

SUMMARY
Regulation of cleavage and polyadenylation (CPA) affects gene expression and polyadenylation site (PAS) choice. Here, we report that the CPA and termination factor PCF11 modulates gene expression on the basis of gene size. Although downregulation of PCF11 leads to inhibition of short gene expression, long genes are upregulated because of suppressed intronic polyadenylation (IPA) enriched in large introns. We show that this regulatory scheme, named PCF11-mediated expression regulation through IPA (PEIPA), takes place in cell differentiation, during which downregulation of PCF11 is coupled with upregulation of long genes with functions in cell morphology, adhesion, and migration. PEIPA targets distinct gene sets in different cell contexts with similar rules. Furthermore, PCF11 is autoregulated through a conserved IPA site, the removal of which leads to global activation of PASs close to gene promotors. Therefore, PCF11 uses distinct mechanisms to regulate genes of different sizes, and its autoregulation maintains homeostasis of PAS usage in the cell.
In Brief
Wang et al. report a gene size-based dichotomic scheme by which the cleavage and polyadenylation factor PCF11 modulates gene expression. Distinct polyA site types are involved to regulate short and long genes. PCF11 expression level is autoregulated and is connected with long gene expression in cell differentiation and across tissues.
Graphical Abstract
INTRODUCTION
Cleavage and polyadenylation (CPA) is an essential step for 3′ end maturation of almost all eukaryotic mRNAs and long noncoding RNAs (lncRNAs) transcribed by RNA polymerase II (RNAPII) (Shi and Manley, 2015). It is also tightly coupled with termination of transcription (Kuehner et al., 2011; Proudfoot, 2016). The site for CPA, also known as polyadenylation (polyA) site or PAS, is defined by surrounding sequence motifs (Tian and Graber, 2012). In mammals, upstream PAS motifs include the A[A/U]UAAA hexamer and other close variants, as well as U-rich and UGUA motifs; downstream motifs include U-rich, GU-rich, and G-rich elements (Hu et al., 2005; Tian and Graber, 2012). Mutations weakening or strengthening the PAS have been shown to alter gene expression in several human diseases (Hollerer et al., 2014), highlighting the importance of PAS for gene expression.
Most eukaryotic genes harbor multiple PASs, leading to expression of alternative cleavage and polyadenylation (APA) isoforms (Derti et al., 2012; Hoque et al., 2013; Shepard et al., 2011). Although the majority of APA sites are located in the last exon of genes (Tian and Manley, 2017), a substantial fraction of PASs are located in upstream regions, mostly in introns (Hoque et al., 2013; Tian et al., 2007). Intronic polyA (IPA) leads to isoforms with different coding sequences (CDSs) or truncated transcripts without apparent functions (Tian and Manley, 2017). APA is highly tissue specific (Derti et al., 2012; Wang et al., 2008; Zhang et al., 2005) and is dynamic in cell proliferation and differentiation (Ji et al., 2009; Mayr and Bartel, 2009; Sandberg et al., 2008). Although still not fully understood, APA has been shown to be regulated by a variety of mechanisms (Tian and Manley, 2017), among which core CPA factor expression level changes appear to be quite potent (Lackford et al., 2014; Li et al., 2015; Martin et al., 2012; Masamha et al., 2014; Yao et al., 2012).
The mammalian CPA complex contains more than 20 core factors (Mandel et al., 2008; Shi and Manley, 2015) and many associated proteins (Shi et al., 2009). Some core factors form sub-complexes, including the CPA specificity factor (CPSF), the cleavage stimulation factor (CstF), cleavage factor I (CFI), and cleavage factor II (CFII). PCF11 is part of CFII and also plays a role in termination of transcription. Although yeast and mammalian PCF11 orthologs differ substantially in size (yeast and mammalian PCF11 contain 626 and ~1,600 amino acids, respectively), several protein domains are well conserved. First, both yeast and mammalian PCF11 proteins contain an N-terminal RNAPII C-terminal domain (CTD)-interacting domain (CID). The CID, which also exists in several other proteins involved in termination of transcription (Larochelle et al., 2017), is believed to mediate coupling of CPA with termination (Meinhart and Cramer, 2004; Zhang and Gilmour, 2006). Domains for interactions with Rna14 and Rna15 in yeast (CstF77 and CstF64 in mammals) and for Clp1 binding are also conserved. In addition, two zinc-binding regions of human PCF11 interact with RNA without strong specificity (Schäfer et al., 2018), and the corresponding regions in yeast PCF11 are critical for CPA (Guéguéniat et al., 2017; Yang et al., 2017). Both yeast and mammalian PCF11 proteins have also been implicated in mRNA nuclear export control (Johnson et al., 2009; Volanakis et al., 2017). Consistent with PCF11’s roles in CPA and termination, we previously showed that conditional ablation of pcf11 in S. pombe and knockdown (KD) of Pcf11 in mouse C2C12 cells lead to global 3′UTR lengthening through APA (Li et al., 2015; Liu et al., 2017b). Here we report that PCF11 level affects expression of genes on the basis of their size. Short and long genes are regulated by PCF11 with distinct mechanisms related to CPA. We show that this regulatory strategy is an integral part of gene expression program in cell differentiation, and PCF11 level is tightly controlled through autoregulation.
RESULTS
PCF11 Regulates Gene Expression Relevant to Cell Proliferation and Differentiation
We previously studied APA regulation by core CPA and splicing factors in mouse C2C12 myoblast cells by individual gene KD and 3′ end sequencing (Li et al., 2015). Analysis of the same data indicated widespread gene expression changes in the KD samples (Figure S1A). Interestingly, gene expression changes in Pcf11 KD cells showed a mild correlation with those in C2C12 differentiation (r = 0.20, Pearson correlation; Figure S1B). Because PCF11 protein is downregulated ~40% at the beginning of C2C12 differentiation (Figure 1A), we wanted to examine to what extent PCF11, a CPA and termination factor, is involved in gene expression changes in C2C12 cell differentiation.
Figure 1. PCF11 Regulates Gene Expression Related to Cell Differentiation.

(A) Western blot analysis of PCF11 expression during C2C12 cell differentiation. Relative levels are based on normalization to GAPDH. MYOG level indicates stage of differentiation.
(B) Top: schematic of experimental design. Bottom: KD efficiency after 32 h of siRNA treatment.
(C) Volcano plot showing expression changes (4sU RNA data only) in siPcf11 versus siCtrl. Significantly regulated genes (fold change > 1.2 and p < 0.05, DESeq analysis) are marked. Gene numbers are indicated.
(D) Scatterplot comparing gene expression changes in 4sU versus total RNA samples. Pearson correlation coefficient (r) is indicated.
(E) Scatterplot comparing gene expression changes in Pcf11 KD cells versus C2C12 differentiation. Pearson correlation coefficient (r) is indicated (gray for all genes, black for significantly regulated genes in either condition). Pro, proliferating cells; Dif, differentiating cells (after 4 days).
(F) Heatmap showing top Gene Ontology (GO) terms associated with regulated genes in Pcf11 KD cells and C2C12 differentiation. Colors represent significance and direction of regulation as indicated.
(G) Cell proliferation of siPcf11 and siCtrl cells as measured using a CFSE assay.
(H) Cell migration analysis of siPcf11 and siCtrl cells by a scratch assay. Error bars are SEM on the basis of ten analyzed regions. **p < 0.05 (t test).
(I) Expression changes of two muscle cell differentiation marker genes, Myh3 and Myog, on the basis of 3′READS data. Error bars are SD.
Both yeast and mammalian PCF11 proteins have been implicated in nuclear export of mRNA (Johnson et al., 2009; Volanakis et al., 2017), raising the possibility that mRNA stability changes resulting from alternation of nuclear export may affect gene expression in Pcf11 KD cells. We thus metabolically labeled cellular RNA with 4-thiouridine (4sU) for 15 min in control and KD cells (two small interfering RNAs [siRNAs], or siPcf11, leading to ~40% of KD at the protein level; Figure 1B) and isolated 4sUlabeled, newly made RNA for 3′READS (3′ region extraction and deep sequencing) analysis, a deep sequencing method specialized for 3′ end analysis of poly(A)+ RNA (Figure 1B) (Zheng et al., 2016). Steady-state total RNA was used for comparison.
As with our previous data, siPcf11 treatment led to widespread mRNA expression changes in 4sU-labeled RNAs, with similar numbers of genes up- or downregulated (1,961 versus 2,030; Figure 1C). Expression changes were largely consistent between newly made and steady-state RNAs (r = 0.89, Pearson correlation; Figure 1D). Interestingly, using transcript abundance ratio between 4sU and total RNAs to reflect RNA decay rate, we found a widespread role of PCF11 in controlling transcript stability (Figure S1C), and there was a mild negative correlation between decay difference and gene expression change (r = −0.1, Pearson correlation; Figure S1D). This result indicates that mRNA stability alteration does play a role, albeit quite minor, in mRNA abundance changes in Pcf11 KD cells.
Consistent with our previous result, gene expression regulation by Pcf11 KD showed a correlation with that in C2C12 differentiation (r = 0.24 overall, r = 0.38 using significantly regulated genes; Figure 1E). This correlation was also supported by Gene Ontology (GO) analysis (Figure 1F), which showed similar significant terms between these two conditions. Downregulated genes were related to cell proliferation, such as “chromosome segregation,” “mitotic nuclear division,” “cell division,” and “chromosome organization” (Figure 1F; see Figure S1E for their relationship). In contrast, genes associated with morphology, adhesion, and migration (named MAM genes for simplicity), such as “cell adhesion,” “locomotion,” “membrane organization,” and “cell morphogenesis,” tended to be upregulated in both conditions (Figure 1F; see Figure S1E for their relationship). Consistent with the gene expression result, we found that KD of Pcf11 in C2C12 cells inhibited cell proliferation on the basis of a CPSE assay (Figure 1G) and led to a mild (~20%) but significant increase in cell migration rate on the basis of a scratch assay (p < 0.05, t test; Figure 1H).
Notably, despite the general correlation of gene expression changes between Pcf11 KD and cell differentiation, muscle cell differentiation marker genes, such as Myog and Myh3, did not show altered expression after Pcf11 KD (Figure 1I). Therefore, PCF11 affects a subset of genes that are regulated in cell differentiation, and its downregulation is not sufficient to induce cell differentiation.
Gene Size Dichotomizes Gene Regulation by PCF11
To examine how PCF11 regulates gene expression, we carried out a regression analysis to identify gene features that correlated with gene expression changes. The gene features we examined included gene size, gene-to-gene distance, splicing features, nucleotide contents of different genic regions, RNA stability, and others (see STAR Methods for detail). Strikingly, the size of the largest intron, overall intron size, and gene size were the most prominent features that correlated with gene expression changes in both steady-state and newly made RNA samples (R2 = 0.13–0.16; Figures 2A and S2A). Note that the top three features were correlated with one another (Figure S2B). Consistently, the largest introns of MAM genes were markedly larger than those of other genes by more than 2-fold (p < 1.0 × 10−9, Wilcoxon test; Figure 2B), and those of cell proliferation genes were substantially shorter by about 50% (p < 1.0 × 10−3, Wilcoxon test; Figure 2B). In addition, genes with large introns (top 10%) showed significantly greater upregulation than MAM genes, and those with short introns (bottom 10%) showed significantly greater downregulation than cell proliferation genes (p < 0.001, Kolmogorov-Smirnov [K-S] test; Figure 2C), indicating that intron and gene size features, rather than gene functions, are the primary reason for mRNA expression changes in Pcf11 KD cells.
Figure 2. PCF11 Regulates Gene Expression through Gene Size.

(A) Top features on the basis of regression analysis of gene features versus gene expression changes in siPcf11 versus siCtrl samples (total RNA). Features are sorted according to individual R2 value. Cumulative R2 value is based on a given feature together with all other features with a better individual R2 value. + and − denote positive and negative correlations, respectively.
(B) The largest intron size of genes in different GO term gene groups. MAM (morphology, adhesion, and migration) and CP (cell proliferation) groups are indicated.
(C) Gene expression changes of different gene groups in siPcf11 versus siCtrl samples. MAM and CP groups correspond to those in (B), respectively. Gene number is indicated in parenthesis, and median value is shown. ***p < 0.001 (K-S test).
(D) Schematic of 3′UTR APA analysis. RED, relative expression difference (pPAS versus dPAS in KD versus control samples).
(E) 3′UTR APA changes in Pcf11 KD cells (total RNA samples). The numbers of genes with significantly lengthened 3′UTRs (red) or shortened 3′UTRs (blue) are indicated.
(F) 3′UTR APA RED of different gene groups in (C). n.s., not significant (K-S test).
(G) Expression changes of genes with different types of 3′UTR regulation in (E). **p < 0.01 (K-S test).
PCF11 was previously shown to regulate 3′UTR size through APA (Li et al., 2015). Indeed, by analyzing the relative expression of the two most abundant 3′UTR APA isoforms of a gene (named proximal PAS [pPAS] and distal PAS [dPAS] isoforms, respectively; Figure 2D), we found that Pcf11 KD led to global 3′UTR lengthening in both steady-state and newly made RNAs (Figures 2E and S2C). The number of genes with lengthened 3′UTRs outnumbered those with shortened 3′UTRs by 2.0- or 2.4-fold (Figures 2E and S2C). However, MAM and cell proliferation genes as well as genes with different intron sizes showed similar levels of 3′UTR lengthening to other genes, as measured using the relative expression difference (RED) score (dPAS versus pPAS isoforms in KD versus control cells; Figure 2F). In addition, genes with lengthened 3′UTRs displayed a similar mRNA abundance change profile to genes without significant 3′UTR size changes (Figure 2G), although, intriguingly, genes with shortened 3′UTRs tended to be downregulated (Figure 2G; see below). Together, these results indicate that 3′UTR regulation does not play a global role in mRNA expression level changes in Pcf11 KD cells.
PAS Strength and Gene Density Are Important for Short Gene Expression
We next reasoned that genes with different PAS strengths might be regulated differently in Pcf11 KD cells. To this end, we compared sequence motifs around the last PASs of upregulated and downregulated genes, respectively, in short and long genes (bottom and top 20%, respectively). Interestingly, we found that upregulated short genes had significantly enriched UGUG motifs (GUGUGU and UGUGUG) in the downstream region of the PAS compared with downregulated short genes (−log10p = 10.7 and 11.9, respectively; Figure 3A). In contrast, albeit consistent, this trend was much less significant with long genes (−log10p = 4.3 and 3.4, respectively; Figure 3A). A similar result was obtained when the most used PAS in the last exon was used for analysis (Figure S3A). Because UGUG motifs are typically associated with strong PASs (Hu et al., 2005), this result indicates that the PAS strength of short genes is important for their gene expression.
Figure 3. Short Genes Depend on PCF11 for Expression.

(A) Enriched motifs around the last PAS of up- or downregulated genes (UP and DN, respectively) in short and long gene groups. Three sub-regions are analyzed, as indicated.
(B) Gene number ratio of upregulated genes to downregulated genes in different gene groups on the basis of gene size and distance to the nearest neighbor. **p < 0.01, ***p < 0.001, and n.s., not significant (Fisher’s exact test).
(C) Pan-stress readthrough genes (pan-stress DoG genes) tend to be downregulated in Pcf11 KD cells. The number of pan-stress DoG genes is indicated. p value is based on K-S test.
(D) Gene number ratio of upregulated genes to downregulated genes in five gene groups on the basis of distance to neighbor. There are four types of relationship between the analyzed gene (colored) and its neighbor, on the basis of relative transcriptional orientation (shown on the right, with arrows indicating direction of transcription). Only short genes are used for analysis.
Because short genes also tend to have short distances to neighboring genes (Figure S3B), one possible explanation for downregulation of short genes in Pcf11 KD cells is transcriptional inference between neighboring genes resulting from unsuccessful CPA, especially when PASs are weak. To explore this, we divided genes into 25 groups on the basis of both gene size and distance to the nearest neighbor (NN) gene and examined gene expression changes in each group (Figure S3C). Interestingly, although distance to the NN did not appear to be relevant to regulation of long genes (top 20%), short genes (bottom 20%) with a long distance to the NN gene were less likely to be downregulated than those with a short distance (Figure S3C). This trend was also apparent when we examined the numbers of upregulated and downregulated genes in each gene group on the basis of gene size and distance to the NN (Figure 3B). In addition, we found that genes previously shown to have transcriptional readthrough in stressed conditions (Vilborg et al., 2017) tended to be downregulated in Pcf11 KD cells (Figure 3C), further supporting the notion that short genes in high-density regions are prone to transcriptional interference among neighboring genes when CPA is weakened.
Interestingly, the relative transcriptional direction between neighboring genes, including head to head, tail to tail, head to tail, and tail to head (head being the transcription start site [TSS] and tail being the last PAS), did not appear to be important for gene expression changes (Figure 3D), suggesting that gene density rather than transcriptional orientation is the major reason for short gene regulation by PCF11. It is thus possible that transcriptional readthrough may go beyond two adjacent genes when it happens. Thismayalso explain why genes with shortened 3′UTRs tend to be downregulated: suppressed CPA at a dPAS would lead to activation of pPAS and transcriptional readthrough at the same time. Taken together, our data indicate that short genes tend to be downregulated when PCF11 level is low, and both gene density and PAS strength are critical for their regulation.
Suppression of IPA Correlates with Upregulation of Long Genes
We next wanted to address why genes with large introns were more likely to be upregulated in Pcf11 KD cells. Because large introns tend to harbor PASs (Tian et al., 2007), we hypothesized that Pcf11 KD might suppress IPA and, consequently, lead to upregulation of gene expression. To test this, we examined the relationship between gene expression changes and frequency of IPA sites in a gene as annotated in the comprehensive PAS database PolyA_DB (Wang et al., 2018). Indeed, genes with higher IPA site frequencies were more likely to be upregulated compared with genes with lower IPA site frequencies (Figure 4A). Using PolyA_DB, we also found that a greater fraction of MAM genes had conserved IPA events than other genes by 25%–50%, whereas cell proliferation genes were similar to other genes (Figure 4B).
Figure 4. Long Genes Are Regulated by PCF11 through Intronic Polyadenylation.

(A) mRNA expression changes of genes with different IPA site frequencies (number of IPA sites per gene) as annotated in PolyA_DB. ***p < 0.001 (Wilcoxon test).
(B) Percentage of genes with conserved IPA sites as annotated in PolyA_DB in different gene groups.
(C) Top: schematic of IPA isoforms; bottom: IPA RED or Δlog2(IPA isoform/TPA isoform) between siPcf11 versus siCtrl samples, for different gene groups. **p < 0.01 and n.s., not significant (K-S test).
(D) IPA RED in C2C12 differentiation. **p < 0.01 (K-S test). Dif, differentiating cells (4 days); Pro, proliferating cells.
(E) IPA distribution maps of suppressed IPA events in Pcf11 KD cells. Color represents ratio (log2) of number of observed (obs) events to number of expected (exp) events.
(F) As in (E), except that suppressed IPA events in C2C12 differentiation are plotted.
(G) An example gene Fgf2, which contains a regulated IPA site. IPA and TPA isoform expression changes on the basis of 3′READS data are indicated.
(H) RT-qPCR analysis of IPA and TPA isoforms of Fgf2 in Pcf11 KD cells. Full-length Pcf11 transcript change is also shown. Error bars represent SD. ***p < 0.01 (t test).
(I) Gene expression changes of different gene groups in C2C12 differentiation. Median value is shown. **p < 0.01 and ***p < 0.001 (K-S test).
(J) Gene expression changes of different gene sets at early stages of C2C12 differentiation (based on GEO: GSE94560). Error bars represent SD.
We next compared IPA isoform abundance with that of isoforms using last exon PASs (named TPA isoforms; Figure 4C, top) in KD and control cells. On the basis of RED scores (Δlog2ratio, IPA isoform versus TPA isoform in KD versus control cells), we found that IPA events were generally suppressed in Pcf11 KD cells (negative values in all groups; Figure 4C, bottom). Importantly, genes with large introns had significantly greater IPA suppression than other genes (p < 0.01, K-S test; Figure 4C, bottom). Although MAM genes as a whole were similar to other genes, those with large introns displayed significantly greater IPA suppression (p < 0.01, K-S test; Figure 4C). This trend is in good agreement with the IPA regulation during C2C12 differentiation, in which large intron-containing genes also had greater IPA suppression than other genes (p < 0.01, K-S test; Figure 4D).
We next asked what type(s) of introns tended to harbor suppressed IPA in Pcf11 KD cells. To this end, we constructed a 2-dimensional IPA distribution map, in which all introns with detectable IPA events by 3′READS were evenly distributed in a 10 × 10 table, on the basis of intron size versus distance from IPA site to the TSS, or 5′ splice site (5′SS) strength, or 3′ splice site (3′SS) strength (Figure 4E). Strikingly, we found that suppressed IPA events were highly enriched in large introns (Figure 4E). Moderately large introns with very weak 5′SS (bottom 10%) also showed some enrichment of IPA suppression (Figure 4E, middle, bottom row). In contrast, 3′SS strength did not show any relevance to IPA suppression by Pcf11 KD (Figure 4E, bottom). Similar patterns could be discerned for IPA suppression in C2C12 cell differentiation (Figure 4F). These results indicate that intron size and 5′SS strength are determinants of IPA suppression in Pcf11 KD cells and C2C12 cell differentiation, with intron size playing a more dominant role in regulation.
An example gene, Fgf2 (encoding fibroblast growth factor 2), is shown in Figure 4G. Fgf2 displayed significant downregulation (3-fold) of an IPA isoform, whose PAS is in a large intron, and upregulation (1.9-fold) of a TPA isoform in Pcf11 KD cells. Using RT-qPCR, we validated Fgf2 IPA and TPA isoform expression changes (Figure 4H). Taken together, our data indicate that decreased expression of PCF11 correlates with IPA suppression and upregulation of genes with large introns. This correlation may be important for cell differentiation, when PCF11 level is downregulated (Figure 1A). Consistent with this view, we found that genes with large introns were substantially upregulated in C2C12 differentiation on the basis of our 3′READS data (Figure 4I) and previous RNA sequencing (RNA-seq) data (Figure 4J; Hamed et al., 2017). Notably, this trend was even more conspicuous in neurogenesis (Figure S4A, note the y-axis scale difference compared with C2C12 differentiation), during which PCF11 level progressively decreased (Figure S4B).
Gene Regulation by PCF11 in Different Cell Contexts
We next asked whether PCF11-mediated gene regulatory scheme can be similarly executed in other cell types. To this end, we knocked down Pcf11 in mouse fibroblast NIH 3T3 and preadipocyte 3T3-L1 cells and carried out 3′READS and RNA-seq analyses, respectively. RNA-seq was used to ensure that our findings were not biased by the sequencing method used. Overall, gene expression changes in these two cell types after Pcf11 KD were correlated with those in C2C12 KD cells (r = 0.46 and r = 0.34, respectively, Pearson correlation; Figure 5A). Interestingly, whereas cell proliferation genes were significantly downregulated in NIH 3T3 KD cells, MAM genes were not substantially upregulated (Figure 5B). In contrast, whereas MAM genes were well upregulated in 3T3-L1 cells, cell proliferation genes were not among the most significantly regulated (Figure 5B). However, importantly, similar to Pcf11 KD in C2C12 cells, largest intron size, overall intron size, and gene size were the top features that correlated with gene expression changes in both NIH 3T3 and 3T3-L1 cells (Figure 5C). Therefore, although distinct sets of genes were regulated by Pcf11 KD, the same regulatory rules applied in different cell types.
Figure 5. Gene Expression Regulation by PCF11 in Different Cell Contexts.

(A) Correlation of gene expression changes by Pcf11 KD in C2C12 cells versus NIH 3T3 (left) or 3T3-L1 (right) cells. NIH 3T3 data are based on 3′READS and 3T3-L1 data on RNA-seq.
(B) Top GO terms associated with up- or downregulated genes in NIH 3T3 and 3T3-L1 Pcf11 KD cells.
(C) Top gene features correlated with gene expression changes by Pcf11 KD in NIH 3T3 and 3T3L1 cells.
(D) mRNA expression changes in NIH 3T3 KD cells for genes with different IPA site frequencies (number of IPA sites per gene) as annotated in PolyA_DB. ***p < 0.001 (Wilcoxon test).
(E) IPA distribution maps for suppressed IPA events in NIH 3T3 KD cells.
As in C2C12 cells, using 3′READS data of NIH 3T3 KD cells, we found that genes with high IPA site frequencies were more likely to be upregulated in KD cells than genes with lower IPA site frequencies (Figure 5D), and IPA events in large introns and moderately large introns with weak 5′SS tended to be regulated in NIH 3T3 KD cells (Figure 5E). Taken together, these data indicate that the same gene size-based rule in PCF11-mediated gene expression regulation applies in different cell contexts.
Pcf11 Expression Is Autoregulated by IPA
From our 3′READS data, we noticed that mouse Pcf11 had several APA sites (Figure 6A), including one located in intron 1 (named IPA site), one located in exon 8 (named internal exonic polyA [EPA] site), and two located in the 3′ terminal exon (named TPA sites). On the basis of PolyA_DB (Wang et al., 2018), these APA sites were all conserved across vertebrates (Figure S5A), and the isoforms were differentially expressed in different tissues (Figure 6A).
Figure 6. Pcf11 Expression Is Regulated by IPA.

(A) Schematic of mouse Pcf11 gene. Multiple PASs are indicated. EPA, internal exonic polyadenylation; IPA, intronic polyadenylation; TPA, 3′ terminal exon polyadenylation. 3′READS data of brain, heart, and testis are shown.
(B) Features of intron 1 compared with other introns in the mouse genome. Each feature has ten bins, and the bin containing intron 1 of Pcf11 is highlighted in red. Distance to TSS is based on all IPA sites in PolyA_DB.
(C) RT-qPCR analysis of IPA and TPA isoform expression in siPcf11 versus siCtrl cells. Error bars are SD on the basis of three replicates.
(D) Top: schematic showing two sgRNAs used to remove a region flanking the IPA site. Bottom: validation of IPA site knockout (IPAPcf11-KO) by genomic DNA PCR using primers flanking the KO region. WT, wild-type 4T1 cells.
(E) RT-qPCR analysis of Pcf11 isoform expression in WT and IPAPcf11-KO cells. Error bars are SD on the basis of three replicates.
(F) Western blot analysis of PCF11 in WT and IPAPcf11-KO cells.
(G) 3′UTR APA changes in IPAPcf11-KO cells.
(H) IPA changes in IPAPcf11-KO cells. IPA isoforms (all combined) were compared with TPA isoforms (all combined) in analysis.
(I) IPA distribution maps of activated IPA events in IPAPcf11-KO cells.
(J) Left: schematic of PASs around the transcription start site (TSS); right: metagene analysis of PAS usage around the TSS in WT and IPAPcf11-KO cells.
(K) Gene expression changes in IPAPcf11-KO cells for different groups of genes on the basis of IPA site frequency in PolyA_DB. ***p < 0.01 (K-S test).
The IPA site was of particular interest because intron 1 of Pcf11 had a much higher conservation level than other introns (Figure 6A), suggesting functional importance of the IPA site. The IPA isoform would have a short open reading frame (ORF) encoding only part of the CID domain. Using a reporter system, we validated the activity of IPA site and found that the site was even stronger than the two TPA sites (Figure S5B).
Interestingly, intron 1 of Pcf11 has a very weak 5′SS (ranked at the 3rd percentile of all introns in the mouse genome), and its size is at the 75th percentile of all introns (Figure 6B). This configuration would make the IPA site regulatable by PCF11 itself (see IPA distribution maps in Figures 4E and 5E). Indeed, using RT-qPCR with primers specifically targeting IPA and TPA isoforms, we found that both TPA and IPA isoform levels were downregulated by 30% and 20%, respectively, in C2C12 cells with Pcf11 KD (Figure 6C). Similar results were obtained using NIH 3T3 cells and mouse breast cancer cell line 4T1, although the degree of downregulation varied (Figure 6C). Because the Pcf11 siRNAs could target TPA isoforms only but not IPA isoforms (STAR Methods), this result suggests that IPA site usage is responsive to PCF11 levels. In other words, the IPA site might be engaged in autoregulation of Pcf11 expression.
To further examine the function of IPA site of Pcf11, we set out to delete the IPA site using the CRISPR/Cas9 method with two single guide RNAs (sgRNAs) targeting flanking regions of the IPA site (Figure 6D). Although we were not able to isolate any clone using C2C12 cells, we confirmed one IPA site knockout clone using 4T1 cells. The clone, named IPAPcf11-knockout (KO), showed homozygous removal of the target region on the basis of genomic PCR (Figure 6D) and Sanger sequencing (Figure S5C). RT-qPCR analysis indicated a 5-fold decrease of the IPA isoform expression and a concomitant 5-fold increase of expression of downstream exons (Figure 6E). Note that some cryptic PASs located in the first intron might be used after removal of the IPA site, leading to some residual transcripts detectable by RT-qPCR using IPA primers. Western blot analysis indicated a 40% increase of PCF11 protein expression in IPAPcf11-KO cells (Figure 6F). Therefore, removal of the IPA site led to substantial upregulation of full-length PCF11 mRNA and protein, supporting the notion that the IPA site plays a negative role in Pcf11 expression.
Increased PCF11 Expression Leads to Global PAS Usage Changes
We next wanted to examine how APA and gene expression changed when PCF11 level was upregulated by 40% through IPA site removal. To this end, we subjected total cellular RNAs from wild-type (WT) and IPAPcf11-KO cells to 3′READS analysis. We first examined 3′UTR APA on the basis of the two most abundant 3′UTR APA isoforms (Figure 6G). We found that genes with shortened 3′UTRs outnumbered genes with lengthened 3′UTRs by 8.2-fold (Figure 6G), indicating substantial global shortening of 3′UTRs in IPAPcf11-KO cells. Strikingly, genes with activated IPA outnumbered those with repressed IPA by 21-fold (Figure 6H), highlighting a very strong effect of PCF11 overexpression on IPA activation.
Interestingly, using IPA distribution maps, we found that in addition to intron size, distance to TSS was a very prominent feature of IPA activation in IPAPcf11-KO cells (Figure 6I, top). Consistently, the first two introns showed much greater activation of IPA than downstream introns (Figure S5D). Therefore, upregulation of PCF11 by 40% could trigger severe shortening of gene transcripts through activation of PASs close to the TSS.
Previous studies indicated widespread expression of upstream antisense RNAs (uaRNAs) near mammalian promoters (Core et al., 2008). Bidirectionality of gene promoters has also been suggested as a driving force for new gene evolution (Wu and Sharp, 2013). Focusing on the ±3 kb region around the TSS, we found that the PASs in both sense and antisense directions around the promoter were activated in IPAPcf11-KO cells (Figure 6J), with the uaRNA PASs being activated to a greater degree. This result indicates that PCF11 has a role in controlling uaRNA expression and potentially contributes to upstream antisense gene evolution.
In line with the notion that IPA regulates gene expression, we found that genes with a high IPA site frequency tended to have greater downregulation of gene expression in IPAPcf11-KO cells compared with genes with a low IPA site frequency (p < 0.001, K-S test; Figure 6K). Notably, IPAPcf11-KO cells had much slower migration and invasion rates than WT 4T1 cells (by 70%; Figures S5E and S5F), which are metastatic cancer cells with high migration and invasion capabilities, suggesting impacts of IPA activation on MAM gene functions.
Taken together, our data based on IPAPcf11-KO cells support a negative role of the IPA site in control of PCF11 level, likely through autoregulation, and indicate that PCF11 is a master regulator of PAS usage genome-wide and modulates long gene expression through IPA.
Systematic Analysis of Long Gene Expression across Tissues
To further explore gene regulation by PCF11 in a broader context, we analyzed Genotype-Tissue Expression (GTEx) RNA-seq datasets (GTEx Consortium, 2017), consisting of 5,032 samples from 27 human tissue types in 545 human individuals. Using the ratio of long gene expression levels to those of all genes as long gene expression index, we found an inverse correlation between long gene expression and PCF11 expression levels across 27 tissue types (r = −0.35, Pearson correlation; Figures 7A). Notably, brain tissues had the lowest PCF11 levels and were among the ones with the highest long gene expression levels (Figure 7A). Using 5,032 RNA-seq samples from the tissues, we detected an overall trend that samples with high long gene expression levels tended to have low PCF11 expression levels (Figure 7B). Conversely, long gene expression levels tended to be low when PCF11 expression levels were high (overall r = −0.30, Pearson correlation; Figure 7B). Therefore, our large-scale GTEx data analysis result supports the notion that PCF11 expression is coordinated with expression of long genes.
Figure 7. Global Analysis of PCF11 Expression versus Long Gene Expression across Human Tissues.

(A) Scatterplot showing correlation between long gene expression and PCF11 expression in 27 human tissues on the basis of GTEx data. Pearson correlation coefficient (r) and its p value are shown.
(B) Heatmap showing correlation between long gene expression and PCF11 expression on the basis of 5,032 GTEx samples. Obs, observed number of samples; Exp, expected number of samples based on the average of all samples.
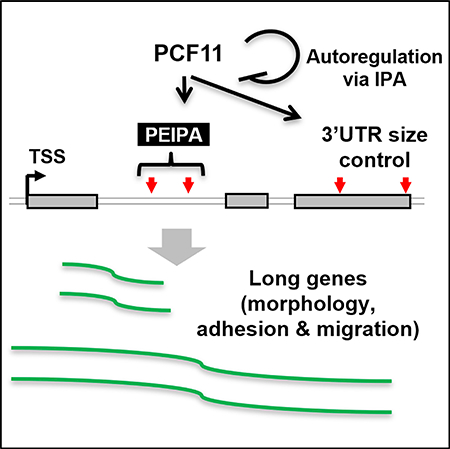
(C) A model summarizing PCF11-mediated gene expression regulation through IPA (PEIPA).
DISCUSSION
In this study, we reveal a dichotomic role of PCF11 in regulation of mRNA expression on the basis of gene size. Short genes, especially those in high-gene density regions and with weak PASs, appear to rely on PCF11 levels for expression. In contrast, long genes are regulated by PCF11 through IPA. This gene regulatory scheme, named PCF11-mediated expression regulation through IPA (PEIPA; Figure 7C), is in play during cell differentiation, when genes with large introns (which tend to have functions in cell morphology, adhesion, and migration) are upregulated. Consistently, long gene expression across tissues is globally coordinated with PCF11 level. Moreover, PCF11 itself is regulated by IPA, which functions to maintain its expression level.
PEIPA
We found that MAM genes are regulated by PEIPA because they tend to have large introns. This trend appears to be conserved in all vertebrates, from humans to zebrafish (Figure S6), indicating that PEIPA is likely to be an evolutionarily conserved mechanism to regulate MAM genes. Notably, we found that genes enriched for neuronal features, such as “neurological system process” and “nervous system process,” were also enriched with large introns, suggesting that PEIPA may have an important role in regulation of neuron-specific genes. This would be consistent with the low IPA isoform expression levels in brain (Liu et al., 2017a; Singh et al., 2018; Zhang et al., 2005) and much more conspicuous upregulation of large intron genes in neurogenesis compared with myogenesis (Figures 4J and S4A). Our migration and invasion data on the basis of 4T1 cells with Pcf11 IPA site KO also suggest a role of PCF11 in cancer development, especially in the metastatic phase when MAM gene expression is substantially changed (Hanahan and Weinberg, 2011). Notably, a recent pan-cancer screen of mutations in non-coding regions identified several single nucleotide variants in the promoter region of PCF11 that are related to cancers (Hornshøj et al., 2018), and low level PCF11 in neuroblastoma was found to be associated with spontaneous tumor regression (Ogorodnikov et al., 2018).
Regulation of PCF11
Our data indicate that PCF11 level is autoregulated through IPA. Given the widespread PAS usage changes in IPAPcf11-KO cells, a tight control of PCF11 level through autoregulation appears critical for the homeostasis of CPA activity in the cell. In a sense, the strong IPA site of Pcf11 coupled with weak 5′SS of intron 1 functions as a general sensor of global IPA activity in the cell. Notably, several other CPA factor genes have been reported to have IPA sites, such as CSTF3 (Luo et al., 2013), RBBP6 (Di Giammartino et al., 2014), and PAPOLA (Zhao and Manley, 1996). Therefore, IPA is a common mechanism regulating CPA factor expression. The rationale may be that IPA bestows rapid regulation of CPA factor gene expression in response to CPA activity through expression of truncated transcripts encoding proteins with no functions or with dominant-negative activities (in the case of RBBP6). Whether these IPA events are coordinately regulated and how they are related to splicing control would be interesting to examine in the future.
Although we show autoregulation of Pcf11 through IPA in this study, our data also indicate other mechanisms modulating PCF11 expression. Interestingly, we found a 4-fold increase of IPA isoform expression after 1 day of C2C12 differentiation (Figure S7A), whose level slightly decreased from 1 to 4 days of differentiation (Figure S7A). By contrast, TPA isoforms (two 3′UTR APA isoforms combined) showed a slight increase at the beginning of C2C12 differentiation followed by a 25% decrease after 4 days of differentiation (Figure S7A). Therefore, the IPA-to-TPA isoform ratio drastically increased at the beginning of differentiation by 2.7-fold and continued to increase to 3.7-fold after 4 days of differentiation (Figure S7A). Because IPA sites were generally suppressed in differentiated cells, opposite to the IPA regulation of Pcf11, other mechanism(s) must be in play for IPA regulation of Pcf11 during differentiation. It is also worth noting that a 5-fold increase of Pcf11 mRNA levels in IPAPcf11-KO cells resulted only in 40% increase of protein expression, indicating additional post-transcriptional regulations.
PCF11 versus Other CPA Factors
In addition to Pcf11 KD, several other CPA factor KD samples also showed positive, albeit milder, correlation with C2C12 differentiation (Figure S1B), raising the possibility that their expression level changes may also contribute to regulation of short and long genes. This is possible because other CPA factors, like PCF11, can also globally regulate PAS usage (Li et al., 2015). One important distinction, however, appears to be that PCF11 regulates PAS usage without apparent sequence preferences (Li et al., 2015; Schäfer et al., 2018). However, in this study, by segregating genes according to size, we found that downstream UGUG motifs are important for short gene expression when PCF11 level is low. Downstream U/G-rich motifs are generally considered as binding sites for CstF-64 (Pérez Cañadillas and Varani, 2003). Whether there is a selective interplay between PCF11 and CstF-64 for CPA of short genes needs to be firmly established in the future.
We found that overexpression of PCF11 in IPAPcf11-KO cells leads to substantial APA changes with opposite directions to those in Pcf11 KD cells. This indicates that PCF11 can singularly regulate PAS usage, and its expression in the cell might be rate limiting. Consistent with this notion, Kamieniarz-Gdula et al. (2019) and colleagues recently found that PCF11 protein is sub-stoichiometric to other CPA factors. Therefore, PCF11 is well suited to be a master regulator of APA. Its role appears more prominent for IPA events, perhaps because of the fast kinetics of splicing sharpens CPA regulation in introns.
PEIPA versus U1 Telescripting
The PEIPA mechanism by PCF11 is reminiscent of the U1 telescripting scheme, by which U1 inhibits intronic PASs to ensure expression of long genes (Berg et al., 2012; Kaida et al., 2010; Oh et al., 2017). We previously showed that functional inhibition of U1 in C2C12 cells activated IPA sites with a 5′ to 3′ polarity (Li et al., 2015), suggesting that IPA sites closer to the TSS are better protected by U1 than downstream ones. Our analysis of IPAPcf11-KO cells indicates that PCF11 overexpression leads to conspicuous activation of IPA sites near the TSS (Figure 6I), suggesting that PCF11 is a potent antagonist of U1. Future studies using PCF11 overexpression at different levels could potentially gauge how different PASs are regulated by these two mechanisms. On the other hand, we found that the IPA site of Pcf11 can substantially be activated by U1 inhibition (Figure S7B), to a greater extent than the IPA site of Cstf3, which we previously identified (Luo et al., 2013). This indicates a crosstalk between these two mechanisms, potentially pegging PCF11 expression with U1 activity and providing a check on U1 telescripting.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bin Tian (btian@rutgers.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
NIH 3T3 cells were cultured in high glucose Dulbecco’s Modified Eagle Medium (DMEM) with 10% calf serum. C2C12 and 3T3-L1 cells were cultured in DMEM with 10% fetal bovine serum. C2C12 cell differentiation was induced by culturing cells in DMEM with 2% horse serum when cell confluency reached > 95%. 4T1-luc2-GFP cells were cultured in RPMI-1640 medium with 10% fetal bovine serum. All culture media were supplemented with 100 I.U./mL penicillin and 100 μg/mL streptomycin.
METHOD DETAILS
siRNA transfection
Transfection with siRNAs was carried out using Lipofectamine 3000 (Thermo Fisher Scientific) according to manufacturer’s recommendations. Transfection was carried out for 32 h. Two siRNAs were used for Pcf11: (A) 5′-CGACAGCUAUUUCAGUAU CAATT/5′-UUGAUACUGAAAUAGCUGUCGTT; (B): 5′-GGGCAAAGAUGAAGAUGUATT/5′-UACAUCUUCAUCUUUGCCCTT. Control siRNAs were 5′-UUCUCCGAACGUGUCACGUTT/5′-ACGUGACACGUUCGGAGAATT.
Pcf11 IPA site knockout by CRISPR/Cas9
To remove the IPA site of Pcf11 gene, two single guide RNAs (sgRNAs) were used, which targeted upstream and downstream regions of the IPA site. sgRNA sequences were designed using a web tool (http://crispr.mit.edu) considering on-target activity and potential off-target sites in the genome. DNA fragments containing sgRNA sequences were cloned into the PX459 plasmid. 4T1-luc2-GFP cells were transfected with the two sgRNA plasmids. Forty-eight hours after transfection, cells were subjected to selection in culture media containing 1.5 μg/ml Puromycin for three days. Live cells were seeded in a 96-well plate to ensure 0.5–1 cell/well. Cell colonies were selected for genomic DNA analysis by PCR. Sanger sequencing of PCR products was carried out to confirm the engineered site. The IPA site knockout clone was named IPAPcf11-KO.
Real-time quantitative PCR (qPCR)
Total RNA was extracted using TRIzol (Thermo Fisher) and treated with Turbo DNase (Thermo Fisher). cDNA was synthesized using oligo(dT) and M-MLV reverse transcriptase with 1.5–2 μg of RNA. cDNA was then mixed with real-time PCR primers and Luna qPCR master mix (NEB). PCR was run on an Applied Biosciences StepOne Plus Real-Time PCR system. Data were analyzed using the ΔΔCt method for gene expression.
Immunoblotting
Total protein was extracted from cells using RIPA buffer. Protein concentration was determined using DC Protein Assay Kit (Bio-Rad). A total of 10–20 μg of protein per sample was resolved by 10% SDS-PAGE, followed by transfer to a PVDF membrane with pore size 0.45 um (Millipore) for immunoblotting. Signals on the blot after adding the ECL reagent were detected by the ChemiDoc Touch Imaging System (Bio-Rad). Quantification was carried out using the Fiji program (Schindelin et al., 2012). Membranes were stripped with ReBlot Plus Strong Antibody Stripping Solution (Millipore) before re-blotting.
Isolation of newly made RNAs
Cells were cultured in medium supplemented with 50 μM of 4-thiouridine (4sU, Sigma) for 15 min before harvest. Total RNA was extracted using TRIzol. 4sU-labeled RNAs were isolated following the protocol described in (Rädle et al., 2013). Briefly, 25 μg of total RNA was biotinylated using biotin-HPDP (1 μg/μl in DMF, Thermo Fisher), and then extracted with chloroform three times and precipitated with ethanol. Biotinylated RNA was captured by Streptavidin C1 Dynabeads. The beads were washed six times before elution with DTT. Eluted RNA was precipitated with ethanol and then used for 3′READS.
3′READS library construction and sequencing
The 3′READS procedure was described in (Zheng et al., 2016). Briefly, Poly(A)+ RNA in 0.1–15 μg of total RNA was captured using 10 μl of oligo(dT)25 magnetic beads in 100 μl 1x binding buffer (10 mM Tris-Cl, pH7.5, 150 mM NaCl, 1 mM EDTA, and 0.05% TWEEN 20) and fragmented on the beads using 1.5 U of RNase III in 30 μl RNase III buffer (10 mM Tris-Cl pH8.3, 60 mM NaCl, 10 mM MgCl2, and 1 mM DTT) at 37°C for 15 min. After washing away unbound RNA fragments with binding buffer, poly(A)+ fragments were eluted from the beads with TE buffer (10 mM Tris-Cl, 1 mM EDTA, pH 7.5) and precipitated with ethanol, followed by ligation to 3 pmol of heat-denatured 5′ adaptor (5′-CCUUGGCACCCGAGAAUUCCANNNN) in the presence of 1 mM ATP, 0.1 μl of SuperaseIn, and 0.25 μl of T4 RNA ligase 1 in a 5 μl reaction at 22°C for 1 h. The ligation products were captured by 10 pmol of biotin-T15-(+TT)5 bound to 12 μl of Dynabeads MyOne Streptavidin C1. After washing with washing buffer (10 mM Tris-Cl pH7.5, 1 mM NaCl, 1 mM EDTA, and 0.05% TWEEN 20), RNA fragments on the beads were incubated with 0.01 U/μl of RNase H at 37°C for 30 min in 30 μl of RNase H buffer (50 mM Tris-Cl pH 7.5, 5 mM NaCl, 10 mM MgCl2, and 10 mM DTT). After washing with RNase H buffer, RNA fragments were eluted from the beads in elution buffer (1 mM NaCl, 1 mM EDTA, and 0.05% TWEEN 20) at 50°C, precipitated with ethanol, and then ligated to 3 pmol of heat-denatured 5′ adenylated 3′ adaptor (5′-rApp/NNNGATCGTCGGACTGTAGAACTCT GAAC/3ddC, where N is a random nucleotide and can be six Ns in some experiments) with 0.25 μl T4 RNA ligase 2 (truncated KQ version) at 22°C for 1 h in a 5 μl reaction containing 15% PEG 8000 and 0.2 μl of SuperaseIn. The ligation products were then precipitated and reverse transcribed using M-MLV reverse transcriptase, followed by PCR amplification using Phusion high-fidelity DNA polymerase and bar-coded PCR primers for 13–18 cycles. PCR products were size-selected twice with AMPure XP beads, using 0.6 volumes of beads (relative to the PCR reaction volume) to remove large DNA molecules and an additional 0.4 volume of beads to remove small DNA molecules. The eluted DNA was selected again with 1 volume of AMPure XP beads to further remove small DNA molecules. The size and quantity of the libraries were measured using a high sensitivity DNA kit on an Agilent Bioanalyzer. Libraries were sequenced on an Illumina NextSeq 500 (1 × 75 bases).
Cell proliferation assay
C2C12 cells in proliferating condition were treated with 0.25% Trypsin-EDTA and counted using Countess II FL Automated Cell Counter (Thermo Fisher). About 5 × 105 cells were stained with CellTrace CFSE (for siPcf11- or siCtrl-treated cells) at the final concentration of 5 μM in 1 mL PBS at 37°C for 20 min. Unabsorbed dye was discarded through cell centrifugation and washing in PBS. For KD experiment, siRNAs were transfected immediately after staining. After 40~48 hours, cells were analyzed in a BD FACScalibur system (BD Biosciences) with an excitation filter of 488 nm and emission filter of 530 nm for CellTrace CFSE.
Cell migration assay
Cells were cultured in growth medium (10% FBS) in a 6-well plate to reach > 95% confluency, followed by starvation in the medium containing 0.1% serum for another 20 h. After washing the cells twice with PBS, scratches were made at the center of the plate with a 200 μl pipette tip. Cells were washed again with PBS to get rid of debris. Cell migration was monitored live for 24 h in an on-stage incubator of EVOS FL Auto Cell Imaging System (Thermo Fisher). Images were quantified and relative migration rate was calculated using the Fiji program (Schindelin et al., 2012).
Transwell-based cell invasion assay
Cells were first cultured in growth medium (10% serum) to reach > 95% confluency, and then in starvation medium containing 0.1% serum for another 20 h. Cells were then trypsinized and placed in a transwell insert pre-coated with 1% Matrigel at 5 × 104 cells per well. Invasion assay was carried out for 92 h. Cells that went through the transwell insert were monitored by an EVOS FL Auto Cell Imaging System at ten random locations. Data were analyzed using the Fiji program (Schindelin et al., 2012).
Fluorescence-activated cell sorting (FACS) analysis
NIH 3T3 cells were transfected with pRiG or related reporter plasmids using Lipofectamine 3000. Cells were trypsinized, washed, and re-suspended in PBS 24 h after transfection. Green and red fluorescence signals for each cell were read at 530 nm and 585 nm, respectively, in a BD FACScalibur system (BD Biosciences). Non-transfected cells were used to determine background fluorescence. Log2(red fluorescence signal/green fluorescence signal) ratio was calculated for each cell.
Processing of 3′READS data
3′READS data were processed and analyzed as previously described (Zheng et al., 2016). Briefly, the sequence corresponding to 5′ adaptor was first removed from raw 3′READS reads using Cutadapt (Martin, 2011). Reads with short inserts (< 23 nt) were discarded. The remaining reads were then mapped to the mouse genome(mm9) using bowtie2 (local mode)(Langmead and Salzberg, 2012). The six random nucleotides at the 5′ end derived from the 3′ adaptor were removed before mapping using the setting “−5 6” in bowtie2. Reads with a mapping quality score (MAPQ) ≥ 10 were kept for further analysis. Reads with ≥ 2 non-genomic 5′Ts after alignment were called PAS reads. PASs within 24 nt from each other were clustered as previously described (Hoque et al., 2013). The PAS read counts mapped to genes were normalized by the median ratio method in DESeq (Anders and Huber, 2010).
Analysis of 3′READS data
For 3′UTR APA analysis, the two most abundant APA isoforms (based on PAS reads) with PASs in the 3′UTR of the last exon were selected. For IPA analysis, all IPA PAS reads were combined and compared to those in the last exon. Significant APA events were those with relative abundance change > 5% and p value < 0.05 (Fisher’s exact test) in at least one replicate and with consist regulation across all replicates. K-S (Kolmogorov–Smirnov) test was used to compare data distribution between samples. The aUTR size was the distance between proximal and dPASs in the 3′UTR. Relative Expression Difference (RED) was calculated as the difference in log2(ratio) of abundances of two PAS isoforms (dPAS v. pPAS) between two samples. For IPA analysis, all intronic PASs in a gene were combined. For gene expression analysis, PAS reads of each gene were summed. The expression fold changes were calculated using DEseq (Anders and Huber, 2010). Significance of difference was assessed by the negative binomial test in DEseq when there were replicates, or otherwise by the Fisher’s exact test. Genes with expression difference greater than 20% and with a p value < 0.05 were considered as signifcanly differentially expressed.
Analysis of RNA-seq data
Raw reads from RNA-seq were first trimmed for adaptor sequences by Cutadapt (Martin, 2011) and then mapped to the mouse genome (mm9) using STAR (v2.5.2) (Dobin et al., 2013) with default parameters. The read count of the coding sequence (CDS) of each gene was summed and then normalized by the median ratio method in DESeq (Anders and Huber, 2010). Only genes with more than five reads in a sample were used for further analysis. GTEx data were downloaded from dbGaP (dbGaP: phs000424.v7)(GTEx Consortium, 2017). A total of 5,032 RNA-seq samples were analyzed. Long genes were the top 10% of genes based on size using RefSeq information. Long gene expression index was based on the ratio of long gene expression (TPM) median to all gene expression median. In tissues-based analysis, only the tissues with more than 20 samples were used.
Gene ontology analysis
Gene ontology (GO) term annotations of genes, including those of human, mouse, chicken, and zebrafish, were obtained from the Gene Ontology database (Gene Ontology Consortium, 2015). Very detailed terms were removed by the program OWLTools (https://github.com/owlcollab/). The Fisher’s exact test was used to derive p values to indicate significance of association between a gene set and a GO term. GO terms associated with more than 1,000 genes were considered too generic and were discarded. To remove redundancy in reporting, each reported GO term was required to have at least 10% of genes that were not associated with another term with a more significant p value.
Gene feature analysis
Gene features were based on RefSeq annotations. RNA stability was based on the 4sU/total RNA ratio (after adjusting the U content in transcript), as we did previously (Zheng et al., 2018). Distance of each gene to its nearest neighbor was defined by RefSeq. Transcriptional orientation of each gene was based on RefSeq. A linear regression model based on the scikit-learn program (Pedregosa et al., 2011) was used to examine correlation between gene features and gene expression changes. The importance and contribution of each feature were assessed by its individual R2 and cumulative R2. The IPA frequency of a gene was obtained from PolyA_DB 3 (Wang et al., 2018), and was calculated as the number of IPA sites divided by the total number of PASs identified for the gene.
Intron feature and IPA analyses
Intron annotations were based on RefSeq database. The strengths of 5′ and 3′ splicing sites were calculated by the MaxEntScan program (Yeo and Burge, 2004). Intron locations were grouped according to relative genic locations as first (+1), second (+2), middle (‘M’, not other groups), the second to the last (−2), and last (−1). Only genes with at least five introns were used for this analysis. For IPA distribution maps, intron size, distance between TSS and IPA site, 5′SS and 3′SS strengths were evenly binned based on all introns with detectable IPA sites in 3′READS samples, and were put into a 10×10 table. The number of introns with regulated IPAs in each cell was normalized to the number of all detected IPA events in the cell.
PAS motif analysis
We defined the ± 100 nt genomic region surrounding each PAS as the PAS region. K-mer frequencies were calculated in three subregions, including −100 to −41 nt, −40 to −1 nt, +1 to +100 nt using Biostrings (https://rdrr.io/bioc/Biostrings/). P values for the comparison of PASs in upregulated versus downregulated genes were based on the Fisher’s exact test. The last 3′-most exon PAS and the most used PAS in the 3′-most exon were defined using the 3′READS data.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all RT-qPCR, WB, and migration, invasion and reporter assay, all grouped data are presented as mean ± s.d.. Student’s t test was used to determine statistical significance between groups, unless specified otherwise. Fisher’s exact test was used to determine the significant level of APA changes. Significance of gene expression difference was assessed by the negative binomial test in DEseq when there were replicates, or otherwise by the Fisher’s exact test. K-S test was used to compare the RED distributions and gene expression distributions in different gene sets. The Mann-Whitney-Wilcoxon test was used to compare expression changes among genes with different IPA frequencies or different features. When exact p values are not indicated, they are represented as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., p > 0.05.
DATA AND SOFTWARE AVAILABILITY
Software
All custom code and scripts used for processing of sequencing data and quantification analysis were written in Python, Perl, or R, and will be provided upon request to the Lead Contact.
Data Resources
Sequencing datasets generated in this study have been deposited into the GEO database under the accession number GEO: GSE115232.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-PCF11 | Santa Cruz Biotechnology | Cat# sc-514158 |
| Mouse anti-Myogenin | Santa Cruz Biotechnology | Cat# sc-12732; RRID: AB_627980 |
| Mouse anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-47724; RRID: AB_627678 |
| Mouse anti-β-tubulin | Developmental Studies Hybridoma Bank (DSHB) | Cat# E7; RRID: AB_528499 |
| m-IgGκ BP-HRP | Santa Cruz Biotechnology | Cat# sc-516102; RRID: AB_2687626 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phosphate buffered saline (PBS) | Lab made | N/A |
| Dulbecco’s Modified Eagle Medium (DMEM) | Lab made | N/A |
| RPMI-1640 | Lab made | N/A |
| Bovine Calf Serum (CS) | Sigma-Aldrich | Cat# 12133C-500ML |
| Fetal Bovine Serum (FBS) | Atlanta biologicals | Cat# S11550 |
| Horse serum | Sigma-Aldrich | Cat# H1138–500ML |
| Penicillin/Streptomycin | GIBCO | Cat# 15140–122 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat# L3000015 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| Oligo d(T)25 Magnetic Beads | NEB | Cat# S1419S |
| Adenosine 5′-Triphosphate (ATP) | NEB | Cat# P0756S |
| SUPERase● In RNase Inhibitor (20 U/μL) | Thermo Fisher Scientific | Cat# AM2694 |
| NEBNext RNase III RNA Fragmentation Module | NEB | Cat# E6146 |
| T4 RNA Ligase 1 | NEB | Cat# M0204S |
| T4 RNA Ligase 2, truncated KQ | NEB | Cat# M0373S |
| TURBO DNA-free Kit | Thermo Fisher Scientific | Cat# AM1907 |
| M-MLV Reverse transcriptase | Promega | Cat# M1705 |
| Phusion High-Fidelity DNA Polymerase | NEB | Cat# M0530S |
| Q5 High-Fidelity DNA Polymerase | NEB | Cat# M0491S |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher Scientific | Cat# 65001 |
| RNase H | Epicenter | Cat# R52250 |
| T4 Polynucleotide Kinase | NEB | Cat# M0201S |
| Shrimp Alkaline Phosphatase (rSAP) | NEB | Cat# M0371S |
| 5′ adaptor: 5′-CCUUGGCACCCGAGAAU UCCANNNN | Sigma-Aldrich | N/A |
| DNA/LNA oligo: biotin-TTTTTTTTTTTTTTT+ TT+TT+TT+TT+TT, where “+T” denotes LNA | Exiqon | N/A |
| 5′ adenylated 3′ blocked 3′ adaptor with degenerated nucleotides: 5′-rApp/NNNNGATCGTCGGACTGTA GAACTCTGAAC/3ddC | Bioo Scientific | N/A |
| Reverse transcription primer: 5′-GTTCAGA GTTCTACAGTCCGACGATC | Sigma-Aldrich | N/A |
| Reverse PCR primer (GX3): 5′-AATGATACGGCGACC ACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA | Sigma-Aldrich | N/A |
| Indexed forward PCR primers (index region in bracket): 5′-CAAGCAGAAGA CGGCATACGAGAT[NNNNNN] GTGACTGGAGTT CCTTGGCACCCGAGAATTCCA | Sigma-Aldrich | N/A |
| AMPure XP beads | Beckman Coulter | Cat# A63881 |
| Agilent RNA 6000 Pico Kit | Agilent Technologies | Cat# 5067–1513 |
| Critical Commercial Assays | ||
| High Sensitivity DNA analysis kit | Agilent Technologies | Cat# 5067–4626 |
| Luna Universal qPCR master mix | NEB | Cat# M3003 |
| NEBNext Magnesium RNA Fragmentation Module | NEB | Cat# E6150S |
| CellTrace CFSE Cell Proliferation Kit, for flow cytometry | Thermo Fisher Scientific | Cat# C34554 |
| Deposited Data | ||
| 3′READS data | This study | GEO: GSE115232 |
| RNA-seq data | This study | GEO: GSE115232 |
| Experimental Models: Cell Lines | ||
| Mouse: NIH 3T3 cells | ATCC | Cat# CRL-1658; RRID: CVCL_0594 |
| Mouse: C2C12 cells | ATCC | Cat# CRL-1772; RRID: CVCL_0188 |
| Mouse: 3T3-L1 cells | ATCC | Cat# CL-173; RRID: CVCL_0123 |
| Mouse: 4T1-luc2-GFP | Perkin Elmer | Cat# 128090; RRID: CVCL_5J28 |
| Mouse: 4T1-IPAPcf11-KO | Generated in this study | N/A |
| Recombinant DNA | ||
| Plasmid: PX459 | Addgene | Cat# 62988; RRID: Addgene_62988 |
| Plasmid: PX459-mPcf11-IPA-g1 | Cloned in this study | N/A |
| Plasmid: PX459-mPcf11-IPA-g2 | Cloned in this study | N/A |
| Plasmid: pRiG | Cloned and stored in lab | N/A |
| Plasmid: pRiG-AD | Cloned and stored in lab | N/A |
| Plasmid: pRiG-Pcf11 Ctrl-seq | Cloned in this study | N/A |
| Plasmid: pRiG- Pcf11 IPA site | Cloned in this study | N/A |
| Plasmid: pRiG- Pcf11 pPAS | Cloned in this study | N/A |
| Plasmid: pRiG- Pcf11 dPAS | Cloned in this study | N/A |
| Oligonucleotides (See Tables S2 and S3) | ||
| Software and Algorithms | ||
| FIJI | Schindelin et al., 2012 | https://imagej.net/Fiji |
| Primer-BLAST | Ye et al., 2012 | https://www.ncbi.nlm.nih.gov/tools/primer-blast/ |
| Prism version 7.0a for Mac | GraphPad Software | N/A |
| Cutadapt | Martin, 2011 | http://cutadapt.readthedocs.io/en/stable/guide.html |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| DESeq & DEXSeq | Anders and Huber, 2010 | https://bioconductor.org/packages/devel/bioc/html/DESeq.html |
| STAR (v2.5.2) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Scikit-learn | Pedregosa et al., 2011 | http://scikit-learn.org/stable/index.html |
| MaxEntScan | Yeo and Burge, 2004 | http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html |
| Other data sets | ||
| KD of core CPA and splicing factors (3′READS) | Li et al., 2015 | GEO: GSE11415 |
| Transcriptional profiling of longitudinal changes during neurogenesis (RNA-seq) | Hubbard et al., 2013 | SRA: SRP017778 |
| Genotype-Tissue Expression Project (GTEx) | GTEx Consortium, 2017 | dbGaP: phs000424.v7 |
| Early stages of C2C12 differentiation (RNA-seq) | Hamed et al., 2017 | GEO: GSE94560 |
| Pan-readthrough genes induced by stress (gene set) | Vilborg et al., 2017 | GEO: GSE98906 |
Highlights.
PCF11 regulates mRNA expression on the basis of gene size
Genes with large introns are regulated by PCF11 through intronic polyadenylation
PCF11 controls its own gene expression through an intronic polyadenylation site
Long gene and PCF11 expression levels are inversely correlated globally
ACKNOWLEDGMENTS
This work was funded by NIH grants R01GM084089, R01GM129069, and P30CA072720 to B.T. We thank Nick Proudfoot, Kinga Kamieniarz-Gdula, and members of the B.T. lab for helpful discussions and Bei You and Weiting Xu for technical assistance at early stages of this work.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes three tables and seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.049.
REFERENCES
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, and Dreyfuss G (2012). U1 snRNP determines mRNA length and regulates isoform expression. Cell 150, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2015). Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, Macisaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, and Babak T (2012). A quantitative atlas of polyadenylation in five mammals. Genome Res. 22, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Li W, Ogami K, Yashinskie JJ, Hoque M, Tian B, and Manley JL (2014). RBBP6 isoforms regulate the human polyadenylation machinery and modulate expression of mRNAs with AU-rich 3′ UTRs. Genes Dev. 28, 2248–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guéguéniat J, Dupin AF, Stojko J, Beaurepaire L, Cianférani S, Mackereth CD, Minvielle-Sébastia L, and Fribourg S (2017). Distinct roles of Pcf11 zinc-binding domains in pre-mRNA 3′-end processing. Nucleic Acids Res. 45, 10115–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamed M, Khilji S, Dixon K, Blais A, Ioshikhes I, Chen J, and Li Q (2017). Insights into interplay between rexinoid signaling and myogenic regulatory factor-associated chromatin state in myogenic differentiation. Nucleic Acids Res. 45, 11236–11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hollerer I, Grund K, Hentze MW, and Kulozik AE (2014). mRNA 3’end processing: a tale of the tail reaches the clinic. EMBO Mol. Med 6, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, and Tian B (2013). Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 10, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornshøj H, Nielsen MM, Sinnott-Armstrong NA, Świtnicki MP, Juul M, Madsen T, Sallari R, Kellis M, Ørntoft T, Hobolth A, and Pedersen JS (2018). Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genom. Med 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Lutz CS, Wilusz J, and Tian B (2005). Bioinformatic identification of candidate cis-regulatory elements involved in human mRNA polyadenylation. RNA 11, 1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard KS, Gut IM, Lyman ME, and McNutt PM (2013). Longitudinal RNA sequencing of the deep transcriptome during neurogenesis of cortical glutamatergic neurons from murine ESCs. F1000Res. 2, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, and Tian B (2009). Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. U S A 106, 7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Cubberley G, and Bentley DL (2009). Cotranscriptional recruitment of the mRNA export factor Yra1 by direct interaction with the 3′ end processing factor Pcf11. Mol. Cell 33, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, and Dreyfuss G (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamieniarz-Gdula K, Gdula MR, Panser K, Nojima T, Monks J, Wiśniewski JR, Riepsaame J, Brockdorff N, Pauli A, and Proudfoot NJ (2019). Selective roles of vertebrate PCF11 in premature and full-length transcript termination. Mol. Cell, Published online February 25, 2019. 10.1016/j.molcel.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehner JN, Pearson EL, and Moore C (2011). Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Biol 12, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi EA, Xie X, Wan J, Xing Y, Freudenberg JM, et al. (2014). Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J. 33, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle M, Hunyadkürti J, and Bachand F (2017). Polyadenylation site selection: linking transcription and RNA processing via a conserved carboxy-terminal domain (CTD)-interacting protein. Curr. Genet 63, 195–199. [DOI] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, and Tian B (2015). Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 11, e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Freitas J, Zheng D, Oliveira MS, Hoque M, Martins T, Henriques T, Tian B, and Moreira A (2017a). Transcription elongation rate has a tissue-specific impact on alternative cleavage and polyadenylation in Drosophila melanogaster. RNA 23, 1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hoque M, Larochelle M, Lemay JF, Yurko N, Manley JL, Bachand F, and Tian B (2017b). Comparative analysis of alternative polyadenylation in S. cerevisiae and S. pombe. Genome Res. 27, 1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Ji Z, Pan Z, You B, Hoque M, Li W, Gunderson SI, and Tian B (2013). The conserved intronic cleavage and polyadenylation site of CstF-77 gene imparts control of 3′ end processing activity through feedback autoregulation and by U1 snRNP. PLoS Genet. 9, e1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel CR, Bai Y, and Tong L (2008). Protein factors in pre-mRNA 3′-end processing. Cell. Mol. Life Sci. 65, 1099–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. [Google Scholar]
- Martin G, Gruber AR, Keller W, and Zavolan M (2012). Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep. 1, 753–763. [DOI] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, and Wagner EJ (2014). CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, and Bartel DP (2009). Widespread shortening of 3’UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhart A, and Cramer P (2004). Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 430, 223–226. [DOI] [PubMed] [Google Scholar]
- Ogorodnikov A, Levin M, Tattikota S, Tokalov S, Hoque M, Scherzinger D, Marini F, Poetsch A, Binder H, Macher-Goeppinger S, et al. (2018). Transcriptome 3ʹend organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun 9, 5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JM, Di C, Venters CC, Guo J, Arai C, So BR, Pinto AM, Zhang Z, Wan L, Younis I, and Dreyfuss G (2017). U1 snRNP telescripting regulates a size-function-stratified human genome. Nat. Struct. Mol. Biol 24, 993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, and Dubourg V (2011). Scikit-learn: machine learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- Pérez Cañadillas JM, and Varani G (2003). Recognition of GU-rich polyadenylation regulatory elements by human CstF-64 protein. EMBO J. 22, 2821–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ (2016). Transcriptional termination in mammals: stopping the RNA polymerase II juggernaut. Science 352, aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rädle B, Rutkowski AJ, Ruzsics Z, Friedel CC, Koszinowski UH, and Dölken L (2013). Metabolic labeling of newly transcribed RNA for high resolution gene expression profiling of RNA synthesis, processing and decay in cell culture. J. Vis. Exp (78) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, and Burge CB (2008). Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer P, Tüting C, Schönemann L, Kühn U, Treiber T, Treiber N, Ihling C, Graber A, Keller W, Meister G, et al. (2018). Reconstitution of mammalian cleavage factor II involved in 3′ processing of mRNA precursors. RNA 24, 1721–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, and Shi Y (2011). Complex and dynamic landscape of RNA polyadenylation revealed by PAS-seq. RNA 17, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Manley JL (2015). The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 29, 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, and Manley JL (2009). Molecular architecture of the human pre-mRNA 3′ processing complex. Mol. Cell 33, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh I, Lee SH, Sperling AS, Samur MK, Tai YT, Fulciniti M, Munshi NC, Mayr C, and Leslie CS (2018). Widespread intronic polyadenylation diversifies immune cell transcriptomes. Nat. Commun 9, 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, and Graber JH (2012). Signals for pre-mRNA cleavage and polyadenylation. Wiley Interdiscip. Rev. RNA 3, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, and Manley JL (2017). Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol 18, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Pan Z, and Lee JY (2007). Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 17, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Sabath N, Wiesel Y, Nathans J, Levy-Adam F, Yario TA, Steitz JA, and Shalgi R (2017). Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. Proc. Natl. Acad. Sci. U S A 114, E8362–E8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volanakis A, Kamieniarz-Gdula K, Schlackow M, and Proudfoot NJ (2017). WNK1 kinase and the termination factor PCF11 connect nuclear mRNA export with transcription. Genes Dev. 31, 2175–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, and Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Nambiar R, Zheng D, and Tian B (2018). PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Res. 46 (D1), D315–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Sharp PA (2013). Divergent transcription: a driving force for new gene origination? Cell 155, 990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Hsu P, Lee SD, Yang W, Hoskinson D, Xu W, Moore C, and Varani G (2017). The C terminus of Pcf11 forms a novel zinc-finger structure that plays an essential role in mRNA 3′-end processing. RNA 23, 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, and Shi Y (2012). Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc. Natl. Acad. Sci. U S A 109, 18773–18778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, and Madden TL (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Gilmour DS (2006). Pcf11 is a termination factor in Drosophila that dismantles the elongation complex by bridging the CTD of RNA polymerase II to the nascent transcript. Mol. Cell 21, 65–74. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lee JY, and Tian B (2005). Biased alternative polyadenylation in human tissues. Genome Biol. 6, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, and Manley JL (1996). Complex alternative RNA processing generates an unexpected diversity of poly(A) polymerase isoforms. Mol. Cell. Biol 16, 2378–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Liu X, and Tian B (2016). 3′READS+, a sensitive and accurate method for 3′ end sequencing of polyadenylated RNA. RNA 22, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Wang R, Ding Q, Wang T, Xie B, Wei L, Zhong Z, and Tian B (2018). Cellular stress alters 3’UTR landscape through alternative polyadenylation and isoform-specific degradation. Nat. Commun 9, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Software
All custom code and scripts used for processing of sequencing data and quantification analysis were written in Python, Perl, or R, and will be provided upon request to the Lead Contact.
Data Resources
Sequencing datasets generated in this study have been deposited into the GEO database under the accession number GEO: GSE115232.