

Abstract
Background and Purpose
The molecular mechanism for the pathogenesis of nonalcoholic fatty liver disease (NAFLD) remains elusive. Both farnesoid X receptor (FXR) signalling and endoplasmic reticulum (ER) stress contribute to the progression of NAFLD; however, it is not clear whether the actions of these two pathways are dependent on each other. Moreover, the pharmacological benefits and mechanism of betulinic acid (BA) in controlling metabolic syndrome and NAFLD are largely unknown.
Experimental Approach
A reporter assay and a time‐resolved FRET assay were used to identify BA as an agonist of the FXR. NAFLD was induced by a methionine and choline‐deficient L‐amino acid diet (MCD) and high‐fat diet (HFD). The pharmacological effects of BA (100 mg·kg−1·day−1) and potential interactions between hepatic FXR activation and ER stress pathways were evaluated by FXR silencing, Western blot and RT‐PCR analyses using control and FXR−/− mice.
Key Results
Activation of the FXR inhibited intracellular PERK/EIF2α/ATF4 and CHOP signalling, thereby alleviating hepatic ER stress, whereas FXR silencing resulted in an opposite effect. Furthermore, we identified BA as an FXR agonist that effectively attenuated the progression of NAFLD and metabolic disorders in both HFD‐ and MCD diet‐fed mice and restored the hepatocellular ER homeostasis by stimulating the FXR signalling pathway and blocking PERK/EIF2α signalling. In contrast, the effects of BA were attenuated in FXR−/− mice.
Conclusions and Implications
Our data demonstrate that pharmacological activation of the FXR by BA reduces hepatocellular ER stress and attenuates NAFLD in an animal model of hepatic steatosis.
Abbreviations
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BA
betulinic acid
- BAT
brown adipose tissue
- CDCA
chenodeoxycholic acid
- CYP7A1
cholesterol 7‐α hydroxylase
- DIO
diet‐induced obesity
- ER
endoplasmic reticulum
- FXR
farnesoid X receptor
- GTT
glucose tolerance test
- HDL‐c
HDL cholesterol
- HFD
high‐fat diet
- IRE1
inositol‐requiring enzyme 1
- ITT
insulin tolerance test
- LCA
lithocholic acid
- LBD
ligand binding domains
- LDL‐c
LDL cholesterol
- MCD
methionine and choline‐deficient
- MS
metabolic syndrome
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OCA
obeticholic acid
- PA
palmitic acid
- PERK
protein kinase RNA‐like ER kinase
- TBA
total bile acids
- TC
total cholesterol
- TCDCA
tauro‐CDCA
- TDCA
tauro‐deoxycholic acid
- TG
triglyceride
- TM
tunicamycin
- TR‐FRET
time‐resolved FRET
- UPLC
ultra‐performance LC
- UPR
unfolded protein response
- WAT
white adipose tissue
What is already known
FXR agonists are the most promising candidate to treat NAFLD.
ER stress is involved in the progress of NAFLD.
What this study adds
Betulinic acid is a novel FXR agonist.
Betulinic acid attenuates NAFLD and alleviates hepatocellular ER stress via stimulating FXR signalling in mice.
What is the clinical significance
Betulinic acid may be developed as a novel therapy for NAFLD.
1. INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common metabolic liver disease encompassing a spectrum of disease states, from hepatic steatosis to nonalcoholic steatohepatitis (NASH) followed by progression to fibrosis, cirrhosis, and hepatocellular carcinoma (Rinella, 2015; Wong et al., 2015). Obesity and insulin resistance are closely associated with NAFLD, and 70% of obese subjects have some form of NAFLD (Huh et al., 2017; Ouchi, Parker, Lugus, & Walsh, 2011). Although multifaceted metabolic factors including insulin resistance (IR), dyslipidaemia, and inflammation have been identified as the important pathogenic events responsible for hepatosteatosis (Rinella, 2015; Utzschneider & Kahn, 2006), the pathogenesis of NAFLD is still obscure. To date, no therapy has been approved for the treatment of NAFLD.
The farnesoid X receptor (FXR) serves as an intracellular bile acid sensor that predominantly exists in the enterohepatic system (Makishima et al., 1999; Matsubara, Li, & Gonzalez, 2013). FXR agonists play an active role in correcting multiple metabolic disorders including cholestasis, dyslipidaemia, IR, inflammation and NAFLD (Stedman et al., 2006; Zhang & Edwards, 2008; Zhang et al., 2006). In contrast, FXR deletion leads to the development of impaired glucose tolerance, insulin sensitivity, dysfunctional lipis metabolism, and severe liver steatosis (Kong, Luyendyk, Tawfik, & Guo, 2009; Sinal et al., 2000). Obeticholic acid (OCA), a well‐known FXR agonist, has been the most promising candidate to treat NAFLD (Musso, Cassader, & Gambino, 2016). Thus, screening for potent selective FXR activators has been the focus of research in the anti‐NAFLD drug development field, and more research into the mechanism by which FXR activation affects NAFLD is imperative.
The endoplasmic reticulum (ER) is a key site for regulating protein synthesis and lipid metabolism (Dara, Ji, & Kaplowitz, 2011; Hotamisligil, 2010; Ozcan et al., 2004). Persistent perturbation of ER homeostasis leads to activation of the unfolded protein response (UPR): component of which are protein kinase RNA‐like ER kinase (PERK), inositol‐requiring enzyme1 (IRE1), activating transcription factor 6 (ATF6), and ultimately induces ER stress (Ron & Walter, 2007; Rutkowski et al., 2008). Previous studies have shown that ER stress is at the intersection of inflammation and metabolic diseases (Hotamisligil, 2010), and in patients, obesity‐associated NAFLD is often accompanied by hepatic ER stress (Dara et al., 2011). In contrast, different inducers of ER stress such as tunicamycin (TM) and palmitic acid (PA) are known to aggravate liver steatosis (Rutkowski et al., 2008). Conversely, an improvement in metabolic homeostasis and fatty liver is observed with alleviators of ER stress, confirming that ER stress has an important role in the pathogenesis of NAFLD (Dara et al., 2011; Ozcan et al., 2006). These findings suggest that there is a link between the UPR and hepatic lipid metabolism. Thus, pharmacological targeting of hepatic ER stress represents a promising therapeutic strategy for NAFLD.
Notably, previous studies have revealed that intrahepatic bile acid metabolism is associated with ER stress (Chung, Kim, Lee, An, & Kwon, 2015), most hepatic ER stress coexists with a lowered expression of FXRs in ageing mice (Lefebvre & Staels, 2014), and multiple FXR agonists have been shown to protect cells from ER stress (Chung et al., 2015; Ozcan et al., 2006). FXR activation has a cytoprotective effect in the kidney, which is mediated by supressing ER stress (Gai et al., 2017). These findings suggest that FXR activation could reduce hepatic ER stress, and this effect may be involved in the anti‐NAFLD effect of FXR agonists. In contrast, in a recent article it was suggested that hepatic FXR activation exacerbates ER stress (Liu et al., 2018). Thus, the role of hepatic FXR activation in regulating the ER stress process remains to be clarified.
The use of natural compounds to treat metabolic syndrome (MS) is attractive to the public. Betulinic acid (BA) is a pentacyclic lupane‐type triterpene that exists widely in food and medicinal herbs such as Semen Zizyphi Spinosae (Csuk, 2014). The biological effects of BA have been well studied for its antihuman immunodeficiency virus, antibacterial, anti‐inflammatory, antinociceptive, immunomodulatory, antiangiogenic, and anticancer properties (S. Y. Lee, Kim, & Park, 2015). However, the effect of BA on MS and NAFLD has not been elucidated.
Here, we show that BA is a novel FXR agonist that inhibits the intrahepatic ER stress in vitro and in vivo. In animal experiments, BA attenuated hyperlipidaemia, insulin resistance, and altered the composition of the liver bile acid pool in high‐fat diet (HFD)‐fed C57BL/6 mice. Additionally, BA treatment significantly inhibited the accumulation of lipids in the liver in HFD‐ and methionine and choline‐deficient (MCD) diet‐fed C57BL/6 mice. Furthermore, we showed that FXR agonism inhibits the intracellular PERK‐EIF2α pathway and attenuates the hepatic ER stress response. Our data shed new light on the role of ER stress and FXR activation in hepatic steatosis.
2. METHODS
2.1. Cell culture and treatment
For the gene mRNA experiment, HepG2 (ATCC) cells (CLS Cat# 300198/p2277_Hep‐G2, RRID:CVCL_0027) were seeded on six‐well plates (1 × 106 cells per well) and grown to 80% confluence with high‐glucose DMEM containing 10% FBS at 37°C in 5% CO2. The following day, cells were treated with DMSO control or BA (25 μM). After 24 hr treatment, the cells were collected for RNA isolation.
For intracellular ER stress induction, HepG2 cells were seeded in six‐well plates and grown to 50% confluence with high‐glucose DMEM followed by 6 hr free‐serum quench. Then, cells were pretreated with OCA (10 μM) or BA (6, 12, 25, and 50 μM) for 12 hr, followed by the stimulation with tunicamycin (TM; 2.5 μg·ml−1) or palmitic acid (PA; 500 μM) for 24 hr. Then cells were collected for gene expression analysis.
2.2. Knockdown of FXR by siRNA
For FXR knockdown, siRNA targeting FXR was chemically synthesized (Gene Pharma, China). The siRNA sequence (5′‐GAGGAUGCCUCAGGAAAUA‐3′) and (5′‐AAAGCGUCUGGAAAAGUCG‐3′) was respectively used for human FXR depletion and control. HepG2 cells were transfected with siRNA using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, USA). The efficiency of knockdown was performed through Western blot analysis.
2.3. Gene reporter luciferase assays
The reporter assay was carried out by the use of a Dual‐Luciferase Reporter Assay System (Promega, USA) as described previously (Huang et al., 2006).
For the nuclear receptor transcription activity assay, the expression plasmids for phFXR, phRXR (retinoid X receptor), and FXR‐dependent reporter (EcRE‐LUC), pCMXGal‐hPPARα, PPARγ, the liver X receptor‐like receptors LXRα and LXRβ, and the pregnane X receptor (PXR)‐LBD and the Gal4 reporter vector MH100 × 4‐TK‐Luc were cotransfected with a reporter constructed so that 1 μg of the relevant plasmid can combine with 1 μg of reporter plasmids and 0.1 μg of pREP7 (Renilla Luciferase) reporter was used to normalize transfection efficiencies. The transfection mixture, which contained 10 μg of total plasmids and 15 μl FuGENE‐HD (Roche) per ml of DMEM, was added to HEK293T cells (ATCC) for 24 hr and then removed. The FXR, PPARα, PPARγ, LXRs, and PXR agonists (GW4064, WY14643, Rosiglitazone, T0901317, and Rifampicin respectively). BA was introduced to fresh media, and the cells were incubated for every other 24 hr to determine luciferase activity.
2.4. Animal experiments
All animal protocols used in this study were approved by and conducted in accordance with the guidelines of the Animal Ethical Committee of Shanghai University of Traditional Chinese Medicine (Approval Number: SZY20150523). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010) and with the recommendations made by the British Journal of Pharmacology (McGrath & Lilley, 2015). Because female mice are known to have a higher expression of retinoic X receptors and higher sensitivity to FXR agonists than males (Kosters et al., 2013), we focused on females in this study. Six‐week‐old female C57BL/6J mice (RRID:IMSR_JAX:000664) were introduced from Jackson Labs by SLAC Laboratory (Shanghai, China), and six‐week‐old female FXR−/− B6.129×1 mice (IMSR_JAX:007214) were purchased from Jackson Labs by Dr. Li Yang. Animals were weighed (20 ± 2 g) and housed under specific pathogen‐free conditions and reared in line with standardized methods, kept under a controlled temperature (22–23°C) and on a 12 hr light, 12 hr dark cycle. Because constant HFD feeding is known to induce obesity, NAFLD, and metabolic disorders in C57BL/6J mice, six‐week‐old female mice were fed a HFD and water ad libitum for 12 weeks to induce obesity, and these mice were then randomly divided into three groups according to body weight: chow group (10% of calories derived from fat), high‐fat group (HF, 60% of calories derived from fat), and BA group (HFD supplemented with BA powder, at a dose of 100 mg.100 g‐1 diet). Mice were then treated for an additional 6 weeks. The food intake amount was measured by recording food weight every 2 days throughout the experiment. The amount of food consumed over a 24 hr period was calculated.
A methionine and choline‐deficient diet can result in the development of severe hepatic steatosis in C57BL/6J mice. Thus, for the MCD‐diet‐fed animal experiments, female C57BL/6J mice were managed using the same methods as in HFD‐fed mice experiments. Eight‐week‐old female mice (20 ± 2 g) were randomly divided into three groups according to body weight: MCS group (fed MCS diet), MCD (fed MCD‐diet), and BA group (MCD diet supplemented with BA powder, at a dose of 100 mg.100 g‐1 diet). Mice were fed corresponding diets and water ad libitum for an additional 6 weeks before the end of the experiment.
At the end of all animal experiments, mice were killed by anaesthetizing them with 20% urethane (Sigma, St. Louis, MO), cardiac blood was obtained, and subsequent liver, adipocyte, and small intestine tissues were harvested for subsequent use in various assays as indicated.
2.5. I.p. glucose tolerance and insulin tolerance tests
For glucose tolerance tests (GTT), at the end of the HFD treatment, mice were fasted overnight (12 hr). The baseline glucose values (0 min), before the injection of glucose (1 g·kg−1 body weight), were measured in blood from the tail vein. Extra blood samples were obtained at regular intervals (15, 30, 60, and 90 min).
For the insulin tolerance tests (ITT), 3 days after the GTT, mice were injected i.p. with 0.75 U·kg−1 body weight of insulin (Sigma, St. Louis, MO, USA). The glucose values (0, 15, 30, 60, 90, and 120 min) were measured in blood samples obtained from the tail vein.
2.6. Rectal temperature measurement
At the end of the treatment period, the rectal temperature of mice was measured using a rectal probe connected to a digital thermometer (Physitemp, Clifton, NJ, USA).
2.7. Serum chemistry analysis
At the end of the experiments, mice were anaesthetized with 20% urethane, and cardiac blood was taken. Levels of serum alanine aminotransferase (ALT), aspartate transaminase (AST), triglyceride (TG), total cholesterol (TC), HDL cholesterol (HDL‐c), and LDL cholesterol (LDL‐c) were measured, in 100 μl of heart blood serum, using a Hitachi 7020 Automatic Analyzer (Hitachi, Ltd., Tokyo, Japan).
2.8. Histochemistry
White adipose tissues (WAT), brown adipose tissue (BAT), and liver tissues were fixed in formalin, and paraffin‐embedded sections were cut at 5 μm. Sections were stained with haematoxylin and eosin (HE) according to a standard procedure. White adipocyte size was calculated from cross‐sectional areas obtained from perimeter tracings using ImageJ software (Macbiophotonics, McMaster University, Hamilton, ON, Canada, RRID:SCR_003070). A pathologist blinded to the experimental conditions evaluated the NAFLD activity score (NAS) according to a simple NAFLD scoring system (Liang et al., 2014).
2.9. Oil Red O staining
Compound‐embedded frozen liver sections, prepared at optimum cutting temperature, were allowed to air dry and were rinsed with 60% isopropanol, followed by staining with fresh 0.5% Oil Red O solution for 15 min. After being stained, the slides were rinsed with 60% isopropanol, washed with distilled water, mounted, and analysed under a light microscope (Zeiss).
2.10. Intracellular lipid content measurement
HepG2 cells were seeded on six‐well plates (1 × 106 cells per well) and grown to 50% confluence with high‐glucose DMEM containing 10% FBS at 37°C in 5% CO2 followed by 6 hr free‐serum quench. Then cells were pretreated with DMSO (0.1%) control or BA (25 μM) for 12 hr, followed by incubation with PA (800 μM) for 24 hr. Then cells were collected and homogenized in lysis buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton), and mixed with an equal volume of chloroform, followed by centrifugation at 18,400× g for 15 min at 4°C. The chloroform layer was separated, dried, and resuspended in 50 μl of isopropyl alcohol to measure the triglyceride (TG) content as previously described (Folch, Lees, & Sloane Stanley, 1957). Protein concentrations were measured using a Bio‐Rad Protein Assay Kit. Intracellular lipid contents in cell lysates were expressed as μg of lipid.mg‐1 cellular protein.
2.11. Liver and faecal lipid content analysis
Liver and faecal lipid contents were measured as described previously (Folch et al., 1957). Briefly, the liver tissue or faeces were homogenized and extracted with equal volume of chloroform–methanol. The chloroform phase was removed to a new tube and dried and was then resuspended in isopropyl alcohol as a total lipid extract sample. The quantities of total cholesterol (TC) and TGs (Kinghawk, China) in liver and faeces were then assayed according to the manufacturers' protocols. Briefly, 300 μl of TC or TG working reagents were mixed with 5 μl of ddH2O, cholesterol, or glycerol standards and the samples respectively. The reaction was incubated at 37°C for 5 min. Absorbance (A) of blank, standards, and samples were recorded at 546 nm. TC and TG concentrations were calculated as (Asample‐Ablank)/(Astandard‐Ablank) × concentration of standards and normalized to the weights of the liver tissue or faeces.
2.12. RNA extraction and quantitative PCR
Total RNA from HepG2 cells or mouse livers was isolated using the TRIzol method (Invitrogen, Carlsbad, CA, 15596018). The first‐strand cDNA was synthesized with a cDNA synthesis kit (Fermentas, Madison, WI, K1671). Quantitative real‐time PCR was carried out using SYBR green PCR Mastermix (Invitrogen, Carlsbad, CA, 4309155). The results were analysed on an ABI StepOnePlus real‐time PCR system (Applied Biosystems, USA) using the 2−ΔΔCt method. Values were normalized to β‐actin. Sequences of primers were listed in Tables S1 and S2.
2.13. Quantification of bile acids in mice
Measurement of total bile acids (TBA) in mouse livers and faeces was performed according to the following. Livers and faeces were weighed and homogenized in 10× volume water. TBA levels were determined in homogenate samples using the TBA measurement kit (KeHua, China) after quantification of intracellular protein concentration. The TBA levels were normalized to intracellular protein content or faecal weight. Protein concentrations were determined by use of a BCA protein assay kit (Sigma, St. Louis, MO).
To measure hepatic BA pool concentration and composition, liver tissue samples were weighed, and TBA was extracted in 5× volumes of acetonitrile followed by centrifugation at 19,200× g for 10 min. The supernatant was dried under nitrogen steam before being redissolved in methanol solution (methanol : water : formic acid = 50:50:0.01) and then subject to centrifugation at 19,200× g for 10 min. Supernatant bile salt species were analysed by ultra‐performance LC (UPLC) triple time of flacid/MS analysis (UPLC‐MS, Waters Co., MA, USA). The relevant parameters were as described in a previous publication.
2.14. Nuclear receptor coactivator assays
The time‐resolved FRET (TR‐FRET) FXR coactivator assay (Invitrogen, Carlsbad, CA, USA) was used to determine the ability of BA to bind with FXR‐LBD according to the manufacturer's protocol. GW4064 was used as a positive control.
2.15. Immunoblotting
The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology. The whole‐tissue lysates from HepG2 cell or livers of diet‐induced obesity (DIO) mice were separated by 10% SDS‐PAGE and transferred to nitrocellulose membranes. Primary antibodies of the insulin receptor β (InsR; Cell Signalling Technology Cat# 3025, RRID:AB_2280448), p‐InsR β (Cell Signalling Technology Cat# 3021, RRID:AB_331578), p‐Akt (Cell Signalling Technology Cat# 9271, RRID:AB_329825), kt(Cell Signalling Technology Cat# 9272, RRID:AB_329827), p‐EIF2α (Cell Signalling Technology Cat# 3398, RRID:AB_2096481), EIF2α (Cell Signalling Technology Cat# 9722, RRID:AB_2230924), p‐PERK (Cell Signalling Technology Cat# 3179, RRID:AB_2095853), BIP (Cell Signalling Technology Cat# 3177, RRID:AB_2119845), SCD1 (Cell Signalling Technology Cat# 2794, RRID:AB_2183099), β‐actin (Proteintech Group Cat# 60008‐1‐Ig, RRID:AB_2289225), PERK (Proteintech Group Cat# 20582‐1‐AP, RRID:AB_10695760), ATF4 (Proteintech Group Cat# 10835‐1‐AP, RRID:AB_2058600), CHOP (Proteintech Group Cat# 15204‐1‐AP, RRID:AB_2292610), ABCC2 (Santa Cruz Biotechnology Cat# sc‐20766, RRID:AB_2242161), SHP (Santa Cruz Biotechnology Cat# sc‐23057, RRID:AB_2154623), CPT1α (Abcam Cat# ab128568, RRID:AB_11141632), and CYP7A1 (Millipore Cat# MABD42, RRID:AB_2756360) were used by 1:1,000 dilution in 5% bovine albumin at 4°C overnight followed by IRDyeTM‐labelled secondary antibodies (1:10,000) for 1 hr at room temperature. Immunoreactive proteins were visualized using LI‐COR Odyssey Infrared Imaging System (LI‐COR Biosciences, Lincoln, Nebraska, USA), according to the manufacturer's instructions. The relative protein levels were normalized to β‐actin by using ImageJ software.
2.16. Statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). All values are expressed as means ± SEM and analysed using the statistical package for the social science (SPSS, version 15.0, Inc., Chicago, IL, USA). Paired or unpaired two‐tailed t tests were used to detect a difference in the mean values of treatment group and control and ANOVA for the difference among more than two groups. ANOVA was employed followed by Dunnett's post hoc test (only in those tests where F achieved the necessary level of statistical significance, P < 0.05), and there was no significant variance inhomogeneity. Differences with P values <0.05 were considered to be statistically significant. Fold change was calculated and expressed as the ratio of the difference between the experimental and the control groups to identify any trends in the effects of BA and control for unwanted sources of variation.
2.17. Materials
BA (Pusi Biotech, Chengdu, China, CID: 64971) powder was dissolved in DMSO to the final concentration of 50 mM for cell culture. GW4064 (CID: 9893571), T0901317 (CID: 447912), rosiglitazone (Rosi, CID: 77999), WY14643 (pirinixic acid; CID: 5694), rifampicin (CID: 5381226), BSA‐conjugated palmitic acid (CID: 985), tunicamycin (CID: 11104835), and all of the bile acids standard references mentioned in this article were purchased from Sigma–Aldrich (St. Louis, MO, USA). FXR siRNA was purchased from Gene Pharma (Shanghai, China). HFDs (HF, 60% of calories derived from fat) and chow diets (chow, 10% of calories derived from fat) were purchased from Research Diet (D12492 and D12450B, New Brunswick, NJ, USA). Methionine and choline‐deficient L‐amino acid diets (MCD, 20% of calories derived from fat) and methionine and choline supplement diet (MCS) were purchased from Research Diet (A02082002B and A02082003B, New Brunswick, NJ, USA).
2.18. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (S. D. Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander, Cidlowski et al., 2017; Alexander, Fabbro et al., 2017a,b; Alexander, Kelly et al., 2017).
3. RESULT
3.1. Identify BA as a natural FXR agonist
To test whether BA (Figure 1a) is able to activate FXR, we performed an FXR gene reporter assay. BA activated the FXR transactivity in a dose‐dependent manner (Figure 1b), whereas it did not show an obvious cytotoxic effect in HepG2 cells (Figure S1). We then analysed the effects of BA on the transactivity of other important nuclear receptors; the results showed that there was no effect of BA on PPARγ, LXRα, β, and PXR transactivities (Figure S2A–D), suggesting that BA activates FXR specifically.
Figure 1.
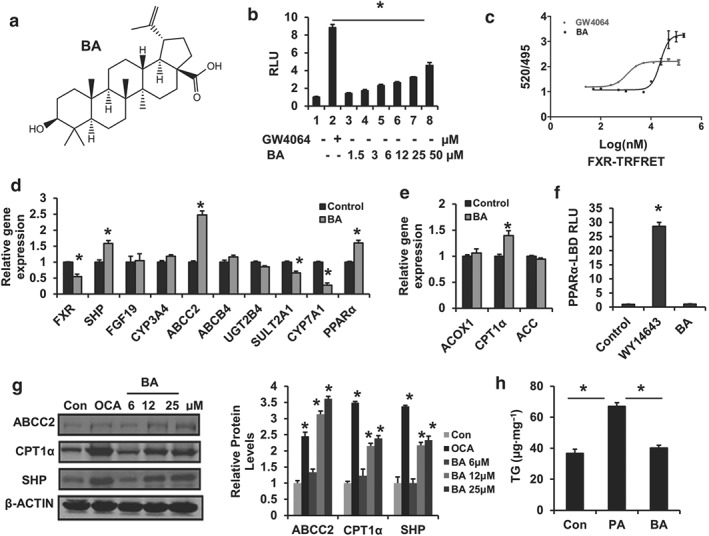
Betulinic acid is a specific FXR agonist. (a) Structure of betulinic acid. (b) FXR transcriptional activity was analysed using transient transfection reporter assays. HEK293T cells were cotransfected with phFXR, phRXR expression plasmids, and FXR‐dependent reporter (EcRE‐LUC) for 24 hr and treated with the FXR agonist GW4064 (10 μM), control (DMSO), betulinic acid (1.5, 3, 6, 12, 25, 50 μM) for another 24 hr. The relative luciferase activities (RLU) were measured by comparison with renilla luciferase activities. Data are presented as means ± SEM (n = 6). *P < 0.05 versus negative control. (c) Binding assays of BA with LBD‐FXR by TR‐FRET assay. GW4064 was used as a positive control. (d, e) The relative gene expression levels of FXR and PPARα targets in betulinic acid‐treated (25 μM) and control (DMSO) HepG2 cells. β‐Actin was used as an internal control for normalizing the mRNA levels and PCR results were analysed using the 2−ΔΔCt method. (f) PPARα transactivity. HEK293T cells were cotransfected with pCMX‐PPARα‐LBD‐Gal4, MH100 × 4‐TK‐Luc expression plasmids for 24 hr and treated with the PPARα agonist WY14643 (10 μM), control (DMSO), and betulinic acid (50 μM) for another 24 hr. The relative luciferase activities (RLU) were measured by comparison with renilla luciferase activities. (g) The Western blotting image for betulinic acid‐treated (12, 25, and 50 μM), OCA‐treated (25 μM), and Con (DMSO) HepG2 cells. The relative protein level was shown and normalized to β‐actin. (h) Determination of total TG content in HepG2 cell challenged with BSA (Con) and PA (BSA‐PA 500 μM) with or without betulinic acid (50 μM) for 24 hr. The results represent three independent experiments, and data are statistically analysed as means ± SEM (n = 6). *P < 0.05 versus vehicle control
To evaluate whether BA directly binds to the FXR, BA was subjected to the LanthaScreen™ TR‐FRET FXR coactivator assay. The result showed that BA triggered TR‐FRET signalling with a dose–response curve similar to the FXR agonist GW4064 (Figure 1c), indicating direct binding of BA to FXR.
FXR agonists have been shown to activate a series of target genes. We, therefore, examined whether BA regulates FXR target gene expression in HepG2 cells. As expected, BA treatment markedly increased the expression of FXR downstream genes such as SHP, ABCC2, SULT2A1, and CYP7A1 (Figure 1d,e,g). Furthermore, PPARα has been identified as a target of FXR in human hepatocytes (Matsubara et al., 2013; Pineda Torra et al., 2003). Similarly, we found PPARα and its target gene CPT1α, which is involved in accelerating fatty acid oxidation, were also significantly increased (Figure 1e) by BA, but BA did not influence PPARα‐LBD reporter activity (Figure 1f), suggesting a stimulation of PPARα signalling that is independent of direct PPARα activation in BA‐treated cells. The results were confirmed by Western blotting, which showed that ABCC2, CPT1α, and SHP protein levels were increased by the BA‐like FXR agonist OCA in HepG2 cells (Figure 1g). Next, we investigated the effect of BA on hepatocellular lipid metabolism in vitro. PA stimulation may result in severe TG accumulation in HepG2 cells. BA treatment blocked TG accumulation in PA‐treated HepG2 cells (Figure 1h). These results suggest a role for BA in regulating intracellular lipid metabolism in vivo.
3.2. BA improves metabolic profiles in DIO mice
To test whether BA exerts therapeutic effects on metabolic disorders in DIO mice, obese C57BL/6J mice induced by a HFD were treated with BA (100 mg BA.100 g‐1 diet) for 6 weeks. BA treatment significantly blocked body weight gain at week 6 (Figure 2a) but did not change the amount of food eaten by the mice (Figure 2b). Next, we tested fasting blood glucose levels, GTTs and ITTs. The HF‐treated mice showed a strikingly higher glucose level compared with chow‐fed lean mice, whereas BA treatment significantly lowered fasting blood glucose levels (Figure 2c). For the GTTs and ITTs, glucose levels at 0, 30, 60, and 90 min (Figure 2d) and 0, 15, and 60 min (Figure 2e) were lowered following an i.p. injection of glucose or insulin in DIO mice, indicating that BA improves glucose homeostasis, glucose intolerance, and insulin resistance in the DIO mice.
Figure 2.
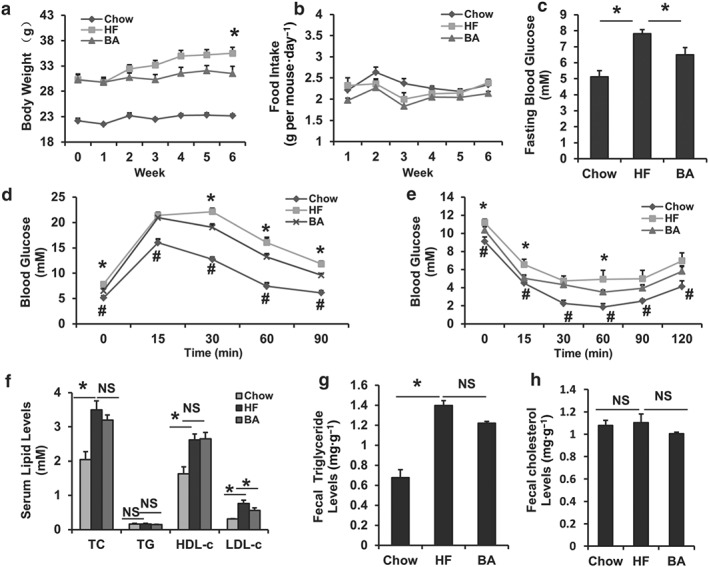
Betulinic acid improves glucose‐lipid homeostasis in DIO C57BL/6J mice. Obese C57BL/6J mice were fed a HFD in the presence and absence of betulinic acid (100 mg·kg−1·day−1) for 6 weeks. (a) Body weight during the 6‐week treatment. (b) Curve for amount of food consumed. (c) Fasting blood glucose. (d) I.p. glucose tolerance test (IPGTT). The mice were fasted overnight, and glucose was injected i.p. (1 g·kg−1 body weight), and blood glucose levels were determined at the indicated time points. (e) I.p. insulin tolerance test (IPITT). The insulin was injected i.p. (0.75 U·kg−1 body weight), and blood glucose levels were determined at the indicated time points. (f) Serum TC, TG, low‐density lipoprotein cholesterol (LDL‐c), HDL‐c levels. (g, h) TG and TC in faeces were extracted and determined. Data are presented as means ± SEM (n = 7). P < 0.05, HF versus chow (*) or HF versus betulinic acid (#) group, NS: no significance
We then assayed lipid profiles in these mice. As expected, the HFD elevated serum TC, HDL‐c, and LDL‐c levels compared with chow controls (Figure 2f). Notably, the 6‐week BA treatment decreased serum LDL‐c levels in DIO mice (Figure 2f) but not TG, TC, or HDL‐c. In faecal lipid content assays, we found the HFD significantly elevated total TG levels but not TC contents in DIO mice compared with chow controls (Figure 2g,h), whereas BA treatment did not change faecal TG contents (Figure 2g), suggesting that BA improves serum lipid dysfunction in the DIO mice independent of an effect on intestinal lipid absorption.
3.3. BA enhances energy expenditure in vivo
To clarify whether the reduction in body weight was due to a decrease in adipose tissue mass, morphological analyses of WAT and BAT from these mice were implemented. Indeed, HFD induced the amount of WAT and BAT to increase compared with chow control mice, whereas BA treatment apparently reduced fat mass in DIO mice according to the HE staining and measurements of WAT size (Figure 3a,b). Subsequently, a lower mRNA expression of the pro‐inflammatory cytokines Il‐6 and Mcp1 (CCL2) was observed in WAT from BA‐treated obese mice (Figure 3b), suggesting the obesity‐associated infiltration of immune cells was reduced in BA‐treated HFD mouse adipocytes compared to HFD controls.
Figure 3.
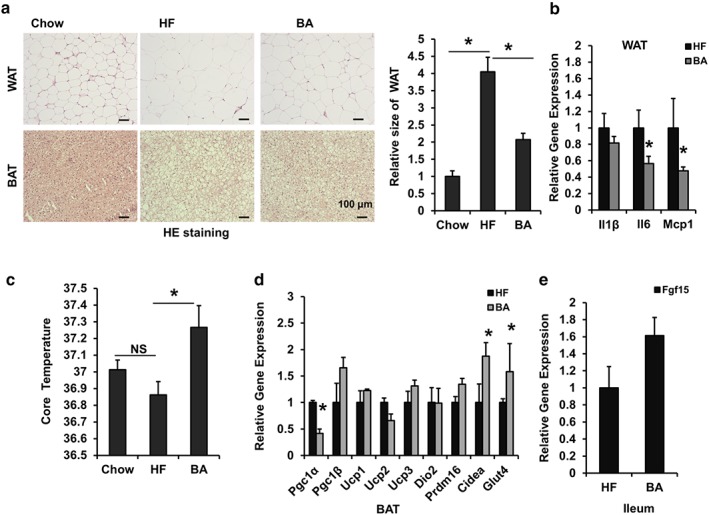
Betulinic acid promotes thermogenesis in DIO C57BL/6J mice. Obese C57BL/6J mice were fed a HFD in the presence and absence of betulinic acid (100 mg·kg−1·day−1) for 6 weeks. (a) After the 6 week betulinic acid treatment, WAT and BAT from mice (n = 7) in the chow, HFD (HF), and betulinic acid groups were subject to HE staining. Scale bars, 100 μm. Relative WAT areas were calculated by comparison with the chow group according to cell diameter. (c) Mice rectal temperature (n = 7). (b, d, e) The relative mRNA levels in WAT, BAT, and ileums from DIO mice were analysed. β‐Actin was used as an internal control for normalizing the mRNA levels. Data are presented as means ± SEM (n = 6). *P < 0.05 versus chow or HF group
Obesity results from an energy imbalance (Spiegelman & Flier, 2001). Because there were no changes in the food intake or fecal lipid contents in BA‐treated DIO mice, we speculated that BA might exert its antiobesity effects by enhancing energy expenditure. To explore this, core body temperature was analysed. Although HF control mice showed a lower, but not significant, core temperature compared with chow control mice, BA treatment significantly elevated the core temperature in DIO mice (Figure 3d), suggesting BA enhanced thermogenesis. BAT is a key tissue for thermogenesis (Inagaki, Sakai, & Kajimura, 2017; Li, 2004). To understand whether the thermogenic effect in BA‐treated DIO mice results from increased BAT activity, we tested functional gene expression in BAT. Cidea and Glut4 were increased, but Pgc1α mRNA levels were decreased in BAT from BA‐treated mice compared with that from HFd controls (Figure 3e), although Pgc1β, UCP1‐3, D2, and Prdm16 levels remained unchanged (Figure 3e). The increased Cidea and Glut4, which are known to mediate thermogenesis and glucose transport (Inagaki et al., 2017; Li, 2004), may explain the thermogenesis and glucose lowering effect of BA. Furthermore, a slight increase in the mRNA expression of Fgf15 in the ileal of BA‐treated DIO mice was found (Figure 3e). Fgf15 is known to be regulated by the FXR, and enterohepatic Fgf15 signalling is reported to enhance energy expenditure in DIO mice (Fang et al., 2015). Thus, the enhanced energy expenditure by BA may be partially related to induction of enterohepatic Fgf15 signalling.
3.4. BA alters bile acid metabolism in vivo
One of the physiological functions of FXR is to maintain bile acid homeostasis (Matsubara et al., 2013). Therefore, the composition of hepatic bile acids in BA‐treated mice was analysed. In contrast to HFD‐fed mice, BA treatment strikingly decreased the fractions of hepatic murine taurocholic acid (TMCA), tauro‐chenodeoxycholic acid (TCDCA), and tauro‐deoxycholic acid (TDCA), which were accompanied by an increase in the levels of chenodeoxycholic acid (CDCA) and lithocholic acid (LCA) in the DIO mice (Figure 4a and Table S3). The BA treatment markedly decreased the total bile acid level (TBA) in livers but caused no changes in the faeces in DIO mice, compared with HFD controls (Figure 4b,c). These results indicate that BA is able to alter the bile acids composition and inhibit the total bile acid accumulation in the livers of DIO mice.
Figure 4.
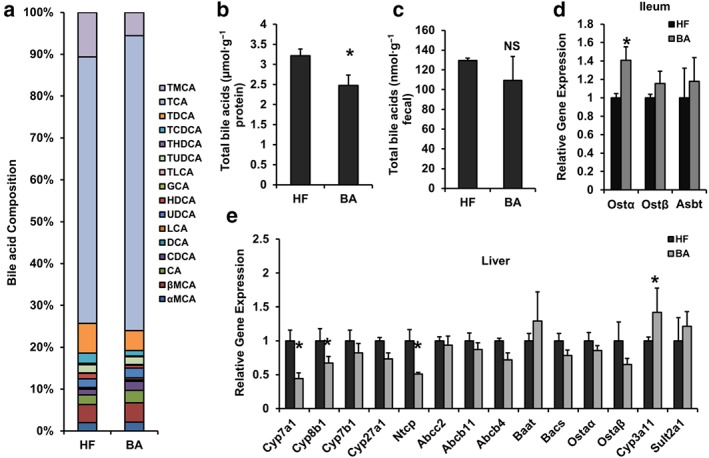
Betulinic acid changes bile acid metabolism in DIO C57BL/6J mice. Obese C57BL/6J mice were fed a HFD in the presence and absence of betulinic acid (100 mg·kg−1·day−1) for 6 weeks. (A) Hepatic BA composition. (b, c) TBA levels in livers and faeces. (d, e) The mRNA expression of bile acids metabolism in ileum (d) and livers (e) from DIO mice were analysed. β‐Actin was used as an internal control for normalizing the mRNA levels. Data are presented as means ± SEM (n = 6). *P < 0.05 versus HF group
To investigate whether the change in bile acid content in DIO mice resulted from an effect on FXR downstream of bile acid metabolism signals in the enterohepatic system, we tested a series of gene expression from the ileum and the liver of the mice. BA treatment significantly increased the expression of Ostα but not Ostβ or Asbt in the ileum (Figure 4d). BA treatment also increased hepatic Cyp3a11 mRNA levels and reduced hepatic Cyp7a1, Cyp8b1, and Ntcp mRNA expression (Figure 4e). Hepatic Ntcp, Cyp3a11, Cyp7a1, Cyp8b1, and intestinal Ostα are key components in the enterohepatic FXR signalling and are essential for bile acid release, detoxification, synthesis, and hydroxylation. These results suggest that the change in bile acid metabolism induced by BA treatment in DIO mice is mediated via an effect on enterohepatic bile acid transport and synthesis signals.
3.5. BA alleviates hepatic steatosis in DIO mice
Currently, the most attractive therapeutic benefit of FXR agonists is the protection from NAFLD and subsequent hepatic inflammation and fibrosis (Bensinger & Tontonoz, 2008; Zhang & Edwards, 2008). FXR‐null mice show apparent liver inflammation and damage (Kong et al., 2009). To determine whether BA could improve hepatic steatosis, liver sections from chow control, HFD control, and BA‐treated mice were subjected to pathological analyses. The HE and Oil Red O staining results show that BA treatment reduced diet‐induced hepatic steatosis (Figure 5a), which was confirmed by reduced lipid content, when compared with HFD‐fed controls (Figure 5b,c). Serum ALT and AST levels, the markers of liver damage, were increased in DIO mice compared with control mice. However, the difference in ALT, but not AST levels, was notably diminished by the BA treatment (Figure 5d,e). These results suggest that BA ameliorates hepatic steatosis in DIO mice. To evaluate the potential mechanisms causing NAFLD resistance in DIO mice, the gene expression in the liver and ileum of BA‐treated mice was analysed. We observed a significant reduction in hepatocyte inflammation associated gene expressions (Mcp1 and Il6) in BA‐treated DIO mice compared with HFD controls, although TNFα and IL‐1β remained unchanged (Figure 5f). Additionally, BA treatment significantly increased the mRNA levels of ileal Shp (Nr0b2; Figure 5g) and hepatic Shp and Pgc1β and decreased Hnf4a, Lxrβ (Nr1h2), Pck1, G6pc, Scd1, and Akr1b7 mRNA levels in the mice (Figure 5h,i). Furthermore, protein analysis results corroborated the change in mRNA levels in the BA‐treated mouse livers (Figure 5j). The data suggest that BA may improve liver lipid metabolism signalling and block the increase in inflammation via the activation of feedback loops involving FXR signalling in DIO mice.
Figure 5.
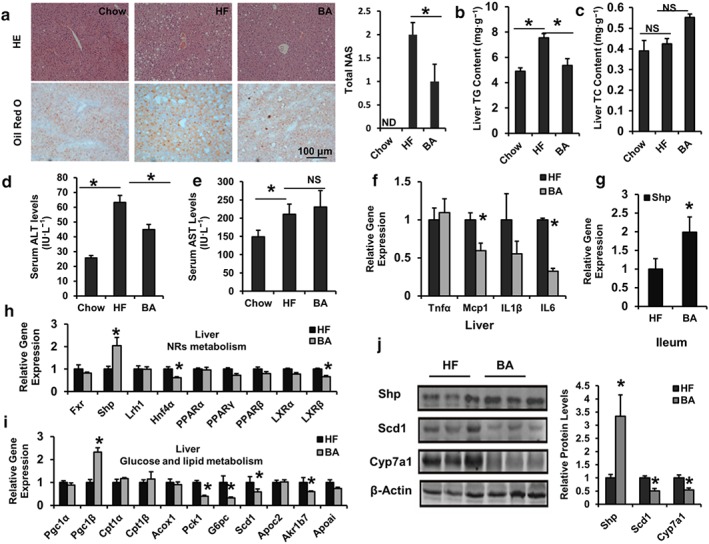
Betulinic acid inhibits lipid accumulation in the liver of DIO C57BL/6J mice. Obese C57BL/6J mice were fed a HFD in the presence and absence of betulinic acid (100 mg·kg−1·day−1) for 6 weeks. (a) The livers from mice were harvested and stained with HE and Oil Red O, and the total NAFLD activity score (NAS) is shown. Scale bars, 100 μm. (b, c) Hepatic TG (b) and TC (c) of chow mice and HFD‐fed mice in the presence or absence of betulinic acid (n = 6 per group). (d, e) Serum ALT and AST activity. (f–i) Relative mRNA expression of FXR signalling associated with hepatic inflammatory factors (f), hepatic nuclear receptor metabolism (h), hepatic glycolipid metabolic genes (i), and Shp in the ileum (g) from DIO mice with or without betulinic acid treating were analysed. (j) Protein levels in the livers were analysed by immunoblotting. The relative protein level was shown. β‐Actin was used as an control for normalizing the mRNA and protein levels. Data are statistically analysed as means ± SEM (n = 6). *P < 0.05 versus chow or HF group. ND: Not detected
3.6. BA ameliorates NASH in MCD diet‐fed mice
The symptoms of NAFLD can be reversed by changing the lifestyle of patients (Zelber‐Sagi, Godos, & Salomone, 2016). MCD‐diet induced NASH mice were used to further assess the antihepatic steatosis effect of BA on NASH. Interestingly, after the 6‐week treatment with BA, we found that the mice showed a substantial resistance to liver steatosis induced by MCD diet according to HE and Oil Red O staining results (Figure 6a). Consistent with the histology phenotype, the hepatic lipid content and serum ALT, AST levels were increased in MCD mice (Figure 6b–e), whereas BA treatment effectively decreased hepatic TG, TC contents (Figure 6b,c), and serum ALT levels, although serum AST levels were not reduced by BA (Figure 6d,e).
Figure 6.
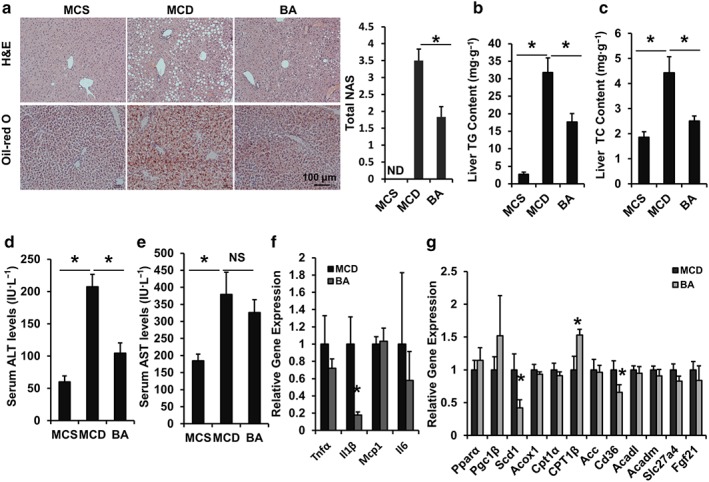
Betulinic acid improves hepatic steatosis in MCD‐diet induced NASH C57BL/6J mice. Eight‐week‐old C57BL/6J mice were fed an MCD diet in the presence and absence of betulinic acid (100 mg·kg−1·day−1) for 6 weeks. (a) The liver sections were stained with HE and Oil Red O and the total NAFLD activity score (NAS) is shown. Scale bars, 100 μm. (b, c) Hepatic TG (b) and TC (c) of MCS mice and MCD mice in the presence or absence of betulinic acid treatment were measured (n = 6 per group). (d, e) Serum ALT and AST activity. (f, g) The mRNA expression of FXR signalling associated with hepatic inflammatory factors (f) and hepatic lipid metabolism (g). β‐Actin was used as an internal control for normalizing the mRNA levels. Data are presented as means ± SEM (n = 6). *P < 0.05 versus MCS or MCD group. ND: Not detected
Moreover, we found the hepatic mRNA levels of Il‐1β (Figure 6f), an inflammation marker, and lipogenesis genes Scd1 and Cd36 were reduced, and the mRNA level of mitochondrial fatty acid β‐oxidation gene Cpt1β was elevated in BA‐treated mice compared with MCD controls (Figure 6g). These data indicated that BA treatment has a protective effect in steatohepatitis mediated via the regulation of hepatic metabolic gene expression.
3.7. BA alleviates liver ER stress
Chronic ER stress has also been implicated in the pathogenesis of metabolic diseases such as diabetes, obesity, inflammation, and fatty liver disease (Hotamisligil, 2010). It has been shown that pharmacological ER stress inducers contribute to hepatic steatosis and subsequent liver inflammation, InsR, and injury (J. S. Lee, Mendez, Heng, Yang, & Zhang, 2012). Considering the therapeutic effect of BA on MS and hepatic steatosis, whether BA could improve liver lipid metabolism through regulating ER homeostasis was tested. Interestingly, BA treatment significantly decreased the expression of hepatic ER stress marker genes, such as Erdj4, Chop, and Bip (Figure 7a) in DIO mice. The protein levels of Chop and Bip were also comfirned by Western blotting (Figure 7b). These results indicate that BA interfers with the progression of liver ER stress during hepatic steatosis.
Figure 7.
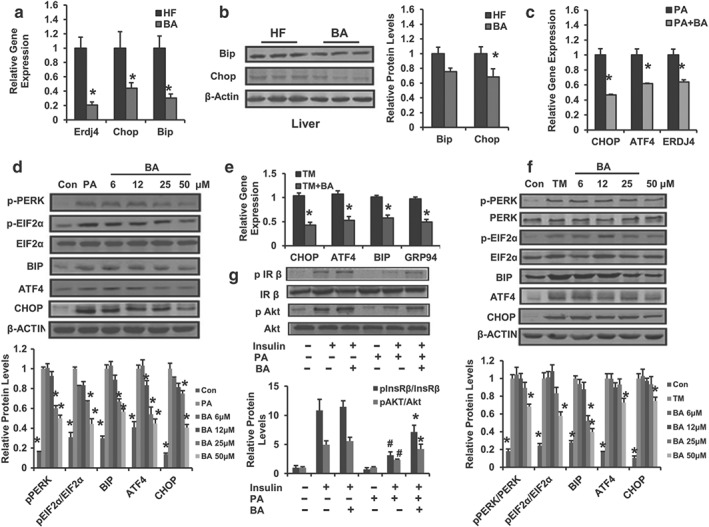
Betulinic acid alleviates hepatocellular ER stress. (a) Relative mRNA expression of hepatic Erdj4, Bip, and Chop in DIO mice with or without betulinic acid treatment. (b) Liver Bip and Chop protein levels were detected with immunoblotting. The relative protein level is shown and normalized to β‐actin. Data are presented as means ± SEM (n = 6). *P < 0.05 versus HFD (HF) group. HepG2 cells were pretreated with betulinic acid at indicated doses for 12 hr, followed by 24 hr stimulation with TM (2.5 μg·ml−1) or PA (500 μM) to induce ER stress, cells treated with DMSO or BSA were used as a negative control. (c, e) Relative mRNA levels of ER stress markers in HepG2 cell in the presence or absence of (TM or PA) challenge were analysed. Cells were incubated with betulinic acid (25 μM) for 24 hr. (d, f) PERK/EIF2α signalling was analysed by immunoblotting with indicated antibody in TM or PA‐challenged HepG2 cell with or without betulinic acid (6, 12, 25, and 50 μM). The relative protein levels are shown and normalized to β‐actin. Data are presented as means ± SEM (n = 6). *P < 0.05 versus PA or TM group. (g) The phosphorylation of insulin receptor β/total insulin receptor β and phosphorylation of Akt/total Akt. The immunoblotting was performed using HepG2 cells in the presence or absence of 24 hr PA and/or betulinic acid (50 μM) pretreatment followed by 30 min DMSO (0.1%) or insulin (100 nM) stimulation. The relative protein level is shown and normalized to total InsRβ or total Akt. Data are statistically analysed as means ± SEM (n = 6). *P < 0.05, insulin + PA + betulinic acid group versus insulin + PA group or # P < 0.05, insulin + PA group versus insulin group. β‐Actin was used for loading control. Data are statistically analysed as means ± SEM (n = 6). The relative protein levels for all western blot images were shown. *P < 0.05 versus HF group. *P < 0.05 versus PA or TM group
To further confirm the action of BA on ER stress and explore the underlying mechanism, we determined the anti‐ER stress effect of BA in HepG2 cells challenged with PA or TM. We found that BA treatment markedly attenuated the expression of intracellular CHOP, ATF4, BIP, and ERDJ4 (Figure 7c), and PERK‐mediated EIF2α phosphorylation (Figure 7d) induced by PA. Next, we observed a similar reduction in the signs of ER stress induced by BA in the TM‐challenged pattern (Figure 7e,f). PERK/EIF2α/ATF4/CHOP signalling is known to mediate ER stress (Cao et al., 2012). Thus, these results suggest that BA reduces the ER stress associated with steatosis by affecting the PERK/EIF2α signalling. Additionally, it has been reported that the PA‐induced ER stress is able to stimulate or impair the hepatic insulin pathway (Wang et al., 2006). As shown in Figure 7g, insulin‐stimulated increase in p‐InsRβ/InsRβ and p‐Akt /Akt protein levels in HepG2 cells was decreased by PA, and this effect of PA was largely reversed by the incubation with BA (Figure 7g). In contrast, BA was not able to increase the insulin sensitivity of HepG2 cells in the absence of PA stimulation (Figure 7g), indicating that BA enhances insulin sensitivity in PA‐insulted HepG2 cells. These results demonstrate that BA alleviates hepatic ER stress through an effect on PERK/EIF2α signalling activity, which is involved in regulating liver energy metabolism.
3.8. The FXR mediates liver ER stress in NAFLD
Hyperactivated hepatic PERK/EIF2α signalling is directly linked to hepatic metabolic dysregulation including impaired liver insulin signalling, accelerated inflammatory damage, and lipid accumulation (H. P. Harding et al., 2001; Hotamisligil, 2010; Ozcan et al., 2004). Conversely, dephosphorylation of EIF2α resulted in lower liver glycogen levels and susceptibility to fasting hypoglycaemia, and glucose tolerance, and diminished hepato‐steatosis in animals fed a HFD (Oyadomari, Harding, Zhang, Oyadomari, & Ron, 2008). However, it is unclear how disrupting the ER influences the metabolic pathways in NAFLD. Considering the largely overlapping effects of FXR agonism and inhibition of the PERK/EIF2α signalling on MS, it is conceivabled that hepatic FXR activation is correlated with the attenuation of ER stress.
To understand whether FXR mediates hepatic ER stress responses, we compared the gene expression levels of ER stress markers between control and FXR‐deleted HepG2 cells treated with PA or TM. Interestingly, deletion of FXR led to a notable increase in the expressions of CHOP, ATF6, and ERDJ4 (Figure 8a) and the protein level of CHOP (Figure 8b). The inhibitory effect of BA on the expression of ER stress markers was largely blunted in FXR‐silenced HepG2 cells challenged with TM (Figure 8c). To further explore whether FXR and ER stress signalling are correlated in vivo, ER stress signalling in the liver of FXR−/− mice fed a normal chow‐diet was tested. Consistent with results from HepG2 cells, expressions of Chop, Atf4, and PERK/EIF2 in FXR−/− mice were also increased, compared with control mice (Figure 8d,e). Finally, in BA‐treated FXR−/− HFD fed mice, the reversal of the diet‐induced hepatosteatosis and liver injury mediated by BA was largely suppressed (Figure 8f and Figure S4E–H). Meanwhile, the inhibitory effects of BA on the expression of ER stress markers (Bip and Chop) were also blocked in FXR‐deficient mouse livers (Figure 8g). These results indicate that a deficiency in FXR signalling may exacerbate the hepatic ER stress response, and FXR is necessary for the anti‐NAFLD effect of BA.
Figure 8.
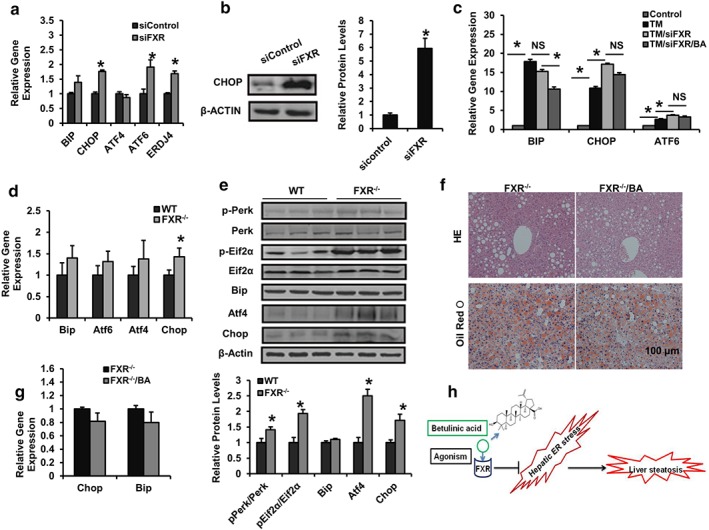
FXR mediates the ameliorative effect of BA on NAFLD and hepatic ER stress. (a, b) At 48 hr after being transfected with siRNA for control or FXR, HepG2 cells were treated with PA (500 μM) for 24 hr and analysed by RT‐PCR and immunoblotting. The relative protein levels are shown. (c) At 48 hr after being transfected with siRNA for control or FXR, HepG2 cells were treated with DMSO or betulinic acid (50 μM) in the presence or absence of TM for 24 hr and analysed by RT‐PCR (d, e) 15‐week‐old wild type female C57BL/6J mice as WT and FXR−/− mice were fed chow diet, livers were harvested and subjected to mRNA and immunoblotting for PERK/EIF2α signalling analysis of ER stress markers. The relative protein levels are shown. Data are presented as means ± SEM (n = 6). *P < 0.05 versus sicontrol or TM group or versus WT group. β‐Actin was used as an internal control for normalizing the mRNA and protein levels. (f, g) Four‐month‐old obese FXR−/− mice were kept on a HFD in the presence and absence of betulinic acid (100 mg·kg−1·day−1) for 6 weeks. Livers from mice were harvested and stained with HE and Oil Red O. Scale bars, 100 μm (f). Relative mRNA expression of hepatic Chop and Bip. Data are presented as means ± SEM (n = 6). *P < 0.05 versus vehicle or FXR−/− group. β‐Actin was used as an internal control for normalizing the mRNA and protein levels (g). (h) The schematic of th epharmacological mechanism by betulinic acid
4. DISCUSSION
The activation of FXR and relief of ER stress are important therapeutic approaches to protect against NAFLD. Here, we demonstrated that BA, a naturally occurring small molecule, is an FXR agonist. We further confirmed the beneficial pharmacological effect of BA on MS, especially its ability to ameliorate NAFLD in DIO and MCD‐induced mice. These effects were achieved by reducing the hepatic ER stress associated with the activation of FXRs.
FXR is regarded as an energy metabolism adaptor that functions to maintain lipid, glucose, and bile acid homeostasis (Makishima et al., 1999). Previous studies have shown that the activation of FXR ameliorates lipid and glucose metabolism in several models of obesity and diabetes (Zhang & Edwards, 2008; Zhang et al., 2006).In contrast, FXR‐deficient mice exhibited marked hypercholesterolaemia, hypertriglyceridaemia, impaired glucose intolerance, and insulin insensitivity (Kong et al., 2009; Matsubara et al., 2013). These phenotypes have been partly attributed to the ability of FXR to control the expression of a series of genes involved in lipid and glucose metabolism, including lipogenesis, lipid uptake, fatty acid oxidation, hepatic gluconeogenesis, and gluconeogenic programmes (Matsubara et al., 2013).
Several studies have reported that BA exerts therapeutic effects on obesity and type 2 diabetes (Kim et al., 2014; Kim, Lee, Kim, & Kim, 2012); however, few studies have focused on NAFLD, and the underlying molecular mechanism of BA is obscure. We found that BA activated FXR transcriptional activity in a ligand‐driven coactivator manner but had no effect on the other nuclear hormone receptors analysed. HepG2 cells (hepatoblastoma) have a high degree of morphological and functional differentiation in vitro; thus, the cells are a suitable model to study the intracellular trafficking and dynamics of bile canaliculi, sinusoidal membrane proteins, lipids in human hepatocytes, liver metabolism and toxicity, and drug targeting in vitro (Mersch‐Sundermann, Knasmuller, Wu, Darroudi, & Kassie, 2004; Moscato, Ronca, Campani, & Danti, 2015). Our results showed that BA blocked the increase in TG content in PA‐challenged HepG2 cells associated with induced expression of fatty acid oxidation genes, PPARα, and CPT1α. The results suggest that BA may exert a positive, ameliorative effect on MS by activating FXR. Notably, several BA analogues such as betulin, epibetulinic acid, ursolic acid, and oleanolic acid, all of which have been well studied as a possible treatment for MS (Csuk, 2014; Szuster‐Ciesielska, Plewka, & Kandefer‐Szerszen, 2011), also displayed FXR agonism (data not shown), supporting that the possibility that the effect of of BA on FXR can be used as an anti‐MS treatment.
Our data showed that BA treatment obviously reduced body weight, fasting blood glucose, and serum LDL‐c levels and improved GTT and ITT in DIO mice. These data indicate that BA increases insulin sensitivity and corrects the glucose and lipid dysfunction in DIO mice. Interestingly, although FXR deletion protected mice from weight gain, liver‐specific deletion of FXR failed to reduce obesity (Prawitt et al., 2011). In comparison, the stimulation of enterohepatic Fgf15 signalling mediated by intestinal FXR agonism has been shown to enhance energy expenditure and reduce the obesity associated with an increase in chenodeoxycholate (CDCA) derivatives in vivo (Fang et al., 2015), suggesting a different role of enterohepatic FXR. Moreover, the FXR agonist OCA has been shown clinically to reduce body weight in NAFLD patients (Matsubara et al., 2013). In this study, we also observed a reduction in fat mass of white adipocytes in BA‐treated obese mice. A similar weight loss was also observed in other BA studies (Kim et al., 2012). Intriguingly, BA treatment elevated the core rectal temperature and the gene expressions of Cidea and Glut4 in BAT in DIO mice, suggesting an enhanced energy expenditure. Furthermore, we found that BA treatment induced a similar change in bile acid metabolism to that evoked by CDCA derivatives, and by acting as as a FXR agonist had a tendency to increase intestinal Fgf15 expression (Fang et al., 2015). Furthermore, various feedforward and feedback loops in FXR signalling may alter the metabolism of bile acids (Makishima et al., 1999). The changes in gene expression of hepatic Shp, Hnf4α, Ntcp, Cyp3a11, Cyp7a1, Cyp8b1, and intestinal Ostα are consistent with lowered hepatic TBA levels and the decreased fraction of TMCA, TCDCA, and TDCA associated with an increased fraction of CDCA and LCA (Figure 5a and Table S3) in BA‐treated DIO mice, indicating that BA inhibits hepatic bile acid synthesis and regulates liver bile acid homeostasis by affecting the enterohepatic Fxr/Shp/Hnf4α feedback network. Taken together, our results indicate that BA specifically activates enterohepatic FXR in vivo.
We successively explore the anti‐hepatosteatosis effect of BA on DIO and MCD‐diet‐induced NASH. Excitingly, BA potently blocked hepatic steatosis and protected liver from damage, suggesting that BA is a potential therapeutic agent for NAFLD and NASH. Consistent with other FXR agonist studies (Fang et al., 2015; Ma, Huang, Yan, Gao, & Liu, 2013), we found BA elevated enterohepatic Shp, and hepatic Pgc1β and Cpt1β mRNAs, but reduced Pck1, G6pc, Scd1, and Cd36 gene expressions. These genes are directly or indirectly regulated by FXR agonism and are involved in hepatic gluconeogenesis, lipogenesis, and fatty acid oxidation. Thus, FXR signalling‐mediated regulation of glucose and lipid metabolism and inflammation genes might account for the improved hepatic steatosis evoked by BA treatment.
Given the diversity of phenotype on glucose and fatty acid homeostasis between liver‐special FXR‐null and global FXR deficient mice (Prawitt et al., 2011), additional mechanisms might also be involved in alleviating NAFLD. Notably, the pathological characteristics associated with chronic ER stress are strikingly similar to hepatic FXR depletion. Furthermore, several studies have demonstrated amelioration of maladaptive ER signalling via FXR agonism in the kidney (Gai et al., 2017; Marquardt et al., 2017). These observations support the notion that FXR agonism may be involved in hepatic ER stress alleviation, although a recent study indicated that FXR activation induces hepatic ER stress via targeting XBP1/IRE1α signalling (Liu et al., 2018).
An attenuation of PERK/EIF2α signalling has been shown to decrease hepatic gene expression of Pck1, G6pc, Scd1, Il1, Il6, and MCP‐1 in mice (Bobrovnikova‐Marjon et al., 2008; Hotamisligil, 2010). Retrospectively, these changes in gene expression are in accordance with the pharmacological action of BA in vivo. In support of this, BA inhibited Bip and Chop mRNA levels in the liver of DIO mice. These results validated the view that BA protects against fat‐associated hepatic ER stress.
Although the mechanism on PERK/EIF2α signalling in ameliorating NAFLD remains to be fully understood, it has been found that the stimulation of PERK/EIF2α signalling leads to persistent ATF4 expression that primarily contributes to the activation of CHOP transcription (Walter & Ron, 2011). As important components of PERK/EIF2α signalling, ATF4 and CHOP are implicated in curtailing ER stress‐induced metabolic dysfunction (Walter & Ron, 2011). ATF4 null mice are resistant to DIO and glucose intolerance (Seo et al., 2009). Moreover, a deficiency in CHOP prevents HFD‐induced insulin resistance, hepatosteatosis, and liver injury (Chikka, McCabe, Tyra, & Rutkowski, 2013). Interestingly, ATF4 and CHOP were severely down‐regulated in BA‐treated hepatocytes. These data further support the notion that BA stabilizes ER stress in the liver.
In a previous study it was reported that ER stress activation may reduce FXR expression in the liver (Lefebvre & Staels, 2014). In contrast, we observed that ER stress did not change the FXR transactivity in PA‐incubated HepG2 cells (Figure S3A). To resolve this discrepancy, we investigated potential variations in UPR controlled by FXR. Loss of hepatic FXR resulted in an enhanced PERK/EIF2α signalling and elevated markers for ER stress, especially CHOP, in both in vitro and in vivo systems, suggesting loss of FXR accelerates hepatic UPR and increases ER stress. Our data are consistent with a recent finding that FXR regulates CHOP expression in steatohepatitis (Fuchs, Claudel, Scharnagl, Stojakovic, & Trauner, 2017), indicating that FXR activation may alleviate hepatic ER stress via inhibiting PERK/EIF2α/ATF4/CHOP signalling. Finally, we found the beneficial, ameliorative effects of BA on obesity‐associated NAFLD and liver ER stress were eliminated in high‐fat‐diet‐induced obese FXR−/− mice (Figure S4A–D), confirming that FXR agonism is directly involved in the effects of BA on NAFLD and crosstalk between FXR and ER stress signalling are implicated in its therapeutic action.
In conclusion, we showed that BA is a specific FXR agonist. BA supplementation lowered fasting blood glucose and serum lipids, reconstituted the bile acid pool, and ameliorated hepatic steatosis and inflammation in DIO and MCD‐diet‐fed mice. The effect of BA was mediated by alleviating hepatic ER stress via FXR‐mediated inhibition of PERK/EIF2α/ATF4/CHOP signalling. Our data suggest that the effects of BA may be used to develop a novel therapy for the management of NAFLD and NASH.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
M.G., Q.T., F.S., Y.B., G.J., and C.H. designed the study. M.G., P.Z., and S.Z. performed the study. M.G., L.Y., and C.H. analysed the data. M.G., Q.T., G.J., and C.H. drafted the manuscript.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
BA nuclear receptor transcription activity assay. (A‐D) Reporter gene assaying of PPARγ, LXRα, LXRβ and PXR. HEK293T cells were co‐transfected with pCMXGal‐hPPARγ, LXRα, β, PXR, the Gal4 reporter vector, PXR reporter vector and MH100×4‐TK‐Luc were respectively co‐transfected into HEK293T cells for 24 h followed by BA (50 μM) and Rosiglitazone, T0901317 or Rifampicin (respectively 20 μM) treating for 24 h, then the cells were subjected to luciferase activity determination. The relative luciferase activities (RLU) were measured by comparison to renilla luciferase activities. The results represent three independent experiments, and data are analysed as means ± SEM. (n = 6). * p < 0.05 vs. vehicle control.
Figure S2. FXR reporter assay under ER stress. hFXR, hRXR expression vectors and FXR‐dependent reporter (EcRE‐LUC) were co‐transfected into HEK293T cells for 24 h followed by TM (5 μg/ml) or PA (500 μM) treating for 24 h, then the cells were subjected to gene reporter assay. The relative luciferase activities (RLU) were measured by comparison to renilla luciferase activities. The results represent three independent experiments, and data are analysed as means ± SEM. (n = 6). * p < 0.05 vs. vehicle control.
Figure S3. BA failed to ameliorate metabolic disorders in FXR‐/‐ mice. 4‐month‐old obese FXR‐/‐ mice were kept on HFD in the presence and absence of BA (100 mg/kg/day) for 6 weeks. (A) Body weight during the 6‐week treatment. (B) Food intake amount curve. (C) Fasting blood glucose level. (D) Serum total cholesterol (TC), triglyceride (TG), low‐density lipoprotein cholesterol (LDL‐c), high‐density lipoprotein cholesterol (HDL‐c) levels. (E and F) Serum ALT and AST enzyme activity. (G and H) Total hepatic lipids were extracted, TG (B) and TC (C) of FXR‐/‐ vehicle mice and BA‐treated FXR‐/‐ mice were measured (n = 6 per group). Data was statistically analysed as means ± SEM (n = 6). *p < 0.05, vs. FXR‐/‐ vehicle mice.
Table S1. Sequences of the human primers used in real time PCR
Table S2. Sequences of the mouse primers used in real time PCR
Table S3. Liver Bile acid composition
ACKNOWLEDGEMENTS
This work was supported by National Natural Science Foundation of China 81620108030 and 81803598 and Natural Science Foundation of Shanghai 18ZR1440000.
Gu M, Zhao P, Zhang S, et al. Betulinic acid alleviates endoplasmic reticulum stress‐mediated nonalcoholic fatty liver disease through activation of farnesoid X receptors in mice. Br J Pharmacol. 2019;176:847–863. 10.1111/bph.14570
Contributor Information
Guang Ji, Email: jiliver@vip.sina.com.
Cheng Huang, Email: chuang@shutcm.edu.cn.
REFERENCES
- Alexander, S. P.H. , Cidlowski, J. A. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The concise guide to pharmacology 2017/18: Nuclear hormone receptors. British Journal of Pharmacology, 174(Suppl 1), S208–S224. 10.1111/bph.13880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. British Journal of Pharmacology, 174, S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , Harding, S. D. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. British Journal of Pharmacology, 174, S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensinger, S. J. , & Tontonoz, P. (2008). Integration of metabolism and inflammation by lipid‐activated nuclear receptors. Nature, 454(7203), 470–477. 10.1038/nature07202 [DOI] [PubMed] [Google Scholar]
- Bobrovnikova‐Marjon, E. , Hatzivassiliou, G. , Grigoriadou, C. , Romero, M. , Cavener, D. R. , Thompson, C. B. , & Diehl, J. A. (2008). PERK‐dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proceedings of the National Academy of Sciences of the United States of America, 105(42), 16314–16319. 10.1073/pnas.0808517105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Dai, D. L. , Yao, L. , Yu, H. H. , Ning, B. , Zhang, Q. , … Yang, Z. X. (2012). Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Molecular and Cellular Biochemistry, 364(1–2), 115–129. 10.1007/s11010-011-1211-9 [DOI] [PubMed] [Google Scholar]
- Chikka, M. R. , McCabe, D. D. , Tyra, H. M. , & Rutkowski, D. T. (2013). C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. The Journal of Biological Chemistry, 288(6), 4405–4415. 10.1074/jbc.M112.432344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, J. , Kim, K. H. , Lee, S. C. , An, S. H. , & Kwon, K. (2015). Ursodeoxycholic acid (UDCA) exerts anti‐atherogenic effects by inhibiting endoplasmic reticulum (ER) stress induced by disturbed flow. Molecules and Cells, 38(10), 851–858. 10.14348/molcells.2015.0094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csuk, R. (2014). Betulinic acid and its derivatives: A patent review (2008‐2013). Expert Opinion on Therapeutic Patents, 24(8), 913–923. 10.1517/13543776.2014.927441 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dara, L. , Ji, C. , & Kaplowitz, N. (2011). The contribution of endoplasmic reticulum stress to liver diseases. Hepatology, 53(5), 1752–1763. 10.1002/hep.24279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, S. , Suh, J. M. , Reilly, S. M. , Yu, E. , Osborn, O. , Lackey, D. , … Evans, R. M. (2015). Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nature Medicine, 21(2), 159–165. 10.1038/nm.3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch, J. , Lees, M. , & Sloane Stanley, G. H. (1957). A simple method for the isolation and purification of total lipides from animal tissues. The Journal of Biological Chemistry, 226(1), 497–509. [PubMed] [Google Scholar]
- Fuchs, C. D. , Claudel, T. , Scharnagl, H. , Stojakovic, T. , & Trauner, M. (2017). FXR controls CHOP expression in steatohepatitis. FEBS Letters, 591(20), 3360–3368. 10.1002/1873-3468.12845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai, Z. , Chu, L. , Xu, Z. , Song, X. , Sun, D. , & Kullak‐Ublick, G. A. (2017). Farnesoid X receptor activation protects the kidney from ischemia‐reperfusion damage. Scientific Reports, 7(1), 9815 10.1038/s41598-017-10168-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, H. P. , Zeng, H. , Zhang, Y. , Jungries, R. , Chung, P. , Plesken, H. , … Ron, D. (2001). Diabetes mellitus and exocrine pancreatic dysfunction in PERK−/− mice reveals a role for translational control in secretory cell survival. Molecular Cell, 7(6), 1153–1163. 10.1016/S1097-2765(01)00264-7 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS guide to pharmacology in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46(D1), D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell, 140(6), 900–917. 10.1016/j.cell.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, C. , Zhang, Y. , Gong, Z. , Sheng, X. , Li, Z. , Zhang, W. , & Qin, Y. (2006). Berberine inhibits 3T3‐L1 adipocyte differentiation through the PPARγ pathway. Biochemical and Biophysical Research Communications, 348(2), 571–578. 10.1016/j.bbrc.2006.07.095 [DOI] [PubMed] [Google Scholar]
- Huh, J. H. , Kim, K. J. , Kim, S. U. , Han, S. H. , Han, K. H. , Cha, B. S. , … Lee, B. W. (2017). Obesity is more closely related with hepatic steatosis and fibrosis measured by transient elastography than metabolic health status. Metabolism, 66, 23–31. 10.1016/j.metabol.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Inagaki, T. , Sakai, J. , & Kajimura, S. (2017). Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nature Reviews. Molecular Cell Biology, 18(8), 527 10.1038/nrm.2017.72 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & NC3Rs Reporting Guidelines Working Group (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Lee, Y. S. , Kim, C. S. , & Kim, J. S. (2012). Betulinic acid has an inhibitory effect on pancreatic lipase and induces adipocyte lipolysis. Phytotherapy Research, 26(7), 1103–1106. 10.1002/ptr.3672 [DOI] [PubMed] [Google Scholar]
- Kim, S. J. , Quan, H. Y. , Jeong, K. J. , Kim, D. Y. , Kim, G. , Jo, H. K. , & Chung, S. H. (2014). Beneficial effect of betulinic acid on hyperglycemia via suppression of hepatic glucose production. Journal of Agricultural and Food Chemistry, 62(2), 434–442. 10.1021/jf4030739 [DOI] [PubMed] [Google Scholar]
- Kong, B. , Luyendyk, J. P. , Tawfik, O. , & Guo, G. L. (2009). Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low‐density lipoprotein receptor‐knockout mice fed a high‐fat diet. The Journal of Pharmacology and Experimental Therapeutics, 328(1), 116–122. 10.1124/jpet.108.144600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosters, A. , Sun, D. , Wu, H. , Tian, F. , Felix, J. C. , Li, W. , & Karpen, S. J. (2013). Sexually dimorphic genome‐wide binding of retinoid X receptor α (RXRα) determines male‐female differences in the expression of hepatic lipid processing genes in mice. PLoS One, 8(8), e71538 10.1371/journal.pone.0071538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. S. , Mendez, R. , Heng, H. H. , Yang, Z. Q. , & Zhang, K. (2012). Pharmacological ER stress promotes hepatic lipogenesis and lipid droplet formation. American Journal of Translational Research, 4(1), 102–113. [PMC free article] [PubMed] [Google Scholar]
- Lee, S. Y. , Kim, H. H. , & Park, S. U. (2015). Recent studies on betulinic acid and its biological and pharmacological activity. EXCLI Journal, 14, 199–203. 10.17179/excli2015-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre, P. , & Staels, B. (2014). Failing FXR expression in the liver links aging to hepatic steatosis. Journal of Hepatology, 60(4), 689–690. 10.1016/j.jhep.2014.01.001 [DOI] [PubMed] [Google Scholar]
- Li, P. (2004). Cidea, brown fat and obesity. Mechanisms of Ageing and Development, 125(4), 337–338. 10.1016/j.mad.2004.01.002 [DOI] [PubMed] [Google Scholar]
- Liang, W. , Menke, A. L. , Driessen, A. , Koek, G. H. , Lindeman, J. H. , Stoop, R. ,… van den Hoek, A. M. (2014). Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One, 9(12), e115922 10.1371/journal.pone.0115922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Guo, G. L. , Kong, B. , Hilburn, D. B. , Hubchak, S. C. , Park, S. , … Green, R. M. (2018). Farnesoid X receptor signaling activates the hepatic X‐box binding protein 1 pathway in vitro and in mice. Hepatology, 68(1), 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, Y. , Huang, Y. , Yan, L. , Gao, M. , & Liu, D. (2013). Synthetic FXR agonist GW4064 prevents diet‐induced hepatic steatosis and insulin resistance. Pharmaceutical Research, 30(5), 1447–1457. 10.1007/s11095-013-0986-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima, M. , Okamoto, A. Y. , Repa, J. J. , Tu, H. , Learned, R. M. , Luk, A. , … Shan, B. (1999). Identification of a nuclear receptor for bile acids. Science, 284(5418), 1362–1365. 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- Marquardt, A. , Al‐Dabet, M. M. , Ghosh, S. , Kohli, S. , Manoharan, J. , ElWakiel, A. , … Isermann, B. (2017). Farnesoid X receptor agonism protects against diabetic tubulopathy: Potential add‐on therapy for diabetic nephropathy. Journal of the American Society of Nephrology, 28(11), 3182–3189. 10.1681/ASN.2016101123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara, T. , Li, F. , & Gonzalez, F. J. (2013). FXR signaling in the enterohepatic system. Molecular and Cellular Endocrinology, 368(1–2), 17–29. 10.1016/j.mce.2012.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. British Journal of Pharmacology, 172(13), 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersch‐Sundermann, V. , Knasmuller, S. , Wu, X. J. , Darroudi, F. , & Kassie, F. (2004). Use of a human‐derived liver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology, 198(1–3), 329–340. 10.1016/j.tox.2004.02.009 [DOI] [PubMed] [Google Scholar]
- Moscato, S. , Ronca, F. , Campani, D. , & Danti, S. (2015). Poly (vinyl alcohol)/gelatin hydrogels cultured with HepG2 cells as a 3D model of hepatocellular carcinoma: A morphological study. Journal of Functional Biomaterials, 6(1), 16–32. 10.3390/jfb6010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso, G. , Cassader, M. , & Gambino, R. (2016). Non‐alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nature Reviews. Drug Discovery, 15(4), 249–274. 10.1038/nrd.2015.3 [DOI] [PubMed] [Google Scholar]
- Ouchi, N. , Parker, J. L. , Lugus, J. J. , & Walsh, K. (2011). Adipokines in inflammation and metabolic disease. Nature Reviews. Immunology, 11(2), 85–97. 10.1038/nri2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari, S. , Harding, H. P. , Zhang, Y. , Oyadomari, M. , & Ron, D. (2008). Dephosphorylation of translation initiation factor 2α enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metabolism, 7(6), 520–532. 10.1016/j.cmet.2008.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan, U. , Cao, Q. , Yilmaz, E. , Lee, A. H. , Iwakoshi, N. N. , Ozdelen, E. , … Hotamisligil, G. S. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science, 306(5695), 457–461. 10.1126/science.1103160 [DOI] [PubMed] [Google Scholar]
- Ozcan, U. , Yilmaz, E. , Ozcan, L. , Furuhashi, M. , Vaillancourt, E. , Smith, R. O. , … Hotamisligil, G. S. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science, 313(5790), 1137–1140. 10.1126/science.1128294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda Torra, I. , Claudel, T. , Duval, C. , Kosykh, V. , Fruchart, J. C. , & Staels, B. (2003). Bile acids induce the expression of the human peroxisome proliferator‐activated receptor α gene via activation of the farnesoid X receptor. Molecular Endocrinology, 17(2), 259–272. 10.1210/me.2002-0120 [DOI] [PubMed] [Google Scholar]
- Prawitt, J. , Abdelkarim, M. , Stroeve, J. H. , Popescu, I. , Duez, H. , Velagapudi, V. R. , … Staels, B. (2011). Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes, 60(7), 1861–1871. 10.2337/db11-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella, M. E. (2015). Nonalcoholic fatty liver disease: A systematic review. JAMA, 313(22), 2263–2273. 10.1001/jama.2015.5370 [DOI] [PubMed] [Google Scholar]
- Ron, D. , & Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews. Molecular Cell Biology, 8(7), 519–529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Rutkowski, D. T. , Wu, J. , Back, S. H. , Callaghan, M. U. , Ferris, S. P. , Iqbal, J. , … Kaufman, R. J. (2008). UPR pathways combine to prevent hepatic steatosis caused by ER stress‐mediated suppression of transcriptional master regulators. Developmental Cell, 15(6), 829–840. 10.1016/j.devcel.2008.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, J. , Fortuno, E. S. 3rd , Suh, J. M. , Stenesen, D. , Tang, W. , Parks, E. J. , … Graff, J. M. (2009). Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes, 58(11), 2565–2573. 10.2337/db09-0335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinal, C. J. , Tohkin, M. , Miyata, M. , Ward, J. M. , Lambert, G. , & Gonzalez, F. J. (2000). Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell, 102(6), 731–744. 10.1016/S0092-8674(00)00062-3 [DOI] [PubMed] [Google Scholar]
- Spiegelman, B. M. , & Flier, J. S. (2001). Obesity and the regulation of energy balance. Cell, 104(4), 531–543. 10.1016/S0092-8674(01)00240-9 [DOI] [PubMed] [Google Scholar]
- Stedman, C. , Liddle, C. , Coulter, S. , Sonoda, J. , Alvarez, J. G. , Evans, R. M. , & Downes, M. (2006). Benefit of farnesoid X receptor inhibition in obstructive cholestasis. Proceedings of the National Academy of Sciences of the United States of America, 103(30), 11323–11328. 10.1073/pnas.0604772103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szuster‐Ciesielska, A. , Plewka, K. , & Kandefer‐Szerszen, M. (2011). Betulin, betulinic acid and butein are inhibitors of acetaldehyde‐induced activation of liver stellate cells. Pharmacological Reports, 63(5), 1109–1123. 10.1016/S1734-1140(11)70630-2 [DOI] [PubMed] [Google Scholar]
- Utzschneider, K. M. , & Kahn, S. E. (2006). Review: The role of insulin resistance in nonalcoholic fatty liver disease. The Journal of Clinical Endocrinology and Metabolism, 91(12), 4753–4761. 10.1210/jc.2006-0587 [DOI] [PubMed] [Google Scholar]
- Walter, P. , & Ron, D. (2011). The unfolded protein response: From stress pathway to homeostatic regulation. Science, 334(6059), 1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wang, X. L. , Zhang, L. , Youker, K. , Zhang, M. X. , Wang, J. , LeMaire, S. A. , … Shen, Y. H. (2006). Free fatty acids inhibit insulin signaling‐stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes, 55(8), 2301–2310. 10.2337/db05-1574 [DOI] [PubMed] [Google Scholar]
- Wong, R. J. , Aguilar, M. , Cheung, R. , Perumpail, R. B. , Harrison, S. A. , Younossi, Z. M. , & Ahmed, A. (2015). Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology, 148(3), 547–555. 10.1053/j.gastro.2014.11.039 [DOI] [PubMed] [Google Scholar]
- Zelber‐Sagi, S. , Godos, J. , & Salomone, F. (2016). Lifestyle changes for the treatment of nonalcoholic fatty liver disease: A review of observational studies and intervention trials. Therapeutic Advances in Gastroenterology, 9(3), 392–407. 10.1177/1756283X16638830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , & Edwards, P. A. (2008). FXR signaling in metabolic disease. FEBS Letters, 582(1), 10–18. 10.1016/j.febslet.2007.11.015 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Lee, F. Y. , Barrera, G. , Lee, H. , Vales, C. , Gonzalez, F. J. , … Edwards, P. A. (2006). Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proceedings of the National Academy of Sciences of the United States of America, 103(4), 1006–1011. 10.1073/pnas.0506982103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
BA nuclear receptor transcription activity assay. (A‐D) Reporter gene assaying of PPARγ, LXRα, LXRβ and PXR. HEK293T cells were co‐transfected with pCMXGal‐hPPARγ, LXRα, β, PXR, the Gal4 reporter vector, PXR reporter vector and MH100×4‐TK‐Luc were respectively co‐transfected into HEK293T cells for 24 h followed by BA (50 μM) and Rosiglitazone, T0901317 or Rifampicin (respectively 20 μM) treating for 24 h, then the cells were subjected to luciferase activity determination. The relative luciferase activities (RLU) were measured by comparison to renilla luciferase activities. The results represent three independent experiments, and data are analysed as means ± SEM. (n = 6). * p < 0.05 vs. vehicle control.
Figure S2. FXR reporter assay under ER stress. hFXR, hRXR expression vectors and FXR‐dependent reporter (EcRE‐LUC) were co‐transfected into HEK293T cells for 24 h followed by TM (5 μg/ml) or PA (500 μM) treating for 24 h, then the cells were subjected to gene reporter assay. The relative luciferase activities (RLU) were measured by comparison to renilla luciferase activities. The results represent three independent experiments, and data are analysed as means ± SEM. (n = 6). * p < 0.05 vs. vehicle control.
Figure S3. BA failed to ameliorate metabolic disorders in FXR‐/‐ mice. 4‐month‐old obese FXR‐/‐ mice were kept on HFD in the presence and absence of BA (100 mg/kg/day) for 6 weeks. (A) Body weight during the 6‐week treatment. (B) Food intake amount curve. (C) Fasting blood glucose level. (D) Serum total cholesterol (TC), triglyceride (TG), low‐density lipoprotein cholesterol (LDL‐c), high‐density lipoprotein cholesterol (HDL‐c) levels. (E and F) Serum ALT and AST enzyme activity. (G and H) Total hepatic lipids were extracted, TG (B) and TC (C) of FXR‐/‐ vehicle mice and BA‐treated FXR‐/‐ mice were measured (n = 6 per group). Data was statistically analysed as means ± SEM (n = 6). *p < 0.05, vs. FXR‐/‐ vehicle mice.
Table S1. Sequences of the human primers used in real time PCR
Table S2. Sequences of the mouse primers used in real time PCR
Table S3. Liver Bile acid composition