

Abstract
7-Methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (2), a promising anticancer lead previously identified by us, inhibited tumor growth by 62% in mice at 1.0 mg/kg without obvious signs of toxicity. Moreover, compound 2 exhibited extremely high antiproliferative activity in the NIH-NCI 60 human tumor cell line panel, with low to subnanomolar GI50 values (10−10 M level). It also showed a suitable balance between aqueous solubility and lipophilicity, as well as moderate metabolic stability in vivo. Mechanistic studies using Mayer’s hematoxylin and eosin and immunohistochemistry protocols on xenograft tumor tissues showed that 2 inhibited tumor cell proliferation, induced apoptosis, and disrupted tumor vasculature. Moreover, evaluation of new synthetic analogues (6a−6t) of 2 indicated that appropriate 2-substitution on the quinazoline ring could enhance antitumor activity and improve druglike properties. Compound 2 and its analogues with a 4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one scaffold thus represent a novel class of tubulin-binding tumor-vascular disrupting agents (tumor-VDAs) that target established blood vessels in tumors.
Graphical Abstract
INTRODUCTION
During the past decade, a novel class of antitumor agents termed tumor-vascular disrupting agents (tumor-VDAs) have been investigated intensively.1,2 Unlike angiogenesis inhibitors (AIs), tumor-VDAs target established tumor blood vessels and exert an almost immediate effect on these vessels, thus leading to a rapid vascular collapse and subsequent tumor cell death via extensive necrosis and apoptosis.3 Due to the morphological vasculature difference between normal tissues and solid tumors, tumor-VDAs preferentially block the blood flow of solid tumors, while the blood flow in normal tissues remains relatively intact.4 Currently, more than a dozen tumor-VDA drug candidates are undergoing clinical trials,5,6 and the most advanced candidate, CA-4P, a phosphate prodrug of combretastatin A-4 (CA-4, Figure 1), is in phase III trials for anaplastic thyroid cancer and Phase II trials for non-small-cell lung cancer (NSCLC).7 Meanwhile, many diverse small molecule tumor-VDAs with high antiproliferative activity have also been reported recently.8,9 Even though the exact mechanism(s) of action of tumor-VDAs are still not known, most of these compounds show common pharmacological charateristics, including inhibition of microtubule polymerization, competitive binding to the colchicine binding site on tubulin, and disruption of the tumor cell cytoskeleton, thus classifing them as tubulin-binding tumor-VDAs,4 such as CA-4.
Figure 1.
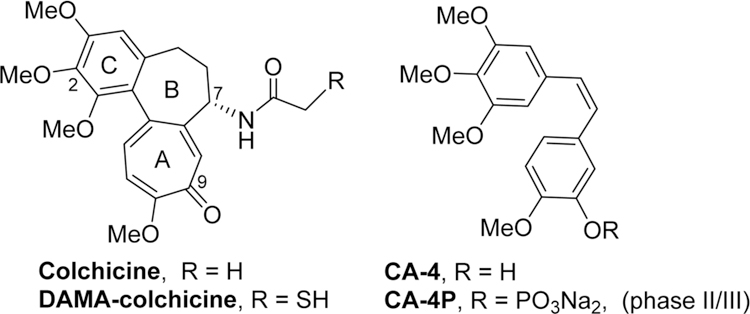
Colchicine and some clinical trial VDA candidates.
In our prior studies on novel antitumor agents,10–12 we discovered a series of new N-aryl-1,2,3,4-tetrahydroquinoline compounds with high antiproliferative activity in cellular assays, exemplified by prior leads 1a and 1b11 shown in Figure 2. Compounds 1a and 1b show high antiproliferative activity, with GI50 values of 1.5−1.7 and 11−18 nM, respectively, and they also inhibit tubulin polymerization with IC50 values of 0.93 and 1.0 μM, respectively. On the other hand, compared with 1a, compound 1b displays improved aqueous solubility, a better log P value, and greater metabolic stability. Subsequent lead optimization on the C-ring (skeleton hopping) led to the discovery of 7-methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (2) as a new lead compound.13 Compared with the corresponding prior lead 1b, compound 2, which has a novel structural scaffold with an altered C-ring, exhibits more potent activities in cytotoxicity and tubulin assays and better drug-like properties (Figure 2), including its log P value (2.97, pH 7.4), aqueous solubility (8.28 μg/mL, pH 7.4), and metabolic stability in vitro (t1/2 55 min in human liver microsomes).13 In the current study, we screened 2 in an extensive cellular panel (NCI-60 cell lines) to confirm the antitumor activity and determine its antitumor spectrum. Moreover, we also evaluated 2 in several in vivo systems, including a H-460 xenograft model and vascular disruption assays. Based on these results, the new scaffold in 2 was confirmed to be important for enhanced antitumor activity. Apart from the scaffold hopping, our prior structure optimizations on lead 1a12 also revealed that the 2-substituent on the quinazoline ring was modifiable to improve druglike properties without loss of antitumor potency, especially with the introduction of an alkylamino side chain. Our prior modeling studies further demonstrated that both 1a and 2 could dock within the tubulin−colchicine binding site and superimposed well with DAMA-colchicine (the bound ligand in the tubulin crystal structure, PDB code 1SA0).12,13 In addition, the docking study showed that the 2-substituent on the quinazoline ring in series-1 analogues overlapped well with the 7-side chain in DAMA-colchicine (Figure 1), thus indicating that the quinazoline 2-position has sufficient chemical space for structural modification. Consequently, in our current study, a series of new 4-(2-substituted quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one derivatives (6a−6t) were designed by maintaining the new scaffold in 2 and introducing various substituents on the quinazoline ring 2-position. Our aims were to simultaneously enhance antitumor activity and improve molecular drug-like properties. All synthesized new compounds were first evaluated in a human tumor cell line (HTCL) panel. Subsequently, new active compounds were further tested in tubulin assembly assays and assessed for their essential drug-like properties.
Figure 2.
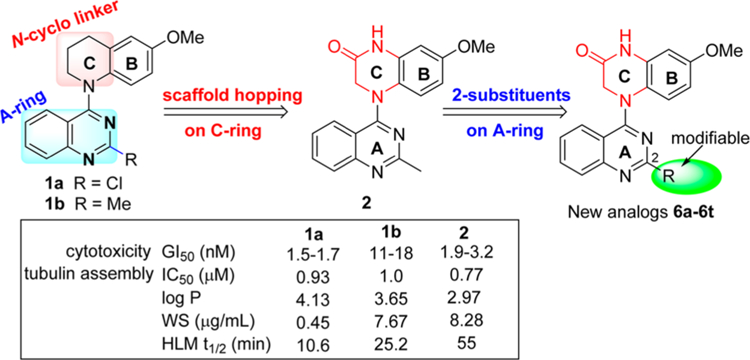
Leads and new derivatives.
CHEMISTRY
New compounds 6a−6p with various 2-alkylamino or 2-alkoxy substituents on the quinazoline ring were synthesized as shown in Scheme 1. The commercially available 2,4-dichloroquinazoline was coupled with 4-methoxy-2-nitroaniline (3) in i-PrOH in the presence of a catalytic amount of HCl to obtain 2-chloro-4-(4-methoxy-2-nitrophenyl)aminoquinazoline (4) in 85% yield. The nitro group was then reduced to an amino group by using zinc powder in acetic acid at 0 °C. Reaction of this intermediate with 2-chloroacetyl chloride produced 5. Next, compound 5 underwent an intramolecular cyclization in DMF in the presence of K2CO3 to yield 4-(2-chloroquinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6a). Displacement of the 2-chloro group on the quinazoline ring of 6a with various amines led to a series of 2-alkylamino-derivatives 6b−6n. The reactions were performed in anhydrous i-PrOH under microwave (MW) heating at less than 120 °C for 20 min in the presence of acidic or weakly basic catalysts, for example, H2SO4 for 6e, 6l, and 6m and K2CO3 for 6h, 6i, 6k, and 6n. Compound 6f was prepared in N,N-dimethylacetamide (DMA) at 155 °C. The syntheses of 2-alkoxy-compounds 6o and 6p were performed in the presence of NaH, used to deprotonate and enhance the nucleophilicity of the alcohol reactant. Moreover, compounds 6q−6t, which have a methylene (CH2) group inserted between the quinazoline ring and the remainder of the 2-side chain, were synthesized as shown in Scheme 2. Using a literature method,14 2-chloromethyl-3H-quinazolin-4-one (8) was prepared from the commercial reagents methyl 2-aminobenzoate and 2-chloroacetonitrile in 85% yield. Treatment of 8 with p-toluenesulfonyl chloride in CH2Cl2 in the presence of trimethylamine and a catalytic amount of dimethylaminopyridine (DMAP) produced the quinazoline sulfonate ester 9 in 88% yield. A substitution reaction between 9 and 3 gave intermediate 10. The 2-chloromethyl on the quinazoline ring in 10 was converted to the 2-acetoxymethyl in intermediate 11. Next, compound 6q was obtained from 11 by successive catalytic hydrogenation, amidation, and intramolecular cyclization. Notably, the cyclization of intermediate 13 was performed in DMA, rather than DMF, in 99% yield. Compound 6q was then hydrolyzed under basic conditions to produce hydroxymethyl compound 6r in 98% yield. Subsequently, compound 6r was reacted with ethyl 4-bromobutanoate15 in the presence of Cs2CO3 to yield 6s with an extended side chain on the quinazoline ring. Finally, compound 6s was treated with hydroxylamine to produce hydroxamide compound 6t. Compounds 6s and 6t were purified by recrystallization from EtOAc.
Scheme 1a.
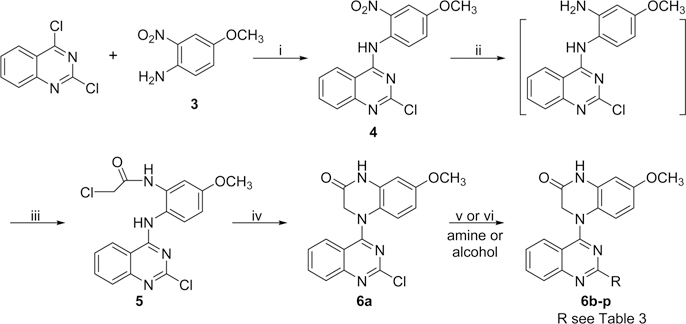
a Reagents and conditions: (i) i-PrOH/HCl, rt, 5−6 h; (ii) Zn/CH3COOH, CH2Cl2, 0.5 h; (iii) ClCH2COCl/K2CO3, 0 °C, 1 h; (iv) K2CO3/DMF, 100 °C, 0.5 h; (v) amine, microwave (MW), i-PrOH, 60−120 °C, 15−30 min (for 6b−d, 6f−g, 6k), or with H2SO4, 120 °C, 20 min (for 6e, 6j, 6l, 6m), or with K2CO3, 60−120 °C, 15−20 min (for 6h, 6i, 6n); (vi) EtOH or cyclopropylcarbinol/NaH at 0 °C, then MW, 95 °C, 5 min (for 6o, 6p).
Scheme 2a.
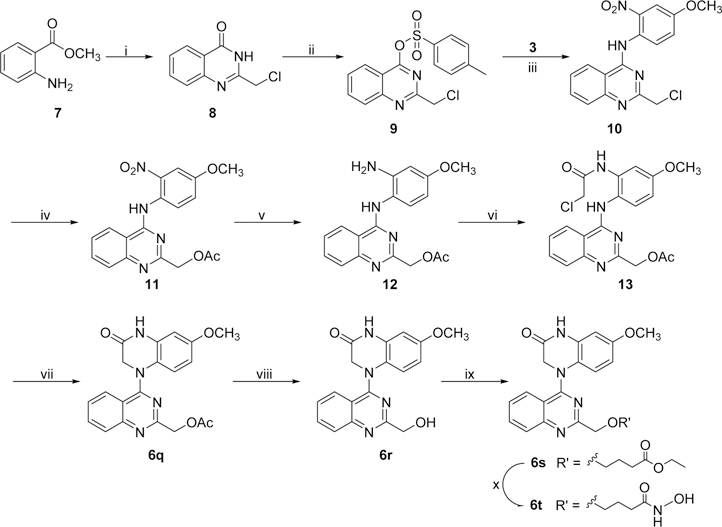
aReagents and conditions: (i) ClCH2CN/HCl (conc), 90 °C, 24 h; (ii) TsCl, DMAP, Et3N, rt, 3 h; (iii) i-PrOH, 45 °C, 6 h; (iv) KOAc, 55 °C, 6 h; (v) Pd/C, H2, rt, 2 h; (vi) ClCH2COCl/K2CO3, acetone, 0 °C, 1 h; (vii) K2CO3/DMF, 100 °C, 1 h; (viii) NaOH (10%), rt, 1 h; (ix) Br(CH2)3COOEt/Cs2CO3, 90 °C, 4 h; (x) NH2OH·HCl, 0 °C, 4 h.
RESULTS AND DISCUSSION
Testing of Compound 2.
NCI-60 Cell Line Screening.
To determine its antitumor spectrum, compound 2 was tested in the National Cancer Institute’s cellular panel of 60 human tumor cell lines (NCI-60). As shown in Table 1, compound 2 exhibited excellent potency with low to subnanomolar GI50 values (at 10−10 M level) against most tumor cell lines, including many multidrug resistant phenotypes, in the standard NCI testing protocol.16 However, the inhibitory potency of 2 was much reduced against six tumor cell lines (HOP-92, MALME-3M, SK-MEL-28, UACC-257, OVCAR-5, and TK-10). Interestingly, MALME-3M and TK10 cell lines were also significantly resistant toward a tubulin polymerization inhibitory 4-substituted-5-methyl-furo[2,3-d]pyrimidine derivative.17 These cell lines may be less sensitive to polymerization inhibitors, due to certain biological properties, such as overexpression or ligand binding site mutations on tubulin. Thus, 2 is an extremely potent antitumor agent with a broad spectrum of activity.
Table 1.
| Leukemia | Colon Cancer | Ovarian Cancer | CNS Cancer | ||||
|---|---|---|---|---|---|---|---|
| CCRF-CEM | 0.551 | COLO205 | 0.322 | IGROV1 | 0.830 | SF-268 | 0.788 |
| HL-60(TB) | 0.361 | HCC-2998 | 0.371 | OVCAR-3 | 0.266 | SF-295 | 0.318 |
| K562 | 0.359 | HCT-116 | 0.369 | OVCAR-4 | 2.43 | SF-539 | 0.298 |
| MOLT-4 | 0.605 | HCT-15 | 0.413 | OVCAR-5 | >100 | SNB-19 | 0.655 |
| RPMI-8226 | 0.603 | HT29 | 0.307 | OVCAR-8 | 0.524 | SNB-75 | 0.360 |
| SR | 0.301 | KM12 | 0.353 | NCI/ADR-RES | 0.301 | U251 | 0.437 |
| Non-Small-Cell Lung Cancer | SW-620 | 0.487 | SK-OV-3 | 0.409 | Prostate Cancer | ||
| A549/ATCC | 0.588 | Melanoma | Renal Cancer | PC-3 | 0.636 | ||
| EKVX | 0.632 | LOX IMVI | 0.526 | 786–0 | 3.46 | DU-145 | 0.409 |
| HOP-62 | 0.690 | MALME-3M | >100 | A498 | 0.376 | Breast Cancer | |
| HOP-92 | >100 | M14 | 0.336 | ACHN | 0.926 | MCF7 | 0.363 |
| NCI-H226 | 3.03 | MDA-MB-435 | 0.208 | CAKI-1 | 0.497 | MDA-MB-231/ATCC | 0.914 |
| NCI-H23 | 0.458 | SK-MEL-28 | >100 | RXF-393 | 0.246 | HS 578T | 0.601 |
| NCI-H322M | 0.904 | SK-MEL-5 | 0.400 | SN12C | 1.01 | BT-549 | 2.10 |
| NCI-H460 | 0.397 | UACC-257 | >100 | TK-10 | >100 | T-47D | c |
| NCI-H522 | 0.294 | UACC-62 | 0.432 | UO-31 | 10.0 | MDA-MB-468 | 0.415 |
The data were provided by the NCI, USA. The TGI and LC50 values are >1.0 × 10−7 M in most tested cell lines in the same assays (see SI).
The concentration that corresponds to 50% growth inhibition (GI50).
Not available.
Antitumor Activity in Vivo.
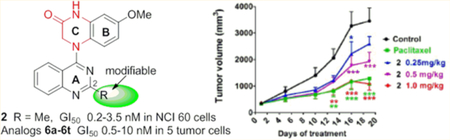
Next, compound 2 was evaluated in vivo due to its remarkable antiproliferative activity in vitro, as well as strong inhibitory effects on tubulin assembly, moderate in vitro metabolic stability (t1/2 55 min in human liver microsomes), and good aqueous solubility and lipophilicity. Pilot experiments showed that 4 mg/kg (one dose treatment) was the lethal dose of 2 in nude mice bearing MCF-7 xenograft tumors.13 This result allowed us to better assess the in vivo pharmacokinetic properties of 2. Consequently, the assessment was performed in SD rats (n = 3) by intravenous (iv) administration at a single-dose of 1.0 mg/kg.18 Compound 2 was relatively stable metabolically in rats with a terminal half-life of 52 h, providing an experimental basis for establishing administration schedules in a subsequent pharmacodynamic study. We established nude mouse subcutaneous xenograft models of NCI-H460 lung cancer and sorted the mice into five groups (n = 8 animals per group) with almost equal mean tumor size (100 mm3). The mice in three groups were treated with 2 by iv injection at doses of 1.0, 0.5, or 0.25 mg/kg every 5 days for 3 weeks. Control mice and paclitaxel-treated mice were treated in the same way, receiving vehicle solution or paclitaxel (15 mg/kg), respectively (Figure 3). All of the animals survived through the entire experiment, and no obvious toxic effects were observed, although there was some effect on animal weights with the two higher doses of 2 and with paclitaxel. At the end of treatment, the mice were sacrificed, and the tumors were recovered and weighed. As shown in Table 2 and Figure 3, compound 2 significantly inhibited the growth of the NCI-H460 xenograft tumors in a dose-dependent manner. The average tumor weights of 2-treated groups were less than those of control mice (statistical significance). The tumor growth inhibition rates were 17.8%, 36.8%, and 61.9% at doses of 0.25, 0.5, and 1.0 mg/kg, respectively. Meanwhile, the reference group treated with paclitaxel at a 15 mg/kg dose showed a tumor inhibition rate of 60.4%, comparable to what was observed with 2 at the 1.0 mg/kg dose. Therefore, compound 2 had significant in vivo antitumor effects.
Figure 3.
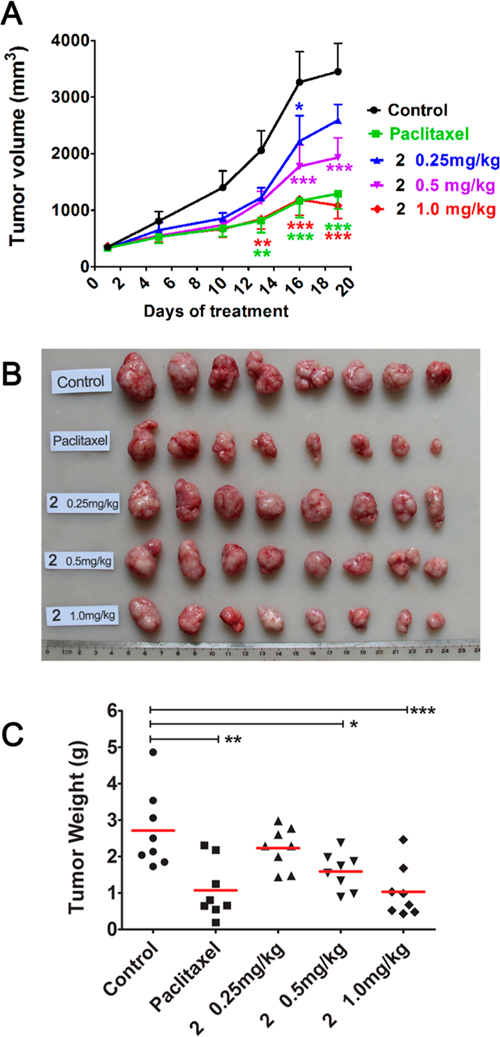
Dose-dependent anticancer effects of compound 2 on NCI-H460 lung cancer xenografts in nu/nu mice. Treatments started when tumors reached a mean volume of around 100 mm3. Mice were treated with 2 at an iv dose of 0.25, 0.5, or 1 mg/kg or paclitaxel at 15 mg/kg as reference or the vehicle as control (n = 8), every 5 days for 3 weeks. (A) Growth difference of tumor volumes at different time points. (B) Images of resected NCI-H460 xenograft tumors. (C) Tumors were resected and weighed at the end of experiment, (●) indicates the weight value of each tumor; (—) indicates average value of tumor weights. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle controls (one-way analysis of variance with Tukey’s post hoc method).
Table 2.
Tumor Growth Inhibition of 2 on Human Lung H460 Xenograft Tumors in Nude Micea
| body weight change |
antitumor activity |
||||||
|---|---|---|---|---|---|---|---|
| dose (mg/kg) | animals start/end | start (g) | end (g) | % | tumor weight (g) | inhibition rate (%) | |
| vehicle | 8/8 | 22.50 ± 1.23 | 26.40 ± 1.43 | 17.42 | 2.71 ± 1.07 | ||
| 2 | 0.25 | 8/8 | 22.44 ± 1.27 | 25.96 ± 1.54 | 15.71 | 2.23 ± 0.57 | 17.8 |
| 2 | 0.5 | 8/8 | 22.28 ± 1.33 | 23.85 ± 1.57 | 7.30 | 1.72 ± 0.78* | 36.8 |
| 2 | 1.0 | 8/8 | 22.13 ± 1.24 | 20.50 ± 3.27 | −7.23 | 1.03 ± 0.71*** | 61.9 |
| paclitaxel | 15 | 8/8 | 22.50 ± 1.29 | 23.59 ± 2.66 | 4.99 | 1.08 ± 0.78** | 60.4 |
Administration by iv. Schedule: Q5D × 4, that is, every 5 days for 3 weeks. Data are presented as the mean ± SD
p < 0.05
p < 0.01
p < 0.001 vs control.
Testing of New Derivatives (6a−6t) of Compound 2.
Antiproliferative Activity in Cellular Assays and SAR Analysis.
The newly synthesized N4-(2-substituted quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one compounds (6a−6t) with the same scaffold as 2 were initially evaluated in a HTCL panel, including A549 (lung carcinoma), KB (originally isolated from epidermoid carcinoma of the nasopharynx), multidrug-resistant (MDR) cell line KB-VIN (vincristine-resistant KB subline), MDA-MB-231 (triple-negative breast cancer), and MCF-7 (estrogen receptor-positive breast cancer),19,20 in parallel with paclitaxel as an experimental control. The activity in the cellular assays was determined using the established sulforhodamine B (SRB) method.21 The antiproliferative activities (GI50) and structures are summarized in Table 3.
Table 3.
Antiproliferative Activity of New Series Compounds (6a−6t) against Human Tumor Cell Lines
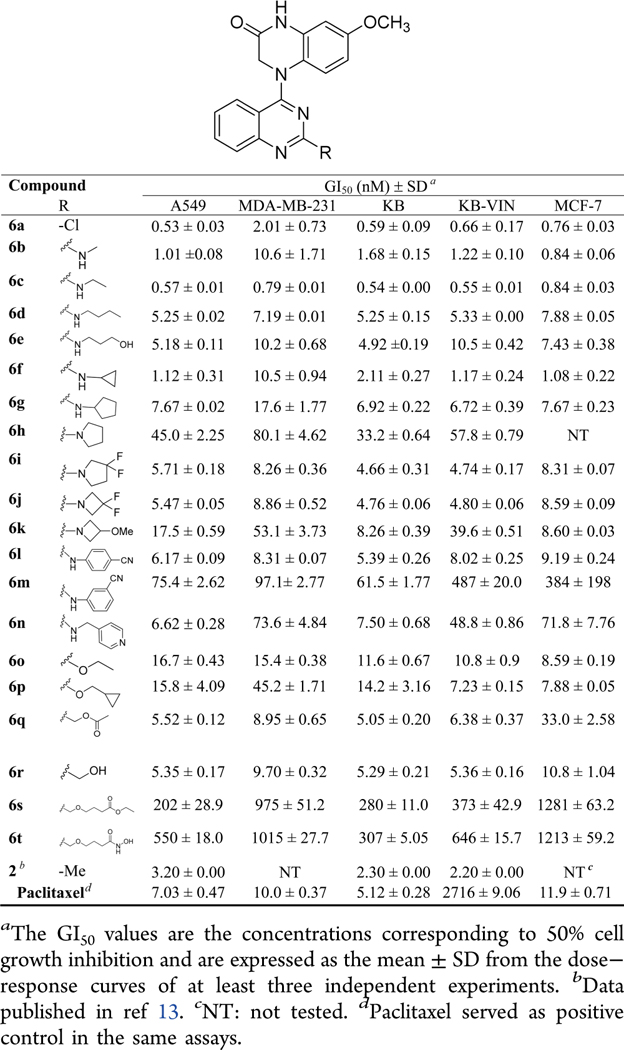 |
As expected, 4-(2-chloroquinazo-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6a) exhibited significant antiproliferative activity with low nanomolar GI50 values of 0.53−2.01 nM (cf, 1a, GI50 1.5−1.7 nM, Figure 2). After the scaffold change in the C-ring from piperidine to piperazin-2-one, the potency increased 3−6-fold (compare prior leads 1a and 1b with new compounds 6a and 2, respectively), which supported our previous hypothesis that the lactam C-ring (3,4-dihydropyrazin-2(1H)-one) might be an important moiety to enhance antiproliferative potency.
In addition, other series-6 compounds with the same new scaffold but different substituents at the quinazoline 2-position showed significant antiproliferative activities in the tested HTCL panel with GI50 values ranging from subnanomolar to submicromolar levels (Table 3). Among them, except for 6h and 6k, most 2-alkylamino-compounds with either a linear side chain (6a−6e) or a small cycloalkylamino substituent (6f, 6g, 6i, and 6j) were highly potent with GI50 values of 0.54−10.6 nM, with comparable or greater potency than paclitaxel in the same assays against chemosensitive cell lines. With GI50 values of >10−80 nM, the 2-(N-pyrrolidinyl) (6h) and 2-(N-(3-methoxyazetidinyl) (6k) compounds exhibited lower potency than the above-mentioned compounds.
Moreover, differing results were obtained when a 2-arylamino substituent was introduced at the quinazoline 2-position. Compound 6l with a 2-(4-cyanophenyl)amino substitutent displayed high potency with low nanomolar GI50 values (<10 nM), but compound 6m with a 2-(3-cyanophenyl)-amino group displayed at least ten times decreased potency (GI50 61.5−384 nM), suggesting that the bulkiness of the 2-substituent, for example, a m-substituted phenyl moiety, might obstruct the small molecule from fitting into the binding pocket on its biological target. Additionally, compound 6n, another 2-arylamino [2-(4-pyridinylmethyl)amino] substituted compound, showed high potency only against A549 and KB cell growth (GI50 6.62 and 7.50 nM, respectively) but decreased potency toward drug-resistant cell lines KB-VIN (GI50 48.8 nM) and MDA-MB-231 (GI50 73.6 nM), as well as MCF-7 (GI50 71.8 nM).
Next, we evaluated the presence of a 2-alkoxy or 2-alkoxymethyl substituent on the quinazoline ring. The corresponding 2-ethoxy (6o), 2-cyclopropylmethoxy (6p), 2-acetoxymethyl (6q), and 2-hydroxymethyl (6r) substituted compounds all exhibited high potency against tumor cell growth, generally showing single to double digit nanomolar GI50 values in the HTCL panel. However, the presence of a longer linear 2-substituent (more than six atoms) reduced antiproliferative potency, see compounds 6s and 6t with GI50 values of 0.2−1.28 and 0.31−1.21 μM, respectively.
In general, most 6-series compounds showed similar potency against A549, MCF-7, KB, and resistant KB-VIN cell lines, but were less potent against the triple-negative breast cancer cell line MDA-MB-231. Because antiproliferative activity was mostly comparable against the KB and MDR KB-VIN cell lines, the new compounds are likely not substrates of P-glycoprotein (Table 3).
Overall, the current results demonstrated that the new structural scaffold, 4-(quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one, is very important for high anticancer activity, while a suitable 2-substituent on the quinazoline ring can enhance or maintain pharmacological activity. The new SAR correlations revealed that (1) linear alkyamino substituents are more favorable than alkoxy and arylamino groups, which is a consistent finding with our prior results,12 (2) H-bond donor groups (i.e., NH or OH) favor high antiproliferative potency, and (3) the length and bulky volume of the 2-substituent should be limited.
Assessments of Physicochemical Properties.
Because potential drug candidates should have a good balance between potency and druglike properties, molecular physicochemical properties of 11 (6a−6g, 6i, 6j, 6q, and 6r) of our highly potent compounds (GI50 ≤ 10 nM) were further assessed in parallel with 2. Aqueous solubility and log P values were measured at pH 7.4 according to methods described previously.22 As shown in Table 4, six compounds (6a, 6b, 6e, 6f, 6q, and 6r) showed moderate aqueous solubility ranging from 8 to 87 μg/mL, which were similar to or improved compared with that of 2 (11 μg/mL). Among them, 6b, 6e, 6f, and 6r have at least one H-bond donor group (NH or OH) in the quinazoline 2-substituent, thus indicating that H-bond donor groups favor increased aqueous solubility. On the other hand, lipophilicity is another major determinant of many ADME/Tox properties, as well as of pharmacological activity. Log P values were also measured at pH 7.4 to estimate molecular lipophilicity. Except for 6d, all compounds in Table 4 showed lower log P values than those of 1a and 1b (4.13 and 3.65, respectively) and fell into a desireable druglike range (1 < log P7.4 < 3), which might lead to increased intestinal absorption. Thus, compound 2 and its new derivatives reach an acceptable balance between solubility and passive diffusion permeability, as well as increased metabolic stability.23 Therefore, our current results indicate that the new structural scaffold in 2 and the series-6 compounds makes a major contribution to reduced molecular lipophilicity, while the changeable quinazoline 2-substituents can enhance anticancer activity and improve aqueous solubility.
Table 4.
Drug-like Properties and Inhibition of Tubulin Polymerization and Colchicine Binding of Active Compounds 6a−6g, 6i−6j, and 6q−6r
| parameters (pH 7.4) |
colchicine binding inhibitionb (%) |
||||
|---|---|---|---|---|---|
| aq. sol. (μg/mL) | log P | tubulin assemblya IC50 (μM) ± SD | 5 μM | 1 μM | |
| 6a | 13.4 ± 0.98 | 2.94 ± 0.15 | 1.60 ± 0.04 | 99 ± 1 | 88 ± 3 |
| 6b | 28.6 ± 7.35 | 2.77 ± 0.40 | 2.80 ± 0.3 | 88 ± 0.9 | 53 ± 5 |
| 6c | 2.74 ± 0.11 | 2.56 ± 0.10 | 0.63 ± 0.09 | 96 ± 1 | 78 ± 4 |
| 6d | 1.34 ± 0.10 | 3.91 ± 0.03 | 0.42 ± 0.01 | 96 ± 1 | 83 ± 0.8 |
| 6e | 80.9 ± 17.8 | 2.60 ± 0.27 | 1.90 ± 0.04 | 77 ± 0.5 | c |
| 6f | 67.8 ± 10.3 | 2.26 ± 0.13 | 2.90 ± 0.3 | 87 ± 0.7 | 47 ± 4 |
| 6g | 1.45 ± 0.12 | 2.33 ± 0.00 | 0.59 ± 0.01 | 70 ± 2 | c |
| 6i | 0.46 ± 0.01 | 2.48 ± 0.17 | 0.97 ± 0.1 | 90 ± 1 | 43 ± 4 |
| 6j | 0.58 ± 0.00 | 2.14 ± 0.01 | 0.48 ± 0.02 | 97 ± 2 | 85 ± 0.3 |
| 6q | 8.07 ± 0.47 | 2.70 ± 0.01 | 0.62 ± 0.05 | 95 ± 0.9 | 67 ± 1 |
| 6r | 19.1 ± 1.58 | 2.14 ± 0.01 | 0.58 ± 0.01 | 97 ± 0.7 | 75 ± 4 |
| 2d | 11.1 ± 0.30 | 2.63 ± 0.00 | 0.77 ± 0.07 | 99 ± 0.02 | 93 ± 0.8 |
| CA-4e | 0.54 ± 0.06 | 98 ± 1 | 82 ± 2 | ||
The tubulin assembly assay measured the extent of assembly of 10 μM tubulin after 20 min at 30 °C.
Inhibition of [3H]colchicine binding was performed by incubating with tested compounds at 5 or 1 μM for 10 min at 37 °C, with tubulin at 1.0 μM.
Not determined.
Tubulin assay data see ref 13.
Reference compound is a drug candidate in phase II/III clinical trials.
Inhibition of Tubulin Polymerization and Colchicine Binding.
To measure effects of the new compounds on microtubule polymerization, the same 11 highly active compounds (6a-g, 6i−j, and 6q−r; GI50 ≤ 10 nM) were further tested in tubulin assembly and colchicine binding assays24 in parallel with CA-4, a clinical trial drug candidate and highly potent competitive inhibitor of the binding of colchicine to tubulin.25 Consistent with our previous results, all of the compounds were highly active in the tubulin assembly assay with low to submicromolar inhibitory IC50 values (0.42−2.90 μM), comparable with those of 2 (0.77 μM) and CA-4 (0.54 μM) in the same assay. However, an obvious difference was seen in the competitive colchicine−tubulin binding assay. With more than 97% and 83% inhibition at 5 and 1 μM, respectively, compounds 6a, 6d, and 6j showed similar potency to CA-4. The eight remaining series-6 compounds were less potent than CA-4. Moreover, compound 2 was more potent (99% at 5 μM and 93% at 1 μM) than CA-4 in the colchicine binding inhibition assay. These results demonstrated that active series-6 compounds, with the same scaffold as 2, also inhibit tubulin polymerization by targeting the tubulin−colchicine binding site; however, different 2-substituents on the quinazoline affect the molecular affinity with tubulin. Next, compounds 6e and 2 were docked at the colchicine binding site with the tubulin crystal structure (PDB code 5LYJ). As shown in Figure 4, molecules 6e and 2 superimposed well with the original ligand CA-4 in the binding site, while the outspread C2-side chain in 6e formed two hydrogen bonds with a.a. Tyr202 and Asp251. These findings support the postulate that, similar to CA-4, this compound series targets the tubulin−colchicine binding site.
Figure 4.
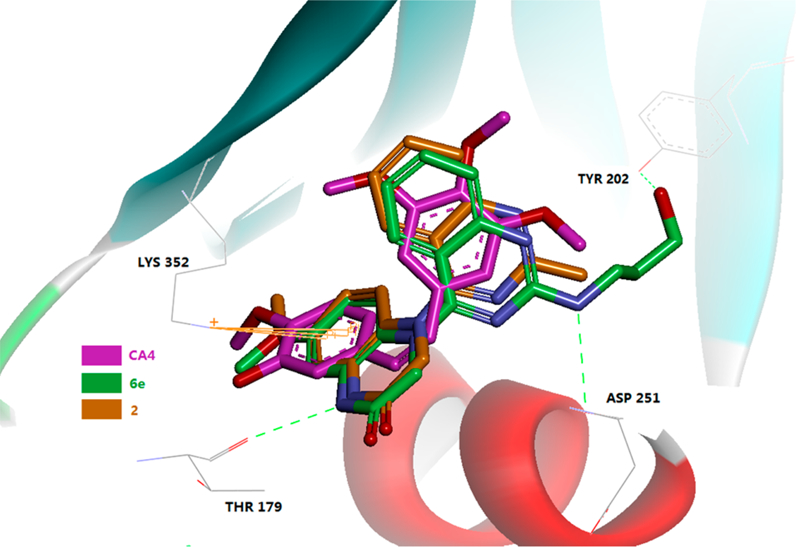
Predicted binding mode of 2 (brown stick) and 6e (green stick) with tubulin (PDB code 5LYJ) and overlapping with CA-4 (pink, the bound of ligand of 5LYJ). Surrounding amino acid side chains are shown in gray stick format and are labeled. Hydrogen bonds are shown by green dashed lines, and the distance between ligands and protein is less than 3 Å.
As representatives, compounds 6d and 6e were further investigated for their effects on the cell cycle (Figure 5). Similarly to CA-4, both compounds induced cell cycle arrest at the G2/M phase. Therefore, this series of 4-(quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one derivatives, including 2 and active series-6, have been identified to have characteristics similar to those of CA-4 and, like CA-4, have potential as new antitumor agents.
Figure 5.
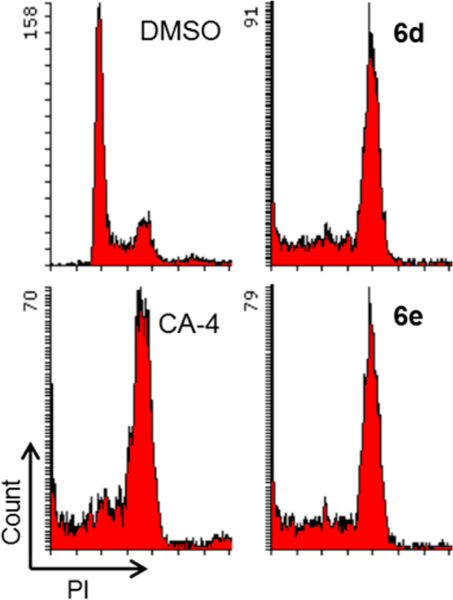
Effects of compounds on cell cycle. A549 cells were treated for 24 h with 6d or 6e at 15 nM. CA-4 (20 nM) or DMSO was used as a tubulin polymerization inhibitor or control, respectively. Fixed and propidium iodide (PI)-stained cells were analyzed by flow cytometry.
Testing of Compound 2 and New Derivatives (6a−t) for Antivascular Effects in Vivo.
Our prior and current studies have revealed that compound 213 and its analogous series-6 derivatives not only exhibit high antiproliferative activity but also inhibit tubulin polymerization and competitive colchicine binding to tubulin (Table 4), as well as arrest cells in the G2/M phase of the cell cycle, indicating that this compound type has characteristics similar to those of CA-4. Thus, we postulated that they might act as new tubulin-binding tumor-VDAs. The antivascular activity of 2 was evaluated on a xenograft tumor tissue section by staining with hematoxylin−eosin (HE) and by immunohistochemistry (IHC) using antibodies to CD31, cleaved caspase-3, and Ki67 (Figure 6). HE staining showed that the nuclei of tumor cells in vehicle controls were large and hyperchromatic, while the nuclei of 2-treated tumor cells became pyknotic. IHC staining of CD31 (endothelial marker) further demonstrated that the numbers and, especially, the sizes of the vessels in the 2-treated groups were reduced (Figure 6). In addition, 2-treated tumors had stronger cleaved caspase-3 (apoptosis marker) staining and weaker Ki67 (proliferation marker) staining, compared to control xenograft tumors. All of these results confirmed that 2 can inhibit proliferation, induce apoptosis, and disrupt tumor vasculature. Therefore, our current study results validate that, like CA-4, compound 2 is a new tubulin-binding tumor-VDA. Most likely, the 6-series compounds, which are analogues of 2, should belong to the same class of new antitubulin agents.
Figure 6.
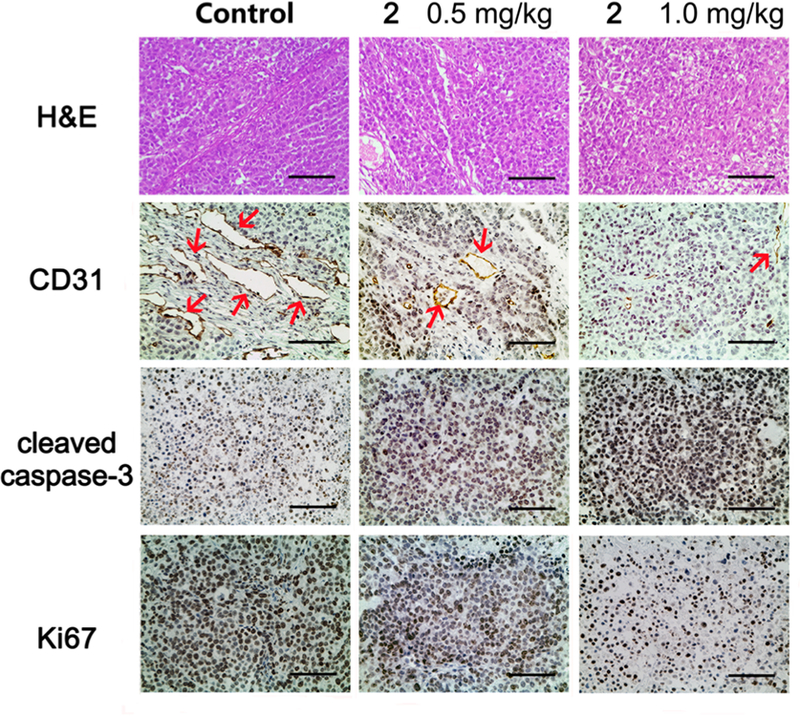
Compound 2 treatment resulted in apoptosis and vascular disruption of NCI-H460 xenograft tumors. Representative images of HE staining and IHC staining of CD31 (endothelial marker), cleaved caspase-3 (apoptosis marker), and Ki67 (proliferation marker) in different treatment groups. Sections were counterstained with hematoxylin. Red arrows in CD31 staining images indicate micro-vessels. Scale bar, 100 μm.
CONCLUSION
In this study, we have identified 7-methoxy-4-(2-methylquinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (2) as a new tubulin-binding tumor-VDA. Compound 2 showed significant dose-dependent antitumor efficacy in vivo in nude mouse H460 subcutaneous xenograft models, suppressing tumor growth by 61.9% at a dose of 1 mg/kg by iv injection every 5 days for 3 weeks without obvious signs of toxicity. It also displayed high antitumor potency in the NIH-NCI60 panel in vitro with low nanomolar GI50 values and reasonable druglike properties, such as an acceptable balance between aqueous solubility and lipophilicity, and moderate metabolic stability in vitro and in vivo. Therefore, compound 2 should be a promising antitumor drug candidate as a new tumor-VDA and merits further development for clinical treatment of various tumors and cancers. With the same scaffold, a new compounds series (6) also exhibited high antiproliferative potency (GI50 < 10 nM) and inhibited tubulin polymerization comparably to paclitaxel and CA-4 in corresponding assays. Thus, these compounds most likely also act as a new class of tubulin-binding tumor-VDAs. New SAR results indicated that (1) the 2-substituent on the quinazoline ring can be modified to enhance antitumor activity and improve druglike properties, (2) the presence of a H-bond donor in the 2-substituent is beneficial to antitumor activity and aqueous solubility, and (3) the length and bulk of 2-substituents should be restricted.
EXPERIMENTAL SECTION
Chemistry.
The proton and carbon nuclear magnetic resonance (1H NMR and 13C NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as internal standard and DMSO as solvent unless otherwise indicated. Mass spectra (MS) and high resolution mass spectra (HRMS) were measured on an API-150 mass spectrometer (ABI, Inc.) and Waters Xevo G2 with an electrospray ionization source, respectively. Melting points were measured with a SGW X-4 Micro-Melting point detector without correction. The MW reactions were performed on a MW reactor from Biotage, Inc. Medium-pressure column chromatography was performed using a CombiFlash Companion system from ISCO, Inc. Thin-layer chromatography (TLC) and preparative TLC were performed on silica gel GF254 plates. Silica gel GF254 was from Qingdao Haiyang Chemical Company. All commercial chemical reagents were purchased from Beijing Chemical Works or InnoChem, Inc. Pooled human liver microsomes (lot no. 20150323H) were purchased from iPhase Bioscience (Beijing) Ltd. Reagents. NADPH, MgCl2, KH2PO4, K2HPO4, reference compounds, and propranolol, as well as HPLC grade acetonitrile (ACN) and methanol (MeOH), were purchased from Sigma-Aldrich. Purities of target compounds were determined by HPLC using the following instruments and conditions: Agilent HPLC-1200 with UV detector and an Agilent Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm), flow rate 0.8 mL/min, UV detection at 254 nm, and injection volume of 15 μL. Mobile elution was conducted with a mixture of solvents A and B (condition 1, ACN/H2O; condition 2, MeOH/H2O). Target compounds were found to be at least 95% pure in the above two HPLC analytical conditions.
2-Chloro-4-(4-methoxy-2-nitro-phenyl)aminoquinazoline (4).
A mixture of 2,4-dichloro-quinazoline (1 g, 5.1 mmol) and 4-methoxy-2-nitroaniline (3, 0.86 g, 5.1 mmol) in anhydrous i-PrOH (50 mL) with a catalytic amount of HCl (conc, 10 drops) was stirred at room temperature (rt) for 5−6 h and monitored by TLC until the reaction was complete. The light yellow solid was removed by filtration and washed with a small amount of i-PrOH. Next, the collected solid was stirred in a saturated aqueous NaHCO3 solution, refiltered, washed with water until pH = 7, and dried to obtain 4 as an orange solid (1.42g, 85% yield). Mp 214−215 °C; 1H NMR δ 3.93 (3H, s, OCH3), 7.44 (1H, dd, J = 9.0 and 2.8 Hz, ArH-5′), 7.57 (1H, d, J = 2.8 Hz, ArH-3′), 7.59 (1H, d, J = 9.0, ArH-6′), 7.65 (1H, t, J = 7.6 Hz, ArH-6), 7.70 (1H, d, J = 7.6 Hz, ArH-5), 7.92 (1H, t, J = 7.6 Hz, ArH-7), 8.46 (1H, d, J = 7.6 Hz, ArH-8), 10.61 (1H, s, NH); MS m/z (%) 331 (M + 1, 100), 333 (M + 3, 33).
4-(2-Chloroacetylamino-4-methoxyphenyl)amino-2-chloroquinazoline (5).
A mixture of 4 (330 mg, 1 mmol) and zinc powder (660 mg, 1 mmol) in 50 mL of CH2Cl2 in the presence of 0.4 mL of HOAc was stirred at 0 °C for 0.5 h. After the solid was removed, the filtrate was concentrated to obtain the amine product. It was immediately dissolved in acetone (25 mL), anhydrous K2CO3 (138 mg, 1 mmol) was added, and the mixture was cooled to 0 °C. Chloroacetyl chloride (0.1 mL, excess) was dropped slowly into the mixture, which was kept at 0 °C with stirring for another 1−2 h. Then the mixture was poured into ice−water. The precipitated solid was removed by filtration, washed successively with water to neutral and brine, and dried to obtain 5 as a pink solid (210 mg, 56% yield). Mp 199−200 °C; 1H NMR δ 3.78 (3H, s, OCH3), 4.23 (2H, s, CH2), 6.83 (1H, dd, J = 8.4 and 2.8 Hz, PhH-5′), 7.34 (1H, d, J = 2.8 Hz, PhH-3′), 7.48 (1H, d, J = 8.4 Hz, PhH-6′), 7.59 (1H, t, J = 7.6 Hz, ArH-6), 7.67 (1H, d, J = 7.6 Hz, ArH-5), 7.83 (1H, t, J = 7.6 Hz, ArH-7), 8.41 (1H, d, J = 7.6 Hz, ArH-8), 9.57 (1H, s, NH), 10.87 (1H, br, NH); MS m/z (%) 377 (M + 1, 100), 379 (M + 3, 58).
4-(2-Chloroquinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6a).
A mixture of 5 (380 mg, 1 mmol) and anhydrous K2CO3 (138 mg, 1 mmol) in DMF (10 mL) was heated to 100 °C for 1 h until the reaction was complete as judged by TLC monitoring. The mixture was poured into ice−water, and the solid product 6a was removed by filtration, washed with water, and dried to give 333 mg in a 98% yield. Yellow solid; mp 250−251 °C; 1H NMR δ 3.74 (3H, s, OCH3), 4.01 (2H, s, CH2), 6.42 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6′), 6.65 (1H, d, J = 2.8 Hz, ArH-8′), 6.84 (1H, d, J = 8.8 Hz, ArH-5′), 7.35 (1H, t, J = 8.4 Hz, ArH-6), 7.41 (1H, d, J = 8.4 Hz, ArH-5), 7.77 (1H, t, J = 8.4 Hz, ArH-7), 7.78 (1H, d, J = 8.4 Hz, ArH-8), 10.87 (1H, s, NH); 13C NMR (DMSO-d6) δ 51.7, 55.9, 102.6, 108.2, 115.6, 122.2, 22.8, 126.3, 126.6, 128.0, 132.7, 134.7, 153.4, 155.8, 157.8, 161.2, 167.8; MS m/z (%) 341 (M + 1, 100), 343 (M + 3, 34); HRMS m/z calcd for C17H14ClN4O2 [M + H]+ 341.0805, found 341.0867, 343.0843; HPLC purity 98.2%.
7-Methoxy-4-(2-(methylamino)quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (6b).
A mixture of 6a (100 mg, 0.29 mmol) and methylamine (1 mL) was heated via MW to 80 °C for 20 min. The mixture was poured into ice−water, and the pH was adjusted to 7 with dilute aq. HCl. The solid was collected, washed with water, and dried to give pure 6b as a yellow solid (59 mg, 61% yield). Mp 274−275 °C; 1H NMR δ 2.85 (3H, d, J = 4.8 Hz, NCH3), 3.67 (3H, s, OCH3), 4.30 (2H, s, CH2), 6.38 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.52 (1H, d, J = 8.8 Hz, ArH-5), 6.61 (1H, d, J = 2.8 Hz, ArH-8), 6.85 (1H, t, J = 8.0 Hz, ArH-6′), 7.05 (1H, q, J = 4.8 Hz, NH-2′), 7.18 (1H, d, J = 8.0 Hz, ArH-5′), 7.39 (1H, d, J = 8.0 Hz, ArH-8′), 7.47 (1H, t, J = 8.0 Hz, ArH-7′), 10.73 (1H, s, NH); 13C NMR (DMSO-d6) δ 28.5, 51.5, 55.8, 102.6, 108.0, 120.8, 124.9, 126.2(x2), 131.7, 133.4 (x2), 154.5, 156.4 (x2), 159.9, 160.5, 168.4; MS m/z (%) 336 (M + 1, 100); HRMS m/z calcd for C18H18N5O2 [M + H]+ 336.1460, found 336.1501; HPLC purity 97.5%.
General Procedure for Preparing Compounds 6c−6n under MW Irradiation.
A mixture of 6a and an amine (excess) in anhydrous i-PrOH was heated under MW irradiation for 15−30 min. The reaction was monitored by TLC or LC-MS until complete. The mixture was poured into ice−water and was neutralized with dilute HCl (ca.10%) to pH 7. Precipitated solid was collected, washed with water, and dried to obtain the corresponding products.
4-(2-(Ethylamino)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6c).
A mixture of 6a (50 mg, 0.14 mmol) and ethylamine (65−75%, 1 mL, excess) in anhydrous i-PrOH (1 mL) was heated to 80 °C under MW irradiation for 30 min to obtain pure 6c as a yellow solid (25 mg, 48% yield). Mp 277−278 °C; 1H NMR δ 1.13 (3H, t, J = 7.2 Hz, CH3), 3.34 (2H, q, J = 7.2 Hz, NCH2), 3.67 (3H, s, OCH3), 4.31 (2H, s, CH2), 6.37 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.51 (1H, d, J = 8.8 Hz, ArH-5), 6.61 (1H, d, J = 2.8 Hz, ArH-8), 6.84 (1H, t, J = 8.0 Hz, ArH-6′), 7.07 (1H, bs, NH), 7.16 (1H, d, J = 8.0 Hz, ArH-5′), 7.36 (1H, d, J = 8.0, ArH-8′), 7.46 (1H, t, J = 8.0 Hz, ArH-7′); 13C NMR (DMSO-d6) δ 15.3, 36.0, 51.5, 55.8, 102.6, 108.0, 112.7, 120.7, 120.8, 124.8, 126.2(x2), 131.7, 133.4, 154.4, 156.4, 159.2, 160.6, 168.4; MS m/z (%) 350 (M + 1, 100); HRMS m/z calcd for C19H20N5O2 [M + H]+ 350.1617, found 350.1626; HPLC purity 96.2%.
4-(2-(n-Butylamino)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6d).
A mixture of 6a (60 mg, 0.18 mmol) and n-butylamine (1 mL, excess) in anhydrous i-PrOH (1 mL) was heated to 120 °C under MW irradiation for 20 min. The mixture was poured into ice−water and was neutralized with dilute HCl (ca.10 mL) to pH 7. The mixture was extracted with EtOAc three times. The combined organic phases were washed with water and brine successively. After solvent was removed under reduced pressure, residue was washed with a small amount of cold acetone to give pure 6d as a yellow solid (20 mg, 29% yield). Mp 204−205 °C; 1H NMR δ 0.87 (3H, t, J = 7.6 Hz, CH3), 1.32 and 1.52 (each 2H, m, J = 7.6 Hz, CH2), 3.31 (2H, m, NCH2), 3.67 (3H, s, OCH3), 4.31 (2H, s, CH2), 6.38 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.51 (1H, d, J = 8.8 Hz, ArH-5), 6.61 (1H, d, J = 2.8 Hz, ArH-8), 6.83 (1H, t, J = 8.0 Hz, ArH-6′), 7.08 (1H, m, NH), 7.16 (1H, d, J = 8.0 Hz, ArH-5′), 7.36 (1H, d, J = 8.0 Hz, ArH-8′), 7.46 (1H, t, J = 8.0 Hz, ArH-7′), 10.73 (1H, s, NH); MS m/z (%) 378 (M + 1, 100); HRMS m/z calcd for C21H24N5O2 [M + H]+ 378.1930, found 378.2017; HPLC purity 97.8%.
4-(2-(3-Hydroxypropylamino)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6e).
A mixture of 6a (100 mg, 0.29 mmol) and 3-aminopropanol (1 mL, excess) in anhydrous i-PrOH (0.5 mL) with one drop of H2SO4 (conc) was heated to 120 °C under MW irradiation for 20 min to obtain 82 mg of 6e (75% yield, white solid), mp 179−180 °C; 1H NMR δ 1.69 (2H, m, CH2), 3.38 and 3.45 (each 2H, m, OCH2 and NCH2), 3.67 (3H, s, OCH3), 4.31 (2H, s, CH2), 4.59 (1H, br, OH), 6.38 (1H, dd, J = 9.2 and 2.8 Hz, ArH-6), 6.53 (1H, d, J = 9.2 Hz, ArH-5), 6.60 (1H, d, J = 2.8 Hz, ArH-8), 6.85 (1H, t, J = 8.0 Hz, ArH-6′), 7.06 (1H, t, J = 5.6 Hz, NH), 7.17 (1H, d, J = 8.0 Hz, ArH-5′), 7.35 (1H, d, J = 8.0 Hz, ArH-8′), 7.47 (1H, t, J = 8.0 Hz, ArH-7′); MS m/z (%) 380 (M + 1, 100); HRMS m/z calcd for C20H22N5O3 [M + H]+ 380.1723, found 380.1730; HPLC purity 99.1%.
4-(2-(Cyclopropylamino)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6f).
A mixture of 6a (100 mg, 0.29 mmol) and cyclopropylamine (1 mL, excess) in DMA (2 mL) was heated to 155 °C under MW irradiation for 20 min to give pure 6f as a yellow solid (68 mg, 65% yield). Mp 269−270 °C; 1H NMR δ 0.49 and 0.65 (each 2H, m, CH2), 2.81 (1H, m, CH), 3.67 (3H, s, OCH3), 4.32 (2H, s, CH2), 6.38 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.54 (1H, d, J = 8.8 Hz, ArH-5), 6.61 (1H, d, J = 2.8 Hz, ArH-8), 6.87 (1H, t, J = 8.0 Hz, ArH-6′), 7.15 (1H, d, J = 8.0 Hz, ArH-5′), 7.29 (1H, m, NH), 7.41 (1H, d, J = 8.0 Hz, ArH-8′), 7.48 (1H, t, J = 8.0 Hz, ArH-7′), 10.72 (1H, s, NH); MS m/z (%) 362 (M + 1, 100); HRMS m/z calcd for C20H20N5O2 [M + H]+ 362.1617, found 362.1658; HPLC purity 95.0%.
4-(2-(Cyclopentylamino)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6g).
A mixture of 6a (100 mg, 0.29 mmol) and cyclopentylamine (1 mL, excess) in i-PrOH (1 mL) was heated to 120 °C under MW irradiation for 30 min to give pure 6g as a yellow solid (70 mg, 62% yield). Mp 268−269 °C; 1H NMR δ 1.51 (4H, m, CH2 × 2), 1.66 (2H, m, CH2), 1.90 (2H, m, CH2), 3.67 (3H, s, OCH3), 4.27 (1H, m, NCH), 4.31 (2H, s, CH2), 6.36 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.51 (1H, d, J = 8.8 Hz, ArH-5), 6.61 (1H, d, J = 2.8 Hz, ArH-8), 6.84 (1H, t, J = 8.0 Hz, ArH-6′), 7.07 (1H, br, NH), 7.16 (1H, d, J = 8.0 Hz, ArH-5′), 7.31 (1H, d, J = 8.0 Hz, ArH-8′), 7.48 (1H, t, J = 8.0 Hz, ArH-7′), 10.72 (1H, br, NH); 13C NMR (DMSO-d6) δ 24.0(x2), 32.9(x2), 51.5, 52.7, 55.8, 102.6, 108.0, 112.5, 120.7, 120.8, 124.9, 126.1(x2), 131.7, 133.4, 154.4, 156.4, 158.9, 160.5, 168.4; MS m/z (%) 390 (M + 1, 100); HRMS m/z calcd for C22H24N5O2 [M + H]+ 390.1930, found 390.1984; HPLC purity 99.1%.
7-Methoxy-4-(2-(pyrrolidin-1-yl)quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (6h).
A mixture of 6a (50 mg, 0.15 mmol), 4-picolylamine (1 mL, excess), and K2CO3 (30 mg, 0.21 mmol) in i-PrOH (2 mL) was heated to 60 °C under MW irradiation for 15 min to obtain 6h as a white solid (42 mg, 58% yield). Mp 217−218 °C; 1H NMR δ 1.90 (4H, m, CH2 × 2), 3.55 (4H, m, NCH2 × 2), 3.67 (3H, s, OCH3), 4.36 (2H, s, CH2), 6.38 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.51 (1H, d, J = 2.8 Hz, ArH-8), 6.62 (1H, d, J = 8.8 Hz, ArH-5), 7.17 (1H, t, J = 8.0 Hz, ArH-6′), 7.19 (1H, d, J = 8.0 Hz, ArH-5′), 7.38 (1H, d, J = 8.0 Hz, ArH-8′), 7.47 (1H, t, J = 8.0 Hz, ArH-7′), 10.72 (1H, s, NH); 13C NMR (DMSO-d6) δ 25.6(x2), 46.9(x2), 51.4, 55.9, 102.6, 107.9, 111.8, 120.7(x2), 124.8, 126.2, 126.3, 131.7, 133.4, 154.7, 154.4, 157.4, 160.2, 168.5; MS m/z (%) 376 (M + 1, 100); HRMS m/ z calcd for C21H22N5O2 [M + H]+ 376.1773, found 376.1784; HPLC purity 95.4% (MeOH/H2O).
4-(2-(3,3-Difluoropyrrolidin-1-yl)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2 (1H)-one (6i).
A mixture of 6a (50 mg, 0.15 mmol) and 3,3-difluoropyrrolidine hydrochloride (30 mg, 0.29 mmol) and K2CO3 (30 mg, 0.21 mmol) in i-PrOH (2 mL) was heated to 120 °C under MW irradiation for 20 min to obtain 6i as a brown solid (52 mg, 85% yield). Mp 247−248 °C; 1H NMR δ 2.51 (2H, m, CH2), 3.68 (3H, s, OCH3), 3.79 (2H, t, J = 7.2 Hz, NCH2), 3.98 (2H, t, JH−F = 12.8 Hz, NCH2CF2), 4.41 (2H, s, 3′-CH2), 6.38 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.56 (1H, d, J = 8.8 Hz, ArH-5), 6.63 (1H, d, J = 2.8 Hz, ArH-8), 6.92 (1H, t, J = 8.0 Hz, ArH-6′), 7.23 (1H, d, J = 8.0 Hz, ArH-5′), 7.45 (1H, d, J = 8.0 Hz, ArH-8′), 7.53 (1H, t, J = 8.0 Hz, ArH-7′), 10.87 (1H, s, NH); 13C NMR (DMSO-d6) δ 33.8, 44.5, 51.4, 53.6, 55.8, 102.7, 108.0, 112.2, 120.9, 121.6, 124.4, 126.2, 126.5, 128.7, 132.0, 133.7, 154.2, 156.6, 157.1, 160.4, 168.4; MS m/z (%) 412 (M + 1, 100); HRMS m/z calcd for C21H20 F2N5O2 [M + H]+ 412.1585, found 412.1664; HPLC purity 95.6%.
4-(2-(3,3-Difluoroazetidin-1-yl)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H) -one (6j).
A mixture of 6a (100 mg, 0.29 mmol) and 3,3-difluoroazetidine hydrochloride (45 mg, 1.1 mmol) in i-PrOH (2 mL) in the presence of one drop H2SO4 (conc) was heated to 120 °C under MW irradiation for 20 min. The crude product was dissolved in MeOH and left uncovered overnight. The precipitated solid was collected to obtain pure 6j as a yellow solid (40 mg, 35% yield). Mp 280−281 °C; 1H NMR δ 3.68 (3H, s, OCH3), 4.41(2H, s, CH2), 4.51 (4H, t, JH−F = 12.8 Hz, NCH2 × 2), 6.39 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.61 (1H, d, J = 8.8 Hz, ArH-5), 6.63 (1H, d, J = 2.8 Hz, ArH-8), 6.99 (1H, t, J = 8.0 Hz, ArH-6′), 7.25 (1H, d, J = 8.0 Hz, ArH-5′), 7.51 (1H, d, J = 8.0 Hz, ArH-8′), 7.58 (1H, t, J = 8.0 Hz, ArH-7′), 10.78 (1H, s, NH); MS m/z (%) 398 (M + 1, 100); HRMS m/z calcd for C20H18 F2N5O2 [M + H]+ 398.1429, found 398.1441 (M + 1, 100); HPLC purity 97.8%.
7-Methoxy-4-(2-(3-methoxyazetidin-1-yl)quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (6k).
A mixture of 6a (50 mg, 0.15 mmol), methoxyazetidine (50 mg, 0.41 mmol), and K2CO3 (50 mg, 0.36 mmol) in anhydrous i-PrOH (2 mL) was heated to 120 °C under MW irradiation for 15 min to obtain pure 6k as a gray solid (23 mg, 39% yield) after recrystallization from MeOH. Mp 221−222 °C; 1H NMR δ 3.23 (3H, s, OCH3), 3.68 (3H, s, OCH3), 3.88 (2H, d, J = 7.2 Hz, NCH2), 4.25 (2H, d, J = 7.2 Hz, NCH2), 4.28 (1H, m, CH), 4.36 (2H, s, CH2), 6.38 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.54 (1H, d, J = 8.8 Hz, ArH-5), 6.62 (1H, d, J = 2.8 Hz, ArH-8), 6.90 (1H, t, J = 8.0 Hz, ArH-6′), 7.20 (1H, d, J = 8.0 Hz, ArH-5′), 7.42 (1H, d, J = 8.0 Hz, ArH-8′), 7.51 (1H, t, J = 8.0 Hz, ArH-7′), 10.72 (1H, s, NH); 13C NMR (DMSO-d6) δ 51.6, 55.8, 55.9, 57.4, 69.8(x2), 102.6, 108.0, 112.5, 120.9, 121.5, 126.2, 127.4, 131.8, 133.6(x2), 154.1, 156.6, 160.0, 160.4, 168.4; MS m/z (%) 392 (M + 1, 100); HRMS m/z calcd for C21H22N5O2 [M + H]+ 392.1723, found 392.1729; HPLC purity 96.0%.
4-(2-(4-Cyanophenyl)aminoquinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6l).
A mixture of 6a (50 mg, 0.15 mmol) and 4-aminobenzonitrile (30 mg, 0.25 mmol) in i-PrOH (2 mL) in the presence of H2SO4 (conc, 1 drop) was heated to 120 °C under MW irradiation for 20 min to obtain pure 6l as a yellow solid (50 mg, 79% yield) after being recrystallized from MeOH. Mp 183−184 °C; 1H NMR δ 3.71 (3H, s, OCH3), 4.57 (2H, s, CH2), 6.47 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.66 (1H, d, J = 8.8 Hz, ArH-5), 6.89 (1H, d, J = 2.8 Hz, ArH-8), 7.16 (1H, t, J = 8.0 Hz, ArH-6′), 7.37 (2H, d, J = 8.2 Hz, 2′-PhH), 7.65 (1H, d, J = 8.0 Hz, ArH-5′), 7.70 (1H, t, J = 8.0 Hz, ArH-7′), 7.75 (2H, d, J = 8.2 Hz, 2′-PhH), 7.95 (1H, d, J = 8.0 Hz, ArH-8′), 10.58 (1H, brs, NH), 10.87 (1H, s, NH); MS m/z (%) 423 (M + 1, 100); HRMS m/z calcd for C24H19N6O2 [M + H]+ 423.1569, found 423.1649; HPLC purity 95.3%.
4-(2-(3-Cyanophenyl)aminoquinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6m).
A mixture of 6a (50 mg, 0.15 mmol) and 3-aminobenzonitrile (30 mg, 0.25 mmol) in i-PrOH (2 mL) in the presence of H2SO4 (conc,1 drop) was heated to 120 °C under MW irradiation for 20 min to obtain 6m as a yellow solid (55 mg, 88% yield). Mp 262−263 °C; 1H NMR δ 3.71 (3H, s, OCH3), 4.57 (2H, s, CH2), 6.50 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.65 (1H, d, J = 2.8 Hz, ArH-8), 7.00 (1H, d, J = 8.8 Hz, ArH-5), 7.22 (1H, m, ArH-6′), 7.41 (1H, s, ArH-3″), 7.56 (2H, m, ArH-5′ and ArH-4″), 7.65 (1H, d, J = 7.6 Hz ArH-8′), 7.76 (1H, t, J = 7.6 Hz ArH-7′), 7.92 (1H, m, ArH-5″), 8.13 (1H, m, ArH-6″), 10.44 (1H, br, NH), 10.84 (1H, s, 1-NH); 13C NMR (DMSO-d6) δ 52.2, 56.0, 102.6, 108.6, 112.3, 119.1, 121.3, 124.1, 124.3, 124.5, 125.2, 127.0, 127.1, 127.3, 128.6, 130.9, 133.3, 135.9, 139.0, 151.9, 152.1, 158.8, 161.6, 167.1; MS m/z (%) 423 (M + 1, 100); HRMS m/z calcd for C24H19N6O2 [M + H]+ 423.1569, found 423.1571; HPLC purity 96.6%.
7-Methoxy-4-(2-((pyridin-4-ylmethyl)amino)quinazolin-4-yl)-3,4-dihydroquinoxalin-2(1H)-one (6n).
A mixture of 6a (35 mg, 0.1 mmol), pyridin-4-ylmethanamine (1 mL, excess), and K2CO3 (30 mg, 0.21 mmol) in i-PrOH (2 mL) was heated to 120 °C under MW irradiation for 1 h to obtain 6n as a yellow solid (20 mg, 47% yield). Mp 219−220 °C; 1H NMR δ 3.71 (3H, s, OCH3), 4.31 (2H, s, 3-CH2), 4.55 (2H, d, J = 6.0 Hz, CH2), 6.36 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.55 (1H, d, J = 8.8 Hz, ArH-5), 6.60 (1H, d, J = 2.8 Hz, ArH-8), 6.67 (1H, t, J = 7.6 Hz, ArH-6′), 7.18 (1H, d, J = 7.6 Hz, ArH-5′), 7.33 (3H, m, ArH-8′ and PyH), 7.47 (1H, t, J = 7.6 Hz, ArH-7′), 7.73 (1H, m, NH), 8.43 (2H, m, PyH), 10.73 (1H, br, 1-NH); MS m/z (%) 413 (M + 1, 100); HRMS m/z calcd for C23H21N6O2 [M + H]+ 413.1726, found 413.1799; HPLC purity 97.8%.
4-(2-Ethoxyquinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6o).
Ethanol (2 mL) was cooled to 0 °C, and NaH (260 mg, 11 mmol) was carefully added in portions with stirring over 1 h. Compound 6a (100 mg, 0.29 mmol) was then added into the solution with stirring and kept at the same temperature for another 0.5 h until hydrogen was no longer formed. The mixture was then heated to 95 °C under MW irradiation for 5 min and monitored by TLC and LC-MS until the reaction was complete. The mixture was poured into ice− water, and the precipitated solid was collected, washed with water, and dried to obtain pure 6o as a brown solid (85 mg, 85% yield). Mp 233− 234 °C; 1H NMR δ 1.34 (3H, t, J = 7.2 Hz, CH3),3.68 (3H, s, 7′-OCH3), 4.41 (4H, m, OCH2 and 3′-CH2), 6.39 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.65 (2H, m, ArH-5 and −8), 7.12 (1H, m, ArH-6′), 7.35 (1H, d, J = 8.0 Hz, ArH-5′), 7.63 (2H, m, ArH-8′, ArH-7′), 10.78 (1H, br s, NH); 13C NMR (DMSO-d6) δ 15.0, 51.6, 55.9, 62.9, 102.7, 108.1, 114.2, 121.4, 123.7, 124.0, 126.2, 127.3, 132.3, 134.0, 153.5, 157.0, 161.9, 162.2, 167.9; MS m/z (%) 351 (M + 1, 100); HRMS m/ z calcd for C19H19N4O3 [M + H]+ 351.1457, found 351.1507; HPLC purity 96.2%.
4-(2-(Cyclopropylmethoxy)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6p).
The preparation was the same as described for 6o. The mixture of cyclopropylcarbinol (2 mL), NaH (260 mg, 11 mmol), and 6a (100 mg, 0.29 mmol) was stirred at 0 °C for about 1.5 h and then heated to 95 °C under MW irradiation for 5 min to obtain pure 6p as a yellow solid (90 mg, 83% yield). Mp 215− 216 °C; 1H NMR δ 0.34 (2H, m, CH2), 0.55 (2H, m, CH2), 0.81 (1H, m, CH), 3.71 (3H, s, OCH3), 4.17 (2H, d, J = 7.2 Hz, OCH2), 4.41 (2H, s, CH2), 6.39 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.64 (2H, m, ArH-5 and −8), 7.13 (1H, m, ArH-6′), 7.35 (1H, d, J = 8.0 Hz, ArH-5′), 7.63 (2H, m, ArH-8′ and −7′), 10.77 (1H, br, NH); 13C NMR (DMSO-d6) δ 3.74(x2), 10.5, 51.6, 55.8, 71.8, 102.6, 108.1, 114.2, 121.4, 123.7, 123.9, 126.2, 127.3, 132.2, 134.0, 153.2, 157.0, 161.9, 162.2, 168.2; MS m/z (%) 377 (M + 1, 100); HRMS m/z calcd for C21H21N4O3 [M + H]+ 377.1614, found 377.1615; HPLC purity 95.1%.
4-(2-Acetoxymethylquinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6q).
A mixture of 13 prepared as described below (390 mg, 0.94 mmol) and anhydrous K2CO3 (260 mg, 1.88 mmol) in DMA (3 mL) was heated at 100 °C for 1 h. After the reaction was complete as judged by LC-MS monitoring, the mixture was poured into ice water and extracted with EtOAc three times. The combined organic phases were washed successively with water and brine and dried over anhydrous Na2SO4. After removal of solvent in vacuo, the residue was purified by flash column chromatography (gradient elution, petroleum ether/EtOAc, 0−30%) to afford 6q as a yellow solid (355 mg, 99% yield). Mp 192−194 °C; 1H NMR (CDCl3) δ 2.26 (3H, s, CH3), 3.80 (3H, s, OCH3), 4.66 (2H, s, NCH2), 5.35 (2H, s, OCH2), 6.40 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.54 (1H, d, J = 2.8 Hz, ArH-8), 6.63 (1H, d, J = 8.8 Hz, ArH-5),7.25 (1H, t, J = 8.0 Hz, ArH-6′), 7.48 (1H, d, J = 8.0 Hz, ArH-5′), 7.72 (1H, t, J = 8.0 Hz, ArH-7′), 7.92 (1H, d, J = 8.0 Hz, ArH-8′), 8.37 (1H, s, NH); 13C NMR (400 Hz, CDCl3) δ 21.0, 51.1, 55.7, 66.4, 102.4, 108.6, 115.7, 120.8, 123.9, 125.6, 125.8, 128.8, 130.4, 133.2, 152.1, 157.2, 159.5, 160.4, 169.3, 170.9; MS m/z (%) 379 (M + 1, 100); HRMS m/z calcd for C20H19N4O4 [M + H]+ 379.1406, found 379.1431; HPLC purity 95.5%.
4-(2-(Hydroxymethyl)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6r).
To a solution of 6q (355 mg, 0.94 mmol) in MeOH (8 mL) was added aqueous NaOH (10%, ca.1.0 mL, excess) at rt. The mixture was stirred for 1.5 h. Then, the pH was adjusted to 7 with aq. HCl (10%), and the mixture was extracted with EtOAc three times. The combined organic layer was washed successively with water and brine and dried over anhydrous Na2SO4. After removal of solvent in vacuo, 6r was obtained as a yellow powder (310 mg, 98% yield). Mp 216−218 °C; 1H NMR (CDCl3) δ 3.80 (3H, s, OCH3), 4.00 (1H, s, OH), 4.67 (1H, s, CH2), 4.85 (1H, s, OCH2), 6.41 (1H, dd, J = 8.8 and 2.8 Hz, ArH-6), 6.57 (1H, d, J = 2.8 Hz, ArH-8), 6.64 (1H, d, J = 8.8 Hz, ArH-5), 7.26 (1H, t, J = 8.0 Hz, ArH-6′),7.51 (1H, d, J = 8.0 Hz, ArH-5′), 7.73 (1H, t, J = 8.0 Hz, ArH-7′), 7.92 (1H, d, J = 8.0 Hz, ArH-8′), 8.82 (1H, s, NH); 13C NMR (400 Hz, CDCl3) δ 51.2, 55.7, 64.4, 102.5, 108.7, 115.9, 120.8, 123.9, 125.6, 125.8, 128.3, 130.4, 133.4, 151.4, 157.2, 159.6, 163.8, 169.2; MS m/z (%) 337 (M + 1, 100); HRMS m/z calcd for C18H17N4O3 [M + H]+ 337.1301, found 337.1290; HPLC purity 96.3%.
4-(2-((4-Ethoxy-4-oxobutoxy)methyl)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6s).
A mixture of 6r (165 mg, 0.5 mmol) and Cs2CO3 (325 mg, 1 mmol) in CH3CN (10 mL) was heated to reflux for 0.5 h. Then, ethyl 4-bromobutyrate (325 mg 1.0 mmol) was added dropwise at the same temperature, and stirring was continued for 3 h. The mixture was passed through Celite, which was washed several times with CH3CN. The combined filtrate was concentrated in vacuo to obtain 6s as a yellow oil, which was used in the next reaction without further purification. However, pure 6s was obtained as a yellow solid by crystallization from EtOAc. Mp 160−161 °C; 1H NMR (CDCl3) δ 1.26 (3H, t, J = 7.2 Hz, CH3), 2.01 (2H, m, CH2), 2.45 (2H, t, J = 7.2 Hz, COCH2), 3.86 (3H, s, OCH3), 4.07 (2H, t, J = 7.2 Hz, OCH2), 4.15 (2H, q, J = 7.2, OCH2), 4.65 (2H, s, CH2), 4.83 (2H, s, ArCH2O), 6.42 (1H, dd, J = 8.8 and 2.4 Hz, ArH-6), 6.66 (1H, d, J = 8.8 Hz, ArH-5), 7.01 (1H, d, J = 2.4 Hz, ArH-8), 7.24 (1H, t, J = 8.0 Hz, ArH-6′), 7.46 (1H, d, J = 8.0 Hz, ArH-5′), 7.72 (1H, t, J = 8.0 Hz, ArH-7′), 7.92 (1H, d, J = 8.0 Hz, ArH-8′); 13C NMR (400 Hz, CDCl3) δ 14.3, 22.1, 31.1, 41.7, 51.7, 55.9, 60.8, 64.5, 103.0, 108.0, 115.9, 121.0, 125.7 (×2), 126.0, 128.4, 133.0, 133.4, 151.5, 157.6, 159.4, 163.9, 167.1, 173.1; MS m/z (%) 451 (M + 1, 100); HRMS m/z calcd for C24H27N4O5 [M + H]+ 451.1981, found 451.2083; HPLC purity 95.7%.
4-(2-((4-(Hydroxyamino)-4-oxobutoxy)methyl)quinazolin-4-yl)-7-methoxy-3,4-dihydroquinoxalin-2(1H)-one (6t).
A solution of 6s (225 mg, 0.5 mmol) in CH2Cl2 and MeOH (9 mL, 1:2, v/v) was cooled to 0 °C. Aqueous hydroxylamine, prepared from hydroxylamine hydrochloride (1.04 g, 1.5 mmol) and NaOH (0.8 g, 2.0 mmol) in 2 mL of water, was added dropwise into the solution of 6s with stirring at 0 °C, and the mixture was stirred for an additional 1 h. After organic solvent was removed under reduced pressure, the water phase was adjusted to pH 7−8 with HOAc and extracted with EtOAc three times. The combined organic phase was washed with water and brine and dried over anhydrous Na2SO4. After solvent was removed in vacuo, the residue was recrystallized from EtOAc to obtain pure 6t as a yellow solid (50 mg), mp 196−198 °C; 1H NMR (CDCl3) δ 1.84 (2H, m, CH2), 2.04 (2H, t, J = 7.2 Hz, C(O)CH2), 3.77 (3H, s, OCH3), 3.95 (2H, t, J = 7.2 Hz, OCH2), 4.60 (2H, s, CH2), 4.61 (2H, s, ArCH2O), 5.20 (1H, s, OH), 6.48 (1H, dd, J = 8.8 and 2.4 Hz, ArH-6′), 6.67 (1H, d, J = 8.8 Hz, ArH-5′), 7.02 (1H, d, J = 2.4 Hz, ArH-8′), 7.31 (1H, t, J = 8.0 Hz, ArH-6), 7.36 (1H, d, J = 8.0 Hz, ArH-5), 7.76 (1H, t, J = 8.0 Hz, ArH-7), 7.83 (1H, d, J = 8.0 Hz, ArH-8), 8.71 (1H, s, NH), 10.38 (1H, s, NH); 13C NMR (400 Hz, DMSO-d6) δ 22.9, 29.6, 40.2, 51.7, 56.1, 65.4, 103.4, 108.4, 115.7, 121.2, 125.9, 126.1, 126.2, 128.6, 133.0, 133.8, 152.0, 157.4, 159.2, 165.3, 167.0, 169.1; MS m/z (%) 438 (M + 1, 100); HRMS m/z calcd for C22H24N5O5 [M + H]+ 438.1777, found 438.1866; HPLC purity 95.1%.
2-Chloromethyl-3H-quinazolin-4-one (8).
Compound 8 was prepared from commercially available methyl 2-aminobenzoate (7) and 2-chloroacetonitrile according to the literature method.26 Mp 240 °C (dec.); 1H NMR δ 4.51 (2H, s, CH2), 7.51(1H, t, J = 8.0 Hz, ArH-6), 7.64 (1H, d, J = 8.0 Hz, ArH-8), 7.80 (1H, t, J = 8.0 Hz, ArH-7), 8.08 (1H, d, J = 8.0 Hz, ArH-5), 12.57 (1H, s, NH); MS m/z (%) 195 (M + 1, 100), 197 (M + 3, 33).
2-(Chloromethyl)quinazolin-4-yl 4-methylbenzenesulfonate (9).
A mixture of 8 (500 mg 2.58 mmol), p-toluenesulfonyl chloride (880 mg, 4.6 mmol), Et3N (521 mg 5.16 mmol), and DMAP (3 mg, 0.026 mmol) in CH2Cl2 (20 mL) was stirred at rt for 3 h until the reaction was complete as judged by TLC monitoring. Solvent was removed under reduced pressure, and the residue was purified by flash column chromatography (gradient elution, EtOAc/petroleum ether, 0−15%) to obtain 9 as a pale yellow solid (790 mg, 88% yield). Mp 149−151 °C; 1H NMR (CDCl3) δ 2.46 (3H, s, CH3), 4.73 (2H, s, CH2), 7.38 (2H, d, J = 8.4 Hz, PhH), 7.68 (1H, t, J = 8.4 Hz, ArH-6), 7.93 (1H, t, J = 8.4 Hz, ArH-7), 7.97 (1H, d, J = 8.4 Hz, ArH-5), 8.18 (2H, d, J = 8.4 Hz, PhH), 8.20 (1H, d, J = 8.4 Hz, ArH-8); MS m/z (%) 349 (M + 1, 100), 351 (M + 3, 33).
2-Chloromethyl-4-(4-methoxy-2-nitrophenyl)aminoquinazoline (10).
A mixture of 9 (500 mg 1.43 mmol) and 3 (240 mg 1.43 mmol) in i-PrOH and CH2Cl2 (15 mL, v/v 2:1) was stirred at 45 °C for 6 h until the reaction was complete as judged by TLC monitoring. After solvent was removed, the residue was dissolved in 50 mL of CH2Cl2, washed with saturated NaHCO3, dried over anhydrous Na2SO4, and concentrated to yield crude product 10, which was used in the next step without further purification. Orange solid; Mp 180−181 °C; 1H MR δ 3.90 (3H, s, OCH3), 4.59 (2H, s, CH2), 7.38 (1H, dd, J = 9.2 and 3.2 Hz, ArH-5′), 7.67 (1H, t, J = 8.0 Hz, ArH-6), 7.77 (1H, d, J = 3.2 Hz, ArH-3′), 7.87 (1H, t, J = 8.0 Hz, ArH-7), 7.95 (1H, d, J = 8.0 Hz, ArH-5), 8.64 (1H, d, J = 8.0 Hz, ArH-8), 9.39 (1H, d, J = 9.2 Hz, ArH-6′), 11.31 (1H, s, NH); MS m/z (%) 345 (M + 1, 100), 347 (M + 3, 33).
2-Acetoxymethyl-4-(4-methoxy-2-nitro-ph e nyl)- aminoquinazoline (11).
A mixture of 10 (500 mg, 1.45 mmol) and KOAc (427 mg, 4.36 mmol) in DMF (2.0 mL) was stirred at 55 °C for 6 h. The mixture was poured into ice−water and extracted with EtOAc. The organic layer was washed with water and brine successively and dried over Na2SO4. After removal of solvent in vacuo, the residue was purified by flash column chromatography (gradient elution, CH2Cl2/ EtOAc, 0−15%) to give 11 as an orange solid (300 mg, 65% yield). Mp 159−160 °C; 1H NMR (CDCl3) δ 2.24 (3H, s, CH3), 3.90 (3H, s, OCH3), 5.35 (2H, s, OCH2), 7.33 (1H, dd, J = 9.6 and 3.2 Hz, ArH-5′), 7.64 (1H, t, J = 8.4 Hz, ArH-6), 7.76 (1H, d, J = 3.2 Hz, ArH-3′), 7.84 (1H, t, J = 8.4 Hz, ArH-7), 7.93 (1H, d, J = 8.4 Hz, ArH-5), 8.04 (1H, d, J = 8.4 Hz, ArH-8), 9.24 (1H, d, J = 9.6 Hz, ArH-6′), 11.24 (1H, s, NH); MS m/z (%) 369 (M + 1, 100).
2-Acetoxymethyl-4-(4-methoxy-2-aminophenyl)-aminoquinazoline (12).
To a solution of 11 (154 mg, 0.54 mmol) in a mixed solvent of EtOAc and EtOH (25 mL, v/v 3:2) was added Pd/C (15 mg, 10% w/w) and hydrogen gas. The mixture was shaken under 40 psi at rt for 2 h. The Pd/C was removed by filtration and washed with EtOAc several times. After removal of the solvent in vacuo, 185 mg of light yellow solid 12 was obtained and used directly in the next step without further purification.
2-Acetoxymethyl-4-(2-chloroacetylamino-4-methoxyphenyl)-aminoquinazoline (13).
Crude 12 (ca. 545 mg, 1.61 mmol) was immediately dissolved in acetone (8 mL), and K2CO3 (667 mg, 4.83 mmol) was added. The mixture was cooled to 0 °C and chloroacetyl chloride (361 mg, 3.22 mmol) was added dropwise. Stirring was continued for an additional 1 h at 0 °C. When the reaction was complete, the mixture was poured into ice water, the pH was adjusted to neutral, and the product was extracted with EtOAc three times. The combined organic phase was washed successively with water and brine, and dried over anhydrous Na2SO4 overnight. After removal of solvent in vacuo, 13 was obtained as a gray solid and used in the synthesis of 6q (400 mg, 60% yield over two steps). Mp 121−122 °C; MS m/z (%) 415 (M + 1, 100), 417 (M + 3, 33).
Aqueous Solubility Studies.
Solubility was measured at pH 7.4 by using an HPLC−UV method. Test compounds were initially dissolved in DMSO at a concentration of 1.0 mg/mL. Ten microliters of this stock solution was added to phosphate buffer (1.0 mL, pH 7.4). The mixture was stirred for 4 h at rt and then centrifuged at 3000 rpm for 10 min. The saturated supernatants were transferred to other vials for analysis by HPLC−UV. Each sample was performed in triplicate. For quantification, a model 1200 HPLC−UV (Agilent) system was used with an Agilent Eclipse XDB-C18 column (150 mm × 4.6 mm, 5 μm), and elution was with 50%−80% ACN in water. The flow rate was 0.8 mL/min, and injection volume was 20 μL. Aqueous concentration was determined by comparison of the peak area of the saturated solution with a standard curve plotted peak area versus known concentrations, which were prepared by solutions of test compound in ACN at 50, 12.5, 3.13, 0.78, and 0.20 μg/mL.
Log P Measurements.
One to two milligrams of test compound was dissolved in 1.0−2.0 mL of n-octanol to obtain a 1.0 mg/mL solution. Next, the same volume of water as n-octanol was added to each vial. The mixture was stirred at rt for 24 h and left without stirring overnight. The aqueous and organic phases of each mixture were transferred to separate vials for HPLC analysis. The instrument and conditions were the same as those for water solubility determinations. The log P was calculated by the peak area ratio in n-octanol and in water.
Cell-Based Assays.
Antiproliferative activities of series-6 compounds were assayed by the SRB method according to procedures described previously.27–29 The panel of cell lines included A-549, MDA-MB-231, KB, KB-VIN (P-gp-overexpressing KB subline), and MCF-7. These cell lines were obtained from the Lineberger Comprehensive Cancer Center (UNC−CH) or ATCC. Cells were propagated in RPMI-1640 (Gibco) supplemented with 10% FBS (Corning), penicillin (100 IU/mL), streptomycin (1 μg/mL), and amphotericin B (0.25 μg/mL) (Corning) and were cultured at 37 °C in a humidified atmosphere of 95% air and 5% CO2. The antiproliferative effects against tumor cell lines were expressed as GI50 values, which represent the molar drug concentrations required to cause 50% tumor cell growth inhibition. Flow cytometry was performed as described previously.30 Briefly, A549 cells were seeded 24 h prior to treatment in a 12-well culture plate at density of 60 000 cells/well. After 24 h treatment with 15 nM 6d, 15 nM 6e, or 20 nM CA-4, cells were fixed and stained with PI containing RNase (BD Biosciences). DMSO (0.001%) was used as a control. Stained cells were analyzed by flow cytometry operated by FACSDiva software (BD LSRFortessa, BD Biosciences). DMSO was used for preparation of compound, and the highest concentration of DMSO for the cell-based assay was less than 0.001% (v/v).
Tubulin Assays.
Tubulin assembly was measured by turbidimetry at 350 nm as described previously.23 Assay mixtures containing 1.0 mg/mL (10 μM) tubulin and varying compound concentrations were preincubated for 15 min at 30 °C without guanosine 5′-triphosphate (GTP). The samples were placed on ice, and GTP to a final concentration of 0.4 mM was added. Reaction mixtures were transferred to 0 °C cuvettes, and turbidity development was followed for 20 min at 30 °C following a rapid temperature jump. Compound concentrations that inhibited increase in turbidity by 50% relative to a control sample were determined. Inhibition of the binding of [3H]colchicine to tubulin was measured as described previously.31 Incubation of 1.0 μM tubulin with 5.0 μM [3H]colchicine and 5.0 or 1.0 μM inhibitor was for 10 min at 37 °C, when about 40−60% of maximum colchicine binding occurred in control samples.
Xenograft Tumor Growth Inhibition in Vivo.
Six-week-old female Balb/c-nu mice were obtained from Vital River (Beijing, China) and housed in specific-pathogen-free conditions in conformity with the Guide for the Care and Use of Laboratory Animals, as adopted and promulgated by Beijing Institute of Radiation Medicine. Human lung cancer NCI-H460 cells (2 × 106) were injected subcutaneously into the right abdominal flanks of the Balb/c-nu mice. Tumor growth was measured with a slide caliper, and volumes were estimated according to the following formula: tumor volume (mm3) = L × W2 × 0.5, where L is length and W is width. When tumor volume reached about 100 mm3, the mice were randomly divided into five groups (n = 8) and compound administration was started on the second day. The control group was dosed intraperitoneally daily with 7 μL/g of vehicle (0.9% NaCl containing 5% polyethylene glycol 400 and 0.5% Tween 80). Compound 2 groups were treated at the doses of 0.25, 0.5, or 1.0 mg/kg body weight, respectively, every 5 days by iv injection for 3 weeks. The reference group received paclitaxel at a dose of 15 mg/kg body weight. Both compound 2 and paclitaxel were dissolved in vehicle solution (5% PEG400/PBS). The body weight and tumor volume were measured at each time point. At the end of the treatment, the mice were sacrificed for autopsy, and the tumors were recovered and weighed. The tumor growth inhibitory rate was calculated as follow: inhibitory rate (%) = [1 − (mean tumor weight of treated group)/(mean tumor weight of control group)] × 100. Data were presented as the mean ± SD. Statistical significance of differences between groups was compared by one-way analysis of variance test followed by Tukey’s post hoc test (GraphPad Prism 5.0; PraphPad Softeare Inc., La Jolla, CA, USA). P < 0.05 was considered to indicate a statistically significant difference. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of the Beijing Institute of Radiation Medicine (Beijing, China) and performed according to the institutional guidelines.
Immunohistochemical Staining.
The tumor tissues were formalin fixed, paraffin embedded, and sectioned into 4 μm slices. The tissue sections were deparaffinized, rehydrated, and treated with 3% H2O2 to quench the endogenous peroxidase activity. Antigen retrieval was performed by boiling slides in sodium citrate buffer (10 mM, pH 6.0). CD31 (Thermo Fisher Scientific; cat. no. PA5–32321), cleaved caspase-3 (Cell Signaling Technology; cat. no. 9661), or Ki67 antibody (Thermo Fisher Scientific; cat. no. MA5–14520) was used as primary antibody, and horseradish peroxidase (HRP)-conjugated antibody was used as secondary antibody. HRP was detected by diaminobenzidine (DAB) (Wuhan Boster Biological Technology, Ltd.). The immunohistochemistry reaction was developed with a DAB substrate kit (Wuhan Boster Biological Technology, Ltd.) prior to counterstaining the slides with hematoxylin. The immunostained sections were examined using an EVOS X1 microscope (Advanced Microscopy Group; Thermo Fisher Scientific, Inc.).
Molecular Modeling.
The molecular modeling study was performed with Discovery Studio 3.0 (Accelrys, San Diego, USA), also used in our prior modeling method.13 The crystal structure of tubulin in complex with CA-4 (PDB code 5LYJ) with 2.4 Å resolution was downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb) for use in the modeling study. LIBDOCK was used to evaluate and predict in silico binding free energy of the inhibitors and for automated docking. The protein model was typed with the CHARMm force field. A binding sphere with radius of 8.5 Å was defined through the original ligand (CA-4) as the binding site for the study. The docked ligand 6e and 2 were refined using in situ ligand minimization with the Smart Minimizer algorithm by standard parameters.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Molecular Pharmacology Branch, DTP, DCTD, NCI, for performing the NCI 60-cell cytotoxicity assay. This investigation was supported by Grants 81120108022 and 30930106 from the Natural Science Foundation of China (NSFC) awarded to L.X. and NIH Grant CA177584 from the National Cancer Institute awarded to K.H.L. This study was also supported in part by the Taiwan Department of Health, China Medical University Hospital Cancer Research Center of Excellence (DOH100-TD-C-111-005), and Grant 81472687 from the Natural Science Foundation of China awarded to L.L. This study was also supported in part by the Eshelman Institute for Innovation, Chapel Hill, North Carolina, awarded to M.G.
ABBREVIATIONS USED
- CA-4
combretastatin A-4
- DAMA-colchicine
N-deacetyl-N-(2-mercaptoacetyl)-colchicine
- DMA
dimethylacetamide
- DMAP
dimethylaminopyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- GI50
concentration that inhibits 50% human tumor cell growth
- HRMS
high resolution mass spectra
- HTCL
human tumor cell line
- IHC
immunohistochemistry
- MW
microwave
- PDB
Protein Data Bank
- SAR
structure−activity relationship
- tumor-VDAs
tumor vascular disrupting agents
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00273.
HPLC purity conditions and data of 6a–6t, HRMS data of 6a–6t, data of 2 in NIH-NCI 60 cell line panel, and representative 13C spectra of target compounds (6a, 6b, 6h, 6q, 6r, 6s) (PDF)
SMILES molecular formula strings (CSV)
Notes
The content of this paper is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
The authors declare no competing financial interest.
REFERENCES
- (1).Perez-Perez MJ; Priego EM; Bueno O; Martins MS; Canela MD; Liekens S Blocking Blood Flow to Solid Tumors by Destabilizing Tubulin: an Approach to Targeting Tumor Growth. J. Med. Chem 2016, 59, 8685–8711. [DOI] [PubMed] [Google Scholar]
- (2).Kaur R; Kaur G; Gill RK; Soni R; Bariwal J Recent Developments in Tubulin Polymerization Inhibitors: an Overview. Eur. J. Med. Chem 2014, 87, 89–124. [DOI] [PubMed] [Google Scholar]
- (3).Tozer GM; Kanthou C; Baguley BC Disrupting Tumor Blood Vessels. Nat. Rev. Cancer 2005, 5, 423–435. [DOI] [PubMed] [Google Scholar]
- (4).Siemann DW The Unique Characteristics of Tumor Vasculature and Preclinical Evidence for Its Selective Disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev 2011, 37, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mason RP; Zhao D; Liu L; Trawick ML; Pinney KG A Perspective on Vascular Disrupting Agents That Interact with Tubulin: Preclinical Tumor Imaging and Biological Assessment. Integr. Biol 2011, 3, 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang XF; Xie L Vascular Disrupting Agents Targeting at Tubulin: a Novel Class of Antitumor Drug. J. Int. Pharm. Res 2012, 39, 445–454. [Google Scholar]
- (7).Siemann DW; Chaplin DJ; Walicke PA A Review and Update of the Current Status of the Vasculature-Disabling Agent Combretastatin-A4 Phosphate (CA4P). Expert Opin. Invest. Drugs 2009, 18, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Galbraith SM; Chaplin DJ; Lee F; Stratford MR; Locke RJ; Vojnovic B; Tozer GM Effects of Combretastatin A4 Phosphate on Endothelial Cell Morphology In Vitro and Relationship to Tumour Vascular Targeting Activity In Vivo. Anticancer Res 2001, 21, 93–102. [PubMed] [Google Scholar]
- (9).Bates D; Feris EJ; Danilov AV; Eastman A Rapid Induction of Apoptosis in Chronic Lymphocytic Leukemia Cells by the Microtubule Disrupting Agent BNC105. Cancer Biol. Ther 2016, 17, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang XF; Ohkoshi E; Wang SB; Hamel E; Bastow KF; Morris-Natschke SL; Lee KH; Xie L Synthesis and Biological Evaluation of N-Alkyl-N-(4-methoxyphenyl)pyridin-2-amines as a New Class of Tubulin Polymerization Inhibitors. Bioorg. Med. Chem 2013, 21, 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang XF; Wang SB; Ohkoshi E; Wang LT; Hamel E; Qian K; Morris-Natschke SL; Lee KH; Xie L Discovery of N-Aryl-6-methoxy-1,2,3,4-tetrahydro-quinolines as a Novel Class of Antitumor Agents Targeting the Colchicine Site of Tubulin. Eur. J. Med. Chem 2013, 67, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang SB; Wang XF; Qin B; Ohkoshi E; Hsieh KY; Hamel E; Cui MT; Zhu DQ; Goto M; Morris-Natschke SL; Lee KH; Xie L Optimization of N-Aryl-6-methoxy-1,2,3,4-tetrahydroquinolines as Tubulin Polymerization Inhibitors. Bioorg. Med. Chem 2015, 23, 5740–5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang XF; Guan F; Ohkoshi E; Guo W; Wang L; Zhu DQ; Wang SB; Wang LT; Hamel E; Yang D; Li L; Qian K; Morris-Natschke SL; Yuan S; Lee KH; Xie L Optimization of 4-(N-Cycloamino)phenylquinazolines as a Novel Class of Tubulin Polymerization Inhibitors Targeting the Colchicine Site. J. Med. Chem 2014, 57, 1390–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lockman JW; Klimova Y; Anderson MB; Willardsen JA Synthesis of Substituted Quinazolines: Application to the Synthesis of Verubulin. Synth. Commun 2012, 42, 1715–1723. [Google Scholar]
- (15).Yang Z; Wang T; Wang F; Niu T; Liu Z; Chen X; Long C; Tang M; Cao D; Wang X; Xiang W; Yi Y; Ma L; You J; Chen L Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem 2016, 59, 1455–1470. [DOI] [PubMed] [Google Scholar]
- (16).Shoemaker RH The NCI-60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- (17).Devambatla RK; Namjoshi OA; Choudhary S; Hamel E; Shaffer CV; Rohena CC; Mooberry SL; Gangjee A Design, Synthesis, and Preclinical Evaluation of 4-Substituted-5-methyl-furo-[2,3-d]pyrimidines as Microtubule Targeting Agents that are Effective Against Multidrug Resistant Cancer Cells. J. Med. Chem 2016, 59, 5752–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).The data were provided by Concord PharmaTech Co., Ltd Nanjing, China. [Google Scholar]
- (19).Pérez-Sayáns M; Somoza-Martín JM; Barros-Angueira F; Diz PG; Rey JMG; García-García A Multidrug Resistance in Oral Squamous Cell Carcinoma: the Role of Vacuolar ATPases. Cancer Lett 2010, 295, 135–143. [DOI] [PubMed] [Google Scholar]
- (20).Hung HY; Ohkoshi E; Goto M; Bastow KF; Nakagawa-Goto K; Lee KH Antitumor Agents. 293. Nontoxic Dimethyl-4,4′-dimethoxy-5,6,5′,6′-dimethylenedioxybiphenyl-2,2′-dicarboxylate (DDB) Analogues Chemosensitize Multidrug-resistant Cancer Cells to Clinical Anticancer Drugs. J. Med. Chem 2012, 55, 5413–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rubinstein LV; Shoemaker RH; Paull KD; Simon RM; Tosini S; Skehan P; Scudiero DA; Monks A; Boyd MR Comparison of In Vitro Anticancer-Drug-Screening Data Generated with a Tetrazolium Assay Versus a Protein Assay Against a Diverse Panel of Human Tumor Cell Lines. J. Natl. Cancer Inst 1990, 82, 1113–1117. [DOI] [PubMed] [Google Scholar]
- (22).Sun LQ; Zhu L; Qian K; Qin B; Huang L; Chen CH; Lee KH; Xie L Design, Synthesis, and Preclinical Evaluations of Novel 4-Substituted 1,5-Diarylanilines as Potent HIV-1 Non-Nucleo-side Reverse Transcriptase Inhibitor (NNRTI) Drug Candidates. J. Med. Chem 2012, 55, 7219–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kerns EH; Di L Drug-like Properties: Concepts, Structure Design and Methods From ADME to Toxicity Optimization; Academic Press: Boston, 2008; pp 43–47. [Google Scholar]
- (24).Hamel E Evaluation of Antimitotic Agents by Quantitative Comparisons of Their Effects on the Polymerization of Purified Tubulin. Cell Biochem. Biophys 2003, 38, 1–21. [DOI] [PubMed] [Google Scholar]
- (25).Lin CM; Ho HH; Pettit GR; Hamel E Antimitotic Natural Products Combretastatin A-4 and Combretastatin A-2: Studies on the Mechanism of Their Inhibition of the Binding of Colchicine to Tubulin. Biochemistry 1989, 28, 6984–6991. [DOI] [PubMed] [Google Scholar]
- (26).Anderson MB Preparation of 4-Arylamino-quinazolines as Activators of Caspases and Inducers of Apoptosis U.S. Pat. Appl. Publ, 20100069383, March 18, 2010. [Google Scholar]
- (27).Boyd MR Status of the NCI Preclinical Antitumor Drug Discovery Screen. In Cancer: Principles and Practice of Oncology Updates; Devita VT, Hellman S, Rosenberg SA, Eds.; Lippincott Williams & Wilkins: Philadelphia, 1989; pp 1–12. [Google Scholar]
- (28).Monks A; Scudiero D; Skehan P; Shoemaker R; Paull K; Vistica D; Hose C; Langley J; Cronise P; Vaigro-Wolff A; Gray-Goodrich M; Campbell H; Mayo J; Boyd M Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst 1991, 83, 757–766. [DOI] [PubMed] [Google Scholar]
- (29).Houghton P; Fang R; Techatanawat I; Steventon G; Hylands PJ; Lee CC The Sulphorhodamine (SRB) Assay and Other Approaches to Testing Plant Extracts and Derived Compounds for Activities Related to Reputed Anticancer Activity. Methods 2007, 42, 377–387. [DOI] [PubMed] [Google Scholar]
- (30).Nakagawa-Goto K; Oda A; Hamel E; Ohkoshi E; Lee KH; Goto M Development of a Novel Class of Tubulin Inhibitor from Desmosdumotin B with a Hydroxylated Bicyclic B-ring. J. Med. Chem 2015, 58, 2378–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Verdier-Pinard P; Lai J-Y; Yoo H-D; Yu J; Maŕquez B; Nagle DG; Nambu M; White JD; Falck JR; Gerwick WH; Day BW; Hamel E. Structure-Activity Analysis of the Interaction of Curacin A, the Potent Colchicine Site Antimitotic Agent, with Tubulin and Effects of Analogs on the Growth of MCF-7 Breast Cancer Cells. Mol. Pharmacol 1998, 53, 62–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.