

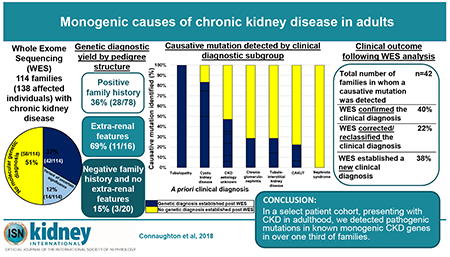
Abstract
Approximately 500 monogenic causes of chronic kidney disease (CKD) have been identified, mainly in pediatric populations. The frequency of monogenic causes among adults with CKD has been less extensively studied. To determine the likelihood of detecting monogenic causes of CKD in adults presenting to nephrology services in Ireland, we conducted whole exome sequencing (WES) in a multi-centre cohort of 114 families including 138 affected individuals with CKD. Affected adults were recruited from 78 families with a positive family history, 16 families with extra-renal features, and 20 families with neither a family history nor extra-renal features. We detected a pathogenic mutation in a known CKD gene in 42 of 114 families (37%). A monogenic cause was identified in 36% of affected families with a positive family history of CKD, 69% of those with extra-renal features, and only 15% of those without a family history or extra-renal features. There was no difference in the rate of genetic diagnosis in individuals with childhood versus adult onset CKD. Among the 42 families in whom a monogenic cause was identified, WES confirmed the clinical diagnosis in 17 (40%), corrected the clinical diagnosis in 9 (22%), and established a diagnosis for the first time in 16 families referred with CKD of unknown etiology (38%). In this multi-centre study of adults with CKD, a molecular genetic diagnosis was established in over one-third of families. In the evolving era of precision medicine, WES may be an important tool to identify the cause of CKD in adults.
Keywords: chronic kidney disease, genetic kidney disease, whole exome sequencing
INTRODUCTION
The estimated global prevalence of chronic kidney disease (CKD) is 11 to 13%.1 CKD is associated with high morbidity and resource utilisation.2 Mounting evidence highlights the urgency for early diagnosis and intervention, to stem the sequelae of elevated cardiovascular risk and to delay progression to end stage kidney disease (ESKD).3 Monogenic causes of CKD in childhood are well established4, whereas very little data exists on monogenic causation of CKD in adults. In 34% of adults with CKD a positive family history is reported which suggests genetic causation.5–7 However, genetic testing for adults is still not routinely performed in clinical practice. Panel sequencing of CKD genes directed towards specific diagnostic groups has revealed a genetic disorder in up to 43% of patients.8 Using whole exome sequencing (WES), a single centre study demonstrated that a monogenic disease-causing gene can be identified in 24% of adults with CKD.9
In this study, we aim to determine the contribution of monogenic CKD genes in an Irish adult cohort with CKD. We hypothesise that genetic causes of CKD in adults are under-recognised, particularly in patients with a positive family history of CKD or presence of extra-renal features. Employing WES in patients with familial nephropathy or extra-renal features may therefore reveal monogenic aetiologic diagnoses in a high percentage of patients.
The estimated prevalence of CKD – aetiology unknown (CKDU) is 10-36% in adults.7, 10 In this setting, patients often present late with bilateral small kidneys that are not amenable to kidney biopsy. Even if a kidney biopsy is obtained, histological examination can still be uninformative, as advanced CKD can result in histological findings that are indistinguishable between multiple diseases.11 We also hypothesise that WES may be especially useful in patients with CKDU. Establishing a molecular diagnosis in patients with CKDU can therefore have resulting consequences for adequate clinical management particularly in the era of “precision medicine”.
RESULTS
A molecular genetic diagnosis was established in 37% of families using WES
We performed WES in 114 families with CKD (138 affected individuals). The median age at time of recruitment was 48 years [range 180-85 years], with a slight male predominance (70/138, 51%, Table 1). We detected a molecular genetic diagnosis in 42 of the 114 families (37%) (Figure 1 A, navy blue segment). The genetic diagnostic rate varied by recruitment group (Figure 2). We detected mutations across a diverse spectrum of known monogenic CKD genes encompassing mutations in 29 different genes (Table 2, Figure 3). These categories included cystic kidney disease genes (8/42 families, Figure 1 B, red segment), syndromic congenital anomalies of the kidney and urinary tract (CAKUT) genes (8/42 families, Figure 1 B, light blue segment), isolated CAKUT genes (6/42 families, Figure 1 B, dark blue segment), chronic glomerulonephritis (GN) genes (5/42 families, Figure 1 B, orange segment), tubulo-interstitial kidney disease (TIKD) genes (4/42 families, Figure 1 B, brown segment), renal tubulopathy genes (4/42, Figure 1 B, purple segment), nephrolithiasis/ nephrocalcinosis (NLNC) genes (4/42 families, Figure 1 B, pink segment), steroid resistant nephrotic syndrome (SRNS) genes (2/42 families, Figure 1 B, green segment), and Fabry disease genes (1/42 families, Figure 1 B, cream segment).
Table 1.
Clinical characteristics of the 138 affected individuals (114 families) with chronic kidney disease that were submitted for whole exome sequencing analysis
| Total cohort | Positive family history | Negative family history but extra-renal features | Negative family history and no extra-renal features | |||||
|---|---|---|---|---|---|---|---|---|
| Individuals (Families) | 138 (114) | 102 (78) | 16 (16) | 20 (20) | ||||
| A priori clinical diagnosis | ||||||||
| Cystic Kidney Disease | 16 | 12% | 9 | 9% | 6 | 38% | 1 | 5% |
| CAKUT | 53 | 38% | 38 | 37% | 3 | 19% | 12 | 60% |
| Chronic GN | 9 | 7% | 7 | 7% | 1 | 6% | 1 | 5% |
| TIKD | 10 | 7% | 10 | 10% | 0 | 0% | 0 | 0% |
| SRNS | 7 | 5% | 4 | 4% | 1 | 6% | 2 | 10% |
| Renal tubulopathy | 2 | 1% | 1 | 1% | 1 | 6% | 0 | 0% |
| CKD aetiology unknown | 41 | 30% | 33 | 32% | 4 | 25% | 4 | 20% |
| Total | 138 | 100% | 102 | 100% | 16 | 100% | 20 | 100% |
| ESKD | ||||||||
| Yes | 90 | 66% | 64 | 65% | 11 | 69% | 15 | 70% |
| No | 48 | 34% | 38 | 35% | 5 | 31% | 5 | 30% |
| Total | 138 | 100% | 102 | 100% | 16 | 100% | 20 | 100% |
| Onset of CKDa(years) | ||||||||
| <18 (childhood onset) | 50 | 36% | 27 | 26% | 9 | 56% | 14 | 70% |
| ≥18 (adult onset) | 85 | 62% | 74 | 73% | 6 | 38% | 5 | 25% |
| Missing data | 3 | 2% | 1 | 1% | 1 | 6% | 1 | 5% |
| Total | 138 | 100% | 102 | 100% | 16 | 100% | 20 | 100% |
| Onset of ESKDb(years) | ||||||||
| <18 (childhood onset) | 21 | 15% | 8 | 8% | 5 | 31% | 8 | 40% |
| ≥18 (adult onset) | 69 | 50% | 56 | 55% | 6 | 38% | 7 | 35% |
| CKD only in adulthood | 48 | 35% | 38 | 37% | 5 | 31% | 5 | 25% |
| Total | 138 | 100% | 102 | 100% | 16 | 100% | 20 | 100% |
| Sex | ||||||||
| Male | 70 | 51% | 49 | 48% | 9 | 56% | 12 | 60% |
| Female | 68 | 49% | 53 | 52% | 7 | 44% | 8 | 40% |
| Total | 138 | 100% | 102 | 100% | 16 | 100% | 20 | 100% |
| Self-reported ethnicity | ||||||||
| Irish | 135 | 98% | 101 | 99% | 14 | 88% | 20 | 100% |
| Other European | 2 | 1% | 1 | 1% | 1 | 6% | 0 | 0% |
| Asian | 1 | 1% | 0 | 0% | 1 | 6% | 0 | 0% |
| Total | 138 | 100% | 102 | 100% | 16 | 100% | 20 | 100% |
A priori clinical diagnosis, the clinical diagnosis of chronic kidney disease defined pre-WES as per the primary nephrologist’s referral; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; ESKD, End Stage Kidney Disease; GN, glomerulonephritis; SRNS, steroid resistant nephrotic syndrome; TIKD, tubulo-interstitial kidney disease
age of 1st presentation to medical services with evidence of CKD
age at commencement of renal replacement therapy i.e. dialysis or kidney transplant
Figure 1. Percentage of the 114 families in Ireland with CKD in whom whole exome sequencing (WES) established a molecular genetic diagnosis (i.e. a pathogenic or likely pathogenic monogenic mutation in a known CKD gene was detected following WES).
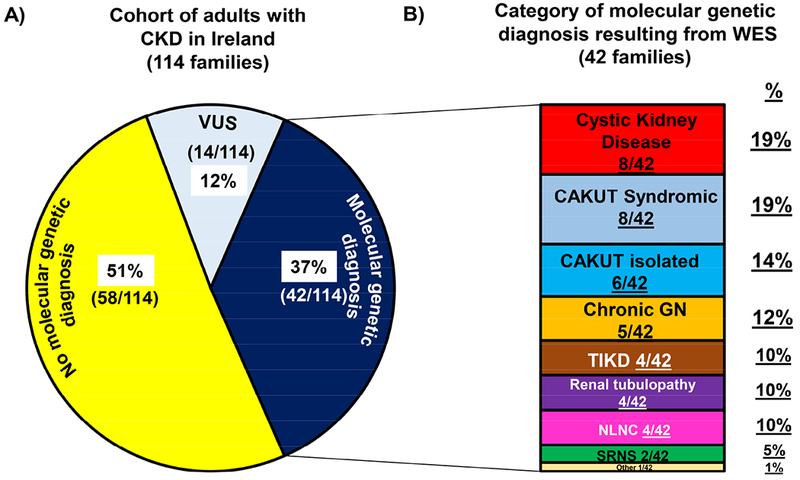
(A) The 37% of families (42/114) in whom a pathogenic or likely pathogenic mutation in a known CKD disease gene was detected (i.e. molecular genetic diagnosis established following WES) is denoted by a navy blue colour. The 12% of families (14/114) in whom a variant of uncertain significance (VUS) in a known CKD gene was detected, is denoted by the light blue colour. Yellow colour indicates that no meaningful genetic variant could be detected in a known CKD gene following WES (i.e. no molecular genetic diagnosis established following WES).
(B) The category and percentage of monogenic mutations detected in the 42 families in whom we identified a pathogenic or likely pathogenic mutation in a known CKD gene (i.e. families in whom we established a molecular genetic diagnosis). Each colour represents a different molecular genetic diagnostic group.
i) Mutations in known cystic kidney disease including nephronophthisis genes (red)
ii) Mutations in known syndromic CAKUT genes (light blue)
iii) Mutations in known isolated CAKUT genes (dark blue)
iv) Mutations in known chronic glomerulonephritis (GN) genes (orange)
v) Mutations in known tubulo-interstitial kidney disease (TIKD) genes (brown)
vi) Mutations in known renal tubulopathy genes (purple)
vii) Mutations in known nephrolithiasis/nephrocalcinosis (NLNC) genes (pink)
viii) Mutations in known steroid resistant nephrotic syndrome (SRNS) genes (green)
ix) Mutations in known rare chronic kidney disease genes (miscellaneous category) (cream) identified
Figure 2. Percentage of the 114 families in Ireland with CKD in whom whole exome sequencing established a molecular genetic diagnosis (i.e. a pathogenic or likely pathogenic monogenic mutation in a known CKD gene was detected following WES) stratified by recruitment group.
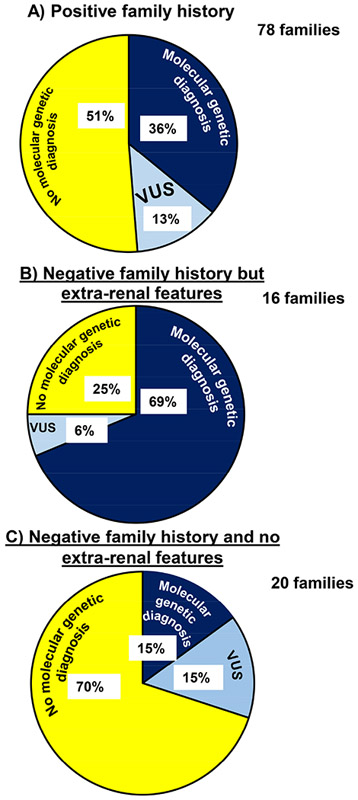
Navy blue colour denotes families in whom a pathogenic or likely pathogenic mutation in a known CKD gene was detected (i.e. molecular genetic diagnosis established following WES). Light blue colour denotes families in whom we identified a variant of uncertain significance (VUS) in a known CKD gene following WES. Yellow colour indicates that no meaningful genetic variant could be detected in a known CKD gene following WES (i.e. no molecular genetic diagnosis established following WES).
A) Positive family history cohort denotes families with CKD who report CKD in either a 1st or 2nd degree relative (78/144 families)
B) Negative family history but extra-renal features cohort (16/114 families)
C) Negative family history and no extra-renal features cohort (20/114 families)
Table 2.
Information on pre-whole exome sequencing a priori clinical diagnosis and post- whole exome sequencing molecular genetic diagnosis in the 42 families in whom pathogenic or likely pathogenic mutations in known monogenic chronic kidney disease genes were identified post WES. All families with a molecular genetic diagnosis were classified as pathogenic or likely pathogenic as per the ACMG guidelines (last column). In families in whom segregation was not possible (column 12) AND the exact mutation was not previously described (last column), the variant was considered a variant of uncertain significance (Supplementary Table S1). In the case of a compound heterozygous mutation, at least one of the alleles were classified as either pathogenic or likely pathogeni
| Fam. ID |
Ind. ID |
A priori
clinical Dx pre- WES (PRD codeŦ) |
Reported clinical phenotype |
Extra-renal features |
Age at 1st Dx of CKD ESKD [years] |
Sex | Inclusion criteriaa |
Molecular genetic Dx post WES #OMIMb |
Genotype (Inheritance) |
WES confirm clinical Dxc |
WES correct clinical Dxc |
WES establish new clinical Dxc |
c. changed
p. changee [evolutionary conservationf] |
Zygosity Segregation |
PP2g
SIFTh MTi |
gnom ADj |
ACMGk
HGMDl ClinVarm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYSTIC KIDNEY DISEASE (Supplementary Table S6) | |||||||||||||||||
| P13 | 65 | NPHP (2836) | Small, cystic kidneys | Retinitis pigmentosa Mild learning disability | 2 27 |
M | Fam Hx | Mainzer-Saldino syndrome # 266920 | IFT140 (AR) | ✓ | c.634G>A p.Gly212Arg (D.m.) | hom Fa=het, Ma=het Aff sibs=hom Unaff sibs=het | 0.917 Del. D.C. | 0/15/277150 | Path. DM Path. | ||
| 60 | NPHP(2836) | Small, cystic kidneys | 5 12 |
F | Fam Hx | Mainzer-Saldino syndrome # 266920 | IFT140 (AR) | c.634G>A p.Gly212Arg (D.m.) | 0.917 Del. D.C. | 0/15/277150 | Path. DM Path. | ||||||
| P80 | 60 | NPHP(2836) | Small, cystic kidneys | / | 21 21 |
F | Fam Hx | Nephron-ophthisis 1, juvenile # 256100 | NPHP1 (AR) | ✓ | c.555_556insA p.Pro186Thrfs*2 | Hom Fa=NA, Ma=NA Aff sibs=hom Unaff sib=het | / | / | Path. DM / | ||
| 61 | NPHP(2836) | Small, cystic kidneys | / | 11 12 |
M | Fam Hx | Nephron-ophthisis 1, juvenile # 256100 | NPHP1 (AR) | c.555_556insA p.Pro186Thrfs*2 | / | / | Path. DM / | |||||
| P389 | 23 | NPHP (2836) | BL echogenic kidneys | Renal tubular acidosis post-transplant | 8 16 |
M | Extrarenal | Nephron-ophthisis 1, juvenile # 256100 | NPHP1 (AR) | ✓ | c.1027G>A p.Gly343Arg (C.i.) | hom Fa=NA, Ma=NA | 1.00 Del. D.C. | 0/32/276716 | Path. DM Path. | ||
| P324 | 12 | NPHP(2836) | BL echogenic kidneys | Intellectual disability Retinitis pigmentosa Diabetes mellitus Obesity | 20 36 |
F | Extrarenal | Bardet-Biedl syndrome 9 # 615986 | BBS9 (AR) | ✓ | c.542C>G p.Pro181Arg (D.m.) | hom Fa=NA, Ma=NA | 0.99 Del./ | 0/1/246048 | Path. DM / | ||
| P231 | 62 | Cystic KD (2794) | Small kidneys with subcortical cysts | / | 8 40 |
M | Fam Hx | Polycystic kidney disease 4 # 263200 | PKHD1 (AR) | ✓ | c.5221 G>A p.Val1741 Met (C.e.) | hom Fa=NA, Ma=NA Aff. sib=hom | 0.76 Del. D.C. | 0/9/276648 | Path. DM Conflicting | ||
| 64 | / | 38 41 |
F | Fam Hx | Polycystic kidney disease 4 # 263200 | PKHD1 (AR) | c.5221 G>A p.Val1741 Met (C.e.) | 0.76 Del. D.C. | 0/9/276648 | Path. DM Conflicting | |||||||
| P317 | 48 | Cystic KD (2794) | Normal size, cystic kidneys | Conqenital Hepatic Fibrosis | 46 52 |
F | Fam Hx | Polycystic kidney disease 4 # 263200 | PKHD1 (AR) | ✓ | c.2702A>C p.Asn901Thr (X.t.) | Comp. het Fa=NA, Ma=NA Unaff. =single het | 0.60 Del. P.M. | 0/5/246108 | VUS Gene/ | ||
| c.107C>T p.Thr36Met (D.r.) | 0.97 Del. D.C. | 0/142/277030 | Path. DM Path. | ||||||||||||||
| P322 | 11 | Unknown (3555) | BSK | Retinitis pigmentosa Dextrocardia Cholestatic liver dysfunction | 62 70 |
F | Extrarenal | Short-rib thoracic dysplasia 3 with or without Polydactyly # 613091 | DYNC2H1 (AR) | ✓ | c.12431C>G p.Pro4144Arg (C.i.) | Comp. het Fa=NA, Ma=NA | 0.97 Del. D.C. | 0/3/276472 | VUS Gene Likely Path. & uncertain / | ||
| DYNC2H1 (AR) | c.10063+2T>G 100% ESS | / | 0/3/227450 | Path. DM at same position Path. | |||||||||||||
| P105 | 58 | Unknown (3555) | BSK | / | 19 19 |
F | Fam Hx | Nephronophthisi s 1, juvenile # 256100 | NPHP1 (AR) | ✓ | c.555_556insA p.Pro186Hisfs*2 | hom Fa=NA, Ma=NA | / | / | Path. DM Path. | ||
| SYNDROMIC CAKUT (Supplementary Table S7) | |||||||||||||||||
| B2330 | 12 | CAKUT (1625) | R RHD Nephrectomy | Seizure disorder | 0 14 |
M | Fam Hx | Syndromic CAKUT # 244200 | PROKR2 (AD) | ✓ | c.332T>G p.Met111 Arg (X.t) | het Fa=NA, Ma=WT (Affection status Fa unknown) | 0.88 Tol. D.C. | 0/1/246266 | Likely Path. DM/ | ||
| B2481 | 83 | CAKUT (1625) | Unilateral RA | Postaxial Polydactyly Inguinal hernia | 0 12 |
M | Extrarenal | Ulnar-mammary syndrome # 181450 | TBX3 (AD) | ✓ | c.915del p.Asp305Glufs*18 | het Fa=NA, Ma=WT (Affection status Fa unknown) | / | / | Path. Gene / | ||
| B2463 | 96 | CAKUT (1673 & 1706) | BL hydronephrosis/ureter, neurogenic bladder R kidney 22cm L kidney 35cm | Height 195cm Joint hypermotility Saddle nose Gum hypertrophy Downslantina palpebral fissures Hiah arch palate Hammer toes Pes planus | 0 CKD only |
F | Extrarenal | Marfan syndrome (Syndromic CAKUT) # 154700 | FBN1 (AD) | ✓ | c.4888C>T p.Gin 1630* | het Fa=NA, Ma=NA | / | / | Path. DM Path. | ||
| B2328 | 44 | NPHP(2836) | BL echogenic kidneys | Craniosynostosis Mild learning disability | 0 18 |
F | Extrarenal | Greig cephalo-polysyndactyly # 175700 | GLI3 (AD) | ✓ | c.539G>A p.Arg180Gln (D.r.) | het Fa=het, Ma=WT (Affection status Fa unknown) | 0.89 Del. D.C. | 0/14/276690 | Likely Path. Gene/ | ||
| B2454 | 13 | NPHP(2836) | PresumedNPHP Renal Bx -TIN | Hypothyroid Retinitis pigmentosa | 30 CKD only |
M | Extrarenal | Di George syndrome # 188400 | TBX1 (AD) | ✓ | c.1309C>T p.Pro437Ser (D.r.) | het Fa=NA, Ma=NA | 1.00 Tol. D.C. | 0/19/211848 | Likely Path. Gene/ | ||
| P320 | 4 | Cystic KD (2794) | Normal size cystic kidneys | Mild intellectualdisability Macrocephalv Hyperkeratosis Lentiaines | 50 CKD only |
M | Fam Hx | Cardiofacio-cutaneous syndrome # 615280 | MAP2K2 (AD) | ✓ | c.692G>T p.Arg231Leu (D.m.) | het Aff Fa=het, Unaff Ma=NA | 1.00 Del. D.C. | / | Likely Path. Gene Likely Path. | ||
| 73 | Cystic KD (2794) | Normal size cystic kidneys plus VUR | 20 CKD only |
F | Fam Hx | MAP2K2 (AD) | c.692G>T p.Arg231 Leu (D.m.) | 1.00 Del. D.C. | / | Likely Path. Gene Likely Path. | |||||||
| P198 | 102 | Unknown (3555) | CKD-aetiology unknown Renal Bx ND | Hypertension Diabetes mellitus Depression | 36 CKD only |
F | Fam Hx | Wolfram-like syndrome, autosomal dominant # 614296 | WFS1 (AD) | ✓ | c.2654C>T p.Pro885Leu (D.m.) | het Fa=NA, Ma=NA Aff.=het | 1.00 Del. D.C. | 0/4/244868 | Likely Path. Gene Likely Path. | ||
| B2479 | 75 | Unknown (3564) | BL small kidneys (renal Bx FSGSquery secondary) | Gout Retinitis Pigmentosa Anemia Diabetes mellitus Pseudotumour cerebri | 2 15 |
M | Extrarenal | Fanconi anemia, complementation group I # 609053 | FANCI (AR) | ✓ | c.217A>T p.lle73Phe(D.r.1) | hom Fa=NA, Ma=NA | 0.81 Del. D.C. | 0/4/277214 | Likely Path. Gene/ | ||
| ISOLATED CAKUT (Supplementary Table S8) | |||||||||||||||||
| P306 | 92 | CAKUT (1618) | VUR R native nephrectomy | / | 3 12 |
F | Fam Hx | Renal cysts and diabetes syndrome # 137920 | HNF1B (AD) | ✓ | c.1333 1334delGC p.Ala445fs*105 | het Aff sibs=het Unaff sibs=WT | / | / | Path. / / | ||
| B2482 | 98 | CAKUT (1687 & 1618) | PUV BL VUR | / | 0 9 |
M | Neither | CAKUT*611559 | UPK3A (AD) | ✓ | c.227C>A p.Ser76* | het Fa=NA, Ma=NA | / | 0/21/246014 | Path. ?DM / | ||
| P69 | 59 | CAKUT (3517) | Unilateral RA | / | 21 37 |
M | Fam Hx | Papillorenal syndrome # 120330 | PAX2 (AD) | ✓ | c.491 C>A p.Thrl 64Asn (D.r.) | het Fa=NA, Ma=NA | 0.03 Del. D.C. | 0/45/223294 | Path. DM/ | ||
| P307 | 77 | CAKUT (1618) | VUR | / | 42 46 |
M | Fam Hx | Papillorenal syndrome # 120330 | PAX2 (AD) | ✓ | c.70G>C p.Gly24Arg (D.m.) | het Aff Fa=het Unaff. Ma=WT Aff sibs=het Unaff sibs=WT | 1.00 Del. D.C | / | Path. DM/ | ||
| 50 | CAKUT (1618) | VUR | / | 18 25 |
F | Fam Hx | PAX2 (AD) | c.70G>C p.Gly24Arg (D.m.) | 1.00 Del. D.C | / | Path. DM/ | ||||||
| 94 | CAKUT (3517) | Unilateral RA | / | 29 29 |
F | Fam Hx | PAX2 (AD) | c.70G>C p.Gly24Arg (D.m.) | 1.00 Del. D.C | / | Path. DM/ | ||||||
| P162 | 99 | CAKUT (1618) | VUR | / | 50 51 |
M | Fam Hx | Fraser Syndrome # 617666 | FREM2 (AR) | ✓ | c.3661 C>T p.Pro1221Ser (C.i.) | Comp. het Aff sib = comp. het, Unaff sib=single het | 1.00 Del. D.C. | 0/16/277168 | Likely Path. Gene/ | ||
| FREM2 (AR) | c.2533C>T p.His845Tyr (D.r.) | 0.01 Del./ | / | Likely Path. Gene/ | |||||||||||||
| B2342 | 44 | TIKD(1897) | Renal Bx -TIN | Diabetes mellitus Annular pancreas | 37 40 |
F | Fam Hx | Renal cysts and diabetes syndrome # 137920 | HNF1B (AD) | ✓ | c.544+3 544+6 del 75% ESS | het Aff sib = het Unaff sib= WT | / | / | Likely Path. Gene/ | ||
| 63 | TIKD(1897) | Renal Bx -TIN | Diabetes mellitus | 42 CKD only |
M | Fam Hx | HNF1B (AD) | c.544+3 544+6 del 75% ESS | / | / | Likely Path. Gene/ | ||||||
| CHRONIC GLOMERULONEPHRITIS (Supplementary Table S9) | |||||||||||||||||
| B2427 | 56 | CAKUT (1625) | Haematuria BLRHD | / | 3 CKD only |
M | Neither | Alport syndrome # 104200 | COL4A3 (AD) | ✓ | c.4981 C>T p.Arg1661Cys (D.m.) | het Fa = NA, Unaff Ma=het | 1.00 Del. D.C. | 1/103/277100 | Path. DM Path. & Likely Path. | ||
| B2347 | 17 | Unknown (3564) | Haematuria Renal Bx indeterminate | / | 12 48 |
F | Fam Hx | Alport syndrome # 104200 | COL4A3 (AD) | ✓ | c.2452G>A p.Gly818Arg (D.m.) | het Fa=NA, Ma=NA | 1.00 Del. D.C | / | Likely Path. DM Path. | ||
| P241 | 63 | Unknown (3564) | Renal Bx indeterminate | / | 33 CKD only |
F | Fam Hx | Alport syndrome # 301050 | COL4A5 (XLD) | ✓ | c.2396G>A p.Gly799Asp (C.e.) | het Fa=NA, Ma=NA | 1.00 Del. D.C. | / | Likely Path. Gene/ | ||
| P58 | 86 | Unknown (3564) | Renal Bx indeterminate, HTN | / | 23 52 |
M | Fam Hx | Alport syndrome # 301050 | COL4A5 (XLD) | ✓ | c.1423+1 G>T 100% ESS | hemi Fa=NA, Ma=NA | / | / | Path. DM Path. | ||
| P100 | 30 | Unknown (3564) | Renal Bx indeterminate | Deafness Glaucoma Recurrent pneumonia | 30 40 |
F | Fam Hx | Alport syndrome # 301050 | COL4A5 (XLD) | ✓ | c.2605G>A p.Gly869Arg (C.e.) | het/hemi Fa=NA, Aff Ma=het Aff sib = hemi | 1.00 Del. D.C. | / | Path. DM Path. | ||
| 16 | Unknown (3564) | Renal Bx indeterminate | Hearina impairment | 17 20 |
M | Fam Hx | COL4A5 (XLD) | c.2605G>A p.Gly869Arg (C.e.) | 1.00 Del. D.C. | / | Path. DM Path. | ||||||
| B2453** | 80 | Microscopic haematuria (3712) | Hypokalemic metabolic alkalosis/Bartter syndrome (3085) | / | 3 CKD only |
M | Extrarenal | Alport syndrome # 301050 | COL4A5 (XLD) | 2 molecular Dx - see purple segment below | c.2692A>G p.Met898Val (D.r.) | hemi Fa=NA, Ma=NA | 0.01 Tol. D.C. | 0/28/57/198228 | Path. DM / | ||
| P640** | 2008 | ChronicGN (1377) | Microscopic hematuria Normal renal Bx | Low complement (C3) levels | 20 Normal renal function |
M | Fam Hx | Susceptibility to atypical haemolytic uraemic syndrome # 612925 | C3 (AD) | 2 molecular Dx - see green segment below | c.4534C>T p.Arg1512Cys (M.m.) | het Aff. Fa=het Aff. sib=het Unaff. Ma=WT Unaff. sib=WT | 0.65 Del. D.C. | 0/2/246248 | Likely Path. Gene/ | ||
| 82 | ChronicGN (3749) | No renal Bx performed | Low complement (C3) levels | 20 30 |
M | Fam Hx | C3 (AD) | c.4534C>T p.Arg1512Cys (M.m.) | 0.65 Del. D.C. | 0/2/246248 | Likely Path. Gene/ | ||||||
| TUBULO-INTERSTITIAL KIDNEY DISEASE (Supplementary Table S6) | |||||||||||||||||
| B2337 | 52 | TIKD(1897) | Renal Bx -TIN | Uveitis Mucosal ulcers | 42 CKD only |
M | Fam Hx | Hyperuricemic nephropathy # 162000 | UMOD (AD) | ✓ | c.317G>A p.Cys106Tyr (X.t.) | het Fa=NA, Ma=NA Aff. sib= het | 1.00 Del. D.C. | / | Path. DM Uncertain | ||
| 53 | TIKD(1897) | Renal Bx -TIN | Hyperuricemia | 42 CKD only | M | Fam Hx | Hyperuricemic nephropathy # 162000 | UMOD (AD) | c.317G>A p.Cys106Tyr (X.t.) | 1.00 Del. D.C. | / | Path. DM Uncertain | |||||
| P193 | 83 | Unknown (3555) | BSK | / | 20 CKD only |
F | Fam Hx | Hyperuricemic nephropathy # 162000 | UMOD (AD) | ✓ | c.280T>C p.Cys94Arg (D.r.) | het Fa=NA, Ma=NA | 1.00 Del. D.C. | / | Likely Path. Gene/ | ||
| P232 | 60 | Unknown (3564) | Renal Bx indeterminate | Hyperuricemia | 8 18 |
M | Fam Hx | Hyperuricemic nephropathy # 162000 | UMOD (AD) | ✓ | c.1382C>A p.Ala461 Glu (X.t.) | het Fa=NA, Ma=NA | 1.00 Tol. D.C. | / | Path. DM Likely Path. | ||
| P88 | 47 | Unknown (3555) | BSK Renal Bx-insufficient tissue | Bronchiectasis Liver dysfunctionNon-melanomatous cancers | 44 45 |
M | Fam Hx | Karyomegalic interstitial nephritis # 614817 | FAN1 (AR) | ✓ | c.2590C>T p.Gln864* | Comp. het Fa=het, Ma=het Aff. sibs=comp het Unaff. sibs=het | / | 0/1/246242 | Path. Gene/ | ||
| c.2774_2775delTT p.Leu925fs | / | 0/10/267846 | Path. Gene/ | ||||||||||||||
| 38 | Unknown (3555) | BSK Renal Bx-insufficient tissue | Metastatic lung cancer | 33 38 |
F | Fam Hx | FAN1 (AR) | c.2590C>T p.Gln864* | / | 0/1/246242 | Path. Gene/ | ||||||
| c.2774_2775delTT p.Leu925fs | / | 0/10/267846 | Path. Gene/ | ||||||||||||||
| RENAL TUBULOPATHY (Suoolementary Table S10) | |||||||||||||||||
| B2457 | 78 | CAKUT (1625 & 2476) | BLRHD BL renal vein thrombosis Hypernatremia/Dehydration | / | 0 10 |
M | Neither | Nephrogenic diabetes insipidus # 125800 | AQP2 (AD) | ✓ | c.782C>T p.Ser261 Leu (X.t.) | het Fa=NA, Ma=NA | 0.99 Del.D.C. | 0/4/228000 | Likely Path. Gene/ | ||
| B2467 | 35 | Unknown (3564) | Hypertension CKD Renal Bx indeterminate | / | 26 CKD only |
F | Fam Hx | Pseudohypo-aldosteronism -hypertensive CKD # 614491 | WNK4 (AD) | ✓ | c.506C>T p.Pro169Leu (D.r.) | het Fa=NA, Ma=NA | 0.69 Del.D.C. | / | Path. DM/ | ||
| B2350** | 32 | Bartter syndrome (3085) | Hypokalemic metabolic alkalosis | Gout | 17 38 |
F | Fam Hx | Bartter syndrome # 607364 | CLCNKB (AR) | ✓ | c.226C>T p.Arg76* | hom Fa=NA, Ma=NA | / | 0/3/246064 | Path. DM/ | ||
| B2453** | 80 | Bartter syndrome (3085) | Hypokalemic metabolic alkalosis | Microscopic hematuria | 3 CKD only |
M | Extrarenal | Bartter syndrome # 607364 | CLCNKB (AR) | ✓ | c.226C>T p.Arg76* | hom Fa=NA, Ma=NA | / | 0/3/246064 | Path. DM/ | ||
| NEPHROLITHIASIS/NEPHROCALCINOSIS (SuDDlementarv Table S11) | |||||||||||||||||
| B2344 | 78 | Unknown (3564) | Renal Bx indeterminate | Gout | 25 CKD only |
M | Extrarenal | Cystinuria # 220100 | SLC3A1 (AD) | ✓ | c.1799G>A p.Gly600Glu (D.m.) | het Fa=NA, Ma=NA Aff. = het | 1.00 Del D.C. | 0/21/276676 | Likely Path. DM/ | ||
| P318 | 50 | Unknown (3555) | BSK | Intellectual disability Finaer and wrist swell inq Seizure disorder | 41 CKD only |
M | Fam Hx | Lowe syndrome # 309000 | OCRL (XL) | ✓ | c.1567G>A p.Asp523Asn (S.c.) | hemi Unaff. Fa=NA Unaff. Ma=het | 1.00 Del. D.C. | / | Path. DM/ | ||
| P182 | 602 | Unknown (3555) | BSK | Hypophosphatemia Bony pain multiple fractures Bony spurs & sclerosis | 51 55 |
M | Fam Hx | Dent disease # 300009 | CLCN5 (XL) | ✓ | c.1938del p.Phe646Leufs*10 | hemi Fa=NA, Ma=NA | / | / | Likely Path. Gene/ | ||
| B2340 | 15 | Unknown (3555) | BSK | (Son has nephrolithiasis) | 60 CKD only |
F | Fam Hx | Dent disease # 300009 | CLCN5 (XL) | ✓ | c.925C>T p.Arg309Cys (D.r.) | het Aff. son=hemi Fa=NA, Ma=NA | 0.92 Del. D.C. | 0/1/6/193605 | Likely Path. Gene/ | ||
| STEROID RESISTANT NEPHROTIC SYNDROME (Supplemental Table S12) | |||||||||||||||||
| KF4 | 16 | Chronic GN (1377) | Renal Bx indeterminate | / | 74 78 |
F | Fam Hx | Focal segmental glomerulosclerosis # 613237 | INF2 (AD) | ✓ | c.653G>A p.Arg218Gln (X.t.) | het Unaff. Fa=NA, Aff. Ma=het | 1.00 Del. D.C. | / | Path. DM Path. | ||
| 15 | Chronic GN (1377) | Renal Bx-indeterminate | Gout | 52 55 |
F | Fam Hx | INF2 (AD) | c.653G>A p.Arg218Gln (X.t.) | 1.00 Del. D.C. | / | Path. DM Path. | ||||||
| P640** | 83 | Chronic GN (1349) | Renal Bx-indeterminate | Normal complement (C3) levels | 20 23 |
F | Fam Hx | Focal segmental glomerulosclerosis # 613237 | INF2 (AD) | ✓ | c.353T>A p.llel 18Asn(D.r.) | het Aff. Fa=het Aff. sib=het Unaff. Ma=WT Unaff. sib=WT | 1.00 Del. D.C. | / | Likely Path. Gene/ | ||
| 82 | ChronicGN (3749) | Renal Bx ND | Low complement (C3) levels | 20 30 | M | Fam Hx | INF2 (AD) | c.353T>A p.llel 18Asn (D.r.) | 1.00 Del. D.C. | / | Likely Path. Gene/ | ||||||
| RARE MONOGENIC CKD genes (MISCELLANEOUS CATEGORY) (Supplementary Table S13) | |||||||||||||||||
| B2327 | 66 | Cystic KD (2794) | Normal size cystic kidneys | Liver dysfunction Unexplained seizures, L facial weakness without definite patholoav noted on brain imaaina, unexplained abdominal symptoms, photophobia | 1 6 |
F | Extrarenal | Fabry Disease # 301500 | GLA (XL) | ✓ | c.352C>T p.Argl 18Cys het(X.t.2) | het Fa=NA, Ma=NA | 0.99 Del. P.M. | 0/15/43/200247 | Likely Path. DM Conflicting | ||
A priori clinical diagnosis, the clinical diagnosis of chronic kidney disease defined pre-WES as per the primary nephrologist’s referral; A, adenine; AD, autosomal dominant; Aff, affected; AR, autosomal recessive; BL, bilateral; BSK, bilateral small kidneys; Bx, biopsy; C, cytosine; c. change, nucleotide change; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; cm, centimeter; comp, compound; conflicting, multiple submitters have provided assertion criteria to the ClinVar database but there are conflicting interpretations; del, deletion; delins, deletion insertion; Del., Deleterious; D.C., Disease Causing; DM, disease mutation; Dx, diagnosis; ESKD, end stage kidney disease; ESS, essential splice site; F, female; Fa, father; Fam. ID, unique family identifier; fs, frameshift mutation; FSGS, focal segmental glomerulosclerosis; G, guanine; GN, glomerulosclerosis; hemi, hemizygous; het, heterozygous; hom, homozygous; HTN, hypertension; Ind. ID, unique individual identifier; ins; insertion; KD, kidney disease; L, left; M, male; Ma, maternal; NA, not available; ND, not done; NPHP, nephronophthisis; p. change, amino acid change; Path., pathogenic; P.M., Polymorphism; PRD, primary renal diagnosis; PUV, posterior urethral valve; R, right; RA, renal agenesis; RHD, renal hypodysplasia; sibs, sibling(s); unknown, CKD - aetiology unknown; T, thymine; TIKD, tubulo-interstitial kidney disease; TIN, tubulo-interstitial nephritis; Tol., Tolerated; VUR, vesico-ureteric reflux; VUS, variant of uncertain significance; WES, whole exome sequencing; WT, wild type; XL, X-linked; XLD, X-linked dominant.
Additional clinical features established post WES, following clinical re-review in full cognizance of the molecular genetic diagnosis are highlighted in bold, underlined text in columns 4 & 5.
indicates additional finding in another category;
nonsense mutation;
Ŧ, ERA-EDTA primary renal disease codes (https://www.era-edta-reg.org/prd.jsp); /, data not available.
Inclusion criteria; Fam Hx, positive family history; Extra-renal, CKD with extra-renal features; Neither, no family history and no extra-renal features
# OMIM, Online Mendelian Inheritance in Man (https://www.omim.org)
Outcome following WES: WES confirm clinical Dx, WES confirmed the clinical diagnosis; WES correct clinical Dx, WES resulted in reclassification/ correction of the clinical diagnosis; WES establish new clinical Dx, WES resulting in establishment of a molecular diagnosis in families with CKD - aetiology unknown.
impact of variant on cDNA level
impact of variant on the amino acid or protein level
evolutionary conservation was assessed across phylogeny over eight species: M.m., Mus musculus; G.g., Gallus gallus; X.t., Xenopus tropicalis; D.r., Danio rerio; C.e., elegans; C.i., Ciona intestinalis; D.m., Drosphilia melanogaster; S.c., Saccharomyces cerevisiae. If conservation is interrupted in one species but otherwise preserved across phylogeny a numerical reference is provided: 1Valine in G. gallus; 2Lysine in M. musculus.
PP2, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2)
SIFT, Sorting intolerant from tolerant (http://sift.jcvi.org/)
MT, Mutation Taster (http://www.mutationtaster.org)
gnom AD, variant frequencies listed for homozygous/ hemizygous (if applicable)/ heterozygous/ total alleles(http://gnomad.broadinstitute.org/)
ACMG, American College of Human Genetics Standards and Guidelines Classification as pathogenic, likely pathogenic or VUS (Richards Genet Med 17(5):405, 2015)
HGMd, Human Gene Mutation Database; https://portal.biobaseinternational.com/hgmd, If the exact variant has previously been reported on HGMD Professional 2017.2 for the reported phenotype and classified as a pathogenic mutations to be disease causing, variant denoted as “DM”. Variant denoted as “?DM” if the variant is a likely pathogenic mutation to be disease causing but where the author indicated some degree of doubt or subsequent evidence calls the deleterious nature of the variant into question. If the gene but not the exact variant has been reported for the corresponding phenotype “Gene” is indicated in this column.
Clin Var, classification if variant has been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar).
Figure 3. Specific mutation category identified by whole exome sequencing in 114 families with chronic kidney disease.
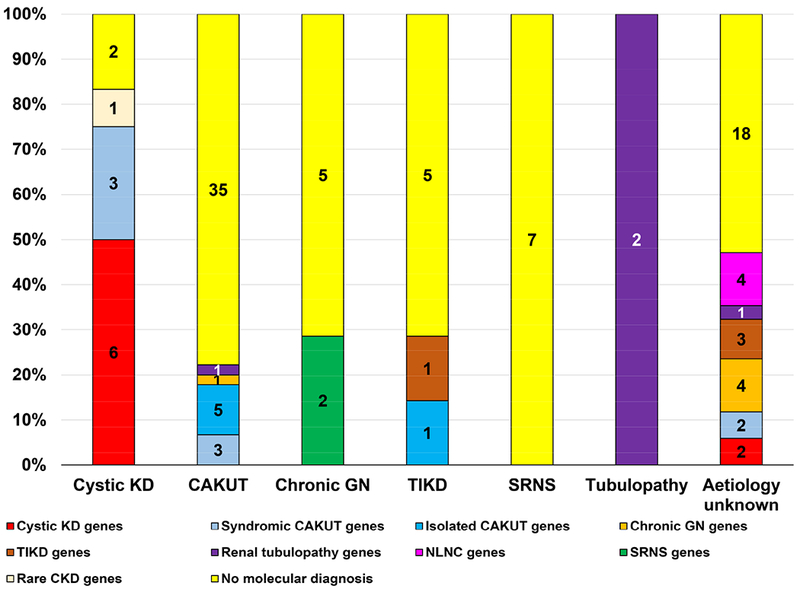
The X-axis displays the 7 a priori clinical diagnostic groups listed horizontally and includes cystic kidney disease (KD), congenital anomalies of the kidney and urinary tract (CAKUT), chronic glomerulonephritis (GN), tubulo-interstitial kidney disease (TIKD), steroid resistant nephrotic syndrome (SRNS), renal tubulopathy (tubulopathy) and CKD - aetiology unknown. The Y-axis displays the molecular genetic diagnosis established following whole exome sequencing and includes the following:
i) Mutations in known cystic kidney disease including nephronophthisis genes (red) identified
ii) Mutations in known syndromic CAKUT genes (light blue) identified
iii) Mutations in known isolated CAKUT genes (dark blue) identified
iv) Mutations in known chronic glomerulonephritis (GN) genes (orange) identified
v) Mutations in known tubulo-interstitial kidney disease (TIKD) genes (brown) identified
vi) Mutations in known renal tubulopathy genes (purple) identified
vii) Mutations in known steroid resistant nephrotic syndrome (SRNS) genes (green) identified
viii) Mutations in known nephrolithiasis/nephrocalcinosis (NLNC) genes (pink) identified
ix) Mutations in known rare chronic kidney disease genes (miscellaneous category) (cream) identified
x) No molecular genetic diagnosis established following WES (yellow)
Detection of a molecular genetic diagnosis in families with an a priori clinical diagnosis of cystic kidney disease
In families with a priori clinical diagnosis of cystic kidney disease (12/114), we detected a pathogenic mutation in ten of 12 families (83%). In six families, the molecular genetic diagnosis confirmed the pre-WES clinical diagnosis, with detection of mutations in cystic kidney disease or nephronophthisis (NPHP) genes (Table 2 red segment, P13, IFT140; P80 and P389, NPHP1; P324, BBS9; P231and P317, PKHD1). In four families, we detected mutations in CKD genes known to phenocopy cystic kidney disease. This pertained mostly to bilateral small kidneys that were thought to represent the phenotype of small cystic kidneys, but in fact represented the CAKUT phenotype of renal hypodysplasia (Table 2, light blue segment, B2328, GLI3; B2454, TBX1; P320, MAP2K2). In one family, WES identified a likely pathogenic mutation in the gene GLA, previously reported in patients with Fabry disease (Table 2, B2327, cream segment).12
Detection of a molecular genetic diagnosis in families with an a priori clinical diagnosis of CAKUT
For families with CAKUT (45/114 families), we detected mutations in ten of 45 families (22%). Five families had mutations in isolated CAKUT genes (Table 2, dark blue segment, P306, HNF1B; B2482, UPK3A; P69 and P307, PAX2; P162, FREM2), while three families had mutations in syndromic CAKUT genes (Table 2, light blue segment, B2330, PROKR2; B2481, TBX3; B2463, FBN1). In three of the families in whom we detected mutations in syndromic CAKUT genes, extra-renal features were present on clinical review that were concordant with the corresponding molecular genetic diagnosis (Table 2, column 5). In two families, we identified mutations in non-CAKUT genes (Table. 2 B2457, AQP2, purple segment and B2427, COL4A3, orange segment). The molecular genetic diagnosis in these two families was discordant with the clinical diagnosis.
Detection of a molecular genetic diagnosis in families with an a priori clinical diagnosis of chronic glomerulonephritis (GN)
In two of the seven families referred with chronic GN (7/114), we detected mutations in genes known to be causative of focal segmental glomerulosclerosis (FSGS) (Table 2, green segment). In both families (KF4 and P640), identification of a pathogenic mutation in the INF2 gene, resulted in the correction of the clinical diagnosis from GN to FSGS.
Detection of a molecular genetic diagnosis in families with an a priori clinical diagnosis of tubulo-interstitial kidney disease (TIKD)
Within the TIKD cohort (7/114), we established a molecular genetic diagnosis in two of seven families (29%). In family B2337, both siblings presented with CKD and gout at 42 years. Examination of renal biopsy specimens in both showed evidence of tubulo-interstitial nephritis. The molecular genetic diagnosis confirmed hyperuricaemic nephropathy with detection of a causative mutation in UMOD (Table 2 B2337, brown segment). In family B2342, the molecular genetic diagnosis facilitated a clinical review of two siblings presenting with CKD and diabetes mellitus in adulthood. Both affected siblings had renal biopsy findings of tubulo-interstitial nephritis, while one sibling (B2342_44) had evidence of pancreatic exocrine dysfunction. Detection of a mutation in the gene HNF1B therefore facilitated reclassification of the clinical diagnosis to renal cyst and diabetes syndrome (Table 2 B2342, dark blue segment).
No molecular genetic diagnosis established in families with an a priori clinical diagnosis of nephrotic syndrome
Of the seven of 114 families referred with nephrotic syndrome, no molecular genetic diagnosis could be established post WES.
Detection of a molecular genetic diagnosis in families with an a priori clinical diagnosis of renal tubulopathy
In two unrelated families with renal tubulopathies (Table 2, purple segment, B2350 and B2453), we detected a pathogenic homozygous mutation in CLCNKB, previously reported as being causative of Bartter syndrome.13 Interestingly, B2453_80 presented both with features of Bartter syndrome and microscopic hematuria. Following WES, we detected a second mutation in the Alport gene COL4A5 (Table 2, purple and orange segment). Patients with this exact mutation are reported to develop late onset microscopic hematuria and renal impairment.14
In summary, in 17 of the 42 solved families (40%), the molecular genetic diagnosis post-WES confirmed the a priori clinical diagnosis. The diagnostic yield varied depending on the a priori clinical diagnosis (Figure 2). In nine of the 42 families (22%), the molecular genetic diagnosis resulted in correction of the clinical diagnosis, while in 16 families with CKD – aetiology unknown” (38%), WES established a new molecular genetic diagnosis (Table 3).
Table 3.
Comparison between the outcomes in the current study and a recent publication
| Current Study n (%) | Lata Study1 n (%) | |
|---|---|---|
| WES confirmed the clinical diagnosis | 17 (40%) | 6 (27%) |
| WES corrected/reclassified the clinical diagnosis | 9 (22%) | 6 (27%) |
| WES established a new clinical diagnosis | 16 (38%) | 7 (32%) |
| Novel candidate gene identified following WES | NA | 3 (14%) |
| Total in whom WES confirmed, corrected, reclassified or established a genetic diagnosis | 42 (100%) | 22 (100%) |
CKD, chronic kidney disease; WES, whole exome sequencing; NA, not applicable.
Lata S et al. Whole-Exome Sequencing in Adults With Chronic Kidney Disease: A Pilot Study. Annals of Internal Medicine 2018; 168: 100–109.
WES corrected the a priori clinical diagnosis
In nine of 42 solved families (22%), WES corrected the clinical diagnosis (Table 3). As an example, patient B2457_78, with an a priori clinical diagnosis of CAKUT, presented with ESKD and a renal ultrasound showing bilateral small kidneys presumed to be due to bilateral renal hypodysplasia. Following WES, we detected a heterozygous AQP2 mutation (Table 2, purple segment). On review post WES, the patient had initially presented as an infant in the 1970s with polyuria, vomiting and hypernatremia and subsequent bilateral renal vein thrombosis. This reverse phenotyping confirmed the molecular genetic diagnosis of nephrogenic diabetes insipidus by WES.
In patient B2427_56, with an a priori clinical diagnosis of CAKUT, we detected a heterozygous mutation in the COL4A3 gene11 (Table 2, orange segment). Due to the lack of a family history and absence of a renal biopsy specimen, the clinical diagnosis of autosomal dominant Alport syndrome had not been suspected initially. This demonstrates the utility of WES in establishing a definitive clinical diagnosis in patients with atypical or indistinct phenotypes.
Family P640 had an initial diagnosis of C3 glomerulonephritis (Table 2, green and orange segment, Supplementary Figure S1). Both affected individuals (P640_82 and P640_83) presented with advanced proteinuric CKD in their twenties. Multiple family members were noted to have low C3 levels but all had normal renal function. Following WES, the molecular genetic diagnosis of FSGS due to a dominant heterozygous mutation in INF2 was established in P640_82 and P640_83. Interestingly, an additional finding of a dominant heterozygous variant in C3 was also identified in P640_82 with ESKD and P640_2008 without ESKD, both of whom were hypocomplementaemic. Mutations in this gene can result in complement dysregulation characterised by low C3 levels thereby increasing susceptibility to atypical haemolytic uraemic syndrome.15
WES established a new clinical diagnosis families with “CKD – aetiology unknown”
In families referred with CKDU (34 of 114 families, 30%), we detected a pathogenic mutation in 16 of 34 families (47%). This represents 38% of the solved cohort (16 of 42 solved families) (Table 4). The molecular genetic diagnoses in these families included cystic kidney disease or NPHP (P322, DYNC2H1; P105, NPHP1; Table 2, red segment), syndromic CAKUT (P198, WFS1 and B2479, FANC1, Table 2, light blue segment), Alport syndrome (B2347 COL4A3, and P241, P58, P100, COL4A5, Table 2, orange segment), TIKD (P193 & P232, UMOD; P88, FAN1, Table 2, brown segment), hypertensive renal disease (B2467, WNK4, Table 2, purple segment), and nephrocalcinosis/nephrolithiasis (B2344, SLC3A1; P318, OCRL; P182 and B2340, CLCN5, Table 2, pink segment). None of the above disease–causing mutations were suspected on clinical grounds before this study, and affected patients were not clinically distinguished from other patients with CKDU. WES therefore facilitated establishment of a molecular genetic diagnosis in families who otherwise would have remained without a formal diagnosis.
Table 4.
Comparison of the clinical characteristics of the 138 affected patients with chronic kidney disease by molecular genetic diagnostic category following whole exome sequencing
| Molecular genetic diagnostic category post WES | Median age in years of onset of ESKD [years] (range) | CKD only in adulthooda (%) | ESKD in adulthoodb n (%) | ESKD in childhoodc n (%) | Total numbers of “solved” individual s n (%) |
|---|---|---|---|---|---|
| Cystic Kidney Disease | 27 (12-70) | 0 | 8 (73) | 3 (27) | 11 (100) |
| CAKUT | 21.5 (9–51) | 6 (33) | 7 (39) | 5 (28) | 18 (100) |
| Chronic GN | 40 (20–52) | 3 (43) | 4 (57) | 0 | 7 (100) |
| TIKD | 38 (18–45) | 3 (50) | 3 (50) | 0 | 6 (100) |
| Renal Tubulopathy | 10, 38 | 2 (50) | 1 (25) | 1 (25) | 4 (100) |
| Nephrolithiasis/Nephrocalcinosis | 55 | 3 (75) | 1 (25) | 0 | 4 (100) |
| Steroid resistant nephrotic syndrome | 37.5 (20-78) | 0 | 4 (100) | 0 | 4 (100) |
| Other disease category | 6 | 0 | 0 | 1 (100) | 1(100) |
| All molecular diagnostic categories | 33 (6-78) | 17 (31) | 28 (51) | 10 (18) | 55 (100) |
CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; ESKD, End Stage Kidney Disease; GN, glomerulonephritis; solved, a pathogenic or likely pathogenic monogenic mutation in a known CKD gene was detected following WES; TIKD, tubulo-interstitial kidney disease.
Adult patients who had CKD at time of analysis (i.e. Not yet progressed to ESKD in adulthood i.e. ≥ 18 years)
Patients who developed ESKD ≥ 18 years of age
Patients who developed ESKD < 18 years of age
Identification of variants of uncertain significance (VUS)
In 12% of families (14/114) we detected a potentially pathogenic mutation in a gene known to cause CKD (Figure 1, light blue segment, Supplementary Table S1), however the identified variants did not meet our a priori criteria for definite confirmation of pathogenicity, either due to lack of clinical evidence to perform adequate genotype- phenotype correlation or lack of additional familial DNA to perform segregation analysis.
Factors associated with obtaining genetic diagnosis
The highest yield in terms of establishing a molecular genetic diagnosis was in families with CKD and extra-renal features (11/16 families, 69%). In families with a positive family history, we obtained a molecular genetic diagnosis in 36% (28/78 families). In families with a negative family history and no extra-renal features, monogenic causation was observed in 15% (3/20 families) (Figure 2). No significant difference was observed in the median age of reaching ESKD in individuals in whom we established a molecular diagnosis (33 years, range 6-78 years, Table 4) versus individuals in whom no molecular diagnosis was established (31 years, range 5-68 years, p=0.955).
DISCUSSION
In this large multicentre study, we systematically evaluated the utility of WES in a cohort of adults with CKD. We established a molecular genetic diagnosis following WES in 42 of 114 (37%) families with CKD attending nephrology services in Ireland. A genetic diagnosis was established in 69% (11/16) of families with extra-renal features, 36% (28/78) of families with familial nephropathy, while in families negative for both family history and extra-renal features, monogenic causation was observed in 15% (3/20). It has previously been estimated that ~10% of all adults with CKD have an underlying genetic aetiology.16 Recently, a higher prevalence of 24% for monogenic causation was reported following WES.9 In this single centre study, Lata et al. recruited 92 patients with either a family history of CKD, undiagnosed CKD or a clinical suspicion of genetic kidney disease due to childhood onset CKD. We observed comparable rates of confirmation, correction, and establishment of a new clinical diagnosis post WES (Table 3). More recently, Mallett demonstrated, using targeted exomic sequencing, a genetic diagnostic rate of 43% in patients with familial renal disease.8 Akin to our findings, the genetic diagnostic rate was similar in those with pediatric onset disease and adult onset disease (Supplementary Table S2). Together, these data provide compelling evidence that monogenic disease causation is under-recognised in adults with CKD, and WES can be utilised to provide a monogenic aetiologic diagnosis in adults with CKD.
Our data highlights that mutations in genes classically described as “childhood” CKD genes can also be identified in adults. We hypothesise that later onset disease is due to allelic heterogeneity with “milder” phenotypes likely attributable to “milder” missense mutations.17 For example, autosomal recessive polycystic kidney disease (ARPKD) has classically been characterised as a childhood onset nephropathy, with few cases of ESKD observed beyond 40 years.18 We identified recessive missense mutations in the PKHD1 gene in two unrelated families with onset of ESKD >40 years (Table 2, red segment, P231 and P317). Patient P317_48, presented with “CKD – aetiology unknown” age 46 years, progressing to ESKD at 52 years. A compound heterozygous missense mutation in PKHD1 was identified by WES (a novel c.2702A>C, p.Asn901Thr variant and a previously reported c.107C>T p.Thr36Met variant18). Recently, it has been shown that compound heterozygous mutations that involve at least one missense mutation of PKHD1 can lead to adult onset disease.19, 20 These data add to the mounting evidence supporting a monogenic causation hypothesis in adults and highlight the utility of WES in the investigation of adults with CKD.
The estimated prevalence of CKDU is 10-36% in adults,7, 10 with a prevalence of 30% (34/114) in the current cohort. By employing WES, we were able to establish a molecular genetic diagnosis in almost half of families with CKDU (16/34, 47%), confirming our hypothesis that CKDU may have a monogenic component. These data are consistent with findings from other groups (Table 3). By employing WES in cases where renal ultrasound and kidney biopsies were uninformative, we detected pathogenic mutations across a diverse spectrum of known monogenic causes of CKD including cystic kidney disease (2/16 families, Table 2, red segment), CAKUT (2/16 families, Table 2, blue segment), chronic GN (4/16 families, Table 2, orange segment), TIKD (3/16 families, Table 2, brown segment), renal tubulopathy (1/16, Table 2, purple segment), and nephrolithiasis/nephrocalcinosis (4/16 families, Table 2, pink segment) (Figure 3). Given that all these patients would have remained without a clinical diagnosis, these findings are significant.
Consistent with prior literature on genetic CKD in childhood, we demonstrated that the likelihood of obtaining a molecular genetic diagnosis in adults increased with the recognition of extra-renal manifestations (69%).21 As demonstrated in our cohort, mild extra-renal features are commonly unrecognised until clinical re-review is performed in full cognizance of the molecular genetic diagnosis (Table 2, Supplementary Table S1).22 This strategy of “reverse phenotyping” has been described extensively in childhood cohorts23, and our data shows that this holds relevance in adults with CKD. Identification of specific pathogenic mutations can also facilitate screening for otherwise undetected extra-renal features. For example, identifications of mutations in the gene HNF1B, has allowed for screening for associated extra-renal features such as diabetes, facilitating early lifestyle intervention strategies and avoidance of pro-diabetic medications in the post-transplant period (P306 and B2342, Table 2, blue segment).
Unnecessary diagnostic interventions can, in certain cases, be avoided following establishment of a molecular genetic diagnosis. This was particularly evident in cases where the pre-test probability of obtaining a diagnosis is low such as occurs in patients presenting with bilateral small kidneys not amenable to biopsy. This was the case for family P88 in whom we detected a pathogenic mutation in the gene FAN1, where multiple attempts to obtain a kidney biopsy were futile (Table 2 brown segment). On retrospective review, WES could have provided an earlier, more precise molecular diagnosis thereby facilitating early institution of anti-proteinuric medication, avoidance of systemic immunosuppression and negate the need for a non-diagnostic kidney biopsy.
Employing WES can allow for establishment of a more precise molecular genetic diagnosis in families with complex clinical presentations. As seen in family P640, with a presumed diagnosis of C3 glomerulonephritis, identification of a pathogenic mutation in INF2 permitted the diagnosis of FSGS (Table 2, P640_83, P640_82, Supplementary Figure S1), while detection of a second variant in the gene C3 may explain the observation of complement dysregulation in other family members (Table 2, P640_2008 and P640_82). In patient B2453_80 with Bartter syndrome, clinical heterogeneity was evident at clinical presentation which remained unresolved prior to WES. This patient presented with microscopic hematuria, a finding which was resolved following identification of a hemizygous mutation in the Alport gene COL4A5 (Table 2, orange segment). Patients with this exact mutation are reported to develop late onset microscopic hematuria and renal impairment.14 Findings such as these can facilitate early intervention with anti-proteinuric medication to stem the progression of CKD. Additionally, given the emerging evidence of increased risk of ESKD in both donors and recipients in families with Alport syndrome24, a molecular genetic diagnosis can help guide physicians when performing risk stratification of potential related donors at live donor assessment.
This study is not without limitations. First, the study was performed on a select population of predominantly Irish ethnicity, thereby reducing generalisability to other populations. Second, our cohort had a higher prevalence of familial CKD (78/114, 68%, Figure 2) compared to the reported prevalence in the general Irish CKD population (629/1840, 34%).7 Finally, although all patients were recruited as adults (median age of recruitment 48 years [range 18-85 years]), some patients developed ESKD in childhood (21/138, 15%, Table 1). Since prior reports suggest a higher prevalence of monogenic causation in childhood4, these findings should be considered when extrapolating to the general CKD population. Interestingly, when comparing the rate of molecular genetic diagnosis in childhood onset CKD versus adult onset CKD, no significant difference was observed in the rate of obtaining a genetic diagnosis (20/50, 40% with childhood onset CKD versus 35/85, 41% with adult onset disease, p=0.893, Supplementary Table S3). Equally, no significant difference in the median age of onset of ESKD was observed in patients, in whom we established a genetic diagnosis (median 33 years, range 6-78 years, Table 4) versus those who remained genetically unsolved following WES (31 years, range 5-68 years, p=0.651). These findings are further supported by other groups, who have demonstrated no significant difference in genetic diagnosis rates in those with peadiatric versus adult onset disease (46% versus 40%, Supplementary Table S2).8
In 58 of 114 families (51%) no molecular genetic diagnosis was established following WES (Supplementary Table S4). In monogenic diseases about 85% of all causative mutations are located within the coding sequence or the adjacent splice sites.25 The remaining 15% are complex deletion-insertion, copy-number variants, or reside within a promotor or other intronic region. As none of these variants can be detected by WES, this technical limitation might explain why some remain without a molecular diagnosis. Furthermore, WES might miss a subset of causative variants due to low coverage in the respective target region. In the current cohort, a mean depth of coverage of 48× was achieved. Specific exonic regions in the 478 known CKD genes, which did not reach this depth of coverage are outlined in Supplementary Table S5. Mutations in these regions may have been missed by WES analysis.
CONCLUSION
In a select patient cohort presenting with CKD in adulthood, we detected pathogenic mutations in known monogenic CKD genes in over one third of families. Detection of monogenic causes of CKD permit molecular genetic diagnosis for patients and families, and open avenues for personalised treatment strategies for CKD.
METHODS
Human subjects
This multi-centre study enroled adult patients with CKD presenting to nephrology services in Ireland in a consecutive manner from January 2014 to July 2017, as previously described.7 Consent for WES was obtained from each individual recruited and approved by the medical ethics boards at each recruitment site in Ireland. Patients with CKD who had either a positive family history of CKD (Supplementary Figure S2 A, 78/114 families) or extra-renal features (Supplementary Figure S2 B, 16/114 families) were recruited. To assess the effect of familial diagnosis and extra-renal features on detection rate of a molecular genetic diagnosis, families with CKD with a negative family history and no extra-renal features were also recruited (Supplementary Figure S2 C, 20/114 families). The clinical diagnosis of CKD was defined pre-WES as per the primary nephrologist’s referral into one of the following a priori clinical diagnoses26:
Cystic kidney disease including nephronophthisis (NPHP), medullary cystic disease, or other renal cystic ciliopathies (12/114 families). Patients with autosomal dominant polycystic kidney disease (ADPKD) were excluded.
Congenital anomalies of the kidney and urinary tract (CAKUT) (45/114 families) defined as any abnormality of number, size, shape, or anatomical position within the kidneys or urinary tract.
Chronic glomerulonephritis (GN) encompassing membranoproliferative GN (MPGN), crescentic GN, and haemolytic uraemic syndrome (7/114 families). Patients with genetically confirmed Alport syndrome and CKD due to systemic vasculitis were excluded.
Tubulo-interstitial kidney disease (TIKD) with biopsy findings of chronic tubulo-intersitital nephritis without an obvious precipitating cause (7/114 families). Patients with confirmed mutations in MUC1 and UMOD were excluded.
Steroid resistant nephrotic syndrome (SRNS), or nephrotic syndrome with biopsy findings of focal segmental glomerulosclerosis (FSGS) (7/114 families).
Renal tubulopathies (2/114 families)
“CKD – aetiology unknown” (CKDU) (34/114 families) where patients had small kidneys bilaterally and/or lacked an informative kidney biopsy.
Whole exome sequencing and variant calling
WES was performed as previously described.23, 27 Genomic DNA was isolated from blood lymphocytes or saliva samples as per standard protocols and subjected to exome capture using Agilent SureSelect™ human exome capture arrays (Life technologies™) followed by next generation sequencing on the Illumina HiSeq™ sequencing platform. Sequence reads were mapped to the human reference genome assembly (NCBI build 37/hg19) using CLC Genomics Workbench™ (version 6.5.2, CLC bio, Aarhus, Denmark). Variants with minor allele frequencies >1% in either dbSNP (version 147), 1,000 Genomes Project, EVS and gnomAD databases were excluded. For patients referred with an a priori clinical diagnosis of nephrotic syndrome, we manually searched for the p.Arg229Gln mutation in the NPHS2 gene, since this allele occurs at a frequency of >1%.28 Synonymous and intronic variants not located within splice site regions were excluded. Retained variants, which included non-synonymous and splice site variants, were then analysed (Supplementary Figure S3 and S4).
Depending on the a priori clinical diagnosis we evaluated WES data for mutations in known CKD genes in the matching disease category (i.e. a priori clinical diagnosis of chronic GN, we examined for mutations in known chronic GN genes, Supplementary Table S6–S13). If the family remained unsolved or the a priori clinical diagnosis was CKDU, we evaluated for mutations in all 478 known CKD genes (Supplementary Figure S5, Supplementary Tables S6–S13). Remaining variants were ranked based on their probable impact on the function of the encoded protein considering evolutionary conservation among orthologues across phylogeny using ENSEMBL Genome Browser and assembled using Clustal Omega, as well as the web-based prediction programmes PolyPhen-2, SIFT, and MutationTaster (Supplementary Table S14). Following filtering using our a priori criteria (Supplementary Figure S3 and S4), each mutation was then classified as per the American College of Medical Genetics and Genomics (ACMG) guidelines.29 In each family in whom we identified a likely causative monogenic mutation, clinical review with the referring physician was conducted to confirm that the phenotype was concordant with previously reported phenotypes, so called “reverse phenotyping”. Each likely causative monogenic mutation was classified as per the ACMG guidelines as pathogenic or likely pathogenic. Variants were classified as variants of uncertain significance (VUS) if there was discordance with previously published phenotypes, the exact variant was not previously reported and additional familial DNA was not available to perform segregation analysis. Remaining mutations were confirmed in original patient DNA by Sanger, with segregation whenever familial DNA was available.
Statistical Analysis
Descriptive statistics were expressed using frequencies and proportions. Age at diagnosis of CKD was defined as age of 1st presentation to a nephrology service with CKD, while age at ESKD was defined as age of commencement of renal replacement therapy (i.e. date of receipt of 1st kidney transplant or date of commencement of dialysis).
Supplementary Material
Supplementary Figure S1. Genetic and clinical data on family P640
Legend:
A. Extended family pedigree of family P640 - male are indicated by a square; female are indicated by a circle.
B. Focus of whole exome sequencing analysis - four individuals were submitted for whole exome analysis.
C. Final evaluation table following WES analysis for family P640.
D. Kidney biopsy of patient P640_83.
Low power light microscopy showing 3 sclerosed glomeruli, 1 partially sclerosed & 1 proliferative glomeruli
Direct immunofluorescence demonstrating a glomerulus with granular, mesangial C3 granular
Electron microscopy demonstrating loss of podocytes - black arrow granular sub-endothelial dense deposits
Supplementary Table S13. 9 genes that represent rare monogenic causes of human chronic kidney disease (miscellaneous category), if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked
Supplementary Table S14. Web Resources.
Supplementary Figure S2. Selection process for the 114 families with CKD for the whole exome sequencing study
Legend: CKD, chronic kidney disease; HSP, Henoch Schonlein Purpura; MUC1, mucin-1; TIKD, tubulo-interstitial kidney disease; UMOD, uromodulin; VHL, von Hippel-Lindau; WES, whole exome sequencing; IKGP, Irish Kidney Gene Project (Connaughton Nephron 2015;130(4):293).
*CKD defined for initial enrollment as per KDOQI guidelines as CKD stage 3 or higher (i.e decreased GFR <60 mL/min/1.73 m2 for ≥ 3 months) OR
Renal imaging showing any abnormality in number, shape, size or location within the kidneys or genitourinary tract (CAKUT) OR
Renal imaging showing chronically increased echogenicity, loss of corticomedullary differentiation, and/or ≥2 renal cysts (Cystic kidney disease or NPHP) OR
Tubulo-interstitial pattern of injury on renal biopsy without obvious precipitating cause (TIKD) OR
Chronic proteinuria resistant to steroid treatment and/or evidence on renal biopsy of FSGS (Nephrotic syndrome) OR
Chronic hematuria and renal biopsy with a pattern of injury consistent with chronic glomerulonephritis OR
Evidence of biochemical abnormalities indicative of renal tubular dysfunction
CKD defined as per the ERA-EDTA primary renal diagnosis coding system (https://www.era-edta-reg.org)
Supplementary Table S15 A. Information on the 14 families in whom pathogenic or likely pathogenic mutations were identified in a known monogenic CAKUT genes were identified post WES.
Legend: AD, autosomal dominant; AR, autosomal recessive; CKD, chronic kidney disease; Dx, diagnosis; ESS, essential splice site; Family ID, unique family identifier; Individual ID, unique individual identifier; fs, frameshift mutation; *nonsense mutation
Supplementary Table S15 B. Distribution of missense and null mutations, stratified by age of first diagnosis of CKD.
Legend: CKD, chronic kidney disease.
Supplementary Figure S3. Variant filtering process for the identification of pathogenic mutations in genes known to cause chronic kidney disease (CKD)
Legend: Schematic overview of the workflow used for filtering of WES data.
(i) Keep rare variants present with a minor allele frequency (MAF) <1% in healthy control cohorts dbSNP147 (https://www.ncbi.nlm.nih.gov/projects/SNP).
(ii) Keep non-synonymous variants and intronic variants that are located within splice sites.
(iii) Applying known gene approach by selecting all variants detected in known CKD genes (Supplementary Table S6–S13).
(iv) Ranking of remaining variants based on their predicted likelihood to be deleterious for the function of the encoded protein using Polyphen 2 (http://genetics.bwh.harvard.edu/pph2, SIFT (http://sift.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org)
(v) Reviewing literature and review with referring physician delineating whether the detected mutation matches the phenotype.
(vi) Cross reference with the ACMG guidelines to determine if pathogenic, likely pathogenic or a variant of uncertain significance.
Supplementary Figure S4. Decision making strategy to pathogenicity of a variant identified by whole exome sequencing
Supplementary Figure S5. Evaluation of whole exome sequencing (WES) data in 114 families with CKD
Legend: Whole exome sequencing (WES) output of the 138 affected individuals was interrogated for mutations in known CKD genes matching the initial referral phenotype in all 114 families with CKD (Supplementary Table S6–S13).
If no likely causative mutation was identified in a known CKD matching the referral phenotype, evaluation for mutations in remaining known CKD genes not matching the referral phenotype was performed. If the a priori clinical diagnosis was CKD of unknown aetiology, evaluation for mutations in all the 478 genes known to cause CKD in humans was performed for the presence of pathogenic or likely pathogenic monogenic mutations was performed (Supplementary Table S6–S13).
CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; fam(s), family/ families; GN, glomerulonephritis; NPHP, nephronophthisis; SRNS, steroid resistant nephrotic syndrome; TIKD, tubulo-interstitial kidney disease.
Supplementary Table S1. Information on pre-whole exome sequencing a priori clinical diagnosis in the 14 families in whom we identified a variant of uncertain significance (VUS) in a known monogenic chronic kidney disease gene.
Legend: A priori clinical diagnosis, the clinical diagnosis of chronic kidney disease defined pre-WES as per the primary nephrologist’s referral; A, adenine; AD, autosomal dominant; AR, autosomal recessive; BL, bilateral; BSK, bilateral small kidneys; Bx, biopsy; C, cytosine; c. change, nucleotide change; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; delins, deletion insertion; Del., Deleterious; D.C., Disease Causing; D.M., disease mutation; Dx, diagnosis; ESKD, end stage kidney disease; F, female; Fa, father; Fam. ID, unique family identifier; G, guanine; het, heterozygous; hom, homozygous; HTN, hypertension; Ind. ID, unique individual identifier; L, left; M, male; Ma, maternal; ND, not done; NA, not available; NPHP, nephronophthisis; p. change, amino acid change; P.M., Polymorphism; PRD, primary renal diagnosis; R, right; RA, renal agenesis; RHD, renal hypodysplasia; RTA, renal tubular acidosis; Unknown, CKD – aetiology unknown; T, thymine; Tol., Tolerated; VUS, variant of uncertain significance; WES, whole exome sequencing; WT, wild type; XL, X-linked; XLD, X-linked dominant. Additional clinical features established post WES, following clinical re-review in full cognizance of the molecular genetic diagnosis are highlighted in bold, underlined text in column 5. ** indicates additional finding in another category; * nonsense mutation; Ŧ, ERA-EDTA primary renal disease codes (https://www.era-edta-reg.org/prd.jsp); /, data not available.
a Inclusion criteria; Fam Hx, positive family history; Extra-renal, CKD with extra-renal features; Neither, no family history and no extra-renal features
b #OMIM, Online Mendelian Inheritance in Man (https://www.omim.org)
c impact of variant on cDNA level, d impact of variant on the amino acid or protein level
e evolutionary conservation was assessed across phylogeny over 8 species: M.m., Mus musculus; G.g., Gallus gallus; X.t., Xenopus tropicalis; D.r., Danio rerio; C.e., elegans; C.i., Ciona intestinalis; D.m., Drosphilia melanogaster; S.c., Saccharomyces cerevisiae. If conservation is interrupted in one species but otherwise preserved across phylogeny a numerical reference is provided: 1Aspartic acid in X. tropicalis
f PP2, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2), g SIFT, Sorting intolerant from tolerant (http://sift.jcvi.org/), h MT, Mutation Taster (http://www.mutationtaster.org)
i gnomAD, variant frequencies listed for homozygous/ hemizygous (if applicable)/heterozygous/ total alleles(http://gnomad.broadinstitute.org/)
j ACMG, American College of Human Genetics Standards and Guidelines Classification as pathogenic, likely pathogenic or VUS (Richards Genet. Med. 17(5):405, 2015)
k HGMD, Human Gene Mutation Database; “Gene” - mutations in this gene but not the exact variant has been reported for the corresponding phenotype (http://portal.biobaseinternational.com/hgmd)
i ClinVar, classification if variant has been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar)
Supplementary Table S2. Comparison between the outcomes in the current study and a recent publication.
Legend: CKD, chronic kidney disease; WES, whole exome sequencing
aAll family members had onset of CKD < 18 years
bAt least 1 family members had onset of CKD ≥ 18 years
1Mallett et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney International 2017; 92:1493 1506.
Supplementary Table S3. Age of onset of end stage kidney disease and chronic kidney disease in the solved cohort (i.e. molecular genetic diagnosis established pre-whole exome sequencing) versus the unsolved cohort (i.e. no molecular genetic diagnosis established post-whole exome sequencing).
Legend: a Solved implies that a molecular genetic diagnosis was established following whole exome sequencing analysis (i.e. 42 families with 55 affected individuals)
b Unsolved implies that a molecular genetic diagnosis was not established following whole exome sequencing analysis (i.e. 72 families with 83 affected individuals)
c age at commencement of renal replacement therapy i.e. dialysis or kidney transplant
d age of 1st presentation to medical services with evidence of CKD
Supplementary Table S5. Information on 16 out of 478 known chronic kidney disease genes which did not achieve a mean coverage of at least 30×.
Legend: Mean coverage, average number of reads that align to the targeted exon; Mean 10× coverage, percentage of bases within each gene that achieve a coverage ≥10×.
Supplementary Table S4. Information on pre-whole exome sequencing a priori clinical diagnosis in the 58 families in whom no variants in known monogenic chronic kidney disease genes were identified.
Legend: BL, bilateral; BSK, bilateral small kidneys; Bx, biopsy; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; cm, centimeter; Dx, diagnosis; ESKD, end stage kidney disease; F, female; Fam. ID, unique family identifier; FSGS, focal segmental glomerulosclerosis; GN, glomerulosclerosis; Ind. ID, unique individual identifier; KD, kidney disease; L, left; M, male; NPHP, nephronophthisis; PRD, primary renal diagnosis; R, right; RA, renal agenesis; RHD, renal hypodysplasia; SIC, self-intermittent catherisation; UPJO, ureteropelvic junction obstruction; TIKD, tubulo-interstitial kidney disease; TIN, tubulo-interstitial nephritis; VUR, vesico-ureteric reflux.
Ŧ, ERA-EDTA primary renal disease codes (https://www.era-edta-reg.org/prd.jsp); /, data not available.
a Inclusion criteria: Fam Hx, positive family history; Extra-renal, CKD with extra-renal features; Neither, no family history and no extra-renal features
Supplementary Table S6. 96 genes that represent monogenic causes of human cystic kidney disease or nephronophthisis, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort)
Legend: AR, autosomal recessive; AD, autosomal dominant; DR, digenic recessive; XLD; X-linked dominant
Supplementary Table S7. 165 genes that represent monogenic causes of human syndromic CAKUT, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort)
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked; Unknown, mode of inheritance not clearly characterized
Supplementary Table S8. 40 genes that represent monogenic causes of human isolated CAKUT, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort)
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked
Supplementary Table S9. 20 genes that represent monogenic causes of human chronic glomerulonephritis, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked
Supplementary Table S10. 51 genes that represent monogenic causes of monogenic human renal tubulopathies, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; DR, digenic recessive; XLD; X-linked recessive
Supplementary Table S11. 38 genes that represent monogenic causes of human nephrolithiasis or nephrocalcinosis, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; DR, digenic recessive; XLD; X-linked recessive
Supplementary Table S12. 59 genes that represent monogenic causes of human nephrotic syndrome, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked; Unknown, mode of inheritance not clearly characterised
ACKNOWLEDGEMENTS
Claire Foley (Dublin, Ireland) and Valerie Logan (Dublin, Ireland).
This research was supported by grants from the National Institutes of Health to FH (DK088767, DK076683, and DK068306). The Yale Center for Mendelian Genomics is funded by U54 HG006504 granted to RPL. DMC was funded by the Health Research Board, Ireland (HPF-206-674), the International Pediatric Research Foundation Early Investigators’ Exchange Program and the Amgen® Irish Nephrology Society Specialist Registrar Research Bursary. NM was supported by funding from the National Institutes of Health T32-DK007726-33 grant at Boston Children’s Hospital and by a Fred Lovejoy Resident Research and Education Award. TMK was supported by a Post-Doctoral Fellowship award from the KRESCENT Program, a national kidney research training partnership of the Kidney Foundation of Canada, the Canadian Society of Nephrology, and the Canadian Institutes of Health Research. Patient recruitment to the Rare Kidney Disease Registry and Biobank was funded by grants from Science Foundation Ireland (11/Y/B2093) to MAL, the Meath Foundation (203170.13161) to PJC and the Beaumont Hospital Department of Nephrology Research Fund. Recruitment of patients was possible through collaboration with the Health Research Board Royal College of Surgeons in Ireland Clinical Research Centre at Beaumont Hospital and the Rare Kidney Disease Registry and Biobank at Trinity College Dublin. The corresponding authors (PJC and FH) had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Funding
Patient recruitment was funded by Science Foundation Ireland and the Meath Foundation. Whole exome sequencing analysis was supported by grants from the National Institute of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE
No conflict of interests
Supplementary information is available at Kidney International's website
REFERENCES
- 1.Hill NR, Fatoba ST, Oke JL, et al. Global Prevalence of Chronic Kidney Disease – A Systematic Review and Meta-Analysis. PLoS One 2016; 11: e0158765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Go AS, Chertow GM, Fan D, et al. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. New England Journal of Medicine 2009; 351: 1296–1305. [DOI] [PubMed] [Google Scholar]
- 3.Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. The Lancet 2013; 382: 339–352. [DOI] [PubMed] [Google Scholar]
- 4.Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol 2016; 12: 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skrunes R, Svarstad E, Reisaeter AV, et al. Familial clustering of ESRD in the Norwegian population. Clin J Am Soc Nephrol 2014; 9: 1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McClellan WM, Satko SG, Gladstone E, et al. Individuals With a Family History of ESRD Are a High-Risk Population for CKD: Implications for Targeted Surveillance and Intervention Activities. American Journal of Kidney Diseases 2009; 53: S100–106. [DOI] [PubMed] [Google Scholar]
- 7.Connaughton DM, Bukhari S, Conlon P, et al. The Irish Kidney Gene Project - Prevalence of Family History in Patients with Kidney Disease in Ireland. Nephron 2015; 130: 293–301. [DOI] [PubMed] [Google Scholar]
- 8.Mallett AJ, McCarthy HJ, Ho G, et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney International 2017; 92: 1493–1506. [DOI] [PubMed] [Google Scholar]
- 9.Lata S MM, Li Y, Fasel DA, Groopman E, Jobanputra V,, Rasouly H MA, Westland R, Verbitsky M, Nestor J, Slater LM, D’Agati V,, Zaniew M M-KA, Lugani F, Caridi G, Rampoldi L, Mattoo A, Newton, et al. Whole-Exome Sequencing in Adults With Chronic Kidney Disease: A Pilot Study. Annals of Internal Medicine 2018; 168: 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neild GH. Primary renal disease in young adults with renal failure. Nephrology Dialysis Transplantation 2010; 25: 1025–1032. [DOI] [PubMed] [Google Scholar]
- 11.Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 2014; 86: 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spada M, Pagliardini S, Yasuda M, et al. High Incidence of Later-Onset Fabry Disease Revealed by Newborn Screening. Am J Hum Genet 2006; 79: 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nozu Kandai, K I, et al. The Pharmacological Characteristics of Molecular-Based Inherited Salt-Losing Tubulopathies. The Journal of Clinical Endocrinology & Metabolism 2010; 95: E511–E518. [DOI] [PubMed] [Google Scholar]
- 14.Barker DF DJ, Atkin CL, Gregory MC. Efficient detection of Alport syndrome COL4A5 mutations with multiplex genomic PCR-SSCP. American Journal of Medical Genetics 2001; 98: 148–160. [DOI] [PubMed] [Google Scholar]
- 15.Frémeaux-Bacchi V et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 2008; 112: 4948–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mallett A, Patel C, Salisbury A, et al. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet Journal of Rare Disease 2014; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohl S HD, Dworschak GC, Hilger AC, Saisawat P, Vivante A, Stajic N, Bogdanovic R, Reutter HM, Kehinde EO, Tasic V, Hildebrandt. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. Journal American Society of Nephrology 2014; 25: 1917–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergmann C, Senderek J, Windelen E, et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int 2005; 67: 829–848. [DOI] [PubMed] [Google Scholar]
- 19.Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nature Genetics 2002; 30: 259. [DOI] [PubMed] [Google Scholar]
- 20.Adeva M, El-Youssef M, Rossetti S, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore) 2006; 85: 1–21. [DOI] [PubMed] [Google Scholar]
- 21.Vivante A, Hwang DY, Kohl S, et al. Exome Sequencing Discerns Syndromes in Patients from Consanguineous Families with Congenital Anomalies of the Kidneys and Urinary Tract. J Am Soc Nephrol 2017; 28: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ven ATvd, Shril S, Ityel H, et al. Whole-Exome Sequencing Reveals FAT4 Mutations in a Clinically Unrecognizable Patient with Syndromic CAKUT: A Case Report. Molecular Syndromology 2018; 8: 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Ven AT, Connaughton DM, Ityel H, et al. Whole-Exome Sequencing Identifies Causative Mutations in Families with Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2018; 29: 2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gross O WM, Fries JW, Müller GA. Living donor kidney transplantation from relatives with mild urinary abnormalities in Alport syndrome: long-term risk, benefit and outcome. Nephrology Dialysis Transplantation 2008; 24: 1626–1630. [DOI] [PubMed] [Google Scholar]
- 25.Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-Genome Sequencing in a Patient with Charcot–Marie–Tooth Neuropathy. New England Journal of Medicine 2010; 362: 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Postorino M LA, Teatini U, Amuso S, Torino C, Di Iorio BR, Martorano C, Marino C, Morosetti M, Santoro A. The new ERA-EDTA codes for primary kidney diseases. G Ital Nefrol 2013; 30: pii: gin/30.32.15. [PubMed] [Google Scholar]
- 27.Gee HY, Otto EA, Hurd TW, et al. Whole exome resequencing distinguishes cystic kidney diseases fromphenocopies in renal ciliopathies. Kidney International 2014; 85: 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machuca E HA, Nevo F, Dantal J, Martinez F, Al-Sabban E, Baudouin V, Abel L, Grünfeld JP, Antignac C. Clinical and epidemiological assessment of steroidresistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney International 2009; 75: 727–735. [DOI] [PubMed] [Google Scholar]
- 29.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015; 17: 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Genetic and clinical data on family P640
Legend:
A. Extended family pedigree of family P640 - male are indicated by a square; female are indicated by a circle.
B. Focus of whole exome sequencing analysis - four individuals were submitted for whole exome analysis.
C. Final evaluation table following WES analysis for family P640.
D. Kidney biopsy of patient P640_83.
Low power light microscopy showing 3 sclerosed glomeruli, 1 partially sclerosed & 1 proliferative glomeruli
Direct immunofluorescence demonstrating a glomerulus with granular, mesangial C3 granular
Electron microscopy demonstrating loss of podocytes - black arrow granular sub-endothelial dense deposits
Supplementary Table S13. 9 genes that represent rare monogenic causes of human chronic kidney disease (miscellaneous category), if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked
Supplementary Table S14. Web Resources.
Supplementary Figure S2. Selection process for the 114 families with CKD for the whole exome sequencing study
Legend: CKD, chronic kidney disease; HSP, Henoch Schonlein Purpura; MUC1, mucin-1; TIKD, tubulo-interstitial kidney disease; UMOD, uromodulin; VHL, von Hippel-Lindau; WES, whole exome sequencing; IKGP, Irish Kidney Gene Project (Connaughton Nephron 2015;130(4):293).
*CKD defined for initial enrollment as per KDOQI guidelines as CKD stage 3 or higher (i.e decreased GFR <60 mL/min/1.73 m2 for ≥ 3 months) OR
Renal imaging showing any abnormality in number, shape, size or location within the kidneys or genitourinary tract (CAKUT) OR
Renal imaging showing chronically increased echogenicity, loss of corticomedullary differentiation, and/or ≥2 renal cysts (Cystic kidney disease or NPHP) OR
Tubulo-interstitial pattern of injury on renal biopsy without obvious precipitating cause (TIKD) OR
Chronic proteinuria resistant to steroid treatment and/or evidence on renal biopsy of FSGS (Nephrotic syndrome) OR
Chronic hematuria and renal biopsy with a pattern of injury consistent with chronic glomerulonephritis OR
Evidence of biochemical abnormalities indicative of renal tubular dysfunction
CKD defined as per the ERA-EDTA primary renal diagnosis coding system (https://www.era-edta-reg.org)
Supplementary Table S15 A. Information on the 14 families in whom pathogenic or likely pathogenic mutations were identified in a known monogenic CAKUT genes were identified post WES.
Legend: AD, autosomal dominant; AR, autosomal recessive; CKD, chronic kidney disease; Dx, diagnosis; ESS, essential splice site; Family ID, unique family identifier; Individual ID, unique individual identifier; fs, frameshift mutation; *nonsense mutation
Supplementary Table S15 B. Distribution of missense and null mutations, stratified by age of first diagnosis of CKD.
Legend: CKD, chronic kidney disease.
Supplementary Figure S3. Variant filtering process for the identification of pathogenic mutations in genes known to cause chronic kidney disease (CKD)
Legend: Schematic overview of the workflow used for filtering of WES data.
(i) Keep rare variants present with a minor allele frequency (MAF) <1% in healthy control cohorts dbSNP147 (https://www.ncbi.nlm.nih.gov/projects/SNP).
(ii) Keep non-synonymous variants and intronic variants that are located within splice sites.
(iii) Applying known gene approach by selecting all variants detected in known CKD genes (Supplementary Table S6–S13).
(iv) Ranking of remaining variants based on their predicted likelihood to be deleterious for the function of the encoded protein using Polyphen 2 (http://genetics.bwh.harvard.edu/pph2, SIFT (http://sift.jcvi.org/) and Mutation Taster (http://www.mutationtaster.org)
(v) Reviewing literature and review with referring physician delineating whether the detected mutation matches the phenotype.
(vi) Cross reference with the ACMG guidelines to determine if pathogenic, likely pathogenic or a variant of uncertain significance.
Supplementary Figure S4. Decision making strategy to pathogenicity of a variant identified by whole exome sequencing
Supplementary Figure S5. Evaluation of whole exome sequencing (WES) data in 114 families with CKD
Legend: Whole exome sequencing (WES) output of the 138 affected individuals was interrogated for mutations in known CKD genes matching the initial referral phenotype in all 114 families with CKD (Supplementary Table S6–S13).
If no likely causative mutation was identified in a known CKD matching the referral phenotype, evaluation for mutations in remaining known CKD genes not matching the referral phenotype was performed. If the a priori clinical diagnosis was CKD of unknown aetiology, evaluation for mutations in all the 478 genes known to cause CKD in humans was performed for the presence of pathogenic or likely pathogenic monogenic mutations was performed (Supplementary Table S6–S13).
CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; fam(s), family/ families; GN, glomerulonephritis; NPHP, nephronophthisis; SRNS, steroid resistant nephrotic syndrome; TIKD, tubulo-interstitial kidney disease.
Supplementary Table S1. Information on pre-whole exome sequencing a priori clinical diagnosis in the 14 families in whom we identified a variant of uncertain significance (VUS) in a known monogenic chronic kidney disease gene.
Legend: A priori clinical diagnosis, the clinical diagnosis of chronic kidney disease defined pre-WES as per the primary nephrologist’s referral; A, adenine; AD, autosomal dominant; AR, autosomal recessive; BL, bilateral; BSK, bilateral small kidneys; Bx, biopsy; C, cytosine; c. change, nucleotide change; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; delins, deletion insertion; Del., Deleterious; D.C., Disease Causing; D.M., disease mutation; Dx, diagnosis; ESKD, end stage kidney disease; F, female; Fa, father; Fam. ID, unique family identifier; G, guanine; het, heterozygous; hom, homozygous; HTN, hypertension; Ind. ID, unique individual identifier; L, left; M, male; Ma, maternal; ND, not done; NA, not available; NPHP, nephronophthisis; p. change, amino acid change; P.M., Polymorphism; PRD, primary renal diagnosis; R, right; RA, renal agenesis; RHD, renal hypodysplasia; RTA, renal tubular acidosis; Unknown, CKD – aetiology unknown; T, thymine; Tol., Tolerated; VUS, variant of uncertain significance; WES, whole exome sequencing; WT, wild type; XL, X-linked; XLD, X-linked dominant. Additional clinical features established post WES, following clinical re-review in full cognizance of the molecular genetic diagnosis are highlighted in bold, underlined text in column 5. ** indicates additional finding in another category; * nonsense mutation; Ŧ, ERA-EDTA primary renal disease codes (https://www.era-edta-reg.org/prd.jsp); /, data not available.
a Inclusion criteria; Fam Hx, positive family history; Extra-renal, CKD with extra-renal features; Neither, no family history and no extra-renal features
b #OMIM, Online Mendelian Inheritance in Man (https://www.omim.org)
c impact of variant on cDNA level, d impact of variant on the amino acid or protein level
e evolutionary conservation was assessed across phylogeny over 8 species: M.m., Mus musculus; G.g., Gallus gallus; X.t., Xenopus tropicalis; D.r., Danio rerio; C.e., elegans; C.i., Ciona intestinalis; D.m., Drosphilia melanogaster; S.c., Saccharomyces cerevisiae. If conservation is interrupted in one species but otherwise preserved across phylogeny a numerical reference is provided: 1Aspartic acid in X. tropicalis
f PP2, PolyPhen 2 (http://genetics.bwh.harvard.edu/pph2), g SIFT, Sorting intolerant from tolerant (http://sift.jcvi.org/), h MT, Mutation Taster (http://www.mutationtaster.org)
i gnomAD, variant frequencies listed for homozygous/ hemizygous (if applicable)/heterozygous/ total alleles(http://gnomad.broadinstitute.org/)
j ACMG, American College of Human Genetics Standards and Guidelines Classification as pathogenic, likely pathogenic or VUS (Richards Genet. Med. 17(5):405, 2015)
k HGMD, Human Gene Mutation Database; “Gene” - mutations in this gene but not the exact variant has been reported for the corresponding phenotype (http://portal.biobaseinternational.com/hgmd)
i ClinVar, classification if variant has been submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar)
Supplementary Table S2. Comparison between the outcomes in the current study and a recent publication.
Legend: CKD, chronic kidney disease; WES, whole exome sequencing
aAll family members had onset of CKD < 18 years
bAt least 1 family members had onset of CKD ≥ 18 years
1Mallett et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney International 2017; 92:1493 1506.
Supplementary Table S3. Age of onset of end stage kidney disease and chronic kidney disease in the solved cohort (i.e. molecular genetic diagnosis established pre-whole exome sequencing) versus the unsolved cohort (i.e. no molecular genetic diagnosis established post-whole exome sequencing).
Legend: a Solved implies that a molecular genetic diagnosis was established following whole exome sequencing analysis (i.e. 42 families with 55 affected individuals)
b Unsolved implies that a molecular genetic diagnosis was not established following whole exome sequencing analysis (i.e. 72 families with 83 affected individuals)
c age at commencement of renal replacement therapy i.e. dialysis or kidney transplant
d age of 1st presentation to medical services with evidence of CKD
Supplementary Table S5. Information on 16 out of 478 known chronic kidney disease genes which did not achieve a mean coverage of at least 30×.
Legend: Mean coverage, average number of reads that align to the targeted exon; Mean 10× coverage, percentage of bases within each gene that achieve a coverage ≥10×.
Supplementary Table S4. Information on pre-whole exome sequencing a priori clinical diagnosis in the 58 families in whom no variants in known monogenic chronic kidney disease genes were identified.
Legend: BL, bilateral; BSK, bilateral small kidneys; Bx, biopsy; CAKUT, congenital anomalies of the kidney and urinary tract; CKD, chronic kidney disease; cm, centimeter; Dx, diagnosis; ESKD, end stage kidney disease; F, female; Fam. ID, unique family identifier; FSGS, focal segmental glomerulosclerosis; GN, glomerulosclerosis; Ind. ID, unique individual identifier; KD, kidney disease; L, left; M, male; NPHP, nephronophthisis; PRD, primary renal diagnosis; R, right; RA, renal agenesis; RHD, renal hypodysplasia; SIC, self-intermittent catherisation; UPJO, ureteropelvic junction obstruction; TIKD, tubulo-interstitial kidney disease; TIN, tubulo-interstitial nephritis; VUR, vesico-ureteric reflux.
Ŧ, ERA-EDTA primary renal disease codes (https://www.era-edta-reg.org/prd.jsp); /, data not available.
a Inclusion criteria: Fam Hx, positive family history; Extra-renal, CKD with extra-renal features; Neither, no family history and no extra-renal features
Supplementary Table S6. 96 genes that represent monogenic causes of human cystic kidney disease or nephronophthisis, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort)
Legend: AR, autosomal recessive; AD, autosomal dominant; DR, digenic recessive; XLD; X-linked dominant
Supplementary Table S7. 165 genes that represent monogenic causes of human syndromic CAKUT, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort)
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked; Unknown, mode of inheritance not clearly characterized
Supplementary Table S8. 40 genes that represent monogenic causes of human isolated CAKUT, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort)
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked
Supplementary Table S9. 20 genes that represent monogenic causes of human chronic glomerulonephritis, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked
Supplementary Table S10. 51 genes that represent monogenic causes of monogenic human renal tubulopathies, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; DR, digenic recessive; XLD; X-linked recessive
Supplementary Table S11. 38 genes that represent monogenic causes of human nephrolithiasis or nephrocalcinosis, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; DR, digenic recessive; XLD; X-linked recessive
Supplementary Table S12. 59 genes that represent monogenic causes of human nephrotic syndrome, if mutated and that were evaluated in WES data of this study.
(* indicates that a variant in this gene was identified in this cohort).
Legend: AR, autosomal recessive; AD, autosomal dominant; XL; X-linked; Unknown, mode of inheritance not clearly characterised