

Abstract
Liability to alcohol dependence (AD) is heritable, but little is known about its complex polygenic architecture or its genetic relationship with other disorders. To discover loci associated with AD and characterize the relationship between AD and other psychiatric and behavioral outcomes, we carried out the largest GWAS to date of DSM-IV diagnosed AD. Genome-wide data on 14,904 individuals with AD and 37,944 controls from 28 case/control and family-based studies were meta-analyzed, stratified by genetic ancestry (European, N = 46,568; African; N = 6,280). Independent, genome-wide significant effects of different ADH1B variants were identified in European (rs1229984; p = 9.8E-13) and African ancestries (rs2066702; p = 2.2E-9). Significant genetic correlations were observed with 17 phenotypes, including schizophrenia, ADHD, depression, and use of cigarettes and cannabis. The genetic underpinnings of AD only partially overlap with those for alcohol consumption, underscoring the genetic distinction between pathological and non-pathological drinking behaviors.
Keywords: Genome-wide association study, alcoholism, psychiatric disorders, alcohol use, pleiotropy
INTRODUCTION
Excessive alcohol use is a leading contributor to morbidity and mortality. One in 20 deaths worldwide is attributable to alcohol consumption, as is 5.1% of the global burden of disease1. Alcohol dependence (AD), as defined by the Fourth Edition of the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-IV)2, is a serious psychiatric disorder characterized by tolerance, withdrawal, loss of control over drinking and excessive alcohol consumption despite negative health and social consequences. Among alcohol drinkers, 12% meet criteria for DSM-IV AD during their lifetimes3. In the United States, only 25% of those with AD ever receive treatment4.
AD is moderately heritable (49% by a recent meta-analysis)5 and numerous genome-wide association studies (GWAS) have aimed to identify loci contributing to this genetic variance (see6 for a review). According to one study, common SNPs are responsible for as much as 30% of the variance in AD7, but few have been identified to date. Variants in the genes responsible for alcohol metabolism, especially ADH1B and ALDH2, have been strongly implicated8–13. The association between AD (and related drinking phenotypes) and rs1229984, a missense SNP (Arg48His) in ADH1B that affects the conversion of alcohol to acetaldehyde, represents one of the largest common-variant effect sizes observed in psychiatry, with the His48 allele accelerating ethanol metabolism and affording approximately 3-fold reduction in likelihood of AD across numerous studies8,10. Another functional polymorphism, rs671 in ALDH2 (Glu504Lys), strongly affects alcohol metabolism by blocking conversion of acetaldehyde to acetate and has an even stronger effect on risk for AD, but is rare except in some Asian populations8,12,13 ADH1B and ALDH2 polymorphisms, however, only explain a small proportion of the heritable variation in AD in populations of European or African ancestry.
In this study, the Substance Use Disorders working group of the Psychiatric Genomics Consortium (PGC-SUD14) compiled the largest numbers of carefully diagnosed alcohol dependent individuals and alcohol-exposed controls to date, from both case-control and family studies. These included substantial numbers of both European ancestry (EU, N = 46,568, including 38,686 unrelated individuals) and admixed African-American ancestry (AA, N = 6,280, including 5,799 unrelated individuals) subjects. AD diagnoses were derived from clinician ratings or semi-structured interviews following DSM-IV2 criteria. Each study was subjected to stringent quality control (QC) before conducting GWAS within each population of each study, followed by a genome-wide meta-analysis. We estimated the SNP-heritability (h2g) of AD and examine the extent to which aggregate genetic variation in AD is related to traits from 45 other GWAS, including continuous measures of alcohol consumption. We also examined whether polygenic risk scores (PRS) derived from these analyses predicted alcohol dependence and related measures of problem drinking in three independent samples.
RESULTS
GWAS meta-analyses:
The trans-ancestral discovery meta-analysis of GWAS of AD in 28 cohorts (Table 1; Supplementary Table S1) identified a genome-wide significant (GWS; p < 5E-8) association in the ADH gene cluster on chromosome 4 (Figure 1; Table 2). Examining this locus in each population (Figure 2), rs1229984 in ADH1B was the strongest associated variant from the analysis in EU (z = −7.13, p = 9.8E-13), while rs2066702, also in ADH1B, was the most significant variant in AA (z = −5.98, p = 2.2E-9). Trans-ancestral modelling reinforced the robust effects of rs1229984 and other ADH1B SNPs on liability to AD across inverse-variance weighted, random effects, and Bayesian models (Supplementary Figure S1, Supplementary Table S2).
Table 1:
Descriptive statistics for cohorts in the meta-analysis of AD.
| European (EU) | African - American (AA) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Male (%) | Ages (years) | N Total | N Unrelated | N Total | N Unrelated | ||||||
| Dataset | PMID | Case | Control | Case | Control | Case | Control | Case | Control | ||
| Case-control: Logistic Regression | |||||||||||
| Comorbidity and Trauma Study (CATS) | 23303482 | 56% | 18-67 | 572 | 817 | 572 | 817 | -- | -- | -- | -- |
| Christchurch Health and Development Study (CHDS) | 23255320 | 48% | 16-30 | 112 | 500 | 112 | 500 | -- | -- | -- | -- |
| Collaborative Study of the Genetics of Alcoholism - case-control cohort (COGA-cc) | 20201924 | 54% | 18-79 | 583 | 363 | 583 | 363 | -- | -- | -- | -- |
| Family Study of Cocaine Dependence (FSCD) | 18243582 | 51% | 18-60 | 266 | 174 | 266 | 174 | 255 | 241 | 255 | 241 |
| German Study of the Genetics of Alcoholism (GESGA) | 19581569 | 65% | 18-84 | 1314 | 2142 | 1314 | 2142 | -- | -- | -- | -- |
| Gene-Environment Development Initiative - Great Smoky Mountains Study (GEDI-GSMS) | 8956679 | 57% | 9-26 | 42 | 565 | 42 | 565 | -- | -- | -- | -- |
| Center on Antisocial Drug Dependence (CADD) | 25637581 | 70% | 13-20 | 400 | 577 | 400 | 577 | 51 | 51 | 51 | 51 |
| Phenomics and Genomics Sample (PAGES) | 28371232 | 57% | 18-74 | 37 | 523 | 37 | 523 | -- | -- | -- | -- |
| Collaborative Study on the Genetics of Nicotine Dependence (COGEND Nico) | 17158188 | 34% | 25-82 | 135 | 272 | 135 | 272 | 46 | 232 | 46 | 232 |
| COGEND - Study of Addiction: Genetics and Environment (COGEND SAGE) | 20202923 | 37% | 18-77 | 311 | 225 | 311 | 225 | 104 | 103 | 104 | 103 |
| Spit For Science | 24639683 | 36% | >18 | 252 | 1863 | 252 | 1863 | 74 | 841 | 74 | 841 |
| National Institute on Alcohol Abuse and Alcoholism Intramural (NIAAA) | n/a | 67% | >18 | 442 | 206 | 442 | 206 | 404 | 110 | 404 | 110 |
| Mayo Clinic Center for the Individual Treatment of Alcohol Dependence (CITA) | 25290263 | 55% | ≥18 | 378 | 646 | 378 | 646 | -- | -- | -- | -- |
| Alcohol Dependence in African Americans (ADAA) | n/a | 57% | 18-69 | -- | -- | -- | -- | 794 | 297 | 794 | 297 |
| Family-based, twins and sibs: Generalized Estimating Equations (GEE) | |||||||||||
| Brisbane Longitudinal Twin Study (BLTS) | 23187020 | 43% | 18-30 | 60 | 938 | 51 | 546 | -- | -- | -- | -- |
| GEDI - Virginia Twin Study on Adolescent Behavioral Development (GEDI-VTSABD) | 9294370 | 38% | 8-32 | 209 | 503 | 188 | 318 | -- | -- | -- | -- |
| Minnesota Center for Twin and Family Research (MCTFR) | 23942779 | 41% | 16-21 | 609 | 2100 | 553 | 1274 | -- | -- | -- | -- |
| Center for Education and Drug Abuse Research (CEDAR) | 21514569 | 63% | 16-34 | 59 | 200 | 54 | 152 | -- | -- | -- | -- |
| Swedish Twin Registry (STR) | 23137839 | 47% | 40-83 | 76 | 8311 | 76 | 6112 | -- | -- | -- | -- |
| Yale-Penn | 24166409 | 58% | 16-79 | 1094 | 301 | 1004 | 252 | -- | -- | -- | -- |
| Family-based, large/complex pedigrees: Logistic Mixed Model | |||||||||||
| Collaborative Study of the Genetics of Alcoholism - family cohort (COGA-fam) | 23089632 | 45% | 12-88 | 605 | 682 | 168 | 138 | -- | -- | -- | -- |
| Australian Alcohol and Nicotine Studies (OZ-ALC-NAG) | 21529783 | 45% | 18-82 | 1571 | 3069 | 1111 | 805 | -- | -- | -- | -- |
| Irish Affected Sib Pair Study of Alcohol Dependence (IASPSAD) | 15770118 | 50% | 17-84 | 721 | 1814 | 436 | 1802 | -- | -- | -- | -- |
| Yale-Penn | 24166409 | 51% | 16-79 | -- | -- | -- | -- | 1607 | 1070 | 1263 | 933 |
| Summary statistics | |||||||||||
| Netherlands Study of Depression and Anxiety / Netherlands Twin Registry (NESDA/NTR) | 18197199 | 31% | >18 | 390 | 1633 | 390 | 1633 | -- | -- | -- | -- |
| Finnish Nicotine Addiction Genetics Project (NAG-Fin) | 17436240 | 52% | 30-92 | 439 | 1137 | 439 | 1137 | -- | -- | -- | -- |
| FinnTwin12 (FT12) | 17254406 | 47% | 20-27 | 88 | 874 | 88 | 874 | -- | -- | -- | -- |
| National Longitudinal Study of Adolescent to Adult Health (Add Health) | 25378290 | 47% | 24-34 | 768 | 2981 | 768 | 2981 | -- | -- | -- | -- |
| Helsinki Birth Cohort Study (HBCS) | 16251536 | 43% | 56-70 | 36 | 1583 | 36 | 1583 | -- | -- | -- | -- |
| Total | 11569 | 34999 | 10206 | 28480 | 3335 | 2945 | 2991 | 2808 | |||
Overview of numbers of alcohol dependent cases and controls from each cohort in the current analysis, including the number of genetically unrelated individuals. Cohorts are listed by study design and analysis method. Sample sizes are listed after QC exclusions and stratified by ancestry group. PubMed identifiers (PMID) are listed for previous publications describing each cohort, along with the percentage of male samples and the age range in the cohort.
Figure 1: Manhattan plot of discovery trans-ancestral meta-analysis showing strong evidence for rs1229984 in ADH1B.
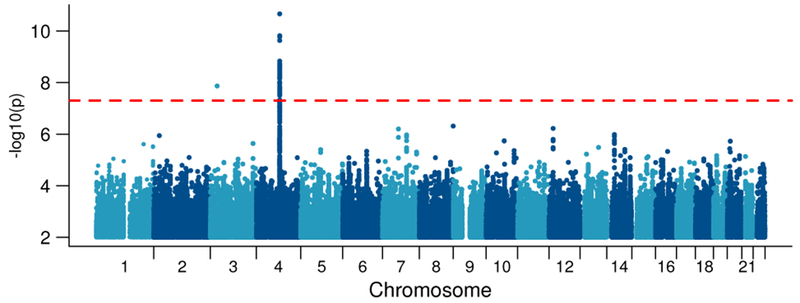
Results from the discovery meta-analysis of all cohorts (Ncase=14,904, Ncontrol=37,944) for association of genome-wide SNPs with AD under a fixed effects meta-analysis weighted by effective sample size. Dashed red reference line indicates genome-wide significance after correction for multiple testing (p < 5E-8).
Table 2:
Top 10 loci from the meta-analyses of alcohol dependence by ancestry
| A1 Allele Freq. | INFO score | Effect size (OR) | Discovery meta-analysis p-value | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | CHR | BP | A1 | A2 | Gene | EU | AA | EU | AA | EU | AA | EU | AA | Trans |
|
Top clumped variants in
trans-ancestral meta-analysis (14,904 cases, 37,944
controls) | ||||||||||||||
| rs7644567* | 3 | 29201672 | A | G | RBMS3 | 0.964 | 0.705 | 0.96 | 1.00 | -- | 1.229 | 3.94E-04 | 6.64E-06 | 1.36E-08 |
| rs2066702 | 4 | 100229017 | A | G | ADH1B | -- | 0.215 | -- | 0.99 | -- | 0.731 | -- | 2.21E-09 | 2.21E-09 |
| rs1229984 | 4 | 100239319 | T | C | ADH1B | 0.040 | 0.014 | 0.90 | 0.91 | 0.486 | 0.912 | 9.79E-13 | 3.48E-01 | 2.18E-11 |
| rs1789912 | 4 | 100263942 | T | C | ADH1C | 0.418 | 0.132 | 1.00 | 1.02 | 1.106 | 1.211 | 1.98E-07 | 1.32E-03 | 1.47E-09 |
| rs6827898 | 4 | 100295863 | A | G | (ADH region) | 0.123 | 0.112 | 0.96 | 0.94 | 1.145 | 1.270 | 5.21E-07 | 9.31E-04 | 2.97E-09 |
| rs894368 | 4 | 100309313 | A | C | (ADH region) | 0.309 | 0.386 | 0.99 | 0.96 | 0.887 | 0.981 | 1.93E-08 | 9.73E-01 | 3.30E-07 |
| rs2461618 | 7 | 68667233 | A | G | -- | -- | 0.088 | -- | 0.98 | -- | 0.669 | -- | 6.30E-07 | 6.30E-07 |
| rs116338421 | 8 | 145761256 | C | G | ARHGAP39 | -- | 0.172 | -- | 0.97 | -- | 0.755 | -- | 4.86E-07 | 4.86E-07 |
| rs79171978 | 12 | 17798824 | C | G | -- | 0.099 | 0.027 | 0.99 | 0.99 | 1.201 | 1.016 | 5.47E-08 | 8.18E-01 | 5.98E-07 |
| rs8017647 | 14 | 32456358 | T | C | -- | 0.792 | 0.565 | 1.00 | 0.99 | 0.901 | 0.923 | 8.05E-06 | 4.71E-02 | 1.03E-06 |
|
Top clumped variants in
African ancestry meta-analysis (3,335 cases, 2,945
controls) | ||||||||||||||
| rs5781337 | 1 | 223883425 | CA | C | -- | 0.263 | 0.212 | 0.98 | 0.93 | 1.007 | 0.664 | 8.85E-01 | 1.62E-07 | 6.59E-02 |
| rs143258048 | 3 | 75982870 | A | AC | ROBO2 | -- | 0.028 | -- | 0.88 | -- | 0.490 | -- | 1.86E-06 | -- |
| rs3857224 | 4 | 100129685 | T | C | ADH6 | 0.315 | 0.585 | 0.99 | 1.00 | 0.970 | 0.814 | 2.40E-01 | 5.86E-07 | 2.36E-03 |
| rs2066702 | 4 | 100229017 | A | G | ADH1B | -- | 0.215 | -- | 0.99 | -- | 0.731 | -- | 2.21E-09 | 2.21E-09 |
| rs2461618 | 7 | 68667233 | A | G | -- | -- | 0.088 | -- | 0.98 | -- | 0.669 | -- | 6.30E-07 | 6.30E-07 |
| rs116338421 | 8 | 145761256 | C | G | ARHGAP39 | -- | 0.172 | -- | 0.97 | -- | 0.755 | -- | 4.86E-07 | 4.86E-07 |
| rs79016499 | 11 | 93010988 | T | C | -- | -- | 0.066 | -- | 0.93 | -- | 1.729 | -- | 1.36E-06 | -- |
| rs10784244 | 12 | 62035165 | G | A | -- | 0.153 | 0.484 | 1.00 | 1.00 | 1.041 | 1.226 | 6.26E-02 | 1.04E-06 | 2.49E-04 |
| rs17199739 | 16 | 25444288 | G | A | -- | 0.176 | 0.096 | 0.99 | 0.96 | 0.994 | 0.693 | 4.25E-01 | 1.11E-06 | 8.66E-03 |
| rs740793 | 17 | 3846353 | G | A | ATP2A3 | 0.453 | 0.350 | 0.97 | 0.97 | 0.996 | 1.370 | 4.66E-01 | 1.48E-06 | 3.44E-01 |
|
Top clumped variants in
European ancestry meta-analysis (11,569 cases, 34,999
controls) | ||||||||||||||
| rs1229984 | 4 | 100239319 | T | C | ADH1B | 0.040 | 0.014 | 0.90 | 0.91 | 0.486 | 0.912 | 9.79E-13 | 3.48E-01 | 2.18E-11 |
| rs3811802 | 4 | 100244221 | G | A | ADH1B | 0.454 | 0.529 | 0.96 | 0.96 | 1.162 | 0.914 | 2.40E-08 | 2.19E-02 | 1.22E-04 |
| rs113659074 | 4 | 100252308 | T | G | ADH1B | 0.068 | 0.093 | 0.98 | 0.95 | 0.800 | 1.166 | 1.54E-06 | 6.63E-02 | 2.99E-04 |
| rs1229863 | 4 | 100252386 | A | T | ADH1B | 0.174 | 0.038 | 0.99 | 0.99 | 1.145 | 1.254 | 7.80E-07 | 4.26E-02 | 9.28E-08 |
| rs1154445 | 4 | 100288521 | G | T | (ADH region) | 0.425 | 0.134 | 0.97 | 0.99 | 1.137 | 1.211 | 1.80E-07 | 2.63E-02 | 1.48E-08 |
| rs6827898 | 4 | 100295863 | A | G | (ADH region) | 0.123 | 0.112 | 0.96 | 0.94 | 1.145 | 1.270 | 5.21E-07 | 9.31E-04 | 2.97E-09 |
| rs894368 | 4 | 100309313 | A | C | (ADH region) | 0.309 | 0.386 | 0.99 | 0.96 | 0.887 | 0.981 | 1.93E-08 | 9.73E-01 | 3.30E-07 |
| rs79171978 | 12 | 17798824 | C | G | -- | 0.099 | 0.027 | 0.99 | 0.99 | 1.201 | 1.016 | 5.47E-08 | 8.18E-01 | 5.98E-07 |
| rs4388946 | 12 | 17935154 | C | A | -- | 0.240 | 0.297 | 0.99 | 0.98 | 1.137 | 0.950 | 7.14E-07 | 1.87E-01 | 7.05E-05 |
| rs34929220 | 15 | 69769635 | T | C | DRAIC | 0.690 | 0.937 | 0.90 | 0.94 | 0.893 | 1.028 | 1.02E-06 | 8.38E-01 | 7.38E-06 |
Top 10 nominally independent variants from the discovery trans-ancestral (Trans.; Ncase=14,904, Ncontrol=37,944) meta-analysis and the discovery meta-analyses in African (AA; Ncase=3,335, Ncontrol=2,945) and European (EU; Ncase=11,569, Ncontrol=34,999) ancestry cohorts, respectively. Independent variants are identified based on clumping for LD (pairwise r2 < 0.1) in 1000 Genomes Project Phase 3 data21. EU results are clumped using European (EUR) ancestry reference samples, AA results are clumped using African ancestry reference samples from the American Southwest (ASW), and trans-ancestral results are clumped using merged EUR and African ancestry (AFR) reference samples. P-values and allele frequencies (Freq.) are reported from two-tailed tests of association with AD in fixed effects meta-analyses weighted by effective sample size. Bold p-values indicate genome-wide significance after correction for multiple testing within the analysis (p < 5E-8). Odds ratios (OR) and INFO scores are reported from the meta-analyses of the subset of unrelated individuals within each ancestry. Variants are sorted by chromosome (CHR) and base pair (BP) position for genome build hg19, with genes annotated by Ensembl VEP49. Allele frequency and OR are given with respect to allele 1 (A1).
SNPs included in the trans-ancestral meta-analysis were not conditioned on being analyzed in both the EU and AA analyses. For instance, a SNP of strong effect in one group may not be sufficiently common or well-imputed for analysis in the other ancestral group (e.g., rs2066702 is not found in non-African populations but is among the top 10 in the trans-ancestral analysis due to strong effects in the AA group). For rs7644567 (denoted with *), the SNP does not passed QC in a sufficient number of cohorts to meet the minimum sample size requirement for inclusion in the EU-only analyses – it is only represented among EU cohorts by summary statistics from two Finnish cohorts – but allele frequency, INFO score, and meta-analyzed p-values from the Finnish summary statistics are reported since they contribute to the trans-ancestral meta-analysis.
Figure 2: Regional plots for the ADH1B locus (rs1229984) in the trans-ancestral discovery, African-American (AA), and European (EU), meta-analyses.

Results of fixed effects meta-analysis with effective sample size weights for the ADH1B locus in (A) all cohorts (Ncase=14,904, Ncontrol=37,944); (B) AA cohorts (Ncase=3,335, Ncontrol=2,945); and (C) EU cohorts (Ncase=11,569, Ncontrol=34,999). Red reference line indicates the genome-wide significance threshold after correction for multiple testing within each analysis (p < 5E-8). Within ancestry, colored points reflect the degree of LD (pairwise r2) to the index variant (indicated by a purple diamond) in 1000 Genomes Project reference data21 for individuals of (B) African or (C) European ancestry, respectively. LD structures in the two ancestries differ, so for the trans-ancestral sample (A) LD is not given, indicted by gray points. Two-tailed tests used for all analyses.
Clumping the ADH locus for linkage disequilibrium (LD; r2 < 0.1 within 500 kb) suggested multiple independent signals in both populations, with the differing leading alleles reflecting different LD structures and allele frequencies in each population (Table 2, Supplementary Figure S2). Conditional analyses controlling for rs1229984 and rs2066702 had limited power, but results showed limited attenuation of effect sizes between marginal and conditional analyses, consistent with the existence of additional independent effects in the region (Supplementary Table S3; Supplementary Figure S3). Suggestive independent signals in the genotyped cohorts included trialleleic variant rs894368 (marginal z = −4.57, p = 4.9E-6; conditional z = −4.53, p = 5.8E-6) and insertion rs112346244 (marginal odds ratio = 0.912, SE = .024, z = −3.81, p = 1.4E-4; conditional odds ratio = 0.883, SE = .025, z = −5.05, p = 4.5E-7; Supplementary Table S3). Several additional variants that were prioritized in the conditional analysis, while not significant, were in moderate to strong LD with rs698 (marginal odds ratio = 1.115, SE = .021, z = 5.19, p = 2.1E-7; conditional odds ratio = 1.084, SE = .021, z = 3.78, p = 1.6E-4), a functional ADH1C variant with a role in AD8,11.
A single novel SNP on chromosome 3, rs7644567, also reached GWS in the meta-analysis (z = 5.68, p = 1.36E-8; Supplementary Figure S4). Potential biological associations with rs7644567, including chromatin contacts (Supplementary Figure S5) and cerebellar expression of RBMS3, are summarized in Supplementary Information A9. However, rs7644567 did not replicate in two independent AA samples (Yale-Penn2 and COGA AAfGWAS) or the independent FINRISK cohort; all three replication cohorts estimating effects of the minor allele in the opposite direction of the discovery meta-analysis (Supplementary Table S4). The SNP is also rare in most EU samples (minor allele frequency [MAF] < 0.01), with the current GWAS results primarily attributable to AA cohorts, along with FinnTwin and NAG-Fin. The EU cohorts in the discovery meta-analysis show no evidence of association of AD with the SNPs in strongest LD with rs7644567 in African (rs13098461; z = 0.27, p = 0.79) or Finnish (rs9854300; z = 0.10, p = 0.92) reference samples (Supplementary Information A9). Based on the clear lack of replication there is insufficient evidence to conclude rs7644567 is associated with AD based on the current results.
There was limited genome-wide evidence for heterogeneity across all cohorts, within ancestry, between ancestries, or between study designs within ancestry (Supplementary Information A8; Supplementary Figures S6–S8). Evidence for inflation from population stratification or other confounding was also limited in the discovery meta-analysis (lambda = 0.962; Supplementary Figure S9) and within EU (lambda = 1.053, LD score regression [LDSR] intercept = 1.018) and AA (lambda = 1.007, LDSR intercept = 0.991-0.997; Supplementary Information A11). Gene-level association testing with MAGMA15 did not identify any additional significant genes in EU or AA (Supplementary Table S5, Supplementary Information A12), likely due to lack of power.
Heritability and genetic correlations:
Liability-scale SNP-heritability of AD was estimated at h2g = 0.090 (SE = 0.019, z = 4.80, p = 8.02E-7) in the meta-analysis of unrelated EU samples. Exclusion of the ADH1B locus did not substantially modify this estimate (h2g = 0.089, SE = 0.0185). Nominally significant polygenic signal for the meta-analysis of unrelated AA individuals was observed based on LDSR with scores computed from 1000 Genomes African populations (z = 2.12, p = 0.017), but the quantitative estimate of h2g was unstable depending on the choice of reference panel, reflecting the challenge of correctly specifying LDSR and robustly modelling LD for the AA population (Supplementary Information A11).
Significant genetic correlation with AD in EU was observed for 17 traits after correction for multiple testing (p < 1.11E-3 for 45 tested traits; Figure 3; Supplementary Table S6). The largest positive correlations were with ever smoking tobacco (rg = 0.708, SE = 0.134, p = 1.3E-7) and lifetime cannabis use (rg = 0.793, SE = 0.217, p = 2.5E-4), and with other psychiatric disorders, especially schizophrenia (rg = 0.357, SE = 0.054, p = 3.2E-11), ADHD (rg = 0.444, SE = 0.097, p = 4.2E-6), and depression (rg = 0.561, SE = 0.085, p = 3.5E-11). Educational attainment (rg = −0.468, SE = 0.066, p = 9.7E-13) and age at first birth (higher values indicate that participants were older when they had their first child; rg = −0.626, SE = 0.104, p = 2.0E-9) showed significant inverse genetic correlation with AD suggesting that liability to AD risk was genetically related to lower educational attainment and lower age at which the participant had his or her first child.
Figure 3: Genetic correlations between 45 traits and alcohol dependence in Europeans.

Genetic correlation results from LD score regression (LDSR) with the meta-analysis of AD in unrelated EU individuals (Ncase=10,206, Ncontrol=28,480). After Bonferroni correction, significant correlations are observed with 17 traits and disorders (p < 1.1E-3; bold), with nominally significant results for 8 additional traits and disorders (p < .05; italics) based on two-tailed tests of the estimated genetic correlation with block jackknife standard errors. Error bars indicate 95% confidence intervals, with arrows indicating intervals extending above 1 or below −1. Vertical gray reference line corresponds to the null hypothesis of no genetic correlation with AD. Phenotypes are organized by research domain.
Unexpected patterns of genetic correlation were observed when comparisons were made to other alcohol-related measures, indicating that those measures reflect aspects of alcohol use that are genetically distinguishable. AD was genetically correlated with alcohol consumption in a meta-analysis of the Alcohol Genome-wide Association (AlcGen) and Cohorts for Aging and Research in Genomic Epidemiology Plus (CHARGE+) consortia16 (rg = 0.695, SE = 0.155, p = 6.9E-6) but only modestly with alcohol consumption from the recent large UK Biobank analysis17 (rg = 0.371, SE = 0.092, p = 5.2E-5). No significant genetic correlation was observed between AD and a recent GWAS of the Alcohol Use Disorders Identification Test (AUDIT) in a 23andMe cohort18 (rg = 0.076, SE = 0.171, p = 0.65), perhaps due to the low levels of drinking and drinking-related problems in that population18. AD is, however, nominally genetically correlated with GWAS of delay discounting in the 23andMe sample19 (rg = 0.487, SE = 0.178, p = 6.0E-3).
Association with ADH1B expression:
Based on the strong observed association with rs1229984 and rs2066702 we examined whether other variants affecting ADH1B expression (eQTLs) were also associated with AD using GTEx V7 results (https://www.gtexportal.org/)20. Three variants, rs11939328 (EU p = 0.78, AA p = 0.98, Trans p = 0.78), rs10516440 (EU p = 3.97E-6, AA p = 1.97E-3, Trans p = 4.72E-8), and rs7664780 (EU p = 0.87, AA p = 0.083, Trans p = 0.405), were selected after LD-informed clumping and the exclusion of variants in LD (r2>0.1) with the GWS coding alleles rs1229984 and rs2066702. Of these, only rs10516440 (AD conditional analyses: EU p = 1.34E-3, AA p = 0.013, Trans p = 7.44E-5) was a significant multi-tissue eQTL in random effects analysis for ADH1B (SFE = 319.4, SHet = 27.6, p = 1.4E-76), ADH1A (SFE = 139.4, SHet = 6.6, p = 6.72E-33), and ADH1C (SFE = 167.3, SHet = 8.9, p = 1.9E-39). Rs10516440 is a LD proxy (r2 > 0.9) of rs6827898 (Table 2) in populations of European and African descent. These variants are both located in an intergenic region in the ADH gene cluster between ADH1C and ADH7. In line with the fact that the protective coding alleles are associated with increased activity of the enzyme encoded by ADH1B, the major allele rs10516440*A was associated with increased ADH1B expression and reduced AD risk.
Associations with other GWS loci:
We examined results for the eight independent variants associated at GWS levels with alcohol consumption in the UK Biobank17 (Supplementary Table S7). Among the UK Biobank findings, three of the four reported variants in the ADH region of chromosome 4 (rs145452708 – a proxy for rs1229984 with D’=1, rs29001570 and rs35081954) were associated in the present study with AD (p ranging from 3.5E-5 – 2.3E-10) with sign concordant effects; the remaining variant was excluded from our analysis due to MAF < 0.01. The UK Biobank lead variant in KLB, rs11940694, was nominally associated with AD (p = 0.0097), though this does not surpass multiple testing correction for the eight GWS alcohol consumption loci. We see little evidence (p > 0.2) for association of AD with the reported loci at GCKR and CADM2, which may be due to differences in power for the given effect size or because these genes exert an influence on liability to consume alcohol but not later problems. The locus on chromosome 18 showed limited regional association with AD, but the index variant was not present in our analysis because it no longer appears in the 1000 Genomes Phase 3 reference panel21.
Polygenic Risk Score (PRS) analyses:
PRS based on our meta-analysis of AD were significantly predictive of AD outcomes in all three tested external cohorts. PRS derived from the unrelated EU GWAS predicted up to 0.51% of the variance in past month alcohol use disorder in ALSPAC (p = 0.0195; Supplementary Figure S10A) and up to 0.3% of problem drinking as indexed by the CAGE screener in GS (p = 7.9E-6; Supplementary Figure S10B). PRS derived from the unrelated AA GWAS predicted up to 1.7% of the variance in alcohol dependence in the COGA AAfGWAS cohort (p = 1.92E-7; Supplementary Figure 10C).
Importantly, PRS derived from the unrelated EU GWAS showed much weaker prediction (maximum R2 = 0.37%, p = 0.01; Supplementary Figure S10D) in the COGA AAfGWAS than the ancestrally-matched AA GWAS-based PRS despite the much smaller discovery sample for AA. In addition, the AD PRS also still yielded significant variance explained after controlling for other genetic factors. Prediction of CAGE scores in GS remained significant and showed minimal attenuation (R2 = 0.29%, p = 1.0E-5) after conditioning on PRS for alcohol consumption derived from UK Biobank results17. In COGA AAfGWAS, the AA PRS derived from our study continued to predict 1.6% of the variance in alcohol dependence after inclusion of rs2066702 genotype as a covariate, indicating independent polygenic effects beyond the lead ADH1B variant (Supplementary Information A14).
Power analysis:
Power analyses indicated that the current meta-analysis is expected to have at least 41% power to detect very common variants (MAF ≥ 0.25) with odds ratios ≥ 1.10 at p < 5E-8 and 63% power for p < 1E-6 (Supplementary Figure S11). Power at p < 1E-6 is relevant because only 5 loci reach that threshold in the current meta-analysis. Power is lower for less common variants (MAF ≤ 0.05) even with odds ratios ≥ 1.20 at p < 1E-6 (60% power) and p < 5E-8 (38% power).
For perspective, power computations using the observed distribution of top effects for other large GWAS of polygenic traits suggest that we observe significantly fewer genome-wide significant loci for AD than would be expected if the loci had true effect sizes and allele frequencies similar to schizophrenia (expected: 25.4 loci, 95% CI 21-30) or obesity (expected: 8.9 loci, 95% CI 6-12), but not fewer than would be expected for effect sizes similar to major depression (Supplementary Information A10, Supplementary Table S8).
DISCUSSION
To our knowledge, this is the largest GWAS of rigorously-defined AD, comprising 14,904 AD individuals and 37,944 controls. We identified known loci in ADH1B that differed between EU and AA, as well as novel genetic correlations between AD and psychiatric disorders (e.g., schizophrenia), tobacco and cannabis use, and social (e.g., socio-economic deprivation) and behavioral (e.g., educational attainment) outcomes. Analyses also revealed a genetic distinction between GWAS results for alcohol consumption and AD. Although larger sample sizes can be amassed by focusing on quantitative measures of consumption, only the upper tail is relevant to AD (as a medical diagnosis) and even that does not capture other aspects of disordered drinking (e.g., loss of control, withdrawal) directly. Conversely, cases derived from electronic medical records (e.g., ICD codes) result in a high rate of false negatives, while self-screening instruments (e.g. AUDIT scores) are best suited to analyses of disordered drinking when a sufficiently high threshold or score cut-off is applied to focus on severity. Our study has the advantage of greater diagnostic precision via use of semi-structured interviews to diagnose AD systematically in a majority of the constituent studies, and therefore greater interpretability in the context of clinically-important AD.
The genome-wide significant SNPs reaffirm the importance of functional variants affecting alcohol metabolism to the risk of AD. The top association in ADH1B, rs1229984, is a missense variant that is amongst the most widely studied in relation to alcohol use, misuse and dependence8–10. The resulting amino acid substitution (Arg48His) increases the rate at which alcohol dehydrogenase 1B oxidizes ethanol to acetaldehyde8. Studies on Asian populations in which the derived allele is common demonstrated strong protection against the development of AD8,9,13. In EU and AA, the protective allele is present at much lower frequencies (EU MAF = 0-4%, AA MAF < 1%), nevertheless, recent large-scale studies have shown an association between this locus and alcohol consumption and problems at GWS levels in EU with similar effect size8–10. The lead variant in AA cohorts, rs2066702 (Arg370Cys), is another functional missense variant in ADH1B, and it also encodes an enzyme with an increased rate of ethanol oxidation8. The allele encoding Cys370 is common in AA, but rare in other populations8. Our results clearly show that these two different functional SNPs in ADH1B both affect risk for alcoholism, with their relative importance dependent upon allele frequency in the population studied. There is a suggestion of additional independent effects in the chromosome 4 region, but larger studies will be needed to evaluate this.
The only other locus to reach significance was rs7644567 on chromosome 3, primarily driven by AA cohorts. The locus failed to replicate in two small, independent AA samples, and in the only European cohort with even a modest allele frequency (FINRISK) the effect was in the opposite direction. There have also been discussions about whether the standard GWAS significance threshold should be applied to the more genetically diverse African-ancestry cohorts22,23 and the possibility of confounding from non-linear relationships between the phenotype and ancestry-informative markers like rs7644567 in admixed samples24, all of which increase our skepticism regarding this finding. There is, therefore, insufficient evidence at this time to conclude that rs7644567 is associated with alcohol dependence. Analyses of much larger samples of African ancestry will be needed to resolve this.
Despite limited SNP-level findings, there is significant evidence for polygenic effects of common variants in both EU and AA cohorts. The estimated h2g = 0.09 for AD in EU is only modestly lower than those recently reported for alcohol consumption (h2g = 0.13)17 and AUDIT scores (h2g = 0.12)18, and comparable to estimates derived for cigarettes-per-day25. Our h2g estimate is lower than a prior report7, likely reflecting a combination of differences in estimation method (GREML vs. LDSR) and greater heterogeneity in ascertainment strategy across samples in the current study (see26–28). The latter is especially relevant in comparing h2g from that prior single cohort to our meta-analysis that included cohorts with a wide range of ages at ascertainment, cultural environments, and ascertainment strategies, including enrichment for other substance use disorders. Similar to other psychiatric disorders (e.g. schizophrenia), a much larger sample size will potentially aid in overcoming across-sample heterogeneity and capture a greater proportion of genetic variance.
Comparing our GWAS to recent GWAS of alcohol consumption measures suggests that the liability underlying normative patterns of alcohol intake and AD are only partially overlapping. Genome-wide, genetic correlations were significantly < 1 with log-scaled alcohol consumption by participants in AlcGen and CHARGE+ Consortia cohorts16 (rg = 0.695) and in the UK Biobank17 (rg = 0.371). We also observe only partial replication of the 8 loci significantly associated with consumption in the UK Biobank, with strongest results from SNPs in the ADH region, including a proxy for rs1229984. In addition there was no significant correlation with GWAS of log-scaled AUDIT scores in 23andMe participants18 (rg = 0.076). Subsequent analyses suggest these estimates are sensitive to sample characteristics, with somewhat higher genetic correlations reported in analysis of alcohol consumption in the full UK Biobank29 (rg = 0.75) and of AUDIT in combined data from 23andMe participants and UK Biobank30 (rg = 0.39). Importantly, initial UK Biobank data inclusion of a subset of participants recruited for a study of smoking and lung function in the first analysis17, which may have resulted in collider bias31 and contributed to the initial lower genetic correlation.
One key factor in interpreting the differences between these traits and AD is that the distribution of consumption levels and AUDIT scores can be highly skewed in population samples, with most individuals at the low (non-pathological) end of the spectrum. This effect may be especially pronounced among the older, healthy volunteers of the UK Biobank cohort32 and in the 23andMe cohort, which is more educated and has higher socioeconomic status than the general US population18. We hypothesize that the variants that affect consumption at lower levels may differ substantively from those that affect very high levels of consumption in alcohol dependent individuals, who are also characterized by loss of control over intake33. This appears to be the case in studies that used specific cut-offs to harmonize AUDIT scores with AD data30,34. The larger of these studies30 reports that the genetic correlation between AD and AUDIT scores is maximized at an AUDIT cutoff ≥ 20 (with controls defined as those scoring ≤ 4; rg = 0.90). Interestingly, that study also found that a score reflecting items related to problem drinking (AUDIT-P) resulted in a stronger genetic correlation (rg = 0.64) than a score related to alcohol consumption alone (rg = 0.33). The strong genetic correlation of AD with lower educational attainment and lower socio-economic status (i.e. higher Townsend deprivation), in contrast to positive genetic correlations of education with consumption17 and AUDIT scores related to consumption30, further underscore this distinction between normative/habitual levels of alcohol intake and diagnosed AD, at least in the respective populations studied.
The current analysis identified robust genetic correlation of AD with a broad variety of psychiatric outcomes. This correlation is strongest for aspects of negative mood, including neuroticism and major depression, as also seen in twin studies35,36 and through recent specific molecular evidence for pleiotropy37,38. Taken together with evidence from other recent genomic studies37, and null correlations for other GWAS of alcohol consumption, but not for measures of problem drinking (e.g., AUDIT-P), these findings suggest that major depression may primarily share genetic liability with alcohol use at pathological levels.
AD was also strongly genetically correlated with poor educational and socioeconomic outcomes, and marginally correlated with measures of risk-taking. Nominally significant genetic correlations with delay discounting (i.e. favoring immediate rewards), risk-taking, and the strong genetic correlation of AD with ADHD, cigarette smoking and cannabis use may similarly reflect a shared genetic factor for risk-taking and reduced impulse control. Common genetic liability to early, risky behaviors is characteristic of both AD39 and age of first birth40. The observed negative genetic correlation with age of first birth is consistent both with risk-taking and with the significant genetic correlations of AD with lower socioeconomic status, as indexed by higher neighborhood Townsend deprivation score, and lower educational attainment. Lower socioeconomic status is correlated with both AD41 and age at first birth42 and the current study suggests that shared genetic liabilities may be one potential mechanism for their observed relationship. However, the question of whether these genetic correlations represent causal processes, horizontal pleiotropy, or the impact of unmeasured confounders should be explored in the future43.
Lower genetic correlations were observed for most biomedical and anthropometric outcomes. Liver enzymes GGT and ALT, once proposed as possible biomarkers for alcohol abuse44, showed only nominal evidence for genetic correlation with AD and neither survived multiple testing correction. Notably, we did not find any association between AD and body-mass index (BMI). Negative genetic correlations with BMI were previously reported for both alcohol consumption17 and AUDIT scores18, but there is prior evidence that BMI has differing underlying genetic architecture in the context of AD and outside of that context45. The negative genetic correlations observed in those studies are consistent with studies of light to moderate drinking, which is also associated with healthier lifestyle behaviors, while heavy and problematic drinking is typically associated with weight gain46.
This study benefits from precision in diagnostic assessment of AD, known alcohol exposure in a majority of the controls, and careful quality control that excluded overlap of individuals between studies. Despite these strengths, our sample size was insufficient to identify additional GWS loci robustly. Power analyses indicate that additional SNPs associated with AD are likely to have small effect sizes, smaller than schizophrenia47 and more consistent with more common psychiatric disorders (e.g. major depression48). This supports the pressing need for collection of large numbers of well characterized cases and controls. The differences between our results and the study of AUDIT scores18 highlight that ascertainment and trait definition are critically important and must be taken into account. Careful study of how screening tools, such as the AUDIT, correlate to genetic liability to AD (as defined by DSM-IV or similar) could substantially boost sample sizes for future AD GWAS. There is also a continued need to characterize the genetic architecture of AD in non-EU populations.
We show a novel genetic distinction between drinking in the pathological range (AD) and habitual drinking that does not cross the threshold into pathology or dependence nor captures behavioral aspects of disordered drinking. Larger future samples will allow us to uncover additional pleiotropy between pathological and non-pathological alcohol use as well as between AD and other neuropsychiatric disorders.
METHODS
Samples:
The Substance Use Disorders working group of the Psychiatric Genomics Consortium (PGC-SUD14) collected individual genotypic data from 14 case/control studies and 9 family-based studies and summary statistics from GWAS of AD from 5 additional cohorts (Table 1). AD was defined as meeting criteria for a DSM-IV2 (or, for one cohort, DSM-IIIR50; a very similar construct; Supplementary Note B1) diagnosis of AD. Diagnoses were derived either from clinician ratings or semi-structured interviews. Excepting three cohorts with population-based controls (N=7,015), all controls were screened for AD. Individuals with no history of drinking alcohol and those meeting criteria for DSM-IV alcohol abuse were excluded as controls where possible (Supplementary Information A1; Life Sciences Reporting Summary). This study was approved by the institutional review board (IRB) of Washington University in St. Louis and was conducted in accordance with all relevant ethical regulations. Each contributing cohort obtained informed consent from their participants and received ethics approvals of their study protocols from their respective review boards in accordance with applicable regulations.
Quality Control and Imputation:
Data for the cohorts that shared raw genotypes were deposited to a secure server for uniform quality control (QC). QC and imputation of the 14 case/control studies was performed using the ricopili pipeline (https://github.com/Nealelab/ricopili). For the 9 family-based cohorts, an equivalent pipeline, picopili (https://github.com/Nealelab/picopili), was developed for QC, imputation, and analysis appropriate for diverse family structures, including twins, sibships and extended pedigrees (Supplementary Information A2).
After initial sample and variant QC, principal components analysis (PCA) was used to identify population outliers for exclusion and to stratify samples in each study by continental ancestry. Identified EU and AA ancestry populations were confirmed by PCA using the 1000 Genomes reference panel21 (Supplementary Figure S12). Ancestry within these 2 groups was accounted for with principal components. Final sample and variant QC, including filters for call rate, heterozygosity, and departure from Hardy-Weinberg equilibrium (HWE), was then performed within each ancestry group in each cohort. Samples were also filtered for cryptic relatedness and for departures from reported pedigree structures (Supplementary Information A3; Life Sciences Reporting Summary).
Each cohort was imputed using SHAPEIT51 and IMPUTE252, using the cosmopolitan (all ancestries) 1000 Genomes reference panel consistent with prior recommendations53 (see also47,54,55). Concordance of minor allele frequencies (MAF) with the reference panel was verified prior to imputation, with SNPs in EU cohorts compared to MAF in European population samples and AA cohorts compared to MAF in African population samples (Supplementary Information A4). Cryptic relatedness between cohorts was excluded after imputation (Supplementary Information A5). Imputed SNPs were then filtered for INFO score > 0.8 and allele frequency > 0.01 prior to analysis.
Association Analysis:
A GWAS of AD status was performed within each ancestry stratum of each sample using an association model appropriate for the study design (Table 1, Supplementary Table S1). For case/control studies, GWAS was performed using logistic regression with imputed dosages. For family-based studies of small, simple pedigrees (e.g., sibships), association with imputed genotypes was tested using generalized estimating equations (GEE). For more complex pedigrees, imputed genotypes were tested using logistic mixed models. Sex was included as a covariate, along with principal components to control for population structure (Supplementary Information A6, Supplementary Note B2, Supplementary Figures S13–S14).
In addition to this primary analysis, subsets of genetically unrelated individuals were selected from each family-based cohort (i.e. the most severe case in each family, by symptom count, was selected, followed by selection of unrelated/married-in controls) and used to perform a conventional case/control GWAS using logistic regression. This was used in place of the family-based GWAS for estimation of effect sizes and LD score regression analyses (Supplementary Table S2).
Genome-wide Meta-Analysis:
The primary discovery meta-analysis of all ancestry-stratified GWAS (Ncase = 14,904; Ncontrol = 37,944) was conducted in METAL56. As the different study designs (family vs. case-control) produced effect sizes that were not comparable, results were combined using weighting by effective sample size (Supplementary Information A7, Supplementary Note B3). Separate ancestry-specific discovery meta-analyses of EU (N = 46,568) and AA (N = 6,280) cohorts were also performed. Heterogeneity was evaluated across all cohorts and between study designs (Supplementary Information A8).
In addition to the discovery meta-analyses, we conducted meta-analyses for two design subsets. First, we performed sample size weighted meta-analysis of the subset of genetically unrelated individuals in EU (N = 38,686) and AA (N = 5,799) cohorts for use in LD score regression (LDSR) analysis. Second, we performed inverse-variance weighted meta-analysis of genetically unrelated individuals in genotyped cohorts to estimate within-ancestry effect sizes for EU (N = 28,757) and AA (N = 5,799). These effect sizes were then used to compare trans-ancestral fine mapping results using inverse-variance weighted fixed effects, random effects57, and Bayesian58 models (Supplementary Information A7). Supplementary Table S2 summarizes all of the meta-analytic models considered in the current analysis.
Replication:
As described below, a novel locus on chromosome 3 was genome-wide significant (GWS) in the trans-ancestral discovery meta-analysis. To seek replication, we examined the association between this locus and DSM-IV AD in two independent AA samples: Yale-Penn 2 (911 cases, 599 controls; tested using GEE) and COGA AAfGWAS (880 cases, 1,814 controls; tested using GWAF59). Association with AD status, broadly defined using hospital and death records, was also examined in the FINRISK cohort (1,232 cases, 22,614 controls) using Firth logistic regression60 (Supplementary Information A1.4; Life Sciences Reporting Summary).
Power Analysis:
Post-hoc power analysis was performed for odds ratios ranging from 1.05 to 1.30 and across allele frequencies using CaTS61 with the estimated effective sample size. Power analysis identifies the range of SNP effect sizes the current study was likely to detected at genome-wide significance if such effects exist. Additionally, we made specific comparisons to the distribution of effects for schizophrenia47, obesity62 and major depression48 as meaningful benchmarks to understand the magnitude of effect sizes plausible for AD (Supplementary Information A10; Life Sciences Reporting Summary).
Heritability and Genetic Correlation Analysis:
LDSR analysis63 was performed to estimate the heritability explained by common SNPs in meta-analyses of unrelated EU and AA samples, respectively. LDSR was performed using HapMap3 SNPs and LD scores computed from 1000 Genomes reference samples corresponding to each population (Supplementary Information A11). Conversion of h2g estimates from observed to liability scale64 was performed assuming population prevalences of 0.159 and 0.111 for AD in alcohol-exposed EU and AA individuals, respectively3. Gene-level enrichments were also tested with MAGMA15 (Supplementary Information A12).
Genetic correlation between AD and 45 traits from LD Hub25 and other published studies16–19,65–71 was examined using LDSR with the same unrelated EU meta-analysis (10,206 cases and 28,480 controls) and precomputed European LD scores. LDSR compares GWAS results for pairs of traits to estimate the correlation in the genetic liabilities explained by all common SNPs in the LD reference panel. To avoid increasing the multiple testing burden, redundant or highly-correlated phenotypes were reduced by manually selecting the version of the phenotype with the greatest predicted relevance to AD, largest sample size, or highest heritability (Supplementary Information A13).
Polygenic Risk Scores:
To test the generalizability of the current GWAS results, polygenic risk scores (PRS) were computed in three external cohorts (Supplementary Information A1.5; Life Sciences Reporting Summary). PRS computed from EU ancestry results were used to predict alcohol dependence in ALSPAC72,73 and COGA AAfGWAS, and CAGE screener scores in Generation Scotland (GS)74. PRS based upon the AA results were used to predict alcohol dependence in COGA AAfGWAS (Supplementary Information A14).
Data availability:
Summary statistics from the genome-wide meta-analyses are available on the Psychiatric Genomics Consortium’s downloads page (http://www.med.unc.edu/pgc/results-and-downloads), including the source data for Figures 1 and 2. Individual-level data from the genotyped cohorts and cohort-level summary statistics will be made available to researchers following an approved analysis proposal through the PGC Substance Use Disorder group with agreement of the cohort PIs; contact the corresponding authors for details. Cohort data are also available from dbGaP except where prohibited by IRB or European Union data restrictions. Expression data used to evaluate variants in ADH1B is available from GTEx (https://gtexportal.org/home/). Hi-C data used to evaluate the chromosome 3 variant can be queried with HUGIn (https://yunliweb.its.unc.edu/hugin/). Publicly available genome-wide summary statistics used for testing genetic correlations are accessible through LD Hub (http://ldsc.broadinstitute.org/), or from the Psychiatric Genomics Consortium (http://www.med.unc.edu/pgc/results-and-downloads), the Social Science Genetic Association Consortium (SSGAC; https://www.thessgac.org/data), Enhancing Neuro Imaging Genetics through Meta Analysis (ENIGMA; http://enigma.ini.usc.edu/research/download-enigma-gwas-results/), and the Neale Lab (http://www.nealelab.is/uk-biobank); for availability of summary statistics from other studies contact the respective authors. The source data for Figure 3 is included in Supplementary Table S6.
Code availability:
Code for GWAS of case/control cohorts with ricopili is available at https://github.com/Nealelab/ricopili. Code for GWAS of family-based cohorts with picopili is available at https://github.com/Nealelab/picopili. Code and reference data for LD score regression analyses are available at https://github.com/bulik/ldsc. Effective sample size calculations were implemented using output from PLINK (https://www.cog-genomics.org/plink2), and GMMAT (https://content.sph.harvard.edu/xlin/software.html#gmmat) and geepack (https://cran.r-project.org/web/packages/geepack/index.html) in R (https://cran.r-project.org/); stand-alone software for this purpose hasn’t been written but example code is available from the first author by request.
Supplementary Material
Acknowledgements
The Psychiatric Genomics Consortium’s Substance Use Disorders (PGC-SUD) Working Group receives support from the National Institute on Drug Abuse and the National Institute of Mental Health via MH109532. We gratefully acknowledge prior support from the National Institute on Alcohol Abuse and Alcoholism. Statistical analyses for the PGC were carried out on the Genetic Cluster Computer (http://www.geneticcluster.org) hosted by SURFsara and financially supported by the Netherlands Scientific Organization (NWO 480-05-003) along with a supplement from the Dutch Brain Foundation and the VU University Amsterdam. We thank Andrew Morris for providing code implementing the MANTRA Bayesian model. We thank Alicia Martin, Alex Bloemendal, and Hilary Finucane for helpful conversations about the analysis protocol for admixed cohorts. We thank Ms. Debra Hughes for her assistance with editorial contributions to the manuscript. We thank Rainer Spanagel for significant contributions as a founding member of GESGA.
We thank the 23andMe research participants and employees for making this work possible.
A.P. is supported by the Academy of Finland, Juselius Foundation; D.E.A. acknowledges 1K01MH093731; R.R. acknowledges AA-12502, AA-09203, AA-00145; M.K. is supported by AA009367, DA005147; M.M. is supported by AA009367, MH066140; R.A.G. is supported by AA017444; K.R. is supported by Academy of Finland; A.C.H. is supported by NIH AA07535, AA07729, AA13320, AA13321, AA11998; A.R.D. is supported by NIH 1K01MH109765-01; A.E.A. is supported by NIH AA011408, AA017828; A.G.W. is supported by NIH 3T32AA7464-38 S1; B.M.N. is supported by NIH U01 MH109539 and R01 MH107649; B.P.R. is supported by NIH AA011408, AA017828, AA022537; B.S.M. is supported by NIH R01DA036525, R01DA039408; B.T.W. is supported by NIH AA011408, AA017828, AA022537; C.Ho. is supported by NIH/NIAAA Intramural program; C.J.H. is supported by NIH DA032555, DA035804, DA011015, DA042755; D.-S.C. is supported by NIH P20AA017830, AA018779; W.I. is supported by NIH DA005147, DA013240, DA024417, DA036216; S.A.B. is supported by NIH DA021905; H.dW. is supported by NIH DA02812; M.C.S. is supported by NIH DA035804; S.V. is supported by NIH DA042755, DA037904, DA040177, HG008983; R.P. is supported by NIH DA12690; J.G. is supported by NIH DA12690; D.B.H. is supported by NIH R01DA036583; D.G. is supported by the NIAAA Intramural program; D.M.D. is supported by NIH R01AA015416, K02AA018755, U10AA008401, P50AA0022537; E.J.C. is supported by NIH DA023026, DA011301, DA024413; E.O.J. is supported by NIH R01 DA044014; N.S. is supported by German Government BMBF #01EB0130; K.M. is supported by German Government BMBF #01EB0410; G.W.M. is supported by an NHMRC fellowship by GNT1078399; N.W. is supported by D.F.G. and B.M.B.F.; N.Da. is supported by D.F.G. and B.M.B.F.; J.D.B. is supported by NIH R01HD060726; M.A.K. is supported by Health Research Council of New Zealand 11/792, 16/600; J.M.Bo. is supported by Health Research Council of New Zealand 11/792, 16/600; J.H. is supported by Health Research Council of New Zealand 11/792, 16/600; J.F.P. is supported by Health Research Council of New Zealand 11/792, 16/600; H.H.M. is supported by NIH DA025109, DA024413, DA016977; J.E.S. is supported by NIH K01 AA024152; J.A.K. is supported by Academy of Finland 265240, 263278, Sigrid Juselius Foundation; J.K.H. is supported by NIH DA011015; J.L.M. is supported by NIH K01DA037914; J.M.Bi. is supported by NIH P20AA017830, AA25214; K.K. is supported by NIH DA011015; K.S.K. is supported by NIH P50AA0022537; L.-S.C. is supported by NIH DA038076; L.J.B. is supported by NIH R01DA036583; L.M.H. is supported by NIH AA011408; AA017828; L.D. is supported by an Australian NHMRC Principal Research Fellowship; M.A.F. is supported by NIH/NIAAA P20AA017830; M.D.R. is supported by CSAT/SAMHSA 1H79T1026423, 1H79T1026446, AHRQ 1R18HS024208, NIH R01DA036628; N.A.G. is supported by NIH R00DA023549; P.-H.S. is supported by NIAAA Intramural Research Program; N.Di. is supported by NIAAA Intramural Research Program; Me.S. is supported by NIAAA Intramural Research Program; R.W. is supported by NSF GRFP DGE 1144083; P.A.F.M. is supported by NIH DA012854, R25DA027995; J.M. is supported by Peter Boris Chair in Addictions Research; K.M.H is supported by NIH R01 HD060726, R01 HD073342, P01 HD031921; M.B.M. is supported by NIH R01HD060726; R.K.W. is supported by NIH U01 MH094432; R.C.C. is supported by NIH R01DA036583; R.E.P. is supported by NIH K01MH113848; R.E.T. is supported by NIH R21DA043735; S.S.-R. is supported by the Frontiers of Innovation Scholars Program (FISP) and the Interdisciplinary Research Fellowship in NeuroAIDS (IRFN), and by NIH P50DA037844; S.-A.B. is supported by NIH AA011408, AA017828, AA022537, AA022717; S.E.M. is supported by NHMRC 1103623; S.M.H. is supported by NIH R21AA024888, K08DA032680; T.B.B. is supported by NIH MH100549; M.M.N. is supported by The BMBF funded e:Med consortium IntegraMent 01ZX1314A, SysMedAlcoholism 01ZX1311A, and DFG-funded Excellence-Cluster ImmunoSensation; S.C. is supported by The Integrated Network IntegraMent 01ZX1314A; Ma.R. is supported by The Integrated Network IntegraMent 01ZX1314G; SysMedAlcoholism 01ZX1311A; T.L.W. is supported by NIH R01 DA021905, R01 DA035804; A.M.G. is supported by NIH U10 AA08401; M.L.-P. is supported by University of Helsinki, Academy of Finland; J.L. is supported by University of Helsinki, Academy of Finland; V.M.K. is supported by NIH P20AA017830, AA25214; W.E.C. is supported by NIH R01HD093651, R01DA036523, P30DA023026; T-K.C. is supported by Wellcome Trust (STRADL) 104036/Z/14/Z; M.J.A. is supported by Wellcome Trust (STRADL) 104036/Z/14/Z; A.M.M. is supported by Wellcome Trust (STRADL) 104036/Z/14/Z. A.A. is supported by NIH K02DA32573.
Funding support for the Comorbidity and Trauma Study (CATS) (dbGAP accession number: phs000277.v1.p1) was provided by the National Institute on Drug Abuse (R01 DA17305); GWAS genotyping services at the CIDR at The Johns Hopkins University were supported by the National Institutes of Health (contract N01-HG-65403). Funding support for the Center for Education and Drug Abuse Research (CEDAR) (dbGAP accession number: phs001649.v1.p1 was provided by the National Institute on Drug Abuse (P50 DA005605). The Christchurch Health and Development Study (CHDS: dbGAP in progress) has been supported by funding from the Health Research Council of New Zealand, the National Child Health Research Foundation (Cure Kids), the Canterbury Medical Research Foundation, the New Zealand Lottery Grants Board, the University of Otago, the Carney Centre for Pharmacogenomics, the James Hume Bequest Fund, US NIH grant MH077874, and NIDA grant ‘‘A developmental model of gene-environment interplay in SUDs’’ (R01DA024413) 2007–2012. The Collaborative Study on the Genetics of Alcoholism (COGA) is supported by NIH Grant U10AA008401 from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) and the National Institute on Drug Abuse (NIDA). Funding support for this GWAS genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research, was provided by the National Institute on Alcohol Abuse and Alcoholism, the NIH GEI (U01HG004438), and the NIH contract “High throughput genotyping for studying the genetic contributions to human disease” (HHSN268200782096C). COGA Principal Investigators: B. Porjesz, V. Hesselbrock, H. Edenberg, L. Bierut, includes eleven different centers: University of Connecticut (V. Hesselbrock); Indiana University (H.J. Edenberg, J. Nurnberger Jr., T. Foroud); University of Iowa (S. Kuperman, J. Kramer); SUNY Downstate (B. Porjesz); Washington University in St. Louis (L. Bierut, J. Rice, K. Bucholz, A. Agrawal); University of California at San Diego (M. Schuckit); Rutgers University (J. Tischfield, A. Brooks); Department of Biomedical and Health Informatics, The Children’s Hospital of Philadelphia; Department of Genetics, Perelman School of Medicine, University of Pennsylvania, Philadelphia PA (L. Almasy), Virginia Commonwealth University (D. Dick), Icahn School of Medicine at Mount Sinai (A. Goate), and Howard University (R. Taylor). Other COGA collaborators include: L. Bauer (University of Connecticut); J. McClintick, L. Wetherill, X. Xuei, Y. Liu, D. Lai, S. O’Connor, M. Plawecki, S. Lourens (Indiana University); G. Chan (University of Iowa; University of Connecticut); J. Meyers, D. Chorlian, C. Kamarajan, A. Pandey, J. Zhang (SUNY Downstate); J.-C. Wang, M. Kapoor, S. Bertelsen (Icahn School of Medicine at Mount Sinai); A. Anokhin, V. McCutcheon, S. Saccone (Washington University); J. Salvatore, F. Aliev, B. Cho (Virginia Commonwealth University); and Mark Kos (University of Texas Rio Grande Valley). A. Parsian are the NIAAA Staff Collaborators. M. Reilly was an NIAAA staff collaborator. We continue to be inspired by our memories of Henri Begleiter and Theodore Reich, founding PI and Co-PI of COGA, and also owe a debt of gratitude to other past organizers of COGA, including Ting-Kai Li, currently a consultant with COGA, P. Michael Conneally, Raymond Crowe, and Wendy Reich, for their critical contributions. The authors thank Kim Doheny and Elizabeth Pugh from CIDR and Justin Paschall from the NCBI dbGaP staff for valuable assistance with genotyping and quality control in developing the dataset available at dbGaP (phs000125.v1.p1; also: phs000763.v1.p1; phs000976.v1.p1). Support for the Study of Addiction: Genetics and Environment (SAGE) was provided through the NIH Genes, Environment and Health Initiative [GEI; U01 HG004422; dbGaP study accession phs000092.v1.p1]. SAGE is one of the genome-wide association studies funded as part of the Gene Environment Association Studies (GENEVA) under GEI. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the GENEVA Coordinating Center [U01 HG004446]. Assistance with data cleaning was provided by the National Center for Biotechnology Information. Support for collection of datasets and samples was provided by the Collaborative Study on the Genetics of Alcoholism [COGA; U10 AA008401], the Collaborative Genetic Study of Nicotine Dependence [COGEND; P01 CA089392; see also phs000404.v1.p1], and the Family Study of Cocaine Dependence [FSCD; R01 DA013423, R01 DA019963]. Funding support for genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research (CIDR), was provided by the NIH GEI [U01HG004438], the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the NIH contract “High throughput genotyping for studying the genetic contributions to human disease” [HHSN268200782096C]. For GESGA, M.R. and M.M.N. were supported by the German Federal Ministry of Education and Research (BMBF) through grants BMBF 01ZX1311A (to MR and MMN), and through grants 01ZX1314A (to MMN) and 01ZX1314G (to MR) within the e:Med research program. The GSMS project (phs000852.v1.p1) was supported by the National Institute on Drug Abuse (U01DA024413, R01DA11301), the National Institute of Mental Health (R01MH063970, R01MH063671, R01MH048085, K01MH093731 and K23MH080230), NARSAD, and the William T. Grant Foundation. We are grateful to all the GSMS and CCC study participants who contributed to this work. The following grants supported data collection and analysis of CADD (dbGAP in progress): DA011015, DA012845, DA021913, DA021905, DA032555, and DA035804. Funding support for Spit for Science (dbGAP in progress) has been provided by Virginia Commonwealth University, P20 AA017828, R37AA011408, K02AA018755, and P50 AA022537 from the National Institute on Alcohol Abuse and Alcoholism, and UL1RR031990 from the National Center for Research Resources and National Institutes of Health Roadmap for Medical Research. We would like to thank the Spit for Science participants for making this study a success, as well as the many University faculty, students, and staff who contributed to the design and implementation of the project. In particular we acknowledge the contributions of the many individuals who have played a critical role data collection, generation, and cleaning, including Amy Adkins, Fazil Aliev, Erin Caraway, Seung Bin Cho, James Clifford, Megan Cooke, Elizabeth Do, Alexis Edwards, Neeru Goyal, Lisa Halberstadt, Sage Hawn, Rebecca Holloway, Jennifer Lent, Mackenzie Lind, Elizabeth Long, Jacquelyn Meyers, John Myers, Ashlee Moore, Arden Moscati, Zoé Neale, Jill Opalesky, Cassie Overstreet, Kimberly Pedersen, Laura Rappa, Brien Riley, Jessica Salvatore, Jeanne Savage, Cuie Sun, Nathaniel Thomas, Bradley Webb & Jia Yan. The NIAAA data (available via: https://btris.nih.gov/) were supported by the National Institute on Alcohol Abuse and Alcoholism Intramural Research Program (NIAAA IRP). Data collection, genotyping and analysis of the Mayo Clinic Center for Individualized Treatment of Addiction (CITA) data was supported by grants from the National Institute on Alcohol Abuse and Alcoholism (P20 AA017830, R21 AA25214) ) as well as the Mayo CTSA Grant Number UL1TR000135 and SC Johnson Genomics of Addiction Program. The Mayo Clinic Biobank, which served as the source of controls for the CITA cases, was supported by Mayo Clinic Center for Individualized Medicine. ADAA was funded by NIH grant R01 AA017444. Brisbane Longitudinal Twin Study (BLTS) was supported by the United States National Institute on Drug Abuse (R00DA023549), and by the Australian Research Council (DP0343921, DP0664638, 464914, 619667, FT110100548). Gene-Environment-Development Initiative -GEDI – Virginia Commonwealth University (VTSABD; dbGAP in progress) was supported by the National Institute on Drug Abuse (U01DA024413, R01DA025109), the National Institute of Mental Health (R01MH045268, R01MH055557, R01MH068521), and the Virginia Tobacco Settlement Foundation grant 8520012. We are grateful to all the VTSABD-YAFU-TSA study participants who contributed to this work. Minnesota Center for Twin and Family Research (MCTFR; phs000620.v1.p1) support contributing to this publication was supported by the National Institutes of Health under award number DA005147, DA013240, DA024417, DA036216, AA009367, MH066140. Funding for TwinGene is a sub-study of the Swedish Twin Registry, managed by Karolinska Institutet and supported by the Swedish Research Council under the grant no 2017-00641. Additional funding was provided by the Swedish Research Council (M-2005-1112), GenomEUtwin (EU/QLRT-2001-01254; QLG2-CT-2002-01254), National Institutes of Health U01-DK 066134, the Swedish Foundation for Strategic Research (SSF), and the Heart and Lung Foundation (20070481). Substance Use Disorder Liability: Candidate System Genes study was supported by R01 DA019157 and P50 DA005605. Yale-Penn (phs000425.v1.p1; phs000952.v1.p1) was supported by National Institutes of Health Grants RC2 DA028909, R01 DA12690, R01 DA12849, R01 DA18432, R01 AA11330, and R01 AA017535 and the Veterans Affairs Connecticut and Philadelphia Veterans Affairs Mental Illness Research, Educational, and Clinical Centers. Australian Alcohol and Nicotine studies (OZ-ALC-NAG; phs000181.v1.p1) were supported by National Institutes of Health Grants AA07535,AA07728, AA13320, AA13321, AA14041, AA11998, AA17688,DA012854, and DA019951; by Grants from the Australian National Health and Medical Research Council (241944, 339462, 389927,389875, 389891, 389892, 389938, 442915, 442981, 496739, 552485,and 552498); by Grants from the Australian Research Council(A7960034, A79906588, A79801419, DP0770096, DP0212016, and DP0343921); and by the 5th Framework Programme (FP-5) GenomEUtwin Project (QLG2-CT-2002-01254). Genome-wide association study genotyping at Center for Inherited Disease Research was supported by a Grant to the late Richard Todd, M.D., Ph.D., former Principal Investigator of Grant AA13320. Irish Affected Sib-Pair Study of Alcohol Dependence (IASPSAD) GWAS data collection and analysis was supported by National Institute on Alcohol Abuse and Alcoholism grants P20-AA-017828 and P50-AA-022537. Sample collection was supported by R01-AA-011408. Control genotyping was supported by National Institute of Mental Health grant R01-MH-083094 and Wellcome Trust Case Control Consortium 2 grant WTCCC-084710. Netherland Twin Register (NTR) and Netherlands Study of Depression and Anxiety (NESDA) funding was obtained from multiple grants from the Netherlands Organization for Scientific Research (NWO) and MagW/ZonMW, including NWO-480-15-001/674: Netherlands Twin Registry Repository: researching the interplay between genome and environment; Geestkracht program of the Netherlands Organization for Health Research and Development (Zon-MW 10-000-1002), Genetic determinants of risk behavior in relation to alcohol use and alcohol use disorder (ZonMW-Addiction-31160008); Biobanking and Biomolecular Resources Research Infrastructure (BBMRI –NL, 184.021.007), Amsterdam Public Health research institute (APH) and Neuroscience Campus Amsterdam (NCA); the European Science Council (ERC-230374 and ERC-284167). Part of the genotyping was funded by NWO/SPI 56-464-14192; the Genetic Association Information Network (GAIN) of the Foundation for the National Institutes of Health, Rutgers University Cell and DNA Repository (NIMH U24 MH068457-06), the Avera Institute for Human Genetics, Sioux Falls, South Dakota (USA) and the National Institutes of Health (NIH R01 HD042157-01A1, MH081802, Grand Opportunity grants 1RC2 MH089951 and 1RC2 MH089995). The Finnish Twin Cohort/Nicotine Addiction Genetics-Finland study was supported by Academy of Finland (grants # 213506, 129680 to JK), NIH DA12854 (PAFM), Global Research Award for Nicotine Dependence / Pfizer Inc. (JK), Wellcome Trust Sanger Institute, UK and the European Community’s Seventh Framework Programme ENGAGE Consortium (HEALTH-F4-2007- 201413). In Finntwin12, support for data collection and genotyping has come from National Institute of Alcohol Abuse and Alcoholism (grants AA-12502, AA-00145, and AA-09203 to RJR and AA15416 and K02AA018755 to DMD), the Academy of Finland (grants 100499, 205585, 118555, 141054 and 264146 to JK) & Wellcome Trust Sanger Institute, UK. This research uses data from Add Health, a program project directed by Kathleen Mullan Harris and designed by J. Richard Udry, Peter S. Bearman, and Kathleen Mullan Harris at the University of North Carolina at Chapel Hill, and funded by grant P01-HD31921 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with cooperative funding from 23 other federal agencies and foundations. Information on how to obtain the Add Health data files is available on the Add Health website (http://www.cpc.unc.edu/addhealth). No direct support was received from grant P01-HD31921 for this analysis. The Helsinki Birth Cohort Study (HBCS) thanks all study participants as well as everybody involved in the Helsinki Birth Cohort Study. Helsinki Birth Cohort Study has been supported by grants from the Academy of Finland, the Finnish Diabetes Research Society, Folkhälsan Research Foundation, Novo Nordisk Foundation, Finska Läkaresällskapet, Juho Vainio Foundation, Signe and Ane Gyllenberg Foundation, University of Helsinki, Ministry of Education, Ahokas Foundation, Emil Aaltonen Foundation. The Avon Longitudinal Study of Parents and Children (ALSPAC) is extremely grateful to all the families who took part in the ALSPAC study, the midwives for their help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. Funding/Support was from The UK Medical Research Council and Wellcome (Grant ref: 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and A.C.E. will serve as guarantor for the contents of this paper. A comprehensive list of grants funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/grant-acknowledgements.pdf). This research was specifically funded by NIH grants AA021399. GWAS data was generated by Sample Logistics and Genotyping Facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. Generation Scotland is grateful to the families who took part in GS, the GPs and Scottish School of Primary Care for their help in recruiting them, and the whole GS team that includes academic researchers, clinic staff, laboratory technicians, clerical workers, IT staff, statisticians and research managers. Generation Scotland received core support from the Chief Scientist Office of the Scottish Government Health Directorates (CZD/16/6) and the Scottish Funding Council (HR03006). Genotyping of the GS samples was carried out by the Genetics Core Laboratory at the Wellcome Trust Clinical Research Facility, Edinburgh, Scotland, and was funded by the Medical Research Council UK and the Wellcome Trust (Wellcome Trust Strategic Award ‘STratifying Resilience and Depression Longitudinally’ (STRADL) Reference 104036/Z/14/Z). LD Hub (http://ldsc.broadinstitute.org/) is grateful to the following GWAS studies, databases and consortia who have kindly made their summary data available: ADIPOGen (Adiponectin genetics consortium), C4D (Coronary Artery Disease Genetics Consortium), CARDIoGRAM (Coronary ARtery DIsease Genome wide Replication and Meta-analysis), CKDGen (Chronic Kidney Disease Genetics consortium), dbGAP (database of Genotypes and Phenotypes), DIAGRAM (DIAbetes Genetics Replication And Meta-analysis), ENIGMA (Enhancing Neuro Imaging Genetics through Meta Analysis), EAGLE (EArly Genetics & Lifecourse Epidemiology Consortium, excluding 23andMe), EGG (Early Growth Genetics Consortium), GABRIEL (A Multidisciplinary Study to Identify the Genetic and Environmental Causes of Asthma in the European Community), GCAN (Genetic Consortium for Anorexia Nervosa), GEFOS (GEnetic Factors for OSteoporosis Consortium), GIANT (Genetic Investigation of ANthropometric Traits), GIS (Genetics of Iron Status consortium), GLGC (Global Lipids Genetics Consortium), GPC (Genetics of Personality Consortium), GUGC (Global Urate and Gout consortium), HaemGen (haemotological and platelet traits genetics consortium), HRgene (Heart Rate consortium), IIBDGC (International Inflammatory Bowel Disease Genetics Consortium), ILCCO (International Lung Cancer Consortium), IMSGC (International Multiple Sclerosis Genetic Consortium), MAGIC (Meta-Analyses of Glucose and Insulin-related traits Consortium), MESA (Multi-Ethnic Study of Atherosclerosis), PGC (Psychiatric Genomics Consortium), Project MinE consortium, ReproGen (Reproductive Genetics Consortium), SSGAC (Social Science Genetics Association Consortium) and TAG (Tobacco and Genetics Consortium), TRICL (Transdisciplinary Research in Cancer of the Lung consortium), UK Biobank. We additionally thank the groups who directly shared GWAS results. We would like to acknowledge all participating groups of the International Cannabis Consortium, and in particular the members of the working group including Sven Stringer, Camelia Minica, Karin Verweij, Hamdi Mbarek, Eske Derks, Nathan Gillespie and Jacqueline Vink. Thanks also to the ENIGMA consortium for providing GWAS results on subcortical brain volumes (available from http://enigma.ini.usc.edu/research/download-enigma-gwas-results/). Finally, we acknowledge the valuable contribution of groups who have publicly released summary statistics from their respective GWAS. Specifically, thanks to researchers from Schumann et al. (2016) including the CHARGE+ and AlcGen consortia (results available at https://grasp.nhlbi.nih.gov/FullResults.aspx) and to all members of Psychiatric Genomics Consortium (PGC; results available for download at http://www.med.unc.edu/pgc/results-and-downloads), in particular the working groups on Attention Deficit/Hyperactivity Disorder (ADHD; chaired by Dr. Stephen Faraone), Autism Spectrum Disorder (ASD; chaired by Drs. Mark Daly and Bernard Devlin), and Eating Disorders (ED; chaired by Drs. Cynthia Bulik and Gerome Breen). Similar thanks to all of the participating groups in the Lundbeck Foundation Initiative for Integrative Psychiatric Research (iPSYCH) consortium for their participation in the ADHD and ASD meta-analyses.
Accession Codes
Comorbidity and Trauma Study (CATS): dbGAP accession phs000277.v1.p1
Center for Education and Drug Abuse Research (CEDAR): dbGAP accession phs001649.v1.p1
Christchurch Health and Development Study (CHDS): dbGAP submission in process
The Collaborative Study on the Genetics of Alcoholism (COGA): dbGaP accession numbers phs000125.v1.p1, phs000763.v1.p1, and phs000976.v1.p1
Study of Addiction: Genetics and Environment (SAGE): dbGAP accession phs000092.v1.p1
Collaborative Genetic Study of Nicotine Dependence (COGEND): dbGAP accession phs000404.v1.p1
Gene-Environment-Development Initiative (GEDI) – Duke University (GSMS): dbGAP accession phs000852.v1.p1
Center on Antisocial Drug Dependence (CADD): dbGAP submission in process
Spit for Science: dbGAP submission in process
NIAAA: available via https://btris.nih.gov/
Gene-Environment-Development Initiative (GEDI) –Virginia Commonwealth University (VTSABD): dbGAP submission in process
Minnesota Center for Twin and Family Research (MCTFR): dbGAP accession phs000620.v1.p1
Yale-Penn: dbGAP accession phs000425.v1.p1 and phs000952.v1.p1
See Data Availability for information on accessing other cohorts.
Author Contributions
R.K.W., H.J.E, J.G. and A.A. conceived the analyses, wrote the first draft and prepared all drafts for submission; R.K.W. conducted primary analyses; R.P., E.C.J. and J.N.M. conducted additional analyses. H.J.E., J.G., A.A. and B.N. supervised all analyses. M.J.A., A.E.A., F.A., S.-A.B., A.B., S.B., J.M.Bi., T.B.B., L-S.C., T.-K.C., Y.-L.C., F.D., A.R.D., A.C.E., P.F., J.C.F., L.F., J.F., I.G., S.G., L.M.H., A.M.H., S.M.H., S.H.-H., S.H., C.Ho., P.H. J.J.H., M.A.K., M.A.-K., B.K., J.L., M.L.-P., D.L., L.L., A.L., B.S.M., H.M., A.M.M., M.B.M., J.L.M., Y.M., T.P., J.F.P., R.E.P., S.R., E.R., N.L.S., J.E.S., S.S-R., Me.S., R.S., F.S., J.S., N.T., J.-C.W., B.T.W., R.W., L.W. A.G.W. prepared individual datasets and, in some cases, conducted analyses and provided summary statistics or results. 23andMe Research Team, J.D.B., D.C., D.-S.C., W.E.C., R.C.C., N.D., L.D., B.W.D., S.L.E., M.A.F., W.G., C.Ha., M.I., M.K., F.K., J.K., S.K., S.L., M.T.L., W.M., K.M., S.M., B.M.-M., A.D.M., J.I.N., A.P., U.P., K.R., M.D.R., Mo.R., N.S., M.A.S., M.S., J.T., S.W, N.W., P.Z. provided critical support regarding phenotypes and data in individual datasets; D.E.A., J.M.Bo., D.I.B., L.J.B., S.A.B., K.K.B., S.C., E.J.C., H.dW., N.D., D.M.D., J.G.E., L.A.F., T.M.F., N.A.G., A.M.G., D.G., R.A.G., D.B.H., K.M.H., A.C.H., V.H., J.K.H., C.J.H., J.H., W.I., E.O.J., J.A.K., V.M.K., K.S.K., H.R.K., K.K., P.L., P.A.L., M.M., J.M., P.A.F.M., H.H.M., P.M., N.G.M., S.E.M., G.W.M., E.C.N., M.M.N., A.A.P., N.L.P., B.W.J.H.P., B.P., J.P.R., Ma.R., B.P.R., R.R., D.R., P-H.S., J.S., M.C.S., R.E.T., M.M.V., S.V., T.L.W., J.B.W., H.Z. as well as H.J.E. & J.G. facilitated data collection and provided critical phenotypic and analytic feedback for individual studies. B.N. and A.A. provided resource support. All authors reviewed the manuscript and approved it for submission.
Competing Interests
L.J.B, A. M. G., J. P. R., J-C. W. and the spouse of N.L.S. are listed as inventors on Issued U.S. Patent 8080,371, “Markers for Addiction” covering the use of certain SNPs in determining the diagnosis, prognosis, and treatment of addiction. N.W. has received funding from the German Research Foundation (DFG) and Federal Ministry of Education and Research Germany (BMBF); he has received speaker’s honoraria and travel funds from Janssen-Cilag and Indivior. He took part in industry sponsored multi-center randomized trials by D&A pharma and Lundbeck. Mo.R. received compensation from Lundbeck Switzerland and Lundbeck institute for advisory boards and expert meeting, and from Lundbeck and Lilly Suisse for workshops and presentations. K.M. received honoraria from Lundbeck, Pfizer, Novartis and AbbVie. K.M. also received Honoraria (Advisory Board) from Lundbeck and Pfizer and speaker fees from Janssen Cilag. H.K. has been an advisory board member, consultant, or continuing medical education speaker for Indivior, Lundbeck, and Otsuka. He is a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was sponsored in the past three years by AbbVie, Alkermes, Amygdala Neurosciences, Arbor Pharmaceuticals, Ethypharm, Indivior, Lilly, Lundbeck, Otsuka, and Pfizer. H.K. and J.G. are named as inventors on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018. P.F., S.L.E. and members of the 23andMe Research Team are employees of 23andMe. M.A.F. has received grant support from Assurex Health, Mayo Foundation, Myriad, National Institute of Alcohol Abuse and Alcoholism (NIAAA), National Institute of Mental Health (NIMH), and Pfizer; he has been a consultant for Intra-Cellular Therapies, Inc., Janssen, Mitsubishi Tanabe Pharma Corporation, Myriad, Neuralstem Inc., Otsuka American Pharmaceutical, Sunovion, and Teva Pharmaceuticals. H. dW. has received support from Insys Therapeutics and Indivior for studies unrelated to this project, and she has consulted for Marinus and Jazz Pharmaceuticals, also unrelated to this project. T.L.W. has previously received funds from ABMRF. J.N. is an investigator for Janssen and Assurex. M.M.N. has received honoraria from the Lundbeck Foundation and the Robert Bosch Stiftung for membership on advisory boards. Mo.R. has received honoraria from Lundbeck Switzerland and the Lundbeck Institute for membership of advisory boards and participation in expert meetings, and from Lundbeck and Lilly Suisse for workshops and presentations. N.S. has received honoraria from Abbvie, Sanofi-Aventis, Reckitt Benckiser, Indivior, Lundbeck, and Janssen-Cilag for advisory board membership and the preparation of lectures, manuscripts, and educational materials. Since 2013, N.S. has also participated in clinical trials financed by Reckitt Benckiser and Indivior. N.W. received speaker’s honoraria and travel expenses from Janssen-Cilag and Indivior; has also participated in industry sponsored multi-center randomized trials conducted by D&A pharma and Lundbeck. W.G. has received symposia support from Janssen-Cilag GmbH, Neuss, Lilly Deutschland GmbH, Bad Homburg, and Servier, Munich, and is a member of the Faculty of the Lundbeck International Neuroscience Foundation (LINF), Denmark. J.A.K. has provided consultations on nicotine dependence for Pfizer (Finland) 2012-15. In the past three years, L.D. has received investigator-initiated untied educational grants for studies of opioid medications in Australia from Indivior, Mundipharma and Seqirus. B.M.N. is a member of the scientific advisory board for Deep Genomics and has consulted for Camp4 Therapeutics Corporation, Merck & Co. and Avanir Pharmaceuticals, Inc. A.A. previously received peer-reviewed funding and travel reimbursement from ABMRF for unrelated research.
Contributors
23andMe Research Team: Michelle Agee17, Babak Alipanahi17, Adam Auton17, Robert K. Bell17, Katarzyna Bryc17, Sarah L. Elson17, Pierre Fontanillas17, Nicholas A. Furlotte17, David A. Hinds17, Karen E. Huber17, Aaron Kleinman17, Nadia K. Litterman17, Jennifer C. McCreight17, Matthew H. McIntyre17, Joanna L. Mountain17, Elizabeth S. Noblin17, Carrie A.M. Northover17, Steven J. Pitts17, J. Fah Sathirapongsasuti17, Olga V. Sazonova17, Janie F. Shelton17, Suyash Shringarpure17, Chao Tian17, Joyce Y. Tung17, Vladimir Vacic17, and Catherine H. Wilson17
References
- 1.World Health Organization. Global status report on alcohol and health, 2014. (World Health Organization, 2014). [Google Scholar]
- 2.American Psychiatric Association. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Am. Psychiatr. Assoc (2000). doi: 10.1176/appi.books.9780890423349 [DOI] [Google Scholar]
- 3.Hasin D, Stinson F, Ogburn E & Grant B Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: Results from the national epidemiologic survey on alcohol and related conditions. Arch. Gen. Psychiatry 64, 830–842 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Dawson DA, Grant BF, Stinson FS & Chou PS Estimating the effect of help-seeking on achieving recovery from alcohol dependence. Addiction 101, 824–834 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Verhulst B, Neale MC & Kendler KS The heritability of alcohol use disorders: a meta-analysis of twin and adoption studies. Psychol. Med 45, 1061–1072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hart AB & Kranzler HR Alcohol dependence genetics: Lessons learned from genome-wide association studies (GWAS) and post-GWAS analyses. Alcohol. Clin. Exp. Res 39, 1312–1327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer RHC et al. Shared additive genetic influences on DSM-IV criteria for alcohol dependence in subjects of European ancestry. Addiction 110, 1922–1931 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurley TD & Edenberg HJ Genes encoding enzymes involved in ethanol metabolism. Alcohol Res 34, 339–344 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li D, Zhao H & Gelernter J Strong association of the alcohol dehydrogenase 1B gene (ADH1B) with alcohol dependence and alcohol-induced medical diseases. Biol. Psychiatry 70, 504–512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bierut LJ et al. ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol. Psychiatry 17, 445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frank J et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addict. Biol 17, 171–180 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li D, Zhao H & Gelernter J Strong protective effect of the aldehyde dehydrogenase gene (ALDH2) 504lys (* 2) allele against alcoholism and alcohol-induced medical diseases in Asians. Hum. Genet 131, 725–737 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luczak SE, Glatt SJ & Wall TJ Meta-analyses of ALDH2 and ADH1B with alcohol dependence in Asians. Psychol. Bull 132, 607–621 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Sullivan PF et al. Psychiatric genomics: an update and an agenda. Am. J. Psychiatry 175, 15–27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol 11, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schumann G et al. KLB is associated with alcohol drinking, and its gene product β-Klotho is necessary for FGF21 regulation of alcohol preference. Proc Natl Acad Sci USA 113, 14372–14377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clarke T-K et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N=112,117). Molecular Psychiatry (2017). doi: 10.1038/mp.2017.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Roige S et al. Genome-wide association study of Alcohol Use Disorder Identification Test (AUDIT) scores in 20,328 research participants of European ancestry. Addict. Biol (2017). doi: 10.1111/adb.12574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Roige S et al. Genome-wide association study of delay discounting in 23,217 adult research participants of European ancestry. Nat. Neurosci 21, 16–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lonsdale J et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudbridge F & Gusnanto A Estimation of significance thresholds for genomewide association scans. Genet. Epidemiol 32, 227–234 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pe’er I, Yelensky R, Altshuler D & Daly MJ Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol 32, 381–385 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Derks EM, Zwinderman AH & Gamazon ER The relation between inflation in Type-I and Type-II error rate and population divergence in genome-wide association analysis of multi-ethnic populations. Behav. Genet 47, 360–368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng J et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics 33, 272–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans LM et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat. Genet 50, 737–745 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wray NR & Maier R Genetic basis of complex genetic disease: The contribution of disease heterogeneity to missing heritability. Curr. Epidemiol. Reports 1, 220–227 (2014). [Google Scholar]
- 29.Polimanti R et al. Evidence of causal effect of major depressive disorder on alcohol dependence: Findings from the Psychiatric Genomics Consortium. bioRxiv (2018). doi: 10.1101/412098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez-Roige S et al. Genome-wide association study meta-analysis of the Alcohol Use Disorder Identification Test (AUDIT) in two population-based cohorts (N=141,958). bioRxiv (2018). doi: 10.1101/275917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munafò MR, Tilling K, Taylor AE, Evans DM & Davey Smith G Collider scope: when selection bias can substantially influence observed associations. Int. J. Epidemiol (2017). doi: 10.1093/ije/dyx206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fry A et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am. J. Epidemiol (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koob GF & Volkow ND Neurobiology of addiction: a neurocircuitry analysis. The Lancet Psychiatry 3, 760–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mbarek H et al. The genetics of alcohol dependence: Twin and SNP-based heritability, and genome-wide association study based on AUDIT scores. Am. J. Med. Genet. Part B Neuropsychiatr. Genet 168, 739–748 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Prescott CA, Aggen S & Kendler KS Sex-specific genetic influences on the comorbidity of alcoholism and major depression in a population-based sample of us twins. Arch. Gen. Psychiatry 57, 803–811 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Kendler KS, Prescott CA, Myers J & Neale MC The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch. Gen. Psychiatry 60, 929–937 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Andersen AM et al. Polygenic scores for major depressive disorder and risk of alcohol dependence. JAMA Psychiatry 74, 1153–1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou HP et al. Genetic risk variants associated with comorbid Alcohol Dependence and Major Depression. JAMA Psychiatry (2017). doi: 10.1001/jamapsychiatry.2017.3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kreek MJ, Nielsen DA, Butelman ER & LaForge KS Genetic influences on impulsivity, risk taking, stress responsivity and vulnerability to drug abuse and addiction. Nature Neuroscience (2005). doi: 10.1038/nn1583 [DOI] [PubMed] [Google Scholar]
- 40.Polimanti R et al. The interplay between risky sexual behaviors and alcohol dependence: Genome-wide association and neuroimaging support for LHPP as a risk gene. Neuropsychopharmacology 42, 598–605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grant BF et al. Epidemiology of DSM-5 Alcohol Use Disorder: Results from the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 72, 757–766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaffee S, Caspi A, Moffitt TE, Belsky J & Silva P Why are children born to teen mothers at risk for adverse outcomes in young adulthood? Results from a 20-year longitudinal study. Dev. Psychopathol (2001). doi: 10.1017/S0954579401002103 [DOI] [PubMed] [Google Scholar]
- 43.Martin J, Taylor MJ & Lichtenstein P Assessing the evidence for shared genetic risks across psychiatric disorders and traits. Psychol. Med 1–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pratt DS & Kaplan MM Evaluation of abnormal liver-enzyme results in asymptomatic patients. N Eng J Med 342, 1266–1271 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Polimanti R et al. Genome-wide association study of body mass index in subjects with alcohol dependence. Addict. Biol 22, 535–549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Traversy G & Chaput J-P Alcohol consumption and obesity: an update. Curr. Obes. Rep 4, 122–130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wray NR et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet 50, 668–681 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zerbino DR et al. Ensembl 2018. Nucleic Acids Res 4, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-Only References
- 50.American Psychiatric Association; Diagnostic and Statistical Manual of Mental Health Disorders, Third Edition, Revised (American Psychiatric Association Press, 1987). [Google Scholar]
- 51.O’Connell J et al. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet 10, e1004234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Howie BN, Donnelly P & Marchini J A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Howie B, Marchini J & Stephens M Genotype imputation with thousands of genomes. G3 Genes, Genomes, Genet 1, 457–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duncan LE et al. Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol. Psychiatry (2018). doi: 10.1038/mp.2017.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hancock DB et al. Assessment of genotype imputation performance using 1000 Genomes in african american studies. PLoS One 7, e50610 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han B & Eskin E Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet 88, 586–598 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris AP Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol 35, 809–822 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen M & Yang Q GWAF: an R package for genome-wide association analyses with family data. Bioinformatics 26, 580–581 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Firth D Bias reduction of maximum-likelihood-estimates. Biometrika 80, 27–38 (1993). [Google Scholar]
- 61.Skol AD, Scott LJ, Abecasis GR & Boehnke M Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet 38, 209–213 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Berndt SI et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat. Genet 45, 501–512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee SH, Wray NR, Goddard ME & Visscher PM Estimating missing heritability for disease from genome-wide association studies. Am. J. Hum. Genet 88, 294–305 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chambers JC et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet 43, 1131–1138 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stringer S et al. Genome-wide association study of lifetime cannabis use based on a large meta-analytic sample of 32 330 subjects from the International Cannabis Consortium. Transl. Psychiatry 6, e769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hancock DB et al. Genome-wide association study across European and African American ancestries identifies a SNP in DNMT3B contributing to nicotine dependence. Mol. Psychiatry (2017). doi: 10.1038/mp.2017.193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Demontis D et al. Discovery of the first genome-wide significant risk loci for ADHD. bioRxiv 145581 (2017). doi: 10.1101/145581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duncan L et al. Significant locus and metabolic genetic correlations revealed in genome-wide association study of anorexia nervosa. Am. J. Psychiatry 174, 850–858 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anney RJL et al. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hibar DP et al. Common genetic variants influence human subcortical brain structures. Nature 520, 224–229 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boyd A et al. Cohort profile: The ‘Children of the 90s’-The index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol 42, 111–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fraser A et al. Cohort profile: The Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol 42, 97–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith BH et al. Cohort profile: Generation Scotland: Scottish family health study (GS: SFHS). The study, its participants and their potential for genetic research on health and illness. Int. J. Epidemiol 42, 689–700 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics from the genome-wide meta-analyses are available on the Psychiatric Genomics Consortium’s downloads page (http://www.med.unc.edu/pgc/results-and-downloads), including the source data for Figures 1 and 2. Individual-level data from the genotyped cohorts and cohort-level summary statistics will be made available to researchers following an approved analysis proposal through the PGC Substance Use Disorder group with agreement of the cohort PIs; contact the corresponding authors for details. Cohort data are also available from dbGaP except where prohibited by IRB or European Union data restrictions. Expression data used to evaluate variants in ADH1B is available from GTEx (https://gtexportal.org/home/). Hi-C data used to evaluate the chromosome 3 variant can be queried with HUGIn (https://yunliweb.its.unc.edu/hugin/). Publicly available genome-wide summary statistics used for testing genetic correlations are accessible through LD Hub (http://ldsc.broadinstitute.org/), or from the Psychiatric Genomics Consortium (http://www.med.unc.edu/pgc/results-and-downloads), the Social Science Genetic Association Consortium (SSGAC; https://www.thessgac.org/data), Enhancing Neuro Imaging Genetics through Meta Analysis (ENIGMA; http://enigma.ini.usc.edu/research/download-enigma-gwas-results/), and the Neale Lab (http://www.nealelab.is/uk-biobank); for availability of summary statistics from other studies contact the respective authors. The source data for Figure 3 is included in Supplementary Table S6.