

Abstract
Triple-negative breast cancer (TNBC), the most aggressive breast cancer subtype, currently lacks effective targeted therapy options. Eicosapentaenoic acid (EPA), an omega-3 fatty acid and constituent of fish oil, is a common supplement with anti-inflammatory properties. Although it is not a mainstream treatment, several preclinical studies have demonstrated that EPA exerts anti-tumor activity in breast cancer. However, against solid tumors, EPA as a monotherapy is clinically ineffective; thus, we sought to develop a novel targeted drug combination to bolster its therapeutic action against TNBC. Using a high-throughput functional siRNA screen, we identified Ephrin type-A receptor 2 (EPHA2), an oncogenic cell-surface receptor tyrosine kinase, as a therapeutic target that sensitizes TNBC cells to EPA. EPHA2 expression was uniquely elevated in TNBC cell lines and patient tumors. In independent functional expression studies in TNBC models, EPHA2 gene-silencing combined with EPA significantly reduced cell growth and enhanced apoptosis compared with monotherapies, both in vitro and in vivo. EPHA2 specific inhibitors similarly enhanced the therapeutic action of EPA. Finally, we identified that therapy-mediated apoptosis was attributed to a lethal increase in cancer cell membrane polarity due to ABCA1 inhibition and subsequent dysregulation of cholesterol homeostasis. This study provides new molecular and pre-clinical evidence to support a clinical evaluation of EPA combined with EPHA2 inhibition in patients with TNBC.
Keywords: Eicosapentaenoic Acid (EPA), EPHA2, Triple-Negative Breast Cancer, Dasatinib, Membrane Dynamics
Introduction
Triple-negative breast cancer (TNBC) is an aggressive disease that comprises 10–20% of all breast cancers. It is a heterogeneous disease that is often characterized by its strong metastatic potential and poor prognosis compared to estrogen receptor (ER)/progesterone receptor (PgR)-positive and HER2-positive breast cancers 1. While conventional chemotherapy is effective in the short term, TNBC often becomes refractory, and the lack of targeted therapy hampers a clinical solution for this disease 2.
Inflammation, a biological process designed to fight infections and heal wounds, can inadvertently support tumor formation and growth by supplying bioactive molecules that facilitate tumor progression and metastasis 3. Pathological assessment of TNBC has identified increased expression of molecular mediators of inflammation, such as prostaglandin G/H synthase 2 (COX2), and prostaglandin E2 (PGE2), representing potential therapeutic targets 4. Recent finding by our laboratory have observed that inhibition of inflammatory pathways through administration of celecoxib (a COX2 inhibitor) 5, or as observed by other using Lovaza (a highly purified, prescription-strength form of the omega-3 acid ethyl esters [O3AEE]: docosahexaenoic acid, DHA, and eicosapentaenoic acid, EPA) 6, can impair the growth of TNBC cells in vitro. Supporting our results, omega-3 fatty acid supplementation has been shown to reduce the growth of rat sarcoma tumors and DMBA-induced mammary tumors in vivo 7–9. Collectively, these studies suggest that the anti-inflammatory action of O3AEE have therapeutic potential. However, the translation of these compounds has been hindered by: 1) inconsistencies in sources, routes of administration and O3AEE composition 9, 2) absence of an established biomarker for therapeutic action, and 3) no definitive subpopulation of breast cancer patients that would benefit from therapy. As a result, there is a critical and unmet need to develop a rational, targeted-approach for the clinical testing of O3AEE in TNBC.
Towards greater clarity regarding the use of O3AEE as a therapeutic, we sought to investigate the anti-tumor effect of highly purified EPA (Vascepa, icosapent ethyl; Amarin Pharma Inc), which was recently approved by the U.S. Food and Drug Administration (FDA) for the treatment of hypertriglyceridemia 10. We demonstrate that EPA has potent tumor suppressive activity in preclinical models of TNBC. However, with no established therapeutic role for EPA in the TNBC patient population, translation of an EPA-based therapy through combination with conventional cancer therapy requires justification.
The main goal of this study was to identify a molecular target that could be targeted in combination with EPA for the effective clinical treatment of TNBC. To this end, we detail a functional genomics-based screen that identified the receptor tyrosine kinase EPHA2 as a therapeutically druggable target that enhances EPA-based therapy in TNBC, and present relevant preclinical studies that establish the rationale for a phase I clinical trial for patients with TNBC.
Results
EPA inhibits the growth of TNBC tumor xenografts
While O3AEE demonstrate anti-tumor effects 8, EPA as a monotherapy in TNBC has not yet been tested. Thus, we initially assessed the anti-tumorigenic potential of EPA in a preclinical xenograft tumor model of TNBC (SUM149PT). EPA therapy was well tolerated at both 0.4 g/kg and 0.8 g/kg doses (equivalent to the human FDA-approved EPA dose; Supplementary Figure S1), with no change in body weight noted (data not shown). EPA levels were readily detectable in the sera obtained from mice undergoing therapy (Figure 1A) and, importantly, were significantly elevated in the cell membrane (phospholipid) fraction of TNBC tumor xenografts (Figure 1B). EPA therapy dose-dependently inhibited the growth of SUM149PT xenografts (Figure 1C), which led to a significant extension in survival (designated as the time required to obtain a 1 500 mm3 tumor) (Figure 1D). These data suggest that EPA is therapeutically active in vivo and can reduce the growth of aggressive TNBC xenografts.
Figure 1. Eicosapentaenoic acid (EPA) reduced tumor growth and prolonged mouse survival in a preclinical xenograft model of TNBC.
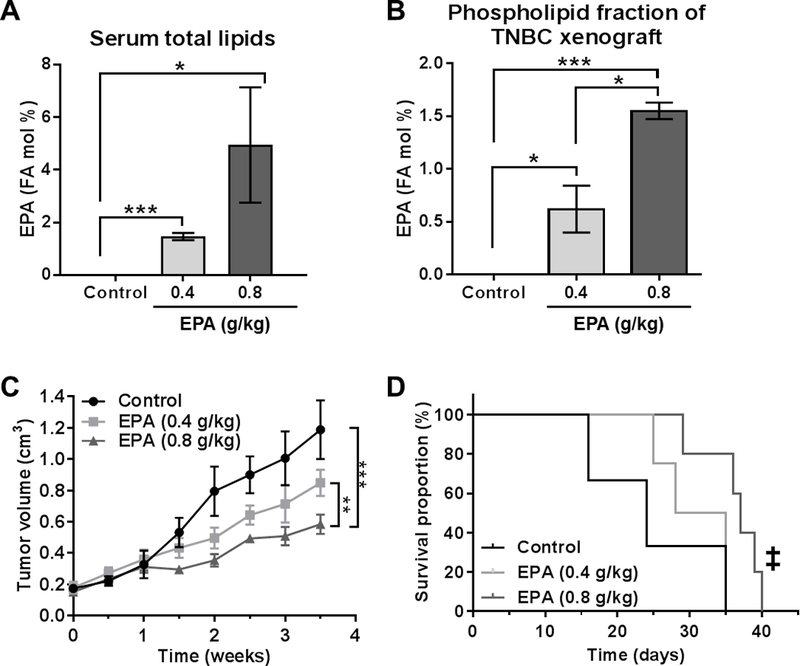
SUM149PT tumor-bearing mice (n = 5) were treated with EPA (0.4 and 0.8 g/kg) or vehicle (control). Gas chromatography-mass spectrometry was used to analyze the fatty acid (FA) mol percentage of EPA in regard to serum total lipids (A) and the tumor phospholipid fraction (B). Mice treated with EPA or control were monitored for (C) tumor growth (volume) and (D) survival (endpoint = 1 500 mm3 tumor size). A-C, Differences between groups were compared by unpaired t-test: *, P < 0.05; **, P < 0.001; ***, P < 0.0001. D, Differences between groups were compared by log-rank test: ‡, P < 0.05 compared to control and EPA (4 g/kg).
EPHA2, a rational target for EPA-based combination therapy in TNBC
As our goal is to incorporate EPA as part of a conventional targeted therapy strategy, we performed a functional genomic, synthetic-lethal siRNA screen in SUM149PT cells to identify potential gene candidates that enhance the sensitivity of TNBC cells to EPA. For this screen, we utilized a library of kinases with known targeting drugs, or ‘druggable’ kinases, to enhance the translational potential of the identified targets. Our screen, consisting of 939 druggable kinase genes, identified 36 targets able to modulate SUM149PT cells’ sensitivity to EPA. Of these 36 targets, 20 genes sensitized SUM149PT cells to EPA, with EPHA2, DUSP4, and EDG2 demonstrating superior therapeutic action in the presence of EPA (Figure 2A). We focused on EPHA2 (ephrin type-A receptor 2), a cell surface receptor tyrosine kinase, because of the known association of its increased expression with cancer progression 11. Independent validation experiments, using three different pooled siRNA sets, confirmed that EPHA2 is a lead target for an EPA-based combination therapy in TNBC (Figure 2B).
Figure 2. EPHA2 is a clinically significant target that enhances TNBC cell sensitivity to EPA.
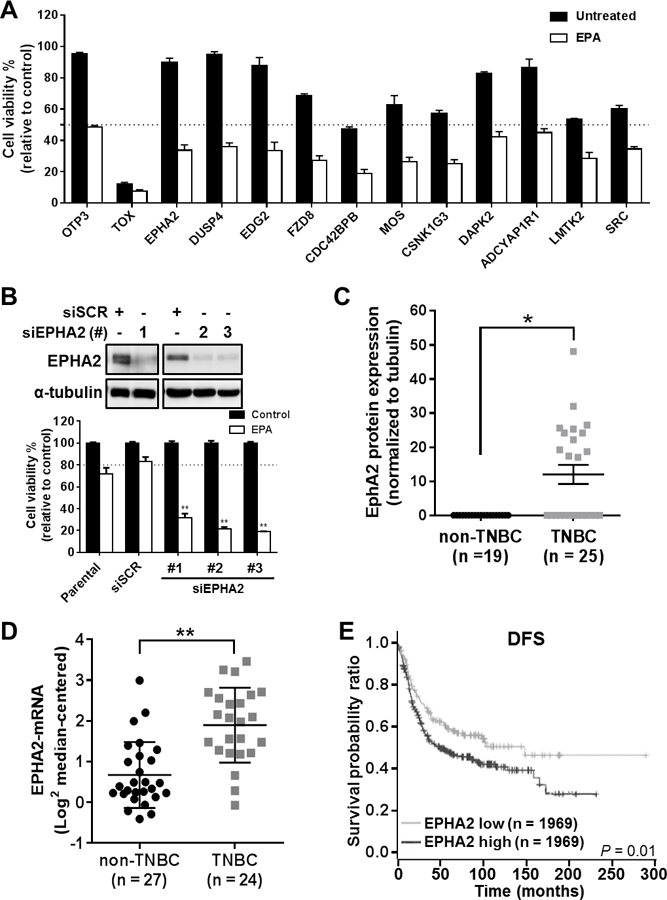
(A) Cell viability readout for the 12 most significant gene siRNAs for sensitizing SUM149PT cells to EPA, identified by high-throughput siRNA synthetic-lethal functional screen. TOX and OTP3 were used as positive and negative transfection controls, respectively. EPHA2 was identified as a top candidate for sensitizing TNBC cells to EPA therapy. (B) EPHA2 inhibition was validated in the cells by immunoblotting analysis with anti-EPHA2 antibody (top) after transfection with three different EPHA2 siRNAs (#1–3) or scrambled control siRNA (siSCR). The effects of EPHA2 inhibition in these cells and in parental (untransfected) cells, alone or in combination with EPA treatment, were confirmed by viability assays (bottom). Data were pooled from three independent experiments and are presented as mean ± SD. (C) EPHA2 protein expression levels were compared in TNBC and non-TNBC cell lines by a capillary-based immunoassay (Simple Western™). The chemiluminescent signal for EPHA2 protein expression was normalized with the signal for tubulin protein expression for each cell line, and these ratios were used to generate the graph; tubulin expression was used as a protein loading control. The mean ± SEM is indicated. (D) EPHA2 mRNA expression levels extracted from a previously published dataset of breast cancer cell lines 12 were compared for TNBC and non-TNBC cell lines. The mean ± SD is indicated. Differences between groups were compared by unpaired t-test: *, P < 0.001; **, P < 0.0001. (E) Kaplan-Meier survival curves were generated for disease-free survival (DFS) of patients with TNBC/basal-like breast cancer classified by EPHA2 mRNA levels in tumors. Data were extracted from the BreastMark mRNA dataset. The log-rank test was used to compare survival curves for high (above median) versus low EPHA2 expression. The initial numbers of patients at risk in each group are indicated in the key.
To evaluate EPHA2 expression in TNBC, we analyzed the protein and mRNA expression of EPHA2 in breast cancer cell lines and in publicly available patient tumor datasets 12. We found that EPHA2 protein levels were significantly higher in TNBC cell lines than in hormone receptor-positive and HER2-positive breast cancer cell lines (Figure 2C and Supplementary Table S1). Similarly, in a previously published collection of 51 breast cancer cell lines, we found that EPHA2 mRNA expression was significantly higher in those with a triple-negative molecular subtype (Figure 2D). Finally, in a population of basal-like (PAM50 molecular classifier) breast cancer patients (60 to 90% TNBC molecular subtype) 13, 14, we discovered that elevated EPHA2 mRNA expression (above the median value) within patients’ tumors was significantly associated with shorter disease-free survival (DFS) (P = 0.01; Figure 2E; hazard ratio = 1.45 [1.08 – 1.94]), and shorter overall survival (OS) which, while approaching significance, is in line with our DFS findings (P = 0.06; Supplementary Figure S2; hazard ratio = 1.563 (0.9712 −2.515). EPHA2 expression did not stratify outcome in hormone receptor-positive or HER2-positive patient samples (data not shown). These results identify EPHA2 as a clinically relevant target in TNBC.
EPA therapy in combination with EPHA2 inhibition synergistically kills TNBC cells through induction of apoptosis
To demonstrate the importance of targeting EPHA2 in our EPA-based therapeutic strategy for TNBC, we utilized two approaches: 1) functional gene silencing and 2) drug-based inhibition studies. We genetically engineered two TNBC cell lines, SUM149PT and BCX010, to stably express a doxycycline (Dox)-inducible EPHA2-shRNA (shEPHA2) or non-targeting-shRNA (vector) cassettes using the pTRIPZ lentiviral system. Specific EPHA2 and p-EPHA2 silencing was observed in both cell lines compared to the non-targeting shRNA construct (vector), while no effects were observed on SRC and p-SRC expression (Figure 3A). In both SUM149PT and BCX010 TNBC cell lines, EPA therapy was more effective when EPHA2 was silenced (P < 0.05) (Figure 3B), and the loss of cell viability was associated with a greater induction of apoptosis (~10% and ~20%, respectively) compared to monotherapies (Figure 3B and Supplementary Figure S3A).
Figure 3. EPHA2 gene silencing or targeted EPHA2 inhibition acts synergistically with EPA against TNBC in vitro in decreasing proliferation and enhancing apoptosis.
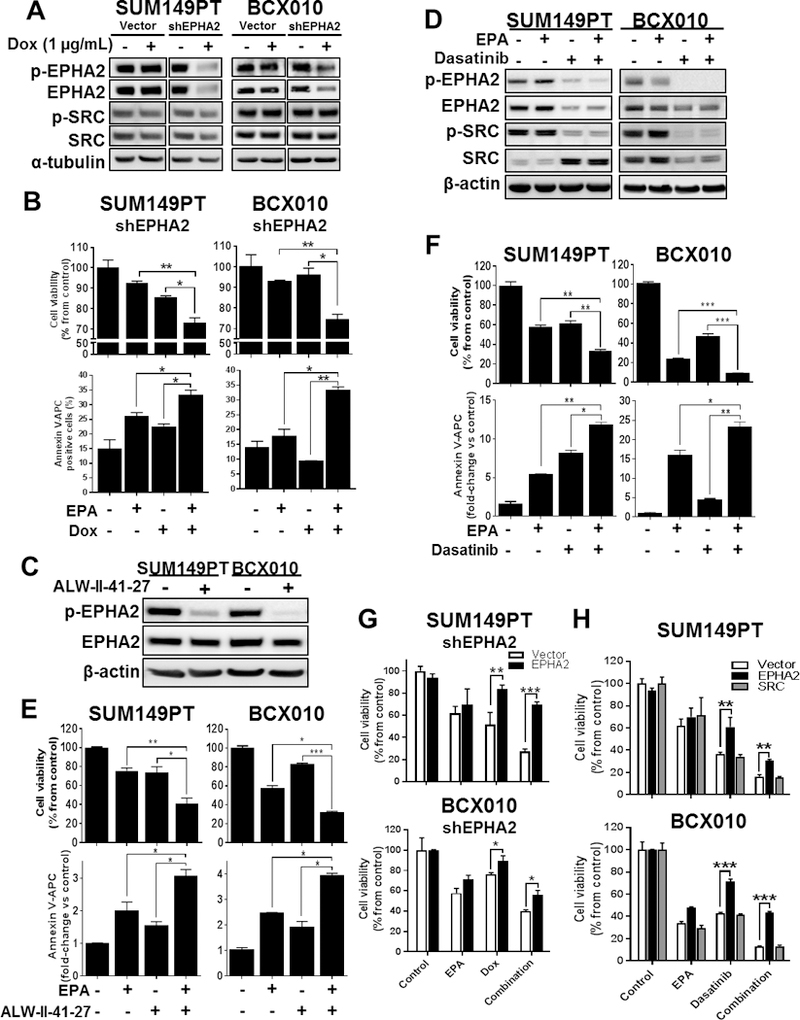
(A) EPHA2 gene silencing in doxycycline (Dox)-inducible SUM149PT-shEPHA2 and BCX010-shEPHA2 knockdown cells not treated (control) or treated with Dox was confirmed by immunoblotting analysis of EPHA2 and phospho-EPHA2 protein expression. SRC and phospho-SRC protein expression were additionally assessed by immunoblotting assay. Expression of α-tubulin was used as a protein loading control. (B) Viability (top) and apoptosis (bottom) were quantified by cell proliferation and Annexin V staining, respectively, in Dox-inducible SUM149PT-shEPHA2 and BCX010-shEPHA2 cells following Dox induction in combination with EPA. (C-D) Effects of EPHA2 inhibitor ALW-II-41–27 (C) and dasatinib (D) on EPHA2, p-EPHA1, SRC and/or p-SRC expression levels in parental SUM149PT and BCX010 TNBC cells were assessed by immunoblotting. Expression of β-actin was used as a protein loading control. (E-F) Viability (top) and apoptosis (bottom) were quantified in SUM149 and BCX010 cells treated with EPA in combination with ALW-II-41–27 (E) or dasatinib (F). (G-H) Viability was quantified by cell proliferation following (G) EPHA2-expression rescue experiments in shEPHA2-inducible TNBC cells, as well as following (H) EPHA2 and SRC-expression rescue in parental TNBC cells that were treated with EPA and/or Dox/dasatinib. For all graphs, data were pooled from three independent experiments and are presented as mean ± SD. Differences between groups were compared by unpaired t-test: *, P < 0.05; **, P < 0.001; ***, P < 0.0001.
In our second approach, we assessed drug-based EPHA2 inhibition in combination with EPA therapy. We selected two drugs: 1) ALW-II-41–27, an ATP-competitive EPHA2-specific inhibitor 15, and 2) dasatinib, an FDA-approved small molecular inhibitor of EPHA2 and SRC. Both ALW-II-41–27 and dasatinib were able to inhibit phospho-EPHA2 levels in SUM149PT and BCX010 cell lines, with dasatinib also inhibiting total EPHA2 and phospho-SRC expression (Figures 3C-D). In combination with EPA, both ALW-II-41–27 and dasatinib displayed synergy against SUM149PT with combination index (CI) scores of 0.8 and 0.2, respectively, and against BCX010 with CI scores of 0.7 and 0.6, respectively. Similar to the results seen with our gene-targeting approach (Figure 3B), the reduced viability of TNBC cells following combination therapy was due to a reduction of proliferative capacity and induction of apoptosis (Figures 3E-F). We observed similar combination therapy synergy in BT549, an EPHA2-positive TNBC cell line. Collectively, these results provide evidence that high EPHA2 expression is required in TNBC cells, regardless of inflammatory breast cancer status, in order for our gene-specific silencing and drug studies to achieve synergistic cytotoxicity after combination therapy. Although ALW-II-41–27 has high specificity to EPHA2, its translational potential is hindered as this drug will not be entering testing in human clinical trials. Thus, dasatinib was selected as our lead drug for translational development.
To determine whether EPHA2 inhibition is critical to combination therapy sensitivity, we performed gain-of-expression studies to assess the response of SUM149PT and BCX010 cells to EPA in combination with Dox-inducible shEPHA2 or dasatinib (Figure 3G-H and Supplementary Figure S3B). Overexpression of EPHA2 significantly blocked sensitivity of cells to EPA in combination with Dox-induced shEPHA2 (Figure 3G) or dasatinib therapy (Figure 3H), reversing cell viability when compared to controls (P < 0.05). Additionally, we tested whether SRC-expression rescue plays a role reversing TNBC cells’ sensitivity to EPA and dasatinib combination therapy (Figure 3H). Following induced SRC expression, we didn’t observe any significant changes in cell viability, suggesting that the observed sensitivity to combination EPA and dasatinib therapy is EPHA2-specific, and does not incorporate EPHA2 signaling associated with SRC. Altogether, these results provide functional evidence that EPA-based combination therapies induce apoptosis in TNBC cells in an EPHA2-inhibition dependent manner.
Combination therapy modifies cell membrane rigidity and cholesterol composition in TNBC cells
Highly lipophilic EPA is known to be incorporated into the plasma membrane of cells, where it can alter membrane structure and protein localization 16. As we demonstrated that 1) EPA could be integrated into the lipid bilayer of tumor xenografts (Figure 1B) and 2) the list of candidate genes identified in our siRNA screen was enriched with those associated with cell membrane biology (as indicated by gene ontology terms GO:0044853, plasma membrane raft; GO:0045121, membrane raft; and GO:0031234, extrinsic component of cytoplasmic side of plasma membrane; P < 0.05), we hypothesized that the effectiveness of the EPA plus EPHA2-inhibition combination therapy was due to alterations within the cell membrane.
Alterations in cellular membrane fluidity, or polarity, can affect the localization of protein receptors, leading to altered signaling pathways, biological processes, and cell viability 16, 17. Following EPA treatment, through mass spectrometry-based content analysis of cell membrane fractions, we observed that EPA was incorporated into the cell membrane (Supplementary Figure S4A). Further, while EPA treatment did not alter total EPHA2 expression within whole-cell lysates, it did reduce the localization of the protein on the cell surface (Supplementary Figure S4B), which may have implications for canonical EPHA2 signaling and suggests a probable change in membrane dynamics. To assess these changes, we measured the general polarization (GP; a surrogate measure of membrane fluidity) index following treatment with EPA and genetic/drug-based inhibition of EPHA2. Strikingly, EPA treatment in combination with either EPHA2 gene silencing or dasatinib treatment led to significant increase in the GP of TNBC cell membranes (P < 0.05) compared to untreated cells or monotherapies (Figure 4A-B), while no significant changes were observed in the membrane polarity of non-targeting vector TNBC cells (Supplementary Figure S3C). This increase in GP, suggesting increased membrane polarity, or rigidity, after treatment with EPA in conjunction with EPHA2 inhibition directly correlates with the synergistic action of our combination therapy in inducing cell death in TNBC cells. Collectively, these results imply that our EPA-based combination therapy may induce cell death in TNBC via modifications of cell membrane polarity.
Figure 4. Combination of EPA with EPHA2 inhibition modifies plasma cell membrane lipid composition, polarity and apoptosis induction via ABCA1 inhibition in TNBC cell lines.
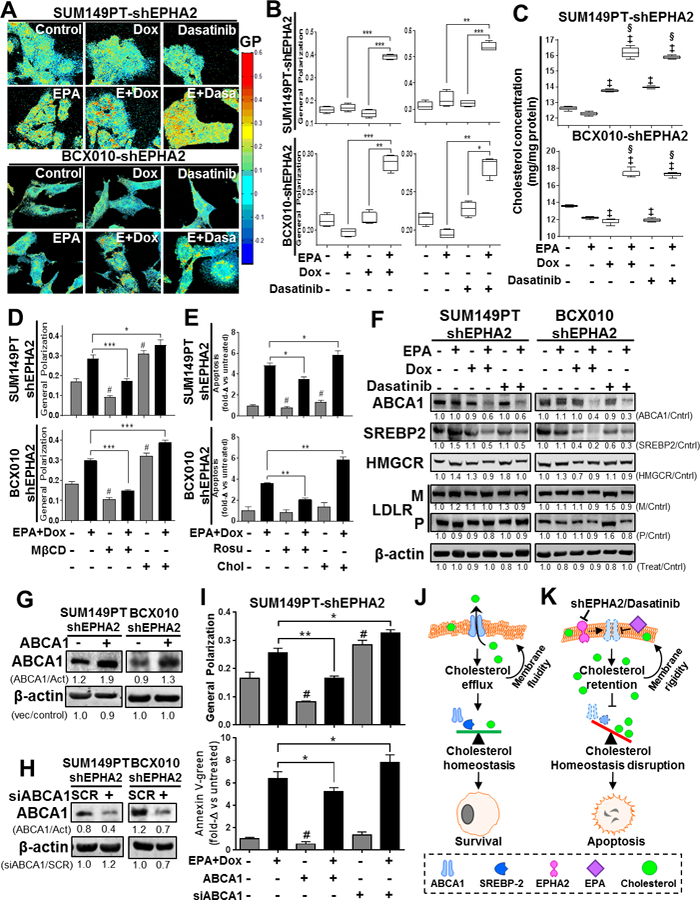
The effects of EPA in combination with EPHA2 inhibition on TNBC cell membranes were determined. (A-B) Cell membrane general polarization (GP, an indicator of polarity or rigidity) was determined in doxycycline (Dox)-inducible SUM149PT-shEPHA2 and BCX010-shEPHA2 cells treated with the indicated combinations of Dox, EPHA2 inhibitor dasatinib, and/or EPA. GP was determined by imaging (A) and quantification (B). Higher GP values indicate greater cell membrane polarization/rigidity. (C) Cholesterol concentrations in the membrane fractions of Dox-inducible SUM149PT-shEPHA2 and BCX010-shEPHA2 cells treated with indicated combinations of Dox, dasatinib, and/or EPA were determined by cholesterol quantification assay. (D-E) Dox-inducible SUM149PT-shEPHA2 and BCX010-shEPHA2 cells were treated with combination Dox and EPA as indicated, with or without cellular cholesterol removal (by methyl-β-cyclodextrin [MβCD] or rosuvastatin [Rosu]) or supplementation (Chol), and general polarization (D) and apoptosis (E) were quantified. Higher general polarization values indicate greater cell membrane polarization/rigidity. (F) Dox-inducible shEPHA2 cells were treated with EPA, Dox, dasatinib, or combinations and subjected to immunoblotting analysis of key cholesterol homeostasis-regulating proteins: ABCA1, mature SREBP2 (55–65 kDa), HMG CoA reductase (HMGCR), and LDL-receptor (LDLR) mature (M) and precursor (P) forms. (G-H) Inducible shEPHA2 cells were transfected with either an ABCA1-expressing (+) or empty control vector (−) (G) or pooled ABCA1 (+) or scrambled (SCR) control siRNA (H) and confirmed by immunoblotting assay. Pixel densities of individual proteins were quantified for each condition, and ratios of protein/β-actin normalized to control, or treated/untreated-control, are shown below the blots. β-actin expression was used as a protein loading control. Act, β-actin; Cntrl, control. (I) Dox-inducible shEPHA2 SUM149PT cells treated with combination EPA and Dox were transfected with ABCA1 expression vector or ABCA1 siRNA (siABCA1). General polarization of the cell membranes (top) and apoptosis (bottom) were quantified relative to untreated controls. Data were pooled from three independent experiments and are presented as mean ± SD, or mean of fold-change (fold-Δ) compared to untreated control ± SD. For all graphs, differences between groups were compared by unpaired t-test: *, P < 0.05; **, P < 0.001; *** P < 0.0001; ‡, P < 0.05 compared to EPA; §, P < 0.0001, compared to dasatinib or Dox; #, P < 0.05 compared to untreated control. (J-K) These schematics show the proposed mechanism of action for apoptosis induced by the combination of EPA and EPHA2-inhibiting therapy, involving increased cholesterol concentration and membrane polarization mediated by ABCA1 inhibition. (J) Cellular cholesterol homeostasis is normally maintained by the balancing effects of the cholesterol efflux channel protein ABCA1 and cholesterol biosynthesis inducer SREBP2. Their activity promotes maintenance of plasma cell membrane polarity and cellular survival. (K) Treatment with combination of EPA with EPHA2 inhibition (shEPHA2 or dasatinib) alters the cell membrane structure, inhibiting the cholesterol-exporting functions of ABCA1. This results in the cellular accumulation of cholesterol and subsequent increase in cell membrane polarity. This aberrant accumulation of intracellular/membrane cholesterol prevents the activity of cholesterol biosynthesis inducer proteins (e.g., SREPB2) and disrupts cellular cholesterol homeostasis, resulting in apoptosis.
Cholesterol is a major regulator of polarity in cell membranes 17. As such, we hypothesized that our EPA-based combination therapy modified cell membrane GP through modulation of cholesterol levels. Indeed, cholesterol accumulated in the membranes of SUM149PT and BCX010 cells following combination therapy, when compared to either monotherapy (P < 0.05) or untreated controls (P < 0.001) (Figure 4C). Collectively, these results suggest that combination EPA and EPHA2-inhibition treatment significantly increased the polarity of TNBC cell membranes through an associated increase in membrane cholesterol content.
Detection of elevated cholesterol levels within TNBC cell membranes prompted us to investigate whether cholesterol homeostasis is a fundamental biological aspect that dictates the efficacy of our EPA-based combination therapy. Initially, we assessed changes in GP in TNBC cells with modulated plasma membrane cholesterol levels through exogenous cholesterol supplementation or cyclodextrin-based cholesterol depletion (Supplementary Figure S5). As expected, cholesterol supplementation elevated the GP of TNBC cells, while depletion of cholesterol using methyl-β-cyclodextrin (MβCD) lowered the GP of TNBC cells treated with the combination of EPA with Dox or dasatinib (Figure 4D and Supplementary Figure S6A). Further, addition of exogenous cholesterol to TNBC cells exacerbated the cytotoxicity of our EPA-based combination therapies (Figure 4E and Supplementary Figure S6B). Conversely, decreased cholesterol content through the use of rosuvastatin (a cholesterol biosynthesis inhibitor) partially blocked the effectiveness of our EPA-based combination therapies (Figure 4E and Supplementary Figure S6B). These data lead us to conclude that EPHA2 inhibition in conjunction with EPA can modify membrane rigidity, and subsequently induce apoptosis, through the accumulation of cholesterol in the plasma membranes of TNBC cells.
Cholesterol exporter protein ABCA1 is a critical mediator of therapeutic action
Cholesterol levels within the cell can be modulated through increased import, reduced export, and/or increased biosynthesis 18, 19. To unravel the molecular events triggered by our combination therapy, we performed a candidate expression screen on four major proteins involved in cholesterol regulation. Compared to single treatment and untreated controls, combination therapy diminished the expression of ABCA1 (ATP-binding cassette sub-family A member 1) and mature SREBP2 (sterol regulatory element-binding protein 2; 55–65 kDa), while LDLR (low-density lipoprotein receptor) and HMGCR (3-hydroxy-3-methylglutaryl-coenzyme A reductase) expression was not altered in TNBC cells (Figure 4F). The reduced levels of ABCA1 (a major cholesterol-exporting protein) would suggest that cholesterol may accumulate within cells, preventing the molecular activation of SREBP2 20, which could subsequently result in an increase in cell membrane polarity and cell death.
To determine whether ABCA1 is critical to maintain cell membrane polarity, we performed gain/loss-of-expression studies to assess the response of SUM149PT and BCX010 cells to combination treatment (Figure 4G-H). Overexpression of ABCA1 significantly blocked EPA and Dox therapy-induced cell membrane polarization (P < 0.001), and subsequently prevented apoptosis (P < 0.05), when compared to controls (Figure 4I and Supplementary Figure S6C). In a reverse-complementary approach, transient knockdown of the ABCA1 gene in TNBC cells further enhanced cell polarization (P < 0.05) and subsequent apoptosis (P < 0.05) following EPA combination therapy (Figure 4I and Supplementary Figure S6C). Altogether, these results provide functional evidence that EPA-based combination therapy induces apoptosis in TNBC cells by impairing cholesterol export from the cell membrane in an ABCA1-dependent manner (Figure 4J-K).
Inhibition of EPHA2 enhances the therapeutic action of EPA in TNBC xenograft models
Our in vitro data suggests that EPA therapy combined with EPHA2 inhibition constitutes a novel and effective therapy preventing TNBC tumor growth. To establish it as a viable combination therapy, we next evaluated whether inhibition of EPHA2, via gene silencing or administration of dasatinib, could enhance the therapeutic effect of EPA in xenograft models of TNBC.
Using a functional genomics approach, mice bearing established SUM149PT-shEPHA2 or BCX010-shEPHA2 Dox-inducible tumor xenografts (Supplementary Figure S7A-B) were size matched and split into four arms to receive: 1) regular diet (control), 2) EPA diet, 3) Dox, or 4) EPA diet and Dox. In mice bearing SUM149PT-and BCX010-non-silencing shRNA (vector) tumors, doxycycline treatment was not associated with significant changes in tumor growth rate (Supplementary Figure S7C). Meanwhile, in both TNBC shEPHA2-inducible model systems monotherapy mildly reduced tumor growth, while the combination of EPHA2 silencing and EPA therapy drastically blocked tumor growth, leading to a significant extension in survival (Figure 5A-B). Reflecting the reduced growth rate, immunohistochemical staining of xenograft tumors revealed reduced levels of Ki67 (proliferation maker) and increased levels of cleaved caspase-3 (apoptosis marker) (Figure 5C-D and Supplementary Figure S8A-B). As expected, our combination therapy inhibited EPHA2 activity (p-EPHA2), leading to decreased expression of cholesterol regulator ABCA1 compared to single treatment and control groups (Figure 5E-F).
Figure 5. Inhibition of EPHA2 acts synergistically with EPA against TNBC tumor xenografts by inhibiting ABCA1.
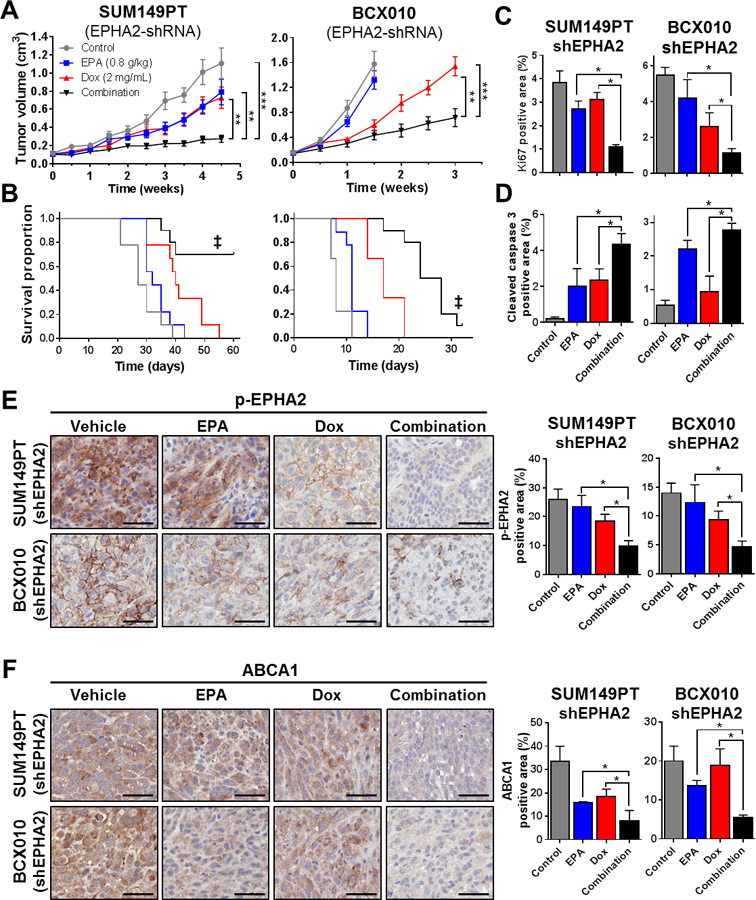
Mouse models of doxycycline (Dox)-inducible SUM149PT-shEPHA2 and BCX010-shEPHA2 TNBC were established, and mice were treated with EPA (0.8 g/kg in diet), Dox (2 mg/mL in water), or both (n = 10 per group). Tumor volumes were monitored over time (A), and Kaplan-Meier survival curves were generated for the endpoint (tumor volume = 1 500 mm3) (B). Levels of proliferation marker Ki67 (C) and apoptosis marker cleaved caspase-3 (D) were determined by immunohistochemical (IHC) staining of paraformaldehyde-fixed, paraffin-embedded sections from three representative tumor samples from each treatment group. Levels of p-EPHA2 (E) and ABCA1 (F) in tumors were determined by IHC staining. Representative images for each treatment group from three independent experiments are shown. Scale bars = 50 µM. Images were analyzed by ImageJ software to quantify Ki-67, cleaved caspase-3, p-EPHA2, and ABCA1. Data are represented as mean ± SD. Differences between groups were compared by unpaired t-test (bar graphs) or log-rank test (survival curves): *, P < 0.05; **, P < 0.001; ***, P < 0.0001; ‡, P < 0.001 combination treatment survival proportion compared to single treatment and untreated control groups.
On the basis of our successful preliminary in vivo study, we next tested our rationally developed EPA/dasatinib combination therapy in mice bearing TNBC tumor xenografts. Mice were split into four arms, receiving either; 1) regular diet (control), 2) EPA diet, 3) dasatinib, or 4) EPA and dasatinib. Similar to the results of our gene-silencing approach, the monotherapies produced modest effects in both SUM149PT and BCX010 tumor model systems, while combination therapy severely prevented TNBC xenograft growth and extended survival (Figure 6A-B). Predictably, TNBC xenografts had a reduction in Ki67 proliferation marker and increase in cleaved caspase-3 apoptosis marker expression following combination therapy (Figure 6C-D). Moreover, we confirmed that dasatinib-containing therapies were able to reduce phospho-EPHA2 levels and that combination therapy reduced the expression of ABCA1 in cancer cells in vivo (Figure 6E-F). Collectively, our in vivo studies confirm that inhibition of EPHA2 sensitizes TNBC tumors to EPA therapy, via an ABCA1-associated mechanism, and has potential for future translational development.
Figure 6. EPHA2 inhibition by dasatinib enhances the efficacy of EPA against TNBC tumor xenografts.
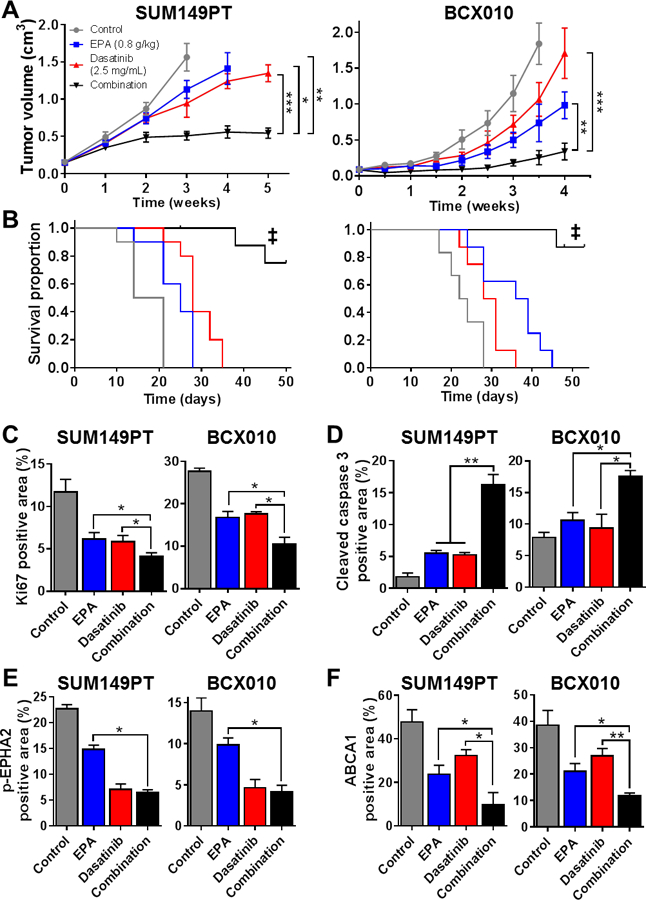
SUM149PT and BCX010 tumor xenograft-bearing mice were fed an EPA diet (0.8 g/kg; ad libitum), treated with dasatinib (2.5 mg/mL intraperitoneal injection), or both (n = 10 per group). Tumor volumes were monitored over time (A) and Kaplan-Meier survival curves to endpoint (tumor volume = 1 500 mm3) were generated (B). Paraformaldehyde-fixed, paraffin-embedded sections from each treatment group were analyzed for proliferation marker Ki67 (C), apoptosis marker cleaved caspase-3 (D), and mechanistic proteins p-EPHA2 (E) and ABCA1 (F) by immunohistochemical (IHC) staining. Three representative tumor samples from each treatment group were analyzed. Images were analyzed by ImageJ software to quantify the individual proteins. Data represent mean ± SD. Differences between groups were compared by unpaired t-test (bar graphs) or log-rank test (survival curves): *, P < 0.05; **, P < 0.001; ***, P < 0.0001; ‡, P < 0.0001 combination treatment survival proportion compared to single treatment and untreated control groups.
Discussion
TNBC remains a disease for which effective therapeutic strategies that can significantly improve patients’ outcomes or survival do not exist. We have successfully demonstrated EPHA2 as a novel and clinically significant therapeutic target against TNBC when combined with EPA therapy. Suppression of EPHA2 through various approaches enhanced the anti-tumor effect of EPA, reducing cancer cell proliferation, and induction of apoptosis in two xenograft models of TNBC. This is the first study to clearly demonstrate that EPA can improve the efficacy of targeted therapy for TNBC.
EPA has been widely tested for its benefits as a dietary supplement or as neoadjuvant therapy in cancer because of its anti-inflammatory qualities and safe toxicity profile 21–23. However, it has never been considered a mainstream approach in cancer treatment. Towards its clinical development, we performed an unbiased and high-throughput functional genomics screen to identify EPHA2, a cell surface tyrosine kinase receptor protein whose inhibition heightens the therapeutic action of EPA towards TNBC.
EPHA2 is an emerging clinical target in aggressive cancers 11. Overexpression of EPHA2 has been associated with poor prognosis in multiple cancers, including colorectal, lung, ovarian, endometrial, and pancreatic cancers 24–29, and has been demonstrated to have a biological role in controlling tumor growth and metastasis 11, 30, 31. We demonstrated that EPHA2 was highly expressed in breast cancer cell lines with a triple-negative molecular subtype. Further, in retrospective analysis of breast cancer patients, we identified specific associations between high EPHA2 expression in tumors and shorter DFS outcomes in patients with the TNBC/basal-like subtype, highlighting the clinical relevance of targeting EPHA2 in TNBC.
Our strategy of targeting EPHA2 in conjunction with EPA therapy was shown to be effective through a novel, membrane-based mechanism that controls cell viability. Cell-surface-localized EPHA2 has been implicated in the control of gap junctions and cell plasma membrane polarity, subsequently contributing to tumor migration and invasion 32, 33. Further, EPA, when incorporated into the plasma membrane, can modify lipid rafts, increase membrane compaction, modify intracellular signaling, and inhibit invasive potential 16, 34, 35. Our findings suggest that EPA combined with EPHA2 inhibition drastically increases the polarity, or rigidity, of the plasma membrane through an ABCA1-dependent accumulation of cholesterol and subsequently activates programmed cell death (Figure 4J-K).
Major regulators of cholesterol homeostasis in cells include the cholesterol exporter ABCA1, the cholesterol importer LDLR, and the cholesterol biosynthesis regulators SREBP2 and HMGCR 18, 20, 36, 37. High cholesterol levels prevent SREBP2 activation, inhibiting the subsequent induction of HMGCR and maturation of LDLR (Figure 4J) 19. Studies in human macrophages have observed that incorporation of EPA into the plasma membrane impairs ABCA1-dependent cholesterol efflux 38, 39, but our study is the first to report this phenomenon in cancer xenograft models. Here, we demonstrated that suppression of ABCA1 activity by the combination of EPA with EPHA2 inhibition induced the accumulation of cellular cholesterol, contributing to increased membrane polarity, inhibition of SREBP2, disruption of cellular lipid homeostasis, and apoptosis (Figure 4K). Supporting our conclusions are recent studies that suggest that inhibition of either ABCA1 or EPHA2 can alter cell membrane polarity, leading to decreased metastatic potential 32, 40, 41. Further, ABCA1 overexpression was associated with poor prognosis in breast, ovarian, and hepatocellular carcinomas 40, 42, 43, which suggests that an EPA-based combination therapy may be effective in other EPHA2-positive cancers which broadens the impact of this study.
Despite the therapeutic benefit of our combination therapy against TNBC in our preclinical testing, a potential challenge for the clinical application of our strategy is the high percentage (49%) of adults in the U.S. population who are currently receiving or are recommended to receive cholesterol biosynthesis inhibitors (i.e. statins) 44, 45. However, due to their pleiotropic effects, statins have been reported to have both tumorigenic and anti-tumorigenic roles 46. Therefore, it is critical for clinical development that our combination therapy be tested in patients who also receive a statin. Due to their functional requirement, our study suggests that future clinical testing should include analysis of ABCA1 and EPHA2 expression and phosphorylation-status as predictive biomarkers of therapeutic response.
In summary, we have identified a novel combination of EPA with EPHA2 inhibition that displays therapeutic efficacy in TNBC. Our study establishes the rationale for clinical testing of a novel EPA/dasatinib combination therapy in EPHA2-positive TNBC and identifies potential markers of therapeutic response. Excitingly, our EPA-based combination therapy may be applicable to other EphA2-positive cancers that include: aggressive lung cancer, invasive ovarian cancer and metastatic melanoma 47, and warrants further exploration.
Materials and Methods
Detailed information regarding the synthetic-lethal siRNA screen, capillary-based immunoassay, breast cancer patient tumor expression analysis, Annexin V apoptosis assays, doxycycline-inducible cell lines, xenograft tumor cell sorting, membrane polarity imaging, fatty acid analysis, and mass spectrometry are included in the online supplementary methods. Detailed information about in vitro cell proliferation assays, transfections, co-immunoprecipitation, and immunohistochemistry (IHC) have been previously described 48, 49.
Cell lines
Human TNBC cell line SUM149PT was purchased from Asterand Bioscience, Inc. (Detroit, MI); human TNBC cell line BCX010 was derived from a patient with TNBC receptor status and inflammatory breast cancer pathological diagnosis, and developed and generously donated for this study by Dr. Funda Meric-Bernstam (MD Anderson Cancer Center) 50. SUM149PT and BCX010 cells were maintained in F12 medium (GIBCO) supplemented with fetal bovine serum (FBS, 5%), penicillin-streptomycin (100 units/mL), insulin (5 μg/mL), and hydrocortisone (1 μg/mL). Both cell lines are negative for estrogen receptor, androgen receptor, and HER2. All cell lines were authenticated by genotyping through MD Anderson Cancer Center’s Characterized Cell Line Core Facility and were routinely tested for mycoplasma contamination using MycoAlert™ (Lonza, Allendale, NJ).
Reagents and antibodies
EPA sodium salt and cholesterol powder bio-reagent suitable for cell culture were purchased from Sigma-Aldrich (St Louis, MO). Doxycycline was purchased from Research Products International (Mount Prospect, IL). ALW-II-41–27 was generously donated for this study by Dr. Nathanael S. Gray (Dana-Farber Cancer Institute). Dasatinib was purchased from Selleck Chemicals (Houston, TX). Methyl-β-cyclodextrin (MβCD) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All in vitro assays were performed after treatments for 24–48 hours with EPA (IC20: 50 µM), doxycycline (Dox, 1–3 µg/mL), ALW-II-41–27 (1 µM), dasatinib (IC20: 0.3 µM), cholesterol (1 mM), MβCD (4 mM), or rosuvastatin (2.5 µM).Combination treatment synergism was calculated by CalcuSyn software (Biosoft, Cambridge, UK). Combination index (CI) was used as a quantitative measure of the degree of drug interaction: < 0.9 = synergy, 0.9 −1.1 = additive effect, > 1.1 = no synergy. We obtained anti-EPHA2 (Cat#: 6997), anti-phosphorylated EPHA2 (Tyr588; Cat#: 12677), anti-SRC (Cat#: 2109), anti-phosphorylated SRC (Cat#: 2105), and anti-cleaved caspase 3 (Cat#: 9661) from Cell Signaling Technology, Beverly, MA; anti-IgG2b-PE (Cat#: 402203), and anti-EPHA2-PE (Cat#: 356803) from BioLegend, San Diego, CA; anti-ABCA1 (Cat#: NB400–105) from Novus Biologicals, Littleton, CO; anti-IgG-APC (Cat#: IC003A), anti-EPHA2-APC (Cat#: FAB3035A), and anti-SREBP2 (Cat#: MAB7119) from R&D Systems, Minneapolis, MN; anti-HMGCR (Cat#: PA5–37367) from Thermo Fisher Scientific, Waltham, MA; anti-Ki67 (Cat#: ab15580) and anti-LDLR (Cat#: ab30532) from Abcam, Cambridge, MA; anti-α-tubulin (Cat#: T5168), and anti-β-actin (Cat#: A5316) from Sigma-Aldrich, St. Louis, MO; and anti-horseradish peroxidase-conjugated antibodies (Thermo Scientific, Rockford, IL).
SiRNAs targeting EPHA2 and ABCA1 were purchased from Sigma-Aldrich (St Louis, MO). For depletion of EPHA2, we used SASI_Hs01_00026514, SASI_Hs01_00026516, and SASI_Hs01_00026517; and for depletion of ABCA1, we used SASI_Hs01_00129036, SASI_Hs01_00129036, and SASI_Hs01_00129038. Knockdown efficacy of pooled siRNAs was tested by immunoblotting. Scrambled siRNA control was purchased from Thermo Fisher Scientific (ON-TARGETplus non-targeting control pool, catalog number D-001810). The following expression vectors were purchased: Human EPHA2 and control (pLOC-GFP) (GE Healthcare Biosciences, Pittsburgh, PA), pCMV3-untagged expression clone ABCA1 (HG11924-UT), and control (pCMV3-GFP) (Sino Biologicals Inc., North Wales, PA). These were transfected into TNBC cells by following the manufacturers’ instructions.
Membrane polarity assay
Membrane polarity, or fluidity, was determined by using the lipophilic fluorescent probe Laurdan (6-dodecanoyl-2-dimethylaminonaphthalene, Thermo Fisher Scientific). Briefly, following treatment for 24 or 48 hours, the cell membranes were extracted by lysis with hypotonic buffer (0.2 mM EDTA, 1 mM NaHCO3) containing protease/phosphatase inhibitors, and cells were allowed to swell for 30 minutes, followed by brief sonication. Remaining intact cells were removed by centrifugation at 800 x g for 10 minutes. The supernatant was collected and subjected to ultra-centrifugation at 100 000 x g for 45 minutes, yielding a crude total membrane pellet; this pellet was re-suspended in buffer (10 mM TRIS and anti-protease cocktail, in PBS) before addition of the Laurdan fluorescent probe and incubation for 1 hour at 37°C. After incubation, the fluorescence measurements were performed using a Quanta-Master model QM3-SS (Photon Technology International) cuvette-based fluorescence spectrometer. Using a Peltier thermoelectric temperature controller, the sample was held at a constant of 37°C and fluorescence measurements (excitation wavelength, 350 nm; emission wavelengths, 440 and 490 nm) were performed. Data were collected from three independent experiments, each containing at least 5 technical replicates, and analyzed using Felix 32 software. General polarization (GP, a measure of polarity) of the membrane was determined by the following equation:
where IF440nm and IF490nm represent the fluorescence intensity detected at the 440 nm and 490 nm wavelengths, respectively. Higher GP values are indicative of increased polarization, or rigidity. Conversely, lower GP values represent a less polarized, more fluid state.
Cholesterol quantification
Total cellular membrane fractions were extracted from treated cells using the Subcellular Protein Fractionation assay (Thermo Scientific) according to the manufacturer’s instructions. After the cell membrane fraction was extracted, cholesterol concentration was quantified by Amplex® Red Cholesterol Assay (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, the extracted membrane fraction (20 µg) was diluted in reaction buffer (50 µL; 0.1 M potassium phosphate, pH 7.4, 0.05 M NaCl, 5 mM cholic acid, 0.1% Triton® X-100 in deionized water), and then Amplex Red working solution (50 µL; 2 U/mL horseradish peroxidase, 2 U/mL cholesterol oxidase, 0.2 U/mL cholesterol esterase, 0.75 μL Amplex Red reagent solution, 0.5 μL horseradish peroxidase (HRP) solution, 0.5 μL cholesterol oxidase solution, and 0.05 μL cholesterol esterase solution in reaction buffer) was added and the tubes were incubated at 37°C for 30 minutes. After incubation, fluorescence was measured in at least three independent experiments in a fluorescence plate reader using excitation at 570 nm and emission detection at 590 nm.
In vivo xenograft animal models
Animal studies were in compliance with ethical regulations approved by the MD Anderson Animal Care and Use Committee. Female immunodeficient NOD SCID gamma (NSG) mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, Jackson Laboratory, Bar Harbor, ME), aged 4–6 weeks, were purchased from MD Anderson’s Department of Experimental Radiation Oncology for xenograft experiments. Mice were housed in pathogen-free conditions and treated in accordance with NIH guidelines. To establish breast cancer xenografts, each mouse was injected with a suspension of SUM149PT (5 × 105 cells/100 µL) or BCX010 (5 × 105 cells/100 µL) into one site in the fourth inguinal mammary fat pad. Tumor incidence rates were 100% for all cell lines. Drug treatments started when the tumors were approximately 100–150 mm3; at this point, mice were randomized among experimental treatment groups (n = 9 or 10 per group) which resulted with each group having a similar average tumor size at beginning of treatments. Investigators were not blinded to group allocation during the experiment. EPA therapy was administered ad libitum through customized AIN-76A mice diets (Research Diets Inc., New Brunswick, NJ) containing Vascepa (FDA-approved purified EPA) at doses equivalent to the maximum (4 g) and half-maximum (2 g) daily recommended doses for humans and equivalent to 6 g and 3 g of Vascepa per kg of food, verified by chromatographic fatty acid analysis (Supplementary Figure S1). These doses are equivalent to EPA daily intakes of 0.8 and 0.4 g/kg for mice, considering an average daily food intake of 3.5 g and average mouse weight of 25 g. Diets were stored at −20°C. Water containing doxycycline (2 mg/mL) and 2% sucrose was provided ad libitum to induce shRNA expression in pTRIPZ-transduced tumor cell lines implanted in mice. Dasatinib (2.5 mg/kg) was administered to mice via intraperitoneal injection 6 days per week. Dasatinib was administered in a solution of 30% PEG 400 (Thermo Fisher Scientific), 1% Tween 80, and 4% dimethyl sulfoxide (Sigma-Aldrich) as drug vehicle. Tumor volume [V = 0.5 x (L x W2)] and body weight were measured twice weekly. Mice tolerated all treatments with no significant adverse events or change in body weight noted. Drug treatments continued until the primary tumor reached 1 500 mm3 or the mouse’s condition reached morbidity. The mice were then euthanized and their tissues harvested for molecular analysis.
Statistical analysis
For experimental outcomes, descriptive statistics (mean and standard deviation) were summarized for each group. An analysis of variance (ANOVA) model was used to compare the mean outcome values among the tested groups. Kaplan-Meier survival curves were generated for disease free survival (DFS) and overall survival (OS) of patients based on EPHA2 mRNA tumor levels (median cut-off), and the log-rank test was used to compare survival curves. Statistical analyses were performed using an unpaired t-test with Prism version 5 (GraphPad Software, La Jolla, CA). P values of < 0.05 were considered statistically significant. Data met normal distribution assumptions. Statistical significance variance is included in the caption of each figure. Synthetic-lethal siRNA screen data analysis was carried out on gene-normalized data using a t-test followed by a beta-uniform mixture model to adjust P values using R statistical computing software (version 3.0.1) 51.
Supplementary Material
Acknowledgments
We thank Geoffrey Bartholomeusz from the Department of Experimental Therapeutics at the MD Anderson Cancer Center for his assistance in the development of the high-throughput siRNA synthetic-lethal screen, and Funda Meric-Bernstam from the Department of Investigational Cancer Therapeutics for generously providing the BCX010 cell line. We also thank Nathanael S. Gray from the Department of Biological Chemistry and Molecular Pharmacology at the Dana-Farber Cancer Institute, Harvard Medical School, for generously providing ALW-II-41–27 for this study, and William Dowhan from the Department of Biochemistry and Molecular Biology at the UTHealth McGovern Medical School for his valuable advice on this project. We also thank Kathryn Hale and Sunita Patterson of the Department of Scientific Publications at MD Anderson Cancer Center for expert editorial assistance. Financial support: This work was supported by the Morgan Welch Inflammatory Breast Cancer Research Program, the State of Texas Rare and Aggressive Breast Cancer Research Program, the National Institutes of Health/National Cancer Institute through grants R01CA123318 and P30CA016672 (used the Characterized Cell Line Core, the Flow Cytometry and Cellular Imaging Facility, and the Bioinformatics Shared Resource at MD Anderson Cancer Center), the Cancer Prevention and Research Institute of Texas through grant RP110532-P3 (used MD Anderson’s siRNA Screening Service), and the John Dunn Research Foundation Endowment awarded to William Dowhan.
Footnotes
Conflict of Interests: All authors declare no potential conflicts of interest.
Data availability: The siRNA synthetic lethal screen data have been deposited in Gene Expression Omnibus (GEO) with accession number: GSE102057.
Supplementary information is available at Oncogene’s website
References
- 1.Bae SY, Kim S, Lee JH, Lee HC, Lee SK, Kil WH et al. Poor prognosis of single hormone receptor-positive breast cancer: similar outcome as triple-negative breast cancer. BMC Cancer 2015; 15: 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le Du F, Eckhardt BL, Lim B, Litton JK, Moulder S, Meric-Bernstam F et al. Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget 2015; 6: 12890–12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 4.Lee AH, Happerfield LC, Millis RR, Bobrow LG. Inflammatory infiltrate in invasive lobular and ductal carcinoma of the breast. Br J Cancer 1996; 74: 796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, Reyes ME, Zhang D, Funakoshi Y, Trape AP, Gong Y et al. EGFR signaling promotes inflammation and cancer stem-like activity in inflammatory breast cancer. Oncotarget 2017; 8: 67904–67917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ford NA, Rossi EL, Barnett K, Yang P, Bowers LW, Hidaka BH et al. Omega-3-Acid Ethyl Esters Block the Protumorigenic Effects of Obesity in Mouse Models of Postmenopausal Basal-like and Claudin-Low Breast Cancer. Cancer Prev Res (Phila) 2015; 8: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos EJ, Middleton FA, Laviano A, Sato T, Romanova I, Das UN et al. Effects of omega-3 fatty acid supplementation on tumor-bearing rats. J Am Coll Surg 2004; 199: 716–723. [DOI] [PubMed] [Google Scholar]
- 8.Laviano A, Rianda S, Molfino A, Rossi Fanelli F. Omega-3 fatty acids in cancer. Curr Opin Clin Nutr Metab Care 2013; 16: 156–161. [DOI] [PubMed] [Google Scholar]
- 9.Fabian CJ, Kimler BF, Hursting SD. Omega-3 fatty acids for breast cancer prevention and survivorship. Breast Cancer Res 2015; 17: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brinton EA, Mason RP. Prescription omega-3 fatty acid products containing highly purified eicosapentaenoic acid (EPA). Lipids Health Dis 2017; 16: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song W, Hwang Y, Youngblood VM, Cook RS, Balko JM, Chen J et al. Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madden SF, Clarke C, Gaule P, Aherne ST, O’Donovan N, Clynes M et al. BreastMark: an integrated approach to mining publicly available transcriptomic datasets relating to breast cancer outcome. Breast Cancer Res 2013; 15: R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB et al. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med 2006; 355: 560–569. [DOI] [PubMed] [Google Scholar]
- 15.Amato KR, Wang S, Hastings AK, Youngblood VM, Santapuram PR, Chen H et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J Clin Invest 2014; 124: 2037–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corsetto PA, Cremona A, Montorfano G, Jovenitti IE, Orsini F, Arosio P et al. Chemical-physical changes in cell membrane microdomains of breast cancer cells after omega-3 PUFA incorporation. Cell Biochem Biophys 2012; 64: 45–59. [DOI] [PubMed] [Google Scholar]
- 17.Jaureguiberry MS, Tricerri MA, Sanchez SA, Finarelli GS, Montanaro MA, Prieto ED et al. Role of plasma membrane lipid composition on cellular homeostasis: learning from cell line models expressing fatty acid desaturases. Acta Biochim Biophys Sin (Shanghai) 2014; 46: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nandi S, Ma L, Denis M, Karwatsky J, Li Z, Jiang XC et al. ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J Lipid Res 2009; 50: 456–466. [DOI] [PubMed] [Google Scholar]
- 19.Miserez AR, Muller PY, Barella L, Barella S, Staehelin HB, Leitersdorf E et al. Sterol-regulatory element-binding protein (SREBP)-2 contributes to polygenic hypercholesterolaemia. Atherosclerosis 2002; 164: 15–26. [DOI] [PubMed] [Google Scholar]
- 20.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002; 109: 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Makarewicz-Wujec M, Parol G, Parzonko A, Kozlowska-Wojciechowska M. Supplementation with omega-3 acids after myocardial infarction and modification of inflammatory markers in light of the patients’ diet: a preliminary study. Kardiologia polska 2017; 75: 674–681. [DOI] [PubMed] [Google Scholar]
- 22.Rose DP, Connolly JM, Coleman M. Effect of omega-3 fatty acids on the progression of metastases after the surgical excision of human breast cancer cell solid tumors growing in nude mice. Clinical cancer research : an official journal of the American Association for Cancer Research 1996; 2: 1751–1756. [PubMed] [Google Scholar]
- 23.Ciftci O, Cetin A, Aydin M, Kaya K, Oguz F. Fish oil, contained in eicosapentaenoic acid and docosahexaenoic acid, attenuates testicular and spermatological damage induced by cisplatin in rats. Andrologia 2014; 46: 1161–1168. [DOI] [PubMed] [Google Scholar]
- 24.Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res 2001; 61: 2301–2306. [PubMed] [Google Scholar]
- 25.Dunne PD, Dasgupta S, Blayney JK, McArt DG, Redmond KL, Weir JA et al. EphA2 expression is a key driver of migration and invasion and a poor prognostic marker in colorecta cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2016; 22: 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinch MS, Moore MB, Harpole DH Jr. Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clinical cancer research : an official journal of the American Association for Cancer Research 2003; 9: 613–618. [PubMed] [Google Scholar]
- 27.Thaker PH, Deavers M, Celestino J, Thornton A, Fletcher MS, Landen CN et al. EphA2 expression is associated with aggressive features in ovarian carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2004; 10: 5145–5150. [DOI] [PubMed] [Google Scholar]
- 28.Kamat AA, Coffey D, Merritt WM, Nugent E, Urbauer D, Lin YG et al. EphA2 overexpression is associated with lack of hormone receptor expression and poor outcome in endometrial cancer. Cancer 2009; 115: 2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mudali SV, Fu B, Lakkur SS, Luo M, Embuscado EE, Iacobuzio-Donahue CA. Patterns of EphA2 protein expression in primary and metastatic pancreatic carcinoma and correlation with genetic status. Clin Exp Metastasis 2006; 23: 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JE, Son AI, Zhou R. Roles of EphA2 in development and disease. Genes (Basel) 2013; 4: 334–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009; 16: 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugiyama N, Gucciardo E, Lehti K. EphA2 bears plasticity to tumor invasion. Cell Cycle 2013; 12: 2927–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang J, Xiao D, Li G, Ma J, Chen P, Yuan W et al. EphA2 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer cells. Oncogene 2014; 33: 2737–2747. [DOI] [PubMed] [Google Scholar]
- 34.Corsetto PA, Montorfano G, Zava S, Jovenitti IE, Cremona A, Berra B et al. Effects of n-3 PUFAs on breast cancer cells through their incorporation in plasma membrane. Lipids Health Dis 2011; 10: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mandal CC, Ghosh-Choudhury T, Yoneda T, Choudhury GG, Ghosh-Choudhury N. Fish oil prevents breast cancer cell metastasis to bone. Biochem Biophys Res Commun 2010; 402: 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osborne TF. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem 2000; 275: 32379–32382. [DOI] [PubMed] [Google Scholar]
- 37.Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS et al. Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Invest 1997; 100: 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fournier N, Tardivel S, Benoist JF, Vedie B, Rousseau-Ralliard D, Nowak M et al. Eicosapentaenoic acid membrane incorporation impairs ABCA1-dependent cholesterol efflux via a protein kinase A signaling pathway in primary human macrophages. Biochim Biophys Acta 2016; 1861: 331–341. [DOI] [PubMed] [Google Scholar]
- 39.Hu YW, Ma X, Li XX, Liu XH, Xiao J, Mo ZC et al. Eicosapentaenoic acid reduces ABCA1 serine phosphorylation and impairs ABCA1-dependent cholesterol efflux through cyclic AMP/protein kinase A signaling pathway in THP-1 macrophage-derived foam cells. Atherosclerosis 2009; 204: e35–43. [DOI] [PubMed] [Google Scholar]
- 40.Zhao W, Prijic S, Urban BC, Tisza MJ, Zuo Y, Li L et al. Candidate antimetastasis drugs suppress the metastatic capacity of breast cancer cells by reducing membrane fluidity. Cancer Res 2016; 76: 2037–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest 2008; 118: 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hou H, Kang Y, Li Y, Zeng Y, Ding G, Shang J. miR-33a expression sensitizes Lgr5+ HCC-CSCs to doxorubicin via ABCA1. Neoplasma 2017; 64: 81–91. [DOI] [PubMed] [Google Scholar]
- 43.Chou JL, Huang RL, Shay J, Chen LY, Lin SJ, Yan PS et al. Hypermethylation of the TGF-beta target, ABCA1 is associated with poor prognosis in ovarian cancer patients. Clin Epigenetics 2015; 7: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pencina MJ, Navar-Boggan AM, D’Agostino RB Sr., Williams K, Neely B, Sniderman AD et al. Application of new cholesterol guidelines to a population-based sample. N Engl J Med 2014; 370: 1422–1431. [DOI] [PubMed] [Google Scholar]
- 45.Hoy SM. Pitavastatin: A review in hypercholesterolemia. Am J Cardiovasc Drugs 2017; 17: 157–168. [DOI] [PubMed] [Google Scholar]
- 46.Cortese F, Gesualdo M, Cortese A, Carbonara S, Devito F, Zito A et al. Rosuvastatin: Beyond the cholesterol-lowering effect. Pharmacol Res 2016; 107: 1–18. [DOI] [PubMed] [Google Scholar]
- 47.Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets 2011; 15: 31–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Torres-Adorno AM, Lee J, Kogawa T, Ordentlich P, Tripathy D, Lim B et al. Histone deacetylase inhibitor enhances the efficacy of MEK inhibitor through NOXA-mediated MCL1 degradation in triple-negative and inflammatory breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2017; 23: 4780–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee J, Bartholomeusz C, Mansour O, Humphries J, Hortobagyi GN, Ordentlich P et al. A class I histone deacetylase inhibitor, entinostat, enhances lapatinib efficacy in HER2-overexpressing breast cancer cells through FOXO3-mediated Bim1 expression. Breast Cancer Res Treat 2014; 146: 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H et al. Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. PLoS One 2015; 10: e0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics 2003; 19: 1236–1242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.