

Abstract
Chloramphenicol nitroreductase (CNR), a drug-modifying enzyme from Haemophilus influenzae, has been shown to be responsible for the conversion of the nitro group into an amine in the antibiotic chloramphenicol (CAM). Since CAM structurally bears a 4-nitrobenzene moiety, we explored the substrate promiscuity of CNR by investigating its nitroreduction of 4-nitrobenzyl derivatives. We tested twenty compounds containing a nitrobenzene core, two nitropyridines, one compound with a vinylogous nitro group, and two aliphatic nitro compounds. In addition, we also synthesized twenty-eight 4-nitrobenzyl derivatives with ether, ester, and thioether substituents and assessed the relative activity of CNR in their presence. We found several of these compounds to be modified by CNR, with the enzyme activity ranging from 1-150% when compared to CAM. This data provides insights into two areas: (i) chemoenzymatic reduction of select compounds to avoid harsh chemicals and heavy metals routinely used in reductions of nitro groups and (ii) functional groups that would aid CAM in overcoming the activity of this enzyme.
Keywords: Antibiotic, Bacterial enzyme, Chemoenzymatic deprotection, Drug-modifying enzyme, Nitroreduction
Graphical Abstract
Chloramphenicol (CAM) (Scheme 1) is an antibiotic with a wide spectrum of activity against Gram-positive and Gram-negative cocci and bacilli.1 It is primarily bacteriostatic and works by binding to specific residues of the 23S rRNA on the 50S subunit of the bacterial ribosome,2, 3 disrupting the action of the peptidyltransferase enzyme and leading to the inhibition of important biological functions such as peptide bond formation,4 termination of translation,5 and translational accuracy.6 However, treatment with CAM may be accompanied by deleterious side effects notably neurotoxicity,7 bone marrow depletion and aplastic anemia,8 and as a result, its use is often limited to topical ophthalmic infections and other serious infections when other suitable drugs are unavailable, such as meningitis caused by Haemophilus influenzae, Streptococcus pneumoniae, and Neisseria meningitides.9 The clinical use of CAM has also been hampered by the rapid emergence of resistant bacterial strains. Mechanisms of resistance include reduced bacterial membrane permeability towards CAM uptake,10, 11 efflux pumps that decrease CAM concentration in the bacterial cell,12 and mutations/modifications of the ribosomal target.13-17 Another common mechanism of resistance to CAM is its enzymatic modification by CAM acetyltransferases (CAT), CAM phosphotransferases (CPT),18-20 CAM hydrolase,21 and CAM nitroreductase (CNR).22, 23
Scheme 1.

Structure of chloramphenicol (CAM) and schematic representation of its reduction by the chloramphenicol nitroreductase (CNR) resistance enzyme.
Antibiotic resistance by drug inactivation/modification is not specific to CAM; it is also present in other families of antibacterials. For instance, aminoglycosides (AGs), another well-known class of antibiotics, have suffered from the emergence of aminoglycoside-modifying enzymes (AMEs).24 Indeed, more than 100 AMEs have been identified,25 threatening the long-term use of AGs. These AMEs include AG acetyltransferases (AACs), nucleotidyltransferases (ANTs), and phosphotransferases (APHs). AACs catalyze the N-acetylation of AGs, while ANTs and APHs catalyze the transfer of phosphate and adenosine phosphate moieties, respectively, to hydroxyl groups of AGs.26 We have previously reported a methodology that couples AACs with analogues of acetyl coenzyme A cosubstrate. Specifically, acyl derivatives of coenzyme A (CoA) were chemically synthesized and served as AAC cosubstrates in the chemoenzymatic generation of N-acylated AG analogues.27 We also expanded the substrate promiscuity of two AACs by exploring their structural modification of a variety of AGs.28, 29 Substrate promiscuity is documented with multiple drug-modifying enzymes including β-lactamases,30, 31 kinases,32, 33 dethiobiotin synthetase,34 acetyltransferases,35-37 and O-nucleotidyltransferases.38, 39
Our group has been engaged in the investigation of CAM resistance enzymes. We have determined the crystal structures of CATI in its unbound and CAM-bound forms, enabling us to further understand the broad substrate specificity of one of the most prevalent types of CATs.40 We have also developed homo- and heterodimers of CAM and examined their susceptibility to enzymatic modifications by CAT and CPT. Although CAT and CPT have been extensively studied, CNR remains underexplored. Because of our long-standing interest in CAM resistance enzymes, we decided to investigate the CNR from H. influenzae,41 recombinantly expressed in Escherichia coli. Here, we investigate its properties at different pHs, at different temperatures, and determine the kinetic parameters of CNR with CAM. Structurally, CAM contains a 4-nitrobenzene moiety, and a recent study has shown that CNR was able to reduce the nitro group of CAM to an amine (Scheme 1). To that end, we (i) examined some commercially available nitro-containing molecules to assess the versatility of this resistance enzyme and (ii) synthesized a series of 4-nitrobenzyl derivatives to explore the promiscuity of the CNR enzyme. A better understanding of the CNR substrate promiscuity is important to (a) give indications as to what modifications of CAM would help alleviate the action of this enzyme, and (b) potentially develop CNR as a chemoenzymatic tool.
Prior to testing any compounds with CNR, we determined the pH and temperature at which the CNR enzyme is most active. The reaction of CAM with CNR was first tested at various pHs ranging from pH 3.0 to pH 9.0 using either citrate, phosphate, or Tris buffer in their appropriate pH ranges (Fig. S57). We established that the CNR enzyme is most active at pH 8.0. From this point on, we used pH 8.0 with 50 mM Tris-HCl as this was one of the reactions with the highest rate of reaction, and Tris-HCl had less variability in the pH range tested (pH 6.8-9.0) than phosphate in the similar pH range (6.0-8.0). With an optimum pH established, we next investigated the optimal temperature for CNR activity (Fig. S58). Again, the reaction rates of CAM with CNR at 21, 25, 30, 37, 42, 50, and 60 °C were determined. From these data we observed that, surprisingly, the optimal temperature for the enzyme is 50 °C, despite the fact that H. influenzae grows optimally at 37 °C. To reconcile this discrepancy, we tested the CNR enzyme at these two temperatures in the next set of experiments.
To establish the effect of temperature on the kinetic parameters of CNR, we next determined the Km and kcat values for CAM with the enzyme at both 37 and 50 °C, the optimal temperature for growth of H. influenzae and that for CNR activity, respectively (Fig. 1). The binding constants (Km) of CAM to CNR at both temperatures were on the same order of magnitude (273 ± 87 μM at 37 °C and 191 ± 48 μM at 50 °C). The catalytic turnover (kcat) values were nearly identical at both temperatures (61 ± 6 s−1 at 37 °C and 65 ± 5 s−1 at 50 °C). As the Km and kcat values were similar at both temperatures, so were the catalytic efficiencies (kcat/Km) (0.22 ± 0.07 s−1μM−1 at 37 °C and 0.34 ± 0.09 s±1μM−1 at 50 °C). Based on these data we decided there was no disadvantage to using 37 °C over 50 °C and therefore performed the rest of the experiments at 37 °C.
Fig. 1.

Michaelis-Menten curves for the kinetic parameters of CNR with CAM as a substrate at A. 37 °C and B. 50 °C.
With the optimized conditions for CNR activity, we determined the initial rate (first 5 min) of nitroreduction of twenty-five commercially available nitro containing compounds (1-25, Fig. 2) by UV-visible assays (Fig. 3A). The compounds consisted of two aliphatic nitro-containing compounds, one vinylogous nitro-containing molecule, twenty nitrobenzene analogues, and two nitropyridine compounds. Most of the aromatic purchased compounds contained the nitro group in para-position to better compare with CAM, which also has its nitro group in that position. A few compounds were selected to be aliphatic or aromatic with their nitro group located in ortho- or meta- of other functionalities in order to preliminarily establish if para-substitution on aromatic compounds was required for CNR activity. The initial rate of CNR was set to 100% for the known substrate CAM for comparison purposes. From these 25 molecules, we found that 8 compounds (n-nitrohexane (1), nitrocyclohexane (2), nitrobenzene (4), 3,4-dichloronitrobenzene (5), 4-nitrobenzyl bromide (12), 4-nitroaniline (18), 4-fluoro-2-nitroaniline (19), and (3,5-dimethyl-4-nitro-2-pyridyl)-1-methanol (25)) were not modified at all by CNR. More promisingly, we also observed that 10 compounds (nitrofurantoin (3), 4-nitrobenzenesulfonamide (7), 4-nitrophenol (8), 2-nitrobenzaldehyde (13), 4-nitrobenzaldehyde (14), 5,5’-dithiobis(2-nitrobenzoic acid) (17), 2-nitro-4,5-difluoroaniline (20), 1-(4-nitrophenyl)piperidine (22), 5-nitroindole (23), and 4-nitropyridine (24)) showed a CNR initial rate between 3.5% and 40% when compared to that of CAM. Additionally, 3 compounds (2-nitrobenzesulfonamide (6), 4-nitrophenylbutyrate (10), and 2-nitro-4,5-dichloroaniline (21)) all had initial rates between 77% and 88% compared to that of CAM. Finally, and more interestingly, 4 compounds (3-(4-nitrophenoxy)propionic acid (9), 4-nitro benzyl alcohol (11), 6-nitrovetraldehyde (15), and 2-nitronaphthaldehyde (16)) reacted with initial rates greater than that of CAM; 157%, 122%, 132%, and 125%, when compared to 100% for CAM, respectively. From these data we can deduce a few criteria for the CNR: (i) the enzyme does not react well with aliphatic nitro compounds, and (ii) inserting a nitrogen atom into the aromatic system significantly harms the ability of the enzyme to reduce the compound effectively. Looking at the substituents located para to the nitro group in three (9-11) of the seven best compounds (6, 9, 10, 11, 15, 16, and 21) in which only a para-substituent exist, we observed that an ether, an ester, or an hydroxymethyl group directly attached to the aryl group allow for the activity of the CNR to be retained. When looking at compounds 6 and 7, we find that a sulfonamide in ortho of the nitro group is well tolerated, whereas the same sulfonamide is not as well tolerated at the para-position. With compounds 15, 16, and 21, we find that having additional substituents at the ortho-, meta-, and para-positions of the aryl ring can be well tolerated by CNR. All combined, these data further confirm the promiscuity of the CNR enzyme. From these data we can also conclude that substituting the phenyl ring of CAM with a pyridyl group could help reduce the action of CNR on the antibiotic. Additionally, increasing the number of substitutents of the phenyl ring of CAM may have a similar effect
Fig. 2:

Structures of commercially available nitro-containing molecules tested with CNR.
Fig. 3.

CNR activity against A. the commercially available nitro-containing molecules tested in this study and B. the p-nitrobenzyl derivatives generated in this study. Note: The exact values used to generate this figure are presented in Table S1.
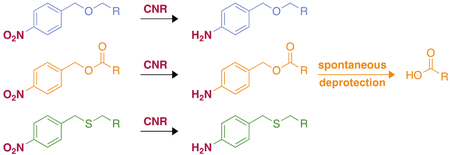
Knowing how the purchased nitro-containing compounds behaved with CNR, we next examined the ability of the reductase to reduce nitro groups on synthetically generated compounds similar to CAM in structure. To investigate the broad application of CNR for nitro reduction and potential deprotection, we synthesized three main classes of 4-nitrobenzyl derivatives, notably 4-nitrobenzyl ethers, esters, and thioethers (Scheme 2 and Fig. S59). With these synthesized compounds, we wanted to establish the effect of a benzylic oxygen atom, either in an ether (compounds from series 26) or ester (compounds from series 27) linkage, as well as that of a benzylic sulfur atom in a thioether linkage (compounds from series 28) on the reduction activity of CNR. Various esters of CAM have previously been designed as potential prodrugs with useful oral administration,42 making this functional group an interesting choice in the design of CAM derivatives. Furthermore, CAM heterodimers, in which CAM was linked to another antibiotic (neomycin B, tobramycin, or clindamycin) through an ether or thioether linkage, exhibited improved antimicrobial properties,23, 43 encouraging us to include ether and thioether functional groups in our study.
Scheme 2.

Synthetic schemes for the preparation of A. 4-nitrobenzyl ether derivatives 26a,b,d-k,o,q, B. 4-nitrobenzyl ester derivatives 27c-i,k-n, and C. 4-nitrobenzyl thioether derivatives 28g-i,k,p.
Briefly, the syntheses of all compounds were performed as follows. The 4-nitrobenzyl ethers 26a,b,d-k,o,q were obtained in 3-71% yields by heating 4-nitrobenzylbromide at reflux with the corresponding alcohols in the presence of silver oxide (Scheme 2A). The alcohols utilized in this study included aliphatic (ethyl and propyl), cyclic (cyclobutylmethyl, cyclopentylmethyl, cyclohexylmethyl, and cyclopentyl), and aromatic (benzyl, 4-bromobenzyl, 4-chlorobenzyl, 4-nitrobenzyl, 4-chloro-2-fluorobenzyl, and phenyl) alcohols. The 4-nitrobenzyl esters 27c-i,k-n, on the other hand, were prepared by heating 4-nitrobenzylbromide with the corresponding carboxylic acids in the presence of potassium carbonate and isolated in 35-88% yields (Scheme 2B). The substituents included propyl, cyclohexylmethyl, cyclobutyl, cyclopentyl, cyclohexyl, 1-adamantyl, phenyl, 4-bromophenyl, 4-chlorophenyl, 4-chloro-2-fluorophenyl, and lithocholyl groups. Finally, refluxing an ethanolic solution of 4-nitrobenzylbromide with benzyl mercaptan, 4-bromobenzyl mercaptan, 4-chlorobenzyl mercaptan, 4-chloro-2-fluorobenzyl mercaptan, or cyclohexanethiol afforded the 4-nitrobenzyl thioethers 28g-i,k,p in 26-73% yields (Scheme 2C). The choice of substituents ranged from a small-sized ethyl group to a bulky litocholyl group allowing us to investigate the steric effect of these substituents on nitroreduction. For complete experimental protocols and characterization of all compounds synthesized, please see the Supporting Information.
As with the 25 purchased compounds (1-25), we evaluated the initial rate (first 5 min) of nitroreduction of compounds 26a,b,d-k,o,q, 27c-i,k-n, and 28g-i,k,p by CNR by UV-visible assays (Fig. 3B and Table S1). The initial rate of CNR was set to 100% for the known substrate CAM for comparison purposes. Prior to performing an in-depth SAR analysis, we first looked for general trends. Overall, compounds 26b, 27e, 27g, 27h, 27k, and 271 showed no modification (0% activity) by CNR at this time interval, while compounds 26d, 26e, 26f, 26g, 26j, 26q, 27c, 27d, 27f, 27i, and 27n were modified at a rate that was less than 50% the rate of CAM (Fig. 3B). With the exception of compounds 26a (56%), 26o (67%), and 28p (62%), all other compounds (26h, 26i, 26k, 27m, 28g, 28h, 28i, and 28k) showed reactions that were similar or faster than that of CAM. Before taking a closer look at the structures of the nitro compounds, it is safe to say that most ester-based compounds did not show a significant conversion in the time monitored with the exception of compound 27m, the adamantly ester. This compound seemed to work slightly better (120%) than CAM.
We next took a close look at the SAR, by comparing the various R groups within a same linkage type (e.g., ether, ester, or thioether). Looking more closely at the R groups of the ethers, propyl (26b, 0%), methylcyclobutyl (26d, 7.5%), methylcyclopentyl (26e, 21%), methylcyclohexyl (26f, 7.0%), benzyl (26g 29%), and 4-nitrobenzyl (26j, 21%) were all very poor substrates. On the contrary ethyl (26a, 56%), 4-bromobenzyl (26h, 116%), 4-chlorobenzyl (26i, 90%), 4-chloro-2-fluorobenzyl (26k, 88%), cyclopentyl (26o, 67%), and phenyl (26q, 45%) were all good to moderate substrates. Based on these data it would seem that any longer or distal cyclic alkyl groups, bulky groups, or electron-withdrawing groups had a significant impact on the reduction activity of the enzyme when reducing the 4-nitrobenzyl ether compounds tested here. In the case of the ester linkage, we interestingly observed that only the adamantyl moiety of 27m resulted in a compound in which the nitro group was easily reduced by CNR. These data indicate that an ester linkage is detrimental to CNR activity. The thioether compounds (28) had less variability with only the cyclohexyl compound (28p, 62%) showing a reduced initial rate compared to CAM.
We finally performed a more in-depth SAR analysis by comparing compounds with the same R groups but different linkage types. By doing so, we wanted to answer the question: Is one type of linkage favored over another when it comes to nitro reduction by CNR? We compared three pairs of ethers and esters (26d and 27d with R = cyclobutyl; 26e and 27e with R = cyclopentyl; 26f and 27f with R = cyclohexyl), and found them to be all poor CNR substrates. In all cases, the reduction of the nitro group in ether-containing molecules (26) was faster than (d and e) or equal to (f) the reduction of the nitro group in ester-containing compounds (27). This is further validation that the ester is not the best linkage for CNR activity. We also compared four groups with identical R chains (g, h, i, and k) in all three families (26, 27, and 28). For the compounds with R = benzyl (g), we observed that the thioether linkage in 28g was much more conducive to CNR activity than the ether linkage in 26g, which itself was better than the ester in 27g. When examining the other three R groups (h, i, and k), we found that both the ether (26) and thioether (28) linkages were conducive to CNR activity, while the ester linkage (27) was not. Overall, we conclude that the best linkage for the reduction of these molecules is the thioether, followed closely by the ether, and lastly by the ester.
Now knowing that CNR could reduce a variety of nitro-containing compounds, we wanted to determine if CNR could reduce the nitro group in the synthesized molecules to an amine, and if the resulting 4-aminoether/thioether/ester would then spontaneously collapse to the corresponding alcohol, thiol, or benzoic acid. Bioreduction of this type has been seen before in an E. coli NADH-dependent nitroreductase.44 To assess the product of the reactions we followed the protocol published for a similar reductase.45 Large-scale reactions were performed using CNR to reduce three compounds (26h, 27h, and 28h), representative of each family synthesized with the same 4-bromophenyl group. The reactions were incubated overnight at 37 °C. After a full 24 h of incubation, the reaction mixtures were extracted into EtOAc to remove NADP(H) and other biomolecules from the reactions. The residues from the extractions were analyzed by LCMS. The LCMS trace was scanned for the possible reduction analogues, the amine, and the free alcohol/thiol/carboxylic acid (reduction and deprotection masses are shown in Table 1). In general, we observed a reduction of the 4-nitrobenzyl ether (26h) and the 4-nitrobenzyl thioether (27h) to the corresponding 4-aminoether and 4-aminothioether (Table 1 and Fig. S60). In the original publication documenting the reduction of CAM,41 the authors predicted that the nitro group went through the nitrosyl (N=O) moiety, and the hydroxylamine (NHOH), along with other intermediates, before reaching the final amine. While analyzing the MS data for the large-scale reaction of compound 26h, in addition to the reduced compound, we were also able to see a peak congruent with the hydroxylamine derivative. This observation confirms the existence of the hydroxylamine intermediate originally proposed for CAM and opens the possibility of this enzyme to reduce hydroxylamines. The 4-nitrobenzyl ester (28h), which did not display detectable initial rate with CNR by UV-visible assays, surprisingly, was not only reduced, but the compound spontaneously collapsed to yield the corresponding benzoic acid. While this data does not agree with the UV-visible assay, the length of incubation is significantly different. To confirm that the observation of the carboxylic acid is real and not an artifact of the mass spectrometry experiments, samples of the reaction were injected onto RP-HPLC at various times during the reaction to monitor the progression. A significant new peak was not seen until 3 h, which agrees with a decrease in the signal from the starting material (Fig. S61). In an effort to confirm that this is a general trend for the esters and not specific to compound 27h, we also investigated by LCMS the 4-chloro-2-fluoro- (27k) and 4-chloro-substituted (27i) derivatives and observed the corresponding benzoic acids for these compounds. The 4-nitrobenzyl group is a common protecting group for carboxylic acids. Traditionally, the 4-nitrobenzyl ester, when used as a protecting group, is removed by electrolysis, SnCl2, sodium dithionite, or catalytic hydrogenation with Pd-C.46 CNR uses no metals or harsh chemicals, just NADPH to reduce this nitro group to the amine. The instability of 4-aminobenzyl esters leads them to spontaneous deprotection. While further studies are needed to fully understand the scope of this CNR enzyme, this is a novel method of completely removing the 4-nitrobenzyl protections on carboxylic acids.
Table 1:
Masses (m/z) observed in the large-scale reactions with CNR for various compounds.
| Compound | m/z observed | m/z calculated for reduction | m/z calculated for deprotection |
|---|---|---|---|
| 26h | 291.0a | 291.0 | 185.0 |
| 27h | 199.0b | 305.0 | 198.9 |
| 27i | 155.1b | 261.1 | 155.0 |
| 27k | 173.0b | 279.1 | 173.0 |
| 28h | 308.0c | 307.0 | 200.9 |
positive mode, M+ ;
negative mode, [M-H]−;
positive mode, [M+H]+.
In conclusion, we have described the synthesis of twenty-eight 4-nitrobenzyl derivatives and evaluated the ability of CNR to modify these compounds and an additional twenty-five commercially available nitro-containing compounds. While the substrate preference of CNR towards the synthesized 4-nitrobenzyl derivatives varied across the range, we found that CNR could reduce many of the tested nitro-containing molecules with different functionalities. We also discovered that CNR could be used to completely remove the 4-nitrobenzyl protecting group from carboxylic acids. This opens the door to the potential use of CNR as a chemoenzymatic tool for the preparation of arylamines or deptrotection of 4-nitrobenzyl protected acids.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (NIH) AI090048 (to S.G.-T.) and by startup funds from the University of Kentucky College of Pharmacy (to S.G.-T.).
Abbreviations
- AG(s)
aminoglycoside(s)
- AAC
aminoglycoside acetyltransferase
- AME
aminoglycoside-modifying enzyme
- ANT
aminoglycoside nucleotidyltransferase
- APH
aminoglycoside phosphotransferase
- CAM
chloramphenicol
- CAT
chloramphenicol acetyltransferase
- CNR
chloramphenicol nitroreductase
- CoA
coenzyme A
- CPT
chloramphenicol phosphotransferase
- EtOAc
ethyl acetate
- Et2o
diethyl ether
- LCMS
liquid chromatography-mass spectrometry
- MeOH
methanol
- NADP(H)
β-nicotinamide-adenine dinucleotide phosphate (reduced)
- NMR
nuclear magnetic resonance
- RP-HPLC
reversed-phase high pressure liquid chromatography
- TLC
thin-layer chromatography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting information available: The Supporting Information is available free of charge. Copies of 1H and 13C NMR spectra for all compounds synthesized are provided in Figs. S1-S56. The graphs for optimization of pH (Fig. S57) and temperature (Fig. S58), chemical structures of the synthesized nitro-containing molecules tested (Fig. S59), mass spectra of crude extract of large-scale CNR reduction reaction (Fig. S60), as well as HPLC traces for time course of large-scale reduction of compound 26h with CNR (Fig. S61) are also provided. A Table (Table S1) of the numerical values used to generate Fig. 3 is also presented.
The authors declare no conflict of interest.
References
- 1.Neu HC; Fu KP In vitro chloramphenicol and thiamphenicol analogs. Antimicrob. Agents Chemother 1980, 18, 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunkle JA; Xiong L; Mankin AS; Cate JH Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci., U. S. A 2010, 107, 17152–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlünzen F; Zarivach R; Harms J; Bashan A; Tocilj A; Albrecht R; Yonath A; Franceschi F Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001, 413, 814–821. [DOI] [PubMed] [Google Scholar]
- 4.Xaplanteri MA; Andreou A; Dinos GP; Kalpaxis DL Effect of polyamines on the inhibition of peptidyltransferase by antibiotics: revisiting the mechanism of chloramphenicol action. Nucleic Acids Res 2003, 31, 5074–5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polacek N; Gomez MJ; Ito K; Xiong L; Nakamura Y; Mankin A The critical role of the universally conserved A2602 of 23S ribosomal RNA in the release of the nascent peptide during translation termination. Mol. Cell 2003, 11, 103–112. [DOI] [PubMed] [Google Scholar]
- 6.Thompson J; O’Connor M; Mills JA; Dahlberg AE The protein synthesis inhibitors, oxazolidinones and chloramphenicol, cause extensive translational inaccuracy in vivo. J. Mol. Biol 2002, 322, 273–279. [DOI] [PubMed] [Google Scholar]
- 7.Tirosh O; Sen CK; Roy S; Packer L Cellular and mitochondrial changes in glutamate-induced HT4 neuronal cell death. Neuroscience 2000, 97, 531–541. [DOI] [PubMed] [Google Scholar]
- 8.Eliakim-Raz N; Lador A; Leibovici-Weissman Y; Elbaz M; Paul M; Leibovici L Efficacy and safety of chloramphenicol: joining the revival of old antibiotics? Systematic review and meta-analysis of randomized controlled trials. J. Antimicrob. Chemother 2015, 70, 979–996. [DOI] [PubMed] [Google Scholar]
- 9.Balbi HJ Chloramphenicol. Pediatr. Rev 2004, 25, 284–288. [DOI] [PubMed] [Google Scholar]
- 10.Burns JL; Hedin LA; Lien DM Chloramphenicol resistance in Pseudomonas cepacia because of decreased permeability. Antimicrob. Agents Chemother 1989, 33, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delcour AH Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 2009, 1974, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daniels C; Ramos JL Adaptive drug resistance mediated by root-nodulation-cell division efflux pumps. Microbiol. Infect 2009, 15 (Suppl. I), 32–36. [DOI] [PubMed] [Google Scholar]
- 13.Kowalak JA; Bruenger E; McCloskey JA Posttranscriptional modification of the central loop of domain V in Escherichia coli 23S ribosomal RNA. J. Biol. Chem 1995, 270, 17758–17764. [DOI] [PubMed] [Google Scholar]
- 14.Persaud C; Lu Y; Vila-Sanjurjo A; Campbell JL; Finley J; O’Connor M Mutagenesis of the modified bases, m5 U1939 and 2504, in Escherichia coli 23S rRNA. Biochem. Biophys. Res. Commun 2010, 392, 223–227. [DOI] [PubMed] [Google Scholar]
- 15.Giessing AMB; Jensen SK; Rasmussen A; Hansen LH; Gondela A; Long K; Vester B; Kirpekar F Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA 2009, 15, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montero CI; Johnson MR; Chou C-J; Conners SB; Geouge SG; Tachdjian S; Nichols JD; Kelly RM Responses of wild-type and resistant strains of hyperthermophilic bacterium Thermotoga maritima to chloramphenicol challenge. Appl. Environ. Microbiol 2007, 73, 5058–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kehrenberg C; Schwarz S; Jacobsen L; Hansen LH; Vester B A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol. Microbiol 2005, 57, 1064–1073. [DOI] [PubMed] [Google Scholar]
- 18.Mosher RH; Camp DJ; Yang K; Brown MP; Shaw WV; Vining LC Inactivation of chloramphenicol by O-phosphorylation. A novel resistance mechanism in Streptomyces venezuelae ISP5230, a chloramphenicol producer. J. Biol. Chem 1995, 270, 27000–27006. [DOI] [PubMed] [Google Scholar]
- 19.Izard T; Ellis J The crystal structures of chloramphenicol phosphotransferase reveal a novel inactivation mechanism. EMBO J. 2000, 19, 2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rajesh T; Sung C; Kim H; Song E; Park HY; Jeon JM; Yoo D; Kim HJ; Kim YH; Choi KY; Song KG; Yang YH Phosphorylation of chloramphenicol by a recombinant protein Yhr2 from Streptomyces avermitilis MA4680. Bioorg. Med. Chem. Lett 2013, 23, 3614–3619. [DOI] [PubMed] [Google Scholar]
- 21.Tao W; Lee MH; Wu J; Kim NH; Kim JC; Chung E; Hwang EC; Lee SW Inactivation of chloramphenicol and florfenicol by a novel chloramphenicol hydrolase. Appl. Environ. Microbiol 2012, 78, 6295–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz S; Kehrenberg C; Doublet B; Cloeckaert A Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol. Rev 2004, 28, 519–542. [DOI] [PubMed] [Google Scholar]
- 23.Berkov-Zrihen Y; Green KD; Labby KJ; Feldman M; Garneau-Tsodikova S; Fridman M Synthesis and evaluation of hetero- and homodimers of ribosome-targeting antibiotics: antimicrobial activity, in vitro inhibition of translation, and drug resistance. J. Med. Chem 2013, 56, 5613–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Labby KJ; Garneau-Tsodikova S Strategies to overcome the action of aminoglycoside-modifying enzymes for treating resistant bacterial infections. Future Med. Chem 2013, 5, 1285–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez MS; Tolmasky ME Aminoglycoside modifying enzymes. Drug Resist. Update 2010, 13, 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fosso MY; Li Y; Garneau-Tsodikova S New trends in the use of aminoglycosides. MedChemComm 2014, 5, 1075–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Green KD; Chen W; Houghton JL; Fridman M; Garneau-Tsodikova S Exploring the substrate promiscuity of drug-modifying enzymes for the chemoenzymatic generation of N-acylated aminoglycosides. ChemBioChem 2010, 11, 119–126. [DOI] [PubMed] [Google Scholar]
- 28.Green KD; Chen W; Garneau-Tsodikova S Effects of altering aminoglycoside structures of bacterial resistance enzymes. Antimicrob. Agents Chemother 2011, 55, 3207–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holbrook SYL; Garneau-Tsodikova S Expanding aminoglycoside resistance enzyme regiospecificity by mutation and truncation. Biochemistry 2016, 55, 5726–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Risso VA; Gavira JA; Majia-Carmona DF; Gaucher EA; Sanchez-Ruiz JM Hyperstability and substrate promiscuity in laboratory resurrections of precambrian β-lactamases. J. Am. Chem. Soc 2013, 135, 2899–2902. [DOI] [PubMed] [Google Scholar]
- 31.Kim Y; Cunningham MA; Mire J; Tesar C; Sacchettini J; Jaochimiak A NDM-1, the ultimate promiscuous enzyme: substrate recognition and catalytic mechanism. FASEB J. 2013, 27, 1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mabanglo MF; Serohijos AW; Poulter CD The Streptomyces-produced antibiotic fosfomycin is a promiscuous substrate for archaeal isopentenyl phosphate kinase. Biochemistry 2012, 51, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fong DH; Berghuis AM Substrate promiscuity of an aminoglycoside antibiotic resistance enzyme via target mimicry. EMBO J. 2002, 21, 2323–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salaemae W; Yap MY; Wegener KL; Booker GW; Wilce MC; Polyak SW Nucleotide triphosphate promiscuity in Mycobacterium tuberculosis dethiobiotin synthetase. Tuberculosis 2015, 95, 259–266. [DOI] [PubMed] [Google Scholar]
- 35.Norris AL; Serpersu EH Antibiotic selection by the promiscuous aminoglycoside acetyltransferase-(3)-IIIb is thermodynamically achieved through the control of solvent rearrangement. Biochemistry 2010, 50, 9309–9317. [DOI] [PubMed] [Google Scholar]
- 36.Green KD; Fridman M; Garneau-Tsodikova S hChAT: a tool for the chemoenzymatic generation of potential acetyl/butyrylcholinesterase inhibitors. ChemBioChem 2009, 10, 2191–2194. [DOI] [PubMed] [Google Scholar]
- 37.Green KD; Porter VR; Zhang Y; Garneau-Tsodikova S Redesign of cosubstrate specificity and identification of important residues for substrate binding to hChAT. Biochemistry 2010, 49, 6219–6227. [DOI] [PubMed] [Google Scholar]
- 38.Porter VR; Green KD; Zolova OE; Houghton JL; Garneau-Tsodikova S Dissecting the cosubstrate structure requirements of the Staphylococcus aureus aminoglycoside resistance enzyme ANT(4'). Biochem. Biophys. Res. Commun 2010, 403, 85–90. [DOI] [PubMed] [Google Scholar]
- 39.Green KD; Garneau-Tsodikova S Domain dissection and characterization of the aminoglycoside resistance enzyme ANT(3")-Ii/AAC(6')-IId from Serratia marcescens. Biochimie 2013, 95, 1319–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas T; Houghton JL; Garneau-Tsodikova S; Tsodikov OV The structural basis for substrate versatility of chloramphenicol acetyltransferase CATI. Protein Sci 2012, 21, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith AL; Erwin AL; Kline T; Unrath WC; Nelson K; Weber A; Howald WN Chloramphenicol is a substrate for a novel nitroreductase pathway in Haemophilus influenzae. Antimicrob. Agents Chemother 2007, 51, 2820–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ambrose PJ Clinical pharmacokinetics of chloramphenicol and chloramphenicol succinate. Clin. Pharmacokinet 1984, 9, 222–238. [DOI] [PubMed] [Google Scholar]
- 43.Kwon M; Kim H-J; Lee J; Yu J Enhanced binding affinity of neomycin-chloramphenicol (or linezolid) conjugates to A-Site model of 16S ribosomal RNA. Bull. Korean Chem. Soc 2006, 27, 1664–1666. [Google Scholar]
- 44.Saneyoshi H; Hiyoshi Y; Iketani K; Kondo K; Ono A Bioreductive deprotection of 4-nitrobenzyl group on thymine base in oligonucleotides for the activation of duplex formation. Bioorg. Med. Chem. Lett 2015, 25, 5632–5635. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen-Tran HH; Zheng GW; Qian XH; Xu JH Highly selective and controllable synthesis of arylhydroxylamines by the reduction of nitroarenes with an electron-withdrawing group using a new nitroreductase BaNTR1. Chem. Commun 2014, 50, 2861–2864. [DOI] [PubMed] [Google Scholar]
- 46.Wuts PGM; Greene TW Greene’s protective groups in organic synthesis, 4th Ed. 4th ed.; Wiley-Interscience: New Jersey, 2007; p 1082. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.