

KDS2010, a MAO-B inhibitor, shows a long-lasting effect in alleviating memory impairment in an Alzheimer’s disease mouse model.
Abstract
Monoamine oxidase–B (MAO-B) has recently emerged as a potential therapeutic target for Alzheimer’s disease (AD) because of its association with aberrant γ-aminobutyric acid (GABA) production in reactive astrocytes. Although short-term treatment with irreversible MAO-B inhibitors, such as selegiline, improves cognitive deficits in AD patients, long-term treatments have shown disappointing results. We show that prolonged treatment with selegiline fails to reduce aberrant astrocytic GABA levels and rescue memory impairment in APP/PS1 mice, an animal model of AD, because of increased activity in compensatory genes for a GABA-synthesizing enzyme, diamine oxidase (DAO). We have developed a potent, highly selective, and reversible MAO-B inhibitor, KDS2010 (IC50 = 7.6 nM; 12,500-fold selectivity over MAO-A), which overcomes the disadvantages of the irreversible MAO-B inhibitor. Long-term treatment with KDS2010 does not induce compensatory mechanisms, thereby significantly attenuating increased astrocytic GABA levels and astrogliosis, enhancing synaptic transmission, and rescuing learning and memory impairments in APP/PS1 mice.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by significant, persistent, and progressive memory loss, usually accompanied by cognitive impairments and personality changes (1, 2). The histopathological hallmarks of AD consist of neurofibrillary tangles, caused by abnormally phosphorylated tau, and amyloid plaques (3, 4). Thus far, the most widely accepted hypothesis for AD pathogenesis has focused on the gradual accumulation of amyloid beta (Aβ) (5, 6). The disruption of this cascade has been the major strategy for drug development (7). About half of the drugs in the AD pipeline target Aβ (8), and their therapeutic mechanisms include the modulation of Aβ production and clearance of Aβ by immunotherapies (9, 10). However, the repeated failures of Aβ antibodies and β-amyloid secretase 1 inhibitors in clinical trials for AD call for a paradigm shift in the current hypothesis for the etiology of this debilitating disease (11–13).
The finding that aberrant levels of γ-aminobutyric acid (GABA), synthesized by monoamine oxidase–B (MAO-B) in reactive astrocytes, causes memory impairments in animal models of AD has suggested the potential of an alternate, Aβ-independent strategy for AD treatment (14, 15). The released astrocytic GABA strongly inhibits synaptic transmission and diminishes the spike probability, leading to an impairment in synaptic plasticity and memory function (14). Suppressing GABA production from reactive astrocytes by inhibiting MAO-B with selegiline, the most broadly used irreversible MAO-B inhibitor, fully restores the impaired spike probability, synaptic plasticity, and learning and memory function in the AD model mice, even in the presence of Aβ (14). This concept introduces MAO-B as a potential therapeutic target for treating memory impairments in AD.
Several clinical studies have reported that short-term treatment with selegiline improves cognitive deficits in patients with moderate AD (16–18), suggesting that selegiline may delay functional deterioration and institutionalization of patients with AD. However, the outcomes of long-term treatments have been disappointing. A meta-analysis of 14 identified clinical trials that involved the long-term treatment with selegiline of patients with AD has reported no statistically significant differences in cognition between selegiline and placebo groups (10, 19, 20). Therefore, there is a pressing need to investigate the direct link between irreversible MAO-B inhibitors such as selegiline and their diminishing effects in long-term treatment. In the previous study, we observed that prolonged treatment with selegiline for 30 days failed to significantly reverse spatial memory impairments in the Morris water maze test in APPswe/PS1dE9 (APP/PS1) mice (14) and suggested that the short-lived action of selegiline was caused by compensatory mechanisms due to the irreversibility of its action (14).
In this study, we investigated the molecular mechanism that underlies the diminishing effect of long-term selegiline treatment in the APP/PS1 mouse model. To overcome the shortcomings of irreversible inhibitors, we have developed a new, potent, selective, and reversible MAO-B inhibitor.
RESULTS
The diminishing effect of long-term selegiline treatment
To corroborate the diminishing effects of selegiline after long-term treatment, we orally administrated selegiline (10 mg/kg per day) for a 4-week period in APP/PS1 mice and monitored the effects on hypertrophied astrocytes and the level of GABA to compare with those of short-term treatment for a 3-day period. At this dose (10 mg/kg per day, orally), selegiline has been shown to specifically inhibit MAO-B activity in the brain (21). Consistent with our previous report (14), we found that aberrant amounts of GABA accumulated in glial fibrillary acidic protein (GFAP)–positive reactive astrocytes in the molecular layer of the dentate gyrus (DG) in 10- to 12-month-old APP/PS1 mice. The aberrant GABA level in APP/PS1 mice was significantly reduced to the level of GABA observed in wild-type (WT) mice after 3 days of selegiline treatment (Fig. 1, A and B). In marked contrast, the 4-week selegiline treatment resulted in the complete return of the aberrant level of GABA to untreated APP/PS1 levels (Fig. 1, A and B). Sholl analysis of individual GFAP-positive astrocytes revealed a significant but incomplete recovery in the complexity of astrocyte branching in both the 3-day and 4-week selegiline-treated groups (Fig. 1, C to E), indicating that selegiline only partially reduces the reactivity of astrocytes in diseased brains. The 1-week selegiline treatment was previously shown to be effective in rescuing memory impairments in the passive avoidance test in APP/PS1 mice (14). We found a similar full recovery with 3-day selegiline treatment (Fig. 1F). However, selegiline treatment for 4 weeks resulted in a diminished recovery of memory impairment (Fig. 1F). These results corroborate previous clinical reports on the diminishing effects of selegiline in long-term treatment.
Fig. 1. The diminishing effect of long-term selegiline treatment.

(A) Immunostaining for GFAP, GABA, and DAPI (4′,6-diamidino-2-phenylindole) after oral administration of selegiline (10 mg/kg for 3 days or 4 weeks) in APP/PS1 mice (n = 4 for each group; both male and female mice aged 8 to 11 months were used). Inset: Magnified images. (B) Mean intensity of GABA in GFAP-positive areas. ****P < 0.0001, Kruskal-Wallis test with Dunnett’s multiple comparisons test. (C) Representative traced astrocytes from images such as those shown in (A) were superimposed over concentric circles for Sholl analysis. (D) Quantification of the total number of intercepts in a single astrocyte. **P < 0.01 and ****P < 0.0001, one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test. (E) Quantification of the ramification index of traced astrocytes. ****P < 0.0001, Kruskal-Wallis test with Dunnett’s multiple comparisons test. (F) Passive avoidance test results of WT and APP/PS1 mice, which either received or did not receive selegiline orally (10 mg/kg for either 3 days or 4 weeks). Left: Experimental protocol for the passive avoidance test. Right: Latency to enter the dark chamber during the passive avoidance test. *P < 0.05, Kruskal-Wallis test with Dunnett’s multiple comparisons test. AU, arbitary unit; APP., APP/PS1 mice; Sele., selegiline; 3D, 3-day treatment; 4W, 4-week treatment. n refers to the number of cells (A and C) or mice (D) analyzed. n.s., not significant. Data are presented as means ± SEM. Bar graphs showing data distribution are presented in fig. S10.
KDS2010 is a new, potent, selective, and reversible MAO-B inhibitor with excellent ADME/Tox profiles
Developing a new drug for central nervous system (CNS) diseases has been particularly challenging with many obstacles to overcome, such as pharmacokinetics (PK) and blood-brain barrier (BBB) permeability, molecular target specificity in the CNS, and CNS safety. We aimed to identify a new MAO-B inhibitor that can act as a CNS drug and replace selegiline. We hypothesized that functionalized amino acids (FAAs) containing a biphenyl moiety would fulfill the requirements of a CNS drug. We designed and synthesized α-amino amide derivatives containing a biphenyl moiety with various functional groups on the phenyl ring B (Fig. 2A and fig. S1). We first introduced various electron-withdrawing groups on phenyl ring B and observed an electron-withdrawing effect of the aryl substituent (X) on MAO-B inhibition with an increasing order of potency (X: CF3 > OCF3 > Cl > F > H). We then systematically placed electron-withdrawing groups at the ortho-, meta-, and para-position and found that the para-position (KDS2010) showed the highest potency among the derivatives (Fig. 2B, fig. S2, and table S1). KDS2010 exhibited higher potency [the half maximal inhibitory concentration (IC50) = 7.6 nM] and much greater selectivity for MAO-A (12,500-fold selectivity) than the irreversible inhibitor, selegiline (Fig. 2, C and D). Furthermore, KDS2010, which is the (S)-stereoisomer, showed a marked enantiomer selectivity (eightfold selectivity) over the (R)-isomer (Fig. 2D). Last, we confirmed reversibility of KDS2010 on MAO-B enzyme activity in a wash-and-recover experiment, as previously described (22). We observed that KDS2010 caused an over 80% recovery of enzyme activity, whereas almost no recovery was observed for selegiline (Fig. 2E and Supplementary Results).
Fig. 2. KDS2010 is a potent, selective, and reversible MAO-B inhibitor.
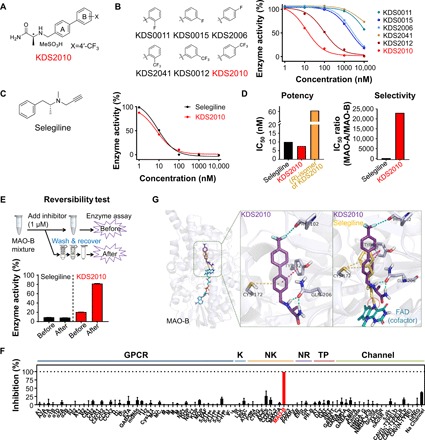
(A) Chemical structure of KDS2010. (B) Chemical structures and concentration-enzyme activity curves of the selected KDS derivatives in the MAO-B enzyme assay (n = 4 assays). (C) Comparison to the well-known irreversible MAO-B inhibitor. Left: Chemical structure of selegiline. Right: Concentration-enzyme activity curves for selegiline and KDS2010 in the MAO-B enzyme assay (n = 4 assays). (D) Potency and selectivity of selegiline and KDS2010 based on IC50 (in nM) levels of MAO-B and the isoform MAO-A. (E) Top: Diagram of the reversibility assay protocol. Bottom: Enzyme activity normalized to the dimethyl sulfoxide–treated group at each “before” and “after” step. (F) The off-target selectivity for 87 primary molecular targets at 1 μM KDS2010. A significant response (≥50% inhibition for biochemical assays) was only observed for MAO-B (>99% inhibition; n = 2 assays; red bar). GPCR, G protein–coupled receptor; K, kinase; NK, non-kinase; NR, nuclear receptor; TP, transporter; Channel, ion channel. See table S4 for the detailed results. (G) Left: Binding position of selegiline (yellow), flavin adenine dinucleotide (FAD) (cyan), and KDS2010 (purple) inside the MAO-B binding pocket. Middle: Molecular interactions of KDS2010 are indicated by the dashed line [halogen bond (cyan), π-sulfur interaction (yellow), π-π T-shaped (magenta), and hydrogen bond (green)]. Right: Selegiline was covalently bound to FAD, an MAO-B cofactor (red dotted circle).
We subsequently evaluated the drug-like properties of KDS2010 through in vitro and in vivo absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) studies. KDS2010 showed low cytochrome P450 inhibitory activity (IC50 > 10 μM for five isotypes), low hERG (human ether-à-go-go-related gene) inhibitory activity (IC50 > 50 μM), and excellent stability in both the liver microsome (92% remaining) and plasma (98% remaining) (table S2 and Supplementary Results). KDS2010 has an excellent pharmacokinetic profile with high bioavailability (>100%) and BBB permeability [brain-to-plasma total drug concentration ratio: 9.2, comparison at 2 hours after oral administration (10 mg/kg)] (table S3 and Supplementary Results). Furthermore, KDS2010 was confirmed to be safe in vivo with no significant toxicity after oral administration of both a single dose of 1000 mg/kg and consecutive doses of 200 mg/kg per day for 14 days (table S2 and Supplementary Results). We also examined the off-target selectivity of KDS2010 with 87 primary molecular targets [G protein–coupled receptors (GPCRs), kinases, non-kinase enzymes, nuclear receptors, transporters, and various ion channels] and 97 kinases (KINOMEscan). We observed an outstanding off-target selectivity at 1 μM KDS2010 (no other target causing >40% inhibition) (Fig. 2F, fig. S3, and tables S4 and S5).
To understand the molecular interaction of KDS2010 with MAO-B, we performed an experiment to investigate the substrate-dependent kinetics of KDS2010 and to investigate the binding modes of KDS2010 using the x-ray crystal structure of MAO-B bound with selegiline [Protein Data Bank (PDB) code: 2BYB] (23). We found that KDS2010 was a competitive inhibitor (fig. S4 and Supplementary Results), targeting the same binding cavity as selegiline, but its interaction with critical residues in the MAO-B binding cavity was more compact and reversible (Fig. 2G, fig. S5, and Supplementary Results). However, unlike selegiline, which formed a covalent bond with the cofactor flavin adenine dinucleotide (FAD), it is possible that the amide tail of KDS2010 does not overlap with the FAD cofactor according to molecular modeling results. Together, we identified KDS2010 as a next-generation drug candidate with excellent drug-like properties and superior pharmacological profiles for the treatment of AD.
Compensatory metabolic pathways during long-term selegiline treatment
We investigated how the aberrant GABA levels are recovered after prolonged treatment with selegiline (Fig. 1A). We measured the mRNA expression level of each enzyme in the metabolic pathway leading to GABA synthesis (Fig. 3A) from cultured astrocytes after treatment with selegiline (100 nM) in the presence of putrescine for 2 weeks (Fig. 3B). We found that the basal levels of diamine oxidase (DAO) mRNA in vehicle-treated astrocytes were lower than those of MAOB, GAD65, and GAD67 (Fig. 3C). The relative mRNA expression level of DAO was significantly higher in astrocytes treated with selegiline for 2 weeks than in vehicle-treated control astrocytes (Fig. 3D). The protein level of DAO was significantly higher in APP/PS1 mice that underwent 4 weeks of selegiline treatment than in APP/PS1 mice that underwent 4 weeks of KDS2010 treatment (Fig. 3, E and F). These findings suggest that prolonged treatment with selegiline reverts the GABA level via compensatory mechanisms involving enhanced GABA production by increased levels of DAO (24, 25). In contrast to selegiline, KDS2010-treated astrocytes did not show any sign of compensatory mechanisms. To determine whether DAO is involved in the tonic GABA increase when MAO-B enzyme activity is completely blocked, we performed whole-cell voltage-clamp recordings from dentate granule cells in the hippocampus. We found that long-term treatment with selegiline significantly reverted aberrant tonic GABA currents in APP/PS1 mice (Fig. 3, G and H). The reverted aberrant tonic GABA currents after long-term selegiline treatment were completely blocked by incubation with the DAO inhibitor aminoguanidine (AG) for 2 hours. These results indicate that DAO is the main compensatory GABA-synthesizing enzyme in long-term selegiline treatment. In contrast, long-term treatment with KDS2010 persistently blocked the aberrant tonic GABA current in APP/PS1 mice (Fig. 3, G and H).
Fig. 3. Compensatory metabolic pathways during long-term selegiline treatment.
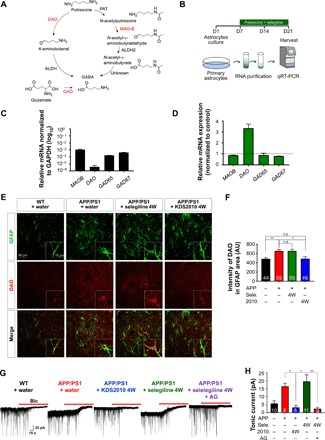
(A) Schematic diagram of the metabolic pathway leading to GABA synthesis. (B) The experimental protocol for measuring mRNA levels by quantitative real-time polymerase chain reaction (qRT-PCR) in primary cultured astrocytes treated with selegiline (100 nM) and putrescine (180 μM) for 2 weeks. (C) Relative mRNA expression normalized to GAPDH (log10). (D) Relative mRNA expression of MAOB (0.88; n = 2), DAO (3.34; n = 2), GAD65 (0.88; n = 2), and GAD67 (0.80; n = 2). Expression levels of MAOB, DAO, GAD65, and GAD67 mRNAs in control naïve astrocytes were used to normalize different conditions. GAPDH mRNA levels were used to normalize the expression of each gene. (E) Immunostaining for GFAP and DAO after the oral administration of either selegiline or KDS2010 (10 mg/kg for 4 weeks) in APP/PS1 mice. Inset: Magnified images. (F) Mean intensity of DAO in GFAP-positive areas. *P < 0.05 and **P < 0.01, Kruskal-Wallis test and Dunnett’s multiple comparisons test. (G) Representative trace of GABAA receptor–mediated currents recorded from granule cells in the DG [n = 10 for WT + water; n = 8 for APP/PS1 + water; n = 8 for APP/PS1 + KDS2010 (10 mg/kg) for 4 weeks; n = 11 for APP/PS1 + selegiline (10 mg/kg) for 4 weeks; n = 9 for APP/PS1 + selegiline (10 mg/kg) for 4 weeks with 100 nM AG; both male and female mice aged 10 to 11 months were used]. The red bar indicates application of the GABAA receptor antagonist bicuculline (BIC) (20 μM). (H) Tonic GABA current generated by bicuculline. 2010, KDS2010. *P < 0.05 and **P < 0.01, Kruskal-Wallis test and Dunnett’s multiple comparisons test. Data are presented as means ± SEM. Bar graphs showing data distribution are presented in fig. S10.
KDS2010 is effective in both short-term and long-term treatments
We subsequently found that treatment with KDS2010 for 3 days significantly ameliorated memory impairments in APP/PS1 mice (10 mg/kg per day) (fig. S6). To determine whether long-term treatment with KDS2010 suppresses GABA production in APP/PS1 mice, we orally administrated KDS2010 (10 mg/kg per day) for 4 weeks. We found that the immunoreactivity for GABA was significantly lower in GFAP-positive astrocytes of KDS2010-treated APP/PS1 mice than in water-treated control mice, and GFAP intensity was significantly reduced by KDS2010 treatment (Fig. 4, A and B). To evaluate the morphological features of astrogliosis, we performed Sholl analysis on GFAP-positive astrocytes. In KDS2010-treated APP/PS1 mice, the number of intercepts and ramifications in astrocytes were similar to those of astrocytes in WT mice (Fig. 4C). To examine the in vivo effect of KDS2010 on learning and memory, we performed passive avoidance tests after long-term treatment with KDS2010. Unlike selegiline, 4-week treatment with KDS2010 fully ameliorated the memory impairments in APP/PS1 mice (Fig. 4, D and E). In addition, 2-week treatment with KDS2010 ameliorated the memory impairments (fig. S7, A and B) and suppressed aberrant tonic GABA currents (fig. S7, C and D) in a dose-dependent manner. We also confirmed the consequence of tonic inhibition on synaptically elicited action potential firing. We performed whole-cell current-clamp recordings and measured the spike probability in DG granule neurons, as previously described (14). Consistent with a previous report, the spike probability was significantly lower in APP/PS1 mice than in WT mice and was not recovered following selegiline treatment for either 2 or 4 weeks (Fig. 4, F and G). In contrast, we found that KDS2010 fully rescued the spike probability after treatment for 2 and 4 weeks (Fig. 4, F and G). These results confirm the strong inverse correlation between tonic GABA currents and spike probability. In the case of high tonic GABA currents (APP/PS1, 4-week selegiline-treated APP/PS1; Fig. 3G), the spike probability was low, whereas in the case of low tonic GABA currents (WT, 4-week KDS2010-treated APP/PS1; Fig. 3G), the spike probability was high. Together, these results indicate that KDS2010 suppresses astrogliosis and aberrant GABA production and restores the spike probability.
Fig. 4. Impairments in spike probability, learning, and memory are fully ameliorated by long-term treatment with KDS2010 but not by selegiline.
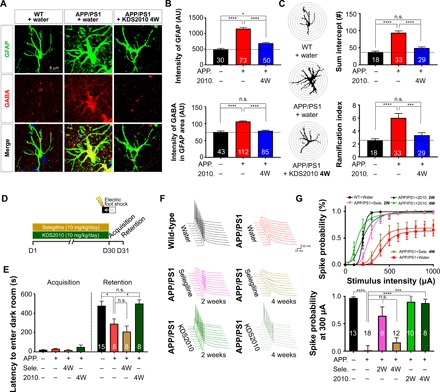
(A) Immunostaining for GFAP and GABA after oral administration of KDS2010 (10 mg/kg for 4 weeks) in APP/PS1 mice (n = 6 for each group; both male and female mice aged 8 to 11 months were used). (B) Mean intensity of GFAP and GABA in GFAP-positive areas. *P < 0.05 and ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test or Kruskal-Wallis test with Dunnett’s multiple comparisons test. (C) Left: Representative traced astrocytes were superimposed over concentric circles for Sholl analysis. Right: Quantification of the total number of intercepts (top) and the ramification index (bottom). ***P < 0.001 and ****P < 0.0001, one-way ANOVA with Tukey’s multiple comparisons test or Kruskal-Wallis test with Dunnett’s multiple comparisons test. (D) Experimental protocol for the passive avoidance test following long-term treatment with either KDS2010 or selegiline. (E) Latency to enter the dark chamber during the passive avoidance test. *P < 0.05, Kruskal-Wallis test with Dunnett’s multiple comparisons test. (F) Evoked spike probability in hippocampal slices obtained from selegiline- or KDS2010-treated mice (10 mg/kg, oral administration) in response to electrical stimulation of the perforant path (0.1 Hz, 100 μs, 100 to 1,000 μA). WT + water, APP/PS1 + water, APP/PS1 + selegiline 2W, and APP/PS1 + selegiline 4W groups include some samples from our previous study (14) (G) Top: Summary graph of spike probability versus stimulus intensity in Fig. 4D. Bottom: Comparison of spike probability at 300-μA stimulation after 2- or 4-week administration of selegiline or KDS2010. ***P < 0.001 and ****P < 0.0001, one-way ANOVA with Kruskal-Wallis test with Dunnett’s multiple comparisons test. 2W, 2-week treatment. n refers to the number of cells (B and C) or mice (D and E) tested. Data are presented as means ± SEM. Bar graphs showing data distribution are presented in fig. S10.
KDS2010 restores spatial learning and memory in the Morris water maze test
We used the Morris water maze test to assess hippocampus-dependent spatial learning and memory in the treated mice (Fig. 5). As previously reported (14), APP/PS1 mice learned slower than WT mice. In the previous study, we observed that selegiline treatment only partially ameliorated the learning and memory deficits in the Morris water maze test (14), which was attributed to the fact that selegiline loses its efficacy when administered over long periods of time (14). First, we performed the Morris water maze test in APP/PS1 mice treated with KDS2010 for 28 days. In contrast to selegiline (14), after treatment with KDS2010 for 28 days, spatial learning and memory significantly improved during the acquisition test in APP/PS1 mice without affecting swimming speed (fig. S8, A to C). We also observed that there was no significant difference in escape latency and swimming speed between WT mice treated with KDS2010 and WT mice that were not treated with KDS2010 (fig. S8, B and C). We performed an additional Morris water maze test in APP/PS1 mice treated with KDS2010 for 37 days (Fig. 5A). We confirmed that spatial learning and memory significantly improved in the acquisition test after treatment with KDS2010 for 37 days without affecting swimming speed (Fig. 5B and fig. S8D). The results of the probe test, which reflect the memory retention of the target location, also showed that the treatment of APP/PS1 mice with KDS2010 significantly restored spatial memory (Fig. 5C). Reversal learning and memory was also fully restored by treatment with KDS2010 for 37 days (Fig. 5D). Together, KDS2010 effectively ameliorated the memory impairments in APP/PS1 mice, regardless of whether they were treated with KDS2010 for 28 days or 37 days.
Fig. 5. KDS2010 significantly reversed spatial learning and memory impairments in the Morris water maze test.
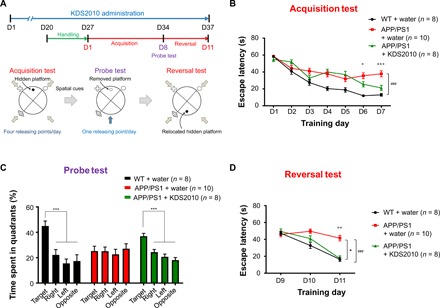
(A) The protocol for the Morris water maze test for APP/PS1 mice that either received or did not receive KDS2010 orally (10 mg/kg per day for 37 days; male mice 10 to 12 months of age). Acquisition tests were performed four times per day for 7 days. The probe test was conducted on the eighth day without the platform. The reversal test was performed four times per day for three consecutive days. (B) Escape latency during acquisition test. ###P < 0.001, compared with WT + water (one-way repeated measures ANOVA). +P < 0.05 and +++P < 0.001, compared with APP/PS1 + water [one-way ANOVA with Fisher’s least significant difference (LSD) analysis for each day]. (C) Time spent in each quadrant during the probe test. ***P < 0.001, compared with target quadrant (one-way ANOVA with Fisher’s LSD). (D) Escape latency during reversal test. *P < 0.05, compared with APP/PS1 + water and ###P < 0.001, compared with WT + water (one-way repeated measures ANOVA). P < 0.05 and ++P < 0.01 compared with APP/PS1 (one-way ANOVA with Fisher’s LSD analysis for each day). Data are presented as means ± SEM. Bar graphs showing data distribution are presented in fig. S10.
DISCUSSION
In this study, we have demonstrated the potential mechanisms that underlie the compensatory up-regulation of alternative astrocytic GABA synthetic enzyme pathways following chronic treatment with an irreversible MAO-B inhibitor but not chronic treatment with a reversible MAO-B inhibitor (fig. S9). In summary, we have delineated that the shortcomings of selegiline is due to a turning-on of the GABA-synthesizing enzyme DAO, resulting in a relapse of the aberrant GABA accumulation in reactive astrocytes. In contrast, the newly developed KDS2010 circumvents the issues of selegiline by significantly attenuating increased astrocytic GABA levels and astrogliosis, enhancing synaptic transmission, and rescuing learning and memory impairments in APP/PS1 mice after both short-term and long-term oral treatments. KDS2010 retains its efficacy for long periods even in the presence of Aβ.
The difference in the long-term efficacy between selegiline and KDS2010 can be explained by how reversible and irreversible inhibitors differentially act on the MAO-B enzyme. Irreversible inhibitors covalently modify the MAO-B enzyme and eventually destroy the enzyme itself. In particular, since the turnover rate of the MAO-B enzyme in the brain is slow [the half-life for brain MAO-B protein is 40 days (26)], permanent damage to the MAO-B enzyme is expected to cause other enzymes to be recruited to compensate for the deficiency in MAO-B enzyme activity. We found that DAO is the major compensatory GABA-synthesizing enzyme because the DAO inhibitor fully blocked the relapsed tonic GABA currents under long-term selegiline treatment. The increased DAO levels might lead to the conversion of putrescine to GABA (27), resulting in a relapse of GABA (Fig. 3A and fig S9). In contrast, reversible inhibitors occupy the active site of MAO-B and compete with substrates, resulting in an intact MAO-B enzyme. Thus, reversible inhibitors may not turn on the compensatory mechanisms because no MAO-B protein is damaged or destroyed. Therefore, irreversible inhibitors in general should be used with caution because they destroy the target molecule, which can, in turn, trigger compensatory mechanisms. In our previous study (14), we also observed that the reversible inhibitor, safinamide, showed prolonged efficacy on spike probability. However, safinamide has other mechanisms of actions (28), including sodium channel inhibition, calcium channel inhibition, and glutamate release reduction, which may cause undesirable side effects during long-term therapy.
To overcome the drawbacks of existing irreversible and reversible MAO-B inhibitors, we have developed a variety of new reversible MAO-B inhibitors, which include benzothiazoles (29), chalcones (22), and FAAs. Among the FAAs, α-amino amides containing a biphenyl moiety have exhibited the highest potency in both in vitro and in vivo model systems and have also shown better ADME/Tox profiles than other chemical scaffolds. The newly developed KDS2010 meets all the prerequisites for preclinical CNS drug candidates with excellent PK and BBB permeability, molecular target specificity, and in vivo safety. According to the molecular modeling of KDS2010 (Fig. 2G), two hydrogen bonds between the two amine protons of α-amino amide and the oxygen atom of GLN206 in the MAO-B active site create a strong binding affinity, resulting in higher potency than that of other irreversible inhibitors. The biphenyl moiety of KDS2010, which has a flat structure and optimal lipophilicity, is most likely the key structure that is responsible for excellent BBB permeability. These conceptual breakthroughs in medicinal chemistry should be further investigated in the future.
Although the recent failure of the reversible MAO-B inhibitor sembragiline in a clinical trial involving patients with AD is discouraging (30), our study offers plausible explanations for sembragiline’s failure. In the aforementioned clinical study, sembragiline was administered to patients with moderate to severe AD who had been taking either donepezil (5 to 10 mg daily) or rivastigmine (4.6 mg daily or 9.5 mg daily patch) for at least 6 months (30). The authors reported that, in their study, they did not observe any treatment effect because the study subjects were already receiving treatment during the study period and that the clinical efficacy of sembragiline may be noted in a monotherapy clinical trial (30). Another selective and reversible MAO-B inhibitor, lazabemide, has been shown to be clinically beneficial for the treatment of AD. It was tested as a monotherapy in patients with mild to moderate AD. Treatment with lazabemide resulted in a 20 to 40% reduction in the decline in cognitive function compared with a placebo during the phase 2 clinical trial. Although the phase 3 studies also provided consistent and statistically significant evidence of cognitive benefit relative to the placebo, the development of lazabemide was discontinued because of liver toxicity in 1999 (30). These results suggest that treatment with reversible MAO-B inhibitors would have been more effective in patients with mild to moderate AD without background treatment. Reviewing the potential success of reversible MAO-B inhibitors in clinical trials may be useful for developing an AD therapy.
Reactive astrocytes are the hallmark of astrogliosis and neuroinflammation, which are commonly observed not only in AD but also in other brain diseases, such as Parkinson’s disease, stroke, epilepsy, and traumatic brain injury (14). A recent report demonstrates that an imbalance in the GABAergic system contributes to neuronal dysfunction in AD (31). In addition, MAO-B has been shown to be elevated in AD brain and regulate Aβ production in neurons via γ-secretase (32). Therefore, these studies support our finding that MAO-B–driven aberrant GABA metabolism induces more severe AD pathologies, proposing MAO-B as the major target for relieving AD symptoms. We consistently observed that MAO-B inhibitors markedly reduce hypertrophy and astrogliosis in addition to astrocytic GABA. For example, selegiline significantly reduced astrocytic reactivity in short-term treatment in AD model mice (14, 33, 34). KDS2010 almost completely reverted astrocytic reactivity to the WT level in both short-term and long-term treatments (Fig. 4, A to C). Our study indicates that MAO-B is a key upstream molecular player of astrogliosis. Furthermore, these results suggest that KDS2010 is an effective pharmacological tool to prevent astrogliosis. Therefore, KDS2010 should be applied to other brain diseases in which reactive astrocytes are implicated and in which there is MAO-B–dependent aberrant GABA production. These exciting possibilities should be examined in future investigations.
MATERIALS AND METHODS
Animals
All mice were kept in a temperature- and humidity-controlled environment with a 12-hour light/12-hour dark cycle, and mice had free access to food and water. Handing and animal care was performed according to the directives of the Animal Care and Use Committee of the Institutional Animal Care and Use Committee of Korea Institute of Science and Technology (KIST) (Seoul, Korea). APP/PS1 mice of B6C3 hybrid background originated from the Jackson Laboratory (USA; stock number 004462) and were maintained as hemizygotes by crossing transgenic mice to B6C3 F1 mice, which were derived from a cross between a C57BL/6 female and a C3H male. This hybrid strain is commonly used in the production of transgenic mice. Both sexes of 8- to 13-month-old transgenic mice and WT littermates were used. In most behavioral experiments, mice were treated by oral administration with each concentration for selegiline or KDS2010 at 1 and 10 mg/kg per day. The amount of each drug (in mg) was calculated according to each mouse’s weight (in kg) and was dissolved in 100 μl of water. Each oral administration was performed once per day. In the case of the Morris water maze experiment, 10 mg of KDS2010 was dissolved in 50 ml of water. The mice were provided with water (control) and KDS2010 solution ad libitum. Behavioral experiments were performed during the light phase of the light-dark cycle at the same time of each day. No statistical method was used to predetermine sample size. When using either sex, different litters, or ages of animals, animals were evenly allocated to each experimental group. All experiments were done with gender- and age-matched controls. Behavioral tests were conducted in a blind manner. The investigator was blinded to the group allocation of the genotype or treatment during the experiment and when assessing all testing.
Cell culture
Primary cortical astrocytes were prepared from 1-day postnatal C57BL/6 mice, as previously described (35). The cerebral cortex was dissected free of adherent meninges, minced, and dissociated into a single-cell suspension by trituration. Dissociated cells were plated onto plates coated with poly-d-lysine (0.1 mg/ml; Sigma). Cells were grown in Dulbecco’s modified Eagle’s medium (Corning) supplemented with glucose (4.5 g/liter), l-glutamine, sodium pyruvate, 10% heat-inactivated horse serum, 10% heat-inactivated fetal bovine serum, and penicillin-streptomycin (1000 U/ml). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. After 3 days, cells were vigorously washed with repeated pipetting using medium, and the medium was replaced to remove debris and other floating cell types.
Real-time polymerase chain reaction
Total RNA was isolated from mouse-cultured astrocytes using an RNA purification kit (GeneAll) according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 1-μg total RNA, and reverse transcription was performed using a SuperScript cDNA synthesis kit (Invitrogen) according to the manufacturer’s instructions. Real-time polymerase chain reaction was performed using a TaqMan gene expression assay (Invitrogen). Primer sets for MAOB, DAO, GAD65, and GAD67 were purchased from Cosmogenetech. GAPDH was used as a housekeeping gene. Primer sequence information is listed in table S6. The delta-delta Ct method was used to calculate fold changes in gene expression (36).
Slice immunostaining for confocal microscopy
For tissue fixation, mice were deeply anesthetized with 2% avertin (20 μl/g, intraperitoneally) and perfused transcardially with 10 ml of heparinized normal saline, followed by ice-cold 4% paraformaldehyde with 0.5% glutaraldehyde solution. Excised brains were postfixed overnight in 4% paraformaldehyde and 0.5% glutaraldehyde at 0.9% saline and immersed in 30% sucrose for 48 hours for cryoprotection. After fixation, brains were mounted into an OCT (optimal cutting temperature compound) embedding solution and frozen at −20° to −80°C. Coronal hippocampal sections were cut at 30 μm in a cryostat and floated in phosphate-buffered saline (PBS). Sections were processed with three additional washes in PBS, incubated for 1 hour in a blocking solution (0.3% Triton X-100 and 4% normal serum in 0.1 M PBS), and then immunostained with a mixture of primary antibodies in a blocking solution at 4°C on a shaker overnight. After washing in PBS three times, sections were incubated with corresponding fluorescent secondary antibodies for 2 hours and then washed with PBS three times. If needed, then 4′,6-diamidino-2-phenylindole (DAPI) staining was done by the addition of DAPI solution (1:1000; Pierce) during the second washing step. Last, sections were mounted with fluorescent mounting medium (Dako) and dried. A series of fluorescent images were obtained with a FV1000 Olympus or an A1 Nikon confocal microscope, and 30-μm Z stack images in 2-μm steps were processed for further analysis using FLUOVIEW (Olympus) or NIS-Elements (Nikon) software and ImageJ (National Institutes of Health) program. Any alterations in brightness or contrast were equally applied to the entire image set. Specificity of primary antibody and immunoreaction was confirmed by omitting primary antibodies or changing fluorescent probes of the secondary antibodies.
Antibodies
The primary antibodies used for immunostaining were as follows: chicken anti-GFAP (1:500; Millipore, ab5541), guinea pig anti-GABA (1:1000; Millipore, ab175), and rabbit anti-ABP1 (amiloride binding protein 1) (DAO) (1:500; Aviva Systems Biology, ARP41908_P050). Fluorescent secondary antibodies were purchased from Invitrogen or Jackson ImmunoResearch and used in 1:200 dilutions.
Tonic GABA recording
Brain slices were prepared from mice transgenic mice and WT littermates aged around 8 to 13 months old. Mice were deeply anesthetized with 2-bromo-2-chloro-1,1,1-trifluoroethane. After anesthetization, the brain was quickly excised from the skull and submerged in ice-cold cutting solution that contained 250 mM sucrose, 26 mM NaHCO3, 10 mM d-(+)-glucose, 4 mM MgCl2, 3 mM myo-inositol, 2.5 mM KCl, 2 mM sodium pyruvate, 1.25 mM NaH2PO4, 0.5 mM ascorbic acid, 0.1 mM CaCl2, and 1 mM kynurenic acid (pH 7.4). All the solutions were gassed with 95% O2 and 5% CO2. Hippocampal slices were obtained by vibrating microtome (Leica VT1000S), and 300-μm-thick coronal hippocampal slices were maintained at room temperature in a submerged chamber with extracellular artificial cerebrospinal fluid (ACSF) solution [126 mM NaCl, 24 mM NaHCO3, 1 mM NaH2PO4, 2.5 mM KCl, 2.5 mM CaCl2, 2 mM MgCl2, and 10 mM d-(+)-glucose (pH 7.4)]. Slices were incubated at room temperature for at least 1 hour before recording with gas bubbling. Slices were transferred to a recording chamber that was continuously perfused with ACSF solution (flow rate, 2 ml/min). The slice chamber was mounted on the stage of an upright Olympus microscope and viewed with a 60× water immersion objective (0.90 numerical aperture) with infrared differential interference contrast optics. Cellular morphology was visualized by a charge-coupled device camera and Imaging Workbench software (INDEC BioSystems). Whole-cell recordings were made from granule cell somata located in the DG. The holding potential was −70 mV. Pipette resistance was typically 6 to 8 megaohms and filled with internal solution [135 mM CsCl, 4 mM NaCl, 0.5 mM CaCl2, 10 mM Hepes, 5 mM EGTA, 2 mM Mg–adenosine triphosphate, 0.5 mM Na2–guanosine triphosphate, and 10 mM QX-314, pH adjusted to 7.2 with CsOH (osmolarity, 278 to 285 mOsm)] (14). Baseline current was stabilized with d-AP5 (50 μM) and 6-cyano-7-nitroquinoxaline-2,3-dione (20 μM) before measuring tonic current. Electrical signals were digitized and sampled at 50-μs intervals with Digidata 1440 A and a MultiClamp 700B amplifier (Molecular Devices) using pCLAMP10.2 software. Data were filtered at 2 kHz. The amplitude of tonic GABA currents was measured by the baseline shift after bicuculline (100 μM) administration using the Clampfit program. Tonic current was measured from the baseline to bicuculline-treated current. Frequency and amplitude of spontaneous inhibitory postsynaptic currents before bicuculline administration were detected and measured by MiniAnalysis (Synaptosoft).
Evoked spike probability
Evoked spike probability recordings were made from horizontal brain slices with a thickness of 300 μm, as described above. Synaptically evoked spikes were triggered by a tungsten bipolar electrode placed in the outer half of the middle third dentate molecular layer. Stimulus intensity was set by 0.1-Hz stimulation of lateral perforant path fibers (100-μs duration; 100- to 1000-mA intensity) via a constant current isolation unit. The evoked EPSPs (excitatory postsynaptic potentials) were recorded using glass pipette electrodes (6 to 8 MW), filled with intracellular solution containing 120 mM potassium gluconate, 10 mM KCl, 1 mM MgCl2, 0.5 mM EGTA, 40 mM Hepes (pH 7.2). Spiking probability was calculated as the ratio of the number of successful (spike-generating) stimulations to the total number of stimulations. Whole-cell patch-clamp recordings, using a Multiclamp7 amplifier (Molecular Devices), were performed from DG granule cells, visually identified with infrared video microscopy and differential interference contrast optics. Data were collected with a MultiClamp 700B amplifier (Molecular Devices) using Clampex10 acquisition software (Molecular Devices) and digitized with Digidata 1322A (Molecular Devices). Raw data were low pass–filtered at 4 kHz and collected for offline analysis at a sampling rate of 10 kHz using pClamp10.2 software.
MAO-A and MAO-B enzyme assay
Evaluation of the IC50 of compound for MAO enzyme activity was based on previously described methods (22). Human recombinant MAO-A (hMAO-A) and MAO-B (hMAO-B) enzymes prepared from insect cells were obtained from Sigma-Aldrich. hMAO-A and MAO-B enzymes (for MAO-A, 6 μl from 5.0 mg/ml solution; for MAO-B, 50 μl from 5.0 mg/ml solution) were diluted in aqueous 50 mM phosphate buffer (10 ml, pH 7.4). The enzyme solution (98 μl; final protein amounts: ~0.3 μg of protein per well for MAO-A and ~2.5 μg of protein per well for MAO-B) was treated with test compound in dimethyl sulfoxide (2 μl; final concentration, 0.1 nM to 10 μM) and then incubated for 15 min at room temperature. The resulting solution (100 μl) was treated with an assay solution (100 μl), which is the mixed solution of 20 mM Amplex Red reagent (200 μl; final concentration, 0.2 mM), 100 mM substrate (200 μl; MAO-A: p-tyramine, MAO-B: benzylamine, final concentration, 1 mM), and horseradish peroxidase (200 U/ml) (100 μl; final concentration, 1 U/ml) in aqueous 50 mM phosphate buffer (9.5 ml, pH 7.4). After 20 min of incubation, the released hydrogen peroxide (H2O2) and the subsequent production of resorufin were quantified using a microplate fluorescence reader (SpectraMax i3 reader, Molecular Devices) with an excitation at 545 nm and an emission at 590 nm.
Reversibility test
The reversibility of MAO-B inhibition was measured using the previous method (22). Briefly, the recombinant hMAO-B enzymes (50 μl from 5.0 mg/ml solution) were diluted in aqueous 50 mM phosphate buffer (2 ml, pH 7.4). The enzyme solution (441 μl) was incubated with the 0.05 mM MAO-B inhibitor (9 μl; final concentration, 2 μM) at room temperature (30 min) and then divided into two aliquots (200 μl) of reaction enzyme solution. An aliquot (200 μl) was examined for hMAO-B activity using the method described above. Another aliquot (200 μl) was used for the washout experiment. For washout experiment, the aliquot was placed in an Amicon Ultra centrifugal filter (3 kDa) (Millipore) and centrifuged (14,000g) at 4°C (10 min). The enzyme retained in the membrane was resuspended in 50 mM phosphate buffer (1 ml) and centrifuged again (under same conditions described above) for three consecutive times. The resulting enzyme was resuspended in aqueous 50 mM phosphate buffer (200 μl, pH 7.4), and the solution was used for determination of the remaining enzyme activity. Control experiments without test compounds were performed with the vehicles to define 100% hMAO-B activity. The corresponding values of percent (%) inhibition were separately determined before and after washout.
Molecular docking study
Ligands were sketched using ChemDraw Professional V15.0 program. X-ray crystal structure of MAO-B bound with selective inhibitor selegiline (PDB code: 2BYB) with a 2.2-Å resolution was taken from the PDB. The protein and ligands were prepared using Protein Prepare Wizard and LigPrep module implement in Maestro (Schrödinger LLC, NY, USA), respectively. The protein was fixed by adding missing side chain and neutralized at pH 7.4. Water was removed with less than one hydrogen bond to non-waters, and only hydrogens were minimized with an OPLS_2005 force field. Ligands were protonated at neutral pH using Epik and energetically minimized with an OPLS_2005 force field. The docking study (37, 38) was performed using Glide with the standard precision scoring function. Figures were rendered with pymol program (www.pymol.org).
Off-target selectivity
To evaluate the activity of KDS2010 on off-targets, the SafetyScreen 87 panel assay was performed at Eurofins Cerep-Panlabs (Taipei, Taiwan) using validated radio-ligand competition binding assays and enzymatic inhibition assays. Experimental conditions for each of these 87 assays are available at the Eurofins web page (www.eurofinspanlabs.com) (see also table S4). To eliminate the possibility of kinase off-target hits, a scanEDGE kinase assay panel was conducted in DiscoverX (CA, USA) using a binding reaction in combination with kinase, liganded affinity beads, and test compounds. Experimental conditions for these 97 assays are available at the DiscoverX web page (www.discoverx.com) (see also table S5).
Passive avoidance test
Before the experiment, mice were handled daily for 7 days. Mice were placed in two-compartment (light and dark) shuttle chambers with a constant current shock generator (MED Associates). On the first experimental day, a mouse was put in the light chamber for the acquisition trial. After 60 s of exploration, the door separating the light and dark compartments was raised, allowing the mouse to freely enter the dark chamber. When the mouse entered a dark chamber with all four paws, the door immediately closed and an electric foot shock (0.5 mA, 2 s duration) was delivered through the floor grid. The mouse was then returned to the home cage, and the retention trial was carried out 24 hours after the acquisition trial. On the second experimental day, the mouse was placed into the light chamber again. After 60 s of exploration, the door was raised to allow the mouse to enter the dark chamber. The step-through latencies of entering the dark chamber before and after the electric shock were measured to a maximum of 600 s.
Morris water maze
Two separate water maze tests were performed using male APP/PS1 mice (28-day treatment and 37-day treatment) (fig. S8 and Fig. 5). The water maze (1.2-m diameter) was filled with water (24° to 25°C) and made opaque by the addition of nontoxic white paint (WeatherTough Forte, Bristol Paints). The water maze was surrounded by a black circular curtain (placed 70 cm away) that held three salient visual cues. Before the water maze test, mice were handled for 7 days. Mice were released and randomly distributed across four quadrants of the water maze, and the mice were allowed a maximum of 60 s to find a hidden platform (10-cm diameter, 1 cm under the water surface). Mice were trained for 7 days (4 trials per day, 10-min intertrial interval). On training day 8, mice were given a 60-s probe test (sans the platform) to test their spatial memory. After the probe test, the hidden platform was placed on the opposite quadrant. Mice were trained an additional 3 days of reversal training. An automated tracking system (EthoVision, Noldus) was used to monitor the animal’s swimming pattern, speed, and the amount of time spent in each of the four quadrants.
Statistical analyses
For comparison of multiple groups, one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test and Dunnett’s multiple comparison test were assessed using Prism software. P values less than 0.05 were considered significant. The significance level is displayed as asterisks (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; n.s. not significant). For all comparisons, samples were not excluded from data except only for tonic GABA current recording; cells with holding currents over −100 pA were excluded, and experiments were stopped after 3 hours from the time of slicing. No animal was excluded from analysis in behavioral experiments. When using either sex, different litters, or ages of animals, animals were evenly allocated to each experimental group, and for slice immunostaining and electrophysiological experiments, we randomly conducted samples in each experimental group. Detailed results for ANOVA are described in table S6.
Supplementary Material
Acknowledgments
We thank H. Hwang for in vitro ADME/Tox tests. We also acknowledge a number of colleagues for critical feedback on the manuscript. Funding: This work was supported by the National Research Council of Science & Technology (NST) grant by the Korean government (MSIP) (no. CRC-15-04-KIST to K.D.P.), the Drug Development Project (KDDF-2105-09-06 to K.D.P.) of Korea Drug Development Fund, the National Research Foundation of Korea (NRF-2018M3A9C8016849 to K.D.P.), the Main Research Program (E0164503-01 to K.D.P.) of the Korea Food Research Institute (KFRI), the Creative Research Initiative Program, the Korean National Research Foundation (2015R1A3A2066619 to C.J.L.), the KIST Institutional Grant (2E26662 to C.J.L.), and the KU-KIST Graduate School of Science and Technology program (R1435281 to C.J.L.). Author contributions: J.-H.P., Y.H.J., C.J.L., and K.D.P. designed the study, analyzed the data, and wrote the manuscript. H.J.S., B.K.J., J.W.C., S.K.Y., S.Y.H., S.W.K., and S.J.C. synthesized and analyzed chemical compounds. J.-H.P., J.W.C., and S.J.S. performed the MAO assay. J.-H.P. performed in vitro ADME studies. J.W. and H.C. performed slice electrophysiology. O.Y. and S.J. performed spike probability test. J.-H.P., Y.H.J., H.J.K., M.P., S.K., Jeongyeon Kim, M.-H.N., and J.C. performed behavior tests and analysis. A.M.L. and A.N.P. contributed to the molecular docking study. All authors contributed to analysis and discussion of the results. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Used research material is available by request from the corresponding author or from commercial sources when applicable.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav0316/DC1
Supplementary Results
Supplementary Materials and Methods
Fig. S1. General procedure for the preparation of KDS compounds.
Fig. S2. Structure-activity relationship of the synthesized compounds.
Fig. S3. The KINOMEscan screening results with 1000 nM KDS2010 for off-target selectivity.
Fig. S4. Mode of KDS2010 binding with MAO-B.
Fig. S5. Three-day and 2-day interactions of selegiline, KDS2010, and KDS0014 inside MAO-B.
Fig. S6. Acute treatment of KDS2010 (3 days) restored memory impairment in APP/PS1 mice.
Fig. S7. Passive avoidance test for learning and memory in APP/PS1 mice with 2-week KDS2010 treatment.
Fig. S8. KDS2010 significantly recovers spatial learning and memory in Morris water maze.
Fig. S9. Model diagrams of long-term treatment of AD with either irreversible or reversible MAO-B inhibitors.
Fig. S10. Data distribution of bar graphs.
Table S1. Inhibitory effects of the synthesized compounds against hMAO enzymes.
Table S2. In vitro and in vivo ADME/Tox profile of KDS2010.
Table S3. In vivo pharmacokinetic parameters of KDS2010.
Table S4. KDS2010 interactions with 87 primary molecular targets including GPCRs, kinases, non-kinase enzymes, nuclear receptors, transporters, and various ion channels.
Table S5. KDS2010 interactions with 97 kinase including TK, TKL, STE, CK1, AGC, CAMK, CMGC, ATYPICAL, LIPID, and Mutant form.
Table S6. Detailed information for statistical analysis.
Table S7. Primer sequences for each enzyme (F: forward primer and R: reverse primer).
REFERENCES AND NOTES
- 1.Alzheimer's Association , 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 8, 131–168 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer's Association , 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 11, 332–384 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Querfurth H. W., LaFerla F. M., Alzheimer’s Disease. N. Engl. J. Med. 362, 329–344 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Heneka M. T., Carson M. J., Khoury J. E., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., Herrup K., Frautschy S. A., Finsen B., Brown G. C., Verkhratsky A., Yamanaka K., Koistinaho J., Latz E., Halle A., Petzold G. C., Town T., Morgan D., Shinohara M. L., Perry V. H., Holmes C., Bazan N. G., Brooks D. J., Hunot S., Joseph B., Deigendesch N., Garaschuk O., Boddeke E., Dinarello C. A., Breitner J. C., Cole G. M., Golenbock D. T., Kummer M. P., Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardy J., Allsop D., Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Selkoe D. J., The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498 (1991). [DOI] [PubMed] [Google Scholar]
- 7.Golde T. E., Schneider L. S., Koo E. H., Anti-Aβ therapeutics in Alzheimer's disease: The need for a paradigm shift. Neuron 69, 203–213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummings J., Lee G., Mortsdorf T., Ritter A., Zhong K., Alzheimer's disease drug development pipeline: 2017. Alzheimers Dement. 3, 367–384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graham W. V., Bonito-Oliva A., Sakmar T. P., Update on Alzheimer's disease therapy and prevention strategies. Annu. Rev. Med. 68, 413–430 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Schneider L. S., Mangialasche F., Andreasen N., Feldman H., Giacobini E., Jones R., Mantua V., Mecocci P., Pani L., Winblad B., Kivipelto M., Clinical trials and late-stage drug development for Alzheimer's disease: An appraisal from 1984 to 2014. J. Intern. Med. 275, 251–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sacks C. A., Avorn J., Kesselheim A. S., The failure of solanezumab—How the FDA saved taxpayers billions. N. Engl. J. Med. 376, 1706–1708 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Doody R. S., Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N. Engl. J. Med. 371, 584 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Mangialasche F., Solomon A., Winblad B., Mecocci P., Kivipelto M., Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 9, 702–716 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Jo S., Yarishkin O., Hwang Y. J., Chun Y. E., Park M., Woo D. H., Bae J. Y., Kim T., Lee J., Chun H., Park H. J., Lee D. Y., Hong J., Kim H. Y., Oh S.-J., Park S. J., Lee H., Yoon B.-E., Kim Y., Jeong Y., Shim I., Bae Y. C., Cho J., Kowall N. W., Ryu H., Hwang E., Kim D., Lee C. J., GABA from reactive astrocytes impairs memory in mouse models of Alzheimer's disease. Nat. Med. 20, 886–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon B.-E., Woo J., Chun Y.-E., Chun H., Jo S., Bae J. Y., An H., Min J. O., Oh S.-J., Han K.-S., Kim H. Y., Kim T., Kim Y. S., Bae Y. C., Lee C. J., Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. Lond. 592, 4951–4968 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sano M., Ernesto C., Thomas R. G., Klauber M. R., Schafer K., Grundman M., Woodbury P., Growdon J., Cotman C. W., Pfeiffer E., Schneider L. S., Thal L. J., A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. N. Engl. J. Med. 336, 1216–1222 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Mangoni A., Grassi M. P., Frattola L., Piolti R., Bassi S., Motta A., Marcone A., Smirne S., Effects of a MAO-B inhibitor in the treatment of Alzheimer disease. Eur. Neurol. 31, 100–107 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Piccinin G. L., Finali G., Piccirilli M., Neuropsychological effects of l-deprenyl in Alzheimerʼs type dementia. Clin. Neuropharmacol. 13, 147–163 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Froestl W., Muhs A., Pfeifer A., Cognitive enhancers (nootropics). Part 2: Drugs interacting with enzymes. Update 2014. J. Alzheimers Dis. 42, 1–68 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Wilcock G. K., Birks J., Whitehead A., Evans J. G., The effect of selegiline in the treatment of people with Alzheimer’s disease: A meta-analysis of published trials. Int. J. Geriatr. Psychiatry 17, 175–183 (2002). [DOI] [PubMed] [Google Scholar]
- 21.Fowler J. S., Volkow N. D., Logan J., Franceschi D., Wang G.-J., MacGregor R., Shea C., Garza V., Pappas N., Carter P., Netusil N., Bridge P., Liederman D., Elkashef A., Rotrosen J., Hitzemann R., Evidence that l-deprenyl treatment for one week does not inhibit mao a or the dopamine transporter in the human brain. Life Sci. 68, 2759–2768 (2001). [DOI] [PubMed] [Google Scholar]
- 22.Choi J. W., Jang B. K., Cho N.-c., Park J.-H., Yeon S. K., Ju E. J., Lee Y. S., Han G., Pae A. N., Kim D. J., Park K. D., Synthesis of a series of unsaturated ketone derivatives as selective and reversible monoamine oxidase inhibitors. Bioorg. Med. Chem. 23, 6486–6496 (2015). [DOI] [PubMed] [Google Scholar]
- 23.De Colibus L., Li M., Binda C., Lustig A., Edmondson D. E., Mattevi A., Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proc. Natl. Acad. Sci. U.S.A. 102, 12684–12689 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caron P. C., Kremzner L. T., Cote L. J., GABA and its relationship to putrescine metabolism in the rat brain and pancreas. Neurochem. Int. 10, 219–229 (1987). [DOI] [PubMed] [Google Scholar]
- 25.Laschet J., Grisar T., Bureau M., Guillaume D., Characteristics of putrescine uptake and subsequent GABA formation in primary cultured astrocytes from normal C57BL/6J and epileptic DBA/2J mouse brain cortices. Neuroscience 48, 151–157 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Fowler J. S., Volkow N. D., Logan J., Wang G.-J., Mac Gregor R. R., Schlyer D., Wolf A. P., Pappas N., Alexoff D., Shea C., Dorflinger E., Kruchowy L., Yoo K., Fazzini E., Patlak C., Slow recovery of human brain MAO B after L-deprenyl (Selegeline) withdrawal. Synapse 18, 86–93 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Kim J.-I., Ganesan S., Luo S. X., Wu Y.-W., Park E., Huang E. J., Chen L., Ding J. B., Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marzo A., Dal Bo L., Monti N. C., Crivelli F., Ismaili S., Caccia C., Cattaneo C., Fariello R. G., Pharmacokinetics and pharmacodynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity. Pharmacol. Res. 50, 77–85 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Nam M.-H., Park M., Park H., Kim Y., Yoon S., Sawant V. S., Choi J. W., Park J.-H., Park K. D., Min S.-J., Lee C. J., Choo H., Indole-substituted benzothiazoles and benzoxazoles as selective and reversible MAO-B inhibitors for treatment of Parkinson's disease. ACS Chem. Neurosci. 8, 1519–1529 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Nave S., Doody R. S., Boada M., Grimmer T., Savola J.-M., Delmar P., Pauly-Evers M., Nikolcheva T., Czech C., Borroni E., Ricci B., Dukart J., Mannino M., Carey T., Moran E., Gilaberte I., Muelhardt N. M., Gerlach I., Santarelli L., Ostrowitzki S., Fontoura P., Sembragiline in moderate Alzheimer's disease: Results of a randomized, double-blind, placebo-controlled phase II trial (MAyflOwer RoAD). J. Alzheimers Dis. 58, 1217–1228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y., Sun H., Chen Z., Xu H., Bu G., Zheng H., Implications of GABAergic neurotransmission in Alzheimer's disease. Front. Aging Neurosci. 8, 31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schedin-Weiss S., Inoue M., Hromadkova L., Teranishi Y., Yamamoto N. G., Wiehager B., Bogdanovic N., Winblad B., Sandebring-Matton A., Frykman S., Tjernberg L. O., Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with gamma-secretase and regulates neuronal amyloid beta-peptide levels. Alzheimers Res. Ther. 9, 57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barres B. A., The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 60, 430–440 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Sofroniew M. V., Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–647 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang E. M., Kim E., Yarishkin O., Woo D. H., Han K.-S., Park N., Bae Y., Woo J., Kim D., Park M., Lee C. J., Park J.-Y., A disulphide-linked heterodimer of TWIK-1 and TREK-1 mediates passive conductance in astrocytes. Nat. Commun. 5, 3227 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Caulfield T. R., Fiesel F. C., Moussaud-Lamodière E. L., Dourado D. F. A. R., Flores S. C., Springer W., Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLOS Comput. Biol. 10, e1003935 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caulfield T. R., Fiesel F. C., Springer W., Activation of the E3 ubiquitin ligase Parkin. Biochem. Soc. Trans. 43, 269–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav0316/DC1
Supplementary Results
Supplementary Materials and Methods
Fig. S1. General procedure for the preparation of KDS compounds.
Fig. S2. Structure-activity relationship of the synthesized compounds.
Fig. S3. The KINOMEscan screening results with 1000 nM KDS2010 for off-target selectivity.
Fig. S4. Mode of KDS2010 binding with MAO-B.
Fig. S5. Three-day and 2-day interactions of selegiline, KDS2010, and KDS0014 inside MAO-B.
Fig. S6. Acute treatment of KDS2010 (3 days) restored memory impairment in APP/PS1 mice.
Fig. S7. Passive avoidance test for learning and memory in APP/PS1 mice with 2-week KDS2010 treatment.
Fig. S8. KDS2010 significantly recovers spatial learning and memory in Morris water maze.
Fig. S9. Model diagrams of long-term treatment of AD with either irreversible or reversible MAO-B inhibitors.
Fig. S10. Data distribution of bar graphs.
Table S1. Inhibitory effects of the synthesized compounds against hMAO enzymes.
Table S2. In vitro and in vivo ADME/Tox profile of KDS2010.
Table S3. In vivo pharmacokinetic parameters of KDS2010.
Table S4. KDS2010 interactions with 87 primary molecular targets including GPCRs, kinases, non-kinase enzymes, nuclear receptors, transporters, and various ion channels.
Table S5. KDS2010 interactions with 97 kinase including TK, TKL, STE, CK1, AGC, CAMK, CMGC, ATYPICAL, LIPID, and Mutant form.
Table S6. Detailed information for statistical analysis.
Table S7. Primer sequences for each enzyme (F: forward primer and R: reverse primer).