

Abstract
Simulation of hypoxic processes in vitro can be achieved through cobalt chloride (CoCl2), which induces strong neurodegeneration. Hypoxia plays an important role in the progression of several retinal diseases. Thus, we investigated whether hypoxia can be reduced by hypothermia. Porcine retinal explants were cultivated for four and eight days and hypoxia was mimicked by adding 300 µM CoCl2 from day one to day three. Hypothermia treatment (30 °C) was applied simultaneously. Retinal ganglion, bipolar and amacrine cells, as well as microglia were evaluated via immunohistological and western blot analysis. Furthermore, quantitative real-time PCR was performed to analyze cellular stress and apoptosis. In addition, the expression of specific marker for the previously described cell types were investigated. A reduction of ROS and stress markers HSP70, iNOS, HIF-1α was achieved via hypothermia. In accordance, an inhibition of apoptotic proteins (caspase 3, caspase 8) and the cell cycle arrest gene p21 was found in hypothermia treated retinae. Furthermore, neurons of the inner retina were protected by hypothermia. In this study, we demonstrate that hypothermia lowers hypoxic processes and cellular stress. Additionally, hypothermia inhibits apoptosis and protects neurons. Hence, this seems to be a promising treatment for retinal neurodegeneration.
Introduction
A deprived oxygen supply in tissues is known as hypoxia and can occur in several retinal diseases, such as glaucoma1. A hallmark for hypoxic processes is the up-regulation of the transcription factor hypoxia inducible factor-1 (HIF-1), especially the stabilization of its oxygen sensitive subunit HIF-1α2. As a result, HIF-1α is translocated into the cell nucleus, where the expression of different hypoxic genes is induced3,4. Although cobalt is important for the neuronal integrity, high concentrations induce cytotoxic mechanisms by binding the oxygen-dependent region of HIF-1α and therefore prevent the degradation process of HIF-1α5. Furthermore, divalent metal ions, such as cobalt, can cause oxidative stress by rupturing the outer cell membrane and disturbing the mitochondrial respiration. These mechanisms of cellular toxicity have been proposed for several neurodegenerative disorders. Through its characteristics as a hypoxia mimicking agent, cobalt chloride is commonly used for the induction of neurodegeneration in different models6–10. In a previous study, we evaluated the effects of different CoCl2 concentrations on porcine retinae and demonstrated that it induced neuronal cell loss, which was associated with increased apoptosis mechanisms11. Further previous performed studies, which evaluated the effect of hypoxia induced by oxygen (O2)-deprivation, point out that always a change of the same parameters in both models was observed12–15. Therefore, hypoxia via CoCl2 is to some extent comparable to hypoxia induced by O2-depriviation.
Hypothermia, described as temperature below 37 °C, seems to have neuroprotective effects, although the underlying molecular mechanism is not completely understood yet16,17. Nevertheless, several neuroprotective effects of hypothermia on the retina were reported. Rat retinae were protected from ischemia/reperfusion induced damage by hypothermia18. Bovine retinal ganglion cells (RGCs) showed prolonged survival under ischemic conditions after hypothermia and RGCs from minipigs were protected from ischemia induced cell loss12,14.
The goal of our study was to investigate possible neuroprotective effects of hypothermia in a CoCl2 induced degeneration model of cultured porcine retinal explants. Hence, hypothermia at 30 °C was applied to retinal explants and hypoxic processes and cellular stress markers were evaluated. Furthermore, the apoptotic conditions of whole retinae and the apoptosis rate of RGCs were analyzed. In addition, bipolar and amacrine cells as well as glial cells were assessed after four and eight days of cultivation.
Here, we prove that hypothermia has neuroprotective effects on CoCl2 treated retinae by reducing hypoxic processes, cellular stress and inhibiting apoptosis. In conclusion, a rescue of neurons, especially RGCs, was achieved.
Results
Hypothermia treatment (30 °C) and hypoxia (300 µM CoCl2) were performed simultaneously (Fig. 1A). After four and eight days, retinal explants were obtained for quantitative real-time PCR (qPCR), histological and western blot analyses. Additionally, pH-measurements were performed after each medium exchange, and reactive oxygen species (ROS) level was evaluated on days one and two.
Figure 1.
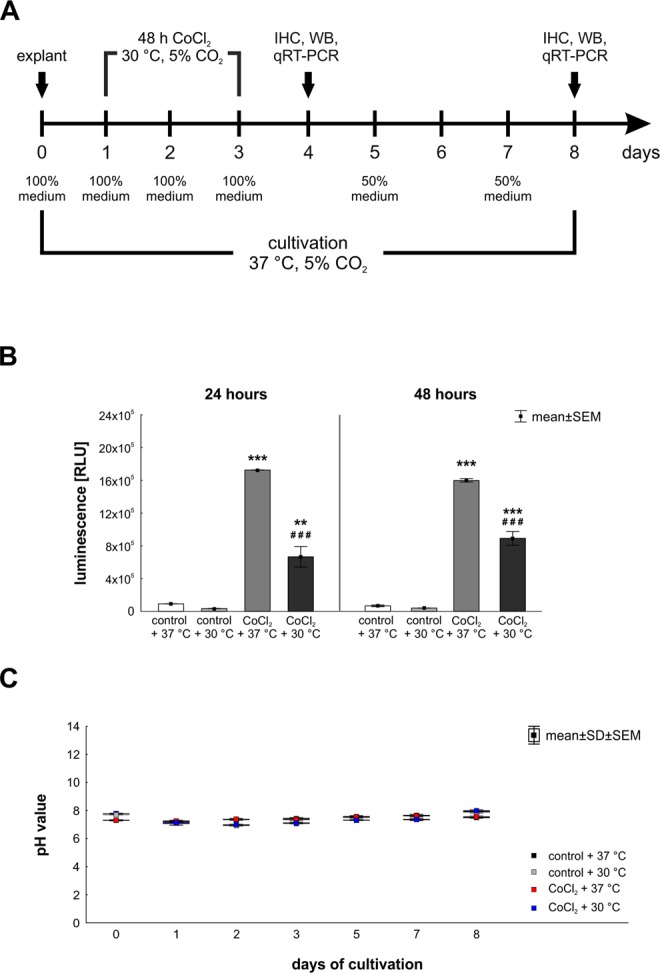
(A) Study timeline. Explants of porcine retinae were prepared at day zero and cultivated for four and eight days. Degeneration processes were induced by adding CoCl2 (300 µM) from day one to day three. Hypothermia treatment (30 °C) was applied simultaneously. Four groups were compared: control + 37 °C, CoCl2 + 37 °C, hypothermia treated control + 30 °C and CoCl2 + 30 °C. At days four and eight retina samples were prepared for immunohistological (IHC), western blot (WB) and qPCR analyses. (B) Hypothermia reduced the ROS-production in cultivated retina. ROS-level was measured 24 and 48 hours after CoCl2-induction. For both points in time, the ROS-level was strongly elevated after CoCl2-treatment. Hypothermia reduced the ROS-production significantly in CoCl2-treated retinae. However, it was still higher than in control + 37 °C retinae. (C) pH-value was measured to assure that degenerative effects were induced by CoCl2 and not by cultivation effects. pH-value was stable at any day of cultivation. B: n = 3/group. C: n = 10/group. **p < 0.01; ###,***p < 0.001.
To assure that degenerative effects were induced by CoCl2, we analyzed the oxidative stress by measuring the level of ROS in cultured retinae 24 and 48 hours after CoCl2-induction (Fig. 1B). In both investigated points in time, the ROS-level of CoCl2 + 37 °C treated retinae (24 h: 1,725,425 ± 3,073.1 RLU; p = 0.0002; 48 h: 1,597,542 ± 18,806.7 RLU; p = 0.0002) was strongly elevated in comparison to control + 37 °C retinae (24 h: 91,389 ± 3,117.6 RLU; 48 h: 67,404 ± 1,008.2 RLU). Interestingly, hypothermia reduced the ROS-level in CoCl2 + 30 °C treated retinae (24 h: 666,153 ± 125,548.3 RLU; 48 h: 891,382 ± 83,562.2 RLU) significantly in contrast to CoCl2 + 37 °C retinae (for both: p = 0.0002; Fig. 1B). Additionally, we measured the pH-value of the media after each medium change. No differences were seen within the groups for each point in time, indicating that degenerative effects were not induced by the cultivation of retinae (Fig. 1C).
For further investigations of the effects of hypothermia on CoCl2, we performed hematoxylin & eosin staining of retinal cross-sections (Sup. Fig. S1A). As described previously11, CoCl2 lead to a reduction of the retinal thickness. To evaluate whether hypothermia inhibited neurodegenerative effects of CoCl2 on porcine retina, retinal thickness was measured. At both investigated points in time, the retinal thickness was reduced significantly through CoCl2 (4 days: p = 0.01; 8 days: 0.03) in comparison to control retinae. For both points in time, four and eight days, hypothermia preserved retinal thickness, that CoCl2 + 30 °C treated retinae were significantly thicker than CoCl2 + 37 °C treated ones (4 days: p = 0.003; 8 days: p = 0.002) and no difference were seen between CoCl2 + 30 °C retinae and control + 37 °C retinae (p > 0.6; Sup. Fig. S1B). These results indicate that hypothermia lowers oxidative stress induced by CoCl2, and preserved retinal thickness, which was reduced by CoCl2-treatment.
Hypothermia inhibited hypoxic processes and reduced cellular stress
Since the accumulation of the transcription factor HIF-1α is a hallmark for hypoxic conditions7, hypoxic cells were stained with anti-HIF-1α antibody (Fig. 2A). To investigate whether CoCl2-treatment indeed induced hypoxic processes in retinae, the amount of HIF-1α+ cells located in the ganglion cell layer (GCL, Fig. 2B) as well as in the whole retina (Fig. 2C) was evaluated at four and eight days. At four days, CoCl2 + 37 °C treated retinae showed three times as many HIF-1α+ cells in the GCL (298.4 ± 51.6% HIF-1α+ cells/GCL; p = 0.0009) and even five times more in the whole retina (494.9 ± 69.8% HIF-1α+ cells/retina; p = 0.0002) in comparison to the control ones (100.0 ± 19.0% HIF-1α+ cells/GCL; 100.0 ± 15.3% HIF-1α+ cells/retina). Hypothermia treatment led to a significantly reduced amount of HIF-1α+ cells in the GCL (150.4 ± 31.8% HIF-1α+ cells/GCL; p = 0.015) as well as in the whole retina (143.4 ± 10.9% HIF-1α+ cells/retina; p = 0.0002) when compared to CoCl2 + 37 °C retinae. No statistical differences were seen between control groups and hypothermia treated CoCl2 + 30 °C groups.
Figure 2.

Reduced hypoxic processes and cellular stress through hypothermia. (A) Representative pictures of hypoxic cells in retinae. Hypoxic cells were stained with anti-HIF-1α (red, arrowheads). DAPI was used to visualize cell nuclei (blue). (B) Statistical evaluation of HIF-1α cell counts showed, that CoCl2 led to a strongly elevated number of HIF-1α+ cells located in the GCL after four and eight days. Hypothermia inhibited hypoxic processes and lowered hypoxia in most of the cells located in the GCL. (C) In regard to the number of hypoxic cells in the whole retina, CoCl2 again led to an increased hypoxia, whereas hypothermia alleviated hypoxic processes. (D) mRNA levels of HIF-1α were evaluated via qPCR. Analyses revealed an increased HIF-1α mRNA expression in the CoCl2 + 37 °C group at both days compared to the control + 37 °C group. At day four, hypothermia led to a control-like HIF-1α expression. (E) mRNA levels of iNOS were analyzed with qPCR. The iNOS mRNA expression was significantly increased by CoCl2 after four days. This effect was counteracted by hypothermia. At day eight, CoCl2 had no effect on iNOS expression, whereas both hypothermia treated groups, showed a reduced iNOS mRNA expression in comparison to the control group. (F) qPCR analysis regarding HSP70. CoCl2 strongly elevated the HSP70 mRNA expression level at days four and eight. At both points in time, hypothermia lowered HSP70 mRNA expression in the CoCl2 stressed retinae. (G) Protein levels of HSP70 (70 kDa) were measured via western blot and normalized against β-actin (42 kDa). (H) At day four, a significantly increased signal intensity of HSP70 was noted in both CoCl2 treated group, irrespectively of the temperature. Hypothermia treatment decreased the HSP70 signal intensity after eight days, causing no difference between the CoCl2 + 30 °C and the control + 37 °C group. Abbreviations: GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; IHC = immunohistochemistry; qPCR = quantitative real-time PCR. Values are mean ± SEM. B, C: n = 9–10/group; D-H: n = 6–7/group. Statistical differences to control + 37 °C group are marked with * and differences to CoCl2 + 37 °C group with #. #,*p < 0.05; ##,**p < 0.01; ###,***p < 0.001. Scale bar = 20 µm.
After eight days, CoCl2 led to a significant increase of HIF-1α+ cells in the GCL (197.1 ± 23.4% HIF-1α+ cells/GCL; p = 0.013) and in the whole retina (264.1 ± 28.8% HIF-1α+ cells/retina; p = 0.0002) in comparison to control retinae (100.0 ± 22.1% HIF-1α+ cells/GCL; 100.0 ± 20.2% HIF-1α+ cells/retina). Interestingly, after hypothermia treatment the amount of HIF-1a+ cells was reduced in the GCL (148.5 ± 19.5% HIF-1α+ cells/GCL; p = 0.42) and significantly decreased in the whole retina (138.5 ± 11.0% HIF-1α+ cells/retina; p = 0.001) to the extent, that no statistical difference was seen between control retinae and hypothermia treated CoCl2 + 30 °C retinae (GCL: p = 0.42; retina: p = 0.58; Fig. 2B,C). To verify that CoCl2 indeed induces hypoxia and hypothermia lowers the number of hypoxic cells, qPCR analyses of the HIF-1α expression level were performed (Fig. 2D). At day four, the HIF-1α expression was significantly increased after CoCl2 induction (2.9 ± 0.8-fold; p = 0.0002) compared to control + 37 °C retinae. As seen in the cell counting, hypothermia treatment reduced the HIF-1α mRNA expression in the CoCl2 + 30 °C group (1.1 ± 0.3-fold; p = 0.0002) compared to the CoCl2 + 37 °C group. Most importantly, no differences between the control + 37 °C and the CoCl2-stressed hypothermia group (p = 0.89) were noted (Fig. 2D), indicating a complete counteraction of the stressor via hypothermia. After eight days, the HIF-1α expression was more prominent in the CoCl2 + 37 °C group (1.65 ± 013-fold) than in the control group (37 °C: 1.1 ± 0.2-fold; p = 0.0007). In contrast to the results at day four and the cell counts, hypothermia treatment had no inhibiting effect on the HIF-1α expression after eight days, as no difference between the CoCl2 + 30 °C (1.6 ± 0.3-fold; p > 0.9) and the CoCl2 + 37 °C group was notable (Fig. 2D).
For further investigations on cellular stress, the iNOS mRNA levels in retinae were analyzed (Fig. 2E). After four days of cultivation, a 1.9 ± 0.6-fold higher iNOS expression was noted in CoCl2-stressed retinae compared to control + 37 °C retinae (p = 0.007). However, hypothermia counteracted the effect of CoCl2 and significantly reduced the iNOS expression (0.5 ± 0.3-fold; p = 0.0002) in comparison to CoCl2 + 37 °C. Again, no differences were notable between hypothermia treated CoCl2-stressed retinae and control ones (p = 0.612; Fig. 2E). After eight days, hypothermia (0.8 ± 0.2-fold; p = 0.015) caused a significantly decreased iNOS expression in comparison to the control + 37 °C group. Interestingly, no alterations were seen comparing CoCl2-stressed retinae (p = 0.570) and control ones. In contrast, a significantly reduced mRNA expression of iNOS was found in the CoCl2 + 30 °C group (0.6 ± 0.2-fold; p = 0.0006) compared to the CoCl2 + 37 °C group (Fig. 2E).
HSP70, a chaperon belonging to the heat shock protein family, is important for the correct folding process of proteins, and accumulates under stress conditions19. To evaluate the effect of CoCl2 and hypothermia on cellular stress HSP70 mRNA expression was analyzed (Fig. 2F). At the early point in time, the HSP70 expression level was significantly higher in the CoCl2 + 37 °C group (29.8 ± 5.7-fold; p = 0.0002) than in the control + 37 °C. CoCl2-stressed retinae treated with hypothermia (8.7 ± 7.4-fold; p = 0.04) still had an increased HSP70 expression compared to control ones, but interestingly, lowering the temperature significantly diminished the HSP70 expression in comparison to the CoCl2-stressed retinae at 37 °C (p = 0.0002). After eight days, the CoCl2 + 37 °C group (8.6 ± 3.2-fold; p = 0.0002) presented a significantly increased mRNA expression level compared to the control + 37 °C group. Hypothermia treatment led to a significant reduction of HSP70 mRNA expression in the CoCl2-stressed hypothermia group (0.8 ± 0.1-fold; p = 0.0002) in comparison to the CoCl2 + 37 °C group. Most importantly, these results prove the complete inhibition of cellular stress after hypothermia treatment since no differences were seen between the CoCl2 + 30 °C group and the control + 37 °C group (p > 0.9; Fig. 2F). Additionally, we performed western blot analyses to evaluate HSP70 protein levels (Fig. 2G,H). A significantly increased signal intensity of HSP70 was noted after four days in the CoCl2 + 37 °C group (231.4 ± 30.8%) in comparison to the control + 37 °C group (100.0 ± 17.8%; p = 0.015). In contrast to the results of qPCR analyses regarding HSP70, the signal intensity of HSP70 was increased in the CoCl2 + 30 °C group (230.5 ± 42.5%) in comparison to the control group (p = 0.044). Nevertheless, western blot analyses of HSP70 after eight days, were in accordance with those results seen in the qPCR. The addition of CoCl2, at 37 °C, led to a strongly increased signal intensity (271.0 ± 60.3%) in comparison to the control + 37 °C group (100.0 ± 19.9%; p = 0.005). Interestingly, hypothermia treatment normalized the signal intensity of HSP70 completely, resulting in no differences between the CoCl2 + 30 °C (139.3 ± 29.1%) and the control + 37 °C group (p = 0.79; Fig. 2H). In summary, these findings indicate a total counteraction of cellular stress and an early prevention of hypoxic processes in CoCl2-stressed retinae after hypothermia treatment.
Neuroprotection via hypothermia
RGCs transfer the electrochemical information via their axons, which build the optic nerve, to the brain. Since RCGs are affected in glaucoma it is important to establish new therapeutic approaches that protect retinal neurons, most of all RGCs.
To evaluate the possible neuroprotective effects of hypothermia on RGCs, we measured the TUBB3 expression level via qPCR analysis (Fig. 3A). At the early point in time, both, the hypothermia alone (1.2 ± 0.2-fold; p = 0.73) and the CoCl2 treated retinae at 37 °C (0.9 ± 0.1-fold; p = 0.32), had a similar TUBB3 expression as control retinae. A 1.3 ± 0.3-fold increased TUBB3 expression level was noted in hypothermia treated CoCl2-stressed retinae (p = 0.013) compared to CoCl2 stressed retinae at 37 °C (Fig. 3A). At day eight, the TUBB3 level in the CoCl2-stressed retinae (0.66 ± 0.2-fold; p = 0.189) was not altered in comparison to the control + 37 °C. In contrast, hypothermia treatment increased the TUBB3 expression in the CoCl2 + 30 °C retinae (1.9 ± 0.4-fold; p = 0.001) compared to control and CoCl2-stressed retinae at 37 °C (p = 0.0002; Fig. 3A).
Figure 3.
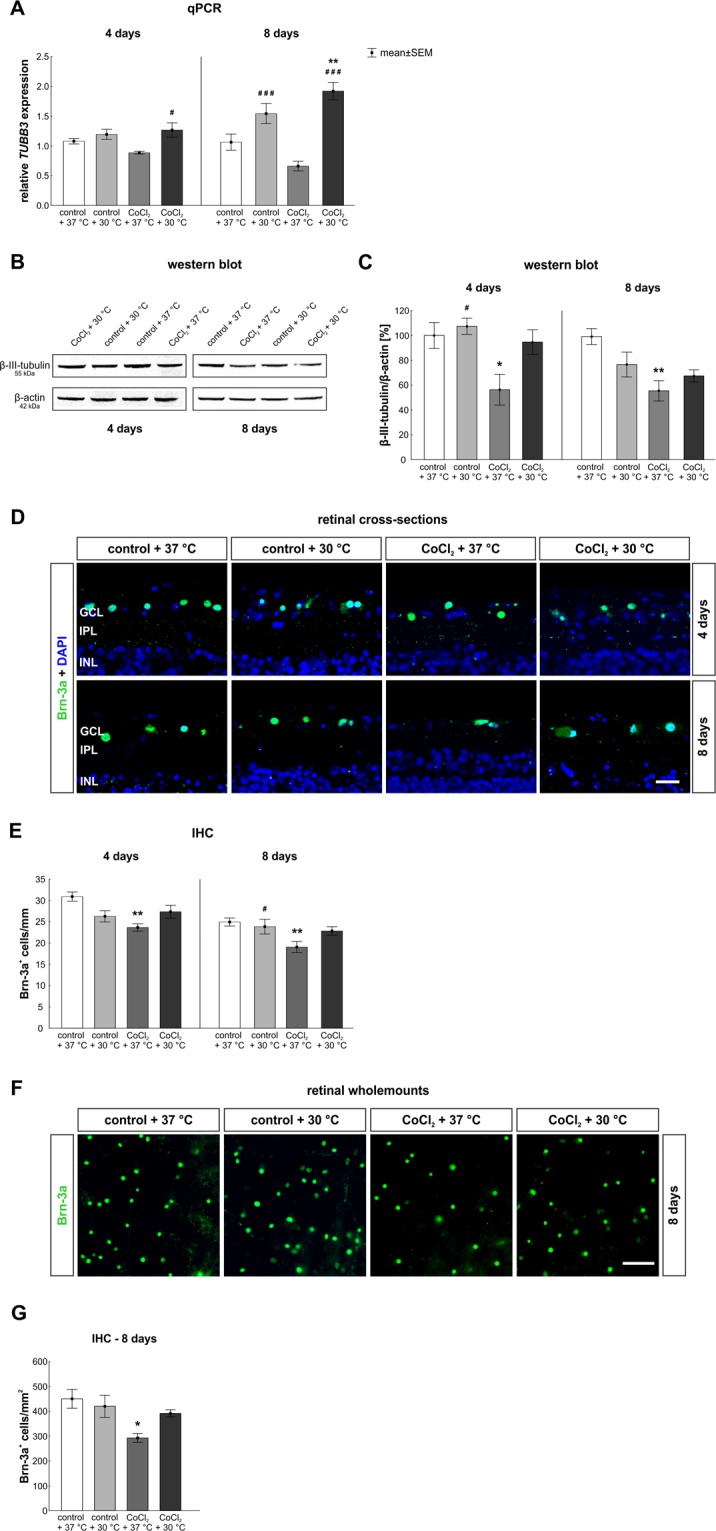
Protection of neurons, especially of retinal ganglion cells (RGCs), after hypothermia. (A) qPCR analysis regarding TUBB3. At day eight, a higher TUBB3 mRNA expression was found in both hypothermia treated groups. (B) Protein levels of β-III-tubulin, at 55 kDA, were measured via western blot and normalized against β-actin, at 42 kDa. (C) A significantly reduced β-III-tubulin protein level was observed after four and eight days via western blot analyses in the CoCl2 + 37 °C group. This effect was counteracted by hypothermia treatment. (D) Representative pictures of the ganglion cell layer. RGCs were stained in retinal cross-sections with anti-Brn-3a (green) at days four and eight. Cell nuclei were labelled with DAPI (blue). (E) Quantification revealed that CoCl2 at 37 °C induced a RGC loss after four and eight days. Degenerative effects of CoCl2 were counteracted via hypothermia treatment at both points in time. (F) Representative images depict RGCs stained in wholemount retinae at eight days using anti-Brn-3a (green). (G) Also in wholemounts, a significant loss was noted in the CoCl2 treated retinae at 37 °C, whereas hypothermia treatment protected RCGs. Abbreviations: GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer. qPCR = quantitative real-time PCR; IHC = Immunohistochemistry. Values are mean ± SEM. A: n = 6–7/group, B,C : n = 4/group; E,G: n = 10/group. Statistical differences to control + 37 °C group are marked with * and differences to CoCl2 + 37 °C group with #. #,*p < 0.05, **p < 0.01, ###p < 0.001. Scale bar = 20 µm (D); scale bar = 50 µm (F).
In addition, the β-III-tubulin protein level was investigated via western blot (Fig. 3B,C). At day four, CoCl2 + 37 °C treated retinae (56.3 ± 12.4%; p = 0.041) presented a significantly diminished signal intensity in comparison to the control + 37 °C (100.0 ± 10.4%). Hypothermia counteracted the harmful effects of CoCl2 on β-III-tubulin and led to a 1.69-fold increased protein level in the CoCl2 + 30 °C group (94.7 ± 9.8%; p = 0.078) in comparison to the CoCl2 + 37 °C group. In accordance with prior results, neuroprotective effects of hypothermia were noted since no differences in the β-III-tubulin intensity were found after hypothermia treatment between the control + 37 °C and CoCl2 + 30 °C group (p > 0.9; Fig. 3C). After eight days, the CoCl2 + 37 °C retinae (55.9 ± 8.2%; p = 0.007) still had an attenuated protein level in contrast to the control + 37 °C ones (100.0 ± 6.4%). Hypothermia diminished the CoCl2 effects on neurons, but nevertheless a slight reduction was seen in the CoCl2 + 30 °C retinae (68.0 ± 4.9%; p = 0.051) compared to the control + 37 °C retinae (Fig. 3C).
For histological evaluation of RGCs, an anti-Brn-3a antibody was used for the staining on retinal cross-sections and retinal wholemounts (Fig. 3D,F). After four days, significantly fewer Brn-3a+ cells were detected in cross-sections of CoCl2 + 37 °C treated retinae (23.7 ± 0.9 Brn-3a+ cells/mm; p = 0.001) than in control retinae (30.9 ± 1.1 Brn-3a+ cells/mm). Some protection of Brn-3a+ RGCs through hypothermia was achieved in the CoCl2 + 30 °C group (27.4 ± 1.5 Brn-3a+ cells; p = 0.158) in comparison to the CoCl2 + 37 °C group (Fig. 3E). At day eight, a significant RGC loss in the CoCl2 + 37 °C group (18.8 ± 1.2 Brn-3a+ cells/mm; p = 0.003) was prevented by hypothermia treatment (CoCl2 + 30 °C: 22.4 ± 0.8 Brn-3a+ cells/mm). In comparison to the CoCl2 + 37 °C group, RGCs were slightly preserved after hypothermia treatment (p = 0.155) and no differences were observed between control + 37 °C (25.1 ± 0.9 Brn-3a+ cells/mm) and CoCl2 + 30 °C groups (p = 0.394; Fig. 3E). To confirm these findings, RGCs were also stained on wholemount retinae (Fig. 3F). Results in wholemounts were in accordance with those seen in retinal cross-sections. A significant loss of RGCs was noted in the CoCl2 + 37 °C group (292.9 ± 18.2 Brn-3a+ cells/mm2; p = 0.031) when compared to control + 37 °C retinae (450.4 ± 38.2 Brn-3a+ cells/mm2; Fig. 3G). Protection of RGCs due to hypothermia were also seen in whole mount retinae, since CoCl2 + 30 °C retinae (391.8 ± 14.5 Brn-3a+ cells/mm2; p > 0.9) had a similar number of Brn-3a+ RGCs as control + 37 °C retinae. The protection via hypothermia was so prominent, that the amount of RGCs in hypothermia treated retinae tended to be higher in CoCl2 + 30 °C retinae than in CoCl2 + 37 °C ones (p = 0.198; Fig. 3G). With these results, we can state that hypothermia completely counteracted the harming effects of CoCl2 on RGCs.
Partial protection of the inner nuclear layer
Based on the previous findings that hypothermia lowers cellular stress and protects RGCs, we were interested if hypothermia also has protective effects on other retinal cell types. To this end, we investigated amacrine (Fig. 4) and bipolar cells (Fig. 5) which are both located in the inner nuclear layer.
Figure 4.
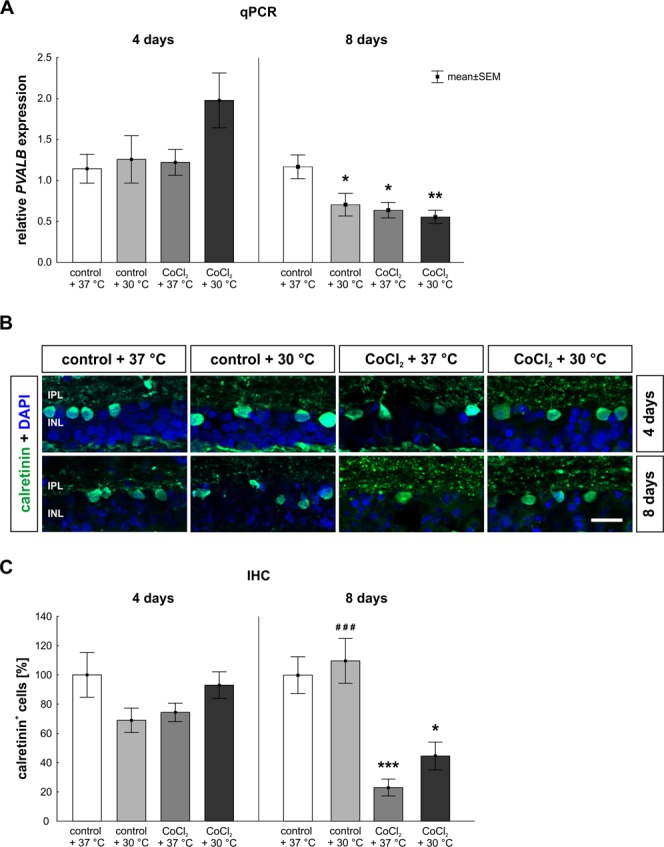
Late loss of amacrine cells. (A) qPCR analysis of PVALB. After eight days, a significantly reduced mRNA expression of PVALB was observed in all groups when compared to the control + 37 °C. (B) Representative pictures of the inner layers. Amacrine cells were stained with anti-calretinin (green) at four and eight days of cultivation. DAPI was used to visualize the cell nuclei (blue). (C) At day eight, a significant loss of amacrine cells was detected in the CoCl2 + 37 °C group. Hypothermia treatment did not rescue the amacrine cells. Abbreviations: IPL = inner plexiform layer; INL = inner nuclear layer; qPCR = quantitative real-time PCR; IHC = immune-histochemistry. Values are mean ± SEM. A: n = 6–7/group, B, C: n = 9–10/group. Statistical differences to control + 37 °C group are marked with * and differences to CoCl2 + 37 °C group with #. *p < 0.05; **p < 0.01; ###,***p < 0.001. Scale bar = 20 µm.
Figure 5.
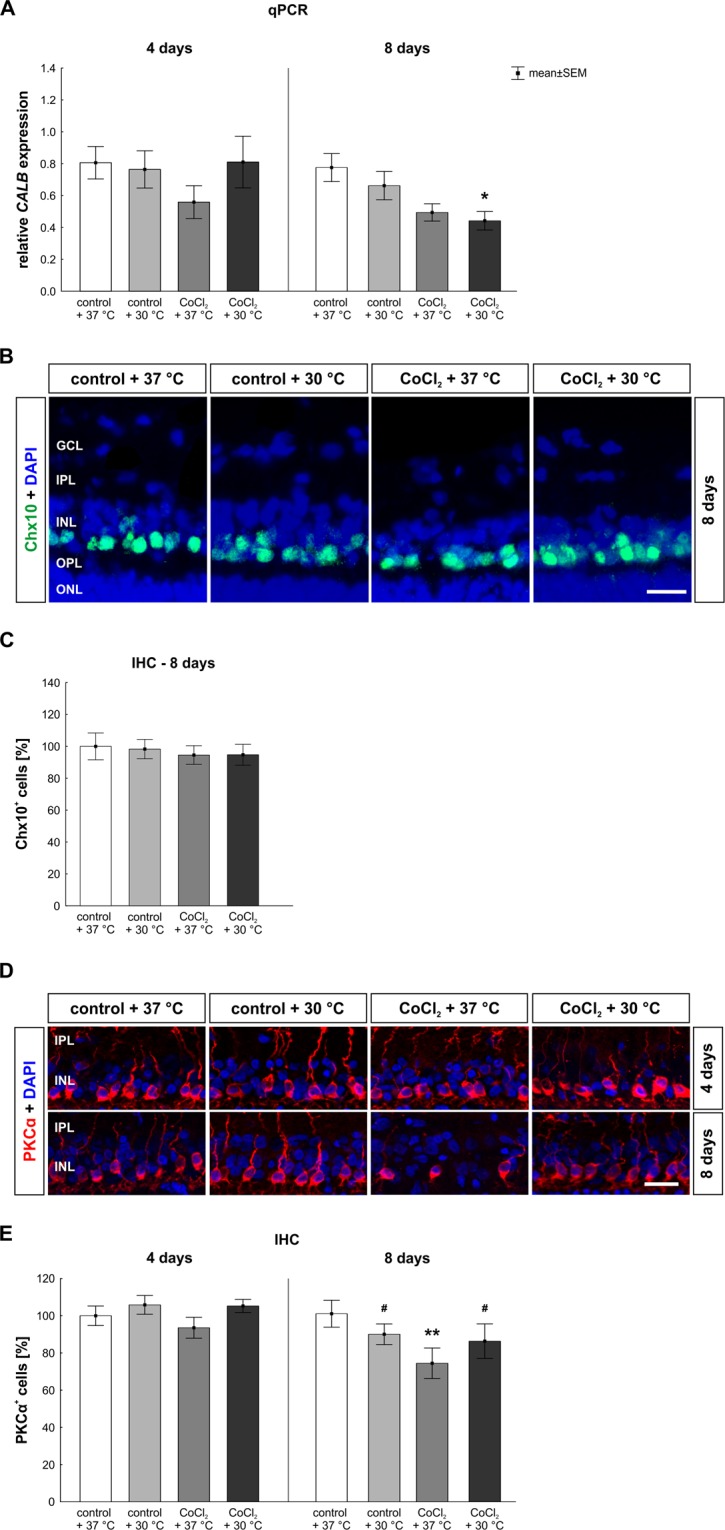
Late loss of bipolar cells was counteracted by hypothermia. (A) CALB mRNA expression was measured via qPCR. After eight days, a significantly decreased CALB mRNA expression was seen in the hypothermia treated CoCl2 + 30 °C retinae. (B) Representative images of bipolar cells stained with anti-Chx10 (green) at eight days. DAPI was used for the visualization of cell nuclei (blue). (C) After eight days, all groups had a similar number of Chx10+ cells. (D) Representative pictures of the inner layers are given. Rod bipolar cells (red) were stained immunohistochemically at days four and eight using an anti-PKCα antibody. Cell nuclei are shown in blue. (E) At day eight, a significant loss of bipolar cells was noted in the CoCl2 + 37 °C. A rescue of PKCα+ cells was achieved by hypothermia. Abbreviations: GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer; ONL = outer nuclear layer; qPCR = quantitative real-time PCR; IHC = immunohistochemistry. Values are mean ± SEM. A: n = 6–7/group; C,E: n = 9–10/group. Statistical differences to control + 37 °C group are marked with * and differences to CoCl2 + 37 °C group with #. #,*p < 0.05; **p < 0.01 Scale bar = 20 µm.
At day four, hypothermia treatment led to a slightly increased PVALB mRNA expression, a gene expressed by amacrine cells (CoCl2 + 30 °C: 2.0 ± 0.8-fold; p = 0.119; Fig. 4A), whereas the other groups presented an unaltered expression compared to the control retinae. At day eight, all groups (control + 30 °C: 0.7 ± 0.4-fold; p = 0.048; CoCl2 + 37 °C: 0.6 ± 0.3-fold; p = 0.019; CoCl2 + 30 °C: 0.6 ± 0.2-fold; p = 0.006) had a lower PVALB expression than the control + 37 °C group (Fig. 4A).
In histological analyses, the number of calretinin labeled amacrine cells was evaluated (Fig. 4B,C). Neither CoCl2 (37 °C: 74.4 ± 6.3% calretinin+ cells; p = 0.311; 30 °C: 93.0 ± 9.1% calretinin+ cells; p > 0.9) nor hypothermia alone (68.9 ± 8.33% calretinin+ cells; p = 0.163) had any impact on the amount of calretinin+ cells compared to control + 37 °C retinae (100.0 ± 15.3% calretinin+ cells; Fig. 4C). In contrast, after eight days, in retinae treated with CoCl2, irrespectively of the temperature (37 °C: 24.3 ± 6.1% calretinin+ cells; p = 0.0005; 30 °C: 47.0 ± 10.0% calretinin+ cells; p = 0.016), significant fewer amacrine cells were counted than in control + 37 °C retinae (100.0 ± 12.5% calretinin+ cells; Fig. 4C).
The mRNA expression of calbindin (CALB), a gene expressed by horizontal cells, was not altered in any of the groups compared (CoCl2 + 37 °C: 0.5 ± 0.3-fold; CoCl2 + 30 °C: 0.8 ± 0.4-fold; p > 0.5) to the control + 37 °C group at day four (Fig. 5A). Only at day eight, a slight reduction was observed in the CoCl2 + 37 °C group (0.5 ± 0.1-fold; p = 0.058) and even a significant reduction of the CALB expression level was noted in the CoCl2 + 30 °C group (0.4 ± 0.2-fold; p = 0.019) in comparison to the control (Fig. 5A).
For investigations of the bipolar cell-population we started with a pan-bipolar cell marker anti-Chx10 after eight days (Fig. 5B). The number of Chx10+ bipolar cells was not altered in any of the groups (control + 30 °C: 98.31 ± 6.08% Chx10+ cells; CoCl2 + 37 °C: 94.57 ± 5.85% Chx10+ cells; CoCl2 + 30 °C: 94.76 ± 6.52% Chx10+ cells) in comparison to the control + 37 °C (100.00 ± 8.40% Chx10+ cells; for all: p > 0.9; Fig. 5C).
Since it is known that CoCl2 leads to a degeneration of rod bipolar cells11, we additionally investigated the amount of PKCα+ rod bipolar cells after four and eight days (Fig. 5D,E). Regarding the amount of PKCα+ rod bipolar cells, the number of bipolar cells was not changed in any of the groups in comparison to the control + 37 °C group (100.0 ± 5.2% PKCα+ cells; Fig. 5E) at day four. However, at day eight, a significant loss of bipolar cells was noted in the CoCl2 + 37 °C group (65.7 ± 5.3% PKCα+ cells; p = 0.006) in comparison to control + 37 °C retinae (100.0 ± 8.0% PKCα+ cells). Even more, a total rescue of PKCα+ cells was achieved by hypothermia (CoCl2 + 30 °C: 94.1 ± 7.1% PKCα+ cells; p = 0.029), resulting in no difference between control + 37 °C and hypothermia treated CoCl2 + 30 °C retinae (p > 0.9; Fig. 5E).
These findings show, that the damaging effect of CoCl2 on amacrine cells was not attenuated by hypothermia. While the total bipolar cell population was not affected neither by CoCl2 nor by hypothermia, the late loss of rod-bipolar cells was totally counteracted after hypothermia.
Apoptotic mechanisms were reduced through hypothermia
To investigate underlying mechanisms that lead to protection of RGCs after hypothermia, we evaluated apoptosis. To this end, we analyzed the expression of several genes that are involved in apoptosis.
The mRNA expression of p21, a well-known regulator of cell cycle arrest under stress conditions, was strongly elevated in the CoCl2 + 37 °C group (5.4 ± 1.5-fold; p = 0.0002; Fig. 6A). This effect was completely counteracted by hypothermia (CoCl2 + 30 °C: 1.1 ± 0.9-fold; p = 0.0002), leading to no differences between CoCl2 + 30 °C and control + 37 °C retinae (p > 0.9). The same effects were seen after eight days. A strong reduction of p21 mRNA expression was noted in control + 30 °C retinae (0.3 ± 0.1-fold) compared to control + 37 °C ones (p = 0.181). Once again, the overexpression of p21 mRNA in CoCl2 + 37 °C stressed retinae (5.9 ± 1.5-fold; p = 0.0002) was successfully reduced after hypothermia (CoCl2 + 30 °C: 1.4 ± 0.2-fold; p = 0.0002), even leading to no differences between the CoCl2 + 30 °C and control + 37 °C retinae (p > 0.9; Fig. 6A).
Figure 6.

Inhibition of apoptotic processes via hypothermia. (A) Expression of the cell arrest gene p21 was evaluated via qPCR. Analysis revealed that p21 gene expression was strongly increased after four and eight days in the CoCl2 + 37 °C group. Hypothermia reduced the expression significantly at both points in time. (B) qPCR analyses of caspase 8. CoCl2 led to a strongly elevated expression of caspase 8 in the CoCl2 + 37 °C group after four and eight days. Again, hypothermia treatment counteracted that effect. (C) Ratio of Bax/Bcl-2 mRNA was measured via qPCR. After eight days, the Bax/Bcl-2 ratio in the CoCl2 + 37 °C group tended to be increased. Hypothermia reduced the Bax/Bcl-2 ratio in the CoCl2 + 30 °C group and no differences were seen in comparison to the control + 37 °C group. (D) Representative apoptotic retinal ganglion cells. RGCs were stained at four and eight days with anti-Brn-3a (RGCs; green) and cl. casp. 3 (apoptosis; red; arrowheads). Cell nuclei were visualized with DAPI (blue). (E) After four and eight days, the amount of apoptotic RGCs was significantly increased in the CoCl2 + 37 °C group. Interestingly, the number of apoptotic RGCs was reduced through hypothermia at four days. However, at eight days, no effects of hypothermia were detectable in the CoCl2 + 30 °C group compared to the CoCl2 + 37 °C. Abbreviation: GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; qPCR = quantitative real-time PCR; IHC = immunohistochemistry. Values are mean ± SEM, A-C: n = 6–7/group; E: n = 10/group. Statistical differences to control + 37 °C group are marked with * and differences to CoCl2 + 37 °C group with #. #,*p < 0.05; ##,**p < 0.01; ###,***p < 0.001. Scale bar = 20 µm.
Apoptosis can be induced in two different ways20. The extrinsic pathway is activated when a ligand binds to a specific “death” receptor. This leads to the activation of caspases, like caspase 8. Therefore, the early extrinsic apoptosis pathway was evaluated via caspase 8 (casp. 8) expression (Fig. 6B). At day four, CoCl2 + 37 °C stressed retinae (4.9 ± 2.1-fold; p = 0.002) showed a significant higher mRNA expression of caspase 8 than control retinae. This effect was lowered by hypothermia treatment (CoCl2 + 30 °C: 3.45 ± 1.69-fold; p = 0.314), but an mRNA increased expression was still observable in comparison to the control + 37 °C group (p = 0.084; Fig. 6B). After eight days, the caspase 8 mRNA expression was significantly increased in the CoCl2 + 37 °C retinae (2.6 ± 1.0-fold expression; p = 0.004). Once again, a reduction of temperature prevented the caspase 8 overexpression in the CoCl2 + 30 °C group (1.9 ± 0.6-fold; p = 0.229) in comparison to the CoCl2 + 37 °C group. Protective effects of hypothermia were seen once again, since no alterations were detectable comparing the expression in both CoCl2 stressed groups (p = 0.244; Fig. 6B).
The other way to induce apoptosis is the intrinsic pathway, in which stress signals lead to a secretion of cytochrome c from the mitochondria into the cytoplasm20. This alters the activation state of several pro- or anti-apoptotic proteins, which then leads to apoptosis. The intrinsic apoptosis pathway was analyzed via the Bax/Bcl-2 ratio (Fig. 6C). After four days, no changes were noted within all groups (Fig. 6C). At day eight, no difference was observable between hypothermia treated CoCl2 + 30 °C retinae (1.14 ± 0.31-fold; p > 0.9) and control + 37 °C ones. In contrast, a significant reduction of the Bax/Bcl-2 ratio, comparing CoCl2 + 30 °C and CoCl2 + 37 °C groups (p = 0.048), was observable (Fig. 6C).
Based on the fact, that we could show that hypothermia lowers apoptotic processes and protects RGCs, we were interested in the amount of apoptotic RGCs. Hence, we performed double immunolabeling of RGCs using Brn-3a and cleaved caspase 3 (Fig. 6D,E). After four days, both CoCl2 treated groups (37 °C: 65.6 ± 3.2% cl. casp. 3+ RGCs; p = 0.0002; 30 °C: 53.2 ± 2.4% cl. casp. 3+ RGCs; p = 0.0006) displayed a significantly increased number of apoptotic RGCs in comparison to the control + 37 °C (36.2 ± 2.6% cl. casp. 3+ RGCs). Nevertheless, hypothermia treatment significantly inhibited apoptotic processes in the CoCl2 + 30 °C group in comparison to the CoCl2 + 37 °C group (p = 0.013; Fig. 6E). After eight days, the addition of CoCl2 led to a significantly elevated apoptosis rate in the CoCl2 + 37 °C group (53.3 ± 3.7% cl. casp. 3+ RGCs; p = 0.002) in comparison to the control + 37 °C (34.4 ± 2.9% cl. casp. 3+ RGCs). At this point in time, hypothermia did not have any inhibiting effects on the apoptosis rate of RGCs, since hypothermia treated CoCl2 + 30 °C retinae (47.9 ± 2.9% cl. casp. 3+ RGCs; p = 0.034) showed a higher apoptosis rate than the control + 37 °C ones (Fig. 6E).
As our results show, hypothermia counteracted the cell cycle arrest of retinal cells triggered by CoCl2. Both, the expression of caspase 8 and the amount of apoptotic RGCs were strongly reduced after hypothermia treatment, which indicates that hypothermia lowered the apoptotic mechanisms.
Rescue of microglia and macroglia
To evaluate microglia, CD11b mRNA expression, a gene which encodes for microglia receptors, was analyzed (Fig. 7A). A 3.3-fold reduction was noted in the CoCl2 + 37 °C group in comparison to the control + 37 °C group (p = 0.098) after four days. In accordance, protective effects of hypothermia were seen, since no alterations were observable between the control + 37 °C and CoCl2 + 30 °C group. After eight days, CoCl2 again induced a 5-fold reduction of CD11b mRNA expression in the CoCl2 + 37 °C group (p = 0.001). Hypothermia completely protected the CD11b mRNA expression in the CoCl2 + 30 °C group. Hence, no differences were notable compared to the control + 37 °C (p = 0.489; Fig. 7A).
Figure 7.
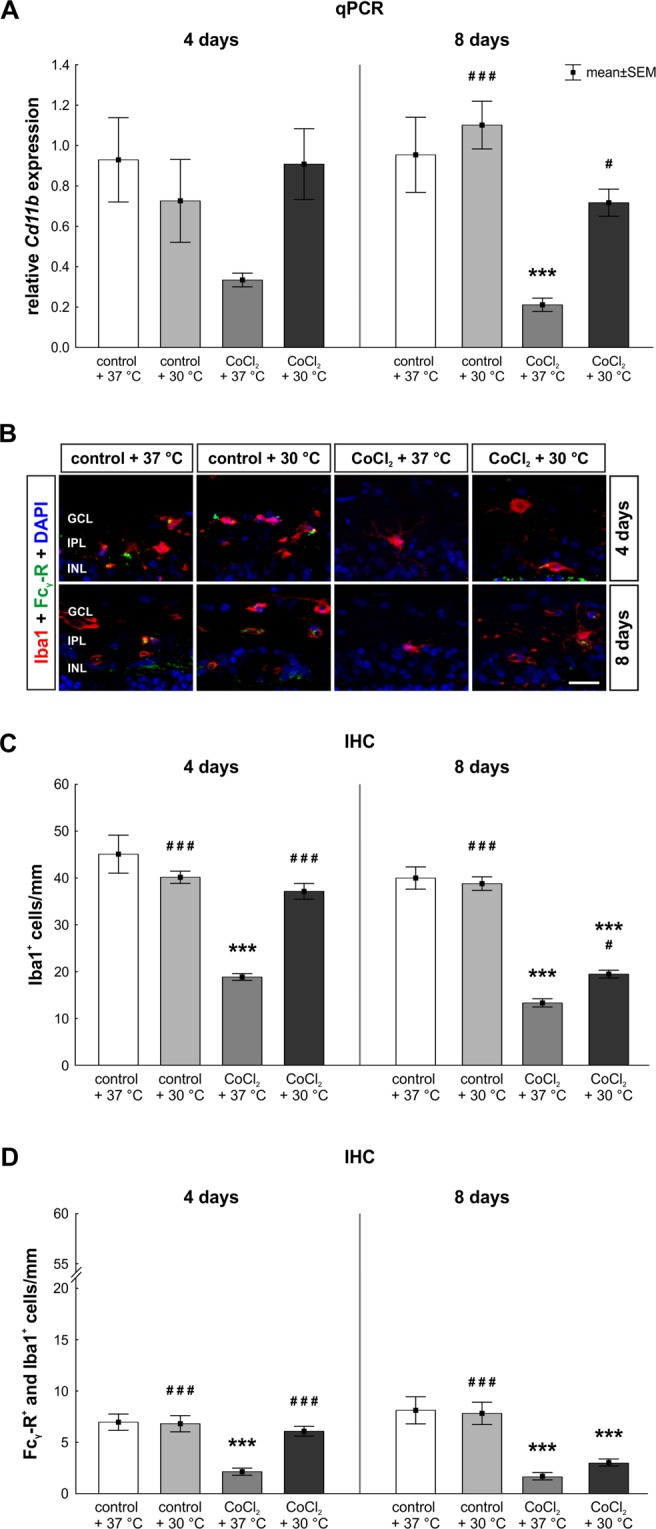
Hypothermia protected microglia. (A) Relative mRNA expression of Cd11b, a gene that is expressed by microglia, was analyzed via qPCR. At day four, a slightly decreased expression of CD11b was noted in the CoCl2 + 37 °C retinae, which was counteracted via hypothermia. After eight days, a significant reduction of CD11b was observed in the CoCl2 + 37 °C group. The damaging effect of CoCl2 was again counteracted by hypothermia in the CoCl2 + 30 °C group. (B) Representative pictures of microglia stained with anti-Iba1 (red) after four and eight days are shown. Anti-Fcγ-R was used as an activity marker for microglia (green). Fcγ-R+ and Iba1+ cells were counted as active microglia. Cell nuclei were visualized with DAPI (blue). (C) The significant loss of microglia due to CoCl2 was counteracted through hypothermia at both points in time. (D) CoCl2 led to a significantly reduced number of active microglia. Hypothermia treatment rescued active microglia at four days, but not at eight days. Abbreviations: GCL = ganglion cell layer; IPL = inner plexiform layer, INL = inner nuclear layer; qPCR = quantitative real-time PCR; IHC = immunohistochemistry. Values are mean ± SEM. A: n = 6–7/group; C, E: n = 10/group. Statistical differences to control + 37 °C group are marked with * and differences to CoCl2 + 37 °C group with #. #p < 0.05; ###,***p < 0.001. Scale bar = 20 µm.
To confirm previous findings, the total number of microglia was analyzed by Iba1 staining of retinal cross-sections (Fig. 7B). In accordance with the qPCR results, a loss of microglia was observable in the CoCl2 + 37 °C group after four days (18.8 ± 0.7 Iba1+ cells/mm; p = 0.0002) in contrast to the control + 37 °C group (45.1 ± 4.1 Iba1+ cells/mm). Hypothermia treatment counteracted the hypoxic effect of CoCl2, which resulted in a total rescue of microglia in the CoCl2 + 30 °C group (37.1 ± 1.7 Iba1 + cells/mm; p = 0.0002) compared to the CoCl2 + 37 °C group. Most importantly, no differences were found comparing the CoCl2 + 30 °C to the control group (Fig. 7C). After eight days, the number of Iba1+ cells was significantly lower in the CoCl2 + 37 °C group (13.3 ± 0.9 Iba1+ cells/mm; p = 0.0002). Again, hypothermia inhibited the impact of CoCl2 on microglia in the hypothermia treated CoCl2-stressed retinae (19.5 ± 0.8 Iba1+ cells/mm; p = 0.033). However, a loss of nearly 50% of the microglia population was still seen when comparing CoCl2 + 30 °C and control + 37 °C retinae (40.0 ± 2.4 Iba1+ cells/mm; p = 0.002; Fig. 7C).
In a next step, the number of active microglia was evaluated via Fcγ-R and Iba1 co-staining (Fig. 7B). A loss of active microglia was noted in the CoCl2 + 37 °C group (2.1 ± 0.3 Fcγ-R+ and Iba1+ cells/mm; p < 0.001) after four days. No differences between the hypothermia treated CoCl2-stressed retinae (CoCl2 + 30 °C: 6.1 ± 0.5 Fcγ-R+ and Iba1+ cells/mm; p = 0.774) and the control retinae (7.0 ± 0.8 Fcγ-R+ and Iba1+ cells/mm) proved a total rescue of active microglia through hypothermia (Fig. 7D). At day eight, the addition of CoCl2 induced a significant reduction of active microglia, irrespectively of the temperature (37 °C: 1.6 ± 0.4 Fcγ-R+ and Iba1+ cells/mm; p = 0.001; 30 °C: 3.0 ± 0.3 Fcγ-R+ and Iba1+ cells/mm; p = 0.003; control + 37 °C: 8.1 ± 1.3 Fcγ-R+ and Iba1+ cells/mm; Fig. 7D).
Investigations of macroglial response revealed a significantly reduced GFAP mRNA expression in both CoCl2 groups at day 4 (37 °C: 0.19 ± 0.20-fold p = 0.001; 30 °C: 0.40 ± 0.30-fold; p = 0.039; Sup. Fig. S2A). At day eight, no differences were observed within all investigated groups (Sup. Fig. S2A). Western blot analyses showed no changes in GFAP signal intensities in any of the groups neither at four nor at eight days (Sup. Fig. S2B,C). The evaluation of GFAP immunoreactivity revealed the same results as seen in qPCR analyzes (Sup. Fig. S2D). Significant lower GFAP signals were seen in the CoCl2 + 37 °C groups in both points in time (4 days: 8.30 ± 0.69 [%]/area; p = 0.029; 8 days: 13.99 ± [%]/area; p = 0.030) compared to the control retinae (4 days: 11.80 ± 0.41 [%]/area; 8 days: 20.12 ± 1.51 [%]/area; CoCl2 + 37 °C). In accordance with the results regarding microglia, hypothermia reduced the effect of CoCl2 also on macroglia. Only a slight reduction was noted comparing the control + 37 °C and the CoCl2 + 30 °C groups (4 days: 9.46 ± 0.34 [%]/area; p = 0.22; 8 days: 16.89 ± 0.94 [%]/area; p = 0.31; Sup. Fig. S2E).
Discussion
The aim of this study was to investigate possible neuroprotective effects of hypothermia (30 °C) on CoCl2-stressed cultured porcine retina explants. We demonstrated that hypoxic damage, such as oxidative stress, due to CoCl2 on retinal cells was diminished through hypothermia. Furthermore, hypothermia had inhibiting effects on the apoptosis and led to an enhanced cell survival.
It has been described that cobalt, like hypoxia, triggers the stabilization of the α-subunit of hypoxia-inducible factor (HIF-1) by preventing its degradation21. Increased levels of HIF-1α activate the expression of certain genes, like iNOS and heat shock proteins (HSPs)22,23. Furthermore, cobalt leads to DNA fragmentation, caspase activation, and to ROS-production through the uncoupling of mitochondrial respiration24,25. Toxic effects of cobalt include a loss of mitochondrial membrane potential, the inhibition of the proteasome degradation, resulting in cell death22,26. Nevertheless, the treatment of CoCl2 can simulate a disease process and can cause symptoms very similar to those of hypoxia22.
Due to the fact, that CoCl2 stabilizes HIF-1α, HIF-1α mRNA expression was investigated to verify that hypoxia was successfully induced in retinae stressed with CoCl2. In our study, CoCl2-stressed hypoxic retinae presented a higher HIF-1α mRNA expression as well as a higher number of hypoxic cells, showing that CoCl2 successfully induced hypoxia. The stabilization of HIF-1α results in a higher expression of genes that encode several proteins, like heat shock proteins (HSPs) and inducible nitric oxide synthase (iNOS), which are essential to manage hypoxic stress27. This effect was successfully seen in CoCl2-stressed retinae, in which the mRNA expression of HSP70 was strongly elevated22. HSPs are chaperons which are important for the defense against cellular stress. They prevent misfolding and protein aggregations. Especially HSP70 is required for the transcriptional activity as well as for the accumulation and function of HIF-1α28. Our results show that hypothermia led to a strong reduction not only of HIF-1α but also of HSP70, pointing out that HIF-1α and HSP70 are strongly linked. The same indirect effect of CoCl2 on HSP70 was also described in other studies, strengthening our suggestions29,30. Nevertheless, our results do not clarify, whether hypothermia first inhibits HIF-1α accumulation which than leads to the reduction of HSP70, or whether hypothermia prevents the HSP70 expression which results in a reduced HIF-1α amount.
Divalent metal ions, such as cobalt, induce a disturbance of the mitochondrial respiration chain and stimulate the rupture of the outer cell membrane, resulting in ROS production and oxidative stress6,8,22. Our results suggest that CoCl2 not only mimics hypoxia through the stabilization of HIF-1α, but also leads to a strongly elevated level of ROS, indicating that it triggers oxidative stress. In accordance with other publications, our results show that hypoxia and oxidative stress trigger apoptotic mechanisms24,25, which then led to the significant loss of RGCs in the retinae. These mechanisms were completely counteracted by hypothermia, which probably first blocked the interaction of CoCl2 and HIF-1α, then HSP70 and iNOS were strongly reduced, leading to decreased oxidative stress and alleviated apoptosis mechanisms.
The prominent loss of RGCs was associated with an increased p21 mRNA expression, a gene which induces cell cycle arrest, and caspase 8, a hallmark for extrinsic apoptosis20. An overexpression of HIF-1α can induce apoptotic processes in different ways: the interaction of HIF-1α and p53, a tumor suppressor gene, leads to the activation of apoptotic mechanisms2. In addition, HIF-1α activates p21 and lowers the cell viability31.
As mentioned before, a HIF-1α overexpression was observed in CoCl2-stressed retinae at both points in time. In the early point in time, hypothermia totally stabilized the expression of HIF-1α to control level. This effect was accompanied by a total reduction of cellular stress, namely control-like HSP70 and iNOS expression- as well as ROS-levels, causing strongly lowered extrinsic apoptosis. After eight days, qPCR analyzes regarding HIF-1α indicate that the effect of hypothermia was lower than after four days. Anyway, the reduced number of HIF-1α+ cells in the retina show that after eight days there was still a positive effect through hypothermia.
In accordance, after eight days, p21 expression was diminished in the hypothermia treated CoCl2-stressed retinae and caspase 8 expression was lower than in CoCl2 retinae. Hence, hypothermia led to an early inhibition of hypoxic processes and reduced the apoptosis. In addition, hypothermia inhibited caspase 3 at the earlier, but not at the later point in time. It is known that hypothermia protects cells through a diminished apoptosis rate by inhibiting the lactate dehydrogenase32 and by reducing the caspase 3 expression33. Moreover, it was shown for retinal explants of mice, that hypothermia treatment during the retinal dissection led to a strongly decreased apoptosis rate in the retina34. Caspase 8 is activated at an early stage of extrinsic apoptosis, whereas caspase 3 is cleaved in later stages of intrinsic apoptosis35. A possible explanation would be that hypothermia rather inhibits the extrinsic than the intrinsic pathway. Furthermore, other upstream proteins, besides caspase 8, might be the reason for the lacking inhibition of caspase 3 after hypothermia at the later point in time.
Bax is a pro-apoptotic protein, which induces the intrinsic apoptosis by opening mitochondrial pores and supporting the secretion of cytochrome c into the cytoplasm. Bcl-2, on the other hand, is an anti-apoptotic protein that inhibits Bax and prevents the secretion of cytochrome c36. In our study, the Bax/Bcl-2 ratio was slightly increased in CoCl2-stressed retinae after eight days, but not in hypothermia treated CoCl2-stressed ones. Hence, hypothermia seems to inhibit the apoptosis via increasing the Bcl-2 expression or decreasing the Bax expression. Several studies revealed that CoCl2-induced hypoxia leads to apoptosis. Kuehn et al. demonstrated that CoCl2-treated porcine retinae showed an increased Bax expression level after four days of cultivation11. Chang et al. observed that hypoxia activates the intrinsic apoptosis via Bax and caspase 3, whereas Lee et al. reported that CoCl2 leads to apoptosis by activating both pathways simultaneously37,38.
In previous studies a loss of calretinin+ amacrine and PKCα+ bipolar cells was noted through CoCl26,11. This is the first study that shows a time dependent damage due to CoCl2 on calretinin+ amacrine cells, parvalbumin (PVALB) expressing displaced amacrine as well as PKCα+ bipolar cells.
Amacrine cells are located in the inner nuclear layer (INL) and play an important role for the modulation of signals to the RGCs39. It is known that they are vulnerable to glaucomatous damage40–42. There is a strong connection between amacrine cells and RGCs via gap-junctions, which leads to a secondary loss of amacrine cells after glaucoma-induced RGC loss42. In good accordance, we observed a delayed loss of amacrine cells at eight days which was induced by CoCl2 and possibly strengthened by the early loss of RGCs. Hypothermia did not protect amacrine cells. We assume that the RGC loss due to CoCl2 led to severe changes in the surrounding tissue, where dendrites of amacrine cells are located and therefore the protection of amacrine cells cannot be achieved.
Bipolar cells are also located in the INL and are connected to rods or cones. We used PKCα to label rod bipolar cells. A time-dependent loss of rod bipolar cells was observed. This was counteracted by hypothermia treatment. This later death is in accordance with affected bipolar cells in different rat glaucoma models41,43. Since hypothermia had a rescue effect on bipolar cells, but not on amacrine cells, we assume that the degeneration process of both cell types is different. To confirm this, further studies are necessary.
Besides HSP70, also the expression of inducible nitric oxide synthase (iNOS) depends on the activity of HIF-1α. CoCl2 has degenerative effects on microglia through increasing the apoptosis rate and diminishing the cell viability via cell arrest11,44. Besides neurons, microglia were also protected by hypothermia. Interestingly, the gene encoding for the enzyme iNOS, which is mainly produced by microglia, was increased by hypoxic stress without a microglia response. HIF-1α might be involved in this pathway, since it is known that HIF-1α increases the iNOS expression by binding the transcription promotor45,46. Therefore, in our model iNOS regulation seems to be independent from a microglia response.
GFAP expression, is a hallmark for gliosis in retinal diseases or injuries. Macroglial signals in the porcine retina are stronger than in rodent retinae47,48. However, we detected a reduced GFAP expression in qPCR and immunohistochemical analyses, while western blot analyses revealed no changes in GFAP signal intensities at any of the investigated points in time. Those results indicate that CoCl2 seems to have toxic effects not only on microglia but also on macroglia. However, our study and other studies reveal that in porcine degeneration models gliosis is not occurring during cultivation48–50. The preparation of retinal explants seems to induce a macroglial response itself. Therefore, it is difficult to detect further changes. Furthermore, in our ex vivo model, retinae are cultivated separate from the optic nerve. Thus, the immigration of astrocytes as a result of macrogliosis is not possible.
Conclusion
Hypoxic processes play a crucial role in several retinal diseases. Cobalt chloride (CoCl2) is known to mimic hypoxic processes in vitro by stabilizing the transcription factor HIF-1α and leading to oxidative stress through a disruption of mitochondrial respiration. These effects were observed in the present study. This hypoxic damage due to CoCl2 was associated with oxidative stress leading to increased apoptosis rates in CoCl2-stressed retinae. We demonstrated that hypothermia completely counteracted these mechanisms by probably disturbing the interaction of CoCl2 and HIF-1α. This led to strongly reduced HSP70 and iNOS synthesis, alleviating oxidative stress and preventing apoptosis. Consequently, most RGCs and bipolar cells were rescued, while amacrine and horizontal cells were not protected.
In conclusion, we demonstrated that our CoCl2-induced hypoxia is a suitable model system to test potential therapies and that hypothermia could be a possible additional treatment for retinal diseases.
Methods
Preparation of retinal explants
Porcine eyes were obtained from the local abattoir and retinae were prepared within three hours from enucleation. The preparation of retinal explants was performed as described previously11,49,51. Briefly, the eyeball was opened to separate anterior parts of the eye from the eye cup. The eye cup was incised four times to produce a cloverleaf-like shape. Next, one retinal explant sample per leaf was punched out in the central part of the retinal quadrant using a dermal punch (Ø = 6 mm, Pfm medical AG). Remaining retinal pigment epithelium was removed by washing retinal explants in Neurobasal-A medium (Life Technologies). Finally, retinal samples were placed on a Millicell culture insert (Millipore) with the GCL facing up and cultured in Neurobasal-A medium (Life Technologies) supplemented with 0.8 mM L-glutamine (Life Technologies), 2% B27 (Life Technologies), 1% N2 (Life Technologies) and 2% penicillin/streptomycin (Sigma-Aldrich), for four and eight days. Medium was exchanged completely at days zero, one, two and three. Additionally, half of the medium volume was replaced at days five and seven (Fig. 1A). The substance most commonly used to simulate a hypoxic environment is CoCl28,9,52,53, hence this was used in the current study. Hypothermia treatment and hypoxia induction via 300 µM CoCl2 (Sigma-Aldrich) were performed simultaneously and took 48 h in total (Fig. 1A). Control groups were cultivated without the stressor and with or without additional hypothermia treatment, so that four groups were compared: control + 37 °C, control + 30 °C, CoCl2 + 37 °C and CoCl2 + 30 °C.
At days four and eight, retinal explants were obtained for quantitative real-time-PCR (qPCR, n = 6–7/group/point in time), histological (n = 9–10/group/point in time) and western blot analyses (n = 4/group/point in time; Fig. 1A).
Measurement of reactive oxygen species (ROS)-level and pH-value
For the measurement of ROS-level in cultured retinae, the non-lytic protocol of ROS-GLO™ H2O2 assay (Promega) was performed. Retinae were cultured as described before and the measurement of ROS-level was performed according to the manufacture’s protocol. For more detailed description please see supplementary part. The measurement of the pH-values was performed with LAQUAtwin B-712. Calibration was performed according to manufactural instructions. After the medium exchange, 200 µl medium was used for the measurement.
Quantitative real-time PCR (qPCR)
The used primer for qPCR analyses are given in Supplementary Table 1. qPCR analyses were performed as described previously11,49,51 for n = 6/group at day four and n = 7/group at days four and eight. All target genes were normalized against housekeeping genes encoding Histone H3 and β-Actin (Sup. Table 1). Ct-Values of both housekeeping genes were not affected. The mean of all samples for β-actin was 18,84 ± 0.8 cycles and for histone H3 20.22 ± 0.9 cycles. Samples having Ct-values 2 cycles higher or lower than the mean were excluded. Geometric mean of the Ct-Values of both genes were calculated and used as reference. For the relative quantification the Δ-ΔCt- algorithm, with efficiency corrected calculation model, based on one sample was used54. All groups were compared to control + 37 °C or CoCl2 + 37 °C groups.
Histological analysis of retinal cells
For immunohistochemical analyses retinal cross-sections (n = 9–10/group/point in time) and flatmounts of retinae (n = 3/group) were prepared as described previously11,49,51. To identify different cell types and proteins specific primary antibodies and matched secondary antibodies were used (Sup. Table 2). For all stainings, 4′,6 diamidino-2-phenylindole (DAPI) was used to visualize the cell nuclei. Cross-sections and flatmounts were blocked with blocking-buffer containing 10–20% donkey or goat serum and 0.1–0.2% TritonX in PBS. Six cross-sections were stained per retina and cells were counted in 24 masked and defined image sections using ImageJ software. Regarding the evaluation of flatmounts, 9 images, including 4 peripheral and 5 central parts of the retina, were counted. For GFAP, the area was measured using an established protocol and an ImageJ macro40,41. For more information please see supplementary part.
Western blot
Western blot analyses were performed (n = 4/group/point in time) as described previously11,49,51. To this end, the primary antibodies (Sup. Table 2), diluted in the blocking solution, were incubated over night at 4 °C. After the washing steps, the secondary antibodies (Sup. Table 2) were applied for 60 min. Protein bands were recorded at 700 and 800 nm and evaluated with the Odyssey infrared imager system 2.1 (LI-COR Bioscience). HSP70, (70 kDa), β-III-tubulin (55 kDa) and GFAP (55 kDa) signal intensities were normalized to β-actin (42 kDa) signal intensities.
Statistical analyses
Groups were compared by one-way ANOVA, followed by Dunnett’s post-hoc test (Statistica V 12; Statsoft). Results are presented as mean ± SEM. A p-value < 0.05 was considered as statistically significant. The level of significance was set to *p < 0.05, **p < 0.01, ***p < 0.001. Statistical differences compared to the control + 37 °C group are shown with *, differences compared to the CoCl2 + 37 °C group are shown with#.
Supplementary information
Acknowledgements
This project is supported in part by the set Stiftung, Germany and FoRUM (Ruhr-University Bochum, Germany). We acknowledge the support by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of University of Tübingen.
Author Contributions
A.M.M. performed experiments, analyzed data and wrote the manuscript, S.K., J.H. and F.H. performed experiments and analyzed data, M.F., K.U.B. and H.B.D. revised the manuscript, S.C.J. and S.S. designed the study and revised the manuscript. All authors have approved the final article.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Stephanie C. Joachim and Sven Schnichels contributed equally.
Contributor Information
Stephanie C. Joachim, Email: stephanie.joachim@rub.de
Sven Schnichels, Email: sven.schnichels@med.uni-tuebingen.de.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-41113-4.
References
- 1.Tezel G, Wax MB. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Archives of ophthalmology. 2004;122:1348–1356. doi: 10.1001/archopht.122.9.1348. [DOI] [PubMed] [Google Scholar]
- 2.Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. The Yale journal of biology and medicine. 2007;80:51–60. [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez-Hernandez B, Cena V, Posadas I. The endoplasmic reticulum stress and the HIF-1 signalling pathways are involved in the neuronal damage caused by chemical hypoxia. British journal of pharmacology. 2015;172:2838–2851. doi: 10.1111/bph.13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hellwig-Burgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W. Review: hypoxia-inducible factor-1 (HIF-1): a novel transcription factor in immune reactions. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2005;25:297–310. doi: 10.1089/jir.2005.25.297. [DOI] [PubMed] [Google Scholar]
- 5.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caltana L, Merelli A, Lazarowski A, Brusco A. Neuronal and glial alterations due to focal cortical hypoxia induced by direct cobalt chloride (CoCl2) brain injection. Neurotoxicity research. 2009;15:348–358. doi: 10.1007/s12640-009-9038-9. [DOI] [PubMed] [Google Scholar]
- 7.Grasselli F, Basini G, Bussolati S, Bianco F. Cobalt chloride, a hypoxia-mimicking agent, modulates redox status and functional parameters of cultured swine granulosa cells. Reproduction, fertility, and development. 2005;17:715–720. doi: 10.1071/RD05059. [DOI] [PubMed] [Google Scholar]
- 8.Zimmerman MA, Biggers CD, Li PA. Rapamycin treatment increases hippocampal cell viability in an mTOR-independent manner during exposure to hypoxia mimetic, cobalt chloride. BMC neuroscience. 2018;19:82. doi: 10.1186/s12868-018-0482-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng Z, et al. A derivative of betulinic acid protects human Retinal Pigment Epithelial (RPE) cells from cobalt chloride-induced acute hypoxic stress. Experimental eye research. 2018;180:92–101. doi: 10.1016/j.exer.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 10.del Olmo-Aguado S, Nunez-Alvarez C, Ji D, Manso AG, Osborne NN. RTP801 immunoreactivity in retinal ganglion cells and its down-regulation in cultured cells protect them from light and cobalt chloride. Brain research bulletin. 2013;98:132–144. doi: 10.1016/j.brainresbull.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Kuehn S, et al. Degenerative effects of cobalt-chloride treatment on neurons and microglia in a porcine retina organ culture model. Experimental eye research. 2017;155:107–120. doi: 10.1016/j.exer.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Schultheiss M, et al. Hypothermia Protects and Prolongs the Tolerance Time of Retinal Ganglion Cells against Ischemia. PloS one. 2016;11:e0148616. doi: 10.1371/journal.pone.0148616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schnichels S, et al. Establishment of a retinal hypoxia organ culture model. Biology open. 2017;6:1056–1064. doi: 10.1242/bio.025429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reinhard K, et al. Hypothermia Promotes Survival of Ischemic Retinal Ganglion. Cells. Investigative ophthalmology & visual science. 2016;57:658–663. doi: 10.1167/iovs.15-17751. [DOI] [PubMed] [Google Scholar]
- 15.Januschowski K, et al. Glutamate and hypoxia as a stress model for the isolated perfused vertebrate retina. Journal of visualized experiments: JoVE. 2015 doi: 10.3791/52270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao QJ, Zhang XG, Wang LX. Mild hypothermia therapy reduces blood glucose and lactate and improves neurologic outcomes in patients with severe traumatic brain injury. Journal of critical care. 2011;26:311–315. doi: 10.1016/j.jcrc.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Antonic A, et al. Hypothermia protects human neurons. International journal of stroke: official journal of the International Stroke. Society. 2014;9:544–552. doi: 10.1111/ijs.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salido EM, et al. Global and ocular hypothermic preconditioning protect the rat retina from ischemic damage. PloS one. 2013;8:e61656. doi: 10.1371/journal.pone.0061656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cellular and molecular life sciences: CMLS. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elmore S. Apoptosis: a review of programmed cell death. Toxicologic pathology. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. The Journal of biological chemistry. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 22.Catalani S, Rizzetti MC, Padovani A, Apostoli P. Neurotoxicity of cobalt. Human & experimental toxicology. 2012;31:421–437. doi: 10.1177/0960327111414280. [DOI] [PubMed] [Google Scholar]
- 23.Karovic O, et al. Toxic effects of cobalt in primary cultures of mouse astrocytes. Similarities with hypoxia and role of HIF-1alpha. Biochemical pharmacology. 2007;73:694–708. doi: 10.1016/j.bcp.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Zou W, et al. Cobalt chloride induces PC12 cells apoptosis through reactive oxygen species and accompanied by AP-1 activation. J Neurosci Res. 2001;64:646–653. doi: 10.1002/jnr.1118. [DOI] [PubMed] [Google Scholar]
- 25.Zou W, et al. Involvement of caspase-3 and p38 mitogen-activated protein kinase in cobalt chloride-induced apoptosis in PC12 cells. J Neurosci Res. 2002;67:837–843. doi: 10.1002/jnr.10168. [DOI] [PubMed] [Google Scholar]
- 26.Araya J, et al. Inhibition of proteasome activity is involved in cobalt-induced apoptosis of human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2002;283:L849–858. doi: 10.1152/ajplung.00422.2001. [DOI] [PubMed] [Google Scholar]
- 27.Sharp FR, et al. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx: the journal of the American Society for Experimental. NeuroTherapeutics. 2004;1:26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mikami H, et al. Requirement of Hsp105 in CoCl2-induced HIF-1alpha accumulation and transcriptional activation. Experimental cell research. 2017;352:225–233. doi: 10.1016/j.yexcr.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 29.Tsuchida S, et al. HIF-1alpha-induced HSP70 regulates anabolic responses in articular chondrocytes under hypoxic conditions. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2014;32:975–980. doi: 10.1002/jor.22623. [DOI] [PubMed] [Google Scholar]
- 30.Yook YJ, et al. Induction of hypoxia-inducible factor-1alpha inhibits drug-induced apoptosis in the human leukemic cell line HL-60. The Korean journal of hematology. 2010;45:158–163. doi: 10.5045/kjh.2010.45.3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carmeliet P, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 32.Bossenmeyer-Pourie C, Koziel V, Daval JL. Effects of hypothermia on hypoxia-induced apoptosis in cultured neurons from developing rat forebrain: comparison with preconditioning. Pediatric research. 2000;47:385–391. doi: 10.1203/00006450-200003000-00017. [DOI] [PubMed] [Google Scholar]
- 33.Zhou T, et al. Mild hypothermia protects hippocampal neurons from oxygen-glucose deprivation injury through inhibiting caspase-3 activation. Cryobiology. 2018;80:55–61. doi: 10.1016/j.cryobiol.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Sardar Pasha SPB, et al. Retinal cell death dependent reactive proliferative gliosis in the mouse retina. Scientific reports. 2017;7:9517. doi: 10.1038/s41598-017-09743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McIlwain, D. R., Berger, T. & Mak, T. W. Caspase functions in cell death and disease. Cold Spring Harbor perspectives in biology7, 10.1101/cshperspect.a026716 (2015). [DOI] [PMC free article] [PubMed]
- 36.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Molecular cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang CY, et al. Roles of microRNA-1 in hypoxia-induced apoptotic insults to neuronal cells. Archives of toxicology. 2016;90:191–202. doi: 10.1007/s00204-014-1364-x. [DOI] [PubMed] [Google Scholar]
- 38.Lee JH, et al. CoCl2 induces apoptosis through the mitochondria- and death receptor-mediated pathway in the mouse embryonic stem cells. Molecular and cellular biochemistry. 2013;379:133–140. doi: 10.1007/s11010-013-1635-5. [DOI] [PubMed] [Google Scholar]
- 39.Nelson R, Kolb H, Robinson MM, Mariani AP. Neural circuitry of the cat retina: cone pathways to ganglion cells. Vision research. 1981;21:1527–1536. doi: 10.1016/0042-6989(81)90028-6. [DOI] [PubMed] [Google Scholar]
- 40.Reinehr S, et al. HSP27 immunization reinforces AII amacrine cell and synapse damage induced by S100 in an autoimmune glaucoma model. Cell and tissue research. 2018;371:237–249. doi: 10.1007/s00441-017-2710-0. [DOI] [PubMed] [Google Scholar]
- 41.Casola C, et al. Specific Inner Retinal Layer Cell Damage in an Autoimmune Glaucoma Model Is Induced by GDNF With or Without HSP27. Investigative ophthalmology & visual science. 2016;57:3626–3639. doi: 10.1167/iovs.15-18999R2. [DOI] [PubMed] [Google Scholar]
- 42.Akopian, A., Kumar, S., Ramakrishnan, H., Viswanathan, S. & Bloomfield, S. A. Amacrine cells coupled to ganglion cells via gap junctions are highly vulnerable in glaucomatous mouse retinas. The Journal of comparative neurology, 10.1002/cne.24074 (2016). [DOI] [PMC free article] [PubMed]
- 43.Hernandez M, Rodriguez FD, Sharma SC, Vecino E. Immunohistochemical changes in rat retinas at various time periods of elevated intraocular pressure. Molecular vision. 2009;15:2696–2709. [PMC free article] [PubMed] [Google Scholar]
- 44.Wang, G. H. et al. Injury effect of CoCl2-induced chemical hypoxia on N9 microglia. Vol. 29 (2013).
- 45.Matrone C, et al. HIF-1alpha reveals a binding activity to the promoter of iNOS gene after permanent middle cerebral artery occlusion. Journal of neurochemistry. 2004;90:368–378. doi: 10.1111/j.1471-4159.2004.02483.x. [DOI] [PubMed] [Google Scholar]
- 46.Jung F, Palmer LA, Zhou N, Johns RA. Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circulation research. 2000;86:319–325. doi: 10.1161/01.RES.86.3.319. [DOI] [PubMed] [Google Scholar]
- 47.Johansson UE, Eftekhari S, Warfvinge K. A battery of cell- and structure-specific markers for the adult porcine retina. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society. 2010;58:377–389. doi: 10.1369/jhc.2009.954933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taylor L, Arner K, Ghosh F. N-methyl-N-nitrosourea-induced neuronal cell death in a large animal model of retinal degeneration in vitro. Experimental eye research. 2016;148:55–64. doi: 10.1016/j.exer.2016.05.023. [DOI] [PubMed] [Google Scholar]
- 49.Kuehn, S. et al. A novel NMDA triggered porcine organ culture induces retinal ganglion cell apoptosis – chances for replacement of animal experiments. ATLA (in press) (2016). [DOI] [PubMed]
- 50.Mollick T, Mohlin C, Johansson K. Human neural progenitor cells decrease photoreceptor degeneration, normalize opsin distribution and support synapse structure in cultured porcine retina. Brain research. 2016;1646:522–534. doi: 10.1016/j.brainres.2016.06.039. [DOI] [PubMed] [Google Scholar]
- 51.Hurst J, et al. A novel porcine ex vivo retina culture model for oxidative stress induced by H(2)O(2) Alternatives to laboratory animals: ATLA. 2017;45:11–25. doi: 10.1177/026119291704500105. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, et al. E2f1 mediates high glucose-induced neuronal death in cultured mouse retinal explants. Cell Cycle. 2017;16:1824–1834. doi: 10.1080/15384101.2017.1361070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang S, Du S, Wu Q, Hu J, Li T. Decorin Prevents Retinal Pigment Epithelial Barrier Breakdown Under Diabetic Conditions by Suppressing p38 MAPK Activation. Invest Ophthalmol Vis Sci. 2015;56:2971–2979. doi: 10.1167/iovs.14-15874. [DOI] [PubMed] [Google Scholar]
- 54.Souaze F, Ntodou-Thome A, Tran CY, Rostene W, Forgez P. Quantitative RT-PCR: limits and accuracy. BioTechniques. 1996;21:280–285. doi: 10.2144/96212rr01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.