

Abstract
Dosage compensation is the process by which transcript levels of the X-chromosome are equalized with those of autosomes. Although diverse mechanisms of dosage compensation evolved across species, these mechanisms all involve distinguishing the X-chromosome from autosomes. Because one chromosome is singled out from other chromosomes for precise regulation, dosage compensation serves as an important model for understanding how specific cis-elements are identified within the highly compacted three-dimensional genome to co-regulate thousands of genes. Recently, multiple genomic approaches have provided key insights into the mechanisms of dosage compensation, extending what we have learned from classical genetic studies. In the future, newer genomic approaches that require very little starting material show great promise to provide an understanding of the heterogeneity of dosage compensation between cells and how it functions in non-model organisms.
Composition of chromatin
Within the eukaryotic cell nucleus, each chromosome consists of a single molecule of double stranded DNA. Frequently thought of in linear space, these DNA molecules are compacted within the confines of the nucleus, as much as 10-thousand-fold, by adopting diverse three-dimensional conformations. Despite extensive compaction of the chromosomes within the nucleus, specific cis-elements need to be precisely identified to regulate genes spatially and temporally.
At the most basic level, chromatin consists of a repeating array of nucleosomes, which are octamers of histone proteins, around which DNA is wrapped. There are two copies of each of the four core histone proteins (H2A, H2B, H3 and H4) within each nucleosome, which is surrounded by 146 bp of DNA [1,2]. Chromatin serves as the substrate for essential cellular processes such as transcription, replication, recombination and cell division [1]. The idea of crumpled polymer globules, or the beads on a string framework for chromatin, was initially introduced forty years ago [3,4]. Surprisingly, chromatin only accounts for roughly half of the available nuclear space [5]; specialized nuclear bodies and diverse chromatin regulators reside in the remaining interchromatin space. Nuclear bodies are dynamic structures that often reflect a cell’s transcriptional state and serve as sites of transcription and/or RNA processing [5,6].
Chromatin can be generalized into two contrasting types: euchromatin, which generally resides in the interior of the nucleus, and heterochromatin, which generally resides close to the nuclear envelope. These types of chromatin differ in their accessibility: euchromatin is regarded as “active” as it contains gene rich regions, and positively correlates with chromatin accessibility and heterochromatin is regarded as “inactive,” is gene poor, and negatively correlates with chromatin accessibility [7–9]. Chromatin may vary dramatically between cell types as histone proteins, histone modifiers (or readers/writers), and other transcription factors are able to recognize and modulate specific loci, further differentiating chromatin compartments and fine-tuning gene expression. Therefore, while chromatin broadly influences gene regulation, it does not fully explain the modulation in activity of individual genes; rather chromatin establishes local environments that are more or less favorable for gene-specific expression [10–13]. In contrast, topologically associated domains (TADs), are physical megabase-sized partitions of the genome. Within an organism, TAD organization is believed to be largely invariant and conserved across cell types [2]. Smaller domains are known by various names, including sub-TADs, chromatin loops, and neighborhoods. These smaller domains may be highly variable between cell types [10,12] and thus play a fundamental role in genomic function and regulation.
Across species, TAD boundaries are enriched for insulator elements, which are bound by a specialized class of insulator proteins that regulate three-dimensional genome organization [9,14]. TAD boundary regions are also enriched for active genes, though recent studies in Drosophila suggest three-dimensional chromatin organization is established during zygotic genome activation, independent of transcription [15]. Few factors, including chromatin regulators and non-coding RNAs, modulate genome organization [9]. Therefore, much remains unknown regarding how specific nuclear domains are established and how domains are targeted to modulate gene regulation.
Overview of dosage compensation
Diverse models of sex chromosome dosage compensation, in which the sex chromosome(s) of one sex is specifically targeted and transcriptionally regulated, serve as model systems in which to study how three-dimensional genome organization regulates the expression of large groups or co-regulated genes. Analyses of dosage compensation across species, described below, have revealed the mechanisms by which large groups of genes are co-regulated within the context of three-dimensional genome organization.
Sex chromosome dosage compensation is an essential—yet diverse—process in many species. There exist many distinct dosage compensation strategies to equalize transcript levels of genes located on the sex chromosome(s) between the sexes [16,17]. A conserved first step in heterogametic XY species, including humans and Drosophila, is distinguishing the X-chromosome from the autosomes. For example, in many mammals, one female X-chromosome is singled out for inactivation. A conserved dosage compensation mechanism, shared across several species including mammals, C. elegans and Drosophila, is upregulation of transcription of genes on the active X-chromosome to equalize their expression with that of autosomal genes [16–18] (see Figure 1).
Figure 1.

Dosage compensation strategies: the dosage compensated sex is represented in red.
In contrast to the X-chromosome, there are no known examples in which the Y-chromosome is targeted for dosage compensation. The Y-chromosome is very small and heterochromatic, though in some species it does carry specialized genes that have male sex-specific functions [19]. X-chromosome upregulation was first proposed by Susumu Ohno; Ohno hypothesized that transcription of the X-chromosome in mammals was upregulated during early evolution of sex chromosomes in order to compensate for degeneration of the Y-chromosome [20]. Furthermore, Ohno theorized X-chromosome upregulation was not limited to males but also occurred in females. Ohno proposed that X-chromosome downregulation evolved in mammals in order to counteract X upregulation in XX females, therefore maintaining equalized dosage with autosomes [20]. This idea is widely known as Ohno’s hypothesis. Data exists that both support and oppose the existence of such a mechanism across species [18,20–22].
Mechanisms of dosage compensation
In the nematode worm, Caenorhabditis elegans, males carry a single X-chromosome and two copies of each autosome (XO, AA), while hermaphroditic females carry two copies of the X-chromosome and each autosome (XX, AA). An unknown mechanism is thought to upregulate expression of the X-chromosome in both sexes. A well-defined dosage compensation complex (DCC) then down regulates gene expression for both X-chromosomes in hermaphrodites to equalize X-linked gene expression to that of males [16,17,23–25] (Figure 1A). The DCC initially localizes to a small number of X-linked “recruitment element on X” (rex) sites, which are located in regions of euchromatin and contain binding motifs that cluster in two and three-dimensional space [25]. The process of C. elegans dosage compensation is believed to occur through X-chromosome compaction as compaction is lost in the absence of DCC activity and the X-chromosome becomes enlarged. Enlargement of the X-chromosome may contribute to upregulation of X-linked genes by a mechanism that likely involves the function of specific X-linked DNA sequences rather than global regulation of all genes on the X-chromosome [22,26]. In the fruit fly Drosophila melanogaster, males carry a single X-chromosome and two copies of each autosome (XY, AA), while females carry two copies of the X-chromosome and each autosome (XX, AA) (Figure 1B). A dosage compensation complex known as the Male Specific Lethal complex (MSL), is specifically assembled in males and upregulates transcription of the single male X-chromosome to approach levels of female X-linked gene expression. Similar to C. elegans, the initial sites of MSL recruitment, “chromatin entry sites” (CES), are located in euchromatin and contain binding motifs that cluster in two and three-dimensional space [14,27,28]. Furthermore, H4K16ac is important for increasing transcript levels on the male X-chromosome [29,30].
In mammals, such as mouse and human, males carry a single X-chromosome and two copies of each autosome (XY, AA), while females carry two copies of the X-chromosome and each autosome (XX, AA) (Figure 1C). In females, one copy of the X-chromosome is randomly inactivated in a process known as X-inactivation (XI), which normalizes female X-linked gene expression with that of males [16]. X-inactivation is characterized by extensive DNA methylation that is thought to maintain the inactive state [31].
In mammals, the inactive female X-chromosome forms two large domains. In C. elegans, compaction along the entire length of both X-chromosomes in the hermaphrodite is associated with reduced transcription levels [26]. In Drosophila, the X-chromosome exhibits enhanced accessibility in both males and females independent of MSL [32]. In the future, three-dimensional techniques could be applied to reveal which species-specific factors are important for setting up and or maintaining the three-dimensional chromosome conformation many dosage compensation complexes use to target an entire chromosome.
Techniques for probing chromatin structural dynamics
Dosage compensation requires the targeting and regulation of an entire chromosome. Therefore, sex chromosomes represent a model that provides great insights into how smaller chromatin domains are targeted. Many improved techniques have had a substantial impact on the study of chromatin structure and function. While most of our understanding of dosage compensation comes from classical genetics and microscopy, which have been very powerful, these approaches do not address the heterogeneity of dosage compensation across cells and tissue types. Further application of the new approaches described below will allow for even greater insight into how this fundamental process functions at high resolution in single cells.
Chromosome conformation capture
The development of chromosome conformation capture (3C) and the subsequent development of the current 3C family of techniques—chromosome conformation capture on chip (4C), chromosome capture carbon-copy (5C), and genome-wide chromosome conformation capture (Hi-C) [33–35] have significantly contributed to our understanding of how three-dimensional genome structure contributes to genomic function (Figure 2A). There have been many recent improvements in this family of techniques. For example, in situ Hi-C may be performed in intact nuclei, which reduces experimental noise due to random ligation events and enables the production of higher resolution data from fewer reads compared to the original technique (dilution Hi-C) [36]. 4C has also been improved by the incorporation of unique molecular identifiers, which reduce the impact of PCR-inflated ligation read counts, allowing the generation of 4C profiles with as few as 100,000 reads per bait [37] compared with 500,000 to 3 million reads for traditional 4C-seq [38].
Figure 2.
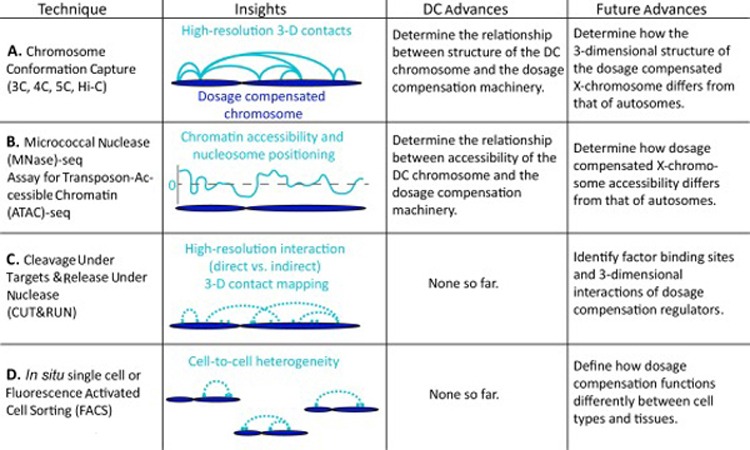
Emerging technologies and their current impacts within the dosage compensation field.
Several groups have used chromosome conformation capture approaches to study dosage compensation in mammals [39–42], C. elegans [25,43], and Drosophila [14,44]. Fluorescent in situ hybridization (FISH) and genome-wide chromosome conformation capture analysis (Hi-C) on C. elegans embryos revealed that the long-range contacts seen between DCC recognition sites are also lost in the absence of dosage compensation [43]. This likely occurs because X-chromosome remodeling that occurs during active dosage compensation is dependent on a condensin IDC subcomplex that is part of the DCC [26,43]. Furthermore, the histone modifiers SET-1, SET-4, and SIR-2.1 are also necessary for dosage compensation X-chromosome remodeling [26,45,46]. Hi-C was performed in several Drosophila cell lines and embryos and discovered that, in contrast to C. elegans, MSL likely takes advantage of a preexisting three-dimensional topology, as long-range contacts between CES are present in both sexes and are independent of dosage compensation [14,26,44]. However, the mechanism by which the long-range contacts between CES are established in both males and females remains unknown.
RNA antisense purification in mouse lung fibroblasts in conjunction with Hi-C analysis revealed that, upon initiation of XI, the long non-coding RNA Xist utilizes the existing three-dimensional topology of the X-chromosome in order to spread from the XIST locus to distant loci [39]. The spreading of Xist leads to structural changes that cause new regions of the chromosome to come in closer proximity to the XIST locus; this then allows the XIST RNA to transfer to these newly proximal sites, eventually coating the entire chromosome [39]. The inactive X-chromosome eventually takes on a unique, highly compacted structure known as a Barr body [47].
The inactive X is enriched for the histone marks H3K9me3 and H3K27me3, although it appears that H3K27me3 is not necessary for XI [31]. The compacted state of the inactive X requires the presence of two proteins, a heterochromatin protein 1 binding protein HBiX1 and structural maintenance of chromosomes hinge domain-containing protein 1 (SMCHD1) [31]. Based on allele-specific Hi-C and RNA-seq experiments, SMCHD1 antagonizes TAD boundary formation and is required for gene silencing during a specific developmental window; it is not required for maintenance, which may occur through other redundant pathways [48].
Using allele-specific Hi-C analysis, it was demonstrated that both X-chromosomes display classical TAD topology prior to XI [40]. However, after XI, the inactive X-chromosome adopts a unique three-dimensional structure, which consists of two very large megabase scale domains, termed super domains, separated by a boundary region [40]. Within these two super domains, classical TAD structure is rare, but does occur at gene clusters that escape XI [40]. Hi-C data demonstrates that these structures are conserved in humans [36]. However, the genomic content of the two domains differs between human and mouse [41]. CRISPR deletions of the boundary locus in mice results in fusion of the two super domains, but the boundary is not necessary for the formation of XI in either mice or humans [36,41,42]. Furthermore, X-inactivation has been studied much more extensively but recent work suggests that X-upregulation also occurs in mammals [18,49].
MNase-seq and ATAC-seq
Micrococcal Nuclease sequencing (MNase-seq) and Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) are two popular methods for probing chromatin accessibility genome-wide [50–52] (Figure 2B). ATAC-seq is a quick, cost-effective, two-step procedure that is able to identify regions of open chromatin (highly accessible regions) and map nucleosome positioning and transcription factor binding sites [50]. The latest version of MNase-seq, which utilizes a titration series, is also able to probe nucleosome positioning as well as relative chromatin accessibility (lowest to highest regions of accessibility) genome-wide [53]. While both techniques offer the ability to determine regions of open chromatin, MNase-seq with titration series offers the added benefit of providing relative chromatin accessibility, allowing diverse regulatory regions throughout the genome to be directly compared.
MNase-seq in Drosophila cultured cells revealed that MSL regulates chromatin accessibility by modulating nucleosome positioning at CES [14]. Furthermore, a non-sex specific dosage compensation protein, Chromatin-Linked Adaptor for MSL Proteins (CLAMP), regulates chromatin accessibility at CES as well as globally across the entire male X-chromosome [32].
CUT&RUN
Cleavage Under Targets and Release Using Nuclease (CUT&RUN), a modification of chromatin immunoprecipitation (ChIP), allows for simultaneous high-resolution chromatin mapping of specific factor localization and probing of the local chromatin environment [54] (Figure 2C). CUT&RUN is performed in situ under native conditions. It requires only a tenth of the sequencing depth of traditional ChIP-seq and has the ability to map histone modifications, even within compacted chromatin [54]. Furthermore, CUT&RUN may be used to map contact sites at near base-pair resolution and differentiate direct protein binding sites versus indirect sites resulting from long-range interactions [54]. This approach has not yet been applied to dosage compensation, however, it has great potential to contribute to better understanding the targeting requirements of various dosage compensation complexes by more accurately identifying binding sites and three-dimensional interactions between dosage compensation regulators across species.
Single cell techniques
Until recently, performing many of the techniques discussed above required large pools of cells. Fluorescence-Activated Cell Sorting (FACS) allows specific marked cell populations to be assayed, but still requires a large number of input cells [55] (Figure 2D). More recently, the modernization of genomic approaches allows them to be performed in situ. This has led to the ability to assay the genomic landscape in single cells. Single cell approaches are powerful; no longer is an average reading recorded from a bulk sample. As a result, cell to cell heterogeneity is uncovered and cells may be clustered into a number of unique groups.
Single cell techniques are currently being used to probe chromatin accessibility [56,57], DNA variation [58], transcription factor binding sites [59], methylation [60] and three-dimensional genome structure [61]. Additionally, microscopy-based approaches such as single-molecule super-resolution microscopy combined with Oligopaints—specialized FISH probes that allow for super-resolution microscopy and visualization of chromatin organization [62]—and assay of transposase accessible chromatin with visualization (ATAC-see)—a modification of ATAC-seq that reveals open chromatin regions targeted by transposons [63]—allow for single cell visualization of chromatin architecture. Single cell methods have not yet been applied to study dosage compensation. However, these approaches have great potential to define how it functions in different tissues and non-model organisms.
Concluding remarks and future perspectives
Dosage compensation is an essential process across species that evolved rapidly to equalize transcript levels from the X-chromosome and autosomes. Dosage compensation systems provide excellent models for defining how a specific part of the genome may be regulated in a context-specific way. Moreover, although diverse dosage compensation mechanisms have evolved, they all share the common feature of specifically distinguishing the X-chromosome from autosomes. In the future, applying the single cell genomic and imaging techniques described above to study how the X-chromosome is distinguished for specific regulation will reveal how heterogeneous this process is across cell types and organisms. Although little is understood about how dosage compensation works beyond classical genetic model organisms, this new frontier of techniques that require little starting material will allow us to study how sex chromosomes differ from autosomes beyond traditional model organisms, which is key to understanding such a rapidly evolving process.
Outstanding questions:
How is the X-chromosome specifically distinguished from autosomes across species?
How heterogeneous is dosage compensation across cell types and tissues?
How does dosage compensation function across a wide array of species including non-model organisms?
Highlights.
Dosage compensation equalizes transcript levels on the X-chromosome with those on autosomes.
Dosage compensation serves as a model for understanding how specific cis-elements are collectively targeted for coordinated regulation within the genome.
Genomic approaches to study chromatin and three-dimensional genome organization have provided new insights into dosage compensation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Widom J (1989) Toward a Unified Model of Chromatin Folding. Annu. Rev. Biophys. Biophys. Chem DOI: 10.1146/annurev.bb.18.060189.002053 [DOI] [PubMed] [Google Scholar]
- [2].Dixon JR et al. (2016) Chromatin domains: the unit of chromosome organization. Mol. Cell 62, 668–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Grosberg AY et al. (1988) The role of topological constraints in the kinetics of collapse of macromolecules. J. Phys 49, 2095–2100 [Google Scholar]
- [4].Grosberg A et al. (1993) Crumpled globule model of the three-dimensional structure of DNA. EPL (Europhysics Lett 23, 373 [Google Scholar]
- [5].Rajapakse I and Groudine M (2011) On emerging nuclear order. J. Cell Biol 192, 711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Aumiller WM Jr et al. (2014) Phase separation as a possible means of nuclear compartmentalization. In International review of cell and molecular biology 307pp. 109–149, Elsevier; [DOI] [PubMed] [Google Scholar]
- [7].Gilbert N et al. (2004) Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell 118, 555–566 [DOI] [PubMed] [Google Scholar]
- [8].Solovei I et al. (2013) LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598 [DOI] [PubMed] [Google Scholar]
- [9].Bonev B and Cavalli G (2016) Organization and function of the 3D genome. Nat. Rev. Genet 17, 661. [DOI] [PubMed] [Google Scholar]
- [10].Dixon JR et al. (2015) Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li L et al. (2015) Widespread Rearrangement of 3D Chromatin Organization Underlies Polycomb-Mediated Stress-Induced Silencing. Mol. Cell 58, 216–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barutcu AR et al. (2015) Chromatin interaction analysis reveals changes in small chromosome and telomere clustering between epithelial and breast cancer cells. Genome Biol 16, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cavalli G and Misteli T (2013) Functional implications of genome topology. Nat. Struct. Mol. Biol 20, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramírez F et al. (2015) High-affinity sites form an interaction network to facilitate spreading of the MSL complex across the X chromosome in Drosophila. Mol. Cell 60, 146–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hug CB et al. (2017) Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228 [DOI] [PubMed] [Google Scholar]
- [16].Ercan S (2015) Mechanisms of X Chromosome Dosage Compensation. J. Genomics 3, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Deng X et al. (2011) Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat. Genet 43, 1179–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Deng X et al. (2014) X chromosome regulation: diverse patterns in development, tissues and disease. Nat. Rev. Genet 15, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Graves JAM (2006) Sex chromosome specialization and degeneration in mammals. Cell 124, 901–914 [DOI] [PubMed] [Google Scholar]
- [20].Veitia RA et al. (2015) X chromosome inactivation and active X upregulation in therian mammals: Facts, questions, and hypotheses. J. Mol. Cell Biol 7, 2–11 [DOI] [PubMed] [Google Scholar]
- [21].Albritton SE et al. (2014) Sex-biased gene expression and evolution of the X chromosome in nematodes. Genetics 197, 865–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wheeler BS et al. (2016) Chromosome-wide mechanisms to decouple gene expression from gene dose during sex-chromosome evolution. Elife 5, e17365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Meyer B (2005) X chromosome dosage compensation. WormBook [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pferdehirt RR et al. (2011) An MLL/COMPASS subunit functions in the C. elegans dosage compensation complex to target X chromosomes for transcriptional regulation of gene expression. Genes Dev 25, 499–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Albritton SE et al. (2017) Cooperation between a hierarchical set of recruitment sites targets the X chromosome for dosage compensation. Elife 6, e23645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lau AC et al. (2014) The C. elegans dosage compensation complex mediates interphase X chromosome compaction. Epigenetics Chromatin 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alekseyenko AA et al. (2008) A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134, 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kuzu G et al. (2016) Expansion of GA dinucleotide repeats increases the density of CLAMP binding sites on the X-chromosome to promote Drosophila dosage compensation. PLoS Genet 12, e1006120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Armstrong RL et al. (2018) Chromatin conformation and transcriptional activity are permissive regulators of DNA replication initiation in Drosophila. Genome Res 28, 1688–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Copur Ö et al. (2018) Sex-specific phenotypes of histone H4 point mutants establish dosage compensation as the critical function of H4K16 acetylation in Drosophila. Proc. Natl. Acad. Sci 115, 13336–13341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nozawa R-S et al. (2013) Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat. Struct. Mol. Biol 20, 566. [DOI] [PubMed] [Google Scholar]
- [32].Urban J et al. (2017) Enhanced chromatin accessibility of the dosage compensated Drosophila male X-chromosome requires the CLAMP zinc finger protein. PLoS One 12, e0186855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dekker J et al. (2002) Capturing chromosome conformation. Science (80-. ) 295, 1306–1311 [DOI] [PubMed] [Google Scholar]
- [34].Simonis M et al. (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nat. Genet 38, 1348. [DOI] [PubMed] [Google Scholar]
- [35].Lieberman-Aiden E et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-. ) 326, 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rao SSP et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schwartzman O et al. (2016) UMI-4C for quantitative and targeted chromosomal contact profiling. Nat. Methods 13, 685. [DOI] [PubMed] [Google Scholar]
- [38].Van De Werken HJG et al. (2012) Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat. Methods 9, 969. [DOI] [PubMed] [Google Scholar]
- [39].Engreitz JM et al. (2013) The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science (80-. ) 341, 1237973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Giorgetti L et al. (2016) Structural organization of the inactive X chromosome in the mouse. Nature 535, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Deng X et al. (2015) Bipartite structure of the inactive mouse X chromosome. Genome Biol 16, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Darrow EM et al. (2016) Deletion of DXZ4 on the human inactive X chromosome alters higher-order genome architecture. Proc. Natl. Acad. Sci 113, E4504–E4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Crane E et al. (2015) Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature DOI: 10.1038/nature14450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Schauer T et al. (2017) Chromosome topology guides the Drosophila Dosage Compensation Complex for target gene activation. EMBO Rep 18, 1854–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vielle A et al. (2012) H4K20me1 contributes to downregulation of X-linked genes for C. elegans dosage compensation. PLoS Genet 8, e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wells MB et al. (2012) Caenorhabditis elegans Dosage Compensation Regulates Histone H4 Chromatin State on X Chromosomes. Mol. Cell. Biol 32, 1710–1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Barr ML and Bertram EG (1949) A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. In Problems of Birth Defects pp. 101–102, Springer; [DOI] [PubMed] [Google Scholar]
- [48].Gdula MR et al. (2019) The non-canonical SMC protein SmcHD1 antagonises TAD formation and compartmentalisation on the inactive X chromosome. Nat. Commun 10, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Deng X et al. (2013) Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Dev. Cell 25, 55–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Buenrostro JD et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang Z and Pugh BF (2011) High-resolution genome-wide mapping of the primary structure of chromatin. Cell 144, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tolstorukov MY et al. (2010) Analysis of the primary structure of chromatin with next-generation sequencing. Epigenomics 2, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mieczkowski J et al. (2016) MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun 7, 11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Skene PJ and Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Griffiths JA et al. (2018) Using single‐cell genomics to understand developmental processes and cell fate decisions. Mol. Syst. Biol 14, e8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cusanovich DA et al. (2015) Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science (80-. ) 348, 910–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Buenrostro JD et al. (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zong C et al. (2012) Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science (80-. ) 338, 1622–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rotem A et al. (2015) Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Farlik M et al. (2015) Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep 10, 1386–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nagano T et al. (2013) Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Beliveau BJ et al. (2015) Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun 6, 7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chen X et al. (2016) ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat. Methods 13, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]