

Abstract
Bioconjugation chemistry has been used to prepare modified biomolecules with functions beyond what nature intended. Central to these techniques is the development of highly efficient and selective bioconjugation reactions that operate under mild, biomolecule compatiable conditions. Methods that form a nucleophile-sp2 carbon bond show promise for creating bioconjugates with new modifications, sometimes resulting in molecules with unparalleled functions. Here we outline and review sulfur, nitrogen, selenium, oxygen, and carbon arylative bioconjugation strategies and their applications to modify peptides, proteins, sugars, and nucleic acids.
Keywords: Bioconjugation, Arylation, Protein, Nucleic Acid
Graphical Abstract
1. Introduction
Bioconjugation processes involve chemical reactions to form either covalent or well-defined non-covalent attachments between two molecular entities, at least one of which is a biomolecule.[1,2] Specifically, the biomolecule involved in a bioconjugation process is usually obtained from natural sources or via genetic means (e.g., protein expression), while bioconjugation reagents are designed and prepared using synthetic chemistry approaches. Similar to Nature’s approach of expanding biomolecule function through modifications (e.g., protein post-translational modifications), the development of novel bioconjugation approaches provides new opportunities for chemical biologists to modulate biomolecule structure and function via synthetic chemistry both in vitro and in living systems. The marriage of biomolecules to synthetic modalities enables the discovery of new drugs,[3–5] the exploration of complex biological processes,[6] the generation of new biomaterials,[7,8] and the invention of novel diagnostic and medical tools.[9]
Bioconjugation reactions that form covalent bonds normally require conditions that are more stringent and milder than those used in small-molecule synthesis. Maintaining low temperatures (≤ 37°C), close to neutral pH, and aqueous buffers help preserve the structure and function of biomolecules. In addition, bioconjugation reactions need to be fast, and reach full conversion in hours with as low as micromolar-to-nanomolar concentrations of substrates. Reaction rates of most bioconjugation reactions are in the range of 1–1000 M−1s−1, with a few fast reactions having rates of more than 1000 M−1s−1.[10,11] In some instances, less stringent conditions (i.e., the use of organic solvents, prolonged reaction time, and heating) can be used for more structurally simple and robust biomolecules such as peptides, oligosaccharides, and oligonucleotides.
Another major challenge for developing efficient bioconjugation processes stems from the intrinsic complexity of biomolecules containing multiple reactive functional groups. Indeed, many bioconjugation reactions utilize nucleophiles in biomolecules (e.g., amines, hydroxyl groups, carboxylates, and thiols). Yet, there are often tens or hundreds of nucleophilic sites in a single biomolecule, making it difficult to control chemoselectivity (e.g., modification of cysteine over other amino acids) and regioselectivity (e.g., reaction with one cysteine among many). Unsurprisingly, many traditional bioconjugation reactions are nonselective and form heterogeneous conjugates. This leads to further challenges for the characterization and purification of products.
The development of bioconjugation protocols has been largely inspired by early work in protein chemistry.[12–14] Procedures to chemically modify proteins were used for practical purposes prior to the scientific conception of bioconjugation and the quest to understand molecular processes in underlying chemical transformations. For example, formaldehyde was used for tanning animal hides long before the discovery of its ability to crosslink proteins.[15] Protein chemists developed reagents to modify amino acids to determine protein composition.[16–19] These reagents were used to covalently bind to active sites of proteins[20] and develop protein sequencing techniques such as Edman degradation.[21] Early work in the area of protein crystallography also relied heavily on the use of various heavy atom-containing reagents (e.g., U, Pb, Hg, Au, Ag, and I) that bind amino acid residues, thereby aiding the structure refinement process.[22]
Site-selectivity is currently recognized as the most important and challenging requirement in devising new bioconjugation processes. The ability to perform bioconjugation in a predictable manner significantly simplifies the purification and characterization of the resulting products. For many bioconjugation reactions, chemoselectivity stems from the differences in the intrinsic reactivity of different functional groups in biomolecules.[1,23] Regioselective modification of a single site (e.g., a particular cysteine thiol among many) is challenging and often achieved through enzyme-mediated reactions.[24–29] More recently, the genetic encoding of unnatural handles (e.g., azides, alkynes, and ketones) enabled bioconjugation using two completely abiotic coupling partners.[30–32] For example, following the introduction of the landmark term “click chemistry” by Sharpless[33] in 1998 to describe high yielding, easy-to-perform, wide-substrate-scope reactions with easy-to-remove byproducts, synthetic chemists quickly started to develop highly efficient and selective reactions for the modification of biomolecules. An even higher selectivity bar was set for bioconjugation reactions used to study biomolecules in a complex biological milieu. Introduced by Bertozzi in 2003, bioorthogonal chemistry provides highly selective bioconjugation reactions that can be applied in living cells.[34]
Arylation reactions generate X–C(sp2) bonds (X is a carbon or a heteroatom) that have significantly different chemical properties compared to other X–C(sp3 or sp) bonds produced using traditional bioconjugation reactions.[35] Until recently, arylation was lagging behind as a bioconjugation strategy due to the lack of well-developed chemistries on biomolecules (Figure 1). This is especially surprising given how routinely the formation of nucleophile–sp2 carbon bonds has been used in small-molecule synthesis over the past decades.[36,37] Recent work has shown that diverse arylation reactions developed for small-molecule synthesis can in fact be successfully utilized in bioconjugation processes (Figure 2). For example, electron-deficient aryl halides react with various nucleophilic groups (e.g., thiols, hydroxyls, amines, and selenols) in biomolecules via nucleophilic aromatic substitution (SNAr, Figure 2A).[38] In addition, nucleophiles in biomolecules react with transition metal-based arylation reagents (Figure 2B).[39,40] Nucleophilic selenols can be oxidized and thereby turned into electrophiles, where the resulting dichalcogenides can be arylated using umpolung approaches with aryl boronic acid-derived nucleophiles (Figure 2C). Finally, palladium-catalyzed cross-coupling reactions[39,40] and C–H activation strategies[41] have been used to forge carbon-carbon(sp2) bonds with biomolecules (Figure 2D).
Figure 1.

The generic arylation process for bioconjugation.
Figure 2.
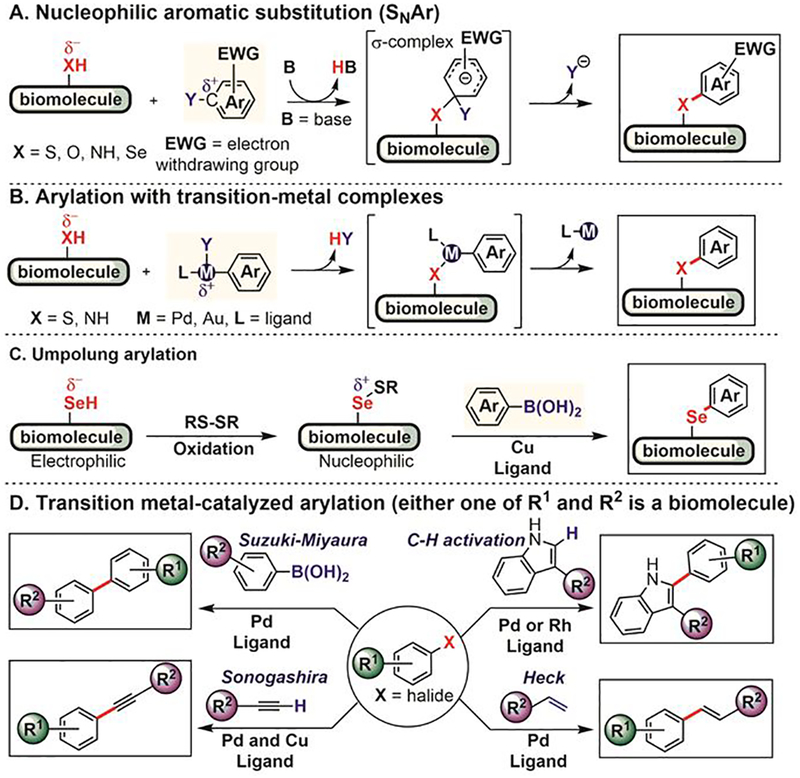
Mechanistically-distinct routes for arylation of biomolecules.
Here we describe processes that result in the formation of X–C(sp2) bonds (X is a carbon or a heteroatom), which we term arylative bioconjugation reactions. Because of the application-oriented nature of bioconjugation, we categorize these reactions according to their target biomolecules in the following sections. (Note that the arylation of small biomolecules, including amino acids, monosaccharides, natural products, and nucleotides is not covered in this review.) The nucleophilic aromatic substitution (SNAr) and metal-mediated arylation of cysteine thiols in peptides and proteins are first discussed. We then describe the N-arylation of the ε-amine of lysine. Arylation of other natural and unnatural amino acid side chains is also discussed with a focus on the umpolung arylation of selenocysteine and the palladium-catalyzed cross-coupling reactions for protein modification. Finally, we discuss metal-mediated arylation chemistry for the post-synthetic modification of nucleic acids. We conclude with a discussion on the limitations of current arylative bioconjugations and future avenues to broaden the scope and application of these transformations.
Throughout this review, we emphasize the fundamental chemistry of arylative bioconjugation reactions. The complex nature of bioconjugates requires well-designed reagents and conditions as well as accurate product characterization prior to employing the resulting conjugates for biological applications. We believe that there is no perfect bioconjugation reaction that fulfills all application needs. This review aims to complement other recent reviews on bioconjugation[42–54] and provide the reader with an expanded understanding of current bioconjugation techniques.
2. Cysteine Arylation
As the only thiol-containing canonical amino acid, cysteine is often used for chemical protein modification.[55,56] In aqueous buffer, the thiol group of cysteine (pKa ≈ 8) is more acidic than other common nucleophiles found in proteins, such as the hydroxyl group of serine (pKa ≈ 13), the phenol group of tyrosine (pKa ≈ 10), or the ε-amine of lysine (pKa ≈ 10 for RNH3+, pKa ≈ 36 for RNH2). Moreover, sulfur is more polarizable (soft) than oxygen or nitrogen. Together, these chemical features make cysteine thiol/thiolate more reactive than other protein nucleophiles towards electrophiles such as maleimides and alkyl halides, enabling chemoselective cysteine modification in the presence of other amino acid side chains. Cysteine is also a relatively rare amino acid within the human proteome, enabling the generation of homogeneous conjugates from target proteins that contain only one chemically accessible cysteine thiol. Commercial maleimide and alkyl halide reagents are routinely used to conjugate fluorescent dyes, affinity labels, and drugs to peptides and proteins. Classic and modern methods for bioconjugation via cysteine residues have been summarized in several recent review articles.[55,56]
A key feature of cysteine arylation is that it generates an S–C(sp2) bond that is different from the S–C(sp3) bonds produced using maleimides or alkyl halides. Electron-deficient aryl halides such as chloronitrobenzenes and perfluoroarenes react with cysteine via a nucleophilic aromatic substitution (SNAr) mechanism. Alternatively, palladium-mediated S-arylation approaches are used to introduce electron-neutral and electron-rich aryls. Compared to SNAr processes, palladium-mediated reactions have a broader substrate scope because they are relatively indifferent to the electronic nature of the aryl group. Through these two complementary approaches, a wide range of aryl groups can be conjugated to cysteine thiols in peptides and proteins, enabling the development of covalent protein modulators, macrocyclic peptides, antibody-drug conjugates, and other hybrid protein-based constructs.
2.1. Cysteine arylation via nucleophilic aromatic substitution (SNAr)
Nucleophilic aromatic substitution (SNAr) reactions generally require strong electron-withdrawing groups on the aromatic ring of the arylation reagents to increase the electrophilicity of the carbon electrophile and to stabilize the negatively-charged σ-complex (Figure 2A). As a result, aryl and heteroaryl electrophiles with strong electron-withdrawing groups such as nitro, fluoro, and sulfonyl are used to arylate cysteine thiols in peptides and proteins. The substitution pattern greatly affects the SNAr reactivity of aryl electrophiles. For example, o- and p-nitroaryl halides are more reactive than m-nitro variants because the resonance effect of o- and p-nitro substituents is absent in the m-nitro substituted analogs.
2.1.1. Chemoselective cysteine SNAr arylation
The chemoselectivity of the SNAr arylation of cysteine is usually high because of the higher nucleophilicity of thiol/thiolate groups compared to other protein nucleophiles. However, the selectivity varies with reaction conditions because arylation reactions with amines and hydroxyl groups can occur competitively in the presence of strong bases. The selectivity of this reaction can also be altered as a function of solvent polarity. In addition, the microenvironment around an amino acid side chain in a protein affects its nucleophilicity, resulting in different reactivity profiles for the same amino acid residue at different protein sites.
Aryl chlorides as cysteine-selective covalent protein modulators
The thiol group of cysteine is utilized by nature to perform a wide range of biochemical functions.[57–59] Many proteins have essential cysteine residues that are either directly involved in catalysis or critical for maintaining the protein structure. This provides an opportunity for the design and discovery of diverse aryl halides as covalent cysteine-selective modulators of protein structure and function.
Thiol-selective aryl halides were initially developed to identify cysteines in the active sites of enzymes. In 1975, Price and Cohn used 4-chloro-7-nitrobenzofurazan (NBD-chloride) to investigate the different environments of two cysteines (Cys-25 and Cys-187) in porcine muscle adenylate kinase (Figure 3).[60] Both cysteine residues were arylated with excess NBD-chloride (Figure 3, top). Surprisingly, the more solvent-exposed Cys-187 reacted about 40 times slower with NBD-chloride (k = 205 M−1s−1) than the buried Cys-25 (k = 8,580 M−1s−1). Regioselective arylation of Cys-25 was achieved using an equimolar amount of NBD-chloride (Figure 3, bottom). In contrast, both cysteine thiols were labeled with similar reaction rates with other thiol-reactive reagents such as Ellman’s reagent (5,5’-dithio-bis-(2-nitrobenzoic acid)). The Cys-25-labeled enzyme was completely inactive, indicating that Cys-25 was in close proximity to the active site of the enzyme. Together, these results highlight the utility of arylation reagents for studies of protein structure and function. Importantly, this early example illustrates how the local environment of an amino acid side chain modulates its reactivity.
Figure 3.
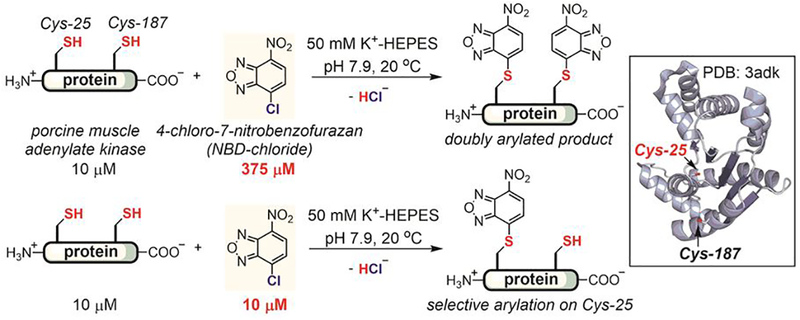
Chemo- and regio-selective arylation of cysteine thiols in porcine muscle adenylate kinase using NBD-chloride.
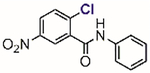
In 2002, Blanchard[61] and Li[62] independently reported two structurally similar covalent antagonists for PPARγ (nuclear hormone receptor peroxisome proliferator-activated receptor gamma) (Table 1, entries 1 and 2). PPARγ regulates several genes involved in adipogenesis and is the molecular target of the thiazolidinedione family of diabetes drugs.[63,64] Both antagonists (GW9662 and T0070907) block ligand binding to PPARγ through selective arylation of Cys-285 located in the orthosteric ligand-binding pocket of PPARγ. Even though the compounds showed activity in vitro and in cell culture, both failed, however, at inhibiting the binding of PPARγ ligands to an allosteric site other than the orthosteric pocket on the ligand binding domain.[65] By enhancing the size of the 2-chloro-5-nitrobenzamidyl scaffold, Kojetin reported in 2017 a dual-site PPARγ antagonist (Table 1, entry 3) that simultaneously blocked the ligand from binding to both the orthosteric pocket and the allosteric site.[66] Together, 2-chloro-5-nitrobenzamidyl-based compounds are useful chemical tools to modulate the PPARγ function in biological processes.
Table 1.
Representative aryl and heteroaryl halides as cysteine-selective covalent protein modulators.
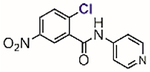
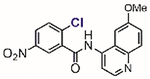
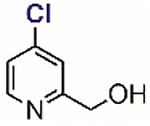
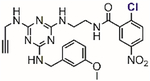
Besides chloronitrobenzenes, 4-halopyridines have also been explored as cysteine-selective covalent protein inhibitors. In 2011, Fast screened a library of 4,000 compounds and found a class of 4-halopyridines that can serve as covalent inhibitors for dimethylarginine dimethylaminohydrolase (DDAH) through the arylation of its active site Cys-249.[67] Inhibition assays and mutation studies indicated that Asp-66 of DDAH was required to inactivate the protein by 4-chloro-2-hydroxylmethylpyridine (Table 1, entry 4). The carboxylate residue of Asp-66 was proposed to stabilize the pyridinium form (computationally predicted pKa = 3.6) of the inhibitor thus promoting the arylation reaction at pH 7.5. This ensures the high selectivity of the compound for DDAH because the free compound in solution resides mainly in the pyridine form and is thus unreactive toward other thiol species. Although computer modeling has supported this proposed mechanism of inactivation, an X-ray structure of inactivated DDAH showed no close contact between the carboxylate of Asp-66 and the pyridine ring of the inhibitor, suggesting possible conformational changes of DDAH after the covalent inhibition event.[68]
Weerapana investigated the reactions of p-chloronitrobenzene-based probes with the HeLa cell lysate through quantitative activity-based protein profiling.[69–71] Using 1,3,5-triazine as the scaffold, different electrophilic probes were prepared to target subsets of the proteome (Figure 4).[69] The reactions between the probes and the proteome were qualitatively assessed using in-gel fluorescence, and then quantitatively measured through enrichment and mass spectrometry. Initial studies with these probes resulted in the discovery of RB-2-cb that covalently arylated Cys-239 of β-tubulin and inhibited tubulin polymerization (Table 1, entry 5).[69]
Figure 4.
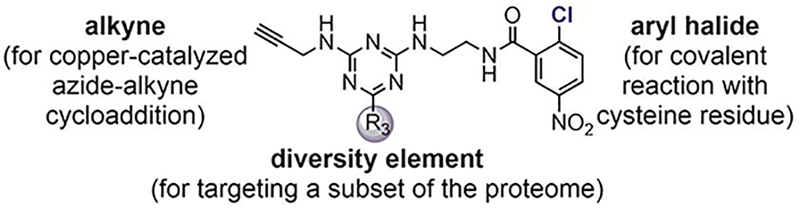
Triazine-derived electrophilic probes for reactions with the human proteome (HeLa cell lysate).
In 2014, using a similar activity-based protein-profiling approach, Weerapana expanded the studies to other aryl halides (Figure 5).[70] Because of the previously mentioned resonance effects, the p-nitro- and o-nitro-chlorobenzene electrophiles (1, 2, and 3) are more reactive towards the proteome than the m-nitro variant (4). This result corroborated the dominant role of p-nitrochlorobenzene in the design of previously reported cysteine-selective covalent protein inhibitors (Table 1). Additionally, p-nitrofluorobenzene (5), chloropyridines (6 and 7), chloropyrimidines (8 and 9), and chlorotriazine (10) were investigated for their reactivity with the proteome. An in-gel fluorescence assay showed that electrophiles 1, 5, and 10 reacted with the proteome at micromolar concentrations, indicating that they might be used for developing more complex covalent protein modifiers. Of importance, mass spectrometry analysis of the labeled proteomes showed that p-nitrochlorobenzene (1) was cysteine-selective while dichlorotriazine (10) favored arylation with lysines. However, electrophile 10 reacted with N- and C-terminal–protected cysteine much faster than electrophile 1. This disparity between reactions with the proteome and with simple amino acids indicates that a subset of the proteome might possess lysine residues that are highly reactive with probe 10. More in-depth structural and mechanistic studies of the labeled proteins identified from mass spectrometry should provide a better understanding of the reactivity and selectivity of these aryl electrophiles with the proteome.
Figure 5.
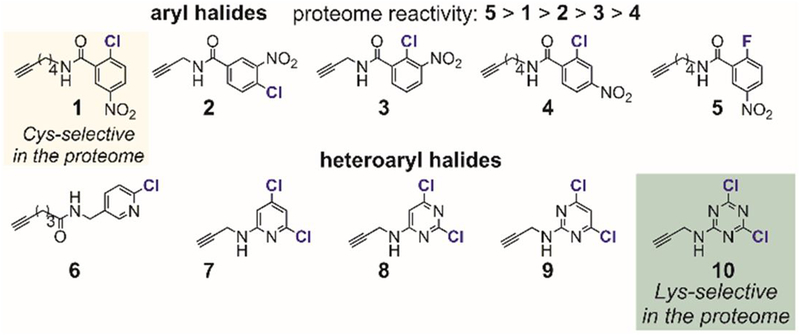
Reactivity and selectivity of aryl halides with the human proteome (HeLa cell lysate).
Dichlorotetrazine for SNAr stapling and photo-triggered unstapling of peptides and proteins
Hochstrasser and Smith identified 1,4-S,S-tetrazine (i.e., a tetrazine with sulfur substituents at positions 1 and 4) as a fast photochemical trigger for generating thiocyanate, enabling the study of the dynamics of peptide and protein folding.[72,73] This S,S-tetrazine unit was incorporated into peptides through either on-resin or in-solution SNAr reactions of 1,4-dichlorotetrazine with cysteine thiols.[74,75]
Based on these early studies, Smith reported in 2015 a method for the rapid stapling and photo-triggered unstapling of peptides and proteins.[76] They used 1,4-dichlorotetrazine to chemoselectively crosslink two cysteine thiols in an unprotected peptide (P1), generating the S,S-tetrazine-stapled peptide macrocycle within one minute (Figure 6A). The resulting stapled peptide (P2) was unstapled under UV irradiation. The cyano groups were removed from the peptide bisthiocyanate (P3) through reactions with free cysteine to restore the original unprotected peptide. While dichlorotetrazine chemoselectively reacts with cysteine thiols in the presence of other nucleophilic amino acid side chains, the reaction is not compatible with the disulfide-reducing reagent tris(2-carboxyethyl) phosphine (TCEP) and therefore requires purification of the reduced dithiol species prior to the stapling step. Of note, the authors showed that S,S-tetrazine can also serve as a bioconjugation handle for the inverse electron demand Diels-Alder reaction (Figure 6B).
Figure 6.
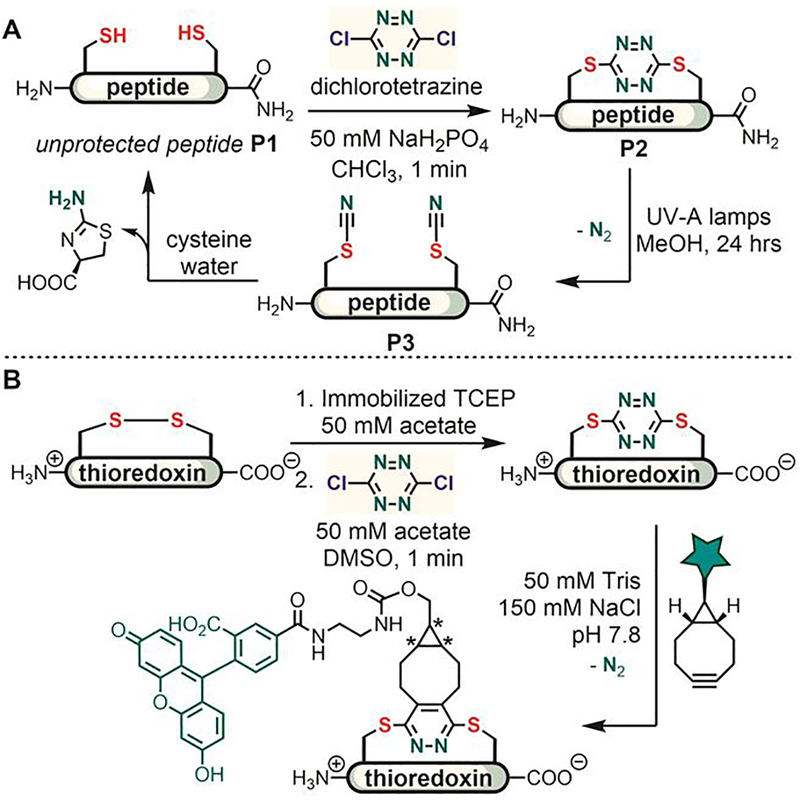
Peptide and protein stapling with dichlorotetrazine. (A) Unprotected peptides with two cysteines were stapled via SNAr reaction with dichlorotetrazine. The resulting tetrazine staple could be removed under photo-triggered conditions. (B) Thioredoxin was first stapled with dichlorotetrazine; the resulting dithio-tetrazine motif was further utilized as a bioconjugation handle for Diels-Alder reaction with a cyclooctyne. *Note that a mixture of diastereomers was formed through cycloaddition from either face of the tetrazine ring.
Heteroaryl methylsulfones for cysteine arylation
Heteroaromatic methylsulfones were developed to generate heteroaryl-thiol linkages in proteins. Xian first reported in 2012 the use of methylsulfonyl benzothiazole (MSBT) as a thiol-selective reagent for blocking cysteines in proteins (Figure 7A).[77] MSBT showed higher product yields in the reaction with N- and C-terminal–protected cysteine than other substituted benzothiazoles including those with halides and diazo groups in C-2 position. Because halides and diazo groups are better leaving groups than the sulfonyl group in SNAr reactions,[78] these more reactive electrophiles could possibly be hydrolyzed in an aqueous solution before they react with the cysteine derivative. MSBT showed excellent chemoselectivity for reaction with the cysteine thiol; no reaction was observed for other protein nucleophiles including those in the side chains of Ser, Tyr, Trp, Met, His, and Lys. The reaction rate correlated with the pH of the solution, consistent with the notion that the thiolate group is more reactive toward MSBT than the thiol group.[77] The labeling reaction was protein-compatible, as demonstrated by the blocking of the active site thiol of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) under mild conditions.
Figure 7.
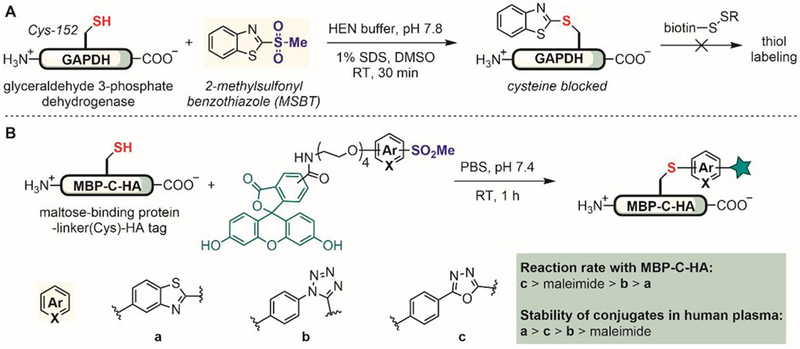
Heteroaryl methylsulfone reagents for protein cysteine arylation. (A) Methylsulfonyl benzothiazole (MSBT) as a protein thiol blocking reagent. RT: room temperature. (B) Stable protein bioconjugates generated from thiol modification with heteroaryl methylsulfonyl reagents.
Inspired by the high chemoselectivity and mild reaction conditions afforded by MSBT, Barbas in 2013 developed a series of heteroaryl methylsulfone reagents and investigated their reactions with cysteine residues in proteins (Figure 7B).[79] These heteroaryl methylsulfone reagents reacted with proteins in a thiol-specific manner, enabling site-selective conjugation of fluorescent dyes and polyethylene glycols (PEGs). The labeling reaction rates were comparable to that of cysteine-maleimide conjugation. Importantly, these heteroaryl-cysteine conjugates are significantly more stable toward acids, bases, and external thiols compared to the corresponding maleimide adducts.
The high rate, selectivity, and stability of these heteroaryl methylsulfone-cysteine reactions enabled their wide application in bioconjugation reactions. Using phenyloxadiazole sulfone linker c (Figure 7B), Barbas prepared site-specific antibody conjugates with improved stability in human plasma compared to the corresponding maleimide conjugates.[80] Mindt prepared 18F-containing phenyloxadiazole sulfone reagents and showed their rapid conjugation to target peptides and proteins for positron emission tomography (PET) applications.[81] Alternatively, 18F-labeled integrin-binding peptides (i.e., RGD peptides) were synthesized by combining the phenyloxadiazole sulfone-cysteine reaction with a fluorinase-catalyzed transhalogenation process.[82] Xian and Furdui developed a new heteroaromatic alkylsulfone, 4-(5-methanesulfonyl-[1,2,3,4]tetrazol-1-yl)-phenol (MSTP), for selective blocking of protein thiols without cross-reaction with sulfenic acids.[83] The high thiol-specificity of these sulfone reagents was further highlighted in selective modifications of complex molecules such as polyacrylamide hydrogels[84] and protein enzymes.[85]
Methylsulfone-based arylation reagents were also developed for thiol conjugation in living cells. In 2016, Fang reported 4-methylsulfonyl-N-n-1,8-naphthalimide (MSBN) as a highly fluorogenic reagent for the specific detection and labeling of thiol species and proteins (Figure 8A).[86] More than a 100-fold increase in fluorescence was observed upon reacting MSBN with thiols, enabling imaging of thiol species and detection of reversible protein thiol modification in living cells.
Figure 8.
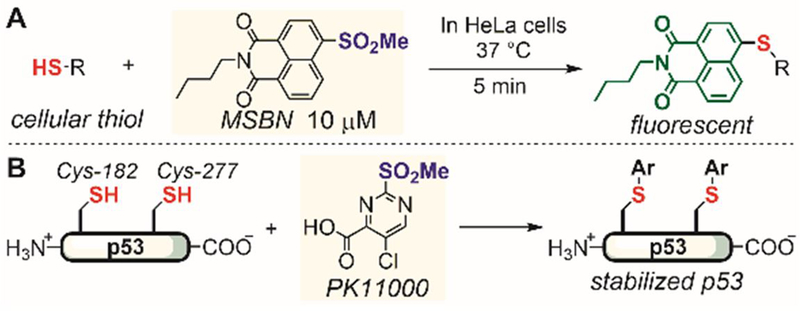
Arylation of thiols in living cells using aryl and heteroaryl methylsulfone reagents.
In 2016, Fersht screened an in-house fragment library and identified 2-sulfonylpyrimidine PK11000 as a cysteine-selective arylation reagent for the oncogenic target p53 (Figure 8B).[87] PK11000 covalently conjugated to Cys-182 and Cys-277 on p53 as shown by mass spectrometry analysis of the labeling reactions of p53 and its cysteine-to-serine mutants. The labeled p53 showed increased stability and retained DNA binding in vitro, which potentially explained the high potency of PK11000 against p53-deactivated or p53-mutated cancer cells.
Perfluoroarenes for cysteine arylation
In the 1960s, several research groups carried out fundamental studies that provided the ground rules for the selectivity, reactivity, and substitution patterns of SNAr reactions between perfluoroarenes and thiolates.[88,89] Tatlow found that the 1,4-disubstituted product predominantly formed when reacting an excess amount of potassium phenylsulfide with hexafluorobenzene in refluxed pyridine (Figure 9A).[88] Later, mechanistic studies by Pitts established the activating effect of p-substitutions on the perfluorobenzene ring.[89] The rate of SNAr reaction with potassium phenylsulfide was more than 1,000 times higher with phenylthiopentafluorobenzene than with pentafluorobenzene (Figure 9B). The thioether substituent activates the phenyl ring toward attack at the o- and p- positions due to the resonance stabilization of the negative charge in the σ-complex intermediate (Figure 9A). Regioselective p-substitution was favored because the o-positions were hindered by the relatively large thiophenyl group.
Figure 9.
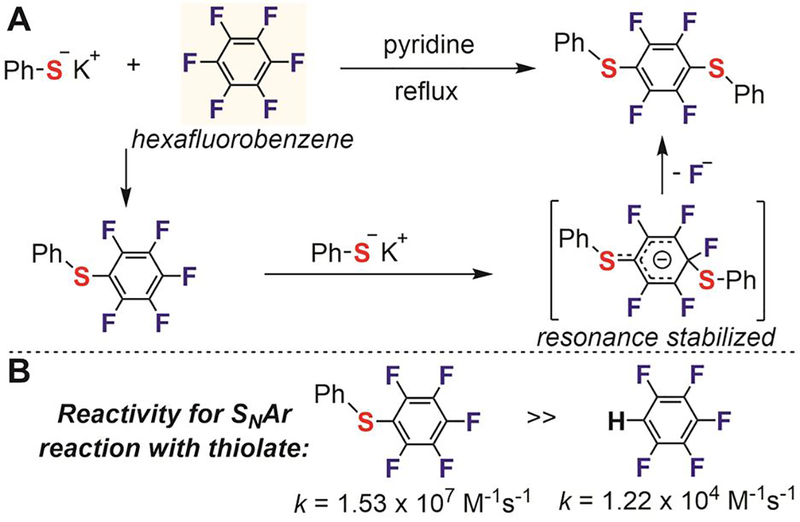
Nucleophilic aromatic substitution reactions between potassium benzenethiolate and hexafluorobenzene.
In the decades following the 1960s, applications of the perfluoroaryl-thiol reaction focused primarily on synthetic molecules, especially modifications of porphyrinoids[90] and synthetic polymers[91–93] (Figure 10). In 1991, Mansuy reported on the SNAr reactions of tetra-(pentafluorophenyl)porphyrin with various nucleophiles.[94] Using triethylamine as the base in refluxing DMF, p-fluorines on the tetra-(pentafluorophenyl)porphyrin were selectively replaced with thiol nucleophiles (Figure 10A). This efficient reaction provided a facile way to functionalize the periphery of different porphyrinoid cores, enabling the synthesis of various modified porphyrinoids for applications in catalysis,[95] materials science,[96] and as drug carriers.[97–99]
Figure 10.

Perfluoroaryl-thiol SNAr reaction for the modification of porphyrins (A) and polymers (B).
In a series of manuscripts from 2003 to 2005, Wooley was first to demonstrate how polymers containing pentafluoroaryl groups can be functionalized and cross-linked via SNAr.[100–102] Following these seminal contributions, Schubert in 2009 developed methods to modify perfluoroaromatic polymers with thiol-containing sugars and suggested that the perfluoroaryl-thiol reaction can be classified as a click process due to its high efficiency and functional group tolerance (Figure 10B).[92,103] In 2015, Dichtel reported a new class of porous polymer materials, which were synthesized via an SNAr reaction between fluorinated arene electrophiles and cyclodextrin precursors.[104] These materials showed unprecedented performance for absorbing various small-molecule pollutants from aqueous media.
In 2016, the perfluoroaryl-thiol click reaction was used by Spokoyny to generate precisely modified boron clusters.[105] Through the peralkylation of boron clusters, Spokoyny created rigid, 3D nanomolecules featuring pentafluorophenyl handles (Figure 11A). These perfluoroaryl boron clusters were used as templates for further diversification with thiol-containing molecules including alkanes, arenes, alcohols, amines, peptides, and sugars (Figure 11B). The conjugation reactions were generally very efficient, quantitatively producing multivalent, homogeneous nanoclusters with defined orientation of the attached ligands. Nanomolecules grafted with glucose exhibited increased binding affinity to concanavalin A (ConA, Figure 11B), and demonstrated how this strategy enables the rapid generation of precise multivalency for molecular recognition. The simplicity of this approach is reminiscent of the assembly of thiol-coated gold nanoparticles (AuNPs), yet it possesses additional advantages such as atomic precision and full covalency.
Figure 11.
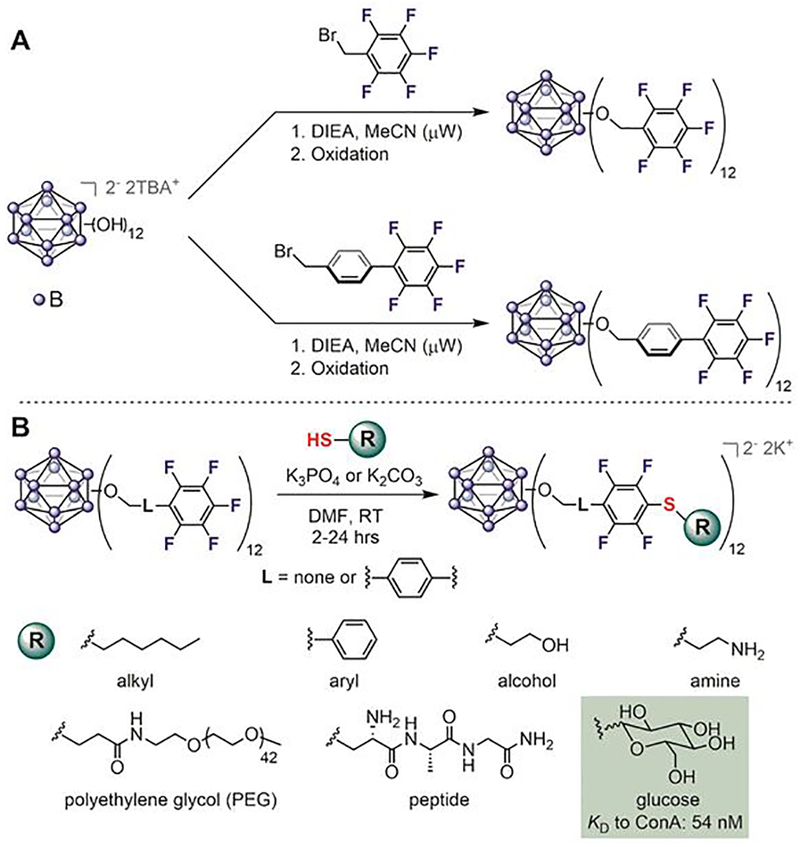
Atomically-precise functionalization of boron clusters via perfluoroaryl-thiol conjugation reactions. (A) Boron clusters were modified with perfluoroarenes. DIEA: diisopropylethylamine. TBA: tetrabutylammonium. μW: microwave. (B) Various chemical structures were attached quantitatively to the perfluoroaryl-linked boron cluster. Sugar-modified boron cluster has significantly increased binding affinity to concanavalin A compared to glucose, presumably because of the multivalency of the modified boron cluster.
Even though the perfluoroaryl-thiol reactions showed good functional group tolerance in the modification of synthetic molecules (Figure 10), their chemoselectivity in a bioconjugation setting had not been extensively studied. Pentelute developed a perfluoroaryl-thiol SNAr reaction platform for bioconjugation.[106–114] As a starting point, Pentelute in 2013 evaluated the reactions between various amino acids and commercial hexafluorobenzene and decafluorobiphenyl reagents.[106] These simple perfluoroarenes were found to react with the thiol group of cysteine but not with other nucleophilic components of amino acid side chains such as the amino group in lysine or the imidazole group in histidine at room temperature in polar organic solvents. Consistent with the previously reported activating effect of the p-thioether group, the 1,4-disubstituted regioisomer was exclusively observed when allowing hexafluorobenzene to react with an excess of cysteine. The monoarylated product was observed only when a large excess (>10-fold) of hexafluorobenzene was used with peptides containing one or several cysteines. A similar substitution pattern was observed for the reaction between decafluorobiphenyl and cysteine. These results prompted further study of this reaction for the modification of peptides.
In further studies, Pentelute found that hexafluorobenzene and decafluorobiphenyl efficiently crosslinked two cysteine thiols in unprotected peptides (Figure 12A).[106] When an excess amount of the perfluoroarene was used, the major product was monosubstituted (Figure 12B). The arylation reactions were fast and usually complete within hours at room temperature in organic solvents such as dimethylformamide (DMF), acetonitrile, and dimethylsulfoxide (DMSO). Many mild organic and inorganic bases such as triethylamine and potassium phosphate could also be employed. Disulfide-reducing reagents such as triphenylphosphine and TCEP were compatible with the reaction and could be added to prevent the oxidation of cysteine thiol. The use of 19F-NMR spectroscopy provides a convenient spectroscopic handle to determine the structural identity of the conjugation products.
Figure 12.
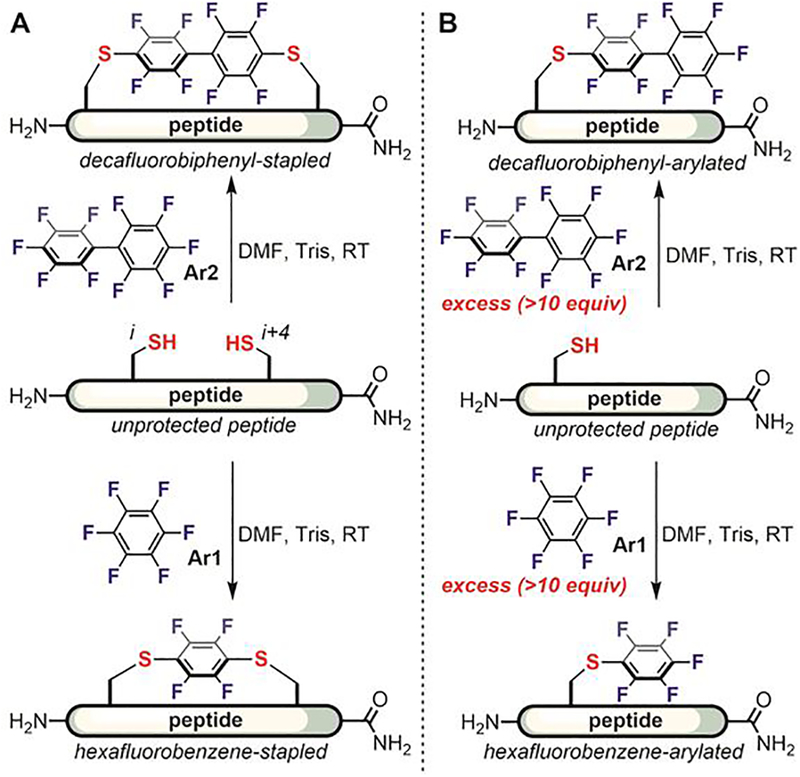
Hexafluorobenzene and decafluorobiphenyl as first-generation perfluoroarenes for peptide modification: (A) Peptide stapling with perfluoroarenes developed by Pentelute. (B) Arylation of peptides containing a single cysteine using >10 equiv of perfluoroarenes.
Based on the initial results using commercial perfluoroarenes, Pentelute designed and synthesized four additional perfluoroaryl linkers, all featuring thioether substituents for p-fluorine activation.[108] These customized perfluoroaryl linkers were used with three commercial perfluoroarenes to cyclize two cysteine thiols positioned at different sites on a peptide chain (Figure 13).[108] Fourteen unprotected peptides with cysteine residues spaced from i, i+1 to i, i+14 were cyclized using seven perfluoroaryl linkers (Figure 13), producing 98 peptide macrocycles. This is the first example of a convergent diversity-oriented macrocyclization scan, which highlighted the generality, efficiency, selectivity, and robustness of the perfluoroaryl-cysteine SNAr reaction for peptide modification.
Figure 13.
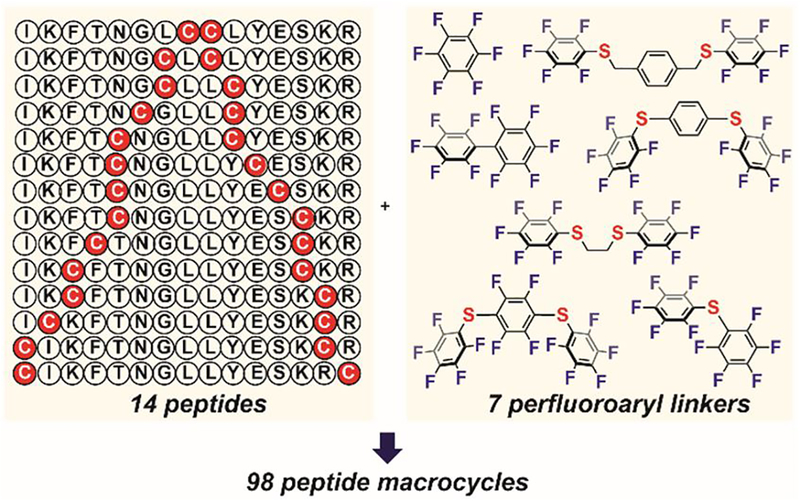
Peptide macrocyclization scan with commercial and customized perfluoroaryl linkers.
The macrocyclic peptide structures produced were further expanded through a two-step peptide macrocyclization strategy (Figure 14).[108] First, both cysteine thiols in an unprotected peptide were arylated using an excess of a simple perfluoroarene to generate a peptide intermediate with perfluoroarylated cysteine residues. These were further crosslinked using commercial dithiol reagents. This strategy shows promise for the synthesis of a range of structurally diverse macrocyclic peptides using commercial dithiols.
Figure 14.
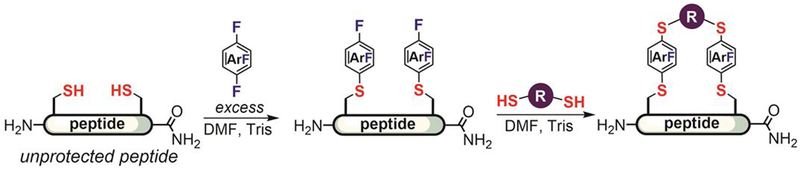
Macrocyclization of perfluoroarylated cysteines with dithiol reagents.
Multi-cycles can be constructed by sequentially arylate cysteine residues within a single peptide chain. Using 2,3,5,6-tetrafluoroterephthalonitrile (4F-2CN) that has sequentially varied cysteine arylation reactivity, Wu in 2017 created multicyclic peptides with interlocked topologies.[115] In 2018, Pentelute synthesized a series of bicyclic cell-penetrating peptides (CPPs) using the strategy outlined in Fig. 14. Compared with their monocycle analogues, these bicyclic CPPs showed improved protease resistance and enhanced ability to deliver antisense oligonucleotide into cells, potentially due to the highly constrained conformation and reduced structural flexibility endowed by the perfluoroaryl bicyclic topologies.[116]
Pentelute and several other groups subsequently applied the perfluoroaryl-cysteine SNAr chemistry to the macrocyclization of bioactive peptides (Table 2). Compared to their linear peptide counterparts, macrocyclic or stapled peptides usually have improved biological properties such as stability against proteases and binding affinity to the target. Pentelute reported a stapled peptide inhibitor of the C-terminal HIV-1 capsid assembly protein (C-CA) that displayed enhanced binding affinity to C-CA and increased stability against trypsin and chymotrypsin digestion (Table 2, entry 1).[106] Importantly, enhanced cellular uptake of the stapled peptide was observed, indicating that macrocyclization using perfluoroaryl linkers can render peptides cell permeable. In 2016, Pun sampled various macrocyclization strategies for a peptide that targets M2 macrophages. Decafluorobiphenyl was found to be the optimum stapling reagent to enhance the binding affinity and the serum stability of the peptide (Table 2, entry 2).[117] Schroeder used hexafluorobenzene to synthesize a macrocyclic peptide as a tumor-targeting reagent and found that the perfluoroaryl motif was not cytotoxic (Table 2, entry 3).[118] Craik cyclized several GLP-1 receptor agonists using perfluoroaryl linkers.[119] However, these macrocyclic peptides showed reduced potency compared to their linear analogs (Table 2, entries 4–6). In 2017, Pentelute used perfluoroarene staples to enhance the penetration of peptides across the blood brain barrier (BBB), both in a BBB spheroid model and in mouse brains (Table 2, entries 7–8).[114] In most cases, perfluoroaryl-cyclized peptides are more stable than their linear counterparts. The effect of perfluoroaryl linkers on the binding affinity of macrocyclic peptides differs as a function of the peptide structure presumably because the linker itself can directly engage or block the peptide from binding to its target. This, however, is not a unique property of perfluoroarene staples; other carbon-based linkers have exhibited these sequence-dependent variabilities as well.[120,121]
Table 2.
Perfluoroaryl-linked macrocyclic bioactive peptides.
| Entry | Peptide sequence | Linker | Function | Properties compared to a linear peptide | Ref. |
|---|---|---|---|---|---|
| 1 | ITFCDLLCYYGKKK | Ar1 Ar2 | Bind HIV-1 capsid assembly protein (C-CA) |
|
106 |
| 2 | CGYEQDPWGVRYWYGCkkk(K-biotin) | Ar2 | Bind M2 macrophage |
|
117 |
| 3 | CGNKRTRGC | Ar1 | Tumor targeting |
|
118 |
| 6[a] | His-aa1-Glu-Gly-Cys-aa2-Thr-Ser-Asp-aa3-aa4-Cys | Ar2 | |||
| 7 | AGYLLGKINLKACAALAKKCL | Ar2 | Penetration across blood-brain barrier (BBB) |
|
114 |
| 8 | IWICQELCRIGDEFNAYYARR | Ar2 | Bind to BCL-2 protein |
|
114 |
aa1: 2-aminoisobutyric acid; aa2: (S)-2-Fluoro-α-methylphenylalanine; aa3: (S)-2-amino-3-(2’ethyl-4-methoxy-[1,1’-biphenyl]4-yl)propanoic acid; aa4: (S)-2-amino-5-phenylpentanoic acid
The perfluoroaryl-cysteine SNAr reaction is also compatible with the chemical synthesis of proteins. Two small proteins, affibody and human relaxin, were modified with perfluoroaryl linkers by combining perfluoroaryl-cysteine SNAr reaction with fast-flow peptide synthesis and native chemical ligation (NCL) techniques (Figure 15).[106,110] Pentelute installed a perfluoroaryl staple on the C-terminal helix of an affibody targeting human epidermal growth factor receptor 2 (HER-2) (Figure 15A).[106] The three segments of affibody were ligated via two sequential NCL reactions. The resulting folded perfluoroaryl-stapled affibody had similar circular dichroism (CD) and binding properties compared to the unmodified affibody. Furthermore, the perfluoroaryl linker was used as an irreversible disulfide surrogate by replacing the disulfide between Cys-10 and Cys-15 on the A-chain of human relaxin (Figure 15B).[110] Upon oxidative folding with the B-chain, a perfluoroaryl relaxin was obtained. Using a cell-based assay, the modified relaxin was found to be active, though with a diminished (100-fold) potency compared to the wild type human relaxin.
Figure 15.
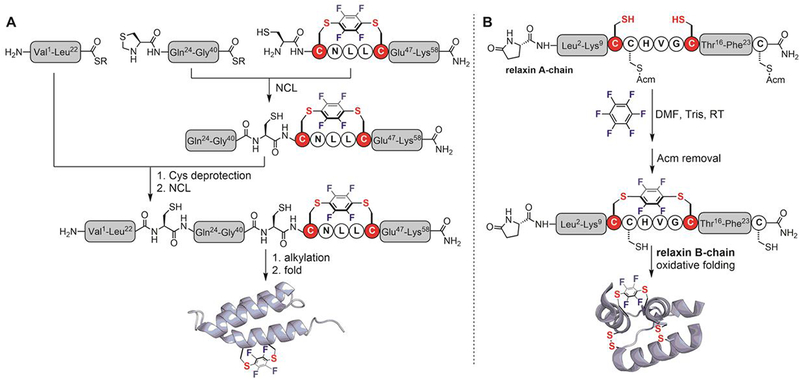
Total chemical synthesis of perfluoroaryl-modified affibody (A) and human relaxin (B) proteins.
The perfluoroaryl-cysteine SNAr reaction is efficient in polar organic solvents, but it often becomes sluggish in aqueous media. Moreover, the low solubility of previously described perfluoroaryl reagents (Figure 13) limits their utility in aqueous solutions. In an attempt to use perfluoroarenes in aqueous media, Derda in 2016 identified decafluorobiphenylsulfone as a very reactive electrophile for the arylation of cysteine thiol under aqueous conditions.[122] A reaction rate of 180 M−1s−1 was observed when treating a pentapeptide (Ser-Trp-Cys-Arg-Cys) with decafluorobiphenylsulfone in Tris buffer (pH 8.5) with 40% acetonitrile. The increased reactivity of decafluorobiphenylsulfone in aqueous media enabled its application for the modification of biomolecules sensitive to organic solvents. Using this reagent, Derda macrocyclized peptide libraries displayed on the surface of the M13 phage with good efficiency (Figure 16). This paved the way to further use this peptide-stapling reaction and phage display to discover bioactive macrocyclic peptides. Consistent with Derda’s observations, Meldal and Diness in 2018 systematically studied the reactivity of fluorobenzene probes and found enhanced arylation reactivity in buffers for probes with increased number of fluorine groups and more electron-withdrawing para-substituents.[123]
Figure 16.
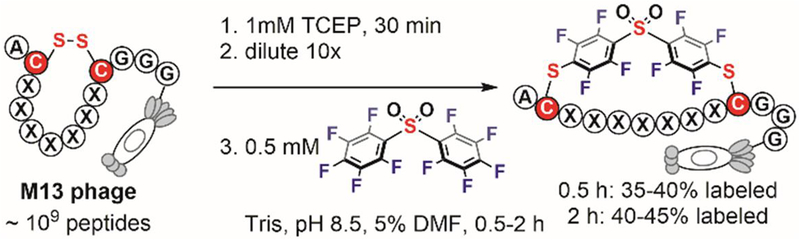
Macrocyclization of phage-displayed peptides via cysteine perfluoroarylation with perfluoroarylsulfone.
Another approach to increase the reactivity of perfluoroarenes in aqueous solutions is through proximity-induced reactions. In 2015, Wang developed a photoswitchable amino acid containing a pentafluorophenyl group for cysteine reaction and an azo bridge for photoswitching (Figure 17).[124] This amino acid was introduced into proteins via genetic code expansion, allowing the pentafluorophenyl group to react with a nearby cysteine to form an azo bridge in situ during protein expression in E. coli. The electron-withdrawing nature of the perfluoroaryl group resulted in a significant separation of absorbance bands corresponding to E and Z isomers, enabling the photoswitch unit to reversibly isomerize using blue and green light irradiation. Applying this strategy to modify calmodulin enabled the photocontrol of calmodulin conformation and binding. The expressed protein was a mixture of the desired azo-bridged protein, the unreacted protein, and a by-product with the perfluoroaryl reacted with glutathione, indicating that further optimization to increase the reaction rate and selectivity is required to generate the desired product in higher yields.
Figure 17.
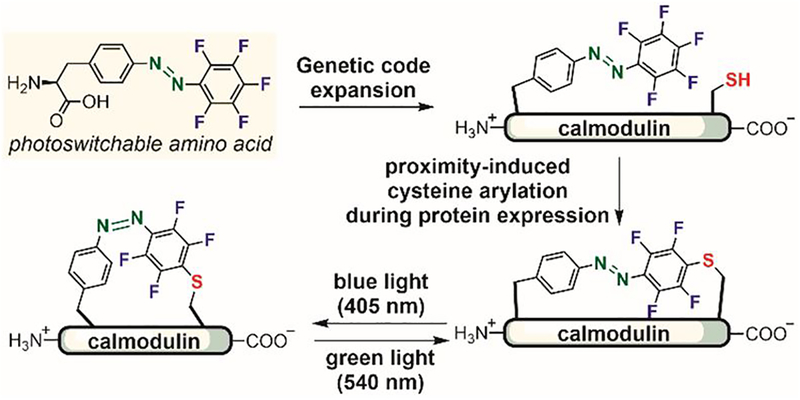
Genetic code expansion and proximity-induced cysteine arylation to introduce photoswitchable azo bridges into proteins.
Cysteine perfluoroarylation was also explored for introducing 18F radiolabels into biomolecules. In 2015, Chen synthesized perfluoroaryl-linked dimers of an integrin-binding peptide using 18F-hexafluorobenzene generated from an 18F exchange with a non-radiolabeled hexafluorobenzene (Figure 18).[125] The 18F-hexafluorobenzene reacted with a thiol-containing cyclic peptide (cyclo(RGDfK)) that binds to cell surface integrin. After a 20 min reaction at room temperature, approximately 50% conversion to the desired 18F-labeled peptide was observed. The 18F-labeled dimeric peptide was obtained with 40% radiochemical yield after HPLC purification. The perfluoroaryl-linked dimeric peptide showed similar binding affinity to the αvβ3 integrin as the original monomer. In addition, this dimer showed good tumor uptake and high tumor-to-background contrast in positron emission tomography (PET) imaging in a mouse tumor xenograft model.
Figure 18.
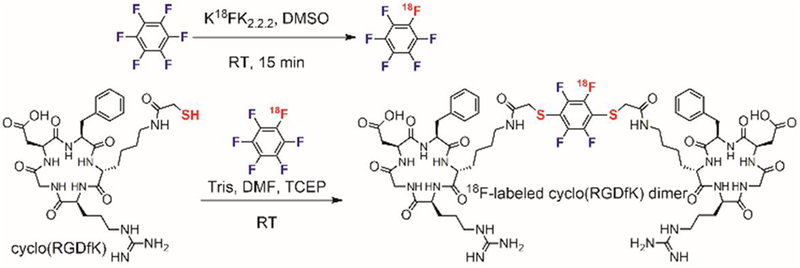
Synthesis of 18F-radiolabelled peptides via cysteine perfluoroarylation.
Chemo- and regio-selective cysteine SNAr arylation
Reactions on proteins that contain several solvent-exposed cysteine sites usually lead to undefined mixtures of products. For example, maleimide was previously used to conjugate drug molecules to reduced interchain cysteines in antibodies.[126] The resulting antibody-drug conjugates (ADCs) form mixtures of products with drug-to-antibody ratio (DAR) ranging from 1 to 8. Even for molecules with the same DAR, the position of drug attachment might differ. Each individual member in this ADC mixture might have different therapeutic properties, which presents a significant challenge for systematic studies of ADCs and calls for the development of site-selective chemistry for ADC synthesis.
Although many chemoselective cysteine bioconjugation methods have been developed, regioselective cysteine conjugations are rare. In 2013, Caddick found that one of the two cysteine residues in superfolder green fluorescent protein can selectively react with 2,5-dibromohexanediamide.[127] In certain instances, the N-terminal cysteine can be selectively modified via native chemical ligation, albeit the final ligation product is an amide bond rather than a modification on the cysteine thiol.[128–131] Other more sophisticated strategies using protecting groups,[132] enzyme catalysts,[133–135] or multiple chemical steps[136] have also been developed for regioselective cysteine conjugation.
Nature uses enzymes to control the chemo- and regio-selectivities of chemical reactions including highly precise post-translational modifications.[137,138] For cysteine arylation, Nature uses glutathione S-transferase (GST) to perform highly selective arylation reactions. GST, a family of promiscuous enzymes, catalyzes the conjugation of electrophilic xenobiotic substrates to the thiol of a tripeptide glutathione (γ-Glu-Cys-Gly, GSH) for detoxification.[139,140] Aryl halides are a known family of electrophiles accepted by GST. In fact, the GST-catalyzed conjugation of 1,4-dinitrochlorobenzene (DNCB) to GSH is routinely used in standard colorimetric assays to assess the enzymatic activity of GSTs (Figure 19A).[141–143] Upon arylation of GSH with DNCB under enzymatic catalysis, the arylated product has an increased UV absorbance at 340 nm, enabling the determination of the enzymatic activity of a broad range of GST isozymes.
Figure 19.
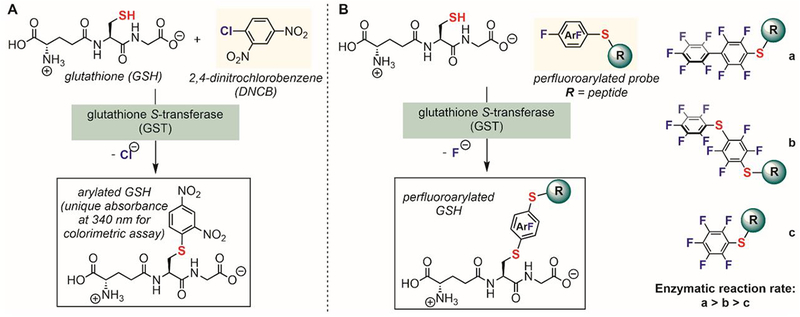
Glutathione S-transferase-catalyzed arylation of cysteine thiol in glutathione. (A) GST catalyzes the arylation of glutathione with 2,4-dinitrochlorobenzene. (B) GST catalyzes the perfluoroarylation of glutathione with various peptide-based perfluoroaryl electrophiles
Because of the substrate promiscuity of natural GSTs, Pentelute hypothesized that these enzymes can be directly hijacked to catalyze the SNAr reaction between perfluoroarenes and glutathione as a means to improve the rate of previously sluggish perfluoroarylation reactions in aqueous solutions (Figure 19B). Perfluoroarenes such as hexafluorobenzene and decafluorobiphenyl have very low solubility in water, however, several water-soluble peptide-based perfluoroaryl electrophiles could be prepared by treating cysteine-containing peptides with more than 10 equivalents of perfluoroarenes (Figure 19B). A similar strategy was used to prepare a wide range of water-soluble perfluoroaryl reagents.[111] These peptide-based perfluoroaryl electrophiles were conjugated to GSH using a mixture of commercial natural GST isozymes.[107] Importantly, no reactions were observed in a control experiment without the enzyme. GSTs purified from different sources displayed similar activity and the decafluorobiphenyl-based electrophile was found to be the most reactive electrophile tested under GST catalysis (Figure 19B). It is worth noting that many glutathione S-transferases contain cysteine residues,[139] therefore, the GST-catalyzed reaction provides a potential approach for selective cysteine modification in the presence of other thiol species.
To further explore GST-catalyzed reactions for protein modification, GSH was installed as a peptide tag on model proteins (Figure 20).[107] Inspired by a crystal structure of GSH bound to GST (Figure 20A),[144] Pentelute hypothesized that appending the protein of interest to the C-termini of GSH might not significantly affect GST’s recognition of GSH. Such a design resulted in an N-terminal GSH tag for protein modification. Pentelute further hypothesized that if GSH was incorporated into the peptide or protein of interest that contained endogenous cysteines, the GST enzyme will catalyze the regioselective conjugation of perfluoroaryl electrophiles to the thiol of GSH in the presence of other competing cysteine residues on the same molecule (Figure 20B). Indeed, it was found that several model peptides with the N-terminal GSH tag efficiently conjugated to perfluoroaryl probes under GST catalysis irrespective of the structure of the amino acid directly linked to the glycine of the GSH tag.
Figure 20.
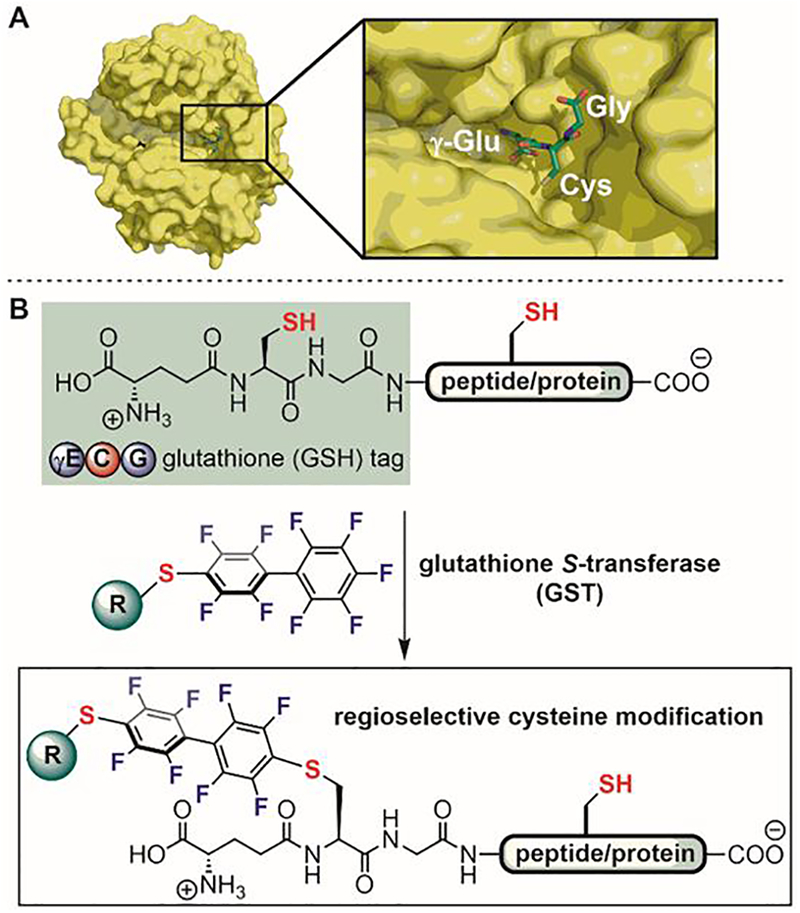
GST-catalyzed regioselective cysteine perfluoroarylation. (A) A crystal structure of GSH bound to GST dimer shows that the Gly moiety of GSH is exposed as a possible site for attaching peptide/protein without compromising the recognition of GSH by GST. (B) Attaching a three-amino acid glutathione (GSH) tag at the N-terminus of the peptide or protein of interest enables recognition by GST and selective conjugation to perfluoroaryl-linked reagents.
The GST-catalyzed regioselective cysteine conjugation enables applications to protecting group-free, dual-protein modification (Figure 21A) and regioselective macrocyclization of unprotected peptides (Figure 21B).[107,109] The GSH tag was installed at the N-terminus of a model protein through native chemical ligation. The resulting protein was dual-labeled with biotin and fluorescein through two sequential reactions. The GSH cysteine was first modified using a perfluoroaryl biotin probe under GST catalysis, after which the other unprotected cysteine was labeled with maleimide fluorescein without the need for additional purification of the monoarylated product (Figure 21A). The GSH tag was also installed at the N-terminus of peptides with C-terminal perfluoroarylated cysteines. Under GST catalysis, macrocyclic peptides with lengths of up to 40-residues were readily synthesized (Figure 21B). In contrast, cyclization reactions without enzymes produced polymers and other side products.
Figure 21.
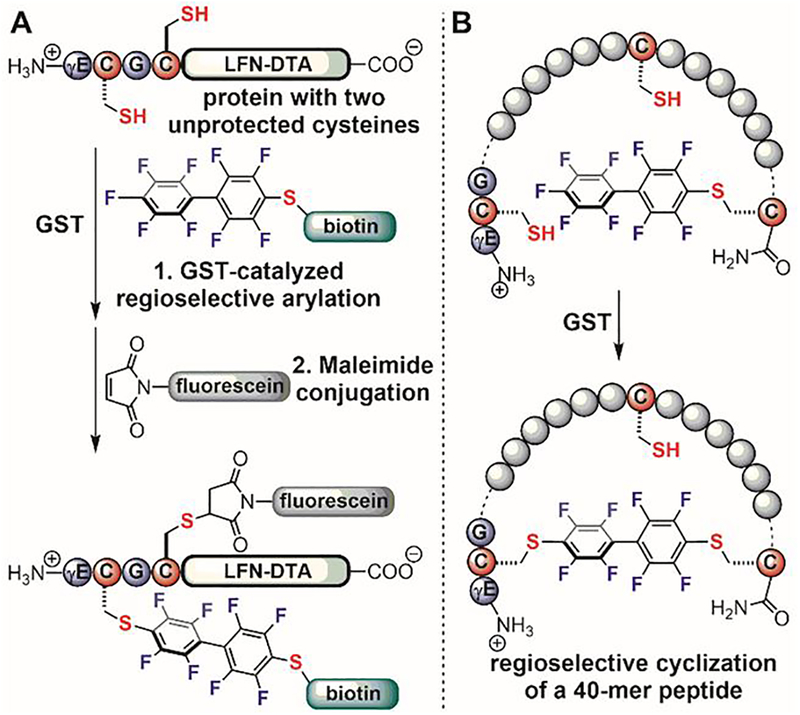
Applications of GST-catalyzed regioselective cysteine perfluoroarylation. (A) Site-selective dual-labeling of an unprotected protein. (B) Enzyme-catalyzed regioselective macrocyclization of a long peptide.
One limitation of the GST-catalyzed cysteine conjugation is the need for a GSH tag with a γ-glutamic acid that is not genetically encoded. To overcome this limitation, a fully genetically encodable peptide tag that promotes cysteine arylation in water is highly desirable. Pioneering work by Tsien in 1998 describes peptide tags containing multiple cysteines to entropically promote selective conjugation to biarsenic reagents.[145,146] In 2015, Davis used arsenic-based reagents to modify two cysteines generated from the reduction of a disulfide.[147] Considering previous observations that different protein microenvironments can lead to different cysteine reactivities,[60,127] Pentelute designed a new strategy for site-selective cysteine arylation that fine-tuned the arylation reactivity of a cysteine within a small peptide sequence.
In 2015, Pentelute discovered that a four-residue peptide sequence (Phe-Cys-Pro-Phe), referred to as the “π-clamp” efficiently promotes cysteine perfluoroarylation in aqueous solutions (Figure 22A).[111] The reaction rate between a π-clamp peptide and a perfluoroaryl probe was 0.76 M−1s−1 which was more than 1000-fold faster than the reaction of a control cysteine-containing peptide. Molecular dynamics (MD) simulations and density functional theory (DFT) calculations showed that the π-clamp sequence promoted the reaction both kinetically (lowering the activation energy) and thermodynamically (generating a more stable product) compared to the control. Systematic mutation studies and structural characterization with solid-state NMR suggest that the hydrophobic side chains in the π-clamp interact with the perfluoroarenes. Importantly, the prolyl amide bond favors trans conformation, as evidenced by both NMR and computational studies.[113]
Figure 22.
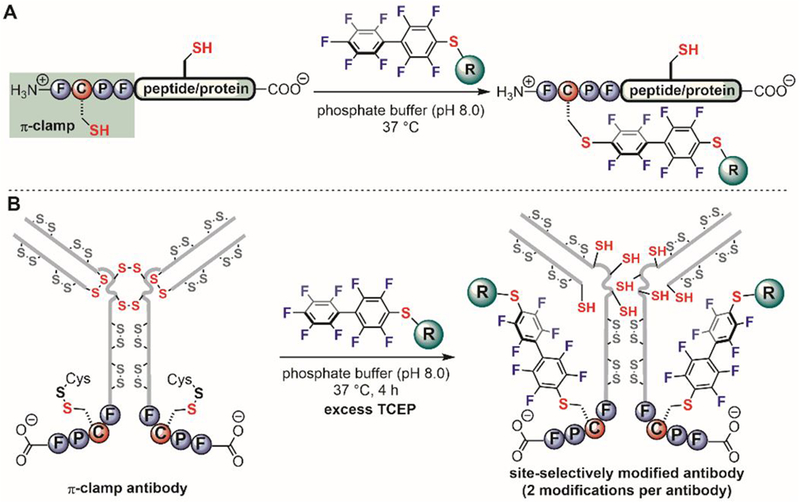
π-Clamp-mediated regioselective cysteine modification. (A) π-Clamp mediates selective arylation on its cysteine residues in the presence of other competing cysteine residues. (B) π-Clamp-mediated site-specific antibody modification.
The π-clamp was applied to site-specific modification of antibodies (Figure 22B).[111] The π-clamp is small and composed entirely of genetically encoded amino acids, enabling the convenient insertion of the π-clamp into the C-terminus of the heavy-chains of antibodies. The expressed π-clamp antibodies readily conjugated to perfluoroaryl-linked probes in a single step under reducing conditions. Only π-clamp cysteine thiols were labeled and other reduced thiols from the antibody interchain disulfides remained intact under the reaction conditions. The resulting site-specific antibody conjugates retained their target-binding affinity, indicating that the insertion of the π-clamp and the reaction conditions did not significantly affect the structure and function of the antibody. Site-specific π-clamp antibody-drug conjugates showed receptor-dependent cell killing. Compared to other site-selective antibody modification methods that require either unnatural amino acids,[148] enzymes,[149–152] or multiple chemical steps,[153,154] the π-clamp provides an enzyme-free method for site-selective antibody modification of a natural amino-acid sequence and paves a foundamentally new route towards next-generation homogeneous antibody-drug conjugates.
While investigating the π-clamp, Pentelute also discovered that adding simple inorganic salts can significantly affect the reaction rate of the bioconjugation.[112] Strikingly, different salts can change the rate constant by over four orders of magnitude for the π-clamp–mediated arylation reaction (Figure 23). The observed trend of this salt effect on the rate constant followed the Hofmeister series,[155,156] a phenomenon that had been known for over 100 years but had never been used for bioconjugation prior to this investigation. Protein-compatible salts including ammonium sulfate were used to greatly enhance bioconjugation reactions such as π-clamp-mediated arylation, enabling quick synthesis of functional antibody drug conjugates. Importantly, the salt effect can be expanded to reactions beyond arylation; Pentelute showed that ammonium sulfate can also enhance the rate of an alkylation reaction, by providing the first example of a site-selective cysteine alkylation mediated by π-clamp. The addition of salts likely tunes the reaction rates by changing the specific hydrophobic interactions between the π-clamp and the perfluoroaryl probe, as supported by computational and mutational studies.[112,113] The advantage of the salt effect is significant and also practical for biomolecule modification, considering that heating and concentrating are generally not compatible with delicate or prone-to-aggregate proteins.
Figure 23.
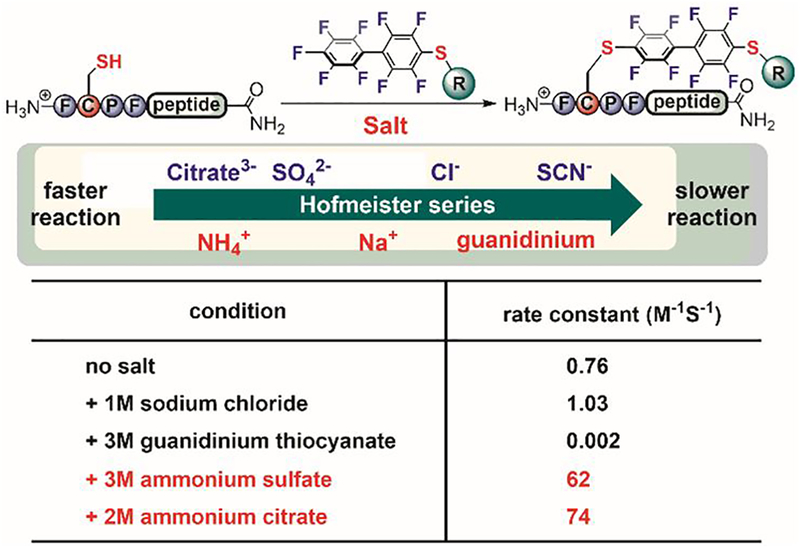
Salt effect on π-clamp mediated cysteine arylation.
2.2. Metal-mediated cysteine arylation
In the past two decades, metal-mediated bioconjugation reactions have emerged as promising tools in chemical biology.[47,52,157–161] Metal-mediated arylation provides a complementary approach to nucleophilic aromatic substitution (SNAr), and expands the scope of aryl modifications for peptides and proteins. A representative selection of the existing organometallic approaches to biomolecule arylation is presented in the following sections.
Chemoselective metal-mediated cysteine arylation
Selective cysteine arylation using organometallic reagents represents an important example of metal-mediated arylative bioconjugation reactions. Metal-catalyzed C–S cross-coupling reactions have been used for small-molecule synthesis by chemists for over three decades.[162–164] Yet, the first report on metal-mediated S-arylation of biomolecules appeared very recently, when in 2014 Wong used gold(III) complexes to transfer aryl groups to thiols on short peptides and bovine serum albumin (BSA, Figure 24).[165] The cyclometalated structure of the gold(III) complex required the use of arylpyridine units, which limited the scope of aryl structures that could be introduced using this method. Moreover, the slow reaction kinetics of the reductive elimination from gold(III) requires prolonged heating that can degrade delicate proteins. To overcome some of these challenges, in 2018 Spokoyny developed an auxiliary-free cysteine arylation method that use ligands that favor the planer structure of gold(III) complexes.[166] These new organometallic gold(III) reagents enabled rapid cysteine bioconjugation (<5 min) at room temperture in buffers with a wide pH range (0.5–14).
Figure 24.
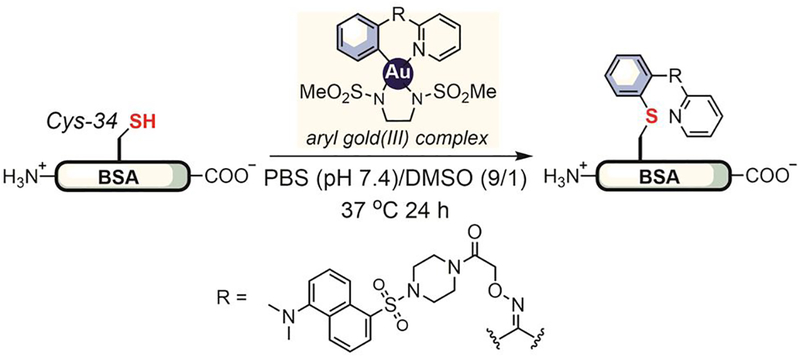
Gold(III)-mediated cysteine arylation.
Shortly after the work of Wong, Buchwald and Pentelute reported in 2015 palladium-based organometallic complexes featuring biarylphosphine ligands that are efficient and versatile reagents for chemoselective cysteine arylation (Figure 25).[167] The reaction proceeded at low micromolar concentrations of both the organometallic complex and the biomolecule and required no heating; only 5% of common organic co-solvent (e.g., CH3CN, DMF, or DMSO) was necessary for quantitative conversions at room temperature within minutes. The organic solvent requirement is due mainly to the low solubility of palladium complexes in water, but this limitation has recently been overcome using water-soluble biarylphosphine ligands.[168] Uniquely, desired products were formed under a wide range of pH (2–10), enabling peptide cysteine residue labeling in water containing 0.1% of trifluoroacetic acid (TFA).
Figure 25.
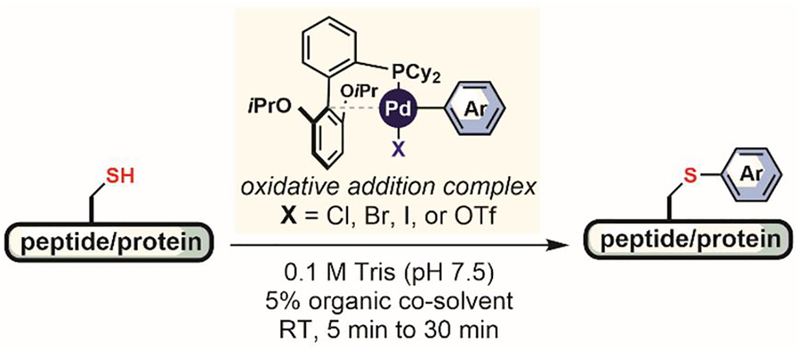
Organometallic palladium(II) reagents for cysteine arylation.
A variety of oxidative-addition complexes can be accessed in high yields (in one step) from commercial aryl halide or trifluoromethanesulfonate precursors (Figure 26A).[167] These storable and air-stable arylation reagents were successfully applied to the cysteine arylation of unprotected peptides, small antibody mimetic proteins, bacterial toxins, and antibodies (Figure 26B). Diverse S-aryl and S-heteroaryl bioconjugates were synthesized to introduce fluorescent dyes, affinity tags, and bioconjugation handles into peptides and proteins (Figure 26). A study of the resulting conjugates comparing maleimide and acetamide analogues showed that S-aryl conjugates are more stable under basic, acidic, and oxidative conditions, as well as in the presence of high concentrations of glutathione. The reaction rate of palladium-mediated S-arylation was comparable to or slightly faster than maleimide at neutral pH and is significantly faster in acidic solutions in which the maleimide reaction generates no product.
Figure 26.
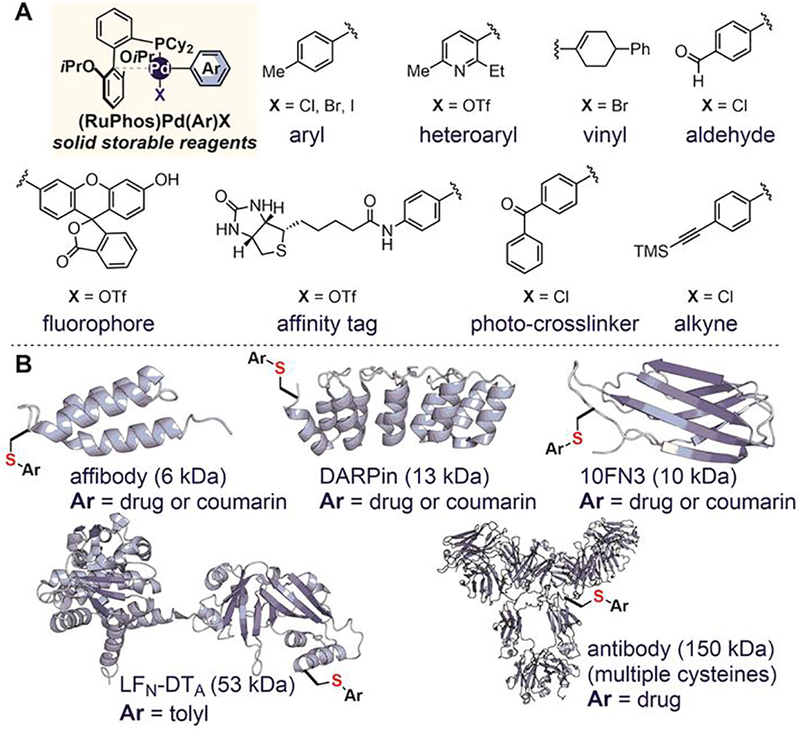
Organometallic palladium complexes (A) and protein targets (B) studied in the palladium-mediated cysteine arylation reaction.
Palladium reagents bearing two metal centers were used by Pentelute and Buchwald in 2016 for peptide stapling and did not form peptide oligomers (Figure 27).[167,169] The high efficiency of this transformation allowed access to a number of aryl-stapled peptides for a comprehensive study on the role of linker hydrophilicity, rigidity, and overall substitution patterns on various peptide properties including lipophilicity, phospholipid affinity, unique volume of distribution, and target binding affinity (Figure 27).
Figure 27.
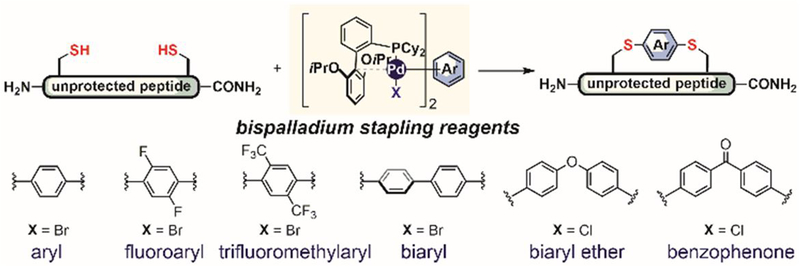
Organometallic palladium reagents for peptide stapling.
One of the advantages of palladium-mediated arylation is its compatibility with TCEP, commonly used to reduce disulfides in proteins and antibodies.[170] This, combined with the mild reaction conditions, enabled the application of this transformation for the efficient synthesis of “linker-free” antibody drug conjugates where the drug molecules were directly attached to cysteine thiols in antibodies (Figure 28).[167] The resulting ADCs have similar target-binding affinity compared to the native antibodies, indicating that the reaction conditions and the use of palladium reagents did not significantly affect antibody function.
Figure 28.
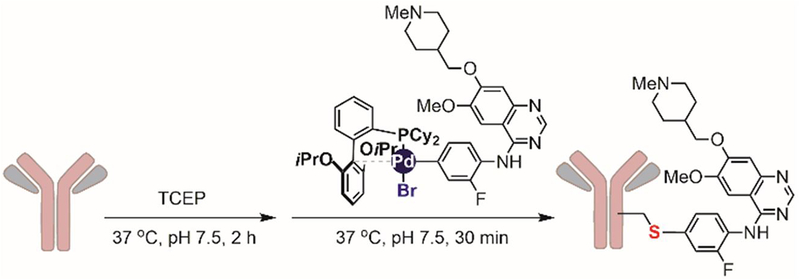
Organometallic palladium reagents for antibody-drug conjugation.
Palladium-mediated arylation can be combined with other chemistry to create tools catered to specific bioconjugation needs. In 2017, Buchwald and Hooker demonstrated the efficient radio-labeling of unprotected peptides using dihaloarene-derived palladium complexes that can first arylate cysteine peptide and then enable [11C]cyanide coupling.[171] Later in 2018, Buchwald and Pentelute created palladium-based bi-functional linkers for both inter- and intra-molecular crosslinking of cysteine and lysine residues in peptides and proteins.[172]
Transition metal-catalyzed arylation reactions have also been developed to cysteine bioconjugation. In 2016, Messaoudi showed that metal-mediated S-arylative bioconjugation reactions can be performed using catalytic amounts of XantPhos-based aminobiphenyl mesylate palladium pre-catalysts (Figure 29).[173] Short unprotected peptides could be converted into the corresponding S-aryl bioconjugates using only 2% of the palladium precatalyst in yields greater than 80%. Trastuzumab antibody was also labeled following pre-reduction with TCEP, however, the efficiency of this catalytic process and the resulting drug-to-antibody ratio still need to be investigated. It is also essential to further optimize the catalyst system to avoid using large amounts of precatalysts in more challenging bioconjugations, as the aryl group on the pre-catalyst can also be transfered to the biomolecule. In 2018, Molander developed Ni/photoredox-catalyzed arylation for cysteine conjugation. Notably, the reaction can be performed at scales ranging from micrograms to grams, enabeling high throughput optimization of reaction conditions as well as scalable arylation of unprotected peptides. It remains to be seen whether this process can be applied more generally in aqueous buffers with more complex protein substrates.[174]
Figure 29.
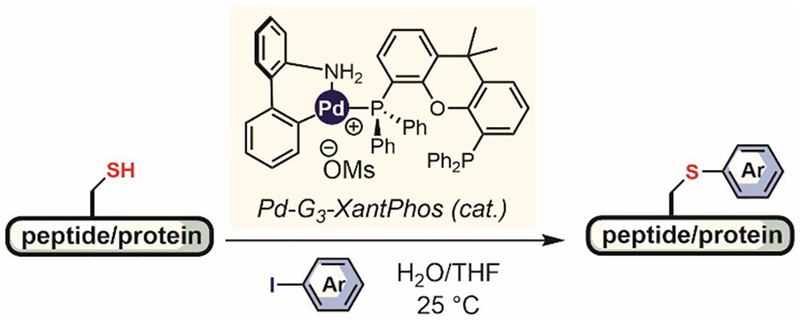
Palladium-catalyzed cysteine arylation.
Chemo- and regio-selective metal site-guided cysteine arylation
An important challenge in the bioconjugation field is to develop efficient reactions with tunable site-selectivity.[44] Organometallic complexes provide a number of promising entry points into the development of site-specific bioconjugation approaches, including the variation of the biomolecule microenvironment, metal/ligand combinations, and leaving groups.
Davis and colleagues used the protein microenvironment for the site-specific arylation of cysteine residues proximal to endogenous metal-binding sites in metalloenzymes.[175] They used a known metalloenzyme — mannosyl-glycerate synthase — to demonstrate that the arylation reaction could be directed with high regioselectivity to the Cys-233 near the endogenous DxD metal-binding motif without affecting the three other cysteine residues on the same protein molecule (Figure 30). While this work highlights the potential of organometallic catalysts for site-specific bioconjugation, the relatively high reaction temperature (65°C) used can lead to protein denaturation and therefore limits the breadth of potential applications of this method.
Figure 30.
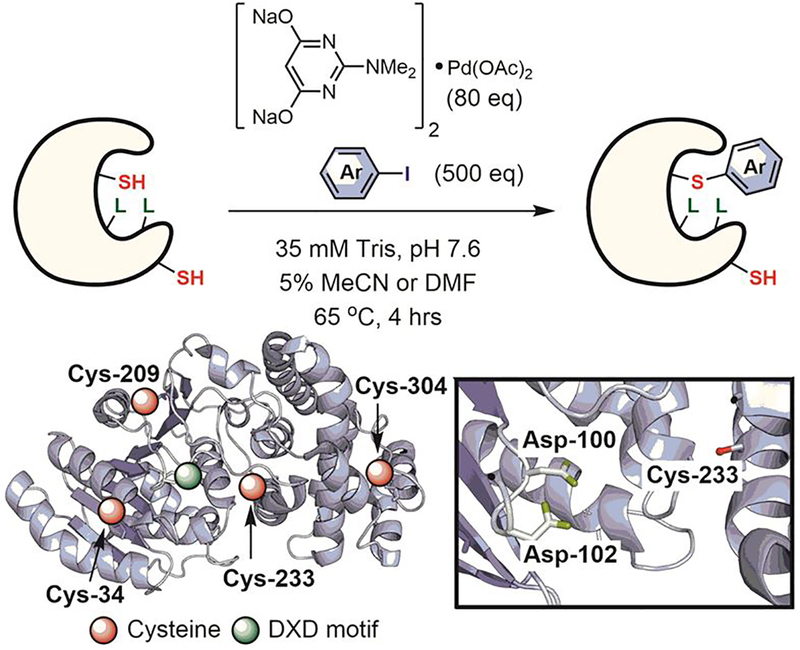
Metal-site guided cysteine arylation.
3. Lysine Arylation
Lysine is one of the most abundant amino acids in the human proteome. Protein surfaces usually have more than one solvent-exposed lysine, making it difficult to site-selectively modify one lysine side chain among many. The lysine ε-amino group is more nucleophilic than the N-terminal amine. However, the protonated form of the lysine ε-amino group (pKa ≈ 10) is less acidic than that of the N-terminal amine (pKa ≈ 8). When both amines are neutral at a higher pH, the lysine ε-amino group can be selectively modified without reaction on the protein N-terminus. At a lower pH, however, the lysine ε-amines are protonated while the free N-terminal amino group can be modified.
Bioconjugation on the ε-amine of lysine is a routine strategy to chemically modify proteins when site-selectivity is not a strict requirement.[35,42] Classic methods use electrophiles such as activated esters, isocyanates, and sulfonyl chlorides. Many lysine-based bioconjugates are used as vaccines and biotherapeutics. For example, the first FDA-approved PEGylated protein therapeutic ADAGEN® was made from conjugation of PEG polymers to lysine ε-amines in adenosine deaminase using succinic esters.[176]
N-arylation is a powerful approach to construct N–C(sp2) bonds in numerous drug molecules and complex natural products. A wide range of electron-deficient aryl halide electrophiles are used to modify amines through nucleophilic aromatic substitution (SNAr) reactions. More routinely used methods for the arylation of amines are transition-metal catalyzed cross-coupling reactions such as the Buchwald-Hartwig reaction,[36,177,178] the Chan-Lam reaction,[179–181] and Ullmann-Goldberg coupling reactions.[182–185] Many aryl halides and aryl boronic acids are commercially available, and provide a wide selection of structures for amine modification using these metal-catalyzed reactions.
Compared to its extensive development for small-molecule synthesis, N-arylation has been much less explored for the modification of large biomolecules. One of the early examples of N-arylation of biomolecules is the use of Sanger’s reagent for protein sequencing (Figure 31).[186] In 1945, Sanger reported the reaction between 2,4-dinitrofluorobenzene (DNFB) and the protein N-terminal amino groups. The DNFB-modified proteins were hydrolyzed under acidic conditions, generating dinitrophenyl-amino acids that were identified through either chromatographic or colorimetric assays.[186] The selectivity of 2,4-dinitrofluorobenzene for the amino group is moderate, as cysteine thiol, histidine imidazole, and tyrosine phenol groups can also react with DNFB.
Figure 31.
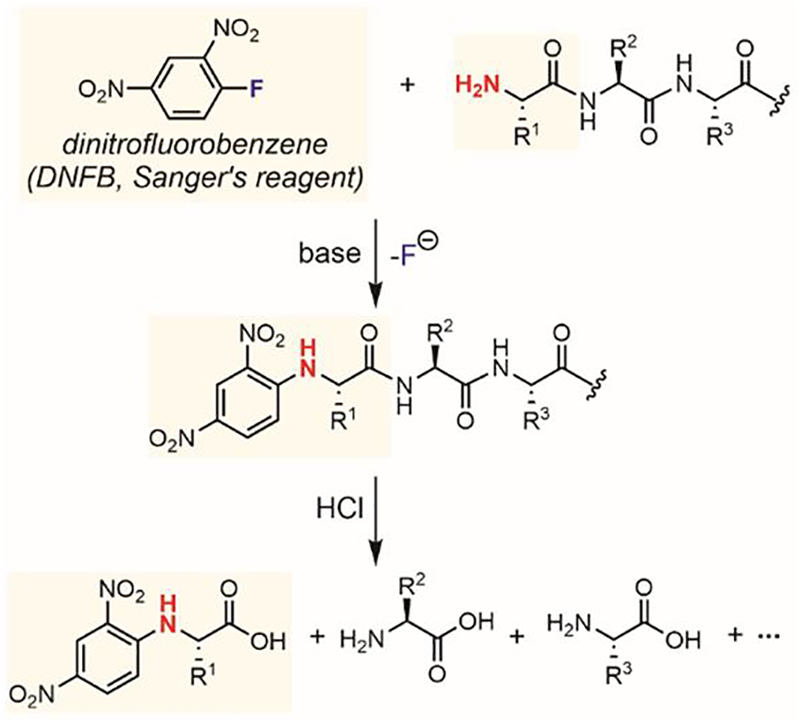
Sanger’s reagent for peptide sequencing.
3.1. Chemoselective lysine arylation via SNAr chemistry
In 1972, Welz used fluoronitrobenzene reagents that are structurally similar to Sanger’s reagent to label lysine ε-amino groups in proteins (Figure 32). These reagents did not distinguish lysine ε-amino groups from the N-terminal amino group, resulting in non-selective labeling of both sets of amines in proteins. Sutton used a trimethylammonium nitrofluorobenzene reagent to arylate bovine insulin, in order to introduce positive charges into the protein aimed at increasing its solubility and crystallizability (Figure 32A).[187] The arylation reagent is not specific to amines because it also arylates two of the four tyrosines in bovine insulin. Ladd developed a similar approach for protein PEGylation (Figure 32B).[188] A PEG derivative of fluoronitrobenzene was conjugated to bovine superoxide dismutase. The resulting PEGylated protein had an average of 8.8 PEG modifications per protein and retained full enzyme activity. These early examples of fluoronitrobenzene-based electrophiles showed promise for the arylation of proteins under mild conditions, albeit with moderate chemoselectivity and poor regioselectivity.
Figure 32.
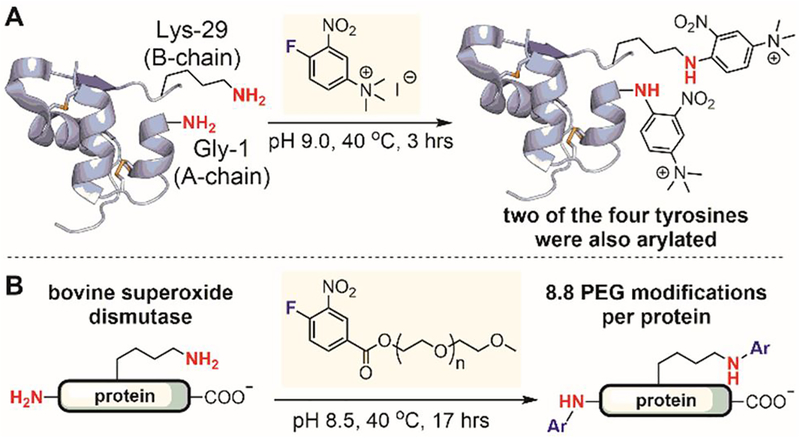
Fluoronitrobenzene reagents for protein lysine modification. (A) Labeling of bovine insulin with 1-fluoro-2-nitro-4-trimethylammoniumbenzene iodide improved the hydrophilicity of the proteinand made it readily crystallizable. (B) PEGylation of proteins using a PEGylated ester of 4-fluoro-3-nitrobenzoic acid.
In 2016, Pentelute reported a series of aryl halides for peptide lysine modification.[189] The SNAr reactivity of five fluorinated aromatic electrophiles and two triazine-based electrophiles (Figure 33) was evaluated for arylation of the lysine ε-amino groups in unprotected peptides. Because of the lower nucleophilicity of amines compared to thiols, a slightly elevated temperature (37°C) and prolonged reaction time (up to 24 hours) were needed to generate the arylated product. The decafluorobiphenylsulfone (Ar8) was found to be the most reactive reagent for lysine arylation, so with it, the desired arylated product was formed in high yields at room temperature within four hours. The electrophile Ar8 is selective for lysine arylation and is not reactive with tyrosine, arginine, and histidine residues under the developed reaction conditions. Cysteine was not compatible with the reaction conditions because the thiol group reacted faster than the lysine amine with the electrophile.
Figure 33.
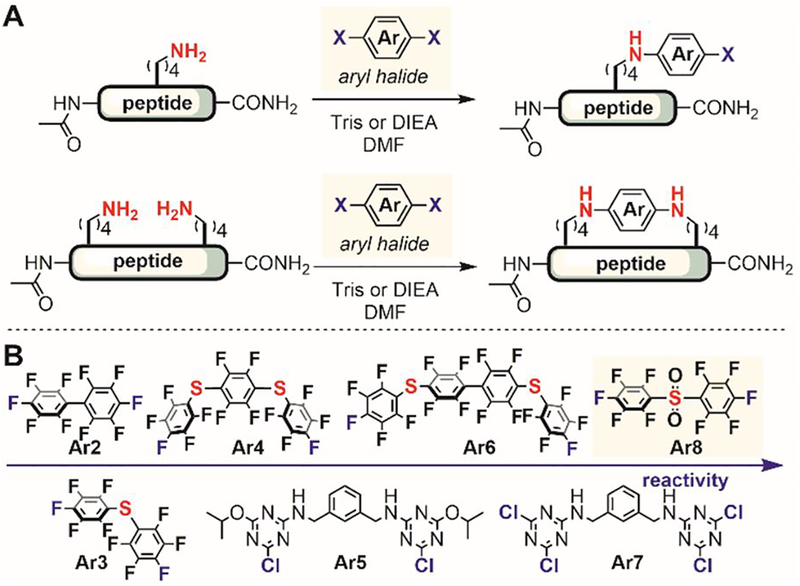
Lysine SNAr arylation for peptide modification. (A) Arylation of a single lysine (top) and cyclization of two lysines (bottom) of unprotected peptides using aryl halides. (B) Arylation reactivity of various aryl halides.
Unprotected peptides containing a range of spacing groups between two lysines were efficiently stapled by Ar8.[189] The lysine-stapled peptides showed improved stability against oxidation and bases compared to the cysteine-stapled counterparts. Furthermore, the stapled peptide variant of a p53-derived peptide inhibitor of MDM2 showed improved stability against proteases, increased binding to MDM2, and improved cellular uptake compared to a linear peptide analog.[189]
3.2. Metal-mediated chemoselective lysine arylation
Transition metal-mediated, cross-coupling reactions are highly useful for constructing C-N bonds in small molecules.[39,40] Importantly, both reactivity and selectivity in such metal-mediated processes can be tuned through rational ligand design. In 2015, Buchwald and Pentelute developed new organometallic palladium complexes for lysine arylation in unprotected peptides (Figure 34) in organic solvents.[190] Comparing oxidative addition complexes derived from several biarylphosphine ligands showed that the t-BuBrettPhos (Figure 34C) was the optimal ligand for efficient arylation of lysine ε-amino groups at room temperature in polar organic solvents. The reaction was chemoselective for lysine; most oxygen and nitrogen protein nucleophiles including those contained in Ser, Tyr, Met, His, Trp and Asn were unreactive under the developed reaction conditions. However, cysteine reacted much faster than lysine under the reported conditions. Some competitive reactivity was seen with peptides containing an Arg or a C-terminal amide. The N-terminal amine was more nucleophilic than the lysine side chain and selective arylation was observed for the N-terminal amine over the lysine ε-amine. Various small molecules were conjugated to peptide lysine side chains using the developed N-arylation approach (Figure 34A), including stapled peptides from bispalladium complexes (Figure 34B). Further optimization of the ligand could potentially enable palladium-mediated N-arylation in aqueous media and expand the scope of this reaction to more complex protein targets.
Figure 34.
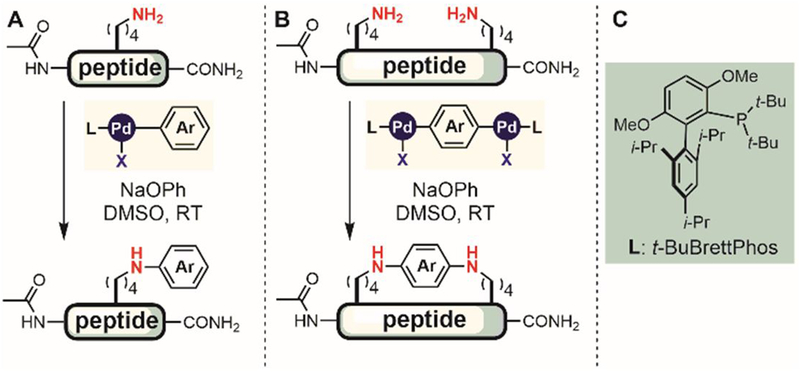
Palladium-mediated lysine arylation.
The challenge of site-selective N-arylation can also be tackled by exploiting recognition and directing effects of protein microenvironments. In 2016, Ball reported a histidine-directed method to site-selectively arylate amide in the protein backbone (Figure 35).[191] Histidine residues in peptides and proteins were found to direct the oxidative coupling of aryl boronic acids and boronates to a proximal backbone amide nitrogen through a Chan-Lam-type cross-coupling reaction. In this study, the reaction proceeded under mild aqueous conditions, enabling backbone modification of histidine-containing proteins. Lysozyme was site-selectively modified with several bioconjugation and affinity handles utilizing its native His-15 as a directing group to promote the arylation of the backbone amide of Arg-14 (Figure 35). This work demonstrates how the native residues in proteins and peptides can be harnessed to direct site-selective chemistry in metal-mediated processes.
Figure 35.
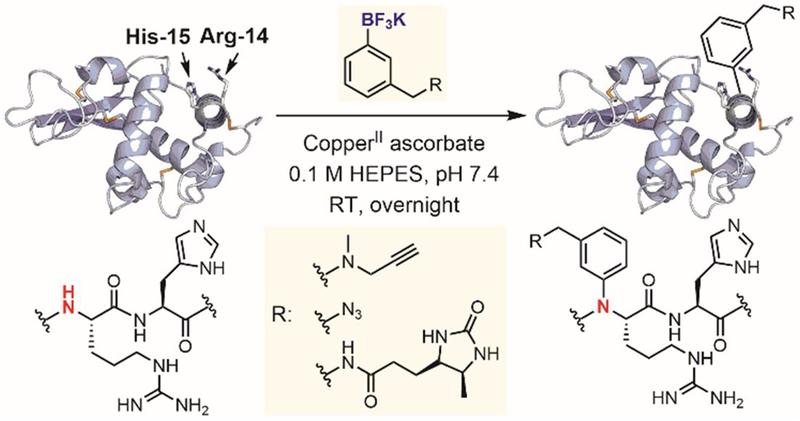
Copper-mediated N-arylation for peptide backbone modification.
4. Arylation of Other Amino Acid Residues
Bioconjugation reactions have been developed to modify amino acids other than cysteine and lysine. The nucleophilic side chains of tyrosine and tryptophan are major targets for arylation reactions on canonical amino acids. Genetic code expansion enables incorporation of various unnatural amino acids into proteins, facilitating various methods for efficient and robust arylation chemistry for site-selective protein modification.
4.1. Arylation of other canonical amino acid residues
Tyrosine
In 2016, Weerapana identified an aryl halide that reacted in a tyrosine-selective manner by profiling the proteome-reactivity and selectivity of a small library of aryl electrophiles (Figure 36).[192] Weerapana identified a dichlorotriazine compound bearing an L-leucine methyl ester moiety to selectively arylate Tyr-108 of glutathione S-transferase P1 (GSTP1). This compound, termed LAS17, selectively inhibited GSTP1 in a concentration-dependent manner in cell lysate. The site of covalent modification was confirmed through extensive mutation studies and mass spectrometry analysis. Weerapana proposed that the covalent arylation on Tyr-108 blocked a GSTP1 glutathione (GSH) binding site. Because GSTP1 was found to promote tumorigenesis and drug resistance in cancer cells, the use of LAS17 as a covalent inhibitor of GSTP1 showed some promise as a chemical probe to further investigate the biological role of GSTP1 and as the basis to develop potential cancer therapeutics.
Figure 36.
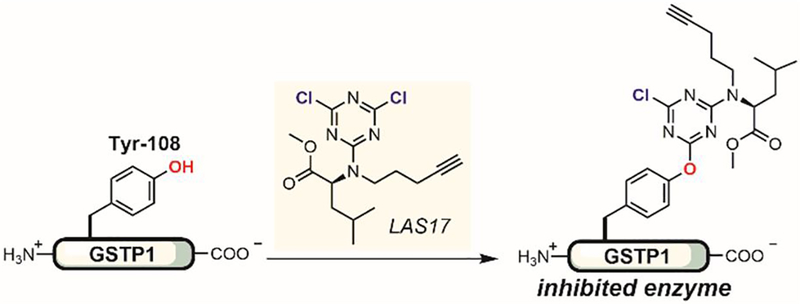
Tyrosine-selective dichlorotriazines as covalent protein inhibitors.
Besides the hydroxyl group, the o-carbons (C-3 and C-5) on the phenol ring of tyrosine can also be used for bioconjugation. In 1995, Kodadek reported that a Ni(II)-tripeptide (NH2-Gly-Gly-His-COOH) complex and oxidants (e.g., monoperoxylphthalic acid) were able to crosslink proteins presumably through oxidative generation of a tyrosyl radical that coupled to another proximal tyrosine residue.[193] Similar protein crosslinking processes can also be mediated by photo-induced single electron transfer with ammonium persulfate and tris(2,2’-bipyridyl)ruthenium(II) or palladium(II) porphyrins.[194,195] The later approaches are promising because they eliminated the use of peracid and associated by-products resulting from non-selective oxidations. In 2004, Finn showed that the tyrosyl radical can be intercepted by cystine analogues, enabling site-selective modification of viral capsid proteins at tyrosine residues.[196]
Heinrich developed a radical-mediated approach to generate 3-aryltyrosine residues in peptides (Figure 37).[197] Using an aryl diazonium salt and titanium(III) chloride under acidic conditions, the tyrosine residue of a neurotensin peptide fragment could be regioselectively arylated. The proposed radical-mediated mechanism is unique as previous approaches using aryl diazonium salts to modify tyrosine residues generated diazo linkages instead.[198] This method enabled quick generation of 3-aryltyrosine analogues of neurotensin for testing their activity against neurotensin receptor subtype 2.
Figure 37.
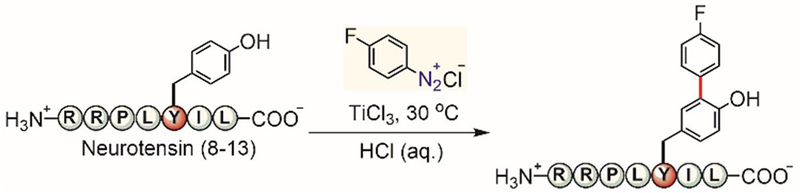
Radical-based arylation of tyrosine residues in peptides.
Other metal-mediated approaches have also been developed to arylate tyrosine residues. The versatility of transition-metal mediated arylation reactions was illustrated in the site-selective arylation of a peptide containing tryptophan, dehydroalanine (Dha), and tyrosine (Figure 38).[199] Willis and Frost reported using palladium and rhodium catalysts to site-selectively label each individual residue in the tripeptide (Trp-Dha-Tyr). Each process showed great chemoselectivity and functional group tolerance. First, they used XantPhos (L1) as the supporting ligand for selective N-arylation of the tryptophan indole moiety. With t-BuXPhos (L2), O-arylation was seen, indicating that fine-tuning of the ligands provides the means to selectively arylate different residues in peptides and possibly proteins. Moreover, rhodium-catalyzed arylation, with racemic 2,2’-bis(diphenylphosphino)-1,1’-binaphthalene (rac-BINAP, L3) as the ligand, was selective for reaction at the dehydroalanine unit, generating a tripeptide product as a single diastereomer.[200]
Figure 38.
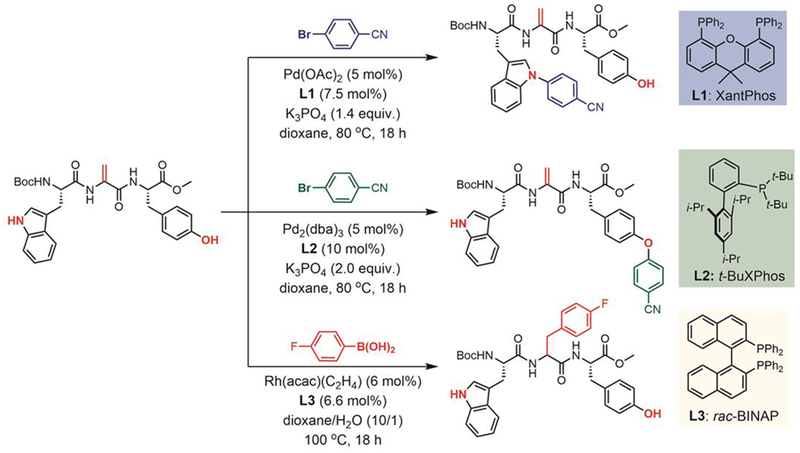
Metal-mediated site-selective arylation of dehydroalanine, tyrosine, and tryptophan.
Tryptophan
Transition metal-mediated C–H activation has become an important tool to introduce carbon-carbon and carbon-heteroatom bonds in complex small molecules.[201] Building upon previous C–H activation strategies for indole arylation,[202] several palladium- and ruthenium-catalyzed C–H activation processes were recently developed to modify the C-2 position of tryptophan residues.[203–209]
In 2010, Albericio and Lavilla first reported the conjugation of a variety of aryl iodides to tryptophan residues in peptides via palladium-catalyzed C–H activation reactions (Figure 39A).[203] The reaction was chemoselective for tryptophan in the presence of other amino acid nucleophiles including Tyr, Arg, His, Lys, Ser, Met, and Gln. This C–H activation strategy was also used to prepare stapled peptides containing Phe-Trp or Tyr-Trp linkages using 3-iodophenylalanine or 2-iodotyrosine (Figure 39D).[204,205] The stapling reactions were performed either on resin or in solution, providing access to a number of structurally diverse stapled peptides.
Figure 39.
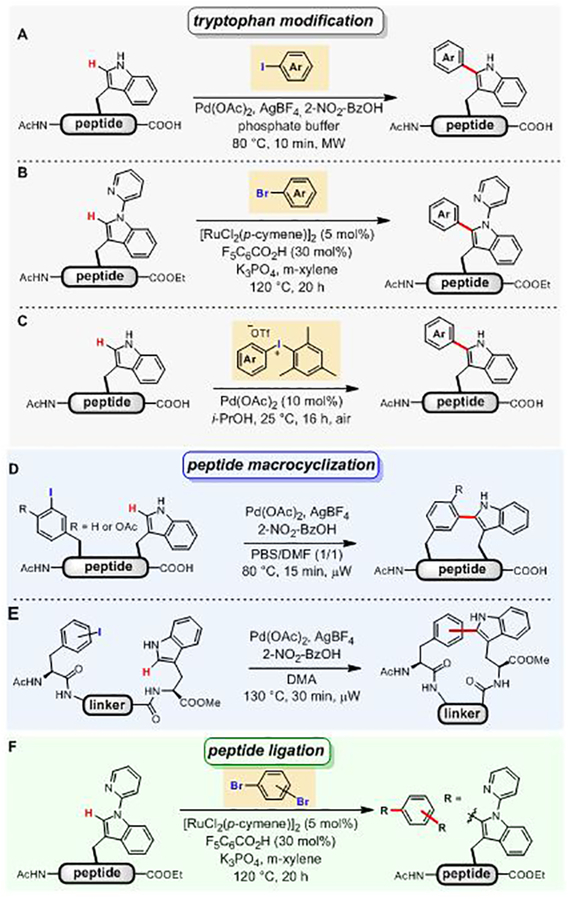
C–H activation for arylation of tryptophan residues in peptides. (A) Palladium-mediated C–H activation for the C-2-arylation of a tryptophan unit using aryl iodides. (B) Diaryliodonium salts as the aryl source for palladium-mediated arylation of tryptophan. (C) Ruthenium-catalyzed C–H activation for the modification of tryptophan in protected peptides using 2-pyridyl as a directing group. (D) Palladium-mediated C–H activation for the synthesis of peptidic macrocycles. (E) C–H activation for the formation of phenylalanine-tryptophan or tyrosine-tryptophan crosslinks. (F) Ruthenium-catalyzed peptide ligation using dibromoaryl linkers.
In 2012, a similar strategy was used by James for the palladium-catalyzed macrocyclization reaction through C–H arylation of a tryptophan side chain with iodophenylalanine (Figure 39E).[206] Macrocycles containing simple aryl and alkyl linkers were synthesized using Pd(OAc)2 as the catalyst, AgBF4 and 2-nitrobenzoic acid (2-NO2-BzOH). Variants with p- and m-iodophenylalanine were readily crosslinked with tryptophan residues. Substrates with o-iodophenylalanine were unreactive under the developed conditions presumably due to steric reasons.
In 2014, a mild and selective palladium-mediated C–H activation reaction was reported by Fairlamb for C-2-arylation of tryptophan residues (Figure 39C).[208,209] The authors used unsymmetrical diaryliodonium salts as the arylation reagent and Pd(OAc)2 as the catalyst to efficiently and selectively generate C-2 arylated tryptophan products at 25°C. The unusually mild conditions are noteworthy as almost all other C–H activation strategies for the arylation of tryptophan require elevated temperatures.
In 2017, Ackermann developed a ruthenium(II)-catalyzed C–H activation process for the C-2 arylation of tryptophan (Figure 39B).[207] A wide range of aryl bromides were coupled to tryptophan using a 2-pyridyl directing group. The reaction showed good chemoselectivity in the presence of added histidine, arginine, tyrosine, aspartic acid, and glutamine. Besides tryptophan modification, this ruthenium (II)-catalyzed reaction was also used for peptide ligation (Figure 39F) to create more complex peptide structures.
4.2. Arylation of oxidized selenocysteine electrophiles
Selenocysteine (Sec) is a close structural analogue of cysteine in which selenium replaces sulfur.[210] Selenols are much more nucleophilic than thiols mainly because of the greater polarizability of selenium compared to sulfur.[211] Moreover, alkyl selenols are quite acidic (pKa ≈ 5),[211] making the selenolate the dominant form of selenocysteine at physiological pH. Both factors make Sec an appealing bioconjugation handle for site-selective protein modification with electrophiles such as maleimides.[212] However, due to the tendency of Sec to undergo facile oxidation resulting in the formation of diselenides, the addition of a strong reducing agent is required.
In 2015, Pentelute and Buchwald reported an umpolung approach to the arylation of selenocysteine in unprotected peptides.[213] Instead of relying on the nucleophilic character of Sec, they explored the conjugation of aryl nucleophiles with oxidized Sec electrophiles (Figure 40). Inspired by previous reports on arylation of selenides in small molecules,[214,215] they devised an approach to arylate oxidized Sec using boronic acid reagents via a copper-mediated process. The reaction was chemoselective for Sec; most amino acids, except cysteine, were compatible with the reaction, enabling arylation of Sec in a complex peptide with a wide range of aryl boronic acids. The utility of this method needs to be further explored by applying it to modifications of large proteins.
Figure 40.
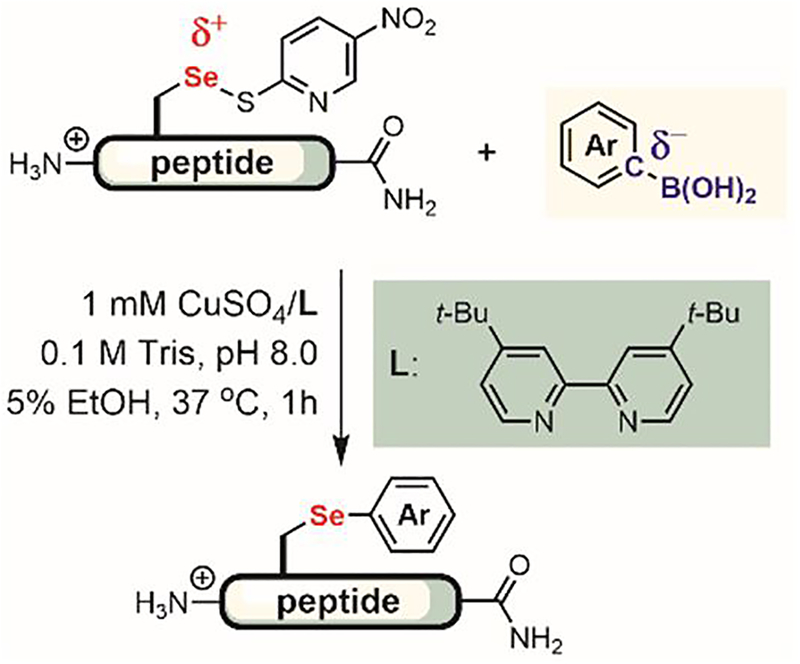
Copper-mediated umpolung arylation of oxidized selenocysteine.
4.3. Arylation of unnatural amino acids
Transition metal-catalyzed coupling reactions are key methods for arylation reactions on unnatural amino acids (Figure 41).[158,159] Palladium-catalyzed Suzuki-Miyaura, Sonogashira, and Heck reactions (Figure 41) have been developed to modify proteins both in the test tube and in living cells. Such metal-mediated bioconjugation processes usually consist of two steps (Figure 42A). First, either one of the coupling partners is installed on the protein of interest as an unnatural amino acid through chemical[216–219] or enzymatic[220] tagging, solid-phase peptide synthesis (SPPS),[221,222] or genetic code expansion[223–228] (Figure 42). Next, the other coupling partner is conjugated to the protein via transition metal-catalyzed coupling reactions.
Figure 41.
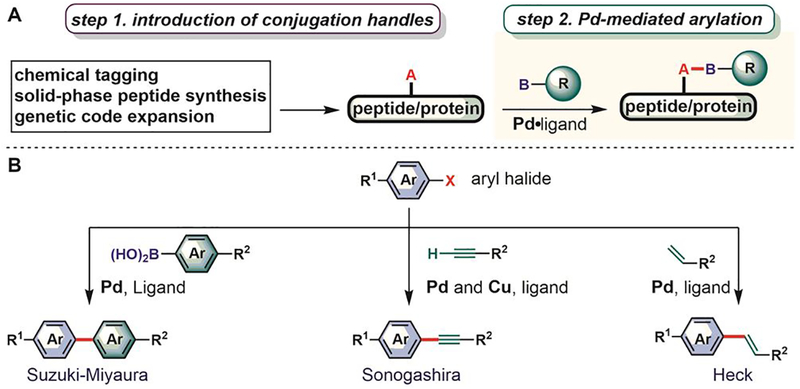
Metal-mediated arylation of unnatural amino acids. (A) A two-step process for palladium-mediated arylation of unnatural amino acids. Functional Groups A and B are partners for Pd-mediated cross-coupling reactions. (B) Key examples of palladium-mediated cross-coupling strategies applied to bioconjugation.
Figure 42.
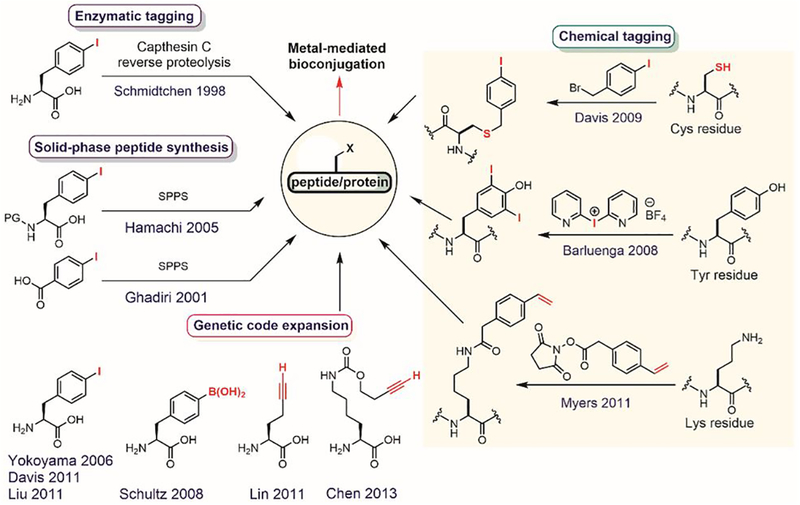
Methods for incorporating unnatural arylation handles into peptides or proteins.
These metal-mediated arylation reactions require the incorporation of unnatural handles (aryl halides, aryl boronic acids, alkynes, or alkenes) into the target protein (Figure 42). Chemical tagging and solid-phase synthesis were early approaches to introduce these handles into proteins. For small peptides amenable to solid-phase synthesis, multiple conjugation handles such as iodophenylalanine and iodotyrosine can be directly incorporated into the peptide chain.[221,222] Canonical amino acid residues in proteins can also be tagged with the desired handles through other conjugation chemistries (e.g., alkylation,[216] iodination,[218] or acylation[217]). Recently, the use of genetic code expansion has enabled the incorporation of these handles as unnatural amino acids.[223–228] Although site-selectivity is ensured in genetic code expansion, the number of unnatural amino acids that can be incorporated into a protein remains limited.
Palladium-ligand combinations determine the reactivity and selectivity of these metal-catalyzed arylation reactions (Figure 43). Initial reports using phosphine-based ligands (Figure 43, L1 and L2)[218,220], Na2PdCl4,[221] or Pd-DBA[224] had limited success because of the low efficiency of these catalysts and the harsh reaction conditions employed. The utilization of non-phosphine ligands (Figure 43, L3-L8)[227,229,230] or Pd(NO3)2 [228] enabled highly efficient arylation of unnatural amino acids on proteins both in vitro and in vivo.
Figure 43.
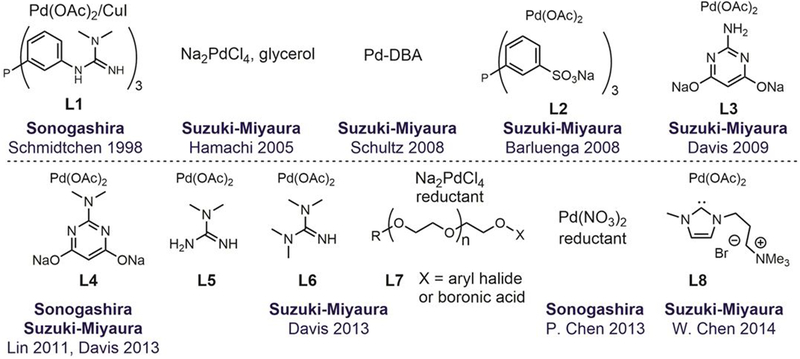
Palladium sources and ligands used for the metal-mediated arylation of unnatural amino acids.
In 1998, Schmidtchen first reported the palladium-catalyzed Sonogashira reaction to modify peptides in aqueous media.[220] A water-soluble guanidino aryl-phosphine ligand (Figure 43, L1) was used in combination with palladium and copper co-catalysts to modify 4-iodophenylalanine enzymatically installed on a small peptide. Later in 2005, Hamachi reported the Suzuki-Miyaura reaction of small chemically synthesized proteins in aqueous buffers using Na2PdCl4 as the catalyst.[221] A significant amount of glycerol (50% v/v) and slightly elevated temperature (40°C) were required to achieve >90% arylation yields. In 2007, Yokoyama and Tachibana first reported the Sonogashira and Heck reactions between terminal alkyne or alkene probes with 4-iodophenylalanine on the protein surface.[219] However, the use of 10% DMSO as a co-solvent was required and low conversions were observed using a Pd(OAc)2-triphenylphosphine-3,3’,3”-trisulfonic acid trisodium salt (TPPTS) catalyst system. Schultz reported the first example of the Suzuki-Miyaura reaction of expressed proteins with 4-boronophenylalanine incorporated via genetic code expansion in 2008.[224] Although the ability to genetically encode such arylation handles greatly broadened the scope of the reaction, the high temperature (70°C) and low efficiency of the Pd-DBA catalyst limited the application of this method. These early examples demonstrated the viability of palladium-mediated bioconjugation reactions but also highlighted the challenge of applying such reactions to complex protein targets.
In 2009, Davis reported a greatly improved Suzuki-Miyaura bioconjugation method using palladium acetate in combination with the sodium salt of 2-amino-4,6-dihydroxypyrimidine (Figure 43, L3) as the ligand (Figure 44). The aryl iodide was introduced into the protein through an alkylation reaction of a cysteine residue. Full conversion (>95%) within 30 minutes was achieved at 37°C in a phosphate buffer (pH 8.0) with a number of aryl, heteroaryl, and vinyl boronic acids.[216] However, the use of a large amount of palladium (50 equivalents) combined with the ability of proteins to ligate metals resulted in poor mass spectrometry data for the conjugates. This problem was later solved by surveying a number of potential metal scavengers for palladium removal, with 3-mercaptopropionic acid identified as the best candidate.[225] The aryl iodide handle was also introduced through genetic encoding of L-4-iodophenylalanine, which set the stage for applying this reaction to label proteins in living systems.[225]
Figure 44.
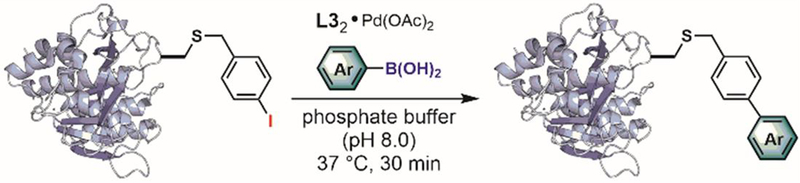
Suzuki-Miyaura arylation of a cysteine mutant of subtilisin Bacillus lentus.
To date, many ligands have been utilized to improve the palladium-mediated Suzuki-Miyaura reactions for protein modification (Figure 43). Ligands including the N,N-dimethyl-2-amino-4,6-dihydroxypyrimidine (L4),[227,229] simple guanidines (L5 and L6),[229] polyethylene glycol (PEG) (L7),[229] and N-heterocyclic carbene (NHC) (L8)[230] were shown to promote Suzuki-Miyaura arylation in water. Unlike other ligands that assist the cross-coupling reaction, the PEG ligand is unique as a self-ligating reagent for protein PEGylation. It is also worth noting that L4 has been shown by Lin to efficiently catalyze the copper-free Sonogashira reaction (Heck alkynylation) between alkynes on a protein surface and aryl iodide probes.[227]
Combining the high efficiency and selectivity of the Suzuki-Miyaura and Sonogashira reactions with genetic code expansion enabled their applications to labeling proteins in living systems. In 2011, Lin applied the copper-free Sonogashira reaction to labeling of ubiquitin in E. coli cells.[227] Around the same time, Davis showed that their Suzuki-Miyaura reactions could be used to couple a fluorescent boronic acid to cell surfaces containing aryl iodides.[231] N-heterocyclic carbenes (NHCs) were also shown by Ma to be viable catalysts for the labeling of membrane proteins through Suzuki-Miyaura coupling with low-catalyst loadings.[230] Further advances were made by Chen who used Pd(NO3)2 to catalyze the arylation of alkyne-containing proteins inside live bacterial cells.[228] This method was used to selectively label and visulize type-III Secretion (T3S) toxin-OspF in live Shigella cells.
Isolated palladium complexes have also been explored as a vehicle to arylate alkynes and alkenes on proteins. Lin reported a series of N-phenylcarbamate palladacycles for conjugation to an alkyne residue in ubiquitin (Figure 45A).[232,233] The reaction rate of this conjugation is very rapid, with the fastest having a rate constant of 19,770 M−1s−1. Mass spectrometry analysis of the trypsin digestion of the labeled ubiquitin showed a modified peptide fragment with a mass consistent with a styrene product (Figure 45A). However, reactions of the palladacycles with a model peptide produced complex reaction mixtures. This disparity between the results of ubiquitin labeling and peptide modification suggests that more in-depth studies of the reaction mechanism is required before further applications of the reaction to other protein targets.
Figure 45.
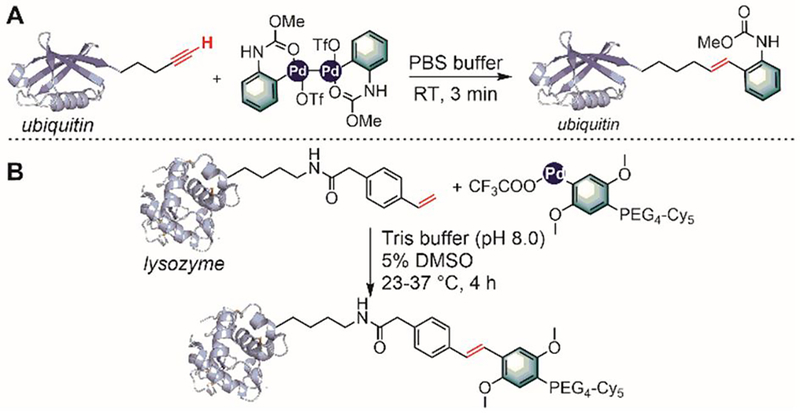
Isolated palladium complexes for the arylation of protein alkynes (A) and alkenes (B).
Heck-type reactions of proteins containing vinyl aryl groups have been reported (Figure 45B) by Myers in 2011.[217] They developed storable palladium complexes formed via decarboxylative palladation of aryl carboxylic acids and demonstrated their utility in bioconjugation of functionalized lysozyme under mild conditions. In 2018, Roelfes reported modification of dehydroalanine in peptides and proteins using aryl boronic acids and Pd(EDTA)(OAc)2 as the precatalyst to generate both Heck-type and conjugate addition products.[234]
5. Arylation of Nucleic Acids
Chemically modified oligonucleotides are widely used for applications including biosensing,[235,236] nanotechnology,[237] medicine,[238] and catalysis[239]. Oligonucleotides have fewer monomeric building blocks compared to proteins. This simplifies the range of chemistry necessary for bioconjugation. Traditional chemical methods have utilized either solid-phase synthesis[240] or enzyme-catalyzed reactions[241] to modify nucleic acid oligomers.
Recently, Suzuki-Miyaura coupling has emerged as a new strategy for post-synthetic modification of nucleic acids.[51] These methods use synthetically incorporated aryl halides as the handle for site-selective conjugation of aryl boronic acids to both single- and double-stranded DNA. The required aryl halides were incorporated through either unnatural phosphoramidites during solid-phase synthesis (Figure 46) or through direct coupling of aryl halides to nucleic acids through amide bond formation (Figure 47).
Figure 46.
Post-synthetic arylation of single-stranded DNA (ssDNA). 8-bromoguanidine (A) and 5-iodouracil (B) were introduced into single-strand DNA (ssDNA) through solid-phase DNA synthesis using designed phosphoramidites.
Figure 47.
Post-synthetic arylation of double-stranded DNA (dsDNA). Aryl or heteroaryl halides were installed on double-stranded DNA (dsDNA) through either amide bond formation (A) or SNAr reaction (B). The aryl halide-dsDNA conjugates were then arylated using a Suzuki-Miyaura coupling reaction.
5.1. Post-synthetic arylation of single-stranded oligonucleotide
In 2011, Manderville first applied Suzuki-Miyaura cross-coupling to the modification of 2’-deoxyguanosine (dG) in single-stranded oligodeoxynucleotides (Figure 46A).[242] The aryl halide conjugation handle was introduced into synthetic DNA using 8-Br-dG phosphoramidite. Using palladium catalysis, a range of aryl boronic acids were site-selectively coupled to the C-8 site of dG. The dG-arylated product was then utilized to study DNA damage caused by radical addition reactions at the C-8 site of dG.
A milder Suzuki-Miyaura arylation procedure was developed by Davis in 2013 to modify 5-iodo-2’-deoxyuridine (5-I-dU) in single-stranded DNA (ssDNA) (Figure 46B).[184] The use of 2-aminopyrimidine-type ligands enabled Suzuki-Miyaura arylation at 37°C under buffered aqueous conditions. Phenyl boronic acid and 3-furyl boronic pinacol ester were used to site-selectively arylate the 2’-deoxyuridine in the ssDNA. Importantly, vinyl boronate esters were also conjugated to the ssDNA under the developed reaction conditions, enabling several handles including a benzophenone, a diazirine, a pyrene, a sugar, and an azobenzene to be appended. The method was used to synthesize an ssDNA probe that contained biotin, diazirine, and 5-hydroxymethylcytosine (5-hmC). This probe was used to identify 5-hmC-binding proteins in the HeLa cell lysate through photo-crosslinking, affinity enrichment, and subsequent mass spectrometry analysis.
In 2018, Srivatsan applied Davis’s catalyst system to the modification of riobonucleic acids (RNAs). Iodouridine-labeled RNA was generated through reverse-transcription using 5-iodouridine triphosphate (IUTP) and T7 RNA polymerase. A wide variety of RNA oligonucleotides were modified with fluorescent and affinity probes as well as fluorogeneic moieties that are sensitive to the nucleoside’s chemical environment within the RNA sequence.[243]
Besides Suzuki-Miyaura cross-coupling, palladium-mediated Buchwald-Hartwig-Migita cross-coupling has also been used by Defrancq and Messaoudi in 2018 to generate glyco-oligonucleotide conjugates.[244] Similar to the conditions developed for their previously reported protein modification method (Fig. 29), G3-XantPhos was used as the ligand to enable the palladium-mediated conjugation of thiolglycosides to oligonucleotides bearing a 5-iodo-2’-deoxyuridine bases.
5.2. Post-synthetic arylation of double-stranded oligonucleotide
In 2015, Suzuki-Miyaura cross-coupling was also used by Ding and Clark to synthesize DNA-encoded small molecule libraries. The authors installed aryl halides on dsDNA by coupling aryl halide carboxylic acids to the terminal amines in dsDNA (Figure 47A).[245] These aryl halide handles were further conjugated to aryl or heteroaryl boronic acids and esters using Pd(PPh3)4 as the catalyst. This catalyst system was applied to the synthesis of a 3.5-million-member DNA-encoded library from which potent hits for phosphoinositide 3-kinase α (PI3Kα) were identified.[246] In 2016, Ding and Clark reported a new catalyst system using [(t-Bu)2P(OH)]2PdCl2 (POPd) as the palladium source and sodium 2’-(dicyclohexylphosphino)-2,6-dimethoxy-[1,1’-biphenyl]-3-sulfonate (sSPhos) as the ligand for the on-DNA Suzuki-Miyaura coupling of challenging phenyl chlorides and pyrimidinyl chlorides (Figure 47B).[247] Increased conjugation yields were observed with this catalyst system for various aryl and heteroaryl boronic acids and esters.
6. Summary and Outlook
The emerging arylative bioconjugation strategies enable formation of nucleophile-sp2 carbon bonds in biomolecules. Compared to the robust and diverse arylation methods available for small molecule synthesis, arylation chemistry for bioconjugation is still in its infancy.
For the existing arylative bioconjugation reactions, further developments are needed to improve their selectivity, reaction rates, and substrate scope. In particular, site-specific arylation reactions are highly desirable to generate homogeneous bioconjugates. The π-clamp[111] shows how the microenvironment within a small peptide sequence can promote site-selective chemistry. We envision extending the same concept to other perfluoroaryl linkers and arylation chemistry beyond perfluoroarylation. Such tags will likely be discovered through screening peptide libraries as shown by recent proof-of-concept studies.[111,248] This will provide a set of small genetically encodable peptide tags for site-specific protein labeling with different aryl modifications. With sufficient selectivity, such peptide tags will provide diverse applications for site-specific labeling of proteins in complex biological environments such as within living cells.
Arylation reactions are desired for natural amino acid residues other than lysine and cysteine. Targeting other nitrogen- and oxygen-based nucleophiles (e.g., residues of His, Ser, Thr, and Tyr) is highly valuable and promising for generating structurally diverse protein conjugates. Such nucleophiles are, in most cases, much less reactive compared to the cysteine thiol group or the lysine ε-amine, presenting significant challenges for developing arylation chemistry targeting these sites. One strategy is to tune the reactivity and selectivity of SNAr arylation reactions by changing the electronic and steric properties of the arylation reagents, as demonstrated by recent reports on arylation of residues other than cysteine using perfluoroheteroaromatic reagents.[249] It is also possible to discover new ligands for copper- or palladium-catalyzed arylation reactions that form C(sp2)-O bonds (for Ser, Thr, and Tyr) and C(sp2)-N(sp2) (for His) bonds on proteins. Another strategy is to utilize protein environments and recognition elements to promote otherwise difficult arylation chemistry, as demonstrated in Ball’s method for arylation of amides in protein backbones.[191]Notably, Chen and Liu recently developed an intramolecular palladium-catalyzed C(sp3)-H arylation strategy for the synthesis of peptide macrocycles, which provided a potential approach to modification of aliphatic chains in amino acid residues.[250]
One obvious challenge in using transition metal-catalyzed or -mediated processes is the removal of the potentially toxic metals following the reaction. While a number of reagents and resin-based techniques have been developed for small-molecule applications, much less is known about their application for protein reactions. Developing catalytic reactions to reduce the amount of metal used should aid the metal removal processes. Notably, recent developments in photoredox catalysis may provide avenues for metal-free arylation processes.[251]
The development of highly selective arylation reactions will enable their application beyond protein and nucleic acid labeling. The modified boron clusters developed by Spokoyny highlighted the utility of selective arylation reactions for creating new types of hybrid nanomaterials. We therefore envision that arylation reactions can be applied to post-synthetic modifications of nanoparticles, surfaces, and polymers.[252,253]
Arylation chemistry provides multiple benefits both in terms of reactivity and properties of the resulting structures as compared to traditional alkylation reactions. For instance, both palladium and gold-based organometallic arylation reagents are found to be reactive to cysteine at low pH (2–5) where traditional alkylation chemistry falls short.[166,167] Moreover, initial studies have shown that certain aryl linkages are more chemically stable against acids, bases, oxidants, and thiol nucleophiles than alkyl-based cysteine conjugates[167], making it attrative to create new antibody-drug conjugates with stable aryl linkers. Lastly, given the metal-mediated arylation chemistry is mechanistically different from many other traditional bioconjugation reactions, it is possible to develop arylation reactions that are orthogonal to traditional reactions in terms of both reactivity and selectivity, providing access to homogeneous bioconjugates with multiple modifications.
The biological benefits (and/or drawbacks) of aryl modifications remain to be further explored. While in one study[117] Pun found that a perfluoroaryl-stapled macrophage-binding peptide have superior binding affinities compared to peptides stapled by other chemistries (e.g., disulfide and amide formations), the effects of staples on the binding properties of peptides need to be investigated on a case by case basis. Although several studies[114,116,254] have successfully applied aryl conjugates to living cells, it remains to be determinted how specific aryl modifications affect the properties of biomolecules in a broader cellular and tissue context. Systematic studies of the biological properties (e.g., stability, toxicity, immunogenicity) of aryl modifications both in vitro and in vivo are necessary to providing guidelines to select aryl linkages for biomolecule modification.
Acknowledgements
We acknowledge National Institutes of Health for support under award number R01-GM58160 and R35-GM122483 for S.L.B., R01-GM110535 for B.L.P., and R35-GM124746 for A.M.S.. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. B.L.P. acknowledges MIT Startup fund, the Sontag Foundation, Alfred P. Sloan Fellwoship, Bristol-Myers Squibb, Amgen, and Novartis for funding. C.Z. is a recipient of the George Büchi Research Fellowship, the MIT Koch Graduate Fellowship in Cancer Research, and the Bristol-Myers Squibb Graduate Fellowship in Synthetic Organic Chemistry. A.M.S. acknowledges the University of California, Los Angeles (UCLA) Department of Chemistry and Biochemistry for start-up funds, 3M for a Non-Tenured Faculty Award, and Alfred P. Sloan Foundation for Research Fellowship in Chemistry. E.V.V. is a Pfizer Fellow of the Life Science Research Foundation. We thank Dr. Andrei Loas for helpful comments and edits. Members of the Buchwald and Pentelute groups are acknowledged for their continuing efforts for developing new bioconjugation chemistries.
Biographies

Chi Zhang received his Ph.D. from MIT in 2017 under the supervision of Prof. Brad Pentelute. He is currently a postdoctoral fellow in the labs of Prof. Ed Boyden and Prof. Brad Pentelute. His research focuses on developing tools for highly multiplexed protein analysis in complex biological systems. Chi is a recipient of the 2015 Bristol-Myers Squibb Fellowship in Synthetic Organic Chemistry, 2016 Regeneron Prize for Creative Innovation, 2016 Genentech Chemical Research Award, 2016 Alfred R. Bader Award for Student Innovation, and 2018 IUPAC-Solvay International Award for Young Chemists.

Ekaterina (Katya) Vinogradova received her Ph.D. from MIT in 2015 under the supervision of Prof. Stephen Buchwald. She is currently pursuing postdoctoral research as a Pfizer Fellow of Life Sciences Research Foundation in the laboratory of Prof. Benjamin Cravatt. Her research interests encompass development of new reagents, probes and chemoproteomic approaches for biomedical applications. Ekaterina is a recipient of 2016 IUPAC-Solvay International Award for Young Chemists, 2015 Division of Inorganic Chemistry of the ACS Young Investigator Award, and 2014 IPMI Bright Futures Graduate Student Award.

Alex Spokoyny is currently an Assistant Professor in Chemistry and Biochemistry at UCLA and a faculty member of the California NanoSystems Institute (CNSI). Prior to this he received a Ph.D. from Northwestern University in inorganic and materials chemistry and conducted a post-doctoral stint at MIT in chemical biology. His group’s research encompasses an interdisciplinary approach focusing on pressing problems in chemistry, biomedicine and materials science. Alex is a recipient of numerous national and international awards including the recent 2017 Alfred P. Sloan Research Fellowship in Chemistry, 2017 NIH/NIGMS Maximizing Investigators Research Award (MIRA) and 2016 Chemical and Engineering News (C&EN) Talented 12.

Bradley L. Pentelute is an Associate Professor of Chemistry at MIT. His reserach group focuses on the use of cysteine arylation to generate abiotic macromolecular proteins, the precision delivery of biomolecules into cells, and the development of fast flow platforms to rapidly produce polypeptides.

Stephen L. Buchwald is the Camille Dreyfus Professor and the Associate Head of the Chemistry Department at MIT. His research group has made contributions in the fields of organic synthesis and organometallic chemistry applied to a number of areas of chemistry.
References
- [1].Hermanson GT, Bioconjugate Techniques, Academic, 2013. [Google Scholar]
- [2].Surís A, Smith J, Powell C, North CS, Ann. Clin. Psychiatry 2013, 25, 33–40. [PMC free article] [PubMed] [Google Scholar]
- [3].Chari RVJ, Miller ML, Widdison WC, Angew. Chemie - Int. Ed 2014, 53, 3796–3827. [DOI] [PubMed] [Google Scholar]
- [4].Heinis C, Nat. Chem. Biol 2014, 10, 696–698. [DOI] [PubMed] [Google Scholar]
- [5].Chudasama V, Maruani A, Caddick S, Nat. Chem 2016, 8, 114–9. [DOI] [PubMed] [Google Scholar]
- [6].Xue L, Karpenko IA, Hiblot J, Johnsson K, Nat. Chem. Biol 2015, 11, 917–923. [DOI] [PubMed] [Google Scholar]
- [7].Cobo I, Li M, Sumerlin BS, Perrier S, Nat. Mater 2015, 14, 143–159. [DOI] [PubMed] [Google Scholar]
- [8].Lutz JF, Zarafshani Z, Adv. Drug Deliv. Rev 2008, 60, 958–970. [DOI] [PubMed] [Google Scholar]
- [9].Kairdolf BA, Qian X, Nie S, Anal. Chem 2017, 89, 1015–1031. [DOI] [PubMed] [Google Scholar]
- [10].Lang K, Chin JW, ACS Chem. Biol 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- [11].Saito F, Noda H, Bode JW, ACS Chem. Biol 2015, 10, 1026–1033. [DOI] [PubMed] [Google Scholar]
- [12].Meanst GE, Feeney RE, Bioconjugate Chem 1990, 1, 2–12. [DOI] [PubMed] [Google Scholar]
- [13].Olcott HS, Fraenkel-Conrat H, Chem. Rev 1947, 41, 151–197. [DOI] [PubMed] [Google Scholar]
- [14].Herriott RM, Adv. Protein Chem 1947, 3, 169–225. [DOI] [PubMed] [Google Scholar]
- [15].Fraenkel-Conrat H, Cooper M, Olcott HS, J. Am. Chem. Soc 1945, 67, 950–954. [Google Scholar]
- [16].Hoare DG, Koshland DE, J. Biol. Chem 1967, 242, 2447–2453. [PubMed] [Google Scholar]
- [17].Barman TE, Koshland DE, J. Biol. Chem 1967, 242, 5771–5776. [PubMed] [Google Scholar]
- [18].Fields R, in Methods Enzymol, 1972, pp. 464–468. [DOI] [PubMed] [Google Scholar]
- [19].Ellman GL, Arch. Biochem. Biophys 1959, 82, 70–77. [DOI] [PubMed] [Google Scholar]
- [20].Baker BR, Annu. Rev. Pharmacol 1970, 10, 35–50. [DOI] [PubMed] [Google Scholar]
- [21].Edman P, Begg G, Eur. J. Biochem 1967, 1, 80–91. [DOI] [PubMed] [Google Scholar]
- [22].Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC, Nature 1958, 181, 662–666. [DOI] [PubMed] [Google Scholar]
- [23].Afagh NA, Yudin AK, Angew. Chemie - Int. Ed 2010, 49, 262–310. [DOI] [PubMed] [Google Scholar]
- [24].Lin CW, Ting AY, J. Am. Chem. Soc 2006, 128, 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wollack JW, Silverman JM, Petzold CJ, Mougous JD, Distefano MD, ChemBioChem 2009, 10, 2934–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 15815–15820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cull MG, Schatz PJ, in Appl. Chimeric Genes Hybrid Proteins Part A Gene Expr. Protein Purif (Ed.: S.D.E.J.N.A. Jeremy Thorner), Academic Press, 2000, pp. 430–440. [Google Scholar]
- [28].Fernández-Suárez M, Baruah H, Martínez-Hernández L, Xie KT, Baskin JM, Bertozzi CR, Ting AY, Nat. Biotechnol 2007, 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Popp MW, Antos JM, Grotenbreg GM, Spooner E, Ploegh HL, Nat. Chem. Biol 2007, 3, 707–708. [DOI] [PubMed] [Google Scholar]
- [30].Wang L, Xie J, Schultz PG, Annu. Rev. Biophys. Biomol. Struct 2006, 35, 225–249. [DOI] [PubMed] [Google Scholar]
- [31].Liu CC, Schultz PG, Annu. Rev. Biochem 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- [32].Lang K, Chin JW, Chem. Rev 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- [33].Kolb HC, Finn MG, Sharpless KB, Angew. Chemie - Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- [34].Sletten EM, Bertozzi CR, Angew. Chemie - Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hermanson GT, in Bioconjugate Tech (Ed.: Hermanson GT), Academic Press, Boston, 2013, pp. 229–258. [Google Scholar]
- [36].Ruiz-Castillo P, Buchwald SL, Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Martin R, Buchwald SL, Acc. Chem. Res 2008, 41, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Burnett JF, Zahler RE, Chem. Rev 1951, 49, 273–412. [Google Scholar]
- [39].Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 2014. [Google Scholar]
- [40].Diederich F, Stang P, Metal-Catalyzed Cross-Coupling Reactions, John Wiley & Sons, 2008. [Google Scholar]
- [41].Zalatan DN, Du Bois J, Yu JQ, Shi Z, C-H Activation, Springer Berlin Heidelberg, Berlin, Heidelberg, 2010. [Google Scholar]
- [42].Boutureira O, Bernardes GJL, Chem. Rev 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- [43].Spicer CD, Davis BG, Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- [44].Hackenberger CPR, Schwarzer D, Angew. Chemie - Int. Ed 2008, 47, 10030–10074. [DOI] [PubMed] [Google Scholar]
- [45].DeGruyter JN, Malins LR, Baran PS, Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vinogradova EV, Pure Appl. Chem 2017, 89, 1619–1640. [Google Scholar]
- [47].Jbara M, Maity SK, Brik A, Angew. Chemie Int. Ed 2017, 56, 10644–10655. [DOI] [PubMed] [Google Scholar]
- [48].Bondalapati S, Jbara M, Brik A, Nat. Chem 2016, 8, 407–418. [DOI] [PubMed] [Google Scholar]
- [49].Krall N, da Cruz FP, Boutureira O, Bernardes GJL, Nat. Chem 2016, 8, 103–113. [DOI] [PubMed] [Google Scholar]
- [50].Koniev O, Wagner A, Chem. Soc. Rev 2015, 44, 5495–551. [DOI] [PubMed] [Google Scholar]
- [51].Messaoudi S, Defrancq E, ChemBioChem 2016, 426–431. [DOI] [PubMed] [Google Scholar]
- [52].Malins LR, Aust. J. Chem 2016, 69, 1360–1364. [Google Scholar]
- [53].Gong Y, Pan L, Tetrahedron Lett 2015, 56, 2123–2132. [Google Scholar]
- [54].Baslé E, Joubert N, Pucheault M, Chem. Biol 2010, 17, 213–227. [DOI] [PubMed] [Google Scholar]
- [55].Chalker JM, Bernardes GJL, Lin YA, Davis BG, Chem. - An Asian J 2009, 4, 630–640. [DOI] [PubMed] [Google Scholar]
- [56].Gunnoo SB, Madder A, ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- [57].Giles NM, Giles GI, Jacob C, Biochem. Biophys. Res. Commun 2003, 300, 1–4. [DOI] [PubMed] [Google Scholar]
- [58].Jacob C, Giles GI, Giles NM, Sies H, Angew. Chemie - Int. Ed 2003, 42, 4742–4758. [DOI] [PubMed] [Google Scholar]
- [59].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin D. a, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Price NC, Cohn M, Schirmer RH, J. Biol. Chem 1975, 250, 644–652. [PubMed] [Google Scholar]
- [61].Leesnitzer LM, Parks DJ, Bledsoe RK, Cobb JE, Collins JL, Consler TG, Davis RG, Hull-Ryde EA, Lenhard JM, Patel L, et al. , Biochemistry 2002, 41, 6640–6650. [DOI] [PubMed] [Google Scholar]
- [62].Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Marc Learned R, et al. , J. Biol. Chem 2002, 277, 19649–19657. [DOI] [PubMed] [Google Scholar]
- [63].Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S, J. Adv. Pharm. Technol. Res 2011, 2, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM, Nat. Med 2013, 99, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hughes TS, Giri PK, de Vera IMS, Marciano DP, Kuruvilla DS, Shin Y, Blayo A-L, Kamenecka TM, Burris TP, Griffin PR, et al. , Nat. Commun 2014, 5, 3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Brust R, Lin H, Fuhrmann J, Asteian A, Kamenecka TM, Kojetin DJ, ACS Chem. Biol 2017, 12, 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Johnson CM, Linsky TW, Yoon DW, Person MD, Fast W, J. Am. Chem. Soc 2011, 133, 1553–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Johnson CM, Monzingo AF, Ke Z, Yoon DW, Linsky TW, Guo H, Robertus JD, Fast W, J. Am. Chem. Soc 2011, 133, 10951–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Banerjee R, Pace NJ, Brown DR, Weerapana E, J. Am. Chem. Soc 2013, 135, 2497–2500. [DOI] [PubMed] [Google Scholar]
- [70].Shannon DA, Banerjee R, Webster ER, Bak DW, Wang C, Weerapana E, J. Am. Chem. Soc 2014, 136, 3330–3333. [DOI] [PubMed] [Google Scholar]
- [71].Qian Y, Weerapana E, Methods Mol. Biol 2017, 1491, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tucker MJ, Courter JR, Chen J, Atasoylu O, Smith AB, Hochstrasser RM, Angew. Chemie - Int. Ed 2010, 49, 3612–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tucker MJ, Abdo M, Courter JR, Chen J, Smith AB, Hochstrasser RM, J. Photochem. Photobiol. A Chem 2012, 234, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Courter JR, Abdo M, Brown SP, Tucker MJ, Hochstrasser RM, Smith AB, J. Org. Chem 2014, 79, 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Abdo M, Brown SP, Courter JR, Tucker MJ, Hochstrasser RM, Smith AB, Org. Lett 2012, 14, 3518–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Brown SP, Smith AB, J. Am. Chem. Soc 2015, 137, 4034–4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zhang D, Devarie-Baez NO, Li Q, Lancaster JR, Xian M, Org. Lett 2012, 14, 3396–3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Burnett JF, Zahler RE, Chem. Rev 1951, 49, 273–412. [Google Scholar]
- [79].Toda N, Asano S, Barbas CF, Angew. Chemie - Int. Ed 2013, 52, 12592–12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Patterson JT, Asano S, Li X, Rader C, Barbas CF, Bioconjug. Chem 2014, 25, 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Chiotellis Aristeidis, ChemComm 2016, 52, 6083–6086. [DOI] [PubMed] [Google Scholar]
- [82].Zhang Q, Dall’Angelo S, Fleming IN, Schweiger LF, Zanda M, O’Hagan D, Chem. - A Eur. J 2016, 22, 10998–11004. [DOI] [PubMed] [Google Scholar]
- [83].Chen X, Wu H, Park C-M, Poole TH, Keceli G, Devarie-Baez NO, Tsang AW, Lowther WT, Poole LB, King SB, et al. , ACS Chem. Biol 2017, 12, 2201–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Farrukh A, Paez JI, Salierno M, Fan W, Berninger B, del Campo A, Biomacromolecules 2017, 18, 906–913. [DOI] [PubMed] [Google Scholar]
- [85].Ema T, Inoue H, Chem. Lett 2015, 44, 1374–1376. [Google Scholar]
- [86].Zhou P, Yao J, Hu G, Fang J, ACS Chem. Biol 2016, 11, 1098–1105. [DOI] [PubMed] [Google Scholar]
- [87].Bauer MR, Joerger AC, Fersht AR, Proc. Natl. Acad. Sci 2016, 113, E5271–E5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Robson P, Smith TA, Stephens R, Tatlow JC, J. Chem. Soc 1963, 3692. [Google Scholar]
- [89].Birchall JM, Green M, Haszeldine RN, Pitts AD, Chem. Commun 1967, 338–339. [Google Scholar]
- [90].Bhupathiraju NVSDK, Rizvi W, Batteas JD, Drain CM, Org. Biomol. Chem 2016, 14, 389–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Ott C, Hoogenboom R, Schubert US, Chem. Commun 2008, 30, 3516–3518. [DOI] [PubMed] [Google Scholar]
- [92].Becer CR, Babiuch K, Pilz D, Hornig S, Heinze T, Gottschaldt M, Schubert US, Macromolecules 2009, 42, 2387–2394. [Google Scholar]
- [93].Noy J-M, Koldevitz M, Roth PJ, Polym. Chem 2014, 6, 436–447. [Google Scholar]
- [94].Battioni P, Brigaud O, Desvaux H, Mansuy D, Traylor TG, Tetrahedron Lett 1991, 32, 2893–2896. [Google Scholar]
- [95].Yang J, Gabriele B, Belvedere S, Huang Y, Breslow R, J. Org. Chem 2002, 67, 5057–5067. [DOI] [PubMed] [Google Scholar]
- [96].Varotto A, Todaro L, Vinodu M, Koehne J, Liu G, Drain CM, Chem. Commun 2008, 1, 4921–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hao E, Friso E, Miotto G, Jori G, Soncin M, Fabris C, Sibrian-Vazquez M, Vicente MGH, Org. Biomol. Chem 2008, 6, 3732–40. [DOI] [PubMed] [Google Scholar]
- [98].Bhupathiraju NVSDK, Vicente MGH, Bioorganic Med. Chem 2013, 21, 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Bhupathiraju NVSDK, Hu X, Zhou Z, Fronczek FR, Couraud PO, Romero IA, Weksler B, Vicente MGH, J. Med. Chem 2014, 57, 6718–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gan D, Mueller A, Wooley KL, J. Polym. Sci. Part A Polym. Chem 2003, 41, 3531–3540. [Google Scholar]
- [101].Cheng C, Wooley KL, Khoshdel E, J. Polym. Sci. Part A Polym. Chem 2005, 43, 4754–4770. [Google Scholar]
- [102].Powell KT, Cheng C, Gudipati CS, Wooley KL, J. Mater. Chem 2005, 15, 5128–5135. [Google Scholar]
- [103].Remzi Becer C, Hoogenboom R, Schubert US, Angew. Chemie - Int. Ed 2009, 48, 4900–4908. [DOI] [PubMed] [Google Scholar]
- [104].Alsbaiee A, Smith BJ, Xiao L, Ling Y, Helbling DE, Dichtel WR, Nature 2015, 529, 190–194. [DOI] [PubMed] [Google Scholar]
- [105].Qian EA, Wixtrom AI, Axtell JC, Saebi A, Jung D, Rehak P, Han Y, Moully EH, Mosallaei D, Chow S, et al. , Nat. Chem 2017, 9, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Spokoyny AM, Zou Y, Ling JJ, Yu H, Lin YS, Pentelute BL, J. Am. Chem. Soc 2013, 135, 5946–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zhang C, Spokoyny AM, Zou Y, Simon MD, Pentelute BL, Angew. Chemie Int. Ed 2013, 52, 14001–14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zou Y, Spokoyny AM, Zhang C, Simon MD, Yu H, Lin Y-S, Pentelute BL, Org. Biomol. Chem 2013, 12, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zhang C, Dai P, Spokoyny AM, Pentelute BL, Org. Lett 2014, 16, 3652–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Lühmann T, Mong SK, Simon MD, Meinel L, Pentelute BL, Org. Biomol. Chem 2016, 14, 3345–3349. [DOI] [PubMed] [Google Scholar]
- [111].Zhang C, Welborn M, Zhu T, Yang NJ, Santos MS, Van Voorhis T, Pentelute BL, Nat. Chem 2016, 8, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Dai P, Zhang C, Welborn M, Shepherd JJ, Zhu T, Van Voorhis T, Pentelute BL, ACS Cent. Sci 2016, 2, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dai P, Williams JK, Zhang C, Welborn M, Shepherd JJ, Zhu T, Van Voorhis T, Hong M, Pentelute BL, Sci. Rep 2017, 7, 7954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fadzen CM, Wolfe JM, Cho C-F, Chiocca EA, Lawler SE, Pentelute BL, J. Am. Chem. Soc 2017, 139, 15628–15631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Liu W, Zheng Y, Kong X, Heinis C, Zhao Y, Wu C, Angew. Chemie Int. Ed 2017, 56, 4458–4463. [DOI] [PubMed] [Google Scholar]
- [116].Wolfe JM, Fadzen CM, Holden RL, Yao M, Hanson GJ, Pentelute BL, Angew. Chemie Int. Ed 2018, 57, 4756–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ngambenjawong C, Pineda JMB, Pun SH, Bioconjug. Chem 2016, 27, 2854–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Conibear AC, Chaousis S, Durek T, Johan Rosengren K, Craik DJ, Schroeder CI, Biopolymers 2016, 106, 89–100. [DOI] [PubMed] [Google Scholar]
- [119].Swedberg JE, Schroeder CI, Mitchell JM, Durek T, Fairlie DP, Edmonds DJ, Griffith DA, Ruggeri RB, Derksen DR, Loria PM, et al. , Eur. J. Med. Chem 2015, 103, 175–184. [DOI] [PubMed] [Google Scholar]
- [120].Hilinski GJ, Kim Y-W, Hong J, Kutchukian PS, Crenshaw CM, Berkovitch SS, Chang A, Ham S, Verdine GL, J. Am. Chem. Soc 2014, 136, 12314–12322. [DOI] [PubMed] [Google Scholar]
- [121].Gunzburg MJ, Kulkarni K, Watson GM, Ambaye ND, Del Borgo MP, Brandt R, Pero SC, Perlmutter P, Wilce MCJ, Wilce JA, Sci. Rep 2016, 6, 27060–27072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kalhor-Monfared S, Jafari MR, Patterson JT, Kitov PI, Dwyer JJ, Nuss JM, Derda R, Chem. Sci 2016, 7, 3785–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Embaby AM, Schoffelen S, Kofoed C, Meldal M, Diness F, Angew. Chemie Int. Ed 2018, 57, 8022–8026. [DOI] [PubMed] [Google Scholar]
- [124].Hoppmann C, Maslennikov I, Choe S, Wang L, J. Am. Chem. Soc 2015, 137, 11218–11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Jacobson O, Yan X, Ma Y, Niu G, Kiesewetter DO, Chen X, Bioconjug. Chem 2015, 26, 2016–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Sun MMC, Beam KS, Cerveny CG, Hamblett KJ, Blackmore RS, Torgov MY, Handley FGM, Ihle NC, Senter PD, Alley SC, Bioconjug. Chem 2005, 16, 1282–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Nathani RI, Moody P, Chudasama V, Smith MEB, Fitzmaurice RJ, Caddick S, Chem. Sci 2013, 4, 3455–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Daly NL, Love S, Alewood PF, Craik DJ, Biochemistry 1999, 38, 10606–10614. [DOI] [PubMed] [Google Scholar]
- [129].Tam JP, Lu YA, Yu Q, J. Am. Chem. Soc 1999, 121, 4316–4324. [Google Scholar]
- [130].Aboye TL, Li Y, Majumder S, Hao J, Shekhtman A, Camarero JA, Bioorganic Med. Chem. Lett 2012, 22, 2823–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Clark RJ, Craik DJ, Biopolymers 2010, 94, 414–422. [DOI] [PubMed] [Google Scholar]
- [132].Puljung MC, Zagotta WN, Biophys. J 2011, 100, 2513–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Gautier A, Juillerat A, Heinis C, Corrêa IR, Kindermann M, Beaufils F, Johnsson K, Chem. Biol 2008, 15, 128–136. [DOI] [PubMed] [Google Scholar]
- [134].Rush JS, Bertozzi CR, J. Am. Chem. Soc 2008, 130, 12240–12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Duckworth BP, Zhang Z, Hosokawa A, Distefano MD, ChemBioChem 2007, 8, 98–105. [DOI] [PubMed] [Google Scholar]
- [136].Kurpiers T, Mootz HD, Angew. Chemie - Int. Ed 2007, 46, 5234–5237. [DOI] [PubMed] [Google Scholar]
- [137].Walsh C, Nature 2001, 409, 226–231. [DOI] [PubMed] [Google Scholar]
- [138].Walsh CT, Garneau-Tsodikova S, Gatto GJ, Angew. Chemie - Int. Ed 2005, 44, 7342–7372. [DOI] [PubMed] [Google Scholar]
- [139].Hayes JD, Flanagan JU, Jowsey IR, Rev. Lit. Arts Am 2005, 45, 51–88. [DOI] [PubMed] [Google Scholar]
- [140].Armstrong RN, Chem. Res. Toxicol 1997, 10, 2–18. [DOI] [PubMed] [Google Scholar]
- [141].Buczyński P, Dijkstra KDB, Mauersberger R, Moroz MD, Odonatologica 2006, 35, 1–13. [Google Scholar]
- [142].Mannervik B, Danielson UH, CRC Crit. Rev. Biochem 1988, 23, 283–337. [DOI] [PubMed] [Google Scholar]
- [143].Wilce MCJ, Parker MW, Biochim. Biophys. Acta 1994, 1205, 1–18. [DOI] [PubMed] [Google Scholar]
- [144].Oakley a J., Lo Bello M, Battistoni a, Ricci G, Rossjohn J, Villar HO, Parker MW, J. Mol. Biol 1997, 274, 84–100. [DOI] [PubMed] [Google Scholar]
- [145].Griffin BA, Adams SR, Tsien RY, Science (80-.) 1998, 281, 269–272. [DOI] [PubMed] [Google Scholar]
- [146].Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY, J. Am. Chem. Soc 2002, 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- [147].Wilson P, Anastasaki A, Owen MR, Kempe K, Haddleton DM, Mann SK, Johnston APR, Quinn JF, Whittaker MR, Hogg PJ, et al. , J. Am. Chem. Soc 2015, 137, 4215–4222. [DOI] [PubMed] [Google Scholar]
- [148].Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, et al. , Proc. Natl. Acad. Sci. U. S. A 2012, 109, 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Zhu Z, Ramakrishnan B, Li J, Wang Y, Feng Y, Prabakaran P, Colantonio S, Dyba MA, Qasba PK, Dimitrov DS, MAbs 2014, 6, 1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Drake PM, Albers AE, Baker J, Banas S, Barfield RM, Bhat AS, De Hart GW, Garofalo AW, Holder P, Jones LC, et al. , Bioconjug. Chem 2014, 25, 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Dennler P, Chiotellis A, Fischer E, Brégeon D, Belmant C, Gauthier L, Lhospice F, Romagne F, Schibli R, Bioconjug. Chem 2014, 25, 569–578. [DOI] [PubMed] [Google Scholar]
- [152].York D, Baker J, Holder PG, Jones LC, Drake PM, Barfield RM, Bleck GT, Rabuka D, BMC Biotechnol 2016, 16, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Junutula JRJR, Raab H, Clark S, Bhakta S, Leipold DDD, Weir S, Chen Y, Simpson M, Tsai SPSP, Dennis MSMS, et al. , Nat. Biotechnol 2008, 26, 925–932. [DOI] [PubMed] [Google Scholar]
- [154].Shen B-Q, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu S-F, Mai E, et al. , Nat. Biotechnol 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- [155].Hofmeister F, Naunyn-Schmiedeberg’s Arch. Pharmacol 1888, 24, 247–260–260. [Google Scholar]
- [156].Kunz W, Henle J, Ninham BW, Curr. Opin. Colloid Interface Sci 2004, 9, 19–37. [Google Scholar]
- [157].Chalker JM, in Chemoselective Bioorthogonal Ligation React, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2017, pp. 231–270. [Google Scholar]
- [158].Yang M, Li J, Chen PR, Chem. Soc. Rev 2014, 43, 6511. [DOI] [PubMed] [Google Scholar]
- [159].Chankeshwara SV, Indrigo E, Bradley M, Curr. Opin. Chem. Biol 2014, 21, 128–135. [DOI] [PubMed] [Google Scholar]
- [160].Ball ZT, Curr. Opin. Chem. Biol 2015, 25, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Antos JM, Francis MB, Curr. Opin. Chem. Biol 2006, 10, 253–262. [DOI] [PubMed] [Google Scholar]
- [162].Kosugi M, Shimizu T, Migita T, Chem. Lett 1978, 7, 13–14. [Google Scholar]
- [163].Migita T, Shimizu T, Asami Y, Shiobara J-I, Kato Y, Kosugi M, Bull. Chem. Soc. Jpn 1980, 53, 1385–1389. [Google Scholar]
- [164].Lee CF, Liu YC, Badsara SS, Chem. - An Asian J 2014, 9, 706–722. [DOI] [PubMed] [Google Scholar]
- [165].Kung KK-Y, Ko H-M, Cui J-F, Chong H-C, Leung Y-C, Wong M-K, Chem. Commun. (Camb) 2014, 50, 11899–902. [DOI] [PubMed] [Google Scholar]
- [166].Messina MS, Stauber JM, Waddington MA, Rheingold AL, Maynard HD, Spokoyny AM, J. Am. Chem. Soc 2018, 140, 7065–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Vinogradova EV, Zhang C, Spokoyny AM, Pentelute BL, Buchwald SL, Nature 2015, 526, 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Rojas AJ, Pentelute BL, Buchwald SL, Org. Lett 2017, 19, 4263–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Rojas AJ, Zhang C, Vinogradova EV, Buchwald NH, Reilly J, Pentelute BL, Buchwald SL, Chem. Sci 2017, 8, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Shafer DE, Inman JK, Lees A, Anal. Biochem 2000, 282, 161–164. [DOI] [PubMed] [Google Scholar]
- [171].Zhao W, Lee HG, Buchwald SL, Hooker JM, J. Am. Chem. Soc 2017, 139, 7152–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kubota K, Dai P, Pentelute BL, Buchwald SL, J. Am. Chem. Soc 2018, 140, 3128–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Al-Shuaeeb RAA, Kolodych S, Koniev O, Delacroix S, Erb S, Nicolaÿ S, Cintrat J-C, Brion J-D, Cianférani S, Alami M, et al. , Chem. - A Eur. J 2016, 22, 11365–11370. [DOI] [PubMed] [Google Scholar]
- [174].Vara BA, Li X, Berritt S, Walters CR, Petersson EJ, Molander GA, Chem. Sci 2018, 9, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Willwacher J, Raj R, Mohammed S, Davis BG, J. Am. Chem. Soc 2016, 138, 8678–8681. [DOI] [PubMed] [Google Scholar]
- [176].Booth C, Gaspar HB, Biologics 2009, 3, 349–358. [PMC free article] [PubMed] [Google Scholar]
- [177].Guram A, Buchwald S, J. Am. Chem. Soc 1994, 116, 7901–7902. [Google Scholar]
- [178].Paul F, Patt J, Hartwig JF, J. Am. Chem. Soc 1994, 116, 5969–5970. [Google Scholar]
- [179].Chan DMT, Monaco KL, Wang RP, Winters MP, Tetrahedron Lett 1998, 39, 2933–2936. [Google Scholar]
- [180].Lam PYS, Clark CG, Saubern S, Adams J, Winters MP, Chan DMT, Combs A, Tetrahedron Lett 1998, 39, 2941–2944. [Google Scholar]
- [181].Lam PYS, in Synth. Methods Drug Discov, 2016, pp. 242–273. [Google Scholar]
- [182].Ullmann F, Berichte der Dtsch. Chem. Gesellschaft 1903, 36, 2382–2384. [Google Scholar]
- [183].Goldberg I, Berichte der Dtsch. Chem. Gesellschaft 1906, 39, 1691–1692. [Google Scholar]
- [184].Lercher L, McGouran JF, Kessler BM, Schofield CJ, Davis BG, Angew. Chemie - Int. Ed 2013, 52, 10553–10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Sambiagio C, Marsden SP, Blacker AJ, McGowan PC, Chem. Soc. Rev 2014, 43, 3525. [DOI] [PubMed] [Google Scholar]
- [186].Sanger F, Biochem. J 1945, 39, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Sutton BDA, Drewes SE, Welz U, Biochem. J 1972, 130, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Ladd DL, Snow RA, Anal. Biochem 1993, 210, 258–261. [DOI] [PubMed] [Google Scholar]
- [189].Lautrette G, Touti F, Lee HG, Dai P, Pentelute BL, J. Am. Chem. Soc 2016, 138, 8340–8343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Lee HG, Lautrette G, Pentelute BL, Buchwald SL, Angew. Chemie Int. Ed 2017, 56, 3177–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Ohata J, Minus MB, Abernathy ME, Ball ZT, J. Am. Chem. Soc 2016, 138, 7472–7475. [DOI] [PubMed] [Google Scholar]
- [192].Crawford L. a, Weerapana E, Mol. Biosyst 2016, 12, 1768–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Brown KC, Yang S-H, Kodadek T, Biochemistry 1995, 34, 4733–4739. [DOI] [PubMed] [Google Scholar]
- [194].Kim K, Fancy DA, Carney D, Kodadek T, J. Am. Chem. Soc 1999, 121, 11896–11897. [Google Scholar]
- [195].Fancy DA, Denison C, Kim K, Xie Y, Holdeman T, Amini F, Kodadek T, Chem. Biol 2000, 7, 697–708. [DOI] [PubMed] [Google Scholar]
- [196].Meunier S, Strable E, Finn MG, Chem. Biol 2004, 11, 319–326. [DOI] [PubMed] [Google Scholar]
- [197].Fehler SK, Pratsch G, Östreicher C, Fürst MCD, Pischetsrieder M, Heinrich MR, Tetrahedron 2016, 72, 7888–7893. [Google Scholar]
- [198].Schlick TL, Ding Z, Kovacs EW, Francis MB, J. Am. Chem. Soc 2005, 127, 3718–3723. [DOI] [PubMed] [Google Scholar]
- [199].Chapman CJ, Matsuno A, Frost CG, Willis MC, Chem. Commun. (Camb) 2007, 3903–3905. [DOI] [PubMed] [Google Scholar]
- [200].Chapman CJ, Hargrave JD, Bish G, Frost CG, Tetrahedron 2008, 64, 9528–9539. [Google Scholar]
- [201].Chen X, Engle KM, Wang D-H, Yu J-Q, Angew. Chemie Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Sandtorv AH, Adv. Synth. Catal 2015, 357, 2403–2435. [Google Scholar]
- [203].Ruiz-Rodriguez J, Albericio F, Lavilla R, Chem. - A Eur. J 2010, 16, 1124–1127. [DOI] [PubMed] [Google Scholar]
- [204].Mendive-Tapia L, Preciado S, García J, Ramón R, Kielland N, Albericio F, Lavilla R, Nat. Commun 2015, 6, 7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Mendive-Tapia L, Bertran A, García J, Acosta G, Albericio F, Lavilla R, Chem. - A Eur. J 2016, 22, 13114–13119. [DOI] [PubMed] [Google Scholar]
- [206].Dong H, Limberakis C, Liras S, Price D, James K, Chem. Commun 2012, 48, 11644–11646. [DOI] [PubMed] [Google Scholar]
- [207].Schischko A, Ren H, Kaplaneris N, Ackermann L, Angew. Chemie Int. Ed 2017, 56, 1576–1580. [DOI] [PubMed] [Google Scholar]
- [208].Williams TJ, Reay AJ, Whitwood AC, Fairlamb IJS, Chem. Commun 2014, 50, 3052–3054. [DOI] [PubMed] [Google Scholar]
- [209].Reay AJ, Williams TJ, Fairlamb IJS, Org. Biomol. Chem 2015, 13, 8298–8309. [DOI] [PubMed] [Google Scholar]
- [210].Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN, Trends Biochem. Sci 2014, 39, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Byun BJ, Kang YK, Biopolymers 2011, 95, 345–353. [DOI] [PubMed] [Google Scholar]
- [212].Hofer T, Thomas JD, Burke TR, Rader C, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 12451–12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Cohen DT, Zhang C, Pentelute BL, Buchwald SL, J. Am. Chem. Soc 2015, 137, 9784–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Taniguchi N, J. Org. Chem 2007, 72, 1241–1245. [DOI] [PubMed] [Google Scholar]
- [215].Zheng B, Gong Y, Xu HJ, Tetrahedron 2013, 69, 5342–5347. [Google Scholar]
- [216].Chalker JM, Wood CSC, Davis BG, J. Am. Chem. Soc 2009, 131, 16346–16347. [DOI] [PubMed] [Google Scholar]
- [217].Simmons RL, Yu RT, Myers AG, J. Am. Chem. Soc 2011, 133, 15870–15873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Vilaró M, Arsequell G, Valencia G, Ballesteros A, Barluenga J, Org. Lett 2008, 10, 3243–3245. [DOI] [PubMed] [Google Scholar]
- [219].Kodama K, Fukuzawa S, Nakayama H, Kigawa T, Sakamoto K, Yabuki T, Matsuda N, Shirouzu M, Takio K, Tachibana K, et al. , ChemBioChem 2006, 7, 134–139. [DOI] [PubMed] [Google Scholar]
- [220].Dibowski H, Schmidtchen FP, Angew. Chemie Int. Ed 1998, 37, 476–478. [DOI] [PubMed] [Google Scholar]
- [221].Ojida A, Tsutsumi H, Kasagi N, Hamachi I, Tetrahedron Lett 2005, 46, 3301–3305. [Google Scholar]
- [222].Bong DT, Ghadiri MR, Org. Lett 2001, 3, 2509–2511. [DOI] [PubMed] [Google Scholar]
- [223].Kodama K, Fukuzawa S, Nakayama H, Sakamoto K, Kigawa T, Yabuki T, Matsuda N, Shirouzu M, Takio K, Yokoyama S, et al. , ChemBioChem 2007, 8, 232–238. [DOI] [PubMed] [Google Scholar]
- [224].Brustad E, Bushey ML, Lee JW, Groff D, Liu W, Schultz PG, Angew. Chemie - Int. Ed 2008, 47, 8220–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Spicer CD, Davis BG, Chem. Commun 2011, 47, 1698–1700. [DOI] [PubMed] [Google Scholar]
- [226].Wang Y-S, Russell WK, Wang Z, Wan W, Dodd LE, Pai P-J, Russell DH, Liu WR, Mol. Biosyst 2011, 7, 714–717. [DOI] [PubMed] [Google Scholar]
- [227].Li N, Lim RKV, Edwardraja S, Lin Q, J. Am. Chem. Soc 2011, 133, 15316–15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Li J, Lin S, Wang J, Jia S, Yang M, Hao Z, Zhang X, Chen PR, J. Am. Chem. Soc 2013, 135, 7330–7338. [DOI] [PubMed] [Google Scholar]
- [229].Dumas A, Spicer CD, Gao Z, Takehana T, Lin YA, Yasukohchi T, Davis BG, Angew. Chemie - Int. Ed 2013, 52, 3916–3921. [DOI] [PubMed] [Google Scholar]
- [230].Ma X, Wang H, Chen W, J. Org. Chem 2014, 79, 8652–8658. [DOI] [PubMed] [Google Scholar]
- [231].Spicer CD, Triemer T, Davis BG, J. Am. Chem. Soc 2012, 134, 800–803. [DOI] [PubMed] [Google Scholar]
- [232].Cheng G, Lim RKV, Li N, Lin Q, Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB, Kaiser ET, et al. , Chem. Commun 2013, 49, 6809. [Google Scholar]
- [233].Cheng G, Lim RKV, Ramil CP, Lin Q, Chem. Commun. (Camb) 2014, 50, 11679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].de Bruijn AD, Roelfes G, Chem. - A Eur. J 2018, 24, 12728–12733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Liu J, Cao Z, Lu Y, Chem. Rev 2009, 109, 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Du Y, Dong S, Anal. Chem 2017, 89, 189–215. [DOI] [PubMed] [Google Scholar]
- [237].Fan C, Ed., DNA Nanotechnology, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013. [Google Scholar]
- [238].Lundin KE, Gissberg O, Smith CIE, Hum. Gene Ther 2015, 26, 475–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Silverman SK, Angew. Chemie Int. Ed 2010, 49, 7180–7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Beaucage SL, Iyer RP, Tetrahedron 1993, 49, 6123–6194. [Google Scholar]
- [241].Jäger S, Rasched G, Kornreich-Leshem H, Engeser M, Thum O, Famulok M, J. Am. Chem. Soc 2005, 127, 15071–15082. [DOI] [PubMed] [Google Scholar]
- [242].Omumi A, Beach DG, Baker M, Gabryelski W, Manderville RA, J. Am. Chem. Soc 2011, 133, 42–50. [DOI] [PubMed] [Google Scholar]
- [243].Walunj MB, Tanpure AA, Srivatsan SG, Nucleic Acids Res 2018, 46, e65–e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Probst N, Lartia R, Théry O, Alami M, Defrancq E, Messaoudi S, Chem. - A Eur. J 2018, 24, 1795–1800. [DOI] [PubMed] [Google Scholar]
- [245].Ding Y, Clark MA, ACS Comb. Sci 2015, 17, 1–4. [DOI] [PubMed] [Google Scholar]
- [246].Ding Y, Franklin GJ, DeLorey JL, Centrella PA, Mataruse S, Clark MA, Skinner SR, Belyanskaya S, ACS Comb. Sci 2016, 18, 625–629. [DOI] [PubMed] [Google Scholar]
- [247].Ding Y, DeLorey JL, Clark MA, Bioconjug. Chem 2016, 27, 2597–2600. [DOI] [PubMed] [Google Scholar]
- [248].Evans ED, Pentelute BL, ACS Chem. Biol 2018, 13, 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Gimenez D, Mooney CA, Dose A, Sandford G, Coxon CR, Cobb SL, Org. Biomol. Chem 2017, 15, 4086–4095. [DOI] [PubMed] [Google Scholar]
- [250].Zhang X, Lu G, Sun M, Mahankali M, Ma Y, Zhang M, Hua W, Hu Y, Wang Q, Chen J, et al. , Nat. Chem 2018, 10, 540–548. [DOI] [PubMed] [Google Scholar]
- [251].Bottecchia C, Rubens M, Gunnoo SB, Hessel V, Madder A, Noël T, Angew. Chemie - Int. Ed 2017, 12702–12707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Wang Z, Cohen SM, Chem. Soc. Rev 2009, 38, 1315–1329. [DOI] [PubMed] [Google Scholar]
- [253].Kango S, Kalia S, Celli A, Njuguna J, Habibi Y, Kumar R, Prog. Polym. Sci 2013, 38, 1232–1261. [Google Scholar]
- [254].Wang Y, Liu J, Cao L, Wang W, Sun Y, Yin Z, Lou Z, ChemBioChem 2018, 19, 1465–1470. [DOI] [PubMed] [Google Scholar]