nuclear cytoplasmic shuttling has important roles in diverse biological processes. Lamin A is a major component of the NM and serves as a physiological anchor for proteins shuttling between the nuclear lamina and nucleoplasm (31). Here, we found that the lamin A C terminus directly binds to the CK2α enzymatic core region, likely to tether it to the NM and subsequently inhibit its kinase activity.
Because tetramers and higher–molecular weight complexes of CK2 differ in kinase activity (32), lamin A might also regulate CK2 activity by modulating the aggregation or dissociation of different CK2 subunits. Previous reports have shown that the NM association of CK2 is enhanced by reagents that stabilize disulfide bonds (33). In support of this notion, we found stronger inhibition of CK2 by prelamin A, which has an additional C661 residue in the CaaX tail that can be farnesylated by a sulfhydryl group. Future work should investigate whether reagents that can break down the disulfide bonds would release CK2 from the NM and enhance kinase activity. Together, lamin A not only provides a platform for CK2 “aggregation” but also buffers CK2 holoenzyme kinase activity. In addition, NM tethering might help transport CK2 in its dormant form to specific subnuclear regions, where it is then released and activated for substrate phosphorylation.
Owing to its oncogenic role, much effort has been made to search for efficient and specific CK2 inhibitors that can be used as cancer therapeutics (14). Most of the currently identified CK2 inhibitors, however, are ATP competitors and thus suffer from nonspecific targeting of multiple ATP kinases. Here, we identified lamin A protein as an endogenous inhibitor of CK2 kinase activity. Specifically, an 18–amino acid peptide corresponding to the prelamin A is enough to suppress CK2 activity in vitro. Continued efforts are now merited to narrow down the essential amino acids required for CK2 inhibition and to examine the effects at the cellular level. Nonetheless, our results indicate that lamin A might function as a tumor suppressor. Supporting this notion, LMNA expression is down-regulated in several malignant and benign neoplasms, including small cell lung carcinoma, colorectal carcinoma, and adenomatous polyps of the colon (34). More evidence for the tumor-suppressive role of lamin A comes from a report showing a direct interaction between the rod domain of lamin A/C and the epigenetic regulator inhibitor of growth protein 1 (35). Cancer incidence, however, has never been reported in patients with HGPS, as prelamin A accumulation prevents cancer invasion and decreases the incidence of infiltrating carcinomas in affected patients (36).
Inhibiting CK2 kinase activity during cancer treatment accelerates senescence, but the mechanism underlying this interesting paradigm is unclear. Here, we reveal a causal function of CK2α in cellular senescence and premature aging. CK2 expression and activity are progressively down-regulated in multiple tissues in physiological aging (fig. S6, A to C). Knocking down CK2α accelerates senescence, and prelamin A accumulation restricts CK2 nuclear distribution and kinase activity, leading to premature senescence. These data extend our understanding of disorganized chromatin remodeling and a defective DDR and DNA repair in premature aging.
Spermidine is a polybasic compound that reacts with an acidic cluster of CK2β to activate the CK2 holoenzyme (37). Notably, we found that long-term exposure to 3 mM spermidine ingested via drinking water activates CK2, restores senescence, and ameliorates premature aging phenotypes in Zmpste24−/− mice without inducing any tumors in the examination period in mutant or WT mice. These findings suggest that spermidine supplementation in mice at the appropriate dosage is likely safe and are in accordance with another recent report regarding spermidine supplementation (38). Data suggest that spermidine delays aging via enhancing autophagy and mitochondrial respiration (39). In Zmpste24−/− cells, spermidine functions in a different way to rapamycin, which is a typical activator of autophagy. Rapamycin enhances LC3II but reduces p62 levels, whereas here, spermidine has a different effect from rapamycin (fig. S6D), suggesting that the anti-aging effects of spermidine are most likely independent of autophagy. Autophagic hyperactivation is observed in Zmpste24−/− progeria mice (39).
Collectively, our findings support an essential role of the lamin A–CK2 axis in deciding cell fate, namely, malignant transformation versus senescence, and in regulating aging and life span. This finding raises the possibility of developing small molecules to shift CK2 activity for anti-aging and cancer suppression simultaneously. These data also highlight the therapeutic potential of spermidine for progeria and age-related diseases by improving CK2 activity.
MATERIALS AND METHODS
Plasmids construction
The CK2 full-length gene and α, β, and α′ isoforms were amplified from their respective cDNAs and subcloned into pKH3-HA, pEGFP-C1, or pET32a vectors. HA-CK2α constructs with V66A, K68A, and E81A amino acid mutations were generated using the QuikChange II site-directed mutagenesis kit (Agilent Technologies), following the manufacturer’s instructions. Lamin A, prelamin A, and progerin were amplified and cloned into DsRedC1, p3xFLAG-CMV-10, or pGEX-4 T-3 vectors. Polymerase chain reaction (PCR)–based deletion of the FLAG–lamin A plasmid template was used to generate the truncated lamin A proteins. Transient transfections of these plasmids were performed using Lipofectamine 3000 (Invitrogen), according to the manufacturer’s protocol. Primer sequences for the amino acid mutations of CK2α were as follows:
V66A-F, 5′-cttttttactggcttgagaattttaacagcaactttttcattatttgtgatgttgat-3′;
V66A-R, 5′-atcaacatcacaaataatgaaaaagttgctgttaaaattctcaagccagtaaaaaag-3′;
K68A-F, 5′-aattttcttcttttttactggcttgagaattgcaacaacaactttttcattatttgtgatgttg-3′;
K68A-R, 5′-caacatcacaaataatgaaaaagttgttgttgcaattctcaagccagtaaaaaagaagaaaatt-3′;
E81A-F, 5′-cctctcaaattctccaaaatctttattgcacgcttaattttcttcttttttactg-3′;
E81A-R, 5′-cagtaaaaaagaagaaaattaagcgtgcaataaagattttggagaatttgagagg-3′.
Antibodies
The following antibodies were used: anti-HA, anti-FLAG, pCDC37Ser13 (SAB4504232), LC3B (L7543), and P62 (P0067) from Sigma-Aldrich; H3K9me3 (ab8898), γH2AX (ab81299), Kap-1 (ab10484), p–Kap-1 (Ser824, ab70369), p16 (ab117443), and pAKTSer129 (133458) from Abcam; lamin A/C (sc-20681), lamin A (sc-293162), p21 (sc-6246), CDC37 (sc-13129), and 53BP1 (sc-10915) from Santa Cruz Biotechnology; γH2AX (05-636) and CK2α from EMD Millipore; and AKT (no. 9272), pCDC37Ser13 (no. 13248), HMGB1 (no. 3935), and cleaved caspase-3 (no. 9661) from Cell Signaling Technology.
Cell lines
HEK293 (CRL-1573) cells were purchased from the American Type Culture Collection. Primary MEFs and HSFs (so-called F2S) were prepared as previously described (8).
Cell culture and drug treatments
Zmpste24−/− MEFs were isolated from 13.5-day-old embryos of Zmpste24+/− intercrossed mice (C57BL background). Littermate-matched Zmpste24+/+ and Zmpste24−/− MEFs were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). Lmna−/− MEFs were obtained from Lmna−/− mice. Primary human F2S fibroblasts were maintained in DMEM (Gibco) supplemented with glucose (4.5 g/liter) and 10% FBS.
To examine the DDR, MEFs or primary human F2S fibroblasts were treated with 4 μM CPT (Sigma-Aldrich, C9911) for 1 hour. CPT is a topoisomerase inhibitor that initiates single- or double-stranded DNA damage. The cells were collected or fixed at the indicated time points after CPT withdrawal, and the medium was changed to normal cell culture conditions. MEFs were treated with 4 μM spermidine for 24 hours and used for analysis.
Immunofluorescence staining
Cells were mounted on glass slides and fixed in 4% paraformaldehyde. The cells were then permeabilized with phosphate-buffered saline (PBS) containing 0.1% Triton X-100 for 15 min and blocked with 1% bovine serum in PBS for 30 min at room temperature. The cells were then incubated with various primary antibodies overnight at 4°C and detected by Alexa Fluor conjugated secondary antibodies (Alexa 488 and Alexa 594; 1:500, Life Technologies) for 1 hour at room temperature in the dark. The cells were then costained with 4′,6-diamidino-2-phenylindole (DAPI; 1:10,000 dilution) to visualize the nuclei. Images were captured under a confocal laser scanning microscope system (Carl Zeiss AG). A representative image for each condition is shown.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using Trizol reagent (Invitrogen, 15596-026). RNA (2 μg) is reverse-transcribed into cDNA using 5× PrimeScript RT Master Mix (Takara). Quantitative real-time PCR was performed using 2× SYBR Green Mix (Takara) in a Bio-Rad detection system. Each sample was run in triplicate and gene expression levels were normalized to β-actin. The primers are listed in table S1.
Western blotting and Co-IP
Cell extracts for Western blotting were prepared in modified radioimmunoprecipitation assay buffer [0.1 M tris-HCl (pH 7.5), 0.1 MNaCl, 1 mM EDTA, 1 mM dithiothreitol, and protease inhibitors], boiled in sodium dodecyl sulfate (SDS) sample loading buffer, resolved on SDS polyacrylamide gels, and transferred to polyvinylidene difluoride membranes (EMD Millipore). The membranes were blocked in 5% milk in tris-buffered saline and Tween 20 [20 mM tris-HCl (pH 7.6), 150 mM NaCl, and 0.05% Tween 20] for 1 hour at room temperature and incubated with primary antibodies overnight at 4°C. The membranes were then probed with the respective horseradish peroxidase–linked secondary antibodies for 1 hour at room temperature. Immunoreactive products were visualized using an enhanced chemiluminescence kit (Pierce, 34095) and a Bio-Rad imaging system.
For immunoprecipitation, cells exposed to the indicated treatments were harvested in lysis buffer [20 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.1 mM EDTA, 10% glycerol, and 0.1% NP-40] containing protease inhibitors (Roche cOmplete). The cell lysates were incubated with 1 μg of the respective antibodies or control immunoglobulin Gs at 4°C overnight. Bead-bound immunoprecipitates were washed with lysis buffer, and the beads were boiled in SDS sample loading buffer and analyzed by Western blotting.
For the peptide pull-down assays, 1 μg of biotinylated peptides with a different amino acid sequence was incubated with 1 μg of His-fused CK2α proteins in binding buffer [50 mM tris-HCl (pH 7.5), 300 mM NaCl, 0.1% NP-40, and 1 mM phenylmethylsulfonyl fluoride] overnight. Streptavidin beads (Amersham) were added to the mixture, and the mixture was incubated for 2 hours with rotation. The beads were then washed five times and analyzed using SDS–polyacrylamide gel electrophoresis and Western blotting.
Protein purification and pull-down assay
GST fusion proteins were expressed in BL21 bacterial cells with isopropyl β-D-1-thiogalactopyranoside and purified using Glutathione Sepharose 4B beads (GE Healthcare) before washing in TEN buffer [20 mM tris-HCl (pH 7.4), 0.1 mM EDTA, and 100 mM NaCl]. The GST fusion proteins (“bait” and “prey” proteins) were purified as detailed above. For GST pull down, 1 μg of GST or GST–lamin A, GST–prelamin A, or lamin A mutant protein was immobilized on Glutathione Sepharose 4B (GE Healthcare) and then incubated with 6× His-tagged CK2α purified from bacterial culture at 4°C overnight in GST binding buffer [50 mM tris-HCl (pH 7.5), 0.2 mM EDTA, 150 mM NaCl, and 0.1% NP-40]. The beads were washed four times with GST binding buffer and then analyzed by Western blotting with the indicated antibodies.
Cell transfection and RNA interference
Plasmid and siRNA transfections were performed with Lipofectamine 3000 (Invitrogen, USA), following the manufacturer’s instructions. Specific custom siRNAs were synthesized by GenePharma (Shanghai, China). The siRNA and short hairpin RNA (shRNA) sequences used in this study are listed in table S2. A scrambled siRNA sequence was used as a control. For stable knockdown, shRNAs were cloned into the pGLVH1 backbone (GenePharma, China) and then the virus was produced in HEK293 cells. Briefly, 10 μg of CK2α lentiviral construct, 10 μg of pSPAX2, and 5 μg of pMD2G were cotransfected into HEK293 cells using Lipofectamine 3000 reagent. The supernatants were collected 48 hours after transfection and filtered through a 0.22-μm membrane (EMD Millipore). Lentiviral infection was performed in the presence of polybrene (5 μg/ml). After 2 days, the infected cells were selected with puromycin (2 μg/ml) (Sigma-Aldrich) to obtain stable cell lines.
Subcellular fractionation
Zmpste24−/− and Lmna−/− MEFs were fractionated as previously described (40). Briefly, 4 × 107 MEFs/ml were collected and suspended in buffer A for cell swelling. The nuclei were separated by centrifugation at 1300g and then lysed in buffer B. The pellet (P2) containing the chromatin and NM proteins and the supernatant (S2) containing the nucleoplasmic proteins were further isolated by centrifugation at 1700g. To obtain NM-associated proteins, the nuclei were resuspended in buffer A plus micrococcal nuclease (Sigma-Aldrich, N3755) before lysis in buffer B to digest the chromatin DNA and release DNA fragments into the S2 fraction. The pellet obtained after digestion consisted of the nuclease-resistant chromatin and NM-associated proteins (P2′) and the supernatant contained the nucleoplasmic and nuclease-susceptible chromatin-associated proteins (S2′). The cell fractions and total cell extracts were analyzed by Western blotting.
CK2 activity assay
CK2 activity assay was performed according to the manufacturer’s protocol, using a CycLex CK2 kinase assay kit (catalog no. CY-1170).
SA-β-gal assay
SA-β-gal activity was assessed using a Senescence β-Galactosidase Staining Kit (Cell Signaling, no. 9860). Briefly, cells were seeded in six-well plates, fixed with 4% paraformaldehyde at room temperature for 10 min, washed with 1× PBS, and stained with 1 ml of freshly prepared 1× β-gal detection solution at 37°C overnight. The cells were washed twice with 1× PBS and overlaid with 70% glycerol/PBS, and images were captured under a microscope. Blue-stained MEFs were counted from >250 randomly chosen cells. A two-tailed t test was applied for P values.
Animal studies
All protocols used for animal studies were approved by the Institutional Animal Care and Use Committee of Shenzhen University (approval ID no. 201412023). CK2α-transgenic mice (CK2α-TG) congenic on a C57BL/6J background were constructed by injecting cloned mCK2α cDNA (NM_007788.3) with a CAG promoter into fertilized eggs. Primers for genotyping the CK2α transgenic allele were as follows: forward, 5′-CTGGTTATTGTGCTGTCTCATCAT-3′; reverse: 5′-TGGCTTCAAACACTTCACTGTA-3′. Zmpste24+/− intercrossed mice and Lmna+/− intercrossed mice (C57BL background) were obtained from Z. Zhongjun (Hong Kong University); Zmpste24+/− mice were then crossed to CK2α-TG mice to generate Zmpste24+/− CK2αTG mice.
Husbandry
All mice were handled in accordance with the Guide for the Care and Use of Laboratory Animals and the Principles for the Utilization and Care of Vertebrate Animals. Heterozygous zinc metallopeptidase STE24 homolog (Zmpste24) knockout mice were intercrossed to produce Zmpste24−/− mice. Littermate Zmpste24+/+ mice were used as controls in all experiments. Lmna−/− mice were obtained as described for Zmpste24+/− mice. All mice were fed a chow diet and housed in a virus-free barrier facility with a 12-hour light/dark cycle.
Analyses
Spermidine (Sigma-Aldrich) was administered in the drinking water at a final concentration of 3 mM/liter from 4 weeks after birth until death. The control mice were provided normal drinking water. Body weight and life span were monitored after spermidine treatment. The survival rate was analyzed by the Kaplan-Meier method, and statistical comparison was performed by log-rank test. Three pairs of 4-month-old spermidine-treated and control mice were placed in a plastic cylindrical holder under anesthesia (3% isoflurane), and the right proximal tibia was scanned by micro-CT (SkyScan 1076, Belgium) at a voltage of 70 keV with a current of 115 μA. Skyscan-1076 micro-CT software was used to analyze bone density.
Statistical analyses
All experiments were carried out with at least three replicates. The data are presented as the means ± SD or means ± SEM (as indicated). The following P values were considered statistically significant: *P < 0.05, **P < 0.01, and ***P < 0.001. Statistical analyses were performed in GraphPad Prism software v.5.03. A two-tailed Student’s t test was used for analyses between two groups. A two-way analysis of variance (ANOVA) was used for body weight and cell growth curves. The log-rank test was used for grip strength and survival. Pearson’s correlation coefficient and the Mann-Whitney U test were used for prelamin A–DAPI colocalization. A one-way ANOVA with Tukey’s or Bonferroni’s post hoc test was used for all other variables.
Supplementary Material
Acknowledgments
Funding: This study was supported by grants from the National Natural Science Foundation of China (81471407, 31501109, 81571374, and 91849208), the Discipline Construction Funding of Shenzhen (2016-1452), and the Shenzhen Municipal Commission of Science and Technology Innovation (JCYJ20140418095735635 and JCYJ20160520170240403). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Author contributions: Y.A., J.Z., B.L., and Z.W. conceived and designed the experiments. Z.Z., W.-G.Z., B.L., and Z.W. contributed new reagents/analytic tools. Y.A., J.Z., Z.L., M.Q., Y.L., Z. Wu, P.S., J. Wu, W.B., J. Wen, X.W., F.L., B.L., and Z.W. analyzed data. Y.A., J.Z., B.L., and Z.W. wrote the manuscript. All authors analyzed and discussed the results and reviewed the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav5078/DC1
Fig. S1. Prelamin A accumulation increases CK2 association in the NM.
Fig. S2. Lamin A interacts with CK2α′ and CK2β.
Fig. S3. CK2 expression is unaffected in Zmpste24−/− MEFs.
Fig. S4. Spermidine rescues CK2 activity and cellular senescence impacted by lamin A.
Fig. S5. Spermidine does not alter CK2α subnuclear localization.
Fig. S6. CK2 expression decreases in progeroid syndrome and during physiological aging.
Table S1. List of primers used in this study.
REFERENCES AND NOTES
- 1.Dechat T., Pfleghaar K., Sengupta K., Shimi T., Shumaker D. K., Solimando L., Goldman R. D., Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 22, 832–853 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon D. N., Wilson K. L., The nucleoskeleton as a genome-associated dynamic 'network of networks. Nat. Rev. Mol. Cell Biol. 12, 695–708 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Pendás A. M., Zhou Z., Cadiñanos J., Freije J. M. P., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodríguez F., Tryggvason K., López-Otín C., Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 31, 94–99 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Eriksson M., Brown W. T., Gordon L. B., Glynn M. W., Singer J., Scott L., Erdos M. R., Robbins C. M., Moses T. Y., Berglund P., Dutra A., Pak E., Durkin S., Csoka A. B., Boehnke M., Glover T. W., Collins F. S., Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fong L. G., Frost D., Meta M., Qiao X., Yang S. H., Coffinier C., Young S. G., A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 311, 1621–1623 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Navarro C. L., Cadiñanos J., Sandre-Giovannoli A. D., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W. J., Wagner A., Giuliano F., Beemer F. A., Freije J. M., Cau P., Hennekam R. C. M., López-Otín C., Badens C., Lévy N., Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 14, 1503–1513 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Olins D. E., Olins A. L., Nuclear envelope-limited chromatin sheets (ELCS) and heterochromatin higher order structure. Chromosoma 118, 537–548 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Liu B., Wang J., Chan K. M., Tjia W. M., Deng W., Guan X., Huang J.-D., Li K. M., Chau P. Y., Chen D. J., Pei D., Pendas A. M., Cadiñanos J., López-Otín C., Tse H. F., Hutchison C., Chen J., Cao Y., Cheah K. S. E., Tryggvason K., Zhou Z., Genomic instability in laminopathy-based premature aging. Nat. Med. 11, 780–785 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Zhang W., Li J., Suzuki K., Qu J., Wang P., Zhou J., Liu X., Ren R., Xu X., Ocampo A., Yuan T., Yang J., Li Y., Shi L., Guan D., Pan H., Duan S., Ding Z., Li M., Yi F., Bai R., Wang Y., Chen C., Yang F., Li X., Wang Z., Aizawa E., Goebl A., Soligalla R. D., Reddy P., Esteban C. R., Tang F., Liu G.-H., Belmonte J. C. I., Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348, 1160–1163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayoub N., Jeyasekharan A. D., Bernal J. A., Venkitaraman A. R., HP1-β mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682–686 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Litchfield D. W., Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 369, 1–15 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seldin D., Leder P., Casein kinase II alpha transgene-induced murine lymphoma: Relation to theileriosis in cattle. Science 267, 894–897 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Landesman-Bollag E., Romieu-Mourez R., Song D. H., Sonenshein G. E., Cardiff R. D., Seldin D. C., Protein kinase CK2 in mammary gland tumorigenesis. Oncogene 20, 3247–3257 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Battistutta R., Sarno S., De Moliner E., Papinutto E., Zanotti G., Pinna L. A., The replacement of ATP by the competitive inhibitor emodin induces conformational modifications in the catalytic site of protein kinase CK2. J. Biol. Chem. 275, 29618–29622 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Götz C., Wagner P., Issinger O. G., Montenarh M., p21WAF1/CIP1 interacts with protein kinase CK2. Oncogene 13, 391–398 (1996). [PubMed] [Google Scholar]
- 16.Schuster N., Prowald A., Schneider E., Scheidtmann K.-H., Montenarh M., Regulation of p53 mediated transactivation by the β-subunit of protein kinase CK2. FEBS Lett. 447, 160–166 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Lee W.-K., Son S. H., Jin B.-S., Na J.-H., Kim S.-Y., Kim K.-H., Kim E. E., Yu Y. G., Lee H. H., Structural and functional insights into the regulation mechanism of CK2 by IP6 and the intrinsically disordered protein Nopp140. Proc. Natl. Acad. Sci. U.S.A. 110, 19360–19365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang B.-D., Watanabe K., Broude E. V., Fang J., Poole J. C., Kalinichenko T. V., Roninson I. B., Effects of p21Waf1/Cip1/Sdi1 on cellular gene expression: Implications for carcinogenesis, senescence, and age-related diseases. Proc. Natl. Acad. Sci. U.S.A. 97, 4291–4296 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J.-H., Lee J.-H., Park J.-W., Kim D.-Y., Hahm J.-H., Nam H. G., Bae Y.-S., Downregulation of protein kinase CK2 activity induces age-related biomarkers in C. elegans. Oncotarget 8, 36950–36963 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H., Roux S. J., Casein kinase II protein kinase is bound to lamina-matrix and phosphorylates lamin-like protein in isolated pea nuclei. Proc. Natl. Acad. Sci. U.S.A. 89, 8434–8438 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tawfic S., Faust R. A., Gapany M., Ahmed K., Nuclear matrix as an anchor for protein kinase CK2 nuclear signalling. J. Cell. Biochem. 62, 165–171 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Tawfic S., Ahmed K., Growth stimulus-mediated differential translocation of casein kinase 2 to the nuclear matrix. Evidence based on androgen action in the prostate. J. Biol. Chem. 269, 24615–24620 (1994). [PubMed] [Google Scholar]
- 23.Raaf J., Issinger O.-G., Niefind K., First inactive conformation of CK2α, the catalytic subunit of protein kinase CK2. J. Mol. Biol. 386, 1212–1221 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Sarno S., Reddy H., Meggio F., Ruzzene M., Davies S. P., Donella-Deana A., Shugar D., Pinna L. A., Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’). FEBS Lett. 496, 44–48 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Zhang P., Davis A. T., Ahmed K., Mechanism of protein kinase CK2 association with nuclear matrix: Role of disulfide bond formation. J. Cell. Biochem. 69, 211–220 (1998). [PubMed] [Google Scholar]
- 26.Goueli B. S., Hsiao K., Tereba A., Goueli S. A., A novel and simple method to assay the activity of individual protein kinases in a crude tissue extract. Anal. Biochem. 225, 10–17 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Di Maira G., Salvi M., Arrigoni G., Marin O., Sarno S., Brustolon F., Pinna L. A., Ruzzene M., Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 12, 668–677 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Miyata Y., Nishida E., Evaluating CK2 activity with the antibody specific for the CK2-phosphorylated form of a kinase-targeting cochaperone Cdc37. Mol. Cell. Biochem. 316, 127–134 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Filhol O., Cochet C., Chambaz E. M., Cytoplasmic and nuclear distribution of casein kinase II: Characterization of the enzyme uptake by bovine adrenocortical nuclear preparation. Biochemistry 29, 9928–9936 (1990). [DOI] [PubMed] [Google Scholar]
- 30.Eisenberg T., Knauer H., Schauer A., Büttner S., Ruckenstuhl C., Carmona-Gutierrez D., Ring J., Schroeder S., Magnes C., Antonacci L., Fussi H., Deszcz L., Hartl R., Schraml E., Criollo A., Megalou E., Weiskopf D., Laun P., Heeren G., Breitenbach M., Grubeck-Loebenstein B., Herker E., Fahrenkrog B., Fröhlich K.-U., Sinner F., Tavernarakis N., Minois N., Kroemer G., Madeo F., Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Ho C. Y., Jaalouk D. E., Vartiainen M. K., Lammerding J., Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boldyreff B., Meggio F., Pinna L. A., Issinger O. G., Protein kinase CK2 structure-function relationship: Effects of the beta subunit on reconstitution and activity. Cell. Mol. Biol. Res. 40, 391–399 (1994). [PubMed] [Google Scholar]
- 33.Ibrahim M. X., Sayin V. I., Akula M. K., Liu M., Fong L. G., Young S. G., Bergo M. O., Targeting isoprenylcysteine methylation ameliorates disease in a mouse model of progeria. Science 340, 1330–1333 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broers J. L., Raymond Y., Rot M. K., Kuijpers H., Wagenaar S. S., Ramaekers F. C., Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am. J. Pathol. 143, 211–220 (1993). [PMC free article] [PubMed] [Google Scholar]
- 35.Han X., Feng X., Rattner J. B., Smith H., Bose P., Suzuki K., Soliman M. A., Scott M. S., Burke B. E., Riabowol K., Tethering by lamin A stabilizes and targets the ING1 tumour suppressor. Nat. Cell Biol. 10, 1333–1340 (2008). [DOI] [PubMed] [Google Scholar]
- 36. Rosa J. , Freije J. M. P., Cabanillas R., Osorio F. G., Fraga M. F., Fernández-García M. S., Rad R., Fanjul V., Ugalde A. P., Liang Q., Prosser H. M., Bradley A., Cadiñanos J., López-Otín C., Prelamin A causes progeria through cell-extrinsic mechanisms and prevents cancer invasion. Nat. Commun. 4, 2268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kreutzer J. N., Olsen B. B., Lech K., Issinger O.-G., Guerra B., Role of polyamines in determining the cellular response to chemotherapeutic agents: Modulation of protein kinase CK2 expression and activity. Mol. Cell. Biochem. 356, 149–158 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Eisenberg T., Abdellatif M., Schroeder S., Primessnig U., Stekovic S., Pendl T., Harger A., Schipke J., Zimmermann A., Schmidt A., Tong M., Ruckenstuhl C., Dammbrueck C., Gross A. S., Herbst V., Magnes C., Trausinger G., Narath S., Meinitzer A., Hu Z., Kirsch A., Eller K., Carmona-Gutierrez D., Buttner S., Pietrocola F., Knittelfelder O., Schrepfer E., Rockenfeller P., Simonini C., Rahn A., Horsch M., Moreth K., Beckers J., Fuchs H., Gailus-Durner V., Neff F., Janik D., Rathkolb B., Rozman J., de Angelis M. H., Moustafa T., Haemmerle G., Mayr M., Willeit P., von Frieling-Salewsky M., Pieske B., Scorrano L., Pieber T., Pechlaner R., Willeit J., Sigrist S. J., Linke W. A., Muhlfeld C., Sadoshima J., Dengjel J., Kiechl S., Kroemer G., Sedej S., Madeo F., Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mariño G., Ugalde A. P., Salvador-Montoliu N., Varela I., Quirós P. M., Cadiñanos J., van der Pluijm I., Freije J. M. P., López-Otín C., Premature aging in mice activates a systemic metabolic response involving autophagy induction. Hum. Mol. Genet. 17, 2196–2211 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Liu J., Yin X., Liu B., Zheng H., Zhou G., Gong L., Li M., Li X., Wang Y., Hu J., Krishnan V., Zhou Z., Wang Z., HP1α mediates defective heterochromatin repair and accelerates senescence in Zmpste24-deficient cells. Cell Cycle 13, 1237–1247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav5078/DC1
Fig. S1. Prelamin A accumulation increases CK2 association in the NM.
Fig. S2. Lamin A interacts with CK2α′ and CK2β.
Fig. S3. CK2 expression is unaffected in Zmpste24−/− MEFs.
Fig. S4. Spermidine rescues CK2 activity and cellular senescence impacted by lamin A.
Fig. S5. Spermidine does not alter CK2α subnuclear localization.
Fig. S6. CK2 expression decreases in progeroid syndrome and during physiological aging.
Table S1. List of primers used in this study.