

Abstract
Chronic activation of the renin‐angiotensin‐aldosterone system (RAAS) promotes and perpetuates the syndromes of congestive heart failure, systemic hypertension, and chronic kidney disease. Excessive circulating and tissue angiotensin II (AngII) and aldosterone levels lead to a pro‐fibrotic, ‐inflammatory, and ‐hypertrophic milieu that causes remodeling and dysfunction in cardiovascular and renal tissues. Understanding of the role of the RAAS in this abnormal pathologic remodeling has grown over the past few decades and numerous medical therapies aimed at suppressing the RAAS have been developed. Despite this, morbidity from these diseases remains high. Continued investigation into the complexities of the RAAS should help clinicians modulate (suppress or enhance) components of this system and improve quality of life and survival. This review focuses on updates in our understanding of the RAAS and the pathophysiology of AngII and aldosterone excess, reviewing what is known about its suppression in cardiovascular and renal diseases, especially in the cat and dog.
Keywords: angiotensin converting enzyme inhibitor, angiotensin receptor blocker, chronic kidney disease, heart failure, mineralocorticoid receptor blocker, proteinuric kidney disease, systemic hypertension
Abbreviations
- 11β‐HSD2
11β‐hydroxysteroid dehydrogenase type 2
- ACE
angiotensin converting enzyme
- ACEI
angiotensin converting enzyme inhibitor
- ACVIM
American College of Veterinary Internal Medicine
- ARB
angiotensin II type‐1 receptor blocker
- AngI
angiotensin I
- AngII
angiotensin II
- AT1R
angiotensin II type‐1 receptor
- AT2R
angiotensin II type‐2 receptor
- CHF
congestive heart failure
- CKCS
Cavalier King Charles Spaniel
- CKD
chronic kidney disease
- DCM
dilated cardiomyopathy
- DRI
direct renin inhibitor
- GFR
glomerular filtration rate
- HCM
hypertrophic cardiomyopathy
- HFpEF
heart failure with preserved ejection fraction
- HFrEF
heart failure with reduced ejection fraction
- HTN
systemic hypertension
- MMVD
myxomatous mitral valve disease
- MR
mineralocorticoid receptor
- MRA
mineralocorticoid receptor antagonist
- RAAS
renin‐angiotensin‐aldosterone system
- ROS
reactive oxygen species
- UAldo:C
urine aldosterone to creatinine ratio
- UP:C
urine protein to creatinine ratio
1. INTRODUCTION
Although renin‐angiotensin‐aldosterone system (RAAS) activation can be compensatory in the early stages of cardiovascular and renal disease, long‐term activation is maladaptive. In patients with heart failure, relative increases in plasma renin activity and the blood aldosterone concentration are considered markers of, and contributors to, the hemodynamic and anatomic derangements of this syndrome.1, 2, 3, 4, 5 The adverse effects of chronic exposure to high concentrations of angiotensin II (AngII) and aldosterone are outlined in Table 1.6, 7, 8, 9, 10, 11, 12, 13, 14 Suppression of RAAS is therefore a key strategy in the treatment of chronic cardiovascular and renal disease and is achieved by the administration of angiotensin converting enzyme inhibitors (ACEI), AngII type‐1 receptor blockers (ARB), and mineralocorticoid receptor antagonists (MRA), alone or in combination. The more we learn about this system, however, the broader and more complex it becomes. Redundancies that bypass our therapeutic blockade or inadvertent suppression of beneficial components of this system might reduce the efficacy of our RAAS suppressive therapies. For example, Angiotensin II and aldosterone levels occasionally rise despite pharmacologic RAAS suppression, and the underlying mechanisms are still not well understood.15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32 Furthermore, Some drugs and strategies used to treat cardiovascular disease, including furosemide, amlodipine, hydralazine and dietary sodium restriction, stimulate the RAAS (Supporting Information Figure S1).5, 33, 34, 35, 36, 37, 38, 39 Continued research into this complex system is necessary to improve medical therapies for cardiovascular and renal diseases, allowing us to more adeptly modulate this system and improve clinical outcomes.
Table 1.
Aldosterone and angiotensin II: harmful cardiovascular and renal effects
| Adverse effect | Direct effects of angiotensin II | Direct effects of aldosterone |
|---|---|---|
| Myocardial remodeling: fibrosis, hypertrophy, necrosis, apoptosis | Yes | Yes |
| Vascular remodeling: hypertrophy, fibrosis | Yes | Yes |
| Increase ROS | Yes | Yes |
| Pro‐inflammatory (cytokines, ROS) | Yes | Yes |
| Arrhythmogenic | Yes | Yes |
| Vascular endothelial dysfunction (ET‐1, vasopressin, acetylcholine‐mediated vasodilatory dysfunctin) | Yes | Yes |
| Systemic hypertension | Yes | Yes |
| Glomerular damage | Yes | Yes |
| Glomerular dysfunction: proteinuria | Yes | Yes |
| Increased intraglomerular pressure | +++ (vasoconstriction) | + (fluid retention & SNS) |
| Tubulointerstitial injury | Yes | Yes |
| Baroreceptor dysfunction➔HR increase | Maybe | Yes |
| Increased SNS tone | Yes | Yes |
| Inotropy | Yes | No |
| Direct HR increase | Yes | No |
| Salt appetite | Yes | Yes |
| Increased thirst | Yes | No |
| Na+ and H2O retention, congestion | Yes | Yes |
| K+ wasting | No | Yes |
Abbreviations: ET‐1, endothelin 1; HR, heart rate; SNS, sympathetic nervous system activation; ROS, reactive oxygen species.
2. CIRCULATING AND TISSUE RAAS
Renin is synthesized as preprorenin in the juxtaglomerular epithelioid cells, cleaved to prorenin, and either released as prorenin or further processed to form active renin, which is stored in granules. Renin granules are then released in a controlled manner, making renin the rate‐limiting step of the renin‐angiotensin‐aldosterone cascade in most species. Conversely, angiotensinogen is constitutively released from the liver (Figure 1) and is usually present in excess when compared to renin. Increased renin synthesis and release occur in situations of low systemic blood pressure, hypovolemia, sodium deprivation, and sympathetic stimulation. In the circulation, renin metabolizes angiotensinogen, liberating angiotensin I (AngI). Angiotensin converting enzyme (ACE), which is released from endothelial cells, converts AngI to AngII. Angiotensin II acts at 2 receptors, the angiotensin type‐1 and type‐2 receptors (AT1R and AT2R). Angiotensin II's actions at the AT1R leads to increased sodium retention, vasoconstriction (including preferential constriction of the efferent arteriole of the kidney), stimulation of thirst and desire for salt, enhanced sympathetic nervous system activity, and aldosterone release from the adrenal gland's zona glomerulosa. The interaction of AngII at this receptor mediates much of the disease change associated with chronic RAAS activation summarized in Table 1. Actions of AT2R stimulation are counter regulatory to those of the AT1R, where type‐2 receptor stimulation leads to anti‐inflammatory, anti‐fibrotic, and vasodilatory effects. The AT2R is the dominant receptor type in the fetus and plays a key role in development, but is less relevant in the normal adult. The AT2R, however, might be upregulated in certain disease states.40, 41, 42
Figure 1.
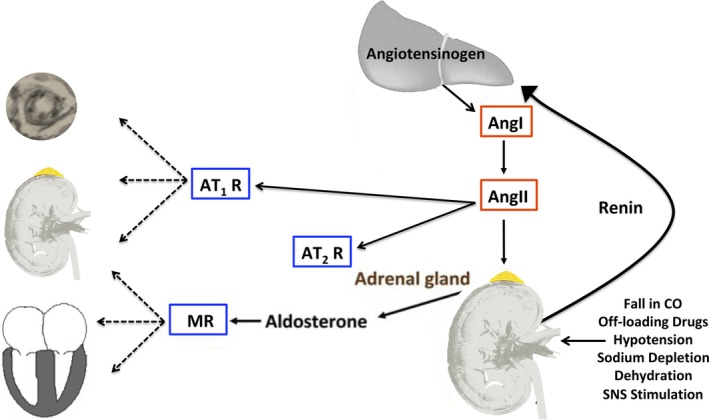
The renin‐angiotensin‐aldosterone system (RAAS) scheme: factors that lead to the release of renin from the juxtaglomerular cells of the kidney and the “target organs” of AngII, for which the actions (both pathophysiologic and pathologic) are primarily mediated by the AT1R. The actions of AngII at the AT2R are thought to counter those of the AT1R. The AT2R is likely of greater importance in the developing fetus, yet might be upregulated in certain disease states in adults. Angiotensin II is also a major secretagogue for aldosterone, which acts via the MR to increase sodium retention in the kidney and also amplifies the pathophysiologic effects of AngII in the heart, kidney, and vasculature. AngI, angiotensin I; AngII, angiotensin II, AT1R, angiotensin type‐1 receptor; AT2R, angiotensin type‐2 receptor; CO, cardiac output; MR, mineralocorticoid receptor; SNS, sympathetic nervous system
Aldosterone, the terminal hormone of RAAS, exerts 90% of the mineralocorticoid activity of adrenal secretions, and is a key regulator of sodium, potassium, and body fluid balance.43, 44 Angiotensin II and increased extracellular K+ concentration, the strongest secretagogues for aldosterone, increase expression of the CYP11B2 gene, which encodes aldosterone synthase.45 Acting via the mineralocorticoid receptor (MR), aldosterone modulates the expression of ion channels, pumps, and exchangers in epithelial tissues (kidney, colon, and salivary and sweat glands). This ultimately leads to an increase in transepithelial Na+ and water reabsorption and K+ excretion. Mineralocorticoid receptors are also found in non‐epithelial tissues such as the retina, brain, myocardium, vascular smooth muscle cells, macrophages, fibroblasts, and adpiocytes.46, 47, 48, 49, 50 Aldosterone's effects are therefore wide spread, extending well beyond its role as a “renal hormone.” Specifically, aldosterone is thought to mediate inflammation and affect energy metabolism in non‐epithelial tissues.50
The unbound MR is primarily located in the cytoplasm, and when bound by ligand, it is shuttled to the nucleus where it acts as a transcription factor.51 Some actions of aldosterone are non‐genomic and occur relatively rapidly. These actions are likely mediated by activation of the small fraction of cell membrane localized MR and MR interaction with other receptors such as the G‐protein coupled estrogen receptor, AT1R, and epidermal growth factor receptor.52 Aldosterone is the primary physiological ligand of the MR, though glucocorticoids such as cortisol have a similar receptor affinity. In epithelial cells and vascular smooth muscle cells, the enzyme 11β‐hydroxysteroid dehydrogenase type 2 (11β‐HSD2) alters the glucocorticoids and prevents their binding to the MR. Thus, more abundant glucocorticoids are prevented from out‐competing mineralocorticoids for the MR. In tissues such as the myocardium where 11β‐HSD2 is scarce, cortisol binding to the MR is likely important.53
Angiotensin peptides and aldosterone are also produced in tissues such as brain, blood vessels, kidneys, and the heart.40, 54, 55, 56, 57 Locally produced RAAS hormones play important roles in normal cardiovascular function and electrolyte‐fluid homeostasis, yet also mediate abnormal remodeling in the tissues. In‐vitro work on cultured vascular smooth muscle cells and cardiomyocytes has shown that aldosterone upregulates components of the RAAS, including ACE activity and AngII‐stimulated signal transduction, leading to increased local (tissue) activity of the RAAS.58, 59, 60, 61, 62 Tissue RAAS components are also modulated by mechanical stretch of the myocardium and vessels, adipocyte secretions, presence of reactive oxygen species (ROS), and inflammation.63, 64 Although the amount of aldosterone produced in the tissues is normally <1% of the amount produced by the adrenal glands,56, 65 locally produced aldosterone likely plays a role in abnormal remodeling (hypertrophy and fibrosis), and tissue RAAS management will likely be part of future pharmacotherapeutic strategy.11, 12, 40
Recently, attention has turned toward the prorenin receptor ((P)RR), which binds both renin and prorenin, and is a multifunctional receptor found in several locations within the kidney, as well as the heart, adipose tissue, and brain.66 The diverse roles of this receptor include maintenance of intra‐ and extracellular pH, central regulation of blood pressure, and sodium homeostasis in the kidney. The role this receptor plays in the pathophysiology of cardiovascular and renal diseases remains unanswered, yet it is known that the (P)RR binds prorenin and renders it active in the tissues, enabling the local generation of angiotensin peptides.54, 66, 67
The quest to find novel/alternative pathways of angiotensin generation and metabolism led to the discovery of angiotensin (1,12) and (1,25), which are found in cardiovascular and renal tissues and serve as precursors for angiotensin peptides such as AngII (Figure 2).68, 69 Chymase, a serine protease, catalyzes the formation of AngII from both angiotensin (1,12) and AngI, allowing ACE‐independent formation of AngII in the tissue, and this pathway is likely the primary generator of tissue AngII.69, 70, 71 In fact, chymase has been labeled the “most efficient AngII‐forming enzyme” and is released from mast cells, cardiac fibroblasts, and vascular endothelial cells during acute and chronic tissue injury and remodeling.72, 73, 74 Chymase activation results in pro‐fibrotic, anti‐fibrotic, or pro‐inflammatory phenotypes, with the exact phenotype or combination of phenotypes depending upon the tissue and the nature and timing of the stress.75 Chymase is an important player in AngII formation (and a pro‐fibrotic phenotype) in cardiac disease.76, 77 Chymase is also hypothesized to exacerbate cell death and mitochondrial injury after cardiac ischemia/reperfusion.78 Additionally, mast cell activation and increased release of mediators such as chymase have been implicated in the pathophysiology of cardiometabolic disease, such as diabetes mellitus and obesity.79 The phenotype of chymase activation differs in canine models of hemodynamic overload and might contribute to both extracellular matrix degradation and fibrosis, contributing to or counter‐balancing ventricular dilatation.80, 81, 82, 83, 84
Figure 2.
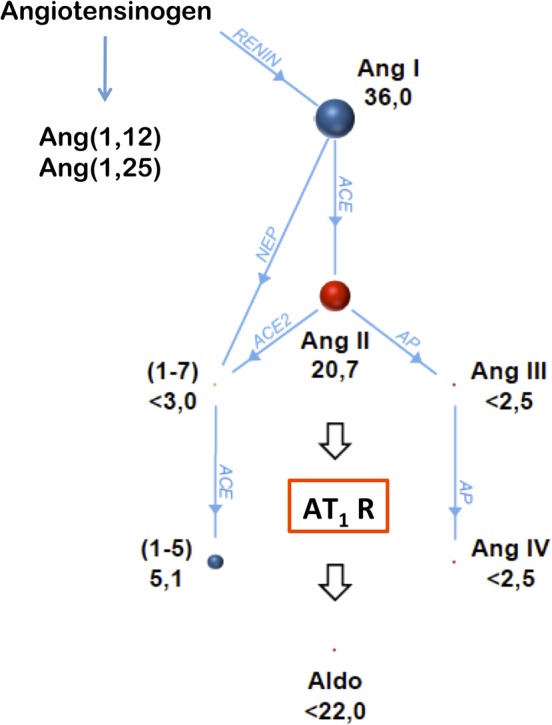
The renin‐angiotensin‐aldosterone system peptide cascade (RAAS Fingerprint) is illustrated as a pedigree starting at angiotensin I. Each intersection represents a specific peptide fragment symbolized by colored spheres; enzymes involved in the reactions are annotated on connecting lines. Size of spheres and numbers beside them represent absolute concentrations of angiotensins (pg/mL, median values) in serum samples from 6 middle‐aged, healthy male Beagles; the concentrations were analyzed by mass spectrometry. Angiotensin (1,7) and (1,5) are breakdown products of both angiotensin I and II. The novel peptides angiotensin (1,12) and (1,25) may be directly derived from angiotensinogen and serve as precursors for angiotensin peptides such as AngII. Aldo, aldosterone; AngI, angiotensin I; AngII, angiotensin II; AngIII, angiotensin III; Ang IV, angiotensin IV; AP, aminopeptidase; AT1R, angiotensin type‐1 receptor; NEP, neutral endopeptidase
The metabolism/degradation of angiotensin peptides is of interest as these pathways can reduce levels of AngI and AngII and result in peptides that are “active” and, in some cases, counter regulatory to RAAS activation. A counter‐regulatory pathway, the ACE2‐Angiotensin(1,7)‐Mas axis, is currently being investigated, as it appears to elicit protective actions, including vasodilation and increased nitric oxide synthesis.40, 54 The generation of Angiotensin(1,7) [Ang(1,7)] in the heart and brain arises from ACE2 processing of AngII, whereas in the circulation and kidney, Ang(1,7) arises from processing of angiotensin I by endopeptidases, such as neprilysin.54, 85 Angiotensin(1,7) and its metabolite Ala1‐Ang(1,7) bind the Mas and Mas‐related G protein‐coupled receptor and elicit the effects noted above.86 The expression of Mas receptors has been most thoroughly explored in rats and mice, where it is expressed in the brain, testis, kidney, heart, and vessels, with expression patterns changing with age.87 Current research is exploring the role of this counter‐regulatory pathway in the pathophysiology of cardiovascular and renal disease.
The actions of other newly discovered angiotensin peptides are also being studied and might reveal pharmacologic targets for both up‐ and downregulation. For example, AngI can also be metabolized by ACE2 to form to Ang(1,9), which appears to bind at the AT2R.88 Also, AngII is metabolized by aminopeptidase A to form angiotensin III, which like its parent peptide is capable of binding both the AT1R and AT2R.89 Angiotensin III can then be metabolized to angiotensin IV via aminopeptidase N. Both of these angiotensin peptides lead to increased atrial stretch‐induced atrial natriuretic peptide secretion in animal models, via the AT2R and the insulin‐regulated aminopeptidase, respectively.90, 91 For a more comprehensive review of tissue RAAS, novel RAAS components, and the production of aldosterone in tissues, the reader is referred elsewhere.40, 42, 54, 55
2.1. Chronobiology of the RAAS
RAAS peptides and antidiuretic hormones oscillate in a circadian (approximately 24 hours) periodicity.92, 93, 94 This leads to day‐night variation in urine flow rate, urinary electrolyte excretion, and blood pressure. The circadian “clock” begins at the cellular level where transcriptional and translational regulators induce expression of multiple genes that ultimately dictate the metabolic rates and homeostasis of an organism.95 Central clocks, residing in the brain, and local cellular clocks respond to environmental and metabolic signals, allowing the organism to adapt to changing circumstances. The time‐variant fluctuation in RAAS hormones in dogs92, 96 is similar to that in humans, and urine flow and sodium excretion have diurnal peaks whereas urine osmolality and potassium excretion have nocturnal peaks. A more recent and detailed characterization of the chronobiology of these peptides shows that, in dogs fed once daily (a.m.) with normal sodium meal, the renin activity and the urinary aldosterone to creatinine ratio (UAldo:C) decrease after feeding, then rise throughout the afternoon, peaking in the evening.97 Systemic blood pressure oscillates in parallel with RAAS peptides, increasing in the first half of the night, decreasing as morning approaches, which is opposite to what is described in healthy adult humans.98 The timing of feeding also affects the circadian rhythm of both the RAAS and systemic blood pressure suggesting that dietary sodium is a key mediator of this modulation.97, 99 An improved understanding of RAAS chronobiology has implications in both the characterization and quantification of RAAS activity and will likely inform the timing of drug administration.100, 101
2.2. RAAS, renal hemodynamics, and tubular function
The principal effects of AngII in the kidney include vasoconstriction of the interlobular artery and afferent and efferent arterioles (greatest effect on the latter), enhanced afferent arteriolar response to tubuloglomerular feedback, and constriction of the glomerular mesangium. The net result of these changes is an increase in filtration fraction. Renin‐angiotensin‐aldosterone system activation enhances sodium reabsorption throughout the renal tubules with AngII effects greatest at the proximal tubule and loop of Henle and aldosterone effects greatest at the distal convoluted tubule and collecting duct. Changes in renal perfusion pressure can also elicit biophysical and paracrine effects, such as increased generation of ATP, nitric oxide, and ROS, and induction of cyclooxygenase enzymes that lead to a pressure natriuresis.102, 103, 104 This allows the kidney to “escape” from the sodium retaining effects of excess AngII (and increased aldosterone production).105 Natriuretic peptides also counterbalance increases in blood volume and are released from both the atria and ventricles in the presence of increased stretch. Recent studies of the chronobiology of the RAAS, as well as studies of experimental sodium loading in dogs, suggest that the circulating and local RAAS might play a more important role than pressure natriuresis in physiological sodium homeostasis in this species.106, 107, 108
2.3. Assessment of RAAS Activity
Commonly evaluated components of the RAAS include plasma renin activity, direct plasma renin levels, plasma or serum aldosterone and AngII concentrations, plasma ACE activity, urine and serum Na+/K+ ratios, 24‐hour urinary aldosterone excretion, and the UAldo:C. More recently, liquid chromatography‐mass spectrometry has been used to create a comprehensive renin‐angiotensin system Fingerprint (Attoquant Diagnostics GmbH, Vienna, Austria) from both blood and tissue samples (Figure 2).109, 110 This comprehensive assessment is beneficial, as looking at only 1 or 2 components of this system might be misleading. For example, assessment of only ACE activity during ACEI treatment might suggest very effective suppression, yet AngII and aldosterone do “break through” this treatment in some patients.15, 17, 31, 106, 111, 112 Additional information regarding aldosterone metabolism and the assessment of RAAS activity in dogs and cats can be found in the Supporting Information of this review.
Genetic profiling of specific populations and individuals will also likely play a role in the development of future RAAS modulation strategies. Renin‐angiotensin‐aldosterone system genotype evaluation has led to the discovery of polymorphisms such as the insertion or deletion of a base pair in intron 16 of the ACE gene in humans and dogs. These insertion and deletion genotypes impact baseline ACE activity and might influence an individual's response to ACEI treatment. A more individualized and comprehensive approach to future RAAS modulation should include an RAAS profile, RAAS genotype, metabolic profile, and diet survey.
3. AngII AND ALDOSTERONE EXCESS
3.1. AngII and aldosterone as cardiovascular and renal toxins
There is a substantial body of evidence that AngII and aldosterone, likely acting in concert, are “cardio‐” and “nephrotoxic” (Table 1).48, 113, 114 This is based on direct (hormone infusion, transgenic animal models) and indirect (RAAS suppression) evidence (Figure 3 and Supporting Information Table S1). These studies link the fibrosis that attends the abnormal remodeling caused by excess AngII and aldosterone to myocardial, vascular, and renal (especially glomerular) dysfunction. The mechanisms by which increased AngII and aldosterone levels lead to fibrosis are multifactorial and likely involve stimulation of fibroblasts, generation of ROS, inflammation, and upregulation of transcription factors such as nuclear factor kappa B, cytokines such as transforming growth factor beta and tumor necrosis factor alpha, and upregulation of molecules such as plasminogen activator inhibitor‐1.12, 115, 116, 117, 118, 119, 120, 121, 122 Ultimately, these changes lead to increased collagen gene expression and synthesis, as well as decreased fibrinolysis. Excess AngII and aldosterone also directly lead to vascular endothelial dysfunction via the vasoconstrictive effects of AngII, increased endothelin expression, inhibition of nitric oxide synthase, cyclooxygenase‐2 activation, enhanced generation of ROS, and non‐genomic effects, such as activation of protein kinase C.11, 123, 124, 125, 126, 127
Figure 3.
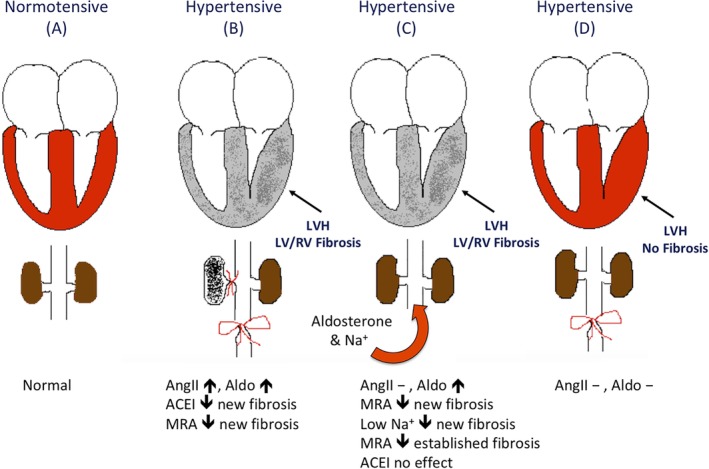
Schematic representation of experiments carried out by Brilla and Weber7, 268, 269, 270, 271 using rats with varying kidney function and experimental hypertension. A, Normotensive control rat heart, aorta, renal arteries, kidneys, and normal angiotensin II and aldosterone production; B, unilateral renal ischemia (unilateral renal artery banding) model with infrarenal aortic banding; C, aldosterone infusion with high‐sodium diet; and D, infrarenal banding. Increased circulating angiotensin II, aldosterone, or both occur in models B and C, which are characterized by interstitial and perivascular fibrosis of both the hypertrophied left and non‐hypertrophied right ventricles. In model B, the angiotensin converting enzyme inhibitor, captopril, prevented interstitial and perivascular fibrosis in rats with renal ischemia, yet did not prevent this remodeling in rats with aldosterone infusion and high sodium diets (model C). When models B and C were treated with spironolactone before and after the induction of hypertension, the rats developed left ventricular hypertrophy as expected, yet had significantly less interstitial and perivascular myocardial fibrosis, when compared to untreated controls. ACEI, angiotensin converting enzyme inhibitor; Aldo, aldosterone; AngII, angiotensin II; LV, left ventricle; LVH, left ventricular hypertrophy; MRA, mineralocorticoid receptor antagonist; RV, right ventricle
Renin‐angiotensin‐aldosterone system activation also contributes to the immune cell infiltration that contributes to the inflammation and fibrosis that attends renal ischemia, myocardial infarction, and systemic hypertension (HTN).128, 129, 130 More specifically, the MR on macrophages has been implicated in polarization of the macrophage population from the anti‐inflammatory M2 subtype to the inflammatory M1 subtype, perpetuating inflammation and tissue remodeling.130, 131, 132, 133 Antagonism or deletion of the macrophage MR is protective against cardiovascular remodeling, even in situations where aldosterone levels are normal.130, 131, 132 Oxidative stress also appears to amplify inflammation and directly activate the MR or allow glucocorticoids to activate the MR.134, 135 Finally, aldosterone excess decreases baroreceptor sensitivity in healthy humans and dogs with experimentally induced heart failure, the result being an undesirable increase in heart rate.8, 136, 137 The mechanisms by which neurohormonal activation decreases baroreceptor sensitivity are not fully understood.136, 138 Decreased baroreceptor‐heart rate sensitivity is a key feature of the heart failure syndrome139 and is an independent predictor of poor prognosis in people with heart failure.140, 141
The deleterious effects of excess aldosterone are augmented when the relationship between sodium and aldosterone is abnormal. In the aforementioned animal studies (Table 1, Supporting Information), sodium was variably unrestricted or given in excess. Under normal physiologic conditions, sodium supplementation leads to the inhibition of aldosterone release, allowing excess sodium to be excreted by the kidney. In the face of high or even normal total body sodium, a high aldosterone level is therefore inappropriate. Further discussion of the interplay among aldosterone, sodium status, metabolic state, inflammation, and oxidative stress can be found in the Supporting Information.
4. SUPPRESSION OF RAAS
Angiotensin converting enzyme inhibitors are widely used in the treatment of cardiovascular and renal diseases in multiple species. The ACEI used most commonly in veterinary medicine have been reviewed and are summarized in Supporting Information Tables S2 and S3.142 Most ACEI are administered as pro‐drugs, requiring esterification in the liver to become active. Drugs in this class inhibit ACE and not only decrease formation of AngII, but also decrease the degradation of the vasodilatory compound bradykinin. The pharmacokinetic properties of ACEI are complex and their disposition during repeated dosing cannot be characterized by a classical noncompartmental model.143 The plasma concentration‐time profile has initial and protracted elimination phases, because of clearance of free drug and release of drug from tissue‐binding sites, respectively.144 Using circulating plasma ACE activity as a surrogate for efficacy, dosages have been determined for several ACEI in animals (Supporting Information Tables S2 and S3). Although circulating ACE activity is significantly suppressed at these recommended dosages, clinical trials are required to determine efficacy and if there are outcome differences between ACEI. Variable efficacy among ACEIs in reducing cardiovascular and renal endpoints might arise from differences in the structure of the active moieties, their lipophilicity and bioavailability, affinity for tissue‐bound ACE, and elimination method. The clinical relevance of these differences, however, is not known. As ACE is anchored to the cell membrane such that its catalytic site faces the extracellular space,145, 146 differences in lipophilicity should be less important, except in the case of certain centrally acting ACEI.147 In humans, differences in the clinical efficacy of ACEI has been hypothesized to be associated with variation in tissue‐ACE affinity57 and this might also hold true for dogs and cats. Chymase has a greater catalytic efficiency than ACE and is thought to serve as the primary generator of AngII in the tissues however, and likely contributes to apparent inefficacy of ACEI.74, 148, 149, 150 Additional explanations for the sporadic failure of ACEI to prevent abnormal remodeling as a class include inadequate aldosterone suppression (aldosterone breakthrough), underdosing, and poor compliance. As previously discussed, novel approaches to target tissue RAAS management is likely to be part of future pharmacotherapeutic strategies.11, 12, 40 The most common adverse effects of ACEI in humans include cough or angioedema, or both, because of the accumulation of bradykinin, as well as increased prostacyclin and nitric oxide formation. These adverse effects are rarely, if at all, recognized in dogs and cats.
Angiotensin II receptor blockers prevent AngII from binding its AT1 receptor and were developed because of the cough and angioedema adverse effects seen with ACE‐inhibition in humans. Theoretical benefits of ARBs include blockade of the actions of AngII at the AT1R, regardless of the pathway of its formation (via ACE or non‐ACE pathways, such as chymase). Also, increased circulating AngII, resulting from AT1R blockade, might stimulate the AT2R, thought to be counter regulatory to the maladaptive actions mediated by the AT1R.151 Commonly used ARBs are summarized in Supporting Information Table S4. As with ACEI, some ARBs are administered as pro‐drugs that require metabolism to an active molecule. Whether ARBs are superior to ACEI in dogs and cats with either cardiovascular or renal disease is not known, though the early experience with telmisartan in cats with hypertension, chronic kidney disease (CKD), and proteinuria suggests that this may be the case (see below). A recent meta‐analysis of studies152, 153, 154, 155 of dual (ACEI and ARB) treatment in patients with heart failure concluded that until further clinical trials are performed, dual treatment should not be routinely administered.156 Controlled clinical trials in dogs and cats evaluating combination of ACEI and ARB treatment have not been performed.
Spironolactone and eplerenone are synthetic steroidal MRA. Mineralocorticoid receptor antagonism in the distal renal tubule cells causes an increase in urinary Na+ excretion and a decrease in K+ excretion. In the normal dog, spironolactone decreases K+ excretion, yet does not significantly increase urine Na+ excretion and urine volume, even at the high dosage of 8 mg/kg/d.157 In dogs with chronic heart failure and chronic aldosterone excess, however, spironolactone likely increases urinary sodium excretion and urine volume. In the dog, spironolactone is quickly absorbed through the gastrointestinal tract into the plasma and is then converted to several active metabolites.158, 159 The bioavailability in the dog is highest when given with food, reaching 80 to 90%.160 During an experimental model of hyperaldosteronism, the spironolactone dosage of 2 mg/kg PO once daily restored the urinary Na+/K+ ratio to near normal, and this is likely the optimal dosage in dogs.160 Because of spironolactone's ability to bind other steroid hormone receptors, adverse effects in people include decreased testosterone production, inhibition of testosterone binding its receptor, and increased estradiol levels, which can cause gynecomastia in men and menstrual abnormalities in women.161, 162, 163 For this reason, the more selective MRA, eplerenone, was developed. Spironolactone is relatively inexpensive and is the most frequently used MRA in veterinary medicine. Additional pharmacological therapies targeting the aldosterone/MR pathway are currently under active investigation. The third‐generation MRA (finerenone) holds promise, as it also modifies co‐regulators of MR signaling.164 Fourth‐generation MRA might provide even greater MR selectivity, targeting the MR in specific tissues and possibly modulating only specific transcriptional functions.
Direct renin inhibitors (DRIs) such as aliskiren theoretically prevent the initiation of the renin‐angiotensin cascade. First‐generation DRIs have not proven to be a panacea for cardiovascular and kidney diseases, however, and next‐generation DRIs have been developed and evaluation of their efficacy is underway.165, 166 There are currently no studies of DRIs in veterinary patients. First‐generation aldosterone synthase inhibitors unfortunately lack selectivity and decrease cortisol production.167 For this reason, more specific next‐generation aldosterone synthase inhibitors are being developed.168
Novel RAAS modulators include neprilysin inhibitors, recombinant human ACE2, and chymase inhibitors. Inhibition of the neutral endopeptidase, neprilysin, decreases the breakdown of endogenous vasoactive peptides, including natriuretic peptides, bradykinin and adrenomedullin. When the neprilysin inhibitor, sacubitril, was combined with an ACEI, the frequency of angioedema and cough in people was unacceptably high.169 A new combination product (Entresto), therefore, pairs sacubitril and the ARB valsartan. This combination was evaluated in a large human clinical trial, where it was superior to ACE inhibition alone in reducing the risk of death and hospitalization for patients with heart failure with reduced ejection fraction (HFrEF).170 This new drug is now a class I recommendation for treatment of HFrEF in humans.171 In dogs with RAAS activation secondary to sodium restriction, Entresto (sacubitril dosed at 15 mg/kg PO q24h) led to a sustained reduction in circulating aldosterone levels when compared to dogs treated with either placebo or valsartan.172 This combination has also been evaluated (sacubitril dosed at 20 mg/kg PO q12h) in a small study of dogs with the American College of Veterinary Internal Medicine (ACVIM) stage B2 myxomatous mitral valve disease (MMVD)173 (Figure 4) and was well tolerated during the 30‐day trial.174 Another novel therapeutic target is the ACE2 enzyme. This enzyme is considered counter regulatory to the vasoconstrictive and fluid retentive aspects of RAAS, as it catalyzes the conversion of AngII to the vasodilatory, anti‐inflammatory, anti‐fibrogenic peptide Ang(1,7) (Figure 2).175, 176, 177, 178 Human recombinant ACE2 has been evaluated in 1 small study in people with acute and chronic heart failure where it was found to normalize AngII levels and increases levels of the beneficial Ang(1,7) metabolite.110 Finally, based on the likely role of chymase in the local generation of angiotensin peptides, such as AngII, chymase inhibitors offer an additional approach to RAAS modulation in cardiovascular and renal diseases. Aside from sacubitril/valsartan, these novel RAAS modulators have not yet been evaluated in veterinary patients.
Figure 4.
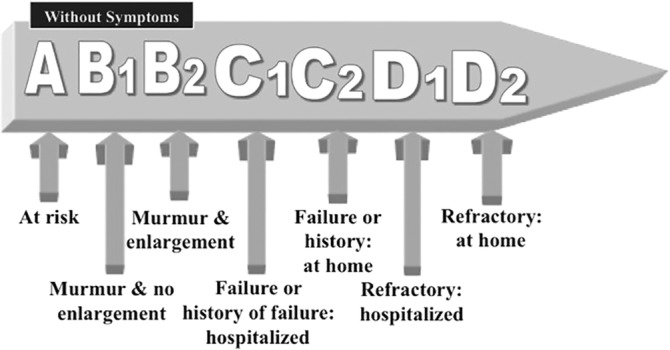
The American College of Veterinary Internal Medicine (ACVIM) classification of cardiac disease. From at risk of heart failure to refractory heart failure173
4.1. Cardiac disease
Studies in humans have shown the benefits of ACEI in the treatment of systolic heart failure, associated with moderate to severe179 and mild to moderate clinical signs.180, 181 Veterinary clinicians have had experience with enalapril, captopril, benazepril, lisinopril, imidapril, alacepril, and ramipril (Supporting Information Tables S2 and S3). Of these, only enalapril and benazepril, and to a lesser degree imidapril and ramipril, have been extensively studied. The benefits of ACE‐inhibition as part of the treatment of heart failure have been demonstrated in multiple placebo‐ or drug‐controlled clinical trials in dogs with heart failure (Figure 5).182, 183, 184, 185, 186 The first randomized, placebo‐controlled, clinical trial evaluating enalapril in dogs with congestive heart failure (CHF) caused by naturally occurring heart disease (either MMVD or dilated cardiomyopathy [DCM]) showed an improvement in short‐term clinical and hemodynamic variables (namely pulmonary capillary wedge pressure and pulmonary edema scores).187 A subsequent study found that enalapril was associated with an improvement in several clinical variables (including heart failure classification, pulmonary edema score, mobility, attitude, and cough frequency).185 Improvement in all of these variables was significant in the DCM group, whereas only improvement in cough and mobility parameters reached significance in the MMVD group.185 Two randomized placebo‐controlled studies in dogs with CHF caused by MMVD or DCM subsequently showed an improvement in survival time and delay in the time to worsening of heart failure.183, 184 This benefit was no longer statistically significant when only the DCM subgroup was analyzed, yet low numbers in the DCM groups in these studies make interpretation of this result difficult and larger studies of RAAS suppression in DCM are needed. Additional studies have found that imidapril is non‐inferior to enalapril and to benazepril.182, 186 In a US FDA pivotal trial (double‐blind and active‐control), the outcome of dogs in CHF caused by MMVD and DCM was compared between those treated with enalapril plus standard therapy versus those receiving standard treatment plus the inodilator, pimobendan (Vetmedin).188 There was no difference between treatment groups in the primary endpoint of treatment success or in the heart insufficiency and pulmonary edema scores. Total deaths caused by CHF at study end (day 56) were identical between groups at 14%. A subsequent European study comparing pimobendan and the ACEI, benazepril, showed modest survival benefit in those receiving pimobendan.189
Figure 5.
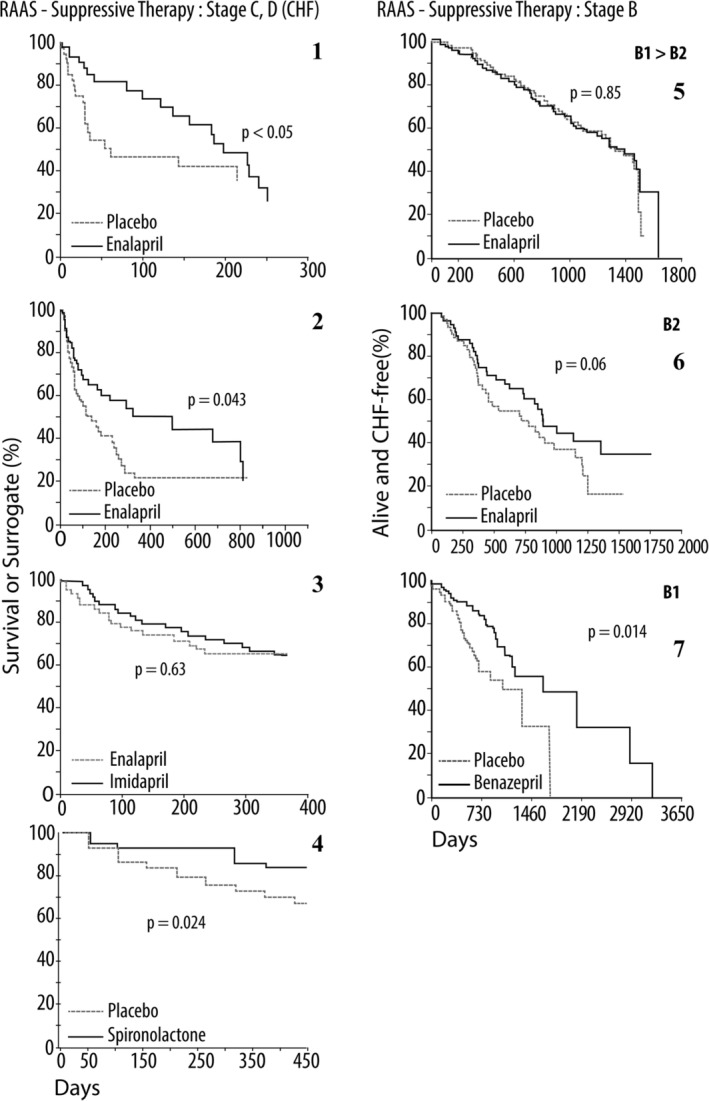
Seven renin‐angiotensin‐aldosterone system (RAAS) suppression clinical trials, 6 of which are of placebo‐controlled, double‐blind, multicenter design, are redrawn from the original publications.37, 182, 183, 184, 185, 186, 190, 191, 192, 210 All studies were funded by pharmaceutical companies.
The first 4 Kaplan‐Meier curves represent trials involving dogs in congestive heart failure (the American College of Veterinary Internal Medicine [ACVIM], Stage C2; congestive heart failure [CHF]), caused by myxomatous mitral valve disease (MMVD) or dilated cardiomyopathy (DCM). Only the 4th panel was composed exclusively of dogs with MMVD. All studies in panels 5 through 7 were made up entirely of dogs with asymptomatic MMVD (ACVIM Stage B). Panels 1 and 2 depict prospective double‐blind clinical trials, involving enalapril. Although the third graph contains data from a retrospective study of dogs treated with benazepril versus those left untreated in ACVIM Stage B1.
Symptomatic (panels 1 through 4)
Panel 1. The LIVE trial was 1 of the first placebo‐controlled double‐blind clinical trials in veterinary cardiology. Significant improvement in survival (death or removal from study) was documented in a population of dogs in CHF (ACVIM, Stage C2), caused by MMVD or DCM, when treated with enalapril plus standard treatment, as compared to placebo plus standard treatment. Placebo plus standard treatment, dashed line. Enalapril plus standard treatment, solid line.
Panel 2. The BENCH Trial compared benazepril to placebo in dogs with CHF caused by MMVD or DCM, and demonstrated significant benefit in survival (time to death or removal from the study for worsening clinical signs), and quality of life, in the dogs receiving benazepril. Placebo plus standard treatment, dashed line. Benazepril plus standard treatment, solid line.
Panel 3. The FIRST (placebo‐controlled, double‐blind) and EFFIC trials (open label) respectively compared imidapril (solid line) or ramipril (not shown) to an established angiotensin converting enzyme inhibitor (ACEI), enalapril (dashed line), in treating dogs with MMVD or DCM, NYHA Stage 2‐4. Data are shown only for the 12‐month placebo‐controlled FIRST trial. Both studies demonstrated comparable survival and quality of life scores between imidapril and the control ACEI.
Panel 4. The CEVA Spironolactone Trial compared spironolactone plus standard treatment (including benazepril) to placebo, in a prospective double‐blind trial involving dogs in ISACH International Small Animal Health Council (ISACHC) classification, II and III, caused by MMVD. In general, these dogs were in relatively mild heart failure. Similar to the RALES trial, significant survival benefit was realized by the dogs receiving spironolactone along with standard treatment. Placebo plus standard treatment, dashed line. Standard treatment plus spironolactone, solid line.
Asymptomatic (panels 5 through 7)
Panel 5. The SVEP trial involved an approximate 50:50 distribution of dogs with ACVIM Stage B1 and Stage B2, (asymptomatic), with treated dogs receiving enalapril, at a dosage of 0.37 mg/kg/d.190 Of these 229 dogs (all Cavalier King Charles Spaniels), approximately 45% reached the defined endpoint, onset of CHF. The 2% benefit seen in the number of days dogs remained in the study, free of CHF, between placebo‐ and enalapril‐treated dogs was not significant (log‐rank test, P = .85). Placebo, dashed line. Enalapril, solid line.
Panel 6. The VETPROOF compared enalapril to placebo, in a double‐blind placebo‐controlled trial of dogs in ACVIM B2. The graph depicting the combined endpoint of survival (all‐cause death and CHF‐free survival), expressed as a Kaplan‐Meier curve, for 124 dogs that met entry requirements is presented. Median times to this combined endpoint in the treatment and placebo groups were 851 and 534 days (59% difference of 317 days [10.6 months] in heart failure and survival benefit), respectively (P = .05). The primary endpoint (time to onset of CHF) was prolonged by enalapril versus placebo, but was not significant (P = .06). However, the numbers of dogs not in CHF were significantly different between groups on days 500 and 1000, as were the curves delineating all‐cause mortality. Placebo, dashed line. Enalapril, solid line.
Panel 7. Kaplan‐Meier survival curves of dogs treated with benazepril (solid line) or untreated (placebo, dashed line), after the initial diagnosis of ISACHC class Ia (ACVIM Stage B1) MMVD, with moderate‐to‐severe mitral regurgitation, demonstrated with echocardiography. Although retrospective, this study demonstrates a delay in the onset of CHF in the treatment group. This raises the possibility that ACEI may be of benefit even before cardiac remodeling is evident and supports the use of ACEI in MMVD, before the onset of congestive heart failure
Although the use of ACEI in canine CHF is well accepted, the data for ACEI are less robust for dogs with cardiac disease, before the onset of CHF (ACVIM Stages A and B; Figure 4). There have been 4 studies addressing this, 2 prospective, double‐blind, placebo‐controlled trials (enalapril versus placebo) and 2 retrospective studies (benazepril versus no treatment). The MMVD studies are summarized in Figure 5. The first of these looked at dogs with MMVD where approximately half were ACVIM Stage B1 and half were Stage B2. Enalapril was dosed at 0.37 mg/kg/d.190 Of these 229 dogs (all Cavalier King Charles Spaniels [CKCS]), 43 and 42% in the treatment and placebo groups, respectively, reached the endpoint, onset of CHF. The second study,191 also a placebo‐controlled double‐blind study in dogs with MMVD, included 23 breeds, with all 124 dogs shown by echocardiography and radiography to be in ACVIM Stage B2 cardiac disease. The treatment group received enalapril at an average dosage of 0.45 mg/kg/d. The result was a significant (P < .02) improvement in all‐cause mortality.191 A retrospective study of dogs with ACVIM Stage B1 MMVD, which had received either an ACEI (benazepril) or no treatment, concluded that dogs receiving benazepril had a significantly longer survival time and fewer cardiac events, including cardiac death.192 A retrospective study of 91 Doberman Pinchers with occult DCM, which had received either an ACEI (benazepril) or no treatment concluded that dogs receiving benazepril had a significantly longer median time to onset of overt DCM.193
In the aforementioned retrospective study,192 the CKCS did not benefit from RAAS suppression, when compared to other breeds. This raises the question of pharmacogenomic differences among dog breeds. Recently, a single nucleotide polymorphism in intron 16 of the ACE gene in a group of 31 dogs (including 10 CKCS) has been described.194 The ACE activity in dogs with the polymorphism was significantly lower than those without, yet both groups had significant suppression of ACE activity after 2 weeks of ACEI treatment. Six of the 10 CKCS carried the polymorphism, which provides support to the concept, held by some, that the RAAS of the CKCS may not truly represent the species as a whole. Further study is necessary to determine whether differences in RAAS phenotypes impact the response to RAAS suppressive treatment and the natural history of cardiac disease in this breed.
In dogs with experimentally created mitral regurgitation, captopril treatment led to a fall in total peripheral resistance index, decrease in regurgitant fraction, and increase in forward ejection fraction as compared to untreated controls.195 Conversely, the use of either an ACEI or ARB was found to exacerbate the left ventricular dilatation and extracellular matrix loss in other studies of experimentally created mitral regurgitation in dogs.82, 83 These acute to subacute models add valuable insight to the pathophysiology of left ventricular volume overload because of mitral regurgitation, yet further study is needed in patients with naturally occurring MMVD. Overall, the effect of ACE‐inhibition in preclinical MMVD might be variable among dogs. Although improvement might result from reduced afterload, decreased size of the regurgitant orifice, and an anti‐fibrotic effect in later stages, detrimental effects might arise from blockade of the inotropic effects of AngII and possible potentiation of extracellular matrix degradation and disruption of myocyte stability in the early stages of remodeling.75
The recently completed placebo‐controlled double‐blind EPIC trial enrolled dogs of various breeds with ACVIM Stage B2 MMVD with radiographic and echocardiographic evidence of cardiomegaly. This study showed that the inodilator pimobendan significantly prolonged the preclinical period in this cohort of dogs.196 Compared to the veterinary enalapril trial to prove reduction in onset of heart failure (VETPROOF) trial, the EPIC trial had more strictly defined cardiomegaly entry criteria and enrolled over 2.5 times as many dogs. The relatively narrow 95% confidence interval of the hazard ratio for the CHF component of the primary endpoint in the EPIC trial supports the benefit of pimobendan in the cohort studied. A similar hazard analysis is not available for the VETPROOF data.
Three large human studies197, 198, 199 have shown a clear benefit from the inclusion of broader RAAS suppressive therapy in the treatment of HFrEF with the addition of an MRA to standard treatment, including an ACEI or ARB. The ground‐breaking randomized aldactone evaluation study (RALES) (ischemic and non‐ischemic heart disease, NYHA III‐IV) and EPHESUS (post‐myocardial infarction) studies evaluated human patients with severe HFrEF. The EMPHASIS‐HF study (ischemic and non‐ischemic heart disease) evaluated patients with mild to moderate HFrEF. In these studies, an MRA (spironolactone or eplerenone) was added to optimal heart failure treatment, including RAAS suppression (either an ACEI or ARB) and beta blockade in the latter 2 studies. Each of these 3 studies showed a reduction in mortality and cardiac‐associated hospitalization. These significantly positive results have led to the recommendation in recent guidelines that MRA be used as a standard therapy in the treatment of HFrEF.200 Furthermore, the clinical benefit associated with addition of the MRA in these studies was seen in people with normal, low, and elevated blood aldosterone levels at study outset. The changes in patient neurohormonal profiles during chronic spironolactone treatment were monitored in a subset of patients in the RALES trial.201 The patients in the spironolactone group had significantly increased circulating AngII and aldosterone levels at 3 and 6 months when compared to people receiving placebo. These increases were hypothesized to result from failure of an aldosterone‐generated negative feedback loop to reduce renin secretion from the juxtaglomerular apparatus, though local tissue production of AngII and aldosterone could also be contributory. As there was a survival benefit associated with spironolactone, the impact of these hormone increases are likely mitigated by antagonism of the MR. It is currently not known if these increased concentrations of circulating AngII and aldosterone secondary to receptor antagonism have any receptor‐independent effects.
On average, plasma aldosterone concentrations in dogs with naturally occurring MMVD and DCM (both asymptomatic and with CHF) are higher than those of controls.35, 202 In dogs with heart failure caused by MMVD, 1 study found that, on average, dogs treated with enalapril had an initial decline in plasma aldosterone concentrations, when first reevaluated after 3 weeks of treatment. This was followed by an increase in average plasma aldosterone concentration, above baseline levels, 6 months after initiation of treatment.37 Another study, however, showed a significant decrease in plasma aldosterone concentrations in all but 1 of 7 dogs with MMVD and CHF, after initiation of captopril.35 Additionally, studies in dogs using pharmacologic RAAS activation and experimentally induced heart failure have found rising aldosterone levels despite ACEI treatment.38, 203, 204, 205, 206, 207 Regardless of its frequency, chronic neurohormonal activation has consequences and has been linked to abnormal remodeling in dogs with naturally occurring heart disease.208 Overall, these data add support for the adjunctive use of an MRA in dogs with heart disease. Although low‐dosage spironolactone (0.5 mg/kg PO q24h) did not significantly impact survival in dogs with advanced CHF caused by either MMVD or DCM, a subsequent double‐blind placebo‐controlled field study in 212 dogs with MMVD and mild to moderate heart failure demonstrated a significant reduction in risk of morbidity and mortality caused by heart disease with the addition of spironolactone (2 mg/kg PO q24h) to “standard therapy” (ACEI, furosemide ± digoxin).209, 210
The role of spironolactone in the treatment of heart failure, caused by ventricular diastolic dysfunction with preserved ejection fraction (HFpEF) is an area of ongoing research. Pathological activation of the RAAS and, in particular, hyperaldosteronism are implicated in the pathologic ventricular remodeling and diastolic myocardial dysfunction in hypertrophic cardiomyopathy (HCM) in humans.122 Small studies in humans with HFpEF have shown that MR blockade, with spironolactone, improves diastolic function.211, 212 The addition of spironolactone to existing heart failure treatment was recently evaluated in a large, placebo‐controlled, double‐blind study of people with symptomatic HFpEF (excluding infiltrative or hypertrophic cardiomyopathies). This study did not result in a significant survival benefit or reduction in hospitalization for heart failure. However, there were substantial differences in patient response among geographic areas, and post hoc subgroup analyses raised concerns about disparities in actual use of spironolactone and heart failure severity (less severe) in the Russian/Georgian patients.213, 214, 215 Overall, the incidence of hospitalization for heart failure was significantly lower in the spironolactone group as a whole, and the use of spironolactone was associated with significant improvement in the quality of life score.213, 216 In a separate study of patients with HFpEF, spironolactone was associated with improved exercise capacity.217 A second clinical trial of spironolactone in the treatment of HFpEF is ongoing (https://clinicaltrials.gov/ct2/show/NCT02901184?term=SPIRRIT&rank=1).
Plasma aldosterone concentration is elevated above normal in Maine Coon cats with asymptomatic HCM.218 Short‐term (4 months) use of spironolactone (2 mg/kg PO q12h) in these asymptomatic cats did not, however, provide reduction in left ventricular mass or improvement in an echocardiographic parameters of diastolic function, and 4 of the 13 cats treated with spironolactone developed ulcerative facial dermatitis.219 A double‐blinded placebo‐controlled clinical trial has more recently evaluated the use of spironolactone (1.7 to 3.3 mg/kg PO q24h) in client‐owned cats with heart failure caused by cardiomyopathy, the majority being afflicted with HCM.220 All cats (n = 20) were also receiving furosemide and an ACEI. Significantly fewer cats receiving spironolactone reached the primary endpoint (cardiovascular death), compared to those receiving placebo. Also, no cat receiving spironolactone experienced ulcerative facial dermatitis. Because of a small sample size and disparities in disease severity between the 2 groups at baseline, the authors concluded that these results are promising, yet additional studies evaluating spironolactone in cats with HCM and heart failure are warranted.
4.2. Systemic hypertension
Suppression of RAAS activity with ACEI, ARB, and MRA alone or in combination has been shown to improve blood pressure control in people with HTN.221, 222, 223 Whereas in people, HTN is usually primary (essential), in dogs and cats it is usually secondary to an underlying renal or endocrine disorder. In dogs, HTN secondary to CKD is often treated with RAAS suppression (ACEI or ARB).224 Hyperadrenocorticism is a common endocrine disease in the dog and is often complicated by HTN, especially in dogs with untreated adrenocortical tumors.225, 226 The mechanisms involved in HTN in dogs with hyperadrenocorticism are multifactorial and the general approach to treatment is an ACEI alone or in combination with amlodipine. Dogs with adrenocortical tumors have significantly higher plasma aldosterone concentrations, when compared to normal dogs and dogs with pituitary‐dependent hyperadrenocorticism,226 and may benefit from the addition of an MRA. The role of hyperaldosteronism in dogs with hypertension, secondary to pituitary dependent hyperadrenocorticism, is unclear,226, 227, 228 as HTN often persists despite medical treatment and control of the hyperadrenocorticism.225 In these cases, the addition of an MRA should be considered.
In cats, HTN is usually secondary to renal disease, endocrine disease, or both. In this species, HTN is a common cause of left ventricular hypertrophy, diastolic dysfunction, and other target organ damage. Plasma aldosterone concentrations and the aldosterone to renin ratio in azotemic hypertensive cats have been shown to be significantly elevated, as compared to normotensive cats.229 In the non‐azotemic hypertensive cats in this study, the average plasma aldosterone concentration was also elevated, but independent of plasma renin activity, which was actually low (suppressed). In this “low‐renin” group, the cats' aldosterone concentrations were not high enough to support primary hyperaldosteronism. Rather, their neurohormonal profile was similar to that of low‐renin hypertension, which is diagnosed disproportionately in human patients of African ancestry. In humans, low‐renin hypertension is most often due to either a low‐renin essential hypertension or primary aldosteronism. In people, the MRA, eplerenone, and the ARB, losartan, are equivalent in lowering blood pressure in the high‐renin patient, yet eplerenone is superior to losartan in the low‐renin patient.230 Despite this, the authors are unaware of studies using MRA treatment in cats with hypertension.
A recent study in normal laboratory cats showed that the ARBs, telmisartan and irbesartan, and the ACEI, benazepril, significantly attenuated an AngI‐induced blood pressure response, whereas losartan did not.231 The effect of telmisartan was significantly greater than that of the other 3 drugs, 90‐minutes after oral administration. Telmisartan and benazepril were also compared 24‐hours after their oral administration and telmisartan, again, led to a more significant attenuation of an AngI‐induced blood pressure rise. A recent clinical field study showed that telmisartan was tolerated and lowered mean arterial blood pressure when compared to placebo in cats with HTN that was either idiopathic or secondary to CKD or hyperthyroidism.232 This study excluded cats with pretreatment blood pressures >200 mm Hg and further study is still needed in this cohort. Finally, circulating RAAS activation in cats is not always associated with the development of hypertension, as demonstrated in studies of cats with hyperthyroidism and CKD.233, 234 Amlodipine, a commonly used antihypertensive agent in both dogs and cats, is known to cause aldosterone levels to rise in normal dogs,38 yet this does not appear to be the case in cats with naturally occurring HTN.229 In summary, the role of RAAS in feline hypertension appears to be complex and, as yet, not adequately defined.
Addition of an MRA is increasingly employed in humans with resistant hypertension, already receiving an ACEI, ARB, or both.221, 222, 223 A recently completed randomized placebo‐controlled clinical trial found that spironolactone was superior to the addition of either a beta blocker or an alpha‐1 blocker in patients with resistant HTN (persistent despite triple treatment with an ACE, ARB, and calcium channel blocker).235 From this study, it appears that spironolactone is the best additional drug for such resistant HTN. Mineralocorticoid receptor antagonist add‐on treatment in dogs and cats with HTN has not yet been evaluated.
4.3. Proteinuric kidney disease
Proteinuria, a marker of kidney injury, is likely a predictor of increased risk for disease progression, and might play a causal role in progression of glomerular disease.236, 237, 238 Clinical trials involving RAAS suppression in people with proteinuric kidney disease are summarized in Supporting Information Table S5. In people, the combination of ACEI and ARB is usually more effective at reducing proteinuria in various forms of kidney disease than either treatment alone. The reduction in proteinuria does not, however, always translate to improved renal outcomes (need for dialysis, increasing creatinine, and fall in estimated glomerular filtration rate [GFR]) and is sometimes associated with increased incidence of hyperkalemia and acute kidney injury when compared to monotherapy.239, 240 The addition of an MRA to an ACE or ARB (alone or together) has also been shown to augment proteinuria reduction in people with either HTN, diabetic nephropathy, or CKD with and without HTN.241, 242 The addition of an MRA to an ACEI or ARB, however, increases the risk of hyperkalemia in patients with CKD.113 The effect of finerenone on cardiovascular and renal outcomes in human patients with diabetic kidney disease is currently being evaluated in 2 large‐scale prospective randomized trials (https://clinicaltrials.gov/ct2/show/NCT02545049?term=figaro&cond=Diabetic+Kidney+Disease&rank=1 and https://clinicaltrials.gov/ct2/show/NCT02540993?term=finerenone&cond=Diabetic+Kidney+Disease&rank=1). Combination RAAS suppression and the addition of an MRA to standard pharmacotherapy has not been studied in dogs and cats with naturally occurring renal disease.
The benefit of RAAS suppressive treatment in canine proteinuric CKD, including idiopathic glomerulonephritis, has been demonstrated in experimental models and small clinical studies.243, 244, 245, 246 In uninephrectomized dogs, with experimentally induced diabetes mellitus and resultant proteinuria, treatment with lisinopril lowered glomerular capillary pressure, attenuated the development of glomerular hypertrophy, and reduced proteinuria significantly, when compared to untreated dogs.244 In a 5/6‐nephrectomy model of CKD, dogs treated with enalapril had, on average, a systolic BP that was 5‐15 mm Hg lower than their untreated counterparts. This difference was significant at 3 months, yet not at 6 months. Furthermore, treated dogs had lower urine protein to creatinine ratios (UP:C) and significantly lower renal tubular and glomerular lesion scores.243 Benazepril significantly reduced azotemia and systemic blood pressure in a short‐term study of experimental renal insufficiency in dogs induced by a 7/8 nephrectomy model of CKD.247 In a double‐blind randomized clinical trial of dogs with biopsy‐proven, membranous, and membranoproliferative glomerulonephritis, treatment with enalapril significantly lowered blood pressure, UP:C, and UP:C corrected for GFR, whereas these indices rose in the placebo group.245 In another study of benazepril versus placebo in dogs with CKD, benazepril treatment was associated with a significant increase in GFR over the 180‐day study, something not seen in the placebo group.246 In that study, the authors hypothesize that the increased GFR could have been because of reduced glomerular capillary hypertension, decreased release of extracellular matrix and collagen from mesangial and tubular cells, and decreased glomerular and interstitial fibrosis, although these factors were not directly evaluated.
The magnitude of proteinuria in dogs newly diagnosed with CKD appears to be related to prognosis,248 as a reduction in proteinuria may prolong survival. Dogs with an UP:C > 1.0 are known to have a relative risk of uremic crisis or death 3 times greater than dogs with an UP:C < 1.0. In a small study of X‐linked, hereditary nephritis, dogs treated with high‐dose enalapril (2 mg/kg PO q12h) had a significantly slower progression of proteinuria and survived significantly longer than those untreated.249 For these reasons, it is now well accepted that ACEI, administered chronically to both human and veterinary patients with naturally occurring proteinuric renal disease, are beneficial.245, 247, 250, 251, 252, 253, 254, 255 Mechanisms for this improvement are postulated to be the antihypertensive effect, reduction of AngII‐induced mesangial cell proliferation, and renal vasodilatory effects of ACEI; the latter resulting in a treatment‐induced fall in renal filtration pressure and proteinuria.250, 252, 253 Therefore, RAAS suppression with an ACEI or, less commonly, an ARB is considered standard of care for dogs with glomerular disease and reduction in proteinuria is considered a surrogate therapeutic endpoint.256 Neither dual RAAS blockade with ACEI and ARB nor the addition of an MRA to standard pharmacotherapy has been studied in dogs with naturally occurring renal disease. Controlled studies are needed to evaluate the effect of these combinations. There is no consensus regarding RAAS suppressive treatment in dogs with non‐proteinuric CKD.
The RAAS is activated in experimental and natural CKD in cats.234, 257 Benazepril has been evaluated in cats with CKD (proteinuric and non‐proteinuric), and it is well tolerated and significantly reduces proteinuria.258 Although this study did not document a benefit of benazepril treatment on renal survival (endpoint of death or euthanasia caused by renal disease or need for parenteral fluid therapy), renal survival times were inversely related to initial UP:C. Importantly, proteinuria has been shown to predict progression of azotemia259 and is negatively associated with survival in cats with chronic renal disease260 and with HTN.261 More recently, the ARB, telmisartan, has been compared to benazepril in cats with CKD and proteinuria.262 Telmisartan was non‐inferior to benazepril in preventing an increase in proteinuria over the 6‐month treatment period and led to a significant reduction in proteinuria at all time points, whereas the reduction seen in the benazepril group did not reach significance. As with dogs, combination RAAS blockade has not been evaluated in cats. Finally, benazepril has been shown to lower blood urea nitrogen, serum creatinine concentration, and blood pressure in cats with polycystic kidney disease.255 This is in stark contradistinction to the findings in human polycystic kidney disease, in which patients so afflicted did not benefit from ACE inhibition.253
4.4. Safety of RAAS suppressive treatment
We have learned through clinical experience with ACEIs that their negative impact on kidney function is minimal, even in the face of severe heart failure. When azotemia is observed, ACEIs are usually being administered in conjunction with diuretics, sodium restriction, and sometimes vasodilators, often with resultant hypotension. Typically, diuretic cessation or reduction in dosage results in the improvement or resolution of azotemia.263 In studies of enalapril in NYHA phase III and IV heart disease (moderate to severe heart failure), caused by MMVD and DCM, there was actually a lower incidence of azotemia in the enalapril‐treated than the placebo‐treated groups.183, 185, 187 Additionally, in a placebo‐controlled double‐blind evaluation of enalapril (average dosage of 0.45 mg/kg/d) in aged, small breed dogs with ACVIM Stage B2 (Figure 4) MMVD, there was no difference between groups in average serum creatinine concentration at any time point, number of dogs developing renal failure, or change in serum creatinine (evaluated every 3 months during the study duration; Supporting Information Figure S2).264
A safety analysis of dogs involved in the study of Bernay and colleagues showed that those receiving spironolactone, in addition to standard treatment (including an ACEI), were not at higher risk for adverse events (such as death from renal disease and abnormalities in serum sodium, potassium, urea nitrogen, and creatinine), when compared with dogs receiving placebo and standard treatment.265 In fact, mortality caused by cardiac disease, kidney disease, or both was lower in the group treated with spironolactone. Importantly, dogs receiving spironolactone in addition to conventional treatment for heart failure (loop diuretic, ACEI, and pimobendan) did not have a greater incidence of hyperkalemia. In a study of Doberman pinschers with occult DCM, significant increases in serum potassium concentration (versus individual baseline values) were seen in dogs receiving both spironolactone and an ACEI.266 Although the authors deemed these changes to be clinically insignificant, the serum potassium concentration exceeded the reference interval in 50% of dogs and regular monitoring of serum electrolytes and renal values (as often as every 3 months) was recommended for Doberman pinschers receiving spironolactone and ACEI. Overall, regular monitoring of serum electrolytes and renal values is prudent in all animals receiving vasodilators and diuretics, including spironolactone and ACEI. Finally, spironolactone in combination with an ACEI appears to be safe, when used to treat dogs with naturally occurring, asymptomatic MMVD, as well those with occult DCM, without preexisting azotemia.266, 267
5. CONCLUSIONS
Sustained RAAS activation adversely affects the heart, vessels, and kidneys. The more we learn about this system, the broader and more complex its web becomes, and redundancies that bypass our therapeutic blockade or inadvertent suppression of beneficial components of this system may reduce the efficacy of our current RAAS suppressive treatments. The future of medical treatment for cardiovascular and kidney diseases will have to be a more adept modulation of this system. This will require a better understanding of how this entire system changes during disease and its treatment.
CONFLICT OF INTEREST DECLARATION
Dr Pitt is a consultant for Bayer, Astra Zeneca, Sanofi, Sarfez, scPharmaceuticals, Relypsa/Vifor, Stealth Peptides, Cytopherx (stock options); Dr Atkins is a consultant for Ceva Sante Animale, Vetoquinol, and Boehringer Ingelheim; Dr Ames is a consultant for Ceva Sante Animale, and Elanco.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Authors declare no IACUC or other approval was needed.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Appendix S1: Supporting Information
Supporting Information Figure S1
Supporting Figure S2.
Supporting Information Table S1 Animal and ex vivo human studies of cardiovascular and kidney diseases providing direct (hormone infusion, transgenic animal models) and indirect (RAAS suppression) evidence of the cardio‐ and nephrotoxicity of chronic and/or excess angiotensin II and aldosterone production. The studies are presented in chronological order.
Supporting Information Table S2 Characteristics of specific ACE‐Inhibitors
Supporting Information Table S3 Pharmacokinetic parameters of ACE‐Inhibitors in dogs and cats
Supporting Information Table S4 Characteristics of specific Angiotensin II Type‐1 Receptor Blockers and an ARB/neprilysin‐inhibitor
Supporting Information Table S5 Clinical trials involving RAAS suppression in people with proteinuric kidney disease. Studies are presented in chronological order
Ames MK, Atkins CE, Pitt B. The renin‐angiotensin‐aldosterone system and its suppression. J Vet Intern Med. 2019;33:363–382. 10.1111/jvim.15454
[Correction added on 04 March 2019, after first online publication: in the Abstract, “RAAS's” was corrected to “RAAS”]
REFERENCES
- 1. Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation. 1990;82(5):1730‐1736. [DOI] [PubMed] [Google Scholar]
- 2. Anker SD, Chua TP, Ponikowski P, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. 1997;96(2):526‐534. [DOI] [PubMed] [Google Scholar]
- 3. Broqvist M, Dahlstrom U, Karlberg BE, et al. Neuroendocrine response in acute heart failure and the influence of treatment. Eur Heart J. 1989;10(12):1075‐1083. [DOI] [PubMed] [Google Scholar]
- 4. Levine TB, Francis GS, Goldsmith SR, Simon AB, Cohn JN. Activity of the sympathetic nervous system and renin‐angiotensin system assessed by plasma hormone levels and their relation to hemodynamic abnormalities in congestive heart failure. Am J Cardiol. 1982;49:1659‐1666. [DOI] [PubMed] [Google Scholar]
- 5. Francis GS, Benedict C, Johnstone DE, et al. Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. Circulation. 1990;82:1724‐1729. [DOI] [PubMed] [Google Scholar]
- 6. Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345(23):1689‐1697. [DOI] [PubMed] [Google Scholar]
- 7. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin‐ angiotensin‐aldosterone system. Circulation. 1991;83(6):1849‐1865. [DOI] [PubMed] [Google Scholar]
- 8. Wang W, McClain JM, Zucker IH. Aldosterone reduces baroreceptor discharge in the dog. Hypertens. 1992;19:270‐277. [DOI] [PubMed] [Google Scholar]
- 9. Brilla CG, Rupp H, Funck R, Maisch B. The renin‐angiotensin‐aldosterone system and myocardial collagen matrix remodelling in congestive heart failure. Eur Heart J. 1995;16(Suppl O):107‐109. [DOI] [PubMed] [Google Scholar]
- 10. Martinez FA. Aldosterone inhibition and cardiovascular protection: more important than it once appeared. Cardiovasc Drugs Ther. 2010;24(4):345‐350. [DOI] [PubMed] [Google Scholar]
- 11. Schiffrin EL. Effects of aldosterone on the vasculature. Hypertension. 2006;47(3):312‐318. [DOI] [PubMed] [Google Scholar]
- 12. Waanders F, de Vries LV, van Goor H, et al. Aldosterone, from (patho)physiology to treatment in cardiovascular and renal damage. Curr Vasc Pharmacol. 2011;9(5):594‐605. [DOI] [PubMed] [Google Scholar]
- 13. Gilbert KC, Brown NJ. Aldosterone and inflammation. Curr Op Endocrinol Diabetes Obes. 2010;17(3):199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Young M, Fullerton M, Dilley R, Funder JW. Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest. 1994;93:2578‐2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Staessen J, Lijnen P, Fagard R, et al. Rise in plasma concentration of aldosterone during long‐term angiotensin II suppression. J Endocrinol. 1981;91(3):457‐465. [DOI] [PubMed] [Google Scholar]
- 16. Girerd N, Pang PS, Swedberg K, et al. Serum aldosterone is associated with mortality and re‐hospitalization in patients with reduced ejection fraction hospitalized for acute heart failure: analysis from the EVEREST trial. Eur J Heart Fail. 2014;15(11):1228‐1235. [DOI] [PubMed] [Google Scholar]
- 17. Lijnen P, Fagard R, Staessen J, Amery A. Effect of chronic diuretic treatment on the plasma renin‐angiotensin‐aldosterone system in essential hypertension. Br J Clin Pharmacol. 1981;12(3):387‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3(9):486‐492. [DOI] [PubMed] [Google Scholar]
- 19. Sato A, Fukuda S. Effect of aldosterone breakthrough on albuminuria during treatment with a direct renin inhibitor and combined effect with a mineralocorticoid receptor antagonist. Hypertens Res. 2013;36(10):879‐884. [DOI] [PubMed] [Google Scholar]
- 20. Sato A, Saruta T. Aldosterone escape during angiotensin‐converting enzyme inhibitor therapy in essential hypertensive patients with left ventricular hypertrophy. J Intern Med. 2001;29:13‐21. [DOI] [PubMed] [Google Scholar]
- 21. Sato A, Hayashi K, Naruse M, Saruta T. Effectiveness of aldosterone blockade in patients with diabetic nephropathy. Hypertension. 2003;41(1):64‐68. [DOI] [PubMed] [Google Scholar]
- 22. Schjoedt KJ, Andersen S, Rossing P, Tarnow L, Parving HH. Aldosterone escape during blockade of the renin‐angiotensin‐aldosterone system in diabetic nephropathy is associated with enhanced decline in glomerular filtration rate. Diabetologia. 2004;47(11):1936‐1939. [DOI] [PubMed] [Google Scholar]
- 23. Tang WHW, Vagelos RH, Yee Y‐G, et al. Neurohormonal and clinicalresponses to high‐ versus low‐dose enalapril therapy in chronic heart failure. J Am Coll Cardiol. 2002;39:70‐78. [DOI] [PubMed] [Google Scholar]
- 24. Cicoira M, Zanolla L, Rossi A, et al. Failure of aldosterone suppression despite angiotensin‐converting enzyme (ACE) inhibitor administration in chronic heart failure is associated with ACE DD genotype. J Am Coll Cardiol. 2001;37:1808‐1812. [DOI] [PubMed] [Google Scholar]
- 25. Bomback AS, Rekhtman Y, Klemmer PJ, Canetta PA, Radhakrishnan J, Appel GB. Aldosterone breakthrough during aliskiren, valsartan, and combination (aliskiren + valsartan) therapy. J Am Soc Hypertens. 2012;6(5):338‐345. [DOI] [PubMed] [Google Scholar]
- 26. Yoneda T, Takeda Y, Usukura M, et al. Aldosterone breakthrough during angiotensin II receptor blockade in hypertensive patients with diabetes mellitus. Am J Hypertens. 2007;20(12):1329‐1333. [DOI] [PubMed] [Google Scholar]
- 27. MacFadyen RJ, Lee AFC, Morton JJ, et al. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart. 1999;82(1):57‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee A, MacFadyen RJ. Neurohormonal reactivation in heart failure patients on chronic ACE inhibitor therapy: a longitudinal study. Eur J Heart Fail. 1999;1(4):401‐406. [DOI] [PubMed] [Google Scholar]
- 29. Horita Y, Taura K, Taguchi T, et al. Aldosterone breakthrough during therapy with angiotensin‐converting enzyme inhibitors and angiotensin II receptor blockers in proteinuric patients with immunoglobulin A nephropathy. Nephrol Ther. 2006;11(5):462‐466. [DOI] [PubMed] [Google Scholar]
- 30. Pitt B. “Escape” of aldosterone production in patients with left ventricular dysfunction treated with an angiotensin converting enzyme inhibitor: implications for therapy. Cardiovasc Drugs Ther. 1995;9(1):145‐149. [DOI] [PubMed] [Google Scholar]
- 31. van de Wal RMA, Plokker HWM, Lok DJA, et al. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int J Cardiol. 2006;106(3):367‐372. [DOI] [PubMed] [Google Scholar]
- 32. Roig E, Perez‐Villa F, Morales M, et al. Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur Heart J. 2000;21(1):53‐57. [DOI] [PubMed] [Google Scholar]
- 33. Aronson D, Burger AJ. Neurohormonal prediction of mortality following admission for decompensated heart failure. Am J Cardiol. 2003;91(2):245‐248. [DOI] [PubMed] [Google Scholar]
- 34. Bayliss J, Norell M, Canepa‐Anson R, Sutton G, Poole‐Wilson P. Untreated heart failure: clinical and neuroendocrine effects of introducing diuretics. Br Heart J. 1987;57(1):17‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Knowlen GG, Kittleson MD, Nachreiner RF, Eyster GE. Comparison of plasma aldosterone concentration among clinical status groups. J Am Vet Med Assoc. 1983;183(9):991‐996. [PubMed] [Google Scholar]
- 36. Lovern CS, Swecker WS, Lee JC, Moon ML. Additive effects of a sodium chloride restricted diet and furosemide administration in healthy dogs. Am J Vet Res. 2001;62(11):1793‐1796. [DOI] [PubMed] [Google Scholar]
- 37. Haggstrom J, Hansson K, Karlberg BE, et al. Effects of long‐term treatment with enalapril or hydralazine on the renin‐angiotensin‐aldosterone system and fluid balance in dogs with naturally acquired mitral valve regurgitation. Am J Vet Res. 1996;57(11):1645‐1652. [PubMed] [Google Scholar]
- 38. Atkins CE, Rausch WP, Gardner SY, et al. The effect of amlodipine and the combination of amlodipine and enalapril on the renin‐angiotensin‐aldosterone system in the dog. J Vet Pharmacol Ther. 2007;30(5):394‐400. [DOI] [PubMed] [Google Scholar]
- 39. Lantis AC, Atkins CE, DeFrancesco TC, et al. Effects of furosemide and the combination of furosemide and the labeled dosage of pimobendan on the circulating renin‐angiotensin‐aldosterone system in clinically normal dogs. Am J Vet Res. 2011;72(12):1646‐1651. [DOI] [PubMed] [Google Scholar]
- 40. De Mello WC, Frohlich ED. Clinical perspectives and fundamental aspects of local cardiovascular and renal Renin‐Angiotensin systems. Front Endocrinol (Lausanne). 2014;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Mello WC. Chemical communication between heart cells is disrupted by intracellular renin and angiotensin ii: implications for heart development and disease. Front Endocrinol (Lausanne). 2015;6:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bader M. ACE2, angiotensin‐(1–7), and Mas: the other side of the coin. Pflugers Arch. 2013;465(1):79‐85. [DOI] [PubMed] [Google Scholar]
- 43. Seelinger E, Wronski T, Ladwig M, et al. The “body fluid pressure control system” relies on the renin‐angiotensin‐aldosterone system: balance studies in freely moving dogs. Clin Exp Pharmacol Physiol. 2005;32(5–6):394‐399. [DOI] [PubMed] [Google Scholar]
- 44. Hall JE, Guyton AC. Role of the kidneys in long‐term control of arterial pressure and in hypertension: the integrated system for arterial pressure regulation Guyton and Hall Textbook of Medical Physiology. Philadelphia, PA: Elsevier Health Sciences; 2010:213‐228. [Google Scholar]
- 45. Beuschlein F. Regulation of aldosterone secretion: from physiology to disease. J Endocrinol. 2013;168(6):R85‐R93. [DOI] [PubMed] [Google Scholar]
- 46. Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242(4878):583‐585. [DOI] [PubMed] [Google Scholar]
- 47. Funder JW, Pearce PT, Smith R, Campbell J. Vascular type I aldosterone binding sites are physiological mineralocorticoid receptors. Endocrinology. 1989;125(4):2224‐2226. [DOI] [PubMed] [Google Scholar]
- 48. Jaisser F, Farman N. Emerging roles of the mineralocorticoid receptor in pathology: toward new paradigms in clinical pharmacology. Pharmacol Rev. 2015;68(1):49‐75. [DOI] [PubMed] [Google Scholar]
- 49. Marzolla V, Armani A, Zennaro M‐C, et al. The role of the mineralocorticoid receptor in adipocyte biology and fat metabolism. Mol Cell Endocrinol. 2012;350(2):281‐288. [DOI] [PubMed] [Google Scholar]
- 50. Marzolla V, Armani A, Feraco A, et al. Mineralocorticoid receptor in adipocytes and macrophages: a promising target to fight metabolic syndrome. Steroids. 2014;91:46‐53. [DOI] [PubMed] [Google Scholar]
- 51. Gomez‐Sanchez E, Gomez‐Sanchez CE. The multifaceted mineralocorticoid receptor. Compr Physiol. 2014;4(3):965‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol. 2013;304(6):C532‐C540. [DOI] [PubMed] [Google Scholar]
- 53. Fuller PJ, Yang J, Young MJ. 30 years of the mineralocorticoid receptor: coregulators as mediators of mineralocorticoid receptor signalling diversity. J Endocrinol. 2017;234(1):T23‐T34. [DOI] [PubMed] [Google Scholar]
- 54. Bader M. Tissue renin‐angiotensin‐aldosterone systems: targets for pharmacological therapy. Annu Rev Pharmacol Toxicol. 2010;50(1):439‐465. [DOI] [PubMed] [Google Scholar]
- 55. Bader M, Ganten D. Update on tissue renin–angiotensin systems. J Mol Med. 2008;86(6):615‐621. [DOI] [PubMed] [Google Scholar]
- 56. Takeda Y, Miyamori I, Yoneda T, et al. Production of aldosterone in isolated rat blood vessels. Hypertension. 1995;25(2):170‐173. [DOI] [PubMed] [Google Scholar]
- 57. Dzau VJ, Bernstein K, Celermajer D, et al. The relevance of tissue angiotensin‐converting enzyme: manifestations in mechanistic and endpoint data. Am J Cardiol. 2001;88(9A):1L‐20L. [DOI] [PubMed] [Google Scholar]
- 58. Harada K, Izawa H, Nishizawa T, et al. Beneficial effects of torasemide on systolic wall stress and sympathetic nervous activity in asymptomatic or mildly symptomatic patients with heart failure: comparison with azosemide. J Cardiovasc Pharmacol. 2009;53(6):468‐473. [DOI] [PubMed] [Google Scholar]
- 59. Ullian ME, Schelling JR, Linas SL. Aldosterone enhances angiotensin II receptor binding and inositol phosphate responses. Hypertension. 1992;20(1):67‐73. [DOI] [PubMed] [Google Scholar]
- 60. Ullian ME, Hutchison FN, Hazen‐Martin DJ, Morinelli TA. Angiotensin II‐aldosterone interactions on protein synthesis in vascular smooth muscle cells. Am J Physiol. 1993;264:C1525‐C1531. [DOI] [PubMed] [Google Scholar]
- 61. Ullian ME, Fine JJ. Mechanisms of enhanced angiotensin. J Cell Physiol. 1994;161(2):201‐208. [DOI] [PubMed] [Google Scholar]
- 62. Harada E, Yoshimura M, Yasue H, et al. Aldosterone induces angiotensin‐converting‐enzyme gene expression in cultured neonatal rat cardiocytes. Circulation. 2001;104(2):137‐139. [DOI] [PubMed] [Google Scholar]
- 63. Rossier MF, Lenglet S, Vetterli L, et al. Corticosteroids and redox potential modulate spontaneous contractions in isolated rat ventricular cardiomyocytes. Hypertension. 2008;52(4):721‐728. [DOI] [PubMed] [Google Scholar]
- 64. Welch WJ. Angiotensin II‐dependent superoxide: effects on hypertension and vascular dysfunction. Hypertension. 2008;52(1):51‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Silvestre JS, Robert V, Heymes C. Myocardial production of aldosterone and corticosterone in the rat physiological regulation. J Biol Chem. 1998;273:4883‐4891. [DOI] [PubMed] [Google Scholar]
- 66. Hennrikus M, Gonzalez AA, Prieto MC. The prorenin receptor in the cardiovascular system and beyond. Am J Phyisol Heart Circ Physiol. 2018;314(2):H139‐H145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109(11):1417‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nagata S, Hatakeyama K, Asami M, et al. Big angiotensin‐25: a novel glycosylated angiotensin‐related peptide isolated from human urine. Biochem Biophys Res Comm. 2013;441(4):757‐762. [DOI] [PubMed] [Google Scholar]
- 69. Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin‐12, a possible component of the renin–angiotensin system. Biochem Biophys Res Comm. 2006;350(4):1026‐1031. [DOI] [PubMed] [Google Scholar]
- 70. Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II‐forming enzyme in the human heart. J Biol Chem. 1990;265(36):22348‐22357. [PubMed] [Google Scholar]
- 71. Urata H, Healy B, Stewart RW, Bumpus FM, Husain A. Angiotensin II‐forming pathways in normal and failing human hearts. Circ Res. 1990;66(4):883‐890. [DOI] [PubMed] [Google Scholar]
- 72. Urata H, Boehm KD, Philip A, et al. Cellular localization and regional distribution of an angiotensin II‐forming chymase in the heart. J Clin Invest. 1993;91(4):1269‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ferrario CM, Ahmad S, Varagic J, et al. Intracrine angiotensin II functions originate from noncanonical pathways in the human heart. 2016;311(2):H404‐H414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase‐dependent generation of angiotensin II from angiotensin‐(1‐12) in human atrial tissue. PLoS One. 2011;6(12):e28501‐e28509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dell'Italia LJ, Collawn JF, Ferrario CM. Multifunctional role of chymase in acute and chronic tissue injury and remodeling. Circ Res. 2018;122(2):319‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin D, Takai S, Sakaguchi M, Okamoto Y, Muramatsu M, Miyazaki M. An antiarrhythmic effect of a chymase inhibitor after myocardial infarction. J Pharmacol Exp Ther. 2004;309(2):490‐497. [DOI] [PubMed] [Google Scholar]
- 77. Matsumoto T. Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation. 2003;107(20):2555‐2558. [DOI] [PubMed] [Google Scholar]
- 78. Zheng J, Wei C‐C, Hase N, et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS One. 2014;9(4):e94732‐e94712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shi G‐P, Bot I, Kovanen PT. Mast cells in human and experimental cardiometabolic diseases. Nat Rev Cardiol. 2015;12(11):643‐658. [DOI] [PubMed] [Google Scholar]
- 80. Stewart JA, Wei C‐C, Brower GL, et al. Cardiac mast cell‐ and chymase‐mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35(3):311‐319. [DOI] [PubMed] [Google Scholar]
- 81. Dillon AR, Dell'Italia LJ, Tillson M, et al. Left ventricular remodeling in preclinical experimental mitral regurgitation of dogs. J Vet Cardiol. 2012;14(1):73‐92. [DOI] [PubMed] [Google Scholar]
- 82. Perry GJ, Wei C‐C, Hankes GH, et al. Angiotensin II receptor blockade does not improve left ventricular function and remodeling in subacute mitral regurgitation in the dog. J Am Coll Cardiol. 2002;39(8):1374‐1379. [DOI] [PubMed] [Google Scholar]
- 83. Dell'Italia LJ, Balcells E, Meng QC, et al. Volume‐overload cardiac hypertrophy is unaffected by ACE inhibitor treatment in dogs. Am J Physiol. 1997;273(Pt 2):H961‐H970. [DOI] [PubMed] [Google Scholar]
- 84. Yamane T, Fujii Y, Orito K, Osamura K, Kanai T, Wakao Y. Comparison of the effects of candesartan cilexetil and enalapril maleate on right ventricular myocardial remodeling in dogs with experimentally induced pulmonary stenosis. Am J Vet Res. 2008;69(12):1574‐1579. [DOI] [PubMed] [Google Scholar]
- 85. Chappell MC. Biochemical evaluation of the renin‐angiotensin system: the good, bad, and absolute? Am J Physiol Heart Circ Physiol. 2016;310(2):H137‐H152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chappell MC. Emerging evidence for a functional angiotensin‐converting enzyme 2‐angiotensin‐(1‐7)‐MAS receptor axis: more than regulation of blood pressure? Hypertension. 2007;50(4):596‐599. [DOI] [PubMed] [Google Scholar]
- 87. Metzger R, Bader M, Ludwig T, Berberich C, Bunnemann B, Ganten D. Expression of the mouse and rat mas proto‐oncogene in the brain and peripheral tissues. FEBS Lett. 1995;357(1):27‐32. [DOI] [PubMed] [Google Scholar]
- 88. Flores‐Muñoz M, Smith NJ, Haggerty C, Milligan G, Nicklin SA. Angiotensin1‐9 antagonises pro‐hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J Physiol. 2011;589(4):939‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Park BM, Gao S, Cha SA, Park BH, Kim SH. Cardioprotective effects of angiotensin III against ischemic injury via the AT2 receptor and KATP channels. Physiol Rep. 2013;1(6):e00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Park BM, Oh Y‐B, Gao S, Cha SA, Kang KP, Kim SH. Angiotensin III stimulates high stretch‐induced ANP secretion via angiotensin type 2 receptor. Peptides. 2013;42:131‐137. [DOI] [PubMed] [Google Scholar]
- 91. Park BM, Cha SA, Han BR, Kim SH. Angiotensin IV stimulates high atrial stretch‐induced ANP secretion via insulin regulated aminopeptidase. Peptides. 2015;63:30‐37. [DOI] [PubMed] [Google Scholar]
- 92. Gordon CR, Lavie P. Day‐night variations in urine excretions and hormones in dogs: role of autonomic innervation. Physiol Behav. 1985;35(2):175‐181. [DOI] [PubMed] [Google Scholar]
- 93. Kawasaki T, Cugini P, Uezono K, et al. Circadian variations of total renin and active renin. Horm Metab Res. 2008;22(12):636‐639. [DOI] [PubMed] [Google Scholar]
- 94. Cugini P, Scavo D, Halberg F, et al. Ageing and circadian rhythm of plasma renin and aldosterone. Maturitas. 1981;3(2):173‐182. [DOI] [PubMed] [Google Scholar]
- 95. Sassone‐Corsi P, Christen Y. The epigenetic and metabolic language of the circadian clock A Time for Metabolism and Hormones. Heidelberg, Germany: Springer Cham; 2016:1‐11. [PubMed] [Google Scholar]
- 96. Reinhardt HW, Seeliger E, Lohmann K, et al. Changes of blood pressure, sodium excretion and sodium balance due to variations of the renin‐angiotensin‐aldosterone system. J Auton Nerv Syst. 1996;57(3):184‐187. [DOI] [PubMed] [Google Scholar]
- 97. Mochel JP, Fink M, Peyrou M, et al. Chronobiology of the renin‐angiotensin‐aldosterone system in dogs: relation to blood pressure and renal physiology. Chronobiol Int. 2013;30(9):1144‐1159. [DOI] [PubMed] [Google Scholar]
- 98. Fernandez JR, Hermida RC, Mojon A. Chronobiological analysis techniques. Application to blood pressure. Philos Trans A Math Phys Eng Sci. 2009;367(1887):431‐445. [DOI] [PubMed] [Google Scholar]
- 99. Mochel JP, Fink M, Bon C, et al. Influence of feeding schedules on the chronobiology of renin activity, urinary electrolytes and blood pressure in dogs. Chronobiol Int. 2014;31(5):715‐730. [DOI] [PubMed] [Google Scholar]
- 100. Martino TA, Tata N, Simpson JA, et al. The primary benefits of angiotensin‐converting enzyme inhibition on cardiac remodeling occur during sleep time in murine pressure overload hypertrophy. J Am Coll Cardiol. 2011;57(20):2020‐2028. [DOI] [PubMed] [Google Scholar]
- 101. Hermida RC, Ayala DE, Mojón A, Fernández JR. Bedtime ingestion of hypertension medications reduces the risk of new‐onset type 2 diabetes: a randomised controlled trial. Diabetologia. 2015;59(2):255‐265. [DOI] [PubMed] [Google Scholar]
- 102. Ivy JR, Bailey MA. Pressure natriuresis and the renal control of arterial blood pressure. J Physiol. 2014;592(18):3955‐3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Guyton AC. Long‐term arterial pressure control: an analysis from animal experiments and computer and graphic models. Am J Physiol. 1990;259(5 Pt 2):R865‐R877. [DOI] [PubMed] [Google Scholar]
- 104. Guyton AC, Coleman TG, Cowley AW, et al. Systems analysis of arterial pressure regulation and hypertension. Ann Biomed Eng. 1972;1(2):254‐281. [DOI] [PubMed] [Google Scholar]
- 105. Hall JE, Granger JP, Hester RL, et al. Mechanisms of escape from sodium retention during angiotensin II hypertension. Am J Physiol. 1984;246(Pt 2):F627‐F634. [DOI] [PubMed] [Google Scholar]
- 106. Mochel JP, Danhof M. Chronobiology and pharmacologic modulation of the renin–angiotensin–aldosterone system in dogs: what have we learned? Reviews of Physiology, Biochemistry and Pharmacology. Vol 169 Cham, Switzerland: Springer; 2015:43‐69. [DOI] [PubMed] [Google Scholar]
- 107. Bie P, Sandgaard NC. Determinants of the natriuresis after acute, slow sodium loading in conscious dogs. Am J Physiol Reg Comp Physiol. 2000;278(1):R1‐R10. [DOI] [PubMed] [Google Scholar]
- 108. Xue C, Siragy HM. Local renal aldosterone system and its regulation by salt, diabetes, and angiotensin II type 1 receptor. Hypertension. 2005;46(3):584‐590. [DOI] [PubMed] [Google Scholar]
- 109. Pavo N, Wurm R, Goliasch G, et al. Renin‐angiotensin system fingerprints of heart failure with reduced ejection fraction. J Am Coll Cardiol. 2016;68(25):2912‐2914. [DOI] [PubMed] [Google Scholar]
- 110. Basu R, Poglitsch M, Yogasundaram H, Thomas J, Rowe BH, Oudit GY. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J Am Coll Cardiol. 2017;69(7):805‐819. [DOI] [PubMed] [Google Scholar]
- 111. Mochel JP, Peyrou M, Fink M, et al. Capturing the dynamics of systemic renin‐angiotensin‐aldosterone system (RAAS) peptides heightens the understanding of the effect of benazepril in dogs. 2012;36(2):174‐180. [DOI] [PubMed] [Google Scholar]
- 112. Ames MK, Atkins CE, Eriksson A, Hess AM. Aldosterone breakthrough in dogs with naturally occurring myxomatous mitral valve disease. J Vet Cardiol. 2017;19(3):218‐227. [DOI] [PubMed] [Google Scholar]
- 113. Bolignano D, Palmer SC, Navaneethan SD, Strippoli GFM. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst Rev. 2014;4:1‐93. [DOI] [PubMed] [Google Scholar]
- 114. van den Berg TNA, Rongen GA, Fröhlich GM, Deinum J, Hausenloy DJ, Riksen NP. The cardioprotective effects of mineralocorticoid receptor antagonists. Pharmacol Ther. 2014;142(1):72‐87. [DOI] [PubMed] [Google Scholar]
- 115. Villarreal FJ, Kim NN, Ungab GD, Printz MP, Dillmann WH. Identification of functional angiotensin II receptors on rat cardiac fibroblasts. Circulation. 1993;88(6):2849‐2861. [DOI] [PubMed] [Google Scholar]
- 116. Lassègue B, Sorescu D, Szöcs K, et al. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II‐induced superoxide formation and redox‐sensitive signaling pathways. Circ Res. 2001;88(9):888‐894. [DOI] [PubMed] [Google Scholar]
- 117. Ni W, Zhan Y, He H, Maynard E, Balschi JA, Oettgen P. Ets‐1 is a critical transcriptional regulator of reactive oxygen species and p47phox gene expression in response to angiotensin II. Circ Res. 2007;101(10):985‐994. [DOI] [PubMed] [Google Scholar]
- 118. Brilla CG, Zhou G, Rupp H, Maisch B, Weber KT. Role of angiotensin II and prostaglandin E2 in regulating cardiac fibroblast collagen turnover. Am J Cardiol. 1995;76(13):8D‐13D. [DOI] [PubMed] [Google Scholar]
- 119. Villarreal FJ, Dillmann WH. Cardiac hypertrophy‐induced changes in mRNA levels for TGF‐beta 1, fibronectin, and collagen. Am J Physiol. 1992;262(Pt 2):H1861‐H1866. [DOI] [PubMed] [Google Scholar]
- 120. Azibani F, Benard L, Schlossarek S, Merval R. Aldosterone inhibits antifibrotic factors in mouse hypertensive heart. Hypertension. 2012;59:1179‐1187. [DOI] [PubMed] [Google Scholar]
- 121. Schreier B, Rabe S, Schneider B, Ruhs S. aldosterone/NaCl‐induced renal and cardiac. Hypertension. 2011;34(5):623‐629. [DOI] [PubMed] [Google Scholar]
- 122. Tsybouleva N, Zhang L, Chen S, et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 2004;109(10):1284‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lariviere R, Thibault G, Schiffrin EL. Increased endothelin‐1 content in blood vessels of deoxycorticosterone acetate‐salt hypertensive but not in spontaneously hypertensive rats. Hypertension. 1993;21(3):294‐300. [DOI] [PubMed] [Google Scholar]
- 124. Xavier FE, Aras‐López R, Arroyo‐Villa I, et al. Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane A2 and prostacyclin. Br J Pharmacol. 2008;154(6):1225‐1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fujita M, Minamino T, Asanuma H, et al. Aldosterone nongenomically worsens ischemia via protein kinase C‐dependent pathways in hypoperfused canine hearts. Hypertension. 2005;46(1):113‐117. [DOI] [PubMed] [Google Scholar]
- 126. Iglarz M, Touyz RM, Viel EC, et al. Involvement of oxidative stress in the profibrotic action of aldosterone: interaction with the renin‐angiotensin system. Am J Hypertens. 2004;17:597‐603. [PubMed] [Google Scholar]
- 127. Ikeda U, Kanbe T, Nakayama I, Kawahara Y, Yokoyama M, Shimada K. Aldosterone inhibits nitric oxide synthesis in rat vascular smooth muscle cells induced by interleukin‐1 beta. Eur J Pharmacol. 1995;290(2):69‐73. [DOI] [PubMed] [Google Scholar]
- 128. Dewald O, Zymek P, Winkelmann K, et al. CCL2/monocyte chemoattractant protein‐1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881‐889. [DOI] [PubMed] [Google Scholar]
- 129. Persy VP, Verhulst A, Ysebaert DK, de Greef KE, de Broe ME. Reduced postischemic macrophage infiltration and interstitial fibrosis in osteopontin knockout mice. Kidney Int. 2003;63(2):543‐553. [DOI] [PubMed] [Google Scholar]
- 130. Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt‐induced cardiac fibrosis and increased blood pressure. Hypertension. 2009;54(3):537‐543. [DOI] [PubMed] [Google Scholar]
- 131. Bienvenu LA, Morgan J, Rickard AJ, et al. Macrophage mineralocorticoid receptor signaling plays a key role in aldosterone‐independent cardiac fibrosis. Endocrinology. 2012;153(7):3416‐3425. [DOI] [PubMed] [Google Scholar]
- 132. Rafatian N, Westcott KV, White RA, Leenen FHH. Cardiac macrophages and apoptosis after myocardial infarction: effects of central MR blockade. Am J Physiol Reg Comp Physiol. 2014;307(7):R879‐R887. [DOI] [PubMed] [Google Scholar]
- 133. Usher MG, Duan SZ, Ivaschenko CY, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120(9):3350‐3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Nagase M, Matsui H, Shibata S, Gotoda T, Fujita T. Salt‐induced nephropathy in obese spontaneously hypertensive rats via paradoxical activation of the mineralocorticoid receptor: role of oxidative stress. Hypertension. 2007;50(5):877‐883. [DOI] [PubMed] [Google Scholar]
- 135. Wang H, Shimosawa T, Matsui H, et al. Paradoxical mineralocorticoid receptor activation and left ventricular diastolic dysfunction under high oxidative stress conditions. J Hypertens. 2008;26(7):1453‐1462. [DOI] [PubMed] [Google Scholar]
- 136. Monahan KD, Leuenberger UA, Ray CA. Aldosterone impairs baroreflex sensitivity in healthy adults. Am J Physiol Heart Circ Physiol. 2006;292(1):H190‐H197. [DOI] [PubMed] [Google Scholar]
- 137. Wang W. Chronic administration of aldosterone depresses baroreceptor reflex function in the dog. Hypertension. 1994;24(5):571‐575. [DOI] [PubMed] [Google Scholar]
- 138. Zucker IH, Pliquett RU. Novel mechanisms of sympatho‐excitation in chronic heart failure. Heart Fail Monit. 2002;3(1):2‐7. [PubMed] [Google Scholar]
- 139. Eckberg DL, Drabinsky M. Defective cardiac parasympathetic control in patients with heart disease. N Engl J Med. 1971;285(16):877‐883. [DOI] [PubMed] [Google Scholar]
- 140. Mortara A, La Rovere MT, Pinna GD, et al. Arterial baroreflex modulation of heart rate in chronic heart failure: clinical and hemodynamic correlates and prognostic implications. Circulation. 1997;96(10):3450‐3458. [DOI] [PubMed] [Google Scholar]
- 141. Osterziel KJ, Hänlein D, Willenbrock R, et al. Baroreflex sensitivity and cardiovascular mortality in patients with mild to moderate heart failure. Br Heart J. 1995;73(6):517‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lefebvre HP, Brown SA, Chetboul V, King J, Pouchelon JL, Toutain P. Angiotensin‐converting enzyme inhibitors in veterinary medicine. Curr Pharm Des. 2007;13(13):1347‐1361. [DOI] [PubMed] [Google Scholar]
- 143. Toutain PL, Lefebvre HP, King JN. Benazeprilat disposition and effect in dogs revisited with a pharmacokinetic/pharmacodynamic modeling approach. J Pharmacol Exp Ther. 2000;292(3):1087‐1093. [PubMed] [Google Scholar]
- 144. MacFadyen RJ, Meredith PA, Elliott HL. Enalapril clinical pharmacokinetics and pharmacokinetic‐pharmacodynamic relationships. An overview. Clin Pharmacokinet. 1993;25(4):274‐282. [DOI] [PubMed] [Google Scholar]
- 145. Bernstein KE, Martin BM, Edwards AS, Bernstein EA. Mouse angiotensin‐converting enzyme is a protein composed of two homologous domains. J Biol Chem. 1989;264(20):11945‐11951. [PubMed] [Google Scholar]
- 146. Rousseau A, Michaud A, Chauvet MT, Lenfant M, Corvol P. The hemoregulatory peptide N‐acetyl‐Ser‐Asp‐Lys‐Pro is a natural and specific substrate of the N‐terminal active site of human angiotensin‐converting enzyme. J Biol Chem. 1995;270(8):3656‐3661. [DOI] [PubMed] [Google Scholar]
- 147. Tan J, Wang J, Leenen F. Inhibition of brain angiotensin‐converting enzyme by peripheral administration of trandolapril versus lisinopril in Wistar rats. Am J Hypertens. 2005;18(2):158‐164. [DOI] [PubMed] [Google Scholar]
- 148. Dell'Italia LJ, Meng QC, Balcells E, et al. Compartmentalization of angiotensin II generation in the dog heart. Evidence for independent mechanisms in intravascular and interstitial spaces. J Clin Invest. 1997;100(2):253‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Wei C‐C, Tian B, Perry G, et al. Differential ANG II generation in plasma and tissue of mice with decreased expression of the ACE gene. Am J Physiol Heart Circ Physiol. 2002;282(6):H2254‐H2258. [DOI] [PubMed] [Google Scholar]
- 150. Uechi M, Tanaka Y, Aramaki Y, et al. Evaluation of the renin‐angiotensin system in cardiac tissues of cats with pressure‐overload cardiac hypertrophy. Am J Vet Res. 2008;69(3):343‐348. [DOI] [PubMed] [Google Scholar]
- 151. Carey RM, Padia SH. Angiotensin AT2 receptors: control of renal sodium excretion and blood pressure. Trends Endocrinol Metab. 2008;19(3):84‐87. [DOI] [PubMed] [Google Scholar]
- 152. Pfeffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM‐Overall programme. Lancet. 2003;362(9386):759‐766. [DOI] [PubMed] [Google Scholar]
- 153. McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation. 1999;100(10):1056‐1064. [DOI] [PubMed] [Google Scholar]
- 154. Maggioni AP, Anand I, Gottlieb SO, et al. Effects of valsartan on morbidity and mortality in patients with heart failure not receiving angiotensin‐converting enzyme inhibitors. J Am Coll Cardiol. 2002;40(8):1414‐1421. [DOI] [PubMed] [Google Scholar]
- 155. McMurray JJV, Östergren J, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and reduced left‐ventricular systolic function taking angiotensin‐converting‐enzyme inhibitors: the CHARM‐Added trial. Lancet. 2003;362(9386):767‐771. [DOI] [PubMed] [Google Scholar]
- 156. Chrysant SG. Current status of dual renin angiotensin aldosterone system blockade for the treatment of cardiovascular diseases. Am J Cardiol. 2010;105(6):849‐852. [DOI] [PubMed] [Google Scholar]
- 157. Jeunesse E, Woehrle F, Schneider M, Lefebvre HP. Effect of spironolactone on diuresis and urine sodium and potassium excretion in healthy dogs. J Vet Cardiol. 2007;9(2):63‐68. [DOI] [PubMed] [Google Scholar]
- 158. Karim A, Kook C, Zitzewitz DJ, Zagarella J, Doherty M, Campion J. Species differences in the metabolism and disposition of spironolactone. Drug Metab Dispos. 1976;4(6):547‐555. [PubMed] [Google Scholar]
- 159. Sadee W, Riegelman S, Jones SC. Plasma levels of spirolactones in the dog. J Pharm Sci. 1972;61(7):1129‐1132. [DOI] [PubMed] [Google Scholar]
- 160. Guyonnet J, Elliott J, Kaltsatos V. A preclinical pharmacokinetic and pharmacodynamic approach to determine a dose of spironolactone for treatment of congestive heart failure in dog. J Vet Pharmacol Ther. 2010;33(3):260‐267. [DOI] [PubMed] [Google Scholar]
- 161. Mosenkis A, Townsend RR. Gynecomastia and antihypertensive therapy. J Clin Hypertens. 2004;6(8):469‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Levy J, Burshell A, Marbach M, et al. Interaction of spironolactone with oestradiol receptors in cytosol. J Endocrinol. 1980;84(3):371‐379. [DOI] [PubMed] [Google Scholar]
- 163. Rose LI, Underwood RH, Newmark SR, Kisch ES, Williams GH. Pathophysiology of spironolactone‐induced gynecomastia. Ann Intern Med. 1977;87(4):398‐403. [DOI] [PubMed] [Google Scholar]
- 164. Amazit L, Le Billan F, Kolkhof P, et al. Finerenone impedes aldosterone‐dependent nuclear import of the mineralocorticoid receptor and prevents genomic recruitment of steroid receptor coactivator‐1. J Biol Chem. 2015;290(36):21876‐21889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Gheorghiade M, Böhm M, Greene SJ, et al. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure. JAMA. 2013;309(11):1125‐1111. [DOI] [PubMed] [Google Scholar]
- 166. McMurray JJV, Krum H, Abraham WT, et al. Aliskiren, enalapril, or aliskiren and enalapril in heart failure. N Engl J Med. 2016;374(16):1521‐1532. [DOI] [PubMed] [Google Scholar]
- 167. Calhoun DA, White WB, Krum H, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double‐blind, placebo‐ and active‐controlled phase 2 trial. Circulation. 2011;124(18):1945‐1955. [DOI] [PubMed] [Google Scholar]
- 168. Hargovan M, Ferro A. Aldosterone synthase inhibitors in hypertension: current status and future possibilities. JRSM Cardiovasc Dis. 2014;3:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Packer M, McMurray JJV. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin‐angiotensin system for the treatment of heart failure. Lancet. 2017;389(10081):1831‐1840. [DOI] [PubMed] [Google Scholar]
- 170. McMurray JJV, Packer M, Desai AS, et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N Engl J Med. 2014;371(11):993‐1004. [DOI] [PubMed] [Google Scholar]
- 171. Writing Committee Members , Yancy CW, Jessup M, et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol. 2016;68(13):1476‐1488. [DOI] [PubMed] [Google Scholar]
- 172. Mochel J, Burkey BF, Fink M, et al. First‐in‐class angiotensin receptor neprilysin inhibitor LCZ696 modulates the dynamics of the renin cascade and natriuretic peptides system with significant reduction. J Am Coll Cardiol. 2014;63:A806. [Google Scholar]
- 173. Atkins C, Bonagura J, Ettinger S, et al. Guidelines for the diagnosis and treatment of canine chronic valvular heart disease. J Vet Intern Med. 2009;23(6):1142‐1150. [DOI] [PubMed] [Google Scholar]
- 174. Newhard DK, Jung S, Winter RL, Duran SH. A prospective, randomized, double‐blind, placebo‐controlled pilot study of sacubitril/valsartan (Entresto) in dogs with cardiomegaly secondary to myxomatous mitral valve disease. J Vet Intern Med. 2018;32(5):1555‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Patel VB, Zhong J‐C, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1–7 axis of the Renin–Angiotensin system in heart failure. Circ Res. 2016;118(8):1313‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhong J, Basu R, Guo D, et al. Angiotensin‐converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122(7):717‐728. [DOI] [PubMed] [Google Scholar]
- 177. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin‐converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822‐828. [DOI] [PubMed] [Google Scholar]
- 178. Mori J, Patel VB, Alrob OA, et al. Angiotensin 1–7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ Heart Fail. 2014;7(2):327‐339. [DOI] [PubMed] [Google Scholar]
- 179. CONSENSUS Trial Study Group . Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316(23):1429‐1435. [DOI] [PubMed] [Google Scholar]
- 180. SOLVD Investigators , Yusuf S, Pitt B, Davis CE, Hood WB Jr, Cohn JN. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325(5):293‐302. [DOI] [PubMed] [Google Scholar]
- 181. Yusuf S, Pepine CJ, Garces C, et al. Effect of enalapril on myocardial infarction and unstable angina in patients with low ejection fractions. Lancet. 1992;340(8829):1173‐1178. [DOI] [PubMed] [Google Scholar]
- 182. Amberger C, Chetboul V, Bomassi E, et al. Comparison of the effects of imidapril and enalapril in a prospective, multicentric randomized trial in dogs with naturally acquired heart failure. J Vet Cardiol. 2004;6(2):9‐16. [DOI] [PubMed] [Google Scholar]
- 183. Ettinger S, Benitz A, Ericsson G, et al. Effects of enalapril maleate on survival of dogs with naturally occurring acquired heart failure. J Am Vet Med Assoc. 1998;213:1573‐1577. [PubMed] [Google Scholar]
- 184. BENCH (BENazepril in Canine Heart Disease) Study Group . The effect of benazepril on survival times and clinical signs of dogs with congestive heart failure: results of a multicenter, prospective, randomized, double‐blinded, placebo‐controlled, long‐term clinical trial. J Vet Cardiol. 1999;1:7‐18. [DOI] [PubMed] [Google Scholar]
- 185. The COVE Study Group . Controlled clinical evaluation of enalapril in dogs with heart failure: results of the Cooperative Veterinary Enalapril Study Group. J Vet Intern Med. 1995;9:243‐252. [DOI] [PubMed] [Google Scholar]
- 186. Besche B, Chetboul V, Lachaud Lefay M‐P, Grandemange E. Clinical evaluation of imidapril in congestive heart failure in dogs: results of the EFFIC study. J Small Anim Pract. 2007;48(5):265‐270. [DOI] [PubMed] [Google Scholar]
- 187. The IMPROVE Study Group . Acute and short‐term hemodynamic, echocardiographic, and clinical effects of enalapril maleate in dogs with naturally acquired heart failure: results of the Invasive Multicenter PROspective Veterinary Evaluation of Enalapril Study. J Vet Intern Med. 1995;9(4):234‐242. [DOI] [PubMed] [Google Scholar]
- 188. FDA New Drug Application . Freedom of Information Summary, VETMEDIN – Pimobendan chewable tablets for dogs. 2007 Apr 30:1‐36.
- 189. Häggström J, Boswood A, O'Grady M, et al. Effect of pimobendan or benazepril hydrochloride on survival times in dogs with congestive heart failure caused by naturally occurring myxomatous mitral valve disease: the QUEST Study. J Vet Intern Med. 2008;22(5):1124‐1135. [DOI] [PubMed] [Google Scholar]
- 190. Kvart C, Häggström J, Pedersen HD, et al. Efficacy of enalapril for prevention of congestive heart failure in dogs with myxomatous valve disease and asymptomatic mitral regurgitation. J Vet Intern Med. 2002;16(1):80‐88. [PubMed] [Google Scholar]
- 191. Atkins CE, Keene BW, Brown WA, et al. Results of the veterinary enalapril trial to prove reduction in onset of heart failure in dogs chronically treated with enalapril alone for compensated, naturally occurring mitral valve insufficiency. J Am Vet Med Assoc. 2007;231(7):1061‐1069. [DOI] [PubMed] [Google Scholar]
- 192. Pouchelon JL, Jamet N, Gouni V, et al. Effect of benazepril on survival and cardiac events in dogs with asymptomatic mitral valve disease: a retrospective study of 141 cases. J Vet Intern Med. 2008;22(4):905‐914. [DOI] [PubMed] [Google Scholar]
- 193. O'Grady MR, O'Sullivan ML, Minors SL, Horne R. Efficacy of benazepril hydrochloride to delay the progression of occult dilated cardiomyopathy in doberman pinschers. J Vet Intern Med. 2009;23(5):977‐983. [DOI] [PubMed] [Google Scholar]
- 194. Meurs KM, Stern JA, Atkins CE, et al. Angiotensin‐converting enzyme activity and inhibition in dogs with cardiac disease and an angiotensin‐converting enzyme polymorphism. J Renin Angiotensin Aldosterone Syst. 2017;18(4):1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Blackford LW, Golden AL, Bright JM, et al. Captopril provides sustained hemodynamic benefits in dogs with experimentally induced mitral regurgitation. Vet Surg. 1990;19(3):237‐242. [DOI] [PubMed] [Google Scholar]
- 196. Boswood A, Haggstrom J, Gordon SG, et al. Effect of pimobendan in dogs with preclinical myxomatous mitral valve disease and cardiomegaly: The EPIC Study‐a randomized clinical trial. J Vet Intern Med. 2016;30(6):1765‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341(10):709‐717. [DOI] [PubMed] [Google Scholar]
- 198. Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. 2003;348(14):1309‐1321. [DOI] [PubMed] [Google Scholar]
- 199. Zannad F, McMurray J, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364(1):11‐21. [DOI] [PubMed] [Google Scholar]
- 200. Butler J, Ezekowitz JA, Collins SP, et al. Update on aldosterone antagonists use in heart failure with reduced left ventricular ejection fraction. Heart Failure Society of America Guidelines Committee. J Card Fail. 2012;18(4):265‐281. [DOI] [PubMed] [Google Scholar]
- 201. Rousseau MF, Gurné O, Duprez D, et al. Beneficial neurohormonal profile of spironolactone in severe congestive heart failure: results from the RALES neurohormonal substudy. J Am Coll Cardiol. 2002;40(9):1596‐1601. [DOI] [PubMed] [Google Scholar]
- 202. Pedersen HD, Olsen LH, Arnorsdottir H. Breed differences in the plasma renin activity and plasma aldosterone concentration of dogs. Zentralbl Veterinarmed A. 1995;42(7):435‐441. [DOI] [PubMed] [Google Scholar]
- 203. Lantis AC, Ames MK, Atkins CE, et al. Aldosterone breakthrough with benazepril in furosemide‐activated renin‐angiotensin‐aldosterone system in normal dogs. J Vet Pharmacol Ther. 2014;38(1):65‐73. [DOI] [PubMed] [Google Scholar]
- 204. Lantis AC, Ames MK, Werre S, Atkins CE. The effect of enalapril on furosemide‐activated renin‐angiotensin‐aldosterone system in healthy dogs. J Vet Pharmacol Ther. 2015;38(5):513‐517. [DOI] [PubMed] [Google Scholar]
- 205. Ames MK, Atkins CE, Lee S, Lantis AC, zumBrunnen JR. Effects of high doses of enalapril and benazepril on the pharmacologically activated renin‐angiotensin‐aldosterone system in clinically normal dogs. Am J Vet Res. 2015;76(12):1041‐1050. [DOI] [PubMed] [Google Scholar]
- 206. Suzuki G. Effects of long‐term monotherapy with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure. Circulation. 2002;106(23):2967‐2972. [DOI] [PubMed] [Google Scholar]
- 207. Cataliotti A. Differential actions of vasopeptidase inhibition versus angiotensin‐converting enzyme inhibition on diuretic therapy in experimental congestive heart failure. Circulation. 2002;105(5):639‐644. [DOI] [PubMed] [Google Scholar]
- 208. Lee J, Mizuno M, Mizuno T, Harada K, Uechi M. Pathologic manifestations on surgical biopsy and their correlation with clinical indices in dogs with degenerative mitral valve disease. J Vet Intern Med. 2015;29(5):1313‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Schuller S, van Israel N, Vanbelle S, et al. Lack of efficacy of low‐dose spironolactone as adjunct treatment to conventional congestive heart failure treatment in dogs. J Vet Pharmacol Ther. 2010;34(4):322‐331. [DOI] [PubMed] [Google Scholar]
- 210. Bernay F, Bland JM, Haggstrom J, et al. Efficacy of spironolactone on survival in dogs with naturally occurring mitral regurgitation caused by myxomatous mitral valve disease. J Vet Intern Med. 2010;24(2):331‐341. [DOI] [PubMed] [Google Scholar]
- 211. Edelmann F, Wachter R, Schmidt AG, et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the Aldo‐DHF randomized controlled trial. JAMA. 2013;309(8):781‐791. [DOI] [PubMed] [Google Scholar]
- 212. Mottram PM, Haluska B, Leano R, Cowley D, Stowasser M, Marwick TH. Effect of aldosterone antagonism on myocardial dysfunction in hypertensive patients with diastolic heart failure. Circulation. 2004;110(5):558‐565. [DOI] [PubMed] [Google Scholar]
- 213. Pitt B, Pfeffer MA, Assmann SF, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15):1383‐1392. [DOI] [PubMed] [Google Scholar]
- 214. de Denus S, O‐Meara E. Spironolactone metabolites in TOPCAT ‐ new insights into regional variation. N Engl J Med. 2017;376(17):1‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bristow MR, Silva Enciso J, Gersh BJ, et al. Detection and management of geographic disparities in the TOPCAT trial. JACC Basic Transl Sci. 2016;1(3):180‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Lewis EF, Kim H‐Y, Claggett B, et al. Impact of spironolactone on longitudinal changes in health‐related quality of life in the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial. Circ Heart Fail. 2016;9(3):e001937. [DOI] [PubMed] [Google Scholar]
- 217. Kosmala W, Rojek A, Przewlocka‐Kosmala M, Wright L, Mysiak A, Marwick TH. Effect of aldosterone antagonism on exercise tolerance in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2016;68(17):1823‐1834. [DOI] [PubMed] [Google Scholar]
- 218. MacDonald KA, Kittleson MD, Larson RF, et al. The effect of ramipril on left ventricular mass, myocardial fibrosis, diastolic function, and plasma neurohormones in maine coon cats with familial hypertrophic cardiomyopathy without heart failure. J Vet Intern Med. 2006;20(5):1093‐1105. [DOI] [PubMed] [Google Scholar]
- 219. MacDonald KA, Kittleson MD, Kass PH. Effect of spironolactone on diastolic function and left ventricular mass in maine coon cats with familial hypertrophic cardiomyopathy. J Vet Intern Med. 2008;22(2):335‐341. [DOI] [PubMed] [Google Scholar]
- 220. James R, Guillot E, Garelli‐Paar C, Huxley J, Grassi V, Cobb M. The SEISICAT study: a pilot study assessing efficacy and safety of spironolactone in cats with congestive heart failure secondary to cardiomyopathy. J Vet Cardiol. 2018;20(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 221. Krum H, Nolly H, Workman D, et al. Efficacy of eplerenone added to renin‐angiotensin blockade in hypertensive patients. Hypertension. 2002;40(2):117‐123. [DOI] [PubMed] [Google Scholar]
- 222. Kawada N, Isaka Y, Kitamura H, Rakugi H, Moriyama T. A pilot study of the effects of eplerenone add‐on therapy in patients taking renin‐angiotensin system blockers. J Renin Angiotensin Aldosterone Syst. 2015;16(2):360‐365. [DOI] [PubMed] [Google Scholar]
- 223. Jansen PM, Danser AHJ, Imholz BP, van den Meiracker AH. Aldosterone‐receptor antagonism in hypertension. J Hypertens. 2009;27(4):680‐691. [DOI] [PubMed] [Google Scholar]
- 224. Brown S, Atkins C, Bagley R, et al. Guidelines for the identification, evaluation, and management of systemic hypertension in dogs and cats. J Vet Intern Med. 2007;21:542‐558. [DOI] [PubMed] [Google Scholar]
- 225. Ortega TM, Feldman EC, Nelson RW, Willits N, Cowgill LD. Systemic arterial blood pressure and urine protein/creatinine ratio in dogs with hyperadrenocorticism. J Am Vet Med Assoc. 1996;209(10):1724‐1729. [PubMed] [Google Scholar]
- 226. Javadi S, Kooistra HS, Mol JA, Boer P, Boer WH, Rijnberk A. Plasma aldosterone concentrations and plasma renin activity in healthy dogs and dogs with hyperadrenocorticism. Vet Rec. 2003;153(17):521‐525. [DOI] [PubMed] [Google Scholar]
- 227. Goy‐Thollot I, Péchereau D, Kéroack S, et al. Investigation of the role of aldosterone in hypertension associated with spontaneous pituitary‐dependent hyperadrenocorticism in dogs. J Small Anim Pract. 2002;43(11):489‐492. [DOI] [PubMed] [Google Scholar]
- 228. Wenger M, Sieber‐Ruckstuhl NS, Müller C, Reusch CE. Effect of trilostane on serum concentrations of aldosterone, cortisol, and potassium in dogs with pituitary‐dependent hyperadrenocorticism. Am J Vet Res. 2004;65(9):1245‐1250. [DOI] [PubMed] [Google Scholar]
- 229. Jepson RE, Syme HM, Elliott J. Plasma renin activity and aldosterone concentrations in hypertensive cats with and without azotemia and in response to treatment with amlodipine besylate. J Vet Intern Med. 2013;28(1):144‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Flack JM, Oparil S, Pratt JH, et al. Efficacy and tolerability of eplerenone and losartan in hypertensive black and white patients. J Am Coll Cardiol. 2003;41(7):1148‐1155. [DOI] [PubMed] [Google Scholar]
- 231. Jenkins TL, Coleman AE, Schmiedt CW, Brown SA. Attenuation of the pressor response to exogenous angiotensin by angiotensin receptor blockers and benazepril hydrochloride in clinically normal cats. Am J Vet Res. 2015;76(9):807‐813. [DOI] [PubMed] [Google Scholar]
- 232. Glaus TM, Elliott J, Herberich E, et al. Efficacy of long‐term oral telmisartan treatment in cats with hypertension: results of a prospective European clinical trial. J Vet Intern Med. 2018;29(Suppl 1):855‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Williams TL, Elliott J, Syme HM. Renin‐Angiotensin‐Aldosterone System Activity in Hyperthyroid Cats with and without Concurrent Hypertension. J Vet Intern Med. 2013;27(3):522‐529. [DOI] [PubMed] [Google Scholar]
- 234. Jensen J, Henik RA, Brownfield M, Armstrong J. Plasma renin activity and angiotensin I and aldosterone concentrations in cats with hypertension associated with chronic renal disease. Am J Vet Res. 1997;58(5):535‐540. [PubMed] [Google Scholar]
- 235. Williams B, MacDonald TM, Morant S, et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug‐resistant hypertension (PATHWAY‐2): a randomised, double‐blind, crossover trial. Lancet. 2015;386:2059‐2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Remuzzi G, Benigni A, Remuzzi A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J Clin Invest. 2006;116(2):288‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Walls J. Relationship between proteinuria and progressive renal disease. Am J Kidney Dis. 2001;37(1):S13‐S16. [DOI] [PubMed] [Google Scholar]
- 238. Geng DF, Sun WF, Yang L, et al. Antiproteinuric effect of angiotensin receptor blockers in normotensive patients with proteinuria: a meta‐analysis of randomized controlled trials. 2014;15(1):44‐51. [DOI] [PubMed] [Google Scholar]
- 239. Kunz R, Friedrich C, Wolbers M, Mann JFE. Meta‐analysis: effect of monotherapy and combination therapy with inhibitors of the renin angiotensin system on proteinuria in renal disease. Ann Intern Med. 2008;148(1):30‐48. [DOI] [PubMed] [Google Scholar]
- 240. Cheng J, Zhang X, Tian J, Li Q, Chen J. Combination therapy an ACE inhibitor and an angiotensin receptor blocker for IgA nephropathy: a meta‐analysis. Int J Clin Pract. 2012;66(10):917‐923. [DOI] [PubMed] [Google Scholar]
- 241. Ando K, Ohtsu H, Uchida S, et al. Anti‐albuminuric effect of the aldosterone blocker eplerenone in non‐diabetic hypertensive patients with albuminuria: a double‐blind, randomised, placebo‐controlled trial. Lancet Diabetes Endocrinol. 2014;2(12):944‐953. [DOI] [PubMed] [Google Scholar]
- 242. Esteghamati A, Noshad S, Jarrah S, Mousavizadeh M, Khoee SH, Nakhjavani M. Long‐term effects of addition of mineralocorticoid receptor antagonist to angiotensin II receptor blocker in patients with diabetic nephropathy: a randomized clinical trial. Nephrol Dial Transplant. 2013;28(11):2823‐2833. [DOI] [PubMed] [Google Scholar]
- 243. Brown SA, Finco DR, Brown CA, et al. Evaluation of the effects of inhibition of angiotensin converting enzyme with enalapril in dogs with induced chronic renal insufficiency. Am J Vet Res. 2003;64(3):321‐327. [DOI] [PubMed] [Google Scholar]
- 244. Brown SA, Walton CL, Crawford P, Bakris GL. Long‐term effects of antihypertensive regimens on renal hemodynamics and proteinuria. Kidney Int. 1993;43(6):1210‐1218. [DOI] [PubMed] [Google Scholar]
- 245. Grauer GF, Greco DS, Getzy DM, et al. Effects of enalapril versus placebo as a treatment for canine idiopathic glomerulonephritis. J Vet Intern Med. 2000;14(5):526‐533. [DOI] [PubMed] [Google Scholar]
- 246. Tenhundfeld J, Wefstaedt P, Nolte IJ. A randomized controlled clinical trial of the use of benazepril and heparin for the treatment of chronic kidney disease in dogs. J Am Vet Med Assoc. 2009;234(8):1031‐1037. [DOI] [PubMed] [Google Scholar]
- 247. Mishina M, Watanabe T. Development of hypertension and effects of benazepril hydrochloride in a canine remnant kidney model of chronic renal failure. J Vet Med Sci. 2008;70(5):455‐460. [DOI] [PubMed] [Google Scholar]
- 248. Jacob F, Polzin DJ, Osborne CA, et al. Evaluation of the association between initial proteinuria and morbidity rate or death in dogs with naturally occurring chronic renal failure. J Am Vet Med Assoc. 2005;226(3):393‐400. [DOI] [PubMed] [Google Scholar]
- 249. Grodecki KM, Gains MJ, Baumal R, et al. Treatment of X‐linked hereditary nephritis in Samoyed dogs with angiotensin converting enzyme (ACE) inhibitor. J Comp Pathol. 1997;117(3):209‐225. [DOI] [PubMed] [Google Scholar]
- 250. Abraham PA, Opsahl JA, Halstenson CE, Keane WF. Efficacy and renal effects of enalapril therapy for hypertensive patients with chronic renal insufficiency. Arch Intern Med. 1988;148(11):2358‐2362. [PubMed] [Google Scholar]
- 251. Brown SA, Brown CA, Jacobs G. Hemodynamic effects of angiotensin converting enzyme inhibition (Benazepril) in cats with chronic renal insufficiency. J Vet Intern Med. 1999;17:716. [Google Scholar]
- 252. Praga M, Hernandez E, Montoyo C, et al. Long‐term beneficial effects of angiotensin‐converting enzyme inhibition in patients with nephrotic proteinuria. Am J Kidney Dis. 1992;20(3):240‐248. [DOI] [PubMed] [Google Scholar]
- 253. Maschio G, Alberti D, Janin G, et al. Effect of the angiotensin‐converting‐enzyme inhibitor benazepril on the progression of chronic renal insufficiency. The Angiotensin‐Converting‐Enzyme Inhibition in Progressive Renal Insufficiency Study Group. N Engl J Med. 1996;334(15):939‐945. [DOI] [PubMed] [Google Scholar]
- 254. Hou FF, Zhang X, Zhang GH, et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N Engl J Med. 2006;354(2):131‐140. [DOI] [PubMed] [Google Scholar]
- 255. Miller RH, Lehmkuhl LB, Smeak DD, DiBartola S, Radin J. Effect of enalapril on blood pressure, renal function, and the renin‐angiotensin‐aldosterone system in cats with autosomal dominant polycystic kidney disease. Am J Vet Res. 1999;60(12):1516‐1525. [PubMed] [Google Scholar]
- 256. IRIS Canine GN Study Group Standard Therapy Subgroup , Brown S, Elliott J, Francey T, Polzin D, Vaden S. Consensus recommendations for standard therapy of glomerular disease in dogs. J Vet Intern Med. 2013;27(s1):S27‐S43. [DOI] [PubMed] [Google Scholar]
- 257. Steele J, Henik R, Stepien R. Effects of angiotensin‐converting enzyme inhibition on plasma aldosterone concentration, plasma renin activity, and blood pressure in spontaneously hypertensive cats with chronic renal disease. Vet Ther. 2002;3(2):157‐166. [PubMed] [Google Scholar]
- 258. King JN, Tasker S, Gunn‐Moore DA, Strehlau G, BENRIC (benazepril in renal insufficiency in cats) Study Group . Prognostic factors in cats with chronic kidney disease. J Vet Intern Med. 2007;21(5):906‐916. [PubMed] [Google Scholar]
- 259. Chakrabarti S, Syme HM, Elliott J. Clinicopathological variables predicting progression of azotemia in cats with chronic kidney disease. J Vet Intern Med. 2012;26(2):275‐281. [DOI] [PubMed] [Google Scholar]
- 260. Syme HM, Markwell PJ, Pfeiffer D, Elliott J. Survival of cats with naturally occurring chronic renal failure is related to severity of proteinuria. J Vet Intern Med. 2006;20(3):528‐535. [DOI] [PubMed] [Google Scholar]
- 261. Jepson RE, Elliott J, Brodbelt D, Syme HM. Effect of control of systolic blood pressure on survival in cats with systemic hypertension. J Vet Intern Med. 2007;21(3):402‐409. [DOI] [PubMed] [Google Scholar]
- 262. Sent U, Gössl R, Elliott J, Syme HM, Zimmering T. Comparison of efficacy of long‐term oral treatment with telmisartan and benazepril in cats with chronic kidney disease. J Vet Intern Med. 2015;29(6):1479‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Wynckel A, Ebikili B, Melin JP, et al. Long‐term follow‐up of acute renal failure caused by angiotensin converting enzyme inhibitors. Am J Hypertens. 1998;11(9):1080‐1086. [DOI] [PubMed] [Google Scholar]
- 264. Atkins CE, Brown WA, Coats JR, et al. Effects of long‐term administration of enalapril on clinical indicators of renal function in dogs with compensated mitral regurgitation. J Am Vet Med Assoc. 2002;221(5):654‐658. [DOI] [PubMed] [Google Scholar]
- 265. Lefebvre HP, Ollivier E, Atkins CE, et al. Safety of spironolactone in dogs with chronic heart failure because of degenerative valvular disease: a population‐based, longitudinal study. J Vet Intern Med. 2013;27(5):1083‐1091. [DOI] [PubMed] [Google Scholar]
- 266. Thomason JD, Rapoport G, Fallaw T, Calvert CA. The influence of enalapril and spironolactone on electrolyte concentrations in Doberman pinschers with dilated cardiomyopathy. Vet J. 2014;202(3):1‐5. [DOI] [PubMed] [Google Scholar]
- 267. Thomason JD, Rockwell JE, Fallaw TK, Calvert CA. Influence of combined angiotensin‐converting enzyme inhibitors and spironolactone on serum K+, Mg2+, and Na+ concentrations in small dogs with degenerative mitral valve disease. J Vet Cardiol. 2007;9(2):103‐108. [DOI] [PubMed] [Google Scholar]
- 268. Brilla CG, Weber KT. mineralocorticoid excess, dietary sodium. J Lab Clin Med. 1992;120(6):893‐901. [PubMed] [Google Scholar]
- 269. Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res. 1990;67(6):1355‐1364. [DOI] [PubMed] [Google Scholar]
- 270. Brilla CG, Matsubara LS, Weber KT. Antifibrotic effects of spironolactone in preventing myocardial fibrosis in systemic arterial hypertension. Am J Cardiol. 1993;71(3):A12‐A16. [DOI] [PubMed] [Google Scholar]
- 271. Jalil JE, Janicki JS, Pick R, Weber KT. Coronary vascular remodeling and myocardial fibrosis in the rat with renovascular hypertension. Response to captopril. Am J Hypertens. 1991;4(1 Pt 1):51‐55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Supporting Information Figure S1
Supporting Figure S2.
Supporting Information Table S1 Animal and ex vivo human studies of cardiovascular and kidney diseases providing direct (hormone infusion, transgenic animal models) and indirect (RAAS suppression) evidence of the cardio‐ and nephrotoxicity of chronic and/or excess angiotensin II and aldosterone production. The studies are presented in chronological order.
Supporting Information Table S2 Characteristics of specific ACE‐Inhibitors
Supporting Information Table S3 Pharmacokinetic parameters of ACE‐Inhibitors in dogs and cats
Supporting Information Table S4 Characteristics of specific Angiotensin II Type‐1 Receptor Blockers and an ARB/neprilysin‐inhibitor
Supporting Information Table S5 Clinical trials involving RAAS suppression in people with proteinuric kidney disease. Studies are presented in chronological order