

Abstract
The peptidoglycan cell wall is essential for the survival and morphogenesis of bacteria.1 For decades it was thought that only class A penicillin-binding proteins (aPBPs) and related enzymes effected peptidoglycan synthesis. Recently, it was shown that RodA, a member of the unrelated SEDS protein family, also acts as a peptidoglycan polymerase.2–4 Not all bacteria require RodA for growth; however, its homologue, FtsW, is a core member of the divisome complex that appears to be universally essential for septal cell wall assembly.5,6 FtsW was previously proposed to translocate the peptidoglycan precursor Lipid II across the cytoplasmic membrane.7,8 We report here that purified FtsW polymerizes Lipid II into peptidoglycan, but show that its polymerase activity requires complex formation with its partner class B PBP (bPBP). We further demonstrate that the polymerase activity of FtsW is required for its function in vivo. Thus, our findings establish FtsW as a peptidoglycan polymerase that works with its cognate bPBP to produce septal peptidoglycan during cell division.
Peptidoglycan is synthesized from the cell wall precursor Lipid II. This lipid-linked disaccharide-peptide is made at the inner face of the cell membrane and then translocated by Lipid II flippases to the extracellular membrane surface where it is polymerized and crosslinked (Fig. 1a).5 The penicillin-binding proteins (PBPs), a major class of enzymes involved in peptidoglycan synthesis, were initially identified as targets of β-lactam antibiotics such as penicillin, which covalently inactivate their crosslinking domains. The PBPs comprise several types of enzymes, of which the most important for peptidoglycan synthesis are the class A PBPs (aPBPs) and the class B PBPs (bPBPs).9 The aPBPs contain a peptidoglycan glycosyltransferase (PGT) domain that polymerizes Lipid II and a transpeptidase (TP) domain that crosslinks the resulting glycan strands (Fig. 1a). The bPBPs consist of a TP domain and a domain of unknown function. Some bacteria also contain monofunctional glycosyltransferases (MGTs) that have homology to the PGT domains of the aPBPs, and these proteins can also polymerize Lipid II.9
Figure 1: FtsW is a peptidoglycan synthase.

a, Peptidoglycan synthesis by the bifunctional class A PBPs (aPBPs). b, Septal peptidoglycan synthesis is directed by the divisome. FtsW may have peptidoglycan polymerase activity like its homologue RodA from the Rod system.2 c, Schematic of the PAGE assay used to detect peptidoglycan generated in vitro. The terminal D-Ala residue of peptidoglycan and Lipid II stem peptide is exchanged with biotin-D-lysine (BDL) using Enterococcus faecalis PBPX or S. aureus PBP4 for western blot detection.16,18 d, S. thermophilus FtsW polymerizes Lipid II in the presence of its cognate bPBP, PBP2x. Asterisk (*) indicates the catalytically inactive variant. Arrowhead (▶) indicates BDL-labeled PBPX formed in the peptidoglycan labeling reaction. e, FtsW requires divalent cations for polymerase activity. f, S. thermophilus FtsW can be activated by a near-cognate bPBP from S. pneumoniae. Asterisk (*) indicates the inactivated TP variant. Representative blots of three (d-e) or two (f) independent experiments are shown.
The β-lactam antibiotics are the most clinically and commercially successful class of antibiotics in history. They continue to be widely used for treating infections, but the emergence of β-lactam resistance in major human pathogens makes the identification of new antibacterial targets a high priority. Proteins that are essential in all bacteria, such as those belonging to the SEDS (shape, elongation, division, and sporulation) family, are particularly appealing therapeutic targets. Phylogenetic analysis has shown that the SEDS proteins RodA and FtsW are more widely conserved than the aPBPs.2 RodA, which serves as a key component of the Rod complex (elongasome) that makes side wall peptidoglycan, is absent or dispensable in some organisms.6,9,10 On the other hand, FtsW is required for the function of the division machinery (divisome) and is essential in most bacteria regardless of their shape.6,9,10
The Rod complex and the divisome each contain an essential bPBP that forms a complex with its cognate SEDS protein within the machinery.11–14 Therefore, the recent discovery that RodA has peptidoglycan polymerase activity has led to the proposal that SEDS-bPBP complexes form two-component peptidoglycan synthases with polymerase and crosslinking activities on separate polypeptides (Fig. 1b).2–4 Accordingly, mutations predicted to disrupt RodA interactions with its cognate elongasome bPBP cause the Rod system to malfunction in vivo.11 However, polymerase activity has thus far only been demonstrated for RodA from Bacillus subtilis.2,11 Whether FtsW, which shares key residues with RodA (Supplementary Fig. 1), also functions as a peptidoglycan polymerase remains unclear.
To address this question, we first expressed and purified FtsW orthologs from the Gram-positive bacteria Streptococcus thermophilus (StFtsW) and Staphylococcus aureus (SaFtsW) in Escherichia coli (Supplementary Fig. 2), but failed to detect polymerase activity (Fig. 1c) when the proteins were incubated with their native Lipid II substrate (Fig. 1d, Supplementary Fig. 3).15,16 Strikingly, when we added the cognate divisome bPBP to the FtsW reactions, peptidoglycan polymers were produced, implying that the bPBP stimulates peptidoglycan polymerase activity (Fig. 1d, Supplementary Fig. 3). The observed polymerase activity was abolished when an invariant aspartate in or near the presumed FtsW active site was mutated to alanine, but not when the TP active site of the bPBP was inactivated (Fig. 1d, Supplementary Figs. 1, 3). Furthermore, the activity of FtsW was insensitive to the phosphoglycolipid antibiotic moenomycin that inhibits aPBPs and MGTs (Supplementary Fig. 4). Another distinction between FtsW and the other class of polymerases is that FtsW polymerase activity requires divalent cations. EDTA abolished StFtsW polymerase activity, but adding excess Mg2+, Ca2+, or Mn2+ to the EDTA-containing reactions restored polymerization (Fig. 1e, Supplementary Fig. 5). Zn2+ did not stimulate polymerase activity (Supplementary Fig. 5). We conclude that FtsW is a peptidoglycan polymerase that requires divalent cations and the presence of a bPBP for activity.
Evolutionary sequence covariation analyses have demonstrated strong evolutionary couplings of residues within the transmembrane (TM) domain of bPBPs and TM 8 and 9 of the SEDS proteins.11,17 This predicted interface is largely hydrophobic in character, suggesting that steric compatibility may determine interaction specificity (Supplementary Fig. 6a). We therefore wondered whether the TM domain of a cognate bPBP is required for the PGT activity of FtsW. To address this question, we first removed the TM domain from StPBP2x and tested the ability of the ΔTM variant (StPBP2x∆™) to activate StFtsW. No peptidoglycan product was detected, suggesting that the TM helix is crucial for promoting FtsW polymerase activity (Fig. 1d). To assess the specificity of the polymerase-promoting activity of divisome bPBPs, we performed StFtsW and SaFtsW reactions in the presence of bPBPs from three different species (Fig. 1f, Supplementary Fig. 7). SaFtsW was only able to polymerize Lipid II when combined with its native bPBP, PBP1 (SaPBP1), but StFtsW was able to polymerize Lipid II when combined either with its cognate bPBP, StPBP2x, or with the divisome bPBP from S. pneumoniae (SpPBP2x), which is 50% identical to its S. thermophilus counterpart and has a similar TM helix (Supplementary Fig. 6b). StFtsW showed no activity in the presence of SaPBP1. We also swapped the TM helices of the elongasome bPBP (PBP2b) and divisome bPBP (PBP2x) from S. thermophilus and S. pneumoniae (Supplementary Fig. 2) and tested whether the resulting chimeras activate StFtsW polymerase activity. We found that StFtsW was activated only by chimeras containing a divisome bPBP TM helix (Supplementary Fig. 8). Finally, as steric complementarity is predicted to play an important role in SEDS-bPBP interactions, we made a S37F substitution in the TM helix of StPBP2x that was expected to destabilize the interaction (Supplementary Fig. 6c). The polymerase activity of FtsW was reduced, but could be restored by a compensatory L345G substitution at the complementary position in FtsW (Supplementary Figs. 6c, 6d). These results, along with the evolutionary covariation analysis, imply that the TM helix of the cognate bPBP interacts with FtsW and is necessary for the effect of this bPBP on FtsW polymerase activity.
To determine if FtsW from a Gram-negative organism could form an active peptidoglycan synthase complex with its cognate bPBP, we co-expressed Pseudomonas aeruginosa FtsW (PaFtsW) with its divisome bPBP (PaPBP3) in E. coli. PaPBP3 co-purified with the affinity-tagged FtsW, and the complex possessed polymerase activity (Fig. 2a, Supplementary Fig. 9). We found that SaFtsW could also be co-expressed and purified as a stable, active complex with SaPBP1* (Fig. 2b, Supplementary Fig. 9). Substitution of the invariant aspartate in or near the presumed active site of FtsW compromised the polymerase activity of both complexes (Figs. 2a, 2b, Supplementary Fig. 1). To assess the in vivo relevance of the FtsW polymerase activity, we compared the effect of overproducing either wild-type FtsW or inactive FtsW variants (designated FtsW*) in P. aeruginosa and S. aureus. Cell growth and division remained normal upon expression of wild-type FtsW (Figs. 2c, 2d, Supplementary Figs. 10, 11, Supplementary Tables 1, 2). However, production of PaFtsW* and SaFtsW* inhibited growth of P. aeruginosa and S. aureus, respectively (Supplementary Fig. 10). Moreover, PaFtsW* induced cell filamentation in P. aeruginosa and SaFtsW* caused septal abnormalities in S. aureus (Figs. 2c, 2d, Supplementary Fig. 11, Supplementary Tables 1, 2). The observed dominant-negative activity of the FtsW* proteins in their host organism most likely results from the defective proteins saturating FtsW binding sites in the divisome complex to disrupt the function of the machinery.
Figure 2: The PGT activity of FtsW is essential for cell division.
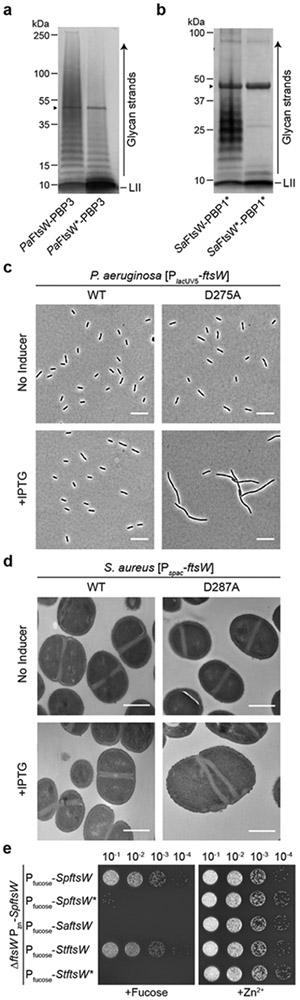
a, In vitro polymerization of Lipid II by co-purified P. aeruginosa FtsW-PBP3 complexes. b, In vitro polymerization of Lipid II by co-purified S. aureus FtsW-PBP1* complexes. Arrowheads (▶) indicate BDL-labeled PBP4 or PBPX formed in the peptidoglycan labeling reaction. c, Phase contrast images of P. aeruginosa cells overexpressing the wild-type FtsW or FtsW*. Scale bar = 10 µm. d, Electron microscopy images of S. aureus cells overexpressing the wild-type FtsW or FtsW*. Scale bar = 500 nm. e, Depletion of FtsW in S. pneumoniae can be rescued by the expression of SpFtsW or StFtsW. Representative data of three (a-b) or two (c-e) independent experiments are shown.
To further investigate the essentiality of FtsW polymerase activity, an FtsW depletion strain of S. pneumoniae was constructed. Expression of SpFtsW or StFtsW from a second locus rescued growth upon SpFtsW depletion (Fig. 2e, Supplementary Fig. 12). However, neither of the corresponding FtsW* nor SaFtsW prevented the lethal SpFtsW depletion phenotype (Fig. 2e, Supplementary Fig. 12). Taken together, the genetic and biochemical results indicate that stable FtsW-bPBP complexes are formed in diverse bacteria and possess peptidoglycan polymerase activity that is required for growth and division.
Having shown that an appropriate bPBP is required for FtsW polymerase activity, we wondered whether FtsW had a reciprocal stimulatory effect on bPBP activity. To address this question, we used an LC-MS based assay to detect peptidoglycan crosslinks. Peptidoglycan synthesized in vitro was digested with mutanolysin (Fig. 3a).16,18 These digestions typically produce three readily detectable muropeptide species: pentapeptide starting material (monomer A), tetrapeptides resulting from hydrolysis of the terminal D-alanine of the pentapeptide (monomer B), and crosslinked units resulting from TP activity (dimer C) (Fig. 3a). When StPBP2x was incubated with StFtsW and Lipid II, we observed an LC-MS peak in the mutanolysin digestions corresponding to the dimeric product (Fig. 3b and Supplementary Fig. 13). Catalytic inactivation of StPBP2x led to the disappearance of this peak, confirming that it resulted from the TP activity of this enzyme. The same dimeric species was observed when StPBP2x was combined with an MGT from S. aureus (SgtB) or with an aPBP with a defective TP active site, S. pneumoniae PBP1a*. We also observed crosslinked products when chimeric StPBPs or StPBP2x∆™ was incubated with SgtB and Lipid II (Supplementary Fig. 14). Because StPBP2x can crosslink peptidoglycan strands produced by other Lipid II polymerases and does not require its TM domain for crosslinking, we conclude that its activity is not dependent on its cognate FtsW partner.
Figure 3: S. thermophilus PBP2x does not require FtsW for crosslinking peptidoglycan.

a, Schematic showing detection of crosslinked muropeptide species by LC-MS in the presence of S. thermophilus PBP2x. b, Representative LC-MS extracted ion chromatograms of three independent experiments showing the products of crosslinking reactions with S. thermophilus PBP2x. Linear peptidoglycan was generated using S. thermophilus FtsW, S. aureus SgtB or TP inactive S. pneumoniae PBP1a*. Crosslinking is detected by the appearance of peak C, a crosslinked muropeptide. Asterisk (*) denotes the inactivated TP variant.
Our studies have conclusively established that FtsW functions as a peptidoglycan polymerase. Although we cannot formally exclude the possibility that FtsW also has Lipid II flippase activity as previously proposed,7,8 a growing body of evidence suggests that this function is largely, if not entirely, carried out by MurJ in vivo.19–21 Unlike the aPBPs and related MGT enzymes, we discovered that the polymerase activity of FtsW requires divalent cations that prefer oxygen ligands as functional cofactors (Supplementary Fig. 5).22 The most likely role of the cation is to anchor the diphosphate of the Lipid II substrate and help neutralize its negative charge, but other functions are possible; structural information is needed to better understand the metal requirement. Similarly, structural information on the peptidoglycan synthase complex would illuminate how the bPBP associates with FtsW and promotes its polymerase activity. Many of the evolutionary couplings observed are between large side chains in one protein and small side chains in the other, and we have shown that disrupting one such interaction hinders polymerization activity (Supplementary Figs. 6c, 6d). An attractive possibility is that these specific interactions between FtsW and bPBP promote the formation of an active conformation of FtsW in the complex. Although B. subtilis RodA had polymerase activity in the absence of its cognate bPBP,2 the activity was weak and RodA may similarly be stimulated by complex formation.
Recent genetic and biochemical results also suggest that the polymerase activity of SEDS proteins requires an activation event beyond complexation with a bPBP partner in vivo.12 Amino acid substitutions in the pedestal domain of PBP2 in E. coli were found to stimulate RodA activity in vivo and in vitro, and these PBP2 variants bypass the requirement for MreC and other components of the shape determining system.12 Similar changes in FtsW and PBP3 from E. coli and Caulobacter crescentus were found to overcome the action of division inhibitors and thus are thought to activate the divisome.23–25 It therefore appears that the synthase activity of SEDS-bPBP complexes from both the Rod system and the divisome may be controlled by similar mechanisms within their respective machineries. The development of peptidoglycan polymerase assays for both FtsW and RodA now sets the stage for the molecular details of this regulation to be uncovered.
Another important outstanding question relates to the division of labor between FtsW-bPBP complexes and aPBPs during septal peptidoglycan biogenesis. In B. subtilis, cells remain viable in the absence of aPBPs, suggesting that the FtsW-bPBP complex plays a major, possibly sufficient, role in this process.26 Septal peptidoglycan synthesis in B. subtilis is directly coupled to treadmilling of FtsZ, which colocalizes with PBP2b, the partner of FtsW.27 However, most bacterial species require at least one aPBP for viability. In S. aureus, the essential aPBP, PBP2, is recruited to the septum and is thought to be crucial for cell division.28 Contrasting with B. subtilis, the initial rate of septal peptidoglycan synthesis in S. aureus is slow and dependent on FtsZ treadmilling, but later in cytokinesis, peptidoglycan synthesis is independent of FtsZ treadmilling.29 Thus, FtsW may serve as the major polymerase during treadmilling-dependent cell division, with PBP2 largely responsible for the subsequent treadmilling-independent phase of peptidoglycan synthesis. Irrespective of their relative roles, both FtsW-bPBP complexes and the aPBPs are clearly critical for proper assembly of peptidoglycan. Further studies of their activity and regulation will therefore pave the way for the discovery of molecules that inhibit their function for future antibiotic development.
Methods
Materials.
Unless otherwise indicated, all chemicals and reagents were purchased from Sigma-Aldrich. Restriction enzymes were purchased from New England Biolabs. Oligonucleotide primers were purchased from Integrated DNA Technologies. Culture media were purchased from Becton Dickinson. S. thermophilus LMG 18311 genomic DNA (gDNA) was purchased from ATCC (ATCC BAA-250D-5). Biotin-D-lysine (BDL) was prepared by de-protecting Fmoc-biotin-D-lysine (Bachem).18 S. aureus Lipid II and E. faecalis Lipid II were isolated from cells as described previously.15,16 S. aureus SgtB, SgtBY181D, PBP4 and E. faecalis PBPX were expressed and purified as previously reported.16,18,30
Bacterial strains, plasmids, oligonucleotide primers and culture conditions.
E. coli strains were grown with shaking at 37 ºC in lysogeny broth (LB), Terrific Broth (TB, Teknova), or on agarized LB plates. S. aureus strains were grown with shaking at 30 ºC or 37 ºC in Tryptic Soy Broth (TSB) or on agarized plates. P. aeruginosa strains were grown with shaking at 30 ºC in LB, M9 broth containing 0.2% casamino acids and 0.2% glucose, or on agarized LB plates. S. pneumoniae strains were grown statically in Todd Hewitt Broth containing 0.5% yeast extract (THY) at 37 ºC in an atmosphere containing 5% CO2. When growth on solid media was required, S. pneumoniae strains were grown on pre-poured Trypticase Soy Agar with 5% Sheep Blood (TSAII 5%SB) plates with a 5 mL overlay of 1% nutrient broth (NB) agar or TSA plates containing 5% defibrinated sheep blood with appropriate additives. The following concentration of antibiotics were used: ampicillin, 50 µg/mL; carbenicillin, 50 µg/mL; chloramphenicol, 35 µg/mL, erythromycin, 0.2 µg/mL (S. pneumoniae) or 5 µg/mL (S. aureus); gentamicin, 30 µg/mL; kanamycin, 50 µg/mL (E. coli) or 250 µg/mL (S. pneumoniae); tetracycline, 0.2 µg/mL. The bacterial strains, plasmids and oligonucleotide primers used in this study are summarized in Supplementary Tables 3-5.
Plasmid construction for protein expression
S. thermophilus FtsW:
The stu0486 [M1-R426] gene encoding FtsW was amplified from S. thermophilus LMG18311 gDNA using primers oAT01 and oAT02. After digestion with NdeI and HindIII, the PCR product was ligated into pET28b(+). The resulting plasmid pATPL107 expresses His6-StFtsW. Plasmid pATPL121, expressing His6-StFtsWD292A (catalytically inactive mutant, StFtsW*), was PCR generated from pATPL107 with primers oAT03 and oAT04 containing the desired mutation. Plasmid pATPL185, expressing His6-StFtsWL345G, was generated from pATPL107 with primers oAT05 and oAT06 via In-Fusion Cloning (Takara Bio).
S. thermophilus PBP2x:
The stu1701 [M1-D755] gene encoding PBP2x was amplified from S. thermophilus LMG18311 gDNA using primers oAT07 and oAT08. After digestion with NdeI and HindIII, the PCR product was ligated into pET22b(+). The resulting plasmid pATPL091 expresses StPBP2x-His6. Plasmid pATPL123, expressing StPBP2xG53-D755-His6 (StPBP2x∆™), was constructed similarly using primers oAT09 and oAT08. Phobius web server was used for the prediction of the transmembrane region.31 Plasmid pATPL112, expressing StPBP2xS343A-His6 (transpeptidase mutant, StPBP2x*), was generated from pATPL091 with primers oAT10 and oAT11 via In-Fusion Cloning. Plasmid pATPL180, expressing StPBP2xS37F-His6, was generated from pATPL091 with primers oAT12 and oAT13 containing the desired mutation.
S. thermophilus PBP2b:
The stu0613 [M1-H704] gene encoding PBP2b was amplified from S. thermophilus LMG18311 gDNA using primers oAT14 and oAT15. After digestion with NdeI and HindIII, the PCR product was ligated into pET22b(+). The resulting plasmid pATPL169 expresses StPBP2b-His6.
S. thermophilus PBP2b™2x∆™:
The stu0613 [M1-M58] gene encoding the transmembrane domain of PBP2b and the stu1701 [G53-D755] gene encoding the extracellular domain of PBP2x were each amplified from S. thermophilus LMG18311 gDNA using the primer pairs oAT14/oAT16 and oAT17/oAT06. The two fragments were combined by overlap extension PCR. The resulting PCR product was digested with NdeI and HindIII before it was ligated into pET22b(+). The resulting plasmid pATPL172 expresses StPBP2bM1-M58StPBP2xG53-D755-His6.
S. thermophilus PBP2x™2b∆™:
The stu1701 [M1-I52] gene encoding the transmembrane domain of PBP2x and the stu0613[Q59-H704] gene encoding the extracellular domain of PBP2b were each amplified from S. thermophilus LMG18311 gDNA using the primer pairs oAT05/oAT18 and oAT19/oAT15. The two fragments were combined by overlap extension PCR. The resulting PCR product was digested with NdeI and HindIII before it was ligated into pET22b(+). The resulting plasmid pATPL173 expresses StPBP2xM1-I52StPBP2bQ59-H704-His6.
S. aureus FtsW:
The SAOUHSC_01063 [K2-N408] gene encoding FtsW was amplified from S. aureus NCTC 8325 gDNA using the primers oAT20 and oAT21. After digestion with NheI and NotI, the PCR product was ligated into pET28b(+). The resulting plasmid pMW2179 expresses His6-SaFtsW. Plasmid pMW2193, expressing His6-SaFtsWD287A (catalytically inactive mutant, SaFtsW*), was PCR generated from pMW2179 with primers oAT22 and oAT23 containing the desired mutation.
S. aureus PBP1:
The SAOUHSC_01145 [A2-D744] gene encoding PBP1 was amplified from S. aureus NCTC 8325 gDNA using the primers oAT24 and oAT25. The PCR product was ligated into NcoI and NotI-cut pET28b(+) via In-Fusion Cloning to produce plasmid pMW2194, which expresses SaPBP1-His6. Plasmid pMW2180, expressing SaPBP1S314A-His6 (transpeptidase mutant, SaPBP1*), was PCR generated from pMW2194 with primers oAT26 and oAT27 containing the desired mutation.
S. pneumoniae PBP2x:
The SPD_0306 [M1-D750] gene encoding PBP2x was amplified from S. pneumoniae D39 ∆cps gDNA using the primers oAT28 and oAT29. After digestion with NdeI and HindIII, the PCR product was ligated into pET22b(+).The resulting plasmid pATPL017 expresses SpPBP2x-His6. Plasmid pATPL067, expressing SpPBP2xS337A-His6 (transpeptidase mutant, SpPBP2x*), was PCR generated from pATPL017 with primers oAT30 and oAT31 containing the desired mutation.
S. pneumoniae PBP2b:
The SPD_1486 [M6-N685] gene encoding PBP2b was amplified from S. pneumoniae D39 ∆cps gDNA using primers oAT32 and oAT33. After digestion with NdeI and HindIII, the PCR product was ligated into pET22b(+). The resulting plasmid pATPL084 expresses SpPBP2b-His6.
S. pneumoniae PBP2b™2x∆™:
The SPD_1486 [M6-Y38] gene encoding the transmembrane domain of PBP2b and the SPD_0306 [T50-D750] gene encoding the extracellular domain of PBP2x were each amplified from S. pneumoniae D39 ∆cps gDNA using the primer pairs oAT28/oAT34 and oAT35/oAT29. The two fragments were combined by overlap extension PCR. The resulting PCR product was digested with NdeI and HindIII before it was ligated into pET22b(+). The resulting plasmid pATPL170 expresses SpPBP2bM6-Y38SpPBP2xT50-D750-His6.
S. pneumoniae PBP2x™2b∆™:
The SPD_0306 [M1-G49] gene encoding the transmembrane domain of PBP2x and the SPD_1486 [M39-N685] gene encoding the extracellular domain of PBP2b were each amplified from S. pneumoniae D39 ∆cps gDNA using the primer pairs oAT28/oAT36 and oAT37/oAT29. The two fragments were combined by overlap extension PCR. The resulting PCR product was digested with NdeI and HindIII before it was ligated into pET22b(+). The resulting plasmid pATPL171 expresses SpPBP2xM1-G49SpPBP2bM39-N685-His6.
S. pneumoniae PBP1a:
The SPD_0336 [M1-P719] gene encoding PBP1a was amplified from S. pneumoniae D39 ∆cps gDNA using the primers oAT38 and oAT39. After digestion with NdeI and XhoI, the PCR product was ligated into pET22b(+). The resulting plasmid pATPL044 expresses SpPBP1a-His6. Plasmid pATPL051, expressing SpPBP1aS370A-His6 (transpeptidase mutant, SpPBP1a*), was PCR generated from pATPL044 with primers oAT40 and oAT41 containing the desired mutation.
P. aeruginosa FtsW-PBP3 co-expression:
The PA4413[M1-R399] and PA4418[M1-G579] genes encoding FtsW and PBP3 were each amplified from P. aeruginosa PAO1 gDNA using the primer pairs oAT42/oAT43 and oAT44/oAT45, respectively. Plasmid pETDuet-FLAG, containing a His6-SUMO-FLAG tag at the first site and a His6 at the second site, was amplified using primers oAT46 and oAT47. This plasmid also encodes a 3C protease site downstream of the FLAG tag, which is followed by a GGSS linker. Next, the intergenic region between the end of the first site and beginning of the second site which contains the T7 promoter and an RBS for the second site, was amplified using primers oAT48 and oAT49. The PCR fragments for PA4413, intergenic region, and PA4418 were assembled and amplified by overlap extension PCR. This insert and the amplified vector backbone were assembled via isothermal assembly. The resulting plasmid pLSM3 expresses His-SUMO-FLAG-PaFtsW and PaPBP3-His6. Plasmid pLSM6, expressing His-SUMO-FLAG-PaFtsWD275A (catalytically inactive mutant, PaFtsW*) and PaPBP3-His6, was generated from pLSM3 using single-oligonucleotide site directed mutagenesis with primer oAT50 containing the desired mutation.
S. aureus FtsW-PBP1 co-expression:
The SAOUHSC_01063 [K2-N408] gene encoding FtsW and SAOUHSC_01145 [A2-D744] gene encoding PBP1 were amplified from S. aureus NCTC 8325 gDNA using the primers oAT51/oAT52 and oAT53/oAT54, respectively. Plasmid pETDuet-FLAG was amplified using primers oAT55 and oAT56 and the DNA fragment ligated with the FtsW insert via isothermal assembly. The resulting plasmid was amplified using primers oAT57 and oAT58 and the DNA fragment ligated with the PBP1 insert via isothermal assembly. The resulting plasmid, pMW2127, expresses His-SUMO-FLAG-SaFtsW and SaPBP1-His6. Plasmid pMW2144, expressing His-SUMO-FLAG-SaFtsW and SaPBP1S314A-His6 (SaPBP1*), was PCR generated from pMW2127 with primers oAT26 and oAT27. Plasmid pMW2176, expressing His-SUMO-FLAG-SaFtsWD287A (SaFtsW*) and SaPBP1S314A-His6 (SaPBP1*), was PCR generated from pMW2144 with primers oAT22 and oAT23.
Protein expression: general procedure.
For expression of Staphylococcus and Streptococcus FtsW and PBPs, E. coli C43(DE3) containing the expression plasmid was grown in 1 L TB supplemented with kanamycin or carbenicillin at 37 ºC with shaking until OD600 was 0.7–0.8. The culture was cooled to 20 ºC before inducing protein expression with 500 µM isopropyl β-D-1-thiogalactopyranoside (IPTG). Cells were harvested 18 h post-induction by centrifugation (4,200 x g, 15 min, 4 ºC) and the pellet was stored at −80 ºC.
Purification of Staphylococcus and Streptococcus FtsW: general protocol.
For purification of FtsW, cells were resuspended in 50 mL lysis buffer A (50 mM HEPES pH 7.5, 0.5 M NaCl) supplemented with 1 tablet of cOmplete EDTA-free Protease Inhibitor Cocktail. Cells were lysed by passaging the resuspended cells through a cell disruptor (EmulsiFlex-C3, Avestin) at 15,000 psi three times. Cell debris was removed by centrifugation (12,000 x g, 5 min, 4 ºC) and the membrane fraction was collected by ultracentrifugation of the supernatant (100,000 x g, 1 h, 4 ºC). The membrane pellet was resuspended in solubilization buffer A (50 mM HEPES pH 7.5, 0.5 M NaCl, 1% n-dodecyl β-D-maltoside (DDM), 20% glycerol) using a glass dounce tissue grinder (Wheaton). The resulting mixture was stirred for 1 h at 4 ºC before ultracentrifugation (100,000 x g, 1 h, 4 ºC). The resulting supernatant was supplemented with 0.5 mL pre-equilibrated Ni-NTA resin (Qiagen) and 20 mM imidazole and stirred for 30 min at 4 ºC. The sample was then loaded onto a gravity column and washed with 25 mL wash buffer A (50 mM HEPES pH 7.5, 0.5 M NaCl, 0.05% DDM, 20% glycerol) containing 20 mM imidazole and 25 mL wash buffer A containing 40 mM imidazole. The protein was then eluted in 10 mL wash buffer A containing 300 mM imidazole. The eluate was further purified by size exclusion chromatography (SEC) with a Superdex 200 10/300 GL column (GE Healthcare) equilibrated in running buffer A (50 mM HEPES pH 7.5, 0.5 M NaCl, 10% glycerol, 0.05% DDM). Fractions containing the target protein were concentrated by centrifugal filtration. The absorbance at 280 nm was measured using a NanoDrop One/One Microvolume UV-Vis Spectrophotometer (ThermoFisher Scientific) and the predicted extinction coefficient (ProtParam) was used to calculate concentration.32 Protein samples were then aliquoted and stored at −80 ºC.
Purification of full-length Staphylococcus and Streptococcus PBPs: general protocol.
PBPs were purified via the same protocol as FtsW, above, with the following modifications. Cells were resuspended in 50 mL lysis buffer A supplemented with 1 mM PMSF. For the solubilization buffer A and wash buffer A, 10% glycerol was used instead of 20%. For SEC, a Superose 6 10/300 GL column (GE Healthcare) was used.
Expression and purification of S. thermophilus PBP2x∆™.
For expression and purification of S. thermophilus PBP2x∆™, E. coli BL21(DE3) containing the expression plasmid was grown in 1 L LB supplemented with carbenicillin at 37 ºC with shaking until OD600 was 0.6. The culture was cooled to 16 ºC before inducing protein expression with 500 µM IPTG. Cells were harvested 18 h post-induction by centrifugation (4,200 x g, 15 min, 4 ºC) and resuspended in 25 mL lysis buffer A supplemented with 1 mM PMSF. Cell were lysed using a cell disruptor and the supernatant was collected after ultracentrifugation (100,000 x g, 30 min, 4 ºC). A pre-equilibrated 0.5 mL Ni-NTA resin and 20 mM imidazole were added and the resulting mixture was stirred for 30 min at 4 ºC. The sample was loaded onto the gravity column and washed with 25 mL lysis buffer A supplemented with 20 mM imidazole followed by 25 mL lysis buffer A supplemented with 40 mM imidazole. The protein was then eluted in 10 mL lysis buffer A containing 300 mM imidazole. The eluate was further purified by SEC with a Superdex 200 10/300 GL column equilibrated in running buffer A, and the fractions containing the target protein were combined, concentrated and stored at −80 ºC.
Co-expression and purification of S. aureus FtsW-PBP1.
For expression of S. aureus FtsW-PBP1 complex, E. coli C43(DE3) containing pAM174 (encodes arabinose-inducible Ulp1 protease) and the expression plasmid was grown in 1 L TB supplemented with carbenicillin and chloramphenicol at 37ºC with shaking until OD600 was 0.7–0.8. The culture was cooled to 20ºC before inducing protein expression with 500 µM IPTG and 0.1% arabinose. Cells were harvested 18 h post-induction by centrifugation (4,200 x g, 15 min, 4 ºC). The FtsW-PBP1 complex was purified via the same protocol as S. aureus FtsW, above, with the following modifications. Lysis, solubilization, and wash buffers for the purification of FLAG-FtsW and PBP1-His6 contained 150 mM NaCl. For the solubilization buffer A and wash buffer A, 10% glycerol was used. After the second ultracentrifugation, the resolubilized protein was applied to 500 µL washed α-FLAG G1 affinity resin (GenScript). The resin was washed with 25 mL wash buffer B (50 mM HEPES pH 7.5, 150 mM NaCl, 0.2% DDM, 10% glycerol) followed by 25 mL wash buffer A. The protein was eluted in 10 mL wash buffer A supplemented with 0.2 mg/mL FLAG peptide (GenScript).
Co-expression and purification of P. aeruginosa FtsW-PBP3.
For expression of P. aeruginosa FtsW-PBP3, E. coli expression strain CAM333 containing pAM174 and the expression plasmid was grown in 2 L TB supplemented with 2 mM MgCl2, ampicillin, and chloramphenicol at 37 ºC with shaking until OD600 was 0.7. The culture was cooled to 20 ºC before inducing protein expression with 1 mM IPTG and 0.1% arabinose. Cells were harvested 18 h post-induction by centrifugation (4,200 x g, 15 min, 4 ºC). To purify FLAG-FtsW and PBP3-His6, the cells were resuspended in lysis buffer B (50 mM HEPES pH 7.5, 150 mM NaCl, 20 mM MgCl2, 0.5 M DTT) and lysed by passage through a cell disruptor (Constant Systems) at 25,000 psi twice. Membranes were collected by ultracentrifugation (100,000 x g, 1 h, 4 ºC). The membrane pellets were resuspended in solubilization buffer B (20 mM HEPES pH 7.0, 0.5 M NaCl, 20% glycerol, and 1% DDM) for 1 h at 4 ºC before ultracentrifugation (100,000 x g, 1 h, 4 ºC). The supernatant was supplemented with 2 mM CaCl2 and loaded onto a homemade M1 ɑ-Flag antibody resin. The resin was washed with 20 column volumes (CVs) of wash buffer C (20 mM HEPES pH 7.0, 0.5 M NaCl, 20% glycerol, 2 mM CaCl2, 0.1% DDM) and the bound protein was eluted from the column with five CVs of elution buffer (20 mM HEPES pH 7.0, 0.5 M NaCl, 20% glycerol, 0.1% DDM, 5 mM EDTA pH 8.0, and 0.2 mg/mL 3 x FLAG peptide). Fractions containing the target protein were concentrated and the protein concentration was measured via Bradford assay. Proteins were then aliquoted and stored at −80 ºC.
Detection of PGT activity via western blot.
The protocol for detecting Lipid II/peptidoglycan was adapted from previously published methods.11,18,33
S. thermophilus FtsW:
Unless otherwise stated, proteins (StFtsW, 0.5 µM; bPBPs, 1 µM; SgtB and SpPBP1a, 0.5 µM) were added to a 1x reaction buffer (50 mM HEPES pH 7.5, 30% DMSO, 2.5 mM MgCl2) containing BDL (2 mM) and E. faecalis Lipid II (10 µM) in a total volume of 10 µL. The samples were incubated at room temperature for 5 min (SgtB and SpPBP1a) or 30 min (StFtsW). The reaction was heat-quenched at 95 ºC for 3 min and cooled to room temperature before the addition of 1 µL E. faecalis PBPX (100 µM) to the reaction mixture to label Lipid II/peptidoglycan with BDL. After 30 min, 11 µL 2x Laemmli sample buffer was added to quench the labeling reaction. The samples were loaded onto a 4–20% gradient polyacrylamide gel (Bio-Rad) and run at 180V. After the products were transferred onto a PVDF membrane (Bio-Rad), the membrane was blocked with SuperBlock TBS blocking buffer (ThermoFisher Scientific) for 30 min. For detection of biotin-labeled products, IRDye 800CW Streptavidin (LI-COR Biosciences) was added at a final concentration of 1:5000 and the membrane was incubated for 1 h. The membrane was washed 3 × 10 min with Tris-buffered saline (TBS) and imaged using an Odyssey CLx imaging system (LI-COR Biosciences).
S. aureus FtsW and FtsW-PBP1*:
SaFtsW and bPBP stocks (50 µM) were combined 1:1 and the mixture was chilled on ice for 30 min. The proteins (SaFtsW, 2.5 µM; bPBPs, 2.5 µM; SgtB, 1 µM) were then added to a 1x reaction buffer (50 mM HEPES pH 7.5, 30% DMSO, 10 mM MgCl2) containing BDL (3 mM), moenomycin (2 µM) and S. aureus Lipid II (10 µM) in a total volume of 10 µL. The samples were incubated at room temperature for 5 min (SgtB) or 1 h (SaFtsW). The reaction was heat-quenched at 95 ºC for 10 min. After cooling, 0.5 µL E. faecalis PBPX (200 µM) was added to the reaction mixture and the samples were incubated for 45 min. Reactions were quenched by the addition of 10.5 µL 2x Laemmli sample buffer. The protocol for Western blot was identical to the one used for StFtsW, described above.
P. aeruginosa FtsW-PBP3:
Proteins (FtsW-PBP3, 0.5 µM; SgtBY181D, 0.5 µM) were added to a 1x reaction buffer (125 mM HEPES pH 7.5, 20 mM MnCl2, 2.5 mM Tween 80, 200 μM cephalexin, 30% DMSO) containing E. faecalis Lipid II (10 µM) in a total volume of 10 μL. The samples were incubated at room temperature for 30 min. Following the incubation, reactions were heat-quenched at 95 °C for 2 min. After cooling, 2 μL BDL (20 mM) and 1 μL S. aureus PBP4 (50 µM) were added to the reaction mixture and the samples were incubated for 1 h. Reactions was quenched by the addition of 13 μL of 2x Laemmli sample buffer and samples were loaded onto a 4–20% polyacrylamide gel. The peptidoglycan product was then transferred onto a PVDF membrane and the membrane was fixed by incubating in 0.4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min. Subsequently, the blot was blocked using SuperBlock blocking buffer. The biotin-labeled products were detected by incubation with IRDye 800CW Streptavidin (1:5,000 dilution). The membrane was then washed four times with TBS with 0.05% Tween-20 (TBST), followed by one wash with PBS prior to imaging.
LC-MS analysis of peptidoglycan crosslinking activity.
This procedure was adapted from prior reports.16,18 E. faecalis Lipid II (20 µM), PGT (0.5 µM), and bPBP (1 µM) were incubated in a 30 µL 1x reaction buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 30% DMSO) at 30 ºC for 1 h unless otherwise indicated. The reactions were heat-quenched at 95 ºC for 3 min and allowed to cool to room temperature. Polymerized material was digested by incubating the mixture at 37 ºC for 3 h after the addition of 65 µL ddH2O and 5 µL mutanolysin (from Streptomyces globisporus, 4000 U/mL). To reduce the muropeptide products, 50 µL sodium borohydride (10 mg/mL) was added and the mixture was incubated at room temperature for 30 min. The pH was adjusted to ~4 with 20% phosphoric acid and the samples were lyophilized to dryness overnight. The samples were resuspended in 20 µL ddH2O and analyzed by LC-MS on an Agilent 6520 Q-TOF mass spectrometer with ESI-MS operating in positive mode. Muropeptide products were separated on a Waters Symmetry Shield RP18 column (5 µm, 3.9 × 150 mm) with the following method: flow rate = 0.5 mL/min, 100% solvent A (H2O, 0.1% formic acid) for 5 min followed by a linear gradient of solvent B (acetonitrile, 0.1% formic acid) from 0 to 40% over 25 min. Molecular ions for the target muropeptide fragments were extracted from the total ion chromatogram.
S. aureus strain construction
The SAOUHSC_01063 [M1-N408] gene encoding FtsW was amplified from S. aureus RN4220 gDNA using primers oAT59 and oAT60. After digestion with SalI and BamHI, the PCR product was ligated into pLOW to produce pLOWftsW, which is an overexpression plasmid for SaftsW. To generate the D287A mutation, primer pairs oAT59/oAT62 and oAT61/oAT60 containing the desired mutation were used to amplify SAOUHSC_01063 fragments. The two fragments were combined by overlap extension PCR, and the PCR product was ligated into pLOW as described above. The resulting plasmid pLOWftsWD287A is an overexpression plasmid for SaftsWD287A.
For transformation of S. aureus, plasmids were electroporated into wild-type RN4220 and the cells were recovered at 30 ºC for 1–2 hrs. Cells were plated on TSB agar containing erythromycin and the transformants were recovered after incubation for 1–2 days.
P. aeruginosa strain construction
The PA4413[M1-R399] genes encoding FtsW and FtsWD275A were amplified from pLSM3 and pLSM6 using primers oAT63 and oAT64. After digestion with SacI and XbaI, the PCR products were ligated into pPSV38 to generate pLSM8 and pLSM9, which are overexpression plasmids for PaftsW and PaftsWD275A, respectively. For transformation of P. aeruginosa, plasmids were electroporated into wild-type PAO1 as previously described.34 Cells were plated on LB agar containing gentamycin and the transformants were recovered after overnight incubation.
S. pneumoniae strain construction
S. pneumoniae R6 and its derivatives were transformed using a previously reported protocol.35 In brief, cells were grown to mid-log phase and back-diluted to OD600 ~0.03 in THY containing 1 mM CaCl2, 0.2% bovine serum albumin, and other appropriate additives. Competence was stimulated by adding 500 pg/mL competence stimulating peptide (CSP-1, AnaSpec) and the cells were incubated for 15 min. To 1 mL culture, 200 ng of DNA product was added and the resulting culture was grown for 1 hr. For selection of transformants, 100 µL of the culture was combined with 5 mL molten 1% NB agar supplemented with the appropriate additives, and the mixture was poured over fresh TSAII 5%SB plates. Transformants were recovered after overnight incubation.
Strain AT202 has a zinc-inducible copy of SpftsW inserted in an intergenic region between cpsO and cpsN described previously.36 To construct AT202, the SPD_0952[M1-K409] gene encoding ftsW was first amplified from D39 ∆cps gDNA using primers oAT65 and oAT66. After digestion with XhoI and BamHI, the PCR product was ligated into pLEM023. The resulting plasmid pATPL139 contains SpftsW under the control of a zinc-inducible Pczc promoter with a consensus RBS (Pzn-SpftsW).37 For integration of this cassette between cpsO and cpsN, the two ~1 kb regions flanking this site were amplified from R6 gDNA using the primer pairs oAT67/oAT68 and oAT69/oAT70. The Pzn-SpftsW cassette and kan gene were amplified from pATPL139 and D39 ∆bgaA::kan gDNA using primer pairs oAT71/oAT72 and oAT73/oAT74, respectively. The four fragments were assembled and amplified by overlap extension PCR. The resulting PCR cassette was transformed into wild-type R6, and the transformants were selected with kanamycin. Integration into the genome was confirmed by diagnostic PCR using primers oAT75 and oAT74.
Strain AT203 was generated by replacing the native ftsW locus of AT202 with an erm cassette. The two ~1 kb regions flanking ftsW were amplified from R6 gDNA using the primer pairs oAT76/oAT77 and oAT78/oAT79. The two fragments, together with the erm gene amplified from S. pneumoniae D39 ∆bgaA::erm genomic DNA with primers oAT73 and oAT74, were assembled and amplified by overlapping PCR. The resulting PCR cassette was transformed into AT202, and the transformants were selected with erythromycin and kanamycin on plates supplemented with 0.2 mM ZnSO4 and 0.02 mM MnCl2.38 Integration into the genome was confirmed by diagnostic PCR using primers oAT80 and oAT74.
Strains AT204-AT208 were constructed by inserting a fucose-inducible copy of ftsW in AT203 bgaA locus. SpftsW, SaftsW and StftsW were amplified using primer pairs oAT65/oAT66, oAT81/oAT82, and oAT83/oAT84, respectively. After digestion with XhoI and BamHI, the PCR product was ligated into pAKF205. The resulting plasmids pATPL137, pATPL141 and pATPL142 contain SpftsW, SaftsW, and StftsW under the control of a fucose-inducible Pfcsk promoter with a consensus RBS (Pfucose-ftsW), respectively.39 Plasmids pATPL138 and pATPL143, containing catalytically inactive SpftsWD289A (SpftsW*) and StftsWD292A (StftsW*) mutants, were PCR generated using primer pairs oAT85/oAT86 and oAT03/oAT04, respectively. Plasmids were transformed into AT203, and the transformants were selected with tetracycline and kanamycin on plates supplemented with 0.2 mM ZnSO4 and 0.02 mM MnCl2. Integration into the genome was confirmed by diagnostic PCR using primers oAT87 and oAT88.
Electron microscopy of S. aureus cells.
Overnight cultures grown in TSB supplemented with erythromycin at 30 ºC were back-diluted 1:100 in fresh TSB supplemented with or without 1 mM IPTG and grown to mid-log phase at 37 ºC. Cells were then fixed by adding a mixture of 1.25% formaldehyde, 2.5% glutaraldehyde, and 0.03% picric acid in 0.1 M sodium cacodylate buffer (pH 7.4). The fixed samples were imaged by electron microscopy (JEOL 1200EX-80Kv, Harvard Medical School EM Facility) as described previously.40
Phase contrast microscopy of P. aeruginosa and S. pneumoniae cells
P. aeruginosa:
Overnight cultures grown in LB supplemented with gentamycin were back-diluted 1:500 in M9 containing 0.2% casamino acids and 0.2% glucose. These cultures were then grown to an OD600 of 0.2 at 30 ºC whereupon 1 mM of IPTG was added to induce expression. The induced cells were grown for 2.5 h. Live cells were imaged using phase contrast microscopy using a Nikon Ti inverted microscope equipped with a 100x Plan Apo 1.45 Oil Ph3 DM objective and an Andor Zyla 4.2 Plus sCMOS camera.
S. pneumoniae:
Cells were grown to mid-log phase in THY supplemented with 0.5 mM ZnSO4 and 0.05 mM MnCl2 and back-diluted to OD600 ~0.02 in THY with the same additives. After 2 h, cells were concentrated by centrifugation, washed with THY lacking any additives, and back-diluted to OD600 ~0.02 in THY containing 1% fucose. The induced cells were grown for 3 h. Cells were then collected by centrifugation and immobilized on 2% agarose pads. Phase contrast imaging was performed on a Nikon TE2000-U inverted microscope equipped with a 100x Plan Apo 1.45 Oil Ph3 DM objective and a Hamamatsu OrcaR2 CCD monochrome camera.
Viability assay of S. pneumoniae strains.
Cultures were grown from glycerol stocks in THY supplemented with 0.2 mM ZnSO4 and 0.02 mM MnCl2 until mid-log phase and normalized to an OD600 of 0.1. The normalized cultures were serially diluted and 5 µL of each dilution was spotted onto TSA 5%SB plates containing 0.5% fucose or 0.2 mM ZnSO4. Plates were imaged after overnight incubation.
Homology modelling.
Homology models of FtsW from S. aureus and S. thermophilus were constructed based on the structure of Thermus thermophilus RodA using MODELLER.41 In brief, since both proteins had low sequence identity (∼28% and ∼27% for S. aureus and S. thermophilus, respectively) to RodA, a multiple sequence alignment of 14 homologues from diverse bacterial taxa was performed (FtsW and RodA from T. thermophilus, B. subtilis, E. coli, S. aureus, S. pneumoniae, S. thermophilus, P. aeruginosa). The resulting alignment was used to guide template alignment in MODELLER, with disordered residues (216–266 and 280–293 for S. thermophilus and 213–256 and 270–283 for S. aureus) omitted from the model generation. A total of ten models were generated and the top-scoring model was chosen for further analysis. Homology models of the TM-H1 portions of PBP1 and PBP2x from S. aureus and S. thermophilus, respectively, were constructed using SWISS-MODEL and were based on the model of E. coli FtsI from Ovchinnikov et al.17,42
Data availability
All data generated or analyzed during this study are available from the corresponding authors upon request.
Supplementary Material
Acknowledgements
We thank the Microscopy Resources on the North Quad (MicRoN) core at the Harvard Medical School for help with imaging and analysis. LC-MS data was acquired on an Agilent 6520 Q-TOF mass spectrometer supported by the Taplin Funds for Discovery Program. Funding for this work was provided by National Institutes of Health grants R01 AI083365 (to T.G.B.), R01 AI099144 (to T.G.B. and S.W.), R01 GM076710 (to D.K. and S.W.), CETR U19 AI109764 (to A.C.K., D.K., T.G.B. and S.W.) and F32 GM123579 (to M.A.W.). A.T. is supported in part by the Funai Overseas Scholarship. W.L. is supported in part by the Charles A. King Trust Postdoctoral Research Fellowship Program.
Footnotes
Competing Interests
The authors declare no competing interest.
References
- 1.Silhavy TJ, Kahne D & Walker S The bacterial cell envelope. Cold Spring Harbor Perspectives in Biology 2, 1–17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meeske AJ et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho H et al. Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat. Microbiol 1, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emami K et al. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat. Microbiol 2, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egan AJF & Vollmer W The physiology of bacterial cell division. Ann. N. Y. Acad. Sci 1277, 8–28 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Otten C, Brilli M, Vollmer W, Viollier PH & Salje J Peptidoglycan in obligate intracellular bacteria. Mol. Microbiol 107, 142–163 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohammadi T et al. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 30, 1425–1432 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohammadi T et al. Specificity of the transport of lipid II by FtsW in Escherichia coli. J. Biol. Chem 289, 14707–14718 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Typas A, Banzhaf M, Gross CA & Vollmer W From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol 10, 123–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santiago M et al. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16, 252 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sjodt M et al. Structure of the peptidoglycan polymerase RodA resolved by evolutionary coupling analysis. Nature 556, 118–121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohs PDA et al. A central role for PBP2 in the activation of peptidoglycan polymerization by the bacterial cell elongation machinery. PLOS Genet. 14, e1007726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weiss DS et al. Localization of the Escherichia coli cell division protein Ftsl (PBP3) to the division site and cell pole. Mol. Microbiol 25, 671–681 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Fraipont C et al. The integral membrane FtsW protein and peptidoglycan synthase PBP3 form a subcomplex in Escherichia coli. Microbiology 157, 251–259 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Qiao Y et al. Lipid II overproduction allows direct assay of transpeptidase inhibition by β-lactams. Nat. Chem. Biol 13, 793–798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welsh MA et al. Identification of a functionally unique family of penicillin-binding proteins. J. Am. Chem. Soc 139, 17727–17730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ovchinnikov S et al. Large-scale determination of previously unsolved protein structures using evolutionary information. Elife 4, 1–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiao Y et al. Detection of lipid-linked peptidoglycan precursors by exploiting an unexpected transpeptidase reaction. J. Am. Chem. Soc 136, 14678–14681 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sham L-T et al. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 719, 220–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rubino FA, Kumar S, Ruiz N, Walker S & Kahne D Membrane potential is required for MurJ function. J. Am. Chem. Soc 140, 4481–4484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruiz N Filling holes in peptidoglycan biogenesis of Escherichia coli. Curr. Opin. Microbiol 34, 1–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng H, Chruszcz M, Lasota P, Lebioda L & Minor W Data mining of metal ion environments present in protein structures. J. Inorg. Biochem 102, 1765–1776 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du S, Pichoff S & Lutkenhaus J FtsEX acts on FtsA to regulate divisome assembly and activity. Proc. Natl. Acad. Sci 113, E5052–E5061 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Modell JW, Hopkins AC & Laub MT A DNA damage checkpoint in Caulobacter crescentus inhibits cell division through a direct interaction with FtsW. Genes Dev. 25, 1328 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Modell JW, Kambara TK, Perchuk BS & Laub MT A DNA damage-Induced, SOS-independent checkpoint regulates cell division in Caulobacter crescentus. PLoS Biol. 12, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McPherson DC & Popham DL Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilis. J. Bacteriol 185, 1423–1431 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bisson-Filho AW et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355, 739–743 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinho MG & Errington J Recruitment of penicillin-binding protein PBP2 to the division site of Staphylococcus aureus is dependent on its transpeptidation substrates. Mol. Microbiol 55, 799–807 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Monteiro JM et al. Peptidoglycan synthesis drives an FtsZ-treadmilling-independent step of cytokinesis. Nature 554, 528–532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rebets Y et al. Moenomycin resistance mutations in Staphylococcus aureus reduce peptidoglycan chain length and cause aberrant cell division. ACS Chem. Biol 9, 459–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Käll L, Krogh A & Sonnhammer ELL Advantages of combined transmembrane topology and signal peptide prediction-the Phobius web server. Nucleic Acids Res. 35, 429–432 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasteiger E et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barrett D et al. Analysis of glycan polymers produced by peptidoglycan glycosyltransferases. J. Biol. Chem 282, 31964–31971 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi KH, Kumar A & Schweizer HPA 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: Application for DNA fragment transfer between chromosomes and plasmid transformation. J. Microbiol. Methods 64, 391–397 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Fenton AK, Mortaji L El, Lau DTC, Rudner DZ & Bernhardt TG CozE is a member of the MreCD complex that directs cell elongation in Streptococcus pneumoniae. Nat. Microbiol 2, 16237 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berg KH, Biørnstad TJ, Straume D & Håvarstein LS Peptide-regulated gene depletion system developed for use in Streptococcus pneumoniae. J. Bacteriol 193, 5207–5215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kloosterman TG, Van Der Kooi-Pol MM, Bijlsma JJE & Kuipers OP The novel transcriptional regulator SczA mediates protection against Zn2+ stress by activation of the Zn2+-resistance gene czcD in Streptococcus pneumoniae. Mol. Microbiol 65, 1049–1063 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Tsui HCT et al. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Mol. Microbiol 100, 1039–1065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan PF et al. Characterization of a novel fucose-regulated promoter (PfcsK) suitable for gene essentiality and antibacterial mode-of-action studies in Streptococcus pneumoniae. J. Bacteriol 185, 2051–2058 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee W et al. The mechanism of action of lysobactin. J. Am. Chem. Soc 138, 100–103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Webb B & Sali A Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinforma 2016, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waterhouse A et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are available from the corresponding authors upon request.