

Abstract
Pancreatic β-cell failure in type 2 diabetes mellitus is a serious challenge that results in an inability of the pancreas to produce sufficient insulin to properly regulate blood glucose levels. Trace amine–associated receptor 1 (TAAR1) is a G protein–coupled receptor expressed by β-cells that has recently been proposed as a potential target for improving glycemic control and suppressing binge eating behaviors. We discovered that TAAR1 is coupled to Gαs-signaling pathways in insulin-secreting β-cells to cause protein kinase A (PKA)/exchange protein activated by cAMP (Epac)–dependent release of insulin, activation of RAF proto-oncogene, Ser/Thr kinase (Raf)–mitogen-activated protein kinase (MAPK) signaling, induction of cAMP response element–binding protein (CREB)–insulin receptor substrate 2 (Irs-2), and increased β-cell proliferation. Interestingly, TAAR1 triggered cAMP-mediated calcium influx and release from internal stores, both of which were required for activation of a MAPK cascade utilizing calmodulin-dependent protein kinase II (CaMKII), Raf, and MAPK/ERK kinase 1/2 (MEK1/2). Together, these data identify TAAR1/Gαs-mediated signaling pathways that promote insulin secretion, improved β-cell function and proliferation, and highlight TAAR1 as a promising new target for improving β-cell health in type 2 diabetes mellitus.
Keywords: diabetes, calcium, pancreas, G protein–coupled receptor (GPCR), insulin secretion
Introduction
Diabetes mellitus is an increasingly prevalent metabolic disease that affects ∼400 million people worldwide, with future projections reaching nearly 600 million by 2035 (1). Type 2 diabetes (T2DM)2 still accounts for 90–95% of cases and is associated with a sedentary lifestyle, poor diet, obesity, metabolic syndrome, and genetic risk factors (2). The pathogenesis of T2DM involves a vicious cycle of escalating insulin insensitivity, elevated blood glucose, and rising insulin levels that eventually exhausts pancreatic β-cells. This results in β-cell dysfunction, decreased insulin output, and β-cell death (3). Despite a multitude of available treatments to bolster insulin levels, new disease-modifying agents that improve the health and function of pancreatic β-cells will be essential to meaningfully reduce overall disease burden and complications of diabetes (4).
One of the more recent trends in T2DM therapies have focused on incretin hormones (5). Stable incretin mimetics targeting the glucagon-like peptide 1 receptor (GLP-1R) have been found to be effective at improving glycemic control in T2DM, with the added benefit of modest weight loss. GLP-1 homologs may confer an advantage over other antihyperglycemic agents such as insulin analogues, sulfonylureas, and thiazolidinediones, which have been associated with weight gain (5, 6). The salutary effects of incretin receptor agonism in β-cells appear to derive from Gαs signaling, which mediates glucose-stimulated insulin secretion (GSIS) and promotes β-cell proliferation while reducing stress-induced apoptosis (7, 8).
Recently it was found that the G protein–coupled trace amine–associated receptor (TAAR1) is expressed in human pancreatic islets and increases insulin secretion and glucose tolerance in mouse models (9). However, the molecular mechanisms by which TAAR1 directly regulates insulin secretion in β-cells are largely unknown, as TAAR1 plays additional roles in gut motility, satiety, and eating behaviors (9, 10). Amine agonists such as octopamine, β-phenylethylamine, tyramine, and amphetamines (11) exhibit neurotransmitter-like activity, and TAAR1 is expressed in the brain, where it has been shown to decrease binge eating behaviors and stimulate weight loss (9, 10). Aside from trace amines, TAAR1 is activated in the periphery by thyroid hormone 3–iodothyronamine (T1AM) (12), and a concerted effort is currently underway to produce selective TAAR1 agonists for use in control of impulsive behaviors (13). Here we delineate the downstream effectors of TAAR1 in pancreatic β-cell lines and provide evidence that TAAR1 agonists trigger beneficial anti-diabetic signaling pathways that could help lead to their use as anti-diabetic agents.
Results
TAAR1 potentiates glucose-stimulated insulin secretion through cAMP-PKA and Epac-dependent signaling in pancreatic β-cells
The primary function of β-cells is to regulate glucose homeostasis by controlling the secretion of insulin. To begin to probe the mechanisms by which TAAR1 may regulate GSIS, we first determined whether TAAR1 utilized adenylyl cyclase signaling pathways. The Ins-1 and Min6 β-cell lines were found to exhibit cAMP-dependent potentiation of GSIS in response to the TAAR1 agonist T1AM. Treatment of cells with MDL-12,330A, an inhibitor of adenylyl cyclase (AC), significantly (p < 0.001) reduced TAAR1 potentiation of GSIS by 77–82% (Fig. 1, A and B). The specificity of T1AM for TAAR1 was confirmed by the use of the selective TAAR1 antagonist EPPTB (14), which significantly (p < 0.001) inhibited (70–82%) potentiation of GSIS by T1AM (Fig. 1, A and B). PKA and Epac are downstream effectors of adenylyl cyclase–cAMP signaling and regulate granule exocytosis. Both H89 (a PKA inhibitor) and HJC-0350 (an Epac inhibitor) caused a significant (p < 0.001) reduction in TAAR1-mediated potentiation of GSIS by 82–93% (Fig. 1A). These data indicate that potentiation of glucose-stimulated insulin secretion by TAAR1 occurs through AC-cAMP dependent signaling pathways and requires both PKA and Epac activity to effect maximal insulin release.
Figure 1.
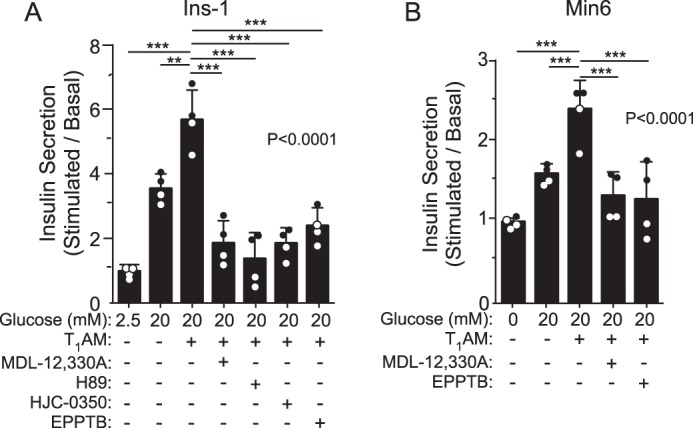
TAAR1 potentiates glucose stimulated insulin secretion through adenylyl cyclase–dependent pathways. A and B, the effect of MDL-12,330A (10 μm, inhibits adenylyl cyclase), H89 (10 μm, inhibits PKA), HJC-0350 (10 μm, inhibits Epac), and EPPTB (10 μm, inhibits TAAR1) on T1AM-potentiated GSIS was examined in the pancreatic β cell lines Ins-1 (A) and Min6 (B). Insulin secretion (2 h) was determined by ELISA (mean ± S.D.) and analyzed by one-way ANOVA (global p values are shown) using Dunnett's multiple comparison post hoc test (n = 4). **, p < 0.01; ***, p < 0.001.
TAAR1 stimulates CREB phosphorylation and the downstream target gene Irs-2 via adenylyl cyclase and PKA
CREB is a key transcription factor that promotes β-cell health (15); therefore, we determined whether phosphorylation of the PKA target CREB could be detected following TAAR1 activation. Because glucose itself can stimulate CREB pathways, a time course for CREB phosphorylation in response to T1AM was first established in Ins-1 cells with or without exogenous glucose (Fig. 2A). T1AM induced peak CREB phosphorylation in both the presence and absence of glucose as early as 5 min and diminished by 30 min (Fig. 2A). The direct adenylyl cyclase activator forskolin gave similar kinetics of activation of pCREB as T1AM with ∼2-fold higher magnitude in the presence of glucose. Treatment of cells with MDL-12,330A (Fig. 2B) as well as H89 (Fig. 2C) to inhibit AC and PKA, respectively, effectively blocked phosphorylation of the CREB transcription factor. Knockdown of the catalytic subunit of PKA with siRNA also caused a reduction in CREB phosphorylation compared with cells treated with control siRNA (Fig. 2D), consistent with results generated using H89. A key CREB target gene, insulin receptor substrate-2 (Irs-2), which promotes pro-proliferative and anti-apoptotic signaling in β cells (15, 16), was significantly (p < 0.001) up-regulated by 50% in cells treated with T1AM (Fig. 2E). Induction of Irs-2 was completely blocked by MDL-12,330A (p < 0.001) but not LY294002, revealing that adenylyl cyclase, but not phosphatidylinositol 3-kinase (PI3K) is required for Irs-2 gene induction by T1AM.
Figure 2.

TAAR1 induces CREB phosphorylation and Irs-2 gene expression in Ins-1 cells. A, Western blots of CREB phosphorylation in response to T1AM (10 μm) and forskolin (0.3 μm) for 5–30 min in the presence (11 mm) or absence of glucose. B–D, addition of 10 μm MDL-12,330A (B), 10 μm H89 (C), or 150 nm PKA Cat-α siRNA (D) inhibits CREB phosphorylation (10 min) induced by T1AM and forskolin. Representative blots of pCREB from one of at least three independent experiments are shown; blots were stripped and reprobed with β-actin as a loading control. E, quantitative PCR of Irs-2 gene expression (1 h) induced by 10 μm T1AM in the presence and absence of MDL-12,330A (10 μm) or LY294002 (10 μm, inhibits PI3K) pretreatment. Data are expressed as mean ΔΔCT (± S.D.) of Irs-2 using Gapdh as the housekeeping gene and were analyzed by one-way ANOVA (global p values are shown) using Dunnett's multiple comparison test, comparing all columns with 10 μm T1AM treatment (n = 3). ***, p < 0.001.
TAAR1 stimulates β-cell proliferation by activation of adenylyl cyclase–dependent Raf-MAPK signaling
Ins-1 cells treated with T1AM and the positive control forskolin exhibited robust ERK1/2 phosphorylation in both the presence and absence of exogenous glucose (Fig. 3A). ERK1/2 phosphorylation appeared at 5 min and peaked at 15 min but mostly faded by 30 min, similar to the kinetics of pCREB (Fig. 2). ERK1/2 phosphorylation induced by both the TAAR1 agonist and forskolin was blocked by MDL-12,330A (Fig. 3B), indicating that adenylyl cyclase is required for TAAR1-mediated ERK1/2 phosphorylation. TAAR1 ERK1/2 phosphorylation was also reduced by siRNA knockdown of the catalytic subunit of PKA (PKA Cat-α) compared with cells treated with control siRNA (Fig. 3C). The Epac inhibitors Esi-09 (Epac1/2 inhibitor) and Esi-05 (Epac 2 inhibitor) also attenuated ERK1/2 phosphorylation downstream of T1AM (Fig. 3D), indicating that both PKA and Epac are involved in TAAR1-mediated ERK1/2 phosphorylation. The suppressive effects of Esi-09 and Esi-05 on ERK1/2 phosphorylation were confirmed by siRNA knockdown of Epac, which again reduced TAAR1 and forskolin-mediated ERK1/2 phosphorylation (Fig. 3E). The MAP Kinase Kinase Kinase Raf has been shown to be regulated by both Epac and PKA (17, 18). Raf and the mitogen-activated protein kinase kinase MEK1/2 were identified as upstream mediators of ERK1/2 phosphorylation, as the pan-Raf inhibitor AZ-628 and the MEK1/2 inhibitor PD98059 completely prevented ERK1/2 phosphorylation in response to both T1AM and forskolin (Fig. 3F). Both the B-Raf and C-Raf isoforms were found to be expressed in Ins-1 β cells with 5-fold higher relative B-Raf expression (Fig. 3G).
Figure 3.

TAAR1 increases proliferation via cAMP-dependent Raf/MEK/ERK signaling in Ins-1 cells. A, Western blots of ERK1/2 phosphorylation in response to T1AM (10 μm) and forskolin (0.3 μm) for 5–30 min in the presence (11 mm) or absence of glucose. B–F, addition of 10 μm MDL-12,330A (B), 150 nm PKA Cat-α siRNA (C), 10 μm Esi-09 and 30 μm Esi-05 (D), 150 nm Epac siRNA (E), or 10 μm AZ-628 (Raf inhibitor) and 50 μm PD98059 (MEK1/2 inhibitor) (F) inhibits ERK1/2 phosphorylation (10 min) induced by T1AM and forskolin. Representative blots of pERK1/2 from one of at least three independent experiments are shown; blots were stripped and reprobed with total ERK1/2 as a loading control. G, quantitative PCR of B-Raf and C-Raf gene expression in Ins-1 β-cells. Data are expressed as relative mean ΔΔCT (± S.D.) compared with B-Raf mRNA levels, using Gapdh as the housekeeping gene, and were analyzed by Student's t test (***, p < 0.001). H, T1AM (10 μm) and forskolin (0.5 μm) increase [3H]thymidine incorporation into Ins-1 cells (24 h), which is blocked by PD-98059 (50 μm) and AZ-628 (10 μm). Data were analyzed using one-way ANOVA (global p values are shown), with Dunnett's multiple comparisons post hoc test to determine significance between relevant groups (n = 6). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
As ERK1/2 phosphorylation is frequently associated with increased rates of cellular proliferation, we sought to determine whether T1AM affected proliferation of Ins-1 cells. Both T1AM and forskolin significantly enhanced radiolabeled thymidine incorporation in insulin-secreting cells (Fig. 3H). AZ-628 and PD98059 completely blocked the increases in radiolabeled thymidine incorporation in response to T1AM, indicating that Raf/MEK1/2/ERK1/2 signaling is required for TAAR1 stimulation of cellular proliferation of Ins-1 cells.
TAAR1-MAPK signaling in insulin-secreting cells requires both calcium influx and intracellular calcium release
Surprisingly, we found that addition of extracellular calcium chelator EGTA to Ins-1 cells completely blocked ERK1/2 phosphorylation in response to T1AM and forskolin (Fig. 4A), indicating that calcium influx was critical for downstream MAPK signaling initiated by Gs-AC/cAMP. Intracellular calcium release from internal stores via IP3 receptors was also required for ERK1/2 phosphorylation, as the 2-APB antagonist completely blocked ERK1/2 phosphorylation to both T1AM and forskolin (Fig. 4B). Likewise, reloading of intracellular calcium stores was partially required for activation of pERK1/2, as blockade of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump with thapsigargin was able to reduce ERK phosphorylation in response to these agents (Fig. 4A). PLC-β, a common mediator of calcium signaling through Gq- and Gi-coupled receptors, however, was not required for TAAR1-initiated ERK1/2 phosphorylation, as the U73122 PLC-β inhibitor had no effect on T1AM or forskolin-mediated ERK1/2 phosphorylation (Fig. 4B). U73122 did, however, effectively inhibit calcium signaling in response to LIGRLO (Fig. S1), an agonist of protease-activated receptor 2 (PAR2), a known Gq-coupled receptor expressed in the pancreas (19), confirming the efficacy of U73122 in these insulin-secreting cells.
Figure 4.

Calcium influx and store release are required for TAAR1-dependent ERK1/2 phosphorylation in Ins-1 cells. A–C, Western blotting of ERK1/2 phosphorylation (10 min) induced by T1AM (10 μm) and forskolin (0.3 μm) in the presence and absence of EGTA (1 mm), thapsigargin (5 μm, inhibits the SERCA calcium pump), 2-APB (50 μm, inhibits IP3R), U73122 (20 μm, inhibits PLC-β), KN-93 (10 μm, inhibits CaMKII), and STO-609 (25 μm, inhibits CaMKKII). Representative blots of pERK1/2 from one of at least three independent experiments are shown, and blots were stripped and reprobed with total ERK1/2 as a loading control. D–G, calcium signaling in Ins-1 cells induced by T1AM (10 μm) was measured in the presence of 1.5 or 0.5 mm extracellular calcium, 1 mm EGTA, 5 μm thapsigargin, 10 μm MDL-12,330A, or 50 μm 2-APB as labeled. H, calcium signaling induced by forskolin (0.3 μm). Representative traces of at least three individual experiments are shown. E and I, calcium flux data induced by T1AM in the presence of various antagonists was quantified by measuring the area under the curve normalized to 100% of the maximum signal and are represented as the mean ± S.D. J–M, calcium signaling in Ins-1 cells induced by the TAAR1 agonist T1AM (10 μm, J and K) or the PAR2 agonist LIGRLO (10 μm, L and M) was measured in the presence of 1.5 mm extracellular calcium with or without the Gq blocker YM-254890 (300 nm). Representative traces of one of three experiments are shown. AUCs were analyzed by one-way ANOVA (global p < 0.0001) using Newman-Keuls multiple comparisons post hoc test to determine significance between groups (n = 3–6). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Ca2+/Calmodulin (CaM) kinase–dependent signaling has been shown previously to regulate Raf/MEK/ERK pathways in certain cell types (20), providing a possible link between intracellular calcium and downstream ERK1/2 phosphorylation. Interestingly, we found that treatment of Ins-1 cells with KN-93, a potent inhibitor of CaM kinase II (CaMKII), blocked ERK1/2 phosphorylation in response to T1AM and forskolin (Fig. 4C). In contrast, the divergent calmodulin-regulated kinase Ca2+/calmodulin-dependent protein kinase kinase II appeared to play a MAPK-suppressive role, as the STO-609 inhibitor enhanced both the basal and agonist-stimulated pERK1/2 signal (Fig. 4C).
TAAR1 activation triggers calcium influx and intracellular calcium release via cAMP-mediated signaling
Consistent with the effects of the various calcium pathway inhibitors on modulating ERK1/2 phosphorylation, we were able to directly detect a significant, rapid, intracellular calcium flux signal in response to T1AM, as well as the cAMP-stimulating positive control forskolin (Fig. 4, D and H). The TAAR1 calcium flux signal depended on extracellular calcium, as reduction of physiologic levels (1.5 mm) of extracellular calcium or introduction of EGTA caused up to a 75% attenuation (p < 0.001) in calcium flux (Fig. 4, D and E). The residual 25% calcium signal could be completely blocked by inclusion of thapsigargin (Fig. 4, D and E), which inhibits reloading of calcium into endoplasmic reticulum stores. This TAAR1-mediated calcium release occurred through cAMP-dependent signaling pathways, as MDL-12,330A significantly (p < 0.001) reduced calcium signaling in response to T1AM (Fig. 4, F and I). Consistent with the effects on ERK1/2 phosphorylation above, 2-APB also significantly (p < 0.001) inhibited the T1AM-induced calcium signal (Fig. 4, G and I), indicating critical involvement of the IP3 receptor in TAAR1-mediated calcium signaling. We ruled out Gq as being involved in the TAAR1-evoked calcium signal, as the potent and selective Gq inhibitor YM-254890 had no effect on the T1AM calcium response (Fig. 4, J and K). In contrast, YM-254890 completely blocked calcium flux in response to LIGRLO (21), an agonist of the Gq-coupled PAR2 (Fig. 4, L and M). Together, these data support a mechanism whereby TAAR1 stimulates Gαs/cAMP-dependent calcium release from internal stores, and this initial calcium flux leads to further calcium influx from external sources.
Small molecule RO5256390 stimulates TAAR1 signaling in insulin-secreting cells
RO5256390 was recently discovered and characterized as a specific agonist of TAAR1 for potential use as a central nervous system–acting drug with potent in vivo activity in rodents and monkeys (13). We sought to determine whether this new small-molecule compound exhibited the same beneficial signaling properties in the β-cell line evoked by the endogenous TAAR1 agonist T1AM. RO5256390 stimulated pCREB and induced downstream target Irs-2 gene expression by 2-fold (p < 0.0001) in Ins-1 cells (Fig. 5, A and B). RO5256390 stimulated ERK1/2 phosphorylation (Fig. 5C), intracellular calcium flux (Fig. 5D), and β-cell line proliferation (Fig. 5E), further validating the TAAR1-mediated signaling pathways with this small-molecule agonist.
Figure 5.

The TAAR1 small-molecule agonist RO5256390 induces pCREB, Irs-2, pERK, calcium signaling, proliferation, and GSIS in Ins-1 cells. A and C, Western blots of CREB phosphorylation (A) or ERK1/2 phosphorylation (C) in Ins-1 cells 10 min after RO5256390 (0–10 μm) or forskolin (0.3 μm). Representative blots of pCREB or pERK1/2 from one of at least three independent experiments are shown, and blots were stripped and reprobed with either β-actin or total ERK1/2 as a loading control. B, quantitative PCR of Irs-2 gene expression (1 h) induced by RO5256390 (10 μm). Data are expressed as mean ΔΔCT (± S.D.) of Irs-2, using Gapdh as the housekeeping gene, and were analyzed by Student's t test (n = 3). ****, p < 0.0001. D, RO5256390 induces calcium flux in Ins-1 cells (1.5 mm extracellular calcium). E, RO5256390 (10 μm) and forskolin (0.3 μm) increase [3H]thymidine incorporation into Ins-1 cells (24 h). F, insulin secretion in response to TAAR1 agonists (RO5256390 and T1AM) or modulators of TAAR1/cAMP-dependent signaling were added to cells at either 3.5 mm (low) or 20 mm (high) glucose. Insulin secretion (2 h) was determined by ELISA (mean ± S.D.) and analyzed by one-way ANOVA (global p values are shown) using Dunnett's multiple comparison post hoc test (E, n = 6) or Newman-Keuls test (F, n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
As shown in Fig. 5F, RO5256390 induced striking potentiation of GSIS compared with T1AM. Neither RO5256390 or T1AM induced insulin secretion under low-glucose conditions (Fig. 5F). This demonstrates that the trace amine receptor behaves similarly as the peptide incretin receptors GLP-1R and GIPR, which potentiate GSIS via Gαs-cAMP signaling but do not trigger insulin secretion under low-glucose conditions. Furthermore, inhibition of cAMP signaling pathways with MDL-12,330A or H89 inhibitors had no significant effect on GSIS in the absence of TAAR1 agonist treatment (Fig. 5F).
TAAR1 stimulates calcium flux, therefore it is possible that it could also have a direct effect on GSIS by increasing calcium-dependent insulin-vesicle release, as has been shown with the Gs-coupled GLP receptor (22). We determined that the IP3 receptor antagonist 2-APB significantly (p < 0.001) reduced TAAR1-dependent insulin secretion in the presence of high glucose (Fig. 5F), underscoring the essential role that calcium flux from internal stores plays in potentiation of GSIS by TAAR1 agonist.
Discussion
T2DM is part of a growing epidemic of metabolic diseases, and current treatments do not adequately meet the needs of this expanding patient population. Recent strategies to improve glycemic control include enhancing GSIS and pancreatic islet viability by targeting a select group of G protein-coupled receptors that couple to Gαs (23, 24). These include receptors for gut peptide incretins such as GIP (gastric inhibitory polypeptide) and GLP-1 (glucagon-like peptide-1), that stimulate cAMP production via AC activation (25, 26). cAMP is a critical second messenger for β-cells, as it activates a tightly regulated and complex signaling network that can induce potentiation of GSIS, protect β-cells from stress-induced apoptosis, and trigger an expansion of β-cell mass to combat β-cell failure (27). One of the biggest hurdles in developing treatments that target Gαs-coupled peptide receptors such as GLP-1R and GIPR is the expense and short t½ of their peptide hormone agonists (28).
Here we present evidence that the trace amine receptor TAAR1 is coupled to Gαs signaling pathways in pancreatic β-cell lines to cause PKA/Epac-dependent release of insulin, activation of Raf-MAPK signaling, induction of CREB-Irs2, and increased proliferation (Fig. 6). We found that TAAR1 potentiates GSIS through adenylyl cyclase–cAMP activation of both PKA and Epac. In β-cells, PKA facilitates vesicle docking and priming for exocytosis by phosphorylation of the key SNARE complex and associated proteins (29, 30). PKA also phosphorylates KATP to reduce channel activity, contributing to membrane depolarization and opening of voltage-gated calcium channels (7). Epac is a cAMP-regulated guanine nucleotide exchange factor that interacts with a variety of intracellular proteins to increase GSIS (31). Epac is recruited to the plasma membrane in response to elevated cAMP in β-cells, where it clusters near secretory vesicles and binds to the exocytosis machinery to increase vesicle priming and fusion at docking sites (32). Epac can interact with Rim2 (Rab3-interacting molecule 2), which, together with Rab3, form a GTP-dependent complex between plasma membranes and docked synaptic vesicles to regulate exocytosis (33). Like PKA, Epac also modulates the activity of ATP regulated potassium channels to increase GSIS (34).
Figure 6.

Mechanism of anti-diabetic signaling of TAAR1 in β-cells. 1), activation of TAAR1-Gαs by amine (pink) ligands leads to generation of cAMP by AC. 2) cAMP then activates Epac and PKA, which are required for potentiation of GSIS by TAAR1. 3) PKA catalytic (c) subunits phosphorylate CREB, leading to induction of the CREB target gene Irs-2. 4) TAAR1 also stimulates cAMP-dependent calcium flux from internal (IP3R-mediated) stores and influx from extracellular sources, leading to CaMKII-dependent activation of Raf/MEK/ERK signaling and increased cellular proliferation in a PKA and Epac-dependent manner.
We found that TAAR1 stimulated PKA-dependent phosphorylation of CREB at Ser-133 in response to T1AM and RO5256390. Induction of TAAR1-CREB signaling by T1AM and RO5256390 led to activation of the Irs-2 gene, which has been shown to coordinate the activation of pro-proliferative and anti-apoptotic signaling pathways in β-cells (15). However, TAAR1 did not utilize PI3K to induce Irs-2 gene expression. IRS-2 is directly phosphorylated by the insulin receptor, leading to the recruitment and activation of other signaling proteins crucial for insulin signaling. Disruption of IRS-2 has been linked to the development of T2DM, and β-cell–specific Irs-2 knockouts have confirmed an essential role in insulin resistance, obesity, β-cell mass, and proliferation (36).
We documented a robust increase in ERK1/2 phosphorylation in response to both TAAR1 agonists that involves AC, PKA, Epac, and Raf/MEK1/2 activation (Fig. 6). Both B- and C-Raf mRNA were expressed, with 5-fold higher levels for B-Raf, and both have been shown to regulate MAPK signaling in β cells. In the case of glucose plus GLP-1R agonism, B-Raf, but not C-Raf, was found previously to be required for ERK1/2 phosphorylation (17, 37). Conversely, C-Raf knockdown increases rather than inhibits ERK1/2 phosphorylation in response to glucose (17). In terms of the roles PKA and Epac may play in Raf activation, Gαs/cAMP-stimulated Epac has been shown to activate Rap1/B-Raf and downstream ERK1/2 in neuronal, endocrine, and other cell types (18, 38). In thyroid cells, PKA has also been shown to directly activate Raf, which leads to ERK1/2 phosphorylation (39).
CaMKII activity was also necessary for activation of the MAPK cascade in the β-cells by both T1AM agonist and cAMP. This is consistent with previous work that showed direct CaMKII association and activation of Raf, which led to ERK1/2 phosphorylation in response to outside–in signaling in thyroid cells (20). Therefore, Raf may likely be an important upstream MAPK signaling node in β-cells that receives input from both calcium-regulated proteins such as CaMKII in addition to the cAMP-regulated proteins PKA and Epac. Although incretins have been shown to stimulate ERK1/2 activity in β-cells, MAPK activation is not required for or related to insulin secretion (40, 41) but is linked to increased cellular proliferation rates, which could play a role in compensatory β-cell hyperplasia in T2DM (42). Indeed, we observed a Raf/MEK-dependent increase in β-cell proliferation in response to TAAR1 agonism, indicating that TAAR1 could potentially improve β-cell mass, although this remains to be shown in vivo. Increased proliferation also occurred in response to the cAMP stimulant forskolin, validating the ability of Gαs/AC pathways to drive β-cell proliferation. ERK1/2 can regulate the insulin gene promoter and coordinate responses to endoplasmic reticulum stress (43), indicating other potential aspects of TAAR1-ERK1/2–mediated anti-diabetic signaling pathways to be explored.
By directly measuring intracellular calcium flux, we were able to observe a rapid and robust TAAR1-mediated calcium signal in the Ins-1 β-cell line. Consistent with the requirement for upstream CaMKII activation within the MAPK cascade, we discovered that both calcium influx from the outside and release from internal calcium stores via IP3 receptors was necessary for activation of ERK1/2 by a TAAR1-cAMP–dependent mechanism (Fig. 6). TAAR1-calcium signaling was not affected by YM-254890 or U73122, confirming that Gαq coupling or PLC-β were not involved. These data support the mechanism in Fig. 6, whereby the calcium signal induced by TAAR1-Gαs/cAMP is initially triggered by intracellular release through IP3 receptors, followed by activation of calcium channels on the plasma membrane, which provide the majority of the total observed calcium signal. Calcium flux has been shown to be a critical second messenger for various Gαs-signaling pathways in β-cells, most importantly insulin granule exocytosis, which depends in large part on the activity of the aforementioned CaMKII (7, 44, 45). CaMKII is localized to the insulin secretory granules and binds to synapsin-1 and MAP-2 proteins, which are involved in exocytosis (46). As 2-APB, an IP3R antagonist, was also found to eliminate potentiation of GSIS by TAAR1, calcium release from intracellular stores induced by TAAR1 activation also plays a role GSIS potentiation by the receptor. In a pathway that diverges from GSIS, however, TAAR1-calcium signaling also induces MAPK activation. This result is akin to what has been observed for glucose and GLP-1R-Gαs/AC coupling, which also require a rise in intracellular calcium to initiate MAPK signaling (47). Although glucose enhanced the ability of TAAR1 to signal pCREB and pERK1/2 pathways in the β-cell lines, we found that TAAR1 could do this independently of exogenous glucose, showing a divergence from the GSIS pathway.
One of the difficulties of studying TAAR1 in the past has been the lack of specific and efficacious agonists of the receptor. The endogenous agonist T1AM is capable of activating the Gi-coupled adrenergic receptor α-2A, albeit with poor affinity (48). Several novel compounds have now been identified as specific, full agonists of TAAR1 (49), including RO5256390 (13). We determined that RO5256390 produced essentially identical but more robust TAAR1 signals as T1AM. In the context of metabolic diseases, it is interesting to note that lisdexamfetamine dimesylate—the first Food and Drug Administration–approved drug for binge eating disorder—is a prodrug that is broken down into the active metabolite d-amphetamine (50, 51), a known TAAR1 agonist (52). Furthermore, in animal studies, inhibition of adrenergic receptors, which are believed to be a target of the prodrug's active metabolite, only blocks some of the effects of lisdexamfetamine dimesylate (53), raising the possibility that this amphetamine exerts some of its effects through TAAR1. In this regard, TAAR1 agonism has been shown to reduce monoaminergic signaling in the brain, reducing binge eating and impulsive behaviors, via TAAR1-mediated down-regulation of dopamine reward circuits (14, 54). The ability of TAAR1 to reduce maladaptive eating behaviors that can contribute to obesity and metabolic disease, coupled with our newly identified beneficial effects of TAAR1 agonism on β-cell lines, highlight the therapeutic potential of TAAR1.
Experimental procedures
Materials
Forskolin, T1AM, H-89, FK506, and thapsigargin were purchased from Cayman Chemical Co. MDL-12,330A, HJC-0350, Esi-05, EPPTB, STO-609, KN-93, and 2-APB were from Tocris. AZ-628, EGTA, U73122, LY-294,002, PD-98059, RO5256390, and Esi-09 were obtained from Sigma. Rat and mouse insulin ELISAs were purchased from Mercodia. RPMI 1640, Dulbecco's modified Eagle's medium, trypsin/EDTA, penicillin/streptomycin, and BAPTA-AM (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid)-acetoxymethyl ester and Fura-2/AM were purchased from Invitrogen. YM-254890, T-PER (tissue protein extraction reagent) lysis buffer, HALT protease/phosphatase inhibitors, and RPMI 1640 were purchased from Fisher Scientific. Antibodies against phospho-ERK1/2 (Thr-202/Tyr-204), ERK1/2, PKA Cat-α, and phospho-CREB (Ser-133) were from Cell Signaling Technology, and Epac and β-actin antibodies were from Santa Cruz Biotechnology. All siRNAs (PKA Cat-α, Epac, and control luciferase) used were from Santa Cruz Biotechnology. [3H]thymidine was from PerkinElmer Life Sciences. 2-Furoyl-LIGRLO-amide (LIGRLO) was synthesized by Oasis Pharmaceuticals.
Cell culture
The clonally derived Ins-1 (832/3) β-cell line that exhibits robust glucose and incretin responsiveness was generously provided by Dr. Christopher Newgard and cultured according to established protocols (55). Briefly, cells were maintained in a humidified incubator at 37 °C in RPMI 1640 medium supplemented with 10% fetal bovine serum, 10 mm HEPES, 2 mm l-glutamine, 1 mm sodium pyruvate, 50 μm 2-mercaptoethanol, 100 units/ml penicillin, and 100 μg/ml streptomycin and subcultured when confluent. The Min6 β-cell line was kindly provided by Dr. Melanie Cobb and grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 55 μm mercaptoethanol.
Insulin release from β-cell lines
Ins-1 or Min6 cells were seeded in 24-well plates and grown until confluent. On the day of the experiment, cells were washed once with Hanks′ Balanced Salt Solution and further incubated for 2 h in HBSS (114 mm NaCl, 4.7 mm KCl, 1.2 mm KH2PO4, 1.16 mm MgSO4, 20 mm HEPES, 2.5 mm CaCl2, 25.5 mm NaHCO3, and 0.2% BSA) with 3.5 mm (basal) D-glucose at 37 °C. Supernatants were sampled for basal insulin secretion levels, after which agonists were incubated with cells for 2 h at 37 °C. Supernatants were harvested, and insulin secretion was measured via insulin ELISA according to the manufacturer's instructions; relative insulin secretion was calculated as stimulated/basal insulin.
Real-time quantitative PCR and RT-PCR
RNA was isolated from cells using the Qiagen RNeasy Mini Kit and reverse-transcribed using Moloney murine leukemia virus reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Real-time PCR analysis was carried out using Lightcycler 480 SYBR Green I (Roche Diagnostics) using the following primers to amplify Irs-2, B-Raf, C-Raf, and Gadph: Irs-2, CGC AAG CAT CGA CTT CTT GTC (forward) and GCC CGC AGC ACT TTA CTC TT (reverse); B-Raf, GGA GCA TAA CCC ACC GTC AA (forward) and AAC AGC TGC TGC TC TCT CTG (reverse); C-Raf, CTG TCG CTG CAC TAC GGG (forward) and TCG TCT TCC AAG CTC CCT GT (reverse); Gapdh, GGC ATC GTG GAA GGA CTC ATG AC (forward) and ATG CCA GTG AGC TTC CCG TTC AGC (reverse). Relative abundance of Irs-2, C-Raf, and B-Raf mRNA was calculated with respect to the housekeeping gene Gapdh.
Western blotting
Ins-1 cells were grown to confluence in 12-well plates, at which point the complete medium was removed and (unless otherwise indicated) replaced with serum- and glucose-free RPMI supplemented with 0.2% BSA. Cells were starved for 2 h at 37 °C, and antagonists were added 30 min prior to addition of agonists. Cells were treated with agonists for the specified times at 37 °C, after which medium was removed, and cells were harvested in ice cold T-Per lysis buffer supplemented with 1× HALT phosphatase and protease inhibitors. Proteins were resolved in 10% SDS-polyacrylamide gels via gel electrophoresis, transferred to nitrocellulose membranes, and blocked in 5% nonfat milk in Tris-buffered saline with 0.05% Tween for 30 min. Membranes were incubated overnight at 4 °C with primary antibodies (phospho-ERK1/2, phospho-CREB) at 1:1000 dilution, followed by horseradish peroxidase–conjugated secondary antibody incubation (1:5000 dilution) for 1 h at room temperature. After detection of phosphoproteins, blots were stripped and reprobed with loading controls (total ERK1/2 and β-actin) for normalization. Several total CREB antibodies were tested (Cell Signaling Technology) for normalization of total CREB in Ins-1 cells, but we were unable to reliably detect CREB, and β-actin was used.
Intracellular Ca2+ measurements
Ins-1 cells were loaded with 2 μm Fura-2/AM in phenol red–free RPMI with 0.5% BSA for 30 min at 37 °C. Cells were washed and resuspended, and calcium flux was assessed by measuring fluorescence emission at a dual excitation of 340 and 380 nm using an LS-50B (PerkinElmer Life Sciences) spectrofluorimeter as before (56). Unless otherwise specified, antagonists were preincubated with cells at room temperature for 30 min before addition of agonists. The area under the curve (AUC) was quantified using Prism 5.0a and standardized to %max AUC using 10 μm T1AM as 100%.
Cellular proliferation
Radiolabeled thymidine incorporation was performed as described previously (35). Briefly, Ins-1 cells were seeded in 24-well plates at 1.5 × 105 cells/ml and allowed to attach overnight in complete medium. After 24 h, medium was aspirated and replaced with RPMI, 0.2% BSA, and 1 mm glucose to induce senescence, and cells were incubated at 37 °C for 24 h. Medium was then aspirated from cells and replaced with RPMI containing 0.2% BSA/4.5 mm glucose. Antagonists were preincubated with cells 30 min prior to addition of agonists, and cells were returned to the incubator for 24 h. During the last 4 h of incubation, 1 μCi of [3H]thymidine was added to each well. Cells were lifted, washed twice, fixed with 6% TCA, and centrifuged. The final pellet was resuspended in 0.2 N NaOH, added to scintillation fluid, and read for [3H]disintegrations per minute (DPM) counts.
Statistical analyses
Data are expressed as means ± S.D. Comparisons between experimental and control cohorts were performed by one-way ANOVA (for which a global p value was calculated) or t test as appropriate, and the mean of individual groups was compared using Dunnett's or Newman-Keuls post hoc correction. Global p values are numerically indicated in the figures, and post-test analyses between individual groups are indicated by asterisks. Analyses were performed using GraphPad Prism 6.0. Statistical significance was defined as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Author contributions
E. S. M., L. C., and A. K. conceptualization; E. S. M. data curation; E. S. M. and A. K. formal analysis; E. S. M. validation; E. S. M. and L. C. investigation; E. S. M. and A. K. visualization; E. S. M., L. C., and A. K. methodology; E. S. M. writing-original draft; E. S. M., L. C., and A. K. writing-review and editing; L. C. and A. K. resources; L. C. and A. K. supervision; L. C. and A. K. funding acquisition; L. C. and A. K. project administration.
Supplementary Material
This study was funded in whole or in part by NIDDK, National Institutes of Health Grant DK101240 (to A. K. and L. C.). Dr. Kuliopulos and Dr. Covic serve as scientific founders of Oasis Pharmaceuticals. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- T2DM
- type 2 diabetes mellitus
- GSIS
- glucose-stimulated insulin secretion
- T1AM
- 3-iodothyronamine
- PKA
- protein kinase A
- AC
- adenylyl cyclase
- CREB
- cAMP response element–binding protein
- PI3K
- phosphatidylinositol 3-kinase
- MAPK
- mitogen-activated protein kinase
- IP3
- inositol 1,4,5-trisphosphate
- LIGRLO
- 2-furoyl-LIGRLO-amide
- CaM
- calmodulin
- MEK
- mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- ERK
- extracellular signal-regulated kinase
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- GLP
- glucagon-like peptide
- AUC
- area under the curve
- ANOVA
- analysis of variance
- IP3R
- inositol 1,4,5-trisphosphate receptor
- EPPTB
- N-(3-ethoxyphenyl)-4-(1-pyrrolidinyl)-3-(trifluoromethyl)benzamide
- pCREB
- phospho CREB
- 2-APB
- 2-aminoethoxydiphenylborane.
References
- 1. Guariguata L., Whiting D. R., Hambleton I., Beagley J., Linnenkamp U., and Shaw J. E. (2014) Global estimates of diabetes prevalence for 2013 and projections for 2035. Diabetes Res. Clin. Pract. 103, 137–149 10.1016/j.diabres.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 2. Alberti K. G., and Zimmet P. Z. (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet. Med. 15, 539–553 [DOI] [PubMed] [Google Scholar]
- 3. Abdul-Ghani M. A., Tripathy D., and DeFronzo R. A. (2006) Contributions of β-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care 29, 1130–1139 10.2337/dc05-2179 [DOI] [PubMed] [Google Scholar]
- 4. Meece J. (2007) Pancreatic islet dysfunction in type 2 diabetes: a rational target for incretin-based therapies. Curr. Med. Res. Opin. 23, 933–944 10.1185/030079906X167336 [DOI] [PubMed] [Google Scholar]
- 5. Waugh N., Cummins E., Royle P., Clar C., Marien M., Richter B., and Philip S. (2010) Newer agents for blood glucose control in type 2 diabetes: systematic review and economic evaluation. Health Technol. Assess. 14, 1–248 [DOI] [PubMed] [Google Scholar]
- 6. Hermansen K., and Mortensen L. S. (2007) Bodyweight changes associated with antihyperglycaemic agents in type 2 diabetes mellitus. Drug Saf. 30, 1127–1142 10.2165/00002018-200730120-00005 [DOI] [PubMed] [Google Scholar]
- 7. Tengholm A. (2012) Cyclic AMP dynamics in the pancreatic β-cell. Ups. J. Med. Sci. 117, 355–369 10.3109/03009734.2012.724732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baggio L. L., and Drucker D. J. (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132, 2131–2157 10.1053/j.gastro.2007.03.054 [DOI] [PubMed] [Google Scholar]
- 9. Raab S., Wang H., Uhles S., Cole N., Alvarez-Sanchez R., Künnecke B., Ullmer C., Matile H., Bedoucha M., Norcross R. D., Ottaway-Parker N., Perez-Tilve D., Conde Knape K., Tschöp M. H., Hoener M. C., and Sewing S. (2016) Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol. Metab. 5, 47–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferragud A., Howell A. D., Moore C. F., Ta T. L., Hoener M. C., Sabino V., and Cottone P. (2017) The trace amine-associated receptor 1 agonist RO5256390 blocks compulsive, binge-like eating in rats. Neuropsychopharmacology 42, 1458–1470 10.1038/npp.2016.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bunzow J. R., Sonders M. S., Arttamangkul S., Harrison L. M., Zhang G., Quigley D. I., Darland T., Suchland K. L., Pasumamula S., Kennedy J. L., Olson S. B., Magenis R. E., Amara S. G., and Grandy D. K. (2001) Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptor. Mol. Pharmacol. 60, 1181–1188 10.1124/mol.60.6.1181 [DOI] [PubMed] [Google Scholar]
- 12. Scanlan T. S. (2009) Minireview: 3-iodothyronamine (T1AM): a new player on the thyroid endocrine team? Endocrinology 150, 1108–1111 10.1210/en.2008-1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Revel F. G., Moreau J. L., Pouzet B., Mory R., Bradaia A., Buchy D., Metzler V., Chaboz S., Groebke Zbinden K., Galley G., Norcross R. D., Tuerck D., Bruns A., Morairty S. R., Kilduff T. S., et al. (2013) A new perspective for schizophrenia: TAAR1 agonists reveal antipsychotic- and antidepressant-like activity, improve cognition and control body weight. Mol. Psychiatry 18, 543–556 10.1038/mp.2012.57 [DOI] [PubMed] [Google Scholar]
- 14. Bradaia A., Trube G., Stalder H., Norcross R. D., Ozmen L., Wettstein J. G., Pinard A., Buchy D., Gassmann M., Hoener M. C., and Bettler B. (2009) The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. U.S.A. 106, 20081–20086 10.1073/pnas.0906522106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jhala U. S., Canettieri G., Screaton R. A., Kulkarni R. N., Krajewski S., Reed J., Walker J., Lin X., White M., and Montminy M. (2003) cAMP promotes pancreatic β-cell survival via CREB-mediated induction of IRS2. Genes Dev. 17, 1575–1580 10.1101/gad.1097103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohanty S., Spinas G. A., Maedler K., Zuellig R. A., Lehmann R., Donath M. Y., Trüb T., and Niessen M. (2005) Overexpression of IRS2 in isolated pancreatic islets causes proliferation and protects human β-cells from hyperglycemia-induced apoptosis. Exp. Cell Res. 303, 68–78 10.1016/j.yexcr.2004.09.011 [DOI] [PubMed] [Google Scholar]
- 17. Duan L., and Cobb M. H. (2010) Calcineurin increases glucose activation of ERK1/2 by reversing negative feedback. Proc. Natl. Acad. Sci. U.S.A. 107, 22314–22319 10.1073/pnas.1016630108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emery A. C., Eiden M. V., Mustafa T., and Eiden L. E. (2013) Rapgef2 connects GPCR-mediated cAMP signals to ERK activation in neuronal and endocrine cells. Sci. Signal. 6, ra51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Michael E. S., Kuliopulos A., Covic L., Steer M. L., and Perides G. (2013) Pharmacological inhibition of PAR2 with the pepducin P2pal-18S protects mice against acute experimental biliary pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G516–G526 10.1152/ajpgi.00296.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Illario M., Cavallo A. L., Bayer K. U., Di Matola T., Fenzi G., Rossi G., and Vitale M. (2003) Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J. Biol. Chem. 278, 45101–45108 10.1074/jbc.M305355200 [DOI] [PubMed] [Google Scholar]
- 21. Shearer A. M., Rana R., Austin K., Baleja J. D., Nguyen N., Bohm A., Covic L., and Kuliopulos A. (2016) Targeting liver fibrosis with a cell-penetrating protease-activated receptor-2 (PAR2) pepducin. J. Biol. Chem. 291, 23188–23198 10.1074/jbc.M116.732743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jacobo S. M., Guerra M. L., and Hockerman G. H. (2009) Cav1.2 and Cav1.3 are differentially coupled to glucagon-like peptide-1 potentiation of glucose-stimulated insulin secretion in the pancreatic β-cell line INS-1. J. Pharmacol. Exp. Ther. 331, 724–732 10.1124/jpet.109.158519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ha T.-Y., Kim Y.-S., Kim C. H., Choi H.-S., Yang J., Park S. H., Kim D. H., and Rhee J.-K. (2014) Novel GPR119 agonist HD0471042 attenuated type 2 diabetes mellitus. Arch. Pharm. Res. 37, 671–678 10.1007/s12272-013-0209-0 [DOI] [PubMed] [Google Scholar]
- 24. Nakagawa Y., Nagasawa M., Yamada S., Hara A., Mogami H., Nikolaev V. O., Lohse M. J., Shigemura N., Ninomiya Y., and Kojima I. (2009) Sweet taste receptor expressed in pancreatic β-cells activates the calcium and cyclic AMP signaling systems and stimulates insulin secretion. PLoS ONE 4, e5106 10.1371/journal.pone.0005106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang H., and Yang L. (2016) Targeting cAMP/PKA signaling pathway for glycemic control and type 2 diabetes therapy. J. Mol. Endocrinol. 57, R93–R108 10.1530/JME-15-0316 [DOI] [PubMed] [Google Scholar]
- 26. Drucker D. J. (2003) Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care 26, 2929–2940 10.2337/diacare.26.10.2929 [DOI] [PubMed] [Google Scholar]
- 27. Lee Y.-S., and Jun H.-S. (2014) Anti-diabetic actions of glucagon-like peptide-1 on pancreatic β-cells. Metabolism 63, 9–19 10.1016/j.metabol.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 28. Mentlein R., Gallwitz B., and Schmidt W. E. (1993) Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7–36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur. J. Biochem. 214, 829–835 10.1111/j.1432-1033.1993.tb17986.x [DOI] [PubMed] [Google Scholar]
- 29. Foster L. J., Yeung B., Mohtashami M., Ross K., Trimble W. S., and Klip A. (1998) Binary interactions of the SNARE proteins Syntaxin-4, SNAP23, and VAMP-2 and their regulation by phosphorylation. Biochemistry 37, 11089–11096 10.1021/bi980253t [DOI] [PubMed] [Google Scholar]
- 30. Chheda M. G., Ashery U., Thakur P., Rettig J., and Sheng Z. H. (2001) Phosphorylation of Snapin by PKA modulates its interaction with the SNARE complex. Nat. Cell Biol. 3, 331–338 10.1038/35070000 [DOI] [PubMed] [Google Scholar]
- 31. Holz G. G., Kang G., Harbeck M., Roe M. W., and Chepurny O. G. (2006) Cell physiology of cAMP sensor Epac. J. Physiology 577, 5–15 10.1113/jphysiol.2006.119644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alenkvist I., Gandasi N. R., Barg S., and Tengholm A. (2017) Recruitment of Epac2A to insulin granule docking sites regulates priming for exocytosis. Diabetes 66, 2610–2622 10.2337/db17-0050 [DOI] [PubMed] [Google Scholar]
- 33. Ozaki N., Shibasaki T., Kashima Y., Miki T., Takahashi K., Ueno H., Sunaga Y., Yano H., Matsuura Y., Iwanaga T., Takai Y., and Seino S. (2000) cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol. 2, 805–811 10.1038/35041046 [DOI] [PubMed] [Google Scholar]
- 34. Leech C. A., Dzhura I., Chepurny O. G., Schwede F., Genieser H.-G., and Holz G. G. (2010) Facilitation of β-cell KATP channel sulfonylurea sensitivity by a cAMP analog selective for the cAMP-regulated guanine nucleotide exchange factor Epac. Islets 2, 72–81 10.4161/isl.2.2.10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sevigny L. M., Austin K. M., Zhang P., Kasuda S., Koukos G., Sharifi S., Covic L., and Kuliopulos A. (2011) Protease-activated receptor-2 modulates protease-activated receptor-1-driven neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 31, e100–e106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kubota N., Terauchi Y., Tobe K., Yano W., Suzuki R., Ueki K., Takamoto I., Satoh H., Maki T., Kubota T., Moroi M., Okada-Iwabu M., Ezaki O., Nagai R., Ueta Y., et al. (2004) Insulin receptor substrate 2 plays a crucial role in β cells and the hypothalamus. J. Clin. Invest. 114, 917–927 10.1172/JCI21484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Trümper J., Ross D., Jahr H., Brendel M. D., Göke R., and Hörsch D. (2005) The Rap-B-Raf signalling pathway is activated by glucose and glucagon-like peptide-1 in human islet cells. Diabetologia 48, 1534–1540 10.1007/s00125-005-1820-5 [DOI] [PubMed] [Google Scholar]
- 38. Luttrell L. M., Wang J., Plouffe B., Smith J. S., Yamani L., Kaur S., Jean-Charles P. Y., Gauthier C., Lee M. H., Pani B., Kim J., Ahn S., Rajagopal S., Reiter E., Bouvier M., et al. (2018) Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 11, eaat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vuchak L. A., Tsygankova O. M., Prendergast G. V., and Meinkoth J. L. (2009) Protein kinase A and B-Raf mediate extracellular signal-regulated kinase activation by thyrotropin. Mol. Pharmacol. 76, 1123–1129 10.1124/mol.109.060129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Panse M., Gerst F., Kaiser G., Teutsch C. A., Dölker R., Wagner R., Häring H. U., and Ullrich S. (2015) Activation of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2) by free fatty acid receptor 1 (FFAR1/GPR40) protects from palmitate-induced β cell death, but plays no role in insulin secretion. Cell Physiol. Biochem. 35, 1537–1545 10.1159/000373969 [DOI] [PubMed] [Google Scholar]
- 41. Khoo S., and Cobb M. H. (1997) Activation of mitogen-activating protein kinase by glucose is not required for insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 94, 5599–5604 10.1073/pnas.94.11.5599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fu Z., Zhang W., Zhen W., Lum H., Nadler J., Bassaganya-Riera J., Jia Z., Wang Y., Misra H., and Liu D. (2010) Genistein induces pancreatic β-cell proliferation through activation of multiple signaling pathways and prevents insulin-deficient diabetes in mice. Endocrinology 151, 3026–3037 10.1210/en.2009-1294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lawrence M., Shao C., Duan L., McGlynn K., and Cobb M. H. (2008) The protein kinases ERK1/2 and their roles in pancreatic β cells. Acta Physiologica 192, 11–17 [DOI] [PubMed] [Google Scholar]
- 44. Prentki M., Glennon M. C., Geschwind J. F., Matschinsky F. M., and Corkey B. E. (1987) Cyclic AMP raises cytosolic Ca2+ and promotes Ca2+ influx in a clonal pancreatic β-cell line (HIT T-15). FEBS Lett. 220, 103–107 10.1016/0014-5793(87)80884-0 [DOI] [PubMed] [Google Scholar]
- 45. Lang J. (1999) Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur. J. Biochem. 259, 3–17 10.1046/j.1432-1327.1999.00043.x [DOI] [PubMed] [Google Scholar]
- 46. Easom R. A. (1999) CaM kinase II: a protein kinase with extraordinary talents germane to insulin exocytosis. Diabetes 48, 675–684 10.2337/diabetes.48.4.675 [DOI] [PubMed] [Google Scholar]
- 47. Frödin M., Sekine N., Roche E., Filloux C., Prentki M., Wollheim C. B., and Van Obberghen E. (1995) Glucose, other secretagogues, and nerve growth factor stimulate mitogen-activated protein kinase in the insulin-secreting β-cell line, INS-1. J. Biol. Chem. 270, 7882–7889 10.1074/jbc.270.14.7882 [DOI] [PubMed] [Google Scholar]
- 48. Zucchi R., Accorroni A., and Chiellini G. (2014) Update on 3-iodothyronamine and its neurological and metabolic actions. Front. Physiol. 5, 402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Galley G., Beurier A., Décoret G., Goergler A., Hutter R., Mohr S., Pähler A., Schmid P., Türck D., Unger R., Zbinden K. G., Hoener M. C., and Norcross R. D. (2016) Discovery and characterization of 2-aminooxazolines as highly potent, selective, and orally active TAAR1 agonists. ACS Med. Chem. Lett. 7, 192–197 10.1021/acsmedchemlett.5b00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ward K., and Citrome L. (2018) Lisdexamfetamine: chemistry, pharmacodynamics, pharmacokinetics, and clinical efficacy, safety, and tolerability in the treatment of binge eating disorder. Expert Opin. Drug Metab. Toxicol. 14, 229–238 10.1080/17425255.2018.1420163 [DOI] [PubMed] [Google Scholar]
- 51. McElroy S. L., Hudson J. I., Mitchell J. E., Wilfley D., Ferreira-Cornwell M. C., Gao J., Wang J., Whitaker T., Jonas J., and Gasior M. (2015) Efficacy and safety of lisdexamfetamine for treatment of adults with moderate to severe binge-eating disorder: a randomized clinical trial. JAMA Psychiatry 72, 235–246 10.1001/jamapsychiatry.2014.2162 [DOI] [PubMed] [Google Scholar]
- 52. Reese E. A., Bunzow J. R., Arttamangkul S., Sonders M. S., and Grandy D. K. (2007) Trace amine-associated receptor 1 displays species-dependent stereoselectivity for isomers of methamphetamine, amphetamine, and para-hydroxyamphetamine. J. Pharmacol. Exp. Ther. 321, 178–186 10.1124/jpet.106.115402 [DOI] [PubMed] [Google Scholar]
- 53. Vickers S. P., Hackett D., Murray F., Hutson P. H., and Heal D. J. (2015) Effects of lisdexamfetamine in a rat model of binge-eating. J. Psychopharmacol. 29, 1290–1307 10.1177/0269881115615107 [DOI] [PubMed] [Google Scholar]
- 54. Revel F. G., Moreau J.-L., Gainetdinov R. R., Bradaia A., Sotnikova T. D., Mory R., Durkin S., Zbinden K. G., Norcross R., Meyer C. A., Metzler V., Chaboz S., Ozmen L., Trube G., Pouzet B., et al. (2011) TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. U.S.A. 108, 8485–8490 10.1073/pnas.1103029108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., and Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 10.2337/diabetes.49.3.424 [DOI] [PubMed] [Google Scholar]
- 56. Covic L., Gresser A. L., and Kuliopulos A. (2000) Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry 39, 5458–5467 10.1021/bi9927078 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.