

SUMMARY
The MEF2 family of transcription factors restricts excitatory synapse number in an activity-dependent fashion during development, yet MEF2 has not been implicated in long-term synaptic depression (LTD), which is thought to initiate synapse elimination. Mutations in MEF2 pathways are implicated in autism spectrum disorders, which include cerebellar dysfunction. Here, we test the hypothesis that cerebellar LTD requires postsynaptic activation of MEF2. Knockdown of MEF2D produces suppression of the transcription-dependent late phase of LTD in cultured Purkinje cells. The late phase of LTD is also completely blocked in Purkinje cells derived from MEF2A+MEF2D null mice and rescued with plasmids that drive expression of MEF2D but not phosphatase-resistant mutant MEF2D S444D. Wild-type Purkinje cells transfected with a constitutively active form of MEF2 show no alterations of synaptic strength. Thus, postsynaptic activation of MEF2 by S444 dephosphorylation is necessary, but not sufficient, for the late phase of cerebellar LTD.
Graphical Abstract
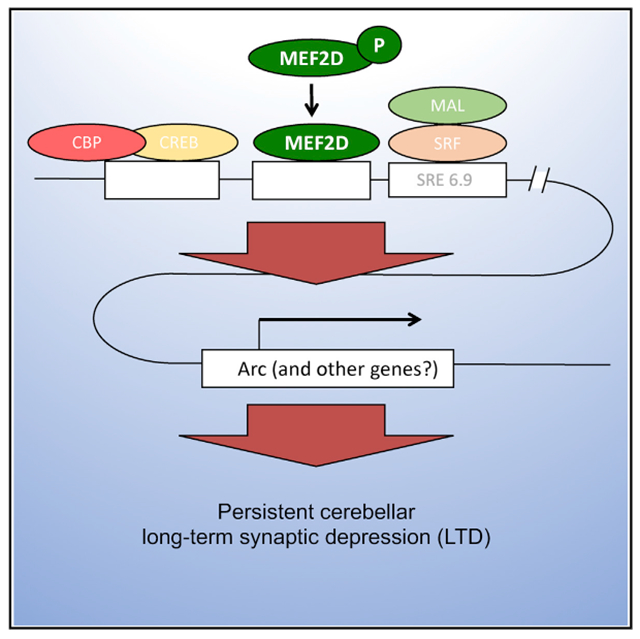
In Brief
Neurodevelopmental disorders can reflect defects in synaptic pruning, which is thought to require activity-dependent weakening of synapses, a process called long-term depression. Andzelm et al. show that MEF2, which is important for neuronal development, is required for the late phase of long-term depression in the cerebellum.
INTRODUCTION
The myocyte enhancer factor 2 (MEF2) family of transcription factors (consisting of MEF2A through D) is highly expressed in the brain where it is activated in response to neuronal activity (Mao and Wiedmann, 1999; Dolmetsch et al., 2001). This is accomplished in part by activation of the Ca-dependent phosphatase calcineurin (Mao and Wiedmann, 1999) and consequent dephosphorylation of MEF2 isoforms (Flavell et al., 2006; Pulipparacharuvil et al., 2008). In hippocampal or striatal neurons, constitutive MEF2 activation produced a strong reduction in the number of excitatory synapses, as indexed by both immunocytochemistry for glutamatergic synaptic markers and recording of miniature excitatory postsynaptic currents (mEPSCs) (Flavell et al., 2006; Pfeiffer et al., 2010; Barylko et al., 2018). Conversely, inhibition of MEF2 activity through knockdown or gene deletion increased the density of excitatory synapses (Flavell et al., 2006; Pfeiffer et al., 2010). In this way, activity-driven MEF2 activation provides a mechanism by which sensory-motor experience can drive programs of gene expression leading to synapse weakening and elimination beginning during the activity-dependent phase of brain development and continuing through adulthood (Chang et al., 2017).
MEF2 target genes are numerous and several of them, including Arc, Syngap, Protocadherin 10, Homer 1a, and ubiquitin protein ligase E3A, act at excitatory synapses (Flavell et al., 2008; Tsai et al., 2012; Wilkerson et al., 2014). In hippocampal pyramidal neurons, it has been shown that synapse elimination triggered by persistent activation of the glutamate receptor mGlu5 acts through MEF2-driven transcription and the subsequent dendritic translation of two different mRNAs. The first is Arc, a synaptic protein that weakens synapses by engaging clathrin and dynamin-mediated endocytosis of AMPA receptors (Wilkerson et al., 2014). The second is protocadherin 10, the translation of which is regulated by the fragile X mental retardation protein (FMRP; Pfeiffer et al., 2010; Tsai et al., 2012). Protocadherin 10 links the synaptic protein PSD-95 to proteasomes, thereby targeting PSD-95 for degradation. When the interaction of protocadherin 10 and PSD-95 was blocked, MEF2-driven synapse elimination was strongly attenuated (Tsai et al., 2012). This is an important confluence of molecular signals because loss-of-function mutations in the genes coding for FMRP (Hallmayer et al., 1994), protocadherin 10 (Morrow et al., 2008), and MEF2C (Mikhail et al., 2011) have all been linked to autism spectrum disorders and the associated failure of excitatory synaptic elimination in early postnatal life.
Long-term depression (LTD) of cerebellar parallel fiber-Purkinje cell synapses is induced postsynaptically through an mGlu1/protein kinase Cα (PKCα) cascade and is initially expressed by PICK1-dependent clathrin and dynamin-mediated endocytosis of GluA2-containing surface AMPA receptors (Steinberg et al., 2006). A late phase of cerebellar LTD in cultured Purkinje cells, beginning 45–60 min after induction, is blocked by chemical transcription or translation inhibitors or by separating the synapses from the nucleus through formation of a stable dendritic outside-out macropatch (Linden, 1996; Murashima and Hirano, 1999). This transcription-dependent late phase does not require continued activation of mGlu1 or PKCα nor does it require continued PICK1-GluA2 interaction (Linden, 2012). It does, however, require persistent clathrin and dynamin-mediated endocytosis driven by the synaptic protein Arc. Arc binds the key endocytotic proteins dynamin and endophilin (Chowdhury et al., 2006) and is expressed in cultured Purkinje cells in response to LTD-inducing stimuli (Smith-Hicks et al., 2010).
While MEF2 activation has been linked to synaptic elimination (as well as synaptic weakening without elimination; Elmer et al., 2013), which proceeds slowly over a period of 12–72 h, it has not been implicated in any form of long-term synaptic depression (LTD), which is induced much more rapidly. This is surprising given that several forms of LTD are triggered by activation of mGlu1 and mGlu5 (Collingridge et al., 2010), and it has been hypothesized that impairment of mGlu-driven LTD underlies the failure of developmental synapse elimination that is associated with autism spectrum disorders (Zoghbi and Bear, 2012; Piochon et al., 2016; Wilkerson et al., 2018). Here, we have sought to test the hypothesis that MEF2 activation is necessary and sufficient for the transcription-dependent late phase of cerebellar LTD.
RESULTS
We used a well-established cell culture model system to investigate the expression mechanism of the late phase of cerebellar LTD (Linden, 1996). Whole-cell voltage-clamp recordings were made from Purkinje cells in cultures derived from embryonic mouse cerebellum. On the first day after culture preparation, cultures were either transfected with plasmids expressing various short hairpin RNAs (shRNAs) or MEF2 proteins or infected with a lentivirus that drives expression of Cre recombinase. Six to eight days later, two iontophoresis electrodes filled with glutamate were used to stimulate separate dendritic branches of a single Purkinje cell in an alternating fashion. Following acquisition of baseline responses to test pulses of glutamate at these two sites, test pulses were halted at the “control” iontophoresis electrode and LTD was induced by applying glutamate and depolarization conjunctive stimulation at the “paired” electrode. This resulted in input-specific induction of LTD as has been previously described (Linden, 1996).
We measured basal MEF2 activity in our cultured Purkinje cells by using a MEF2-activity reporter consisting of a triple repeat of the MEF2-response element (MRE) fused to a minimal fos promoter (Flavell et al., 2006) driving destabilized EGFP. The reporter was introduced after 3 days in culture. Only 3/100 unstimulated Purkinje cells were EGFP-positive (Table 1). When Purkinje cells were whole-cell voltage clamped with a somatic electrode at a command potential of −70 mV for 40 min, 2/20 were EGFP-positive (the n is lower because it takes much more time and effort to patch clamp Purkinje cells than to just find them in a culture dish). After glutamate and depolarization paired stimulation (the protocol that would normally be used to evoke LTD), nearly all of the cells were EGFP-positive (19/20). However, neither repeated depolarization alone nor repeated glutamate pulses alone produced substantial MEF2 activation (3/20 cells for each condition). Thus, in our culture conditions, under either unrecorded or voltage-clamp conditions, basal MEF2 activity was low but could be activated by an LTD induction protocol.
Table 1.
Both LTD Induction by Glutamate + Depolarization Pairing and Transfection with a Tamoxifen-Inducible MEF2 Construct Activated MEF2 Transcriptional Activity as Measured Using an MEF2 Reporter Plasmid
| Manipulation | MEF2 Reporter Positive Purkinje Cells (>4× Background) at t = 40 Min |
|---|---|
| Patched Cells | |
| Vhold = −70 mV | 2/20 |
| Depolarization | 3/20 |
| Glutamate pulses | 3/20 |
| Depolarization + glutamate pulses | 19/20 |
| Vhold = −70mV (MEF2A+2D DKO) | 1/20 |
| Depolarization + glutamate pulses (MEF2A+2D DKO) | 0/20 |
| Non-patched Cells | |
| No treatment | 3/100 |
| MEF2-VP16-ER, 16–26 h tamoxifen | 100/100 |
| MEF2-ΔDBD-VP16-ER, 16–26 h tamoxifen | 2/100 |
| Tamoxifen, 16–26 h | 3/100 |
| Phorbol-12,13-diacetate, 200 nM, 10 min | 97/100 |
| MEF2D S444A | 2/100 |
A MEF2-activity reporter was created consisting of a triple repeat of the MEF2-response element (MRE) fused to a minimal promoter of the fos gene (that does not itself confer glutamate or depolarization responsiveness; Flavell et al., 2006) inserted in front of destabilized EGFP. Using a gene gun, this reporter plasmid was delivered to Purkinje cells, together with a separate dsRedexpress2 plasmid as a marker, 3 days after the culture was prepared. Measurements were then made 7–10 days after transfection.
This MEF2 reporter construct cannot distinguish between MEF2 isoforms; multiple family members are capable of activating it. However, MEF2A/MEF2D brain-specific double knockout mice have impaired locomotion as assessed by rotarod performance (Akhtar et al., 2012), while MEF2A-only brain-specific knockout mice or MEF2C-only brain-specific knockout mice displayed normal motor activity (Akhtar et al., 2012; Barbosa et al., 2008). Because defects in locomotor activity have been associated with defects in Purkinje cell plasticity (Vinueza Veloz et al., 2015), we reasoned MEF2A and MEF2D could be the MEF2 family members required for Purkinje cell LTD. We examined the expression of MEF2A and MEF2D in the cerebellum and confirmed that both family members were expressed in Purkinje cells (Figure S1A and data not shown).
To test the hypothesis that MEF2A and MEF2D are required for the late phase of LTD, Purkinje cells were first transfected with a plasmid expressing a MEF2A-degrading shRNA (Figure 1A). As a control, Purkinje cells were transfected with an empty vector. The empty vector and MEF2A shRNA Purkinje cells showed similar expression of the early phase of input-specific LTD (control pathway: empty vector, 103% ± 6.4% of baseline, n = 7; MEF2A shRNA, 102% ± 7.6%, n = 6; paired pathway: empty vector; 54% ± 7.1%; MEF2A shRNA, 50% ± 6.5%, all at t = 40 min; mean ± SEM). Starting at about t = 65 min, the late phase of LTD was slightly attenuated by MEF2A shRNA treatment, and this persisted until the end of the recording (paired pathway: empty vector; 51% ± 5.4%; MEF2A shRNA, 68% ± 6.6% at t = 120 min). When Purkinje cells were transfected with a plasmid expressing MEF2D-degrading shRNA, normal early-phase LTD was seen and the late phase of LTD was substantially, but not completely, attenuated (54% ± 5.9% at t = 40 min and 82% ± 6.5% at t = 120 min, n = 8) when compared with either an empty vector control (54% ± 7.0% at t = 40 min and 51% ± 5.4% at t = 120 min, n = 7) or a MEF2D shRNA with point mutations that fails to target MEF2D (58% ± 8.0% at t = 40 min and 57% ± 7.2% at t = 120 min, n = 7). When Purkinje cells were transfected with both MEF2A and MEF2D shRNAs, a nearly complete suppression of the late phase of LTD was produced (99% ± 7.7% at t = 120 min, n = 8). None of the shRNAs used in this experiment altered morphological characteristics of cultured Purkinje cells, including somatic area and dendritic spine density (Figure S2; Table S1).
Figure 1. MEF2A and MEF2D Knockdown with shRNA Blocks the Late Phase of Cerebellar LTD, and This Blockade Is Completely Rescued by the Co-expression of shRNA-Resistant MEF2D.
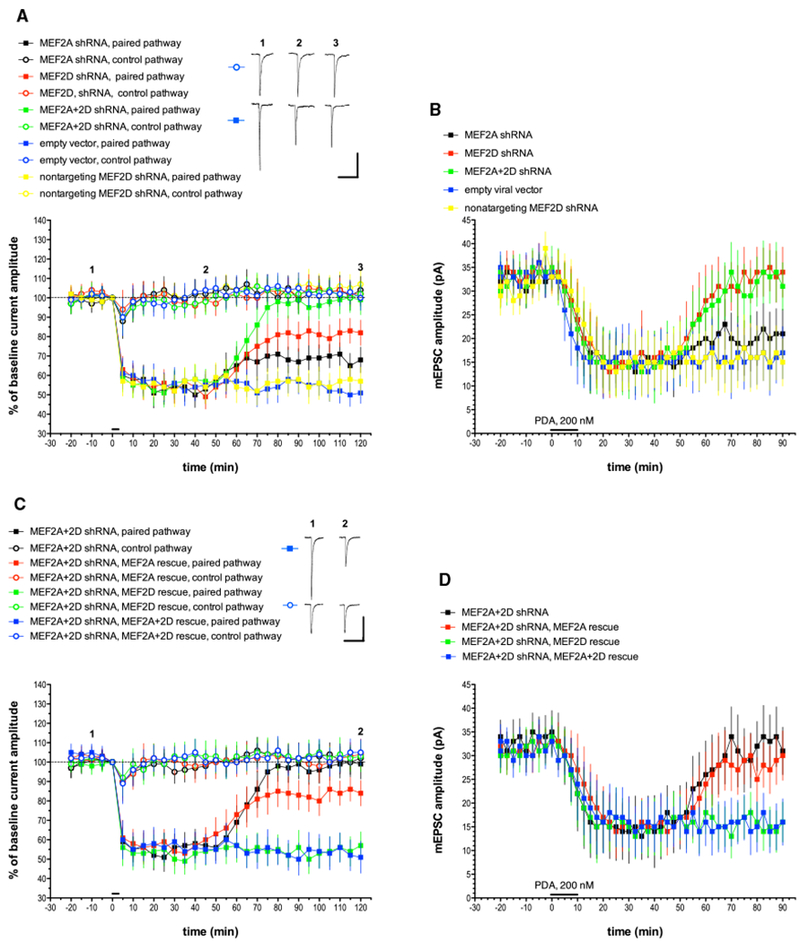
(A) Purkinje cells were transfected with plasmids designed to express various shRNAs, and recordings were made 1–3 days later. Test pulses of glutamate were applied to two non-overlapping sites on the Purkinje cell dendrite. Pulses were alternated at 10 s intervals. To induce LTD, at t = 0 min, six 3-s-long depolarizing commands to 0 mV were coupled with glutamate pulses delivered only to the paired pathway at t = 0 min, as indicated by the horizontal bar. The control pathway received only somatic step depolarization at t = 0 min. Alternate test pulses were then resumed for the duration of the experiment. Exemplar traces are single (unaveraged) responses, and they correspond to the points indicated on the time course graph. Plot points indicate the mean ± SEM in this and all subsequent graphs. MEF2A shRNA, n = 8; MEF2D shRNA, n = 8; MEF2A+2D shRNA, n = 8; empty vector, n = 7; nontargeting MEF2D shRNA, n = 7. Scale bars represent 2 s, 100 pA. When compared with the empty vector paired pathway, the late phase of LTD measured at t = 120 min was significantly different from the paired pathway responses in the MEF2A shRNA group (p < 0.05), the MEF2D shRNA group (p < 0.01), and the MEF2+2D shRNA group (p < 0.001) but not the nontargeting MEF2D shRNA group (p > 0.20). Statistics by Mann-Whitney U test with Bonferroni correction for multiple comparisons in this and all following LTD figures.
(B) Purkinje cells were treated with shRNAs as described above and recordings of mEPSCs were made in the presence of tetrodotoxin (TTX) and picrotoxin. After measuring baseline mEPSCs, a global, chemical form of LTD was produced by a 10-min-long bath application of the synthetic PKC activator phorbol-12,13-diactate (PDA, 200 nM, indicated by the horizontal bar), and mEPSC recording was continued. n = 10 cells/group. When compared with the empty vector pathway, the late phase of chemical LTD measured at t = 90 min was significantly different from the paired pathway responses in the MEF2D shRNA group (p < 0.05) and the MEF2A+2D shRNA group (p < 0.05) but not the nontargeting MEF2D shRNA group (p > 0.20) or the MEF2A shRNA group (p > 0.20).
(C) Exemplar traces are single (unaveraged) responses, and they correspond to the points indicated on the time course graph. MEF2A+2D shRNA, n = 8(these are the same data shown in Figure 1A, re-plotted here to allow for comparison with rescue treatments); empty vector, n = 7; nontargeting MEF2D shRNA, n = 7; MEF2A+2D shRNA, MEF2A rescue, n = 7; MEF2A+2D shRNA, MEF2D rescue, n = 6; MEF2A+2D shRNA, MEF2A+2D rescue, n = 7. Scale bars represent 2 s, 100 pA. When compared with the MEF2A+2D shRNA group with no rescue plasmid, paired pathway late LTD responses were significantly different from the MEF2D rescue group (p < 0.001) and the MEF2A+2D rescue group (p < 0.001) but not the MEF2A rescue group (p > 0.10).
(D) mEPSC recordings and chemical LTD induction by PDA were performed as indicated for Figure 1B. n = 10 cells/group. When compared with the MEF2A+2D shRNA group with no rescue plasmid, late chemical LTD responses were significantly different from the MEF2D rescue group (p < 0.05) and the MEF2A+2D rescue group (p < 0.05) but not the MEF2A rescue group (p > 0.20).
See also Figures S1 and S2 and Table S1.
The response to iontophoretic pulses of glutamate cannot give an accurate measure of basal postsynaptic strength. To assess the baseline level of postsynaptic function in MEF2A and MEF2D shRNA-treated cells, we recorded miniature excitatory postsynaptic currents (mEPSCs), which are mediated by AMPA-type glutamate receptors. After measuring baseline mEPSCs, a global, chemical form of LTD was produced by a 10-min-long bath application of the synthetic PKC activator phorbol-12,13-diacetate (PDA, 200 nM) (Figure 1B). The results were mostly consistent with those in which glutamate pulse and depolarization pairing was used to induce LTD. Either MEF2D shRNA alone or MEF2A + MEF2D shRNA dual treatment produced Purkinje cells with normal basal mEPSC amplitude (MEF2D shRNA: 33 pA ± 3.5 pA; MEF2A+2D shRNA: 35 pA ± 4.5 pA at t = 0 min, mean ± SEM, n = 10 cells/group) and chemical LTD with an early phase but no late phase (MEF2D shRNA: 16 pA ± 5.5 pA at t = 40 min, 34 pA ± 5.3 pA at t = 90 min; MEF2A+2D shRNA: 14 pA ± 7.6 pA at t = 40 min, 31 pA ± 5.0 pA at t = 90 min). MEF2A shRNA alone produced a small attenuation of the late phase (21 pA ± 5.2 pA at t = 90 min, n = 10) that was not substantially different from either the empty vector control (17 pA ± 5.5 pA at t = 90 min, n = 10) or the MEF2D nontargeting shRNA control (15 pA ± 5.0 pA at t = 90 min, n = 10).
As a further test of the hypothesis that MEF2 activation is required for the late phase of cerebellar LTD, Purkinje cells were transfected with both MEF2A and MEF2D shRNA-expressing plasmids together with rescue by plasmids designed to drive expression of MEF2A and MEF2D (Figures 1C; Figure S1B). These plasmids were engineered with mismatches so that their mRNA products would not be degraded by the corresponding shRNAs (Flavell et al., 2006). Rescue with either MEF2D plasmid or MEF2D and MEF2A plasmids together produced a complete rescue of the late phase of LTD as measured by either glutamate and depolarization pairing (MEF2D rescue: 57% ± 7.1% at t = 120 min, n = 6; MEF2A+2D rescue: 51% ± 8.2% at t = 120 min, n = 7) or chemical LTD induction with PDA (MEF2D rescue: 16 pA ± 4.2 pA at t = 90 min, n = 10; MEF2A+2D rescue: 16 pA ± 4.9 pA at t = 90 min, n = 10). By contrast, rescue with the MEF2A plasmid alone produced only a small late phase as measured with pairing (84% ± 6.5% at t = 120 min, n = 7) and no rescue of the late phase evoked with PDA (30 pA ± 5.2 pA at t = 90 min; n = 10).
We generated MEF2A+MEF2D double conditional mice by crossing previously made MEF2D conditional mice (Andzelm et al., 2015) with newly generated MEF2A conditionally targeted mice (Figure S3). Cerebellar cultures were prepared from MEF2A+MEF2D double conditional mice, which were then infected by lentivirus expressing Cre (MEF2A+2D double knockout [DKO]; Figures 2A and 2B). These mice have normal basal mEPSC amplitude (31 pA ± 3.8 pA at t = 0 min, n = 10) and show normal early-phase LTD but a complete abolition of late-phase LTD evoked by either glutamate and depolarization pairing (100% ± 7.9% at t = 120 min, n = 8) or PDA (34 pA ± 5.5 pA at t = 90 min, n = 10). In these MEF2A+2D knockout Purkinje cells, the late phase of both forms of LTD was completely rescued by transfection with a MEF2D plasmid (pairing: 50% ± 7.1% at t = 120 min, n = 7; PDA: 16 pA ± 6.0 pA at t = 90 min, n = 10) but not an empty control plasmid (pairing: 104% ± 5.4% at t = 120 min, n = 6; PDA: 36 pA ± 6.7 pA at t = 90 min, n = 10). Transfection with a MEF2A plasmid failed to produce substantial rescue of the late phase of LTD evoked by either pairing or PDA (pairing: 90% ± 6.4% at t = 120 min, n = 8; PDA: 33 pA ± 5.9 pA at t = 90 min, n = 10). Taken together with the shRNA experiments, these results indicate that MEF2D’s transcription factor activity is necessary for the late phase of LTD but that MEF2A has only a minor role in this process.
Figure 2. The Late Phase of LTD Is Abolished in Cre-lentivirus-Infected Purkinje Cells Derived from MEF2A+2D Double Conditional Mice and Is Completely Rescued by a WT MEF2D Expression Construct but Not a Phosphomimetic Point Mutant, MEF2D S444D.
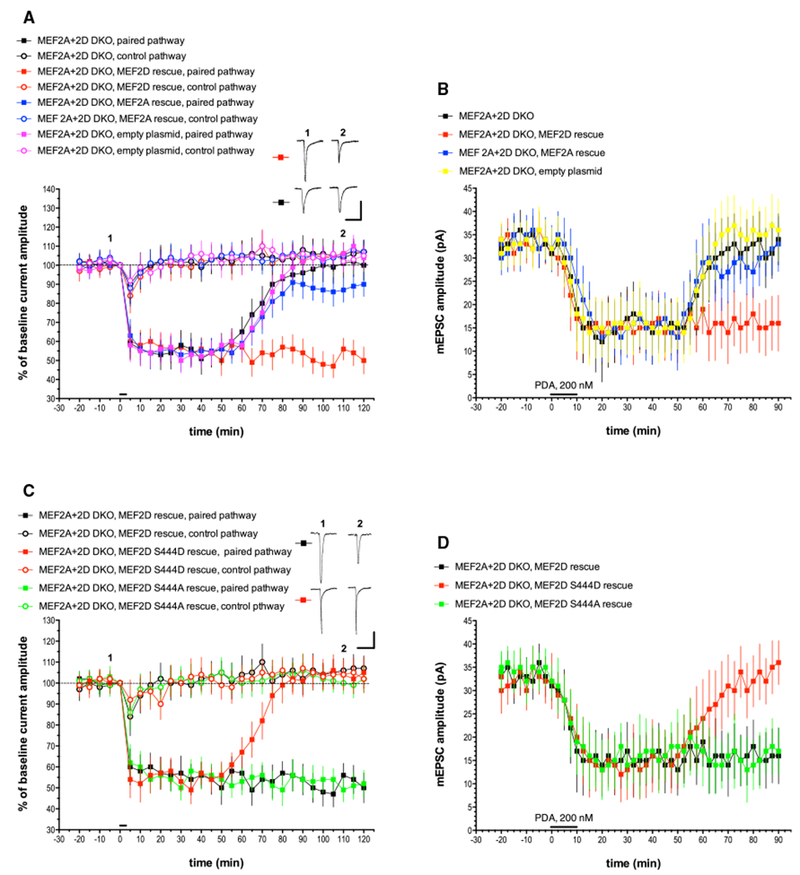
(A) Exemplar traces are single (unaveraged) responses, and they correspond to the points indicated on the time course graph. MEF2A+2D DKO, n = 8; MEF2A+2D DKO, MEF2D rescue, n = 7; MEF2A+2D DKO, MEF2A rescue, n = 8; MEF2A+2D DKO, empty plasmid, n = 7. Scale bars represent 2 s, 100 pA. When compared with the MEF2A+2D DKO paired pathway, the late phase of LTD measured at t = 120 min was significantly different from the paired pathway responses in the MEF2D rescue group (p < 0.001) but not the MEF2A rescue group (p > 0.10) or the empty plasmid control group (p > 0.20).
(B) mEPSC recordings and chemical LTD induction by PDA were performed as indicated for Figure 1B. n = 10 cells/group. When compared with the MEF2A+2D DKO paired pathway, the late phase of chemical LTD measured at t = 90 min was significantly different from the responses in the MEF2D rescue group (p < 0.05) but not the MEF2A rescue group (p > 0.20) or the empty plasmid control group (p > 0.20).
(C) Exemplar traces are single (unaveraged) responses, and they correspond to the points indicated on the time course graph. MEF2A+2D DKO, MEF2D rescue, n = 7 (these are the same data shown in Figure 2A, re-plotted here to allow for comparison with the point mutants); MEF2A+2D DKO, MEF2D S444D rescue, n = 6; MEF2A+2D DKO, MEF2D S444A rescue, n = 6. Scale bars represent 2 s, 100 pA. When compared with the MEF2A+2D DKO, MEF2D rescue paired pathway, the late phase of LTD was significantly different from the paired pathway responses in the MEF2D S444D rescue group (p < 0.001) but not the MEF2D S444A rescue group (p > 0.10) or the empty plasmid control group (p > 0.20).
(D) mEPSC recordings and chemical LTD induction by PDA were performed as indicated for Figure 1B. n = 10 cells/group. When compared with the MEF2A+2D DKO, MEF2D rescue group, the late phase of chemical LTD was significantly different from the paired pathway responses in the MEF2D S444D rescue group (p < 0.01) but not the MEF2A S444A rescue group (p > 0.20).
See also Figures S1–S3 and Table S1.
The activation of MEF2 isoforms by excitatory neuronal activity requires dephosphorylation of a particular serine by the Ca-activated phosphatase calcineurin (Mao and Wiedmann, 1999; Flavell et al., 2006), which is known to be expressed in Purkinje cells (Usuda et al., 1996). For the MEF2D isoform, the key residue is serine 444 (ser-444) (Pulipparacharuvil et al., 2008). To test the hypothesis that dephosphorylation of this calcineurin target is important for the late phase of LTD, we used two mutant MEF2D plasmids, one harboring the mutation S444D, which is phosphomimetic, and the other harboring the mutation S444A, which cannot be phosphorylated (Figures 2C and 2D). When MEF2A+2D DKO Purkinje cells were transfected with S444A plasmid, this produced a complete rescue of the late phase of pairing and PDA-evoked LTD similar to that seen with wild-type MEF2D transfection (pairing: 52% ± 6.4% at t = 120 min, n = 6; PDA: 17 pA ± 4.9 pA at t = 90 min, n = 10). However, when the phosphomimetic S444D plasmid was used, there was a complete failure to rescue the late phase of both pairing and PDA-evoked LTD (pairing: 105% ± 7.5% at t = 120 min, n = 6; PDA: 36 pA ± 4.6 pA at t = 90 min, n = 10). Thus, dephosphorylation of MEF2D ser-444 is necessary for the late phase of LTD.
Is MEF2 activation sufficient to produce LTD in Purkinje cells? To address this question, we used a plasmid that drives expression of tamoxifen-inducible constitutively active MEF2 (Flavell et al., 2006). In this experiment, wild-type Purkinje cells were transfected with the MEF2-VP16-ER plasmid followed by tamoxifen treatment 3 days later. Recordings were made 16–26 h after tamoxifen (Figure 3). We validated this approach by transfecting Purkinje cells with our MEF2 reporter (Table 1) and found that MEF2-VP16-ER transfection followed by tamoxifen treatment activated the MEF2 EGFP reporter in 100/100 cells. When tamoxifen treatment was delivered alone, only 3/100 Purkinje cells responded. Likewise, when a mutant form of MEF2-VP16-ER lacking the crucial DNA-binding domain was delivered, followed by tamoxifen (MEF2-ΔDBD-VP16-ER), only 2/100 Purkinje cells were EGFP-positive.
Figure 3. MEF2 Activation Is Not Sufficient to Produce LTD in Purkinje Cells.
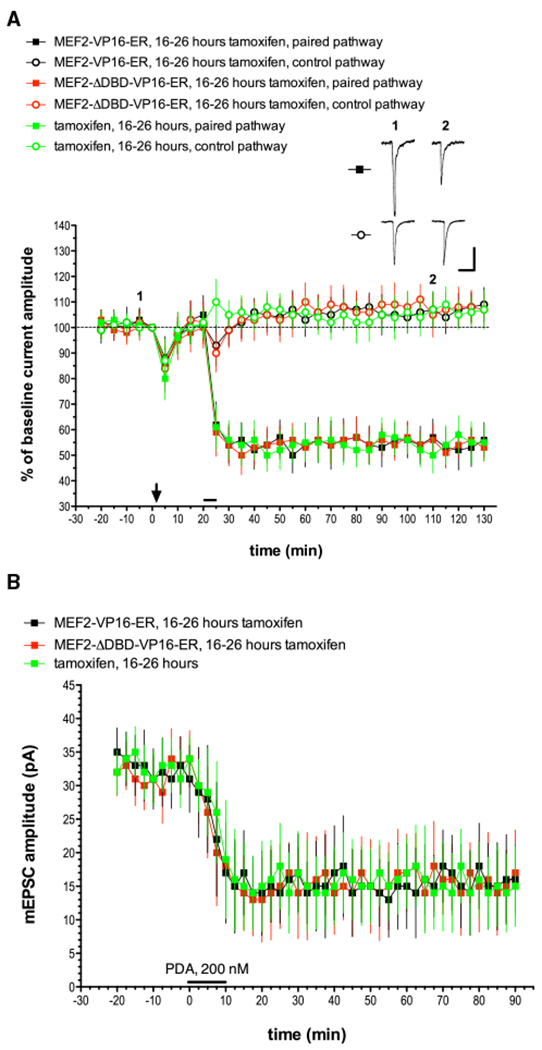
Cultured Purkinje cells were transfected with a plasmid that drives expression of tamoxifen-inducible constitutively active MEF2 (MEF2-VP16-ER) or an inactive version that fails to bind DNA (MEF2-VP16-ER-ΔDBD). Wild-type Purkinje cells were transfected with the MEF2-VP16-ER plasmid and then received tamoxifen treatment 3 days later. Recordings were then made 16–26 h after the application of tamoxifen.
(A) Exemplar traces are single (unaveraged) responses, and they correspond to the points indicated on the time course graph. The downward arrow at t = 0 min indicates the delivery of depolarizing pulses alone, without paired glutamate pulses at either pathway. The horizontal bar at t = 20 min indicates glutamate and depolarization pairing delivered to the paired pathway. MEF2-VP16-ER, 16–26 h tamoxifen, n = 8; MEF2-ΔDBD-VP16-ER, 16–26 h tamoxifen, n = 7; tamoxifen alone, 16–26 h, n = 6. Scale bars represent 2 s, 100 pA. When compared with the MEF2-VP16-ER + tamoxifen paired pathway, the late phase of LTD measured at t = 120 min was not significantly different from the paired pathway responses in the MEF2-ΔDBD-VP16-ER + tamoxifen group (p > 0.20) or the tamoxifen-alone control group (p > 0.20).
(B) mEPSC recordings and chemical LTD induction by PDA were performed as indicated for Figure 1B. n = 10 cells/group. When compared with the MEF2-VP16-ER + tamoxifen paired pathway, the late phase of LTD measured at t = 90 min was not significantly different from the responses in the MEF2-ΔDBD-VP16-ER + tamoxifen group (p > 0.20) or the tamoxifen-alone control group (p > 0.20).
In MEF2-VP16-ER-transfected Purkinje cells treated with tamoxifen, delivery of depolarization alone (at t = 0 min), failed to evoke LTD (105% ± 7.6% at t = 20 min, n = 8) as is the case in untreated control Purkinje cells. When pairing stimulation was applied 20 min after depolarization alone, normal LTD was evoked with a robust early and late phase (54% ± 8.6% at t = 60 min and 56% ± 6.8% at t = 120 min). Similar responses were seen in Purkinje cells treated with the transcriptionally inactive DNA-binding domain mutant construct MEF2-ΔDBD-VP16-ER plus tamoxifen (53% ± 5.1% at t = 120 min, n = 7) or tamoxifen alone (55% ± 7.4% at t = 120 min, n = 6). Basal mEPSC amplitude was normal in MEF2-VP16-ER plus tamoxifen-treated Purkinje cells (34 56% ± 6.8% at t = 120 min 4.2 pA at t = 0 min, n = 10), indicating that MEF2 activation is not sufficient to produce an LTD-like effect nor did this treatment alter the subsequent chemical induction of LTD by PDA application (17 pA ± 6.4 pA at t = 90 min, n = 10). Similar results were seen in the tamoxifen-alone control group (15 pA ± 6.0 pA at t = 90 min, n = 10) and in cells transfected with MEF2-ΔDBD-VP16-ER and later treated with tamoxifen (16 ± 5.1 pA at t = 90 min, n = 10). These experiments indicate that MEF2 activation is not sufficient for induction of either the early or late phase of LTD nor can it replace either depolarization or glutamate pulses in the induction protocol.
Might various treatments that block or activate MEF2 have side effects on the immediate signals necessary for LTD induction like depolarization-evoked Ca influx and glutamate-evoked mGlu1 activation? There are two lines of evidence that suggest that these processes are not impaired when MEF2 activity is perturbed. First, chemical LTD induction by PDA directly activates PKC and thereby bypasses the requirements for mGlu1 activation and Ca influx. Yet the effects of the MEF2 reagents used here are nearly identical for pairing and PDA-evoked LTD. Second, we have measured Ca transients evoked by depolarization and by application of the mGlu1 agonist (S)-3,5-Dihydroxyphenylglycine (DHPG). Both of these were normal for all of the manipulations herein (Table S1).
DISCUSSION
The main findings of this study are that the induction of cerebellar LTD in cultured Purkinje neurons is associated with stimulation of MEF2 family transcription factor activity in these cells and that manipulations designed to interfere with MEF2D selectively blocked the late phase of cerebellar LTD. In MEF2A+MEF2D null Purkinje cells, the late phase of LTD could not be rescued by a mutant phosphomimetic form of MEF2D, MEF2D S444D, which interferes with the dephosphorylation and subsequent activation of MEF2D by calcineurin. The effects of all of these MEF2 manipulations were nearly identical for pairing-induced and chemically induced LTD. Taken together, these results suggest that the late phase of cerebellar LTD in cultured Purkinje cells requires MEF2D transcription factor activity that is triggered by dephosphorylation of ser-444.
Constitutive activation of MEF2 alone neither produced an LTD-like phenomenon nor did it interact with subsequent LTD induction. Thus, activation of MEF2D transcription factor activity appears to be necessary, but not sufficient, for the induction of the late phase of cerebellar LTD. In other neuronal types, MEF2 family transcription factors have been implicated in slow developmental synaptic pruning (Flavell et al., 2006; Chang et al., 2017) through the transcriptional activation of Arc (Wilkerson et al., 2014) and protocadherin 10 (Tsai et al., 2012). However, to our knowledge, this is the first time that a MEF2 family transcription factor has been implicated in any form of LTD in any location in the nervous system.
There are several caveats that should be considered in interpreting these results. First, these experiments were conducted in a cell culture model system, and it is possible that the requirements for induction of cerebellar LTD in the intact brain of a behaving animal are different. Second, there is a possibility that MEF2A may have a minor role in the late phase of LTD. MEF2A knockdown produced a small attenuation of the late phase in pairing-induced and chemically induced LTD, but this effect did not achieve statistical significance. Similarly, in MEF2A+MEF2D null Purkinje cells, transfection with wild-type (WT) MEF2A produced a small, partial rescue of the late phase of pairing-induced, but not chemically induced, LTD. Third, while it is has been shown in biochemical assays that calcineurin is the relevant phosphatase for dephosphorylation of ser-444 and the consequent activation of MEF2D transcription factor activity, we have not directly demonstrated the involvement of calcineurin in the late phase of LTD. Calcineurin inhibitors have basal effects on the strength of parallel fiber-Purkinje cell synapses (Ajima and Ito, 1995; Eto et al., 2002) and are reported to block cerebellar LTP as well (Belmeguenai and Hansel, 2005), and so interpreting their potential effects on the late phase of LTD is not straightforward.
MEF2 transcription factors upregulate and downregulate hundreds of genes, many of which function at synapses (Flavell et al., 2008). Perhaps the most important MEF2-upregulated gene for activity-dependent synaptic weakening is Arc. Arc binds the key endocytotic proteins dynamin and endophilin (Chowdhury et al., 2006) and is expressed in cultured Purkinje cells in response to LTD-inducing stimuli (Smith-Hicks et al., 2010). Knockdown or deletion of Arc in cerebellar Purkinje cells produces a selective blockade of the late phase of cerebellar LTD, similar to that seen here.
Overexpression of Arc in Purkinje cells drives endocytosis of surface AMPA receptors, thereby producing synaptic depression. This depression occludes subsequent pairing and chemically induced LTD (an effect similar to that seen in hippocampal neurons: Plath et al., 2006; Rial Verde et al., 2006; Waung et al., 2008). Overexpression of mutant forms of Arc that were deficient in dynamin or endophilin binding failed to produce this LTD-like effect (Smith-Hicks et al., 2010). There are two possible interpretations of this result. One is that Arc expression is indeed sufficient to produce the late phase of LTD. The other is that the LTD-like effect produced by Arc is an artifact of Arc overexpression and would not be seen with natural levels of Arc at the synapse. It will be of interest to determine if overexpression of Arc rescues the defect in late LTD that is observed in the absence of MEF2s.
The promoter region of the Arc gene has a cluster of cis-regulatory elements, binding sites for the transcription factors CREB, MEF2, and SRF, within a short stretch of DNA located approximately 6.9 kb upstream of the transcriptional start site. Kawashima et al. (2009) have confirmed that these three transcription factors do indeed bind at these clustered sites. They have named this region the synaptic activity response element (SARE) and have shown that it can confer regulated, activity-dependent expression upon a fluorescent reporter and that its structure is well conserved across mammalian species.
Previous work has shown that in addition to being blocked by inhibition of MEF2, the late phase of cerebellar LTD is selectively blocked by expression of a dominant-negative form of CREB (Ahn et al., 1999) or by either knockdown or deletion of SRF in Purkinje cells. Furthermore, mutation of the SRF-binding site, called SRE6.9, within the SARE region of the Arc promoter in Purkinje cells can also selectively block the late phase of LTD (Smith-Hicks et al., 2010). We propose that convergent MEF2D, CREB, and SRF binding in this SARE region is required for Arc transcription in Purkinje cells and that, as a result, this triple binding is necessary and possibly sufficient for the late protein synthesis-dependent phase of cerebellar LTD.
Cerebellar motor signs are seen in approximately 80% of children with autism spectrum disorder (Fournier et al., 2010). In addition, alterations in cerebellar morphology such as hypoplasia of the vermis are among the most reliable neuroanatomical correlates of autism spectrum disorders (Courchesne, 1997). A copy number variation model of autism in the mouse (patDp/+) showed a deficit in the early phase of parallel fiber LTD and a concomitant impairment of many-to-one climbing fiber maturation in early postnatal life (Piochon et al., 2014)
How might disruption of the MEF2-dependent late phase of LTD result in these alterations of cerebellar structure and function? One likely possibility is that disruption of MEF2-mediated persistent LTD results in developmental miswiring of the cerebellum (see Piochon et al., 2016 for a review of this general hypothesis). For example, when the key MEF2 target gene, Arc, was knocked down only in cerebellar Purkinje cells, this resulted in a failure of activity-dependent maturation of the climbing fiber-Purkinje cell synapse, resulting in persistent multiple innervation, ataxia, and the failure of perisomatic synapses to translocate to the distal dendrite (Mikuni et al., 2013). LTD of both climbing fiber and parallel fiber synapses on Purkinje cells are mGlu1 (Hansel and Linden, 2000; Linden et al., 1991; Conquet et al., 1994) and Arc dependent (Smith-Hicks et al., 2010), and it is likely that a MEF2-dependent deficit in persistent LTD would impair both parallel fiber and climbing fiber functional connectivity in the activity-dependent early postnatal phase of cerebellar development.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Linden (dlinden@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mutant Mice
Targeting of the Mef2a locus was carried out by sequential targeting of ES cells with homologous recombination (Figure S3). First, the second exon of Mef2a was flanked with loxP sites and these mice were successfully generated. Then, ES cells from these mice were obtained and a loxP site was inserted at the 3′ end of the Mef2a coding sequence. Targeting was verified by Southern blotting. Successfully targeted ES cells were transfected with Cre to remove the neomycin selection cassette and then injected into pseudo-pregnant females, and subsequent progeny were assessed for successful germline transmission of the targeted allele. Homozygous conditional Mef2a mice were generated and crossed to Nestin-cre for immunoblot validation of the targeting approach. The floxed mice (without Cre) were then crossed with the previously described Mef2d conditional mice (Andzelm et al., 2015) to generate the double conditional knockout lines used in this study.
For routine experimentation, animals were genotyped using a PCR-based strategy. Conditional knockout animals were genotyped for the presence of the loxP site, which shifts the size of the PCR product. All experiments described here were performed using animals derived from a sv129/C57BL/6 hybrid genetic background, with the mutation backcrossed in the C57BL/6 background (Charles River Laboratories) between 3 and 8 generations. The Institutional Animal Care and Use Committees at Harvard University and Johns Hopkins University approved all of the experiments in this study. Embryos of both sexes were used to derive the tissue for cell culture experiments
METHOD DETAILS
Recording and Ca Imaging
Cell culture and whole-cell patch-clamp recording were performed as previously described (Smith-Hicks et al., 2010). Briefly, patch electrodes were filled with a solution containing (in mM) 135 CsCl, 10 HEPES, 0.5 EGTA, 4 Na2-ATP, and 0.4 Na-GTP, adjusted to pH 7.35 with CsOH. Cells were bathed in 140 NaCl, 5 KCl, 2 CaCl2, 0.8 MgCl2, 10 HEPES, 10 glucose, 0.0005 TTX, and 0.3 picrotoxin, adjusted to pH 7.35 with NaOH, which flowed at a rate of 0.5 ml/min. Electrodes were pulled from N51A glass and polished to yield a resistance of 2 – 4 MΩ. Iontophoresis electrodes (1 μm tip diameter) were filled with 10 mM glutamate (in 10 mM HEPES, pH 7.1) and were positioned ~20 μm away from large-caliber dendrites. Test pulses were delivered using negative current pulses (300–900 nA, 30–70 msec duration). LTD-inducing pairing stimuli consisted of six, 3 s-long depolarizations to 0 mV, each delivered together with a test pulse of glutamate.
Membrane currents were recorded with an Axopatch 200A amplifier, and digitized at 10 kHz for exogenous glutamate responses and 20 kHz for mEPSCs. Signals were lowpass filtered at 5 kHz (1 kHz for mEPSCs) and acquired using Axograph X software (Axograph Scientific, Sydney, Australia). Recordings in which Rinput or Rseries varied by more than 15% were excluded from the analysis. For analysis of mEPSCs, Axograph X mini analysis software was used. This detected events based on closeness of fit of the mEPSC to a sliding template. Events smaller than −4 pA were discarded. A separate template was created for each recording by averaging 30 of its most unambiguous mEPSCs as selected by eye.
Bis-fura-2 ratio imaging of intracellular free Ca2+ in the dendrites of cultured Purkinje cells was accomplished by measuring the background corrected fluorescence ratio at 340 and 380 nm excitation using a cooled CCD camera system as described (Linden, 1996). In these experiments, EGTA was removed from the internal saline and replaced with 100 μM bis-fura-2 (Invitrogen).
Viruses, Plasmids and Drugs
Lentiviral infections of the FUGW-Cre lentivirus were performed on the day after cultures were prepared, which was applied at 0.55 μg/ml (1.1 μg/culture plate).
The MRE-d2EGFP plasmid was generated by replacing luciferase by destabilized EGFP (Kawashima et al., 2009) in a previously described MRE-luciferase plasmid (Flavell et al., 2006). The MEF2D expression plasmids (WT-RNAi Resistant (RIR), S444A-RIR, S444D-RIR) were generated by subcloning previously used plasmids (Pulipparacharuvil et al., 2008) into the pCAG-ires-GFP vector (Matsuda and Cepko, 2004). To determine expression levels pCAG (empty), pCAG-MEF2D-ires-GFP, pCAG-MEF2DS444A-ires-GFP and pCAG-MEF2DS444D-ires-GFP were transfected into 293t cells using calcium phosphate.
For plasmid delivery of MEF2A/MEF2D shRNAs, MEF2 gene expression constructs or the MEF2 reporter, MRE-d2EGFP, a gold/glycerol mixture was first prepared as follows: 20 mg of 0.6 μm gold microcarriers were washed once in 70% ethanol, 3 times in sterile deionized, distilled water, and resuspended in 0.5 mL of sterile 50% glycerol. For each transfection into Purkinje cell cultures, 5 μg of plasmid cDNA, 1 μg of pCAG-DsRed2 cDNA, 50 μL of 2.5 M CaCl2, and 10 μL of 1 M spermidine were sequentially added to 50 μL of the gold/glycerol mixture. The preparation was gently vortexed for 15 minutes, after which the gold particles were centrifuged down and washed once with 70% ethanol and once with 100% ethanol. Washed gold particles were resuspended in 60 μL of 100% ethanol and pipetted onto macrocarrier disks (9 μL/disk). Following vortexing and an ethanol wash, microcarrier–containing solution was evaporated on the surface of six macrocarrier discs and delivered to the cerebellar cultures (at 4–7 DIV) using the Helios Gene Gun System (Bio–Rad) operating at a pressure of 450 psi and a vacuum of 20 in–Hg. Conditioned medium was aspirated from the surface of the cell culture immediately before transfection and was returned immediately thereafter. Cultures were then returned to the incubator prior to electrophysiological recording. Transfected Purkinje neurons were identified by imaging dsRed signals with 543 nm illumination.
The compound (S)-3,5-Dihydroxyphenylglycine (DHPG) was purchased from Tocris. Glutamate, tetrodotoxin, 4-OH-tamoxifen and the various salts were purchased from Sigma. 4-OH-tamoxifen (Sigma H7904) was applied 18–22 hours before recording. It was prepared as a 10mM stock in EtOH and used at 1/10,000 dilution to achieve a final concentration of 1uM in the culture media.
Immunoblotting and Immunohistochemistry
For immunoblotting of MEF2A, brains from Mef2a brain-specific knockout mice and littermate controls without cre were homogenized in RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 10 mM NaF, 1 mM sodium orthovanadate, complete protease inhibitor cocktail tablet (Roche)) and equal volumes of protein sample were loaded on a gel. Conventional western blotting used enhanced chemiluminescence and HRP-conjugated secondary antibodies. A previously described anti-MEF2A antibody was used to detect MEF2A (Andzelm et al., 2015) as well as an anti-beta actin antibody as a loading control (Abcam).
For the MEF2D overexpression constructs, 293t cells were harvested 2 days after transfection and processed using NuPAGE sample buffer (Life Technologies). Equal volumes of protein sample were loaded on a NuPAGE gel (Life Technologies). Fluorescence based immunoblotting was performed using Odyssey (Li-Cor). Antibodies used were mouse anti-MEF2D (BD Biosciences) and chicken anti-GFP (Aves).
For immunohistochemistry (IHC) of MEF2, mice were perfused with 4% formaldehyde in phosphate buffered saline (PBS), their brains were removed, subjected to successive sucrose gradients and finally frozen in a 1:1 solution of 30% sucrose in 1×PBS:Tissue Tek O.C.T (Sakura). 20 μm sagittal cryo-sections were generated on a Leica CM1950 cryostat and mounted on slides. Sections were incubated in block solution (10% goat serum and 0.25% Triton X-100 in 1XPBS)for 1 hour and then incubated with primary antibodies in block solution for 2 hours at room temperature or 4°C overnight. Alexa dye-conjugated secondary antibodies were used at 1:500 dilutions in block solution (Life Technologies). Primary antibodies were anti-MEF2D (BD biosciences), anti-GFP (Aves), anti-Calbindin (Swant) and anti-MEF2A (rabbit 1:1000; Andzelm et al., 2015). DAPI was used to stain nuclei (Life Technologies).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was assessed for the endpoints of LTD experiments were performed using the Mann-Whitney U test with Bonferroni correction for multiple comparisons as implemented in GraphPad Prism.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-calbindin-D28k | Swant | Cat# CB38; RRID:AB_10000340 |
| Chicken anti-GFP | Aves Labs | Cat# GFP1202; RRID:AB_2734732 |
| Rabbit anti-MEF2A | M.E. Greenberg lab | Andzelm et al., 2015 |
| Mouse anti-MEF2D | BD Biosciences | Cat# 610774; RRID:AB_398095 |
| Rabbit anti-beta-actin | Abcam | Cat# 1854-1; RRID:AB_764434 |
| Bacterial and Virus Strains | ||
| FUGW Cre-lentivirus | R. Huganir Lab | N/A |
| Experimental Models: Organisms/Strains | ||
| Mef2d flox/flox mice | M.E. Greenberg lab | Andzelm et al., 2015 |
| Mef2a flox/flox mice | M.E. Greenberg lab | This paper |
| Oligonucleotides | ||
| MEF2A shRNA | M.E. Greenberg lab | Flavell et al., 2008 |
| MEF2D shRNA | M.E. Greenberg lab | Flavell et al., 2008 |
| Recombinant DNA | ||
| MRE-d2EGFP plasmid | M.E. Greenberg lab | This paper |
| MEF2D-ires-GFP RNAi resistant expression plasmid | M.E. Greenberg lab | This paper |
| MEF2D-S444A-ires-GFP RNAi resistant expression plasmid | M.E. Greenberg lab | This paper |
| MEF2D-S444D-ires-GFP RNAi resistant expression plasmid | M.E. Greenberg lab | This paper |
| pCAG-dsRed2 plasmid | Addgene | Plasmid #15777 |
| MEF2-VP16-ER plasmid | M.E. Greenberg lab | Flavell et al., 2006 |
| MEF2-ΔDBD-VP16-ER plasmid | M.E. Greenberg lab | Flavell et al., 2006 |
| Software and Algorithms | ||
| ImageJ | https://imagej.net/ | RRID:SCR_003070 |
| Axograph X | https://axograph.com | N/A |
| GraphPad Prism | https://www.graphpad.com | RRID:SCR_002798 |
Highlights.
Knockdown or deletion of postsynaptic MEF2 blocks the late phase of cerebellar LTD
Purkinje cells transfected with active MEF2 show normal synaptic strength
Activation of MEF2 is necessary, but not sufficient, for the late phase of LTD
ACKNOWLEDGMENTS
Thanks to members of the Linden and Worley labs for helpful suggestions. Thanks to Olof Lagerlof and Richard Huganir for providing FUGW-Cre lentivirus. This work has been supported by USPHS NIH R01 NS095901 and P50 MH084020 to D.J.L., as well as a Stuart H.Q. & Victoria Quan Fellowship (M.M.A.), the Ruth L. Kirschstein NIH Training Grant T32GM007753 to M.M.A., and NIH grant NS028829 to M.E.G.
Footnotes
DECLARATION OF INTERESTS
M.E.G. is on the Board of Directors of and holds equity in Allergan.
REFERENCES
- Ahn S, Ginty DD, and Linden DJ (1999). A late phase of cerebellar long-term depression requires activation of CaMKIV and CREB. Neuron 23, 559–568. [DOI] [PubMed] [Google Scholar]
- Ajima A, and Ito M (1995). A unique role of protein phosphatases in cerebellar long-term depression. Neuroreport 6, 297–300. [DOI] [PubMed] [Google Scholar]
- Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, Bassel-Duby R, Olson EN, Kavalali ET, and Monteggia LM (2012). In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS ONE 7, e34863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andzelm MM, Cherry TJ, Harmin DA, Boeke AC, Lee C, Hemberg M, Pawlyk B, Malik AN, Flavell SW, Sandberg MA, et al. (2015). MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron 86, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, and Olson EN (2008). MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. USA 105, 9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barylko B, Wilkerson JR, Cavalier SH, Binns DD, James NG, Jameson DM, Huber KM, and Albanesi JP (2018). Palmitoylation and membrane binding of Arc/Arg3.1: a potential role in synaptic depression. Biochemistry 57, 520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmeguenai A, and Hansel C (2005). A role for protein phosphatases 1, 2A, and 2B in cerebellar long-term potentiation. J. Neurosci 25, 10768–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-W, Wilkerson JR, Hale CF, Gibson JR, and Huber KM (2017). Distinct stages of synapse elimination are induced by burst firing of CA1 neurons and differentially require MEF2A/D. eLife 6, e26278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, and Worley PF (2006). Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, and Wang YT (2010). Long-term depression in the CNS. Nat. Rev. Neurosci 11, 459–473. [DOI] [PubMed] [Google Scholar]
- Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, et al. (1994). Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature 372, 237–243. [DOI] [PubMed] [Google Scholar]
- Courchesne E (1997). Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr. Opin. Neurobiol 7, 269–278. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, and Greenberg ME (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339. [DOI] [PubMed] [Google Scholar]
- Elmer BM, Estes ML, Barrow SL, and McAllister AK (2013). MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J. Neurosci 33, 13791–13804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M, Bock R, Brautigan DL, and Linden DJ (2002). Cerebellar longterm synaptic depression requires PKC-mediated activation of CPI-17, a myosin/moesin phosphatase inhibitor. Neuron 36, 1145–1158. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, and Greenberg ME (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, and Greenberg ME (2008). Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier KA, Hass CJ, Naik SK, Lodha N, and Cauraugh JH (2010). Motor coordination in autism spectrum disorders: a synthesis and meta-analysis. J. Autism Dev. Disord 40, 1227–1240. [DOI] [PubMed] [Google Scholar]
- Hallmayer J, Pintado E, Lotspeich L, Spiker D, McMahon W, Petersen PB, Nicholas P, Pingree C, Kraemer HC, Wong DL, et al. (1994). Molecular analysis and test of linkage between the FMR-1 gene and infantile autism in multiplex families. Am. J. Hum. Genet 55, 951–959. [PMC free article] [PubMed] [Google Scholar]
- Hansel C, and Linden DJ (2000). Long-term depression of the cerebellar climbing fiber–Purkinje neuron synapse. Neuron 26, 473–482. [DOI] [PubMed] [Google Scholar]
- Kawashima T, Okuno H, Nonaka M, Adachi-Morishima A, Kyo N, Okamura M, Takemoto-Kimura S, Worley PF, and Bito H (2009). Synaptic activity-responsive element in the Arc/Arg3.1 promoter essential for synapse-to-nucleus signaling in activated neurons. Proc. Natl. Acad. Sci. USA 106, 316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ (1996). A protein synthesis-dependent late phase of cerebellar long-term depression. Neuron 17, 483–490. [DOI] [PubMed] [Google Scholar]
- Linden DJ (2012). A late phase of LTD in cultured cerebellar Purkinje cells requires persistent dynamin-mediated endocytosis. J. Neurophysiol 107, 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden DJ, Dickinson MH, Smeyne M, and Connor JA (1991). A longterm depression of AMPA currents in cultured cerebellar Purkinje neurons. Neuron 7, 81, 59. [DOI] [PubMed] [Google Scholar]
- Mao Z, and Wiedmann M (1999). Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J. Biol. Chem 274, 31102–31107. [DOI] [PubMed] [Google Scholar]
- Matsuda T, and Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA 101, 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, Korf BR, and Carroll AJ (2011). Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am. J. Med. Genet. A 155A, 2386–2396. [DOI] [PubMed] [Google Scholar]
- Mikuni T, Uesaka N, Okuno H, Hirai H, Deisseroth K, Bito H, and Kano M (2013). Arc/Arg3.1 is a postsynaptic mediator of activity-dependent synapse elimination in the developing cerebellum. Neuron 78, 1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashima M, and Hirano T (1999). Entire course and distinct phases of day-lasting depression of miniature EPSC amplitudes in cultured Purkinje neurons. J. Neurosci 19, 7326–7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, and Huber KM (2010). Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kloth AD, Grasselli G, Titley HK, Nakayama H, Hashimoto K, Wan V, Simmons DH, Eissa T, Nakatani J, et al. (2014). Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat. Commun 5, 5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piochon C, Kano M, and Hansel C (2016). LTD-like molecular pathways in developmental synaptic pruning. Nat. Neurosci 19, 1299–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, et al. (2006). Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444. [DOI] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, et al. (2008). Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron 59, 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rial Verde EM, Lee-Osbourne J, Worley PF, Malinow R, and Cline HT (2006). Increased expression of the immediate-early gene arc/arg3.1 reduces AMPA receptor-mediated synaptic transmission. Neuron 52, 461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Hicks C, Xiao B, Deng R, Ji Y, Shepherd JD, Kuhl D, Huganir RL, Ginty DD, Worley PF, and Linden DJ (2010). SRF binding to SRE 6.9 in the Arc promoter is essential for the late phase of cerebellar LTD. Nat. Neurosci 13, 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg JP, Takamiya K, Shen Y, Xia J, Rubio ME, Yu S, Jin W, Thomas GM, Linden DJ, and Huganir RL (2006). Targeted in vivo mutations of the AMPA receptor subunit GluR2 and its interacting protein PICK1 eliminate cerebellar long-term depression. Neuron 49, 845–860. [DOI] [PubMed] [Google Scholar]
- Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, and Huber KM (2012). Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151, 1581–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuda N, Arai H, Sasaki H, Hanai T, Nagata T, Muramatsu T, Kincaid RL, and Higuchi S (1996). Differential subcellular localization of neural isoforms of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin) in central nervous system neurons: immunohistory on formalin-fixed paraffin sections employing antigen retrieval by microwave irradiation. J. Histochem. Cytochem 44, 13–18. [DOI] [PubMed] [Google Scholar]
- Vinueza Veloz MF, Zhou K, Bosman LWJ, Potters JW, Negrello M, Seepers RM, Strydis C, Koekkoek SKE, and De Zeeuw CI (2015). Cerebellar control of gait and interlimb coordination. Brain Struct. Funct 220, 3513–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, and Huber KM (2008). Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JR, Tsai NP, Maksimova MA, Wu H, Cabalo NP, Loerwald KW, Dictenberg JB, Gibson JR, and Huber KM (2014). A role for dendritic mGluR5-mediated local translation of Arc/Arg3.1 in MEF2-dependent synapse elimination. Cell Rep. 7, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JR, Albanesi JP, and Huber KM (2018). Roles for Arc in metabotropic glutamate receptor-dependent LTD and synapse elimination: Implications in health and disease. Semin. Cell Dev. Biol 77, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, and Bear MF (2012). Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol 4, a009886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.